Submitted:
15 May 2024
Posted:
16 May 2024
You are already at the latest version
Abstract
Background. Our knowledge regarding the epidemiology of pediatric cardiomyopathy is based on large national population studies reporting an annual incidence of 1 case per 100 000 children with a higher incidence observed in infancy and among selected populations. The aim is to document the epidemiology of pediatric cardiomyopathy in a Mediterranean population. Methods. Children younger than 18 years of age, living on the Mediterranean island of Crete, Greece and evaluated during 20 years since the establishment of tertiary pediatric cardiology services (2002-2022) were included in this retrospective study. Results. A total of 40 children were included, corresponding to an average annual incidence of pediatric cardiomyopathy of 1.59 cases (95% CI: 1.4-2,3) and a prevalence of 26 cases per 100.000 children. In decreasing order of frequency most cases corresponded to dilated cardiomyopathy (50%) followed by hypertrophic (42.5%), arrhythmogenic (5%) and restrictive (2.5%). An etiology was identified in 40%, including a genetic diagnosis in 22.5%. Conclusion. The incidence of pediatric cardiomyopathy in the Mediterranean island of Crete is higher compared with that reported previously for other Caucasian populations. Further study is needed to investigate the exact prevalence and specific genetic factors associated with the epidemiology of pediatric cardiomyopathy in Mediterranean populations.
Keywords:
Cardiomyopathy
; Childhood
; Pediatric Cardiomyopathy
; Epidemiology
; Crete
; Mediterranean
1. Introduction
Cardiomyopathies (CMPs) are defined as myocardial disorders that manifest with various structural and functional phenotypes, in the absence of other known cardiovascular disorders (congenital, valvular, coronary artery disease) [1]. The spectrum of CMPs in children differs from that in adults, being heterogeneous in origin. Although sarcomeric and cytoskeleton mutations can also cause pediatric cardiomyopathies, syndromic, metabolic or neuromuscular disorders are also relevant [2,3]. The most common morphological phenotypes are dilated (DCM) and hypertrophic cardiomyopathy (HCM), whereas arrhythmogenic (ACM), restrictive (RCM) and LV non-compaction cardiomyopathy (LVNC) occur less frequently [2].
Our knowledge regarding the epidemiology of pediatric CMPs is based on limited North American [4], Australian [5] and European registries [6]. These population studies and registries estimate the overall annual incidence of primary CMP in children around 1 per 100 000 children, with a higher incidence during the first 2 years of life while ethnic/ geographical differences are also present [4,5,6]. Our knowledge about the epidemiology of pediatric CMP in Mediterranean region is limited. The aim of our study was to determinate the prevalence and the annual incidence of pediatric CMP in pediatric Mediterranean population of the island of Crete. To the best of our knowledge this represents the first study regarding the epidemiology of pediatric CMPs in the Southeastern Mediterranean region.
2. Materials and Methods
This is a retrospective study including children younger than 18 years of age, living in the region of Crete Island, with the diagnosis of CMP, who were presented to the Pediatric Cardiology Unit, University Hospital in Heraklion, Crete during 20 years since its first establishment (1.2002-12.2022). The cumulative prevalence at the last year of enrollment and the overall annual incidence rate of pediatric CMP in the pediatric population of Crete Island during the whole period of enrolment was estimated.
This study was performed in line with the principles of the Declaration of Helsinki. University Hospital Heraklion, Ethics Committee approval (1007, 33/2.12.2020) was obtained for collection and processing of data from medical records of each study patient. Data collection included demographic characteristics, brief family history, clinical findings and quantitative echocardiographic measurements. All personal data were anonymized.
The classification of CMPs in our study was based on the structural and functional phenotype using a classification based on morphofunctional echocardiographic characteristics [2] and following the European Society of Cardiology approach [1,7]. Clearly defined inclusion morphological criteria in accordance with previous population-based studies on epidemiology of pediatric CMPs [4,5,8] were used for enrollment in our study. Patients with any of the secondary causes of CMPs (congenital heart disease, hypertension, endocrine and pulmonary diseases, toxic exposure, drugs), the cohorts of asymptomatic pediatric patients with incomplete disease expression (subclinical forms) or children phenotypically negative but genotypically positive were excluded [2,8].
