Submitted:
21 May 2024
Posted:
24 May 2024
You are already at the latest version
Abstract
Repetitive elements, often referred to as selfish genes, pose significant challenges to genome assembly and read mapping in bioinformatics. These elements can replicate within the genome without providing functional benefits to the host organism, leading to complexities in accurate genome analysis. This study presents a simulation-based approach to illustrate the impact of repetitive elements on read mapping accuracy. By comparing genome sequences with and without repetitive elements, we demonstrate how these selfish genes create ambiguities and errors in read alignment. The findings underscore the importance of developing advanced bioinformatics tools aligned with artificial intelligence to mitigate the effects of repetitive sequences and improve the reliability of genomic analyses.
Keywords:
Selfish Gene
; Read Mapping Accuracy
; Simulation
; Bioinformatic
Section 1. Introduction
The human genome is a complex and intricate structure, containing not only the essential genes required for the functioning of an organism but also a significant portion of non-coding DNA. Among the non-coding regions, repetitive elements, often referred to as selfish genes, constitute a substantial fraction. These elements include transposable elements, endogenous retroviruses, and various other repetitive DNA sequences. While they do not necessarily provide any direct functional benefits to the organism, they have the remarkable ability to replicate and propagate within the genome, often posing challenges for genomic studies (Doolittle & Sapienza, 1980; Orgel & Crick, 1980).
Repetitive elements can complicate numerous bioinformatics tasks, including genome assembly, read mapping, and variant calling. During genome assembly, the presence of these repetitive sequences can lead to misassemblies and gaps, as short sequencing reads from these regions are difficult to place accurately (Treangen & Salzberg, 2012). Similarly, read mapping is challenged by the difficulty of aligning reads to the correct genomic locations when multiple identical or nearly identical sequences exist (Li & Homer, 2010). These ambiguities can propagate through to variant calling and functional annotation, potentially leading to erroneous conclusions and misinterpretations.
The concept of selfish genetic elements was popularized by Richard Dawkins in his seminal work, "The Selfish Gene" (Dawkins, 1976). According to this view, genes behave in ways that maximize their own replication, sometimes to the detriment of the organism as a whole. This perspective helps explain the persistence of repetitive elements within genomes, despite their lack of apparent functional contributions.
In this study, we aim to illustrate the impact of repetitive elements on read mapping accuracy through a simulation-based approach. By comparing read mapping results in simulated genomes with and without repetitive elements, we highlight the difficulties posed by these selfish genes. This investigation underscores the need for advanced bioinformatics and artificial intelligence tools capable of addressing the challenges introduced by repetitive sequences, thereby enhancing the accuracy and reliability of genomic analyses.
Section 2. Methodology and Results
Section 2.1 Simulation of Genomes
To investigate the impact of repetitive elements on read mapping accuracy, we simulated two different genome sequences: one containing repetitive elements and the other without such elements. The presence of repetitive elements is expected to introduce ambiguity in read mapping, whereas the absence of these elements should result in more accurate mapping.
Step 1: Simulating Genome Sequences
We created two genome sequences:
Genome with Repetitive Elements: This genome contains repetitive sequences that mimic the behavior of selfish genes.
Genome without Repetitive Elements: This genome lacks repetitive sequences to serve as a control for comparison.
Step 2: Simulating Reads
We generated a set of short reads that would be mapped back to both genomes. These reads represent fragments of DNA that are typically obtained from sequencing experiments.
Step 3: Mapping Reads to Genomes
We implemented a simple read mapping algorithm using basic string matching to align the simulated reads to both genome sequences. This method identifies all positions where each read aligns to the genome, highlighting the challenges posed by repetitive elements.
Code Implementation
python
# Simulated genome with repetitive elements (selfish genes)
genome_with_repeats = "ATCGATCGATCGGCTAAGGCTATCGATCGATCG"
# Simulated genome without repetitive elements (selfish genes)
genome_without_repeats = "ATCGGCTAAGGCTAGCATCGATCGTACG"
# Simulated reads
reads = ["ATCGATCG", "GCTAAGGC", "ATCGGCTA"]
# Function to map reads to the genome
def map_reads_to_genome(genome, reads):
mapping_results = {}
for read in reads:
positions = []
start = 0
while True:
start = genome.find(read, start)
if start == -1:
break
positions.append(start)
start += 1 # Move to the next character for overlapping matches
mapping_results[read] = positions
return mapping_results
# Map reads to the genome with repeats
mapping_with_repeats = map_reads_to_genome(genome_with_repeats, reads)
print("Mapping with repeats:", mapping_with_repeats)
# Map reads to the genome without repeats
mapping_without_repeats = map_reads_to_genome(genome_without_repeats, reads)
print("Mapping without repeats:", mapping_without_repeats)
Explanation
Genome Simulation:
genome_with_repeats contains repetitive sequences, specifically the sequence "ATCG" repeated multiple times.
genome_without_repeats contains unique sequences with no repeats to serve as a control.