Inclusion criteria were based on echocardiographic measurements of the LV end-diastolic dimension (LVEDD), LV posterior wall and septal thicknesses, LV end-systolic dimension (LVESD), fractional shortening (FS) and ejection fraction (EF). All measurements were expressed as z-scores > 2 standard deviations to adjust for patient size. Boston Pediatric Echo Z-Score database was used as reference data resource [9]. The diagnosis of DCM was based on LV end-diastolic diameter (LVEDD) and LV end systolic diameter (LVESD) greater than 2 standard deviations (SD) from the mean value for the population corrected for body surface area (z-scores > 2) with decreased systolic function, characterized by reduced measures of FS < 28% and EF < 55%. The diagnosis of HCM was made if regional of global maximal LV wall thickness was greater than 2 standard deviations above the body surface area–corrected mean (z-scores > 2) with both normal and reduced LV systolic function. The diagnosis of RCM was based on specific patterns: both atria enlarged relative to ventricles of normal or small size with evidence of impaired diastolic filling and the absence of significant valvular heart disease. The diagnosis of ACM was based on positive diagnostic criteria. Inclusion criterion was considered the autopsy report (SCD as a presenting symptom).
The evaluation of patients included medical history, physical evaluation, standard blood chemical analysis, chest radiography and echocardiography in all cases. CMR was performed in selected patients. The investigation for inborn errors of metabolism was performed in the majority of patients diagnosed in infancy. Genetic evaluation was performed in a subset of patients. The prevalence and incidence rates were calculated accordingly to the age specific population at risk between 2002 and 2022 in the Island of Crete. The pediatric population data were obtained from the Hellenic Statistical Authority Office [10]. The data that support the findings of this study are available from the corresponding author on reasonable request.
Statistical analysis. The incidence rates in various subgroups (year, sex, age, type of cardiomyopathy) were compared using the Fisher exact probability test and exact 95 percent confidence intervals. Data were summarized as frequencies and percentages for categorical data and as medians and means for the age of presentation. All reported P values were based on two-sided tests.
3. Results
Over the period of 20 years (January 2002 –December 2022) the total number of 40 new cases of pediatric CMP was diagnosed. The total average island population in this period was 614.000, including a total of 119.343 children less than 18 years of age. The average annual incidence of new CMP pediatric cases (<18 yrs) on Crete Island was estimated at 1,59 per 100.000 (95% CI: 1,4 to 2,3). There was no evidence of change over time in the incidence rate of any type of CMP. The cumulative prevalence of pediatric CMP in Crete increased in the first years following the establishment of pediatric cardiology services, reaching a plateau at the last year of enrollment, corresponding to a cumulative prevalence of 26 cases per 100.000 children at the last year of enrolment. Figure 1
The incidence of DCM was estimated at 0,79 /100000/year, a total of 20 cases of DCM (50%). The incidence of HCM was estimated at 0,67/100000/year, a total of 17 cases of HCM (42,5%). The incidence of other types of CMP was 0,11/100000/year corresponding to 2 cases of ACM (5%) and 1 case of RCM (2,5%). Table 1 Of the total number of 40 children with CMP 27 patients were male (67,5%) and 13 patients were female (32,5%, p = 0.001). The association between the type of CMP and sex was significant only in DCM patients (male 80%, p < .0001). In HCM male and female distribution was at nearly equal percentage (9 male and 8 female).The other CMP were very rare and the sex dominance was not observed.