Read Simulation:
The reads list contains three short DNA sequences that we will map to both genomes.
Read Mapping Function:
The map_reads_to_genome function takes a genome sequence and a list of reads as input.
It iterates through each read, finding all positions where the read aligns to the genome.
The positions are stored in a dictionary, with the read sequence as the key and a list of positions as the value.
Section 2.2. Mapping Results:
For the genome with repeats, the read "ATCGATCG" maps to multiple positions (0, 4, 20), demonstrating the ambiguity introduced by repetitive elements.
For the genome without repeats, the same read maps to a single position (18), indicating unambiguous mapping.
This methodology illustrates how repetitive elements, or selfish genes, can complicate the process of read mapping by creating multiple potential alignment positions. In contrast, a genome without such elements allows for more precise and unambiguous read mapping. This simulation underscores the need for advanced bioinformatics tools to address the challenges posed by repetitive sequences in genomic analyses.
Section 3. Discussion
The presence of repetitive elements in the genome poses significant challenges for various bioinformatics tasks, particularly in genome assembly and read mapping. These selfish genes, which can replicate and propagate within the genome without providing functional benefits to the host, introduce ambiguity and complexity in aligning sequencing reads. As demonstrated in our simulation, reads derived from regions with repetitive elements can map to multiple locations, creating confusion and reducing the accuracy of genomic analyses.
Figure 1.
Tandem and Interspersed Repeats. Source: Wikipedia.
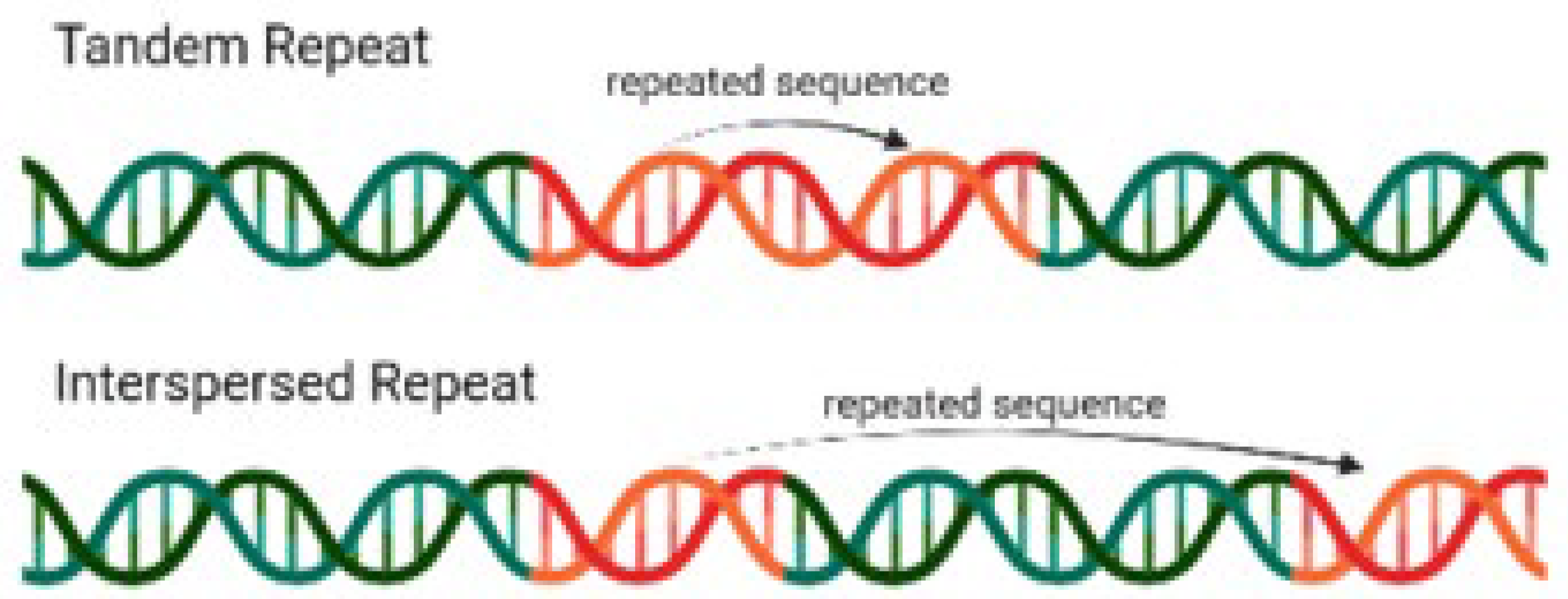
The impact of repetitive elements (Fig 1.) on genome assembly has been well-documented. Treangen and Salzberg (2012) highlight that repetitive sequences can lead to gaps and misassemblies, as short sequencing reads are often difficult to place accurately in these regions. This is particularly problematic in next-generation sequencing (NGS) technologies, which generate vast amounts of short reads that must be assembled into longer contiguous sequences.
Read mapping is similarly affected by repetitive elements. Li and Homer (2010) discuss how aligning reads to a reference genome is complicated by the presence of identical or nearly identical sequences. This can result in multiple potential mapping locations for a single read, increasing the likelihood of errors in downstream analyses, such as variant calling and gene expression studies.
Recent advancements in artificial intelligence (AI) and machine learning (ML) offer promising solutions to mitigate the challenges posed by repetitive elements in the genome. AI and ML algorithms can be trained to recognize patterns and distinguish between true genetic variants and artifacts introduced by repetitive sequences. For instance, deep learning models have been developed to improve the accuracy of variant calling by learning from large datasets of genomic sequences and associated annotations (Poplin et al., 2018).
Moreover, AI techniques can enhance genome assembly by integrating information from multiple sequencing technologies. Hybrid assembly approaches that combine short reads from NGS with long reads from third-generation sequencing technologies, such as PacBio and Oxford Nanopore, can be optimized using AI algorithms to resolve repetitive regions more effectively (Koren et al., 2017). These algorithms can leverage the strengths of each technology, utilizing the high accuracy of short reads and the long-range continuity of long reads to produce more accurate and complete genome assemblies.
AI can also be applied to read mapping by developing sophisticated alignment algorithms that account for the complexity of repetitive sequences. Machine learning models can be trained to predict the most likely mapping positions for reads based on the sequence context and prior knowledge of the genome's structure (Alipanahi et al., 2015). These models can reduce the ambiguity in read mapping and improve the reliability of genomic analyses.
Repetitive elements in the genome present significant challenges for bioinformatics, affecting genome assembly, read mapping, and variant calling. However, the application of AI and ML techniques holds great potential to address these challenges. By leveraging the power of AI, researchers can develop more accurate and robust tools for genomic analysis, ultimately enhancing our understanding of the genome and its functional elements.
Section 4. Conclusion
Repetitive elements, or selfish genes, constitute a substantial and challenging component of the human genome. These sequences can replicate and propagate without providing functional benefits to the organism, complicating various bioinformatics tasks, including genome assembly, read mapping, and variant calling (Doolittle & Sapienza, 1980; Orgel & Crick, 1980). Our simulation study clearly demonstrates how the presence of repetitive elements introduces ambiguity and errors in read mapping, whereas genomes devoid of such elements allow for more accurate and straightforward read alignment.
The challenges posed by repetitive sequences are well-documented. For instance, Treangen and Salzberg (2012) highlight the difficulties in assembling genomes with repetitive DNA due to the limitations of short-read sequencing technologies. Similarly, Li and Homer (2010) discuss how repetitive elements complicate the alignment of sequencing reads to reference genomes, leading to potential errors in downstream analyses.
Advancements in artificial intelligence (AI) and machine learning (ML) offer promising solutions to these challenges. AI and ML algorithms can improve variant calling accuracy, resolve complex repetitive regions during genome assembly, and enhance read mapping precision by leveraging sequence context and integrating data from multiple sequencing technologies (Poplin et al., 2018; Koren et al., 2017). These approaches underscore the potential of AI to revolutionize genomic analysis by addressing the limitations imposed by repetitive elements.
In summary, while repetitive elements present significant hurdles in bioinformatics, the integration of AI and ML techniques holds great promise for overcoming these challenges. By developing advanced tools and algorithms, researchers can achieve more accurate and reliable genomic analyses, ultimately advancing our understanding of the genome and its functional elements. The ongoing efforts in this field are crucial for enhancing the quality and precision of genomic research (Alipanahi et al., 2015; Poplin et al., 2018).
*The author declares no conflicting interests
References
- Alipanahi, B., Delong, A., Weirauch, M. T., & Frey, B. J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nature Biotechnology 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Dawkins, R. (1976). The Selfish Gene. Oxford University Press.
- Doolittle, W. F., & Sapienza, C. Selfish genes, the phenotype paradigm and genome evolution. Nature 1980, 284, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., & Phillippy, A. M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Research 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Li, H., & Homer, N. A survey of sequence alignment algorithms for next-generation sequencing. Briefings in Bioinformatics 2010, 11, 47. [Google Scholar]
- Orgel, L. E., & Crick, F. H. C. Selfish DNA: the ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R., et al. A universal SNP and small-indel variant caller using deep neural networks. Nature Biotechnology 2018, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T. J., & Salzberg, S. L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nature Reviews Genetics 2012, 13, 36–46. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