Clinical presentation of CMP was symptomatic in 34 (85%) children. (Table 1) Congestive heart failure symptoms were dominant in clinical symptomatology of 32,5% pediatric CMP patients. Heart failure symptoms were present in all DCM patients diagnosed in infancy (13 infants of the total 20 DCM cases). Murmur and exercise intolerance were present in all HCM patients diagnosed in childhood and adolescence (12 children from the total 17 HCM cases). Congenital malformations were present in 5 infants with CMP. Syncope/seizures episodes were presenting symptoms in 3 children (infant HCM, child HCM, adolescent ACM). Exercise syncope was an initial symptom in one adolescent with ACM. Sudden cardiac death (SCD) was the first manifestation of ACM in a single adolescent patient, based on autopsy findings. A total of 6 (15%) children were completely asymptomatic at the time of CMP diagnosis with 4 cases (10%) being evaluated during cascade family screening and 2 cases (5%) during preparticipation screening for sports. The age distribution was not uniform. Proportionally most cases (20 infants) were diagnosed in the first two years of life (50 % of patients).The median age at diagnosis was 2 years (mean 4,5 years). One third of patients (13 children) were diagnosed at school age, while in adolescence 7 patients (17,5%).There was an association between the age of diagnosis and the type of CMP. 65% (13 infants) DCM patients were diagnosed at <1 year of age (p = 0.05).The median age at diagnosis of DCM patients was 6 months of age (mean 2,5 years). In our study there were two age groups with an increased frequency of HCM diagnosis. The first peak of HCM diagnosis was the first year of life – 5 cases (29,5% of total HCM cases), the other peak was late childhood-adolescence with 8 cases (47% of total HCM cases). All patients with ACM and RCM were diagnosed in adolescence.
The etiology of CMP patients enrolled in our study was identified in 16 (40%) of 40 children (Table 1). The evaluation for inborn errors of metabolisms was performed in 9 cases (22,5% of examined children). Positive result was found in nearly 50 % of evaluated patients. Specifically it revealed 1 infant HCM phenotype with glycogen storage disease type 3 and 3 school aged children with disorders of fatty-acid metabolism – 2 children HCM phenotype (different families, no relatives) with primary carnitine deficiency and 1 child DCM phenotype with LCAD (Long chain Acyl-CoA Dehydrogenase deficiency). In our cohort there was only 1 patient with HCM phenotype with genetically confirmed diagnosis of Noonan syndrome. Other patients with malformations and/or syndromic clinical features without confirmed genetic diagnosis were not included in this etiologic subgroup. One adolescent DCM patient with the diagnosis of Duchenne’s muscular dystrophy (neuromuscular disease) was enrolled in our study. Acute myocarditis was the initial diagnosis in 1 child with DCM (the diagnosis was established by CMR imaging). Genetic evaluation was performed in 13 cases (32,5%children), in patients with different forms of CMP. Genetic cause was identified in 29 cases (2,5% children). Positive results, i.e. pathogenic variants, were found in 9 cases (69% of those tested). In 4 cases (30% of those tested) DNA analysis revealed pathogenic sarcomeric variant: 2 children with HCM phenotype (pathogenic variant of MYH7 gene) and 2 sibling children with DCM phenotype double heterozygous (MYH7/ TTN).
The distribution of pediatric CMP cases in Crete is not homogenous but there is a significant concentration of children with CMP in the western regions of the island. The comparison of the incidence of pediatric CMP on the island of Crete compared to previous studies is presented on Table 2.
The outcome of CMP patients was variable ranging from radical improvement in 1 DCM patient to 1 DCM patient enlisted for heart transplantation. Medical treatment was indicated in 95% patients. ICD was implanted to 7 patients (4 children with HCM, 2 children with DCM and 1 child with RCM). Death was the outcome for 5 children (12,5% of the total) - 2 DCM cases, 1 HCM case, 2 ACM cases.

Table 2.
Characteristics of pediatric CMP epidemiologic studies.
| Arola Am J Epid 1997 |
Nugent NEJM 2003 |
Lipshulz NEJM 2003 |
Crete 2023 | |
|---|---|---|---|---|
|
Pro/ retrospective |
retrospective | retrospective | prospective | retrospective |
|
Duration (years) |
12 (1980-1991) |
10 (1987-1996) |
3 (1996-1999) |
20 (2002-2022) |
| Country | Finland | Australia | USA | Greece-Crete |
| Ages | 0-20 | 0-10 | 0-18 | 0-18 |
| Cases | 118 | 314 | 467 | 40 |
|
CMP incidence/ 100000/year |
0,65-0,74 | 1,24 | 1,13 | 1,59 |
| 95%C.I | 0,65-0,74 | 1,11-1,38 | 1,03-1,23 | 1,4-2,3 |
| HCM (%) | 37 | 25 | 42 | 42,5 |
| DCM (%) | 52 | 58 | 51 | 50 |
Abbreviations: CMP – cardiomyopathy, DCM – Dilated cardiomyopthy, HCM – Hypertrophic cardiomyopathy.
4. Discussion
The Mediterranean region is characterized by increased prevalence of many hereditary diseases. Historical populations’ movements that occurred along the Mediterranean Sea in the past had a crucial impact on the variability in prevalence and in genetic structure of these diseases in different ethnic groups of the Mediterranean population. The hemoglobinopathies and glucose-6-phosphate dehydrogenase (G6PD) deficiency are the commonest single-gene disorders encountered in the region [11]. Familial Mediterranean Fever (FMF), the most frequent autoinflammatory disease, is common in the Eastern Mediterranean area but rare elsewhere [12]. Increased prevalence of specific hereditary diseases, including inherited CMPs, is typical for historically isolated and differentiated populations living in Mediterranean islands [13]. Aegean Islands and the Island of Crete are geographical regions with increased prevalence of ARVC (Naxos disease) and hereditary transthyretin – related amyloidosis (hATTR) cardiomyopathy [14], respectively. While the epidemiology of adult CMP in the Mediterranean region is described and monitored by European and ethnic registries [15], the knowledge of pediatric CMP epidemiology in Mediterranean region is limited [16].
We aimed to record the epidemiologic data of pediatric CMPs in the Island of Crete and to compare the variability that we found with the results of the other population based studies [4,5,6]. The island of Crete is a clearly defined geographic area with mixed population. The pediatric population in Crete is in more than 97% Caucasian living partly in urban areas but also in relatively isolated rural mountain areas. In these communities increase rate of parental consanguinity was documented and it may lead to a high prevalence of private and ultra-rare AR disorders [17]. A limited number of children, nearly 3% belong to other ethnic groups (Arabs, Asians) [10].
Regional differences in the incidence of CMP were found. Τhere was an increased concentration of children with CMP in the central-western rural, mountain regions of the island with the highest incidence of pediatric CMPs all years of the study (incidence 3,2 per 100000/years vs 1,59 per 100000/year). A similar interesting distribution of genotypes in an east –west direction among native Cretans has been previously described, possibly reflecting diverse patterns of island colonization in the past from different Mediterranean regions [18].
In comparison with previous studies, the annual incidence of primary CMP was estimated at 1,59 per 100 000 children younger than 18 years of age. There was no significant difference in the frequency of the diagnosis according to the year. The annual incidence in our study was estimated higher than incidences reported in other population-based studies in the North America 1,13/100000/year [4], Australia 1,24/100000/year [5], and Finland 0,65–0,74/100000/year [6].
The most frequent phenotype in our study was DCM (50 %) which correlates with the results of other studies [4,5]. The incidence of DCM was estimated at 0,79 per 100.000 children/year, which correlates with data of the large prospective study undertaken in the United Kingdom and Ireland [19], where the authors reported an incidence of 0,76 symptomatic DCM cases per 100000 children under 16 years but is higher than data from multicentre North American studies [6,20], where incidence of 0,57 DCM cases/100000/year was reported. In the first year of life 65% of children with DCM were diagnosed, while nearly 75 % of them in the first 2 years of life. Congestive heart failure symptoms were dominant in all children < 2 years of age diagnosed with DCM. In multicentre North American study [4] as well as in the National Australian Childhood Cardiomyopathy Study [5] the highest annual incidence of DCM was observed during the first year of life. A Finnish study demonstrated an 11-fold higher incidence of DCM during the first year of life [6].
HCM was the second most frequent type of CMP in our study (42,5%). The incidence of HCM was estimated at 0,67/100.000/year. The pattern of two peaks of increased incidence was typical for patients with HCM. The first early peak of incidence of HCM was in infancy with the second peak in late childhood and adolescence. This pattern of “two peaks” incidence in HCM cases was presented in reports of other investigators [21]. The data from a multicentre cohort of Marston et al [22], indicate that cases of phenotypic HCM tends to cluster during infancy and adolescence, including high proportions of patients with RASopathies and inborn errors of metabolism in infancy [23].
The only patient of our study with RCM was diagnosed in early adolescent age. RCM is a rare form of pediatric CMP, became more frequent with increasing age [24], frequently with positive genetic findings [25].
The diagnosis of ACM was established in both patients at early adolescence confirming bibliography resources [26]. The phenotypic features of ACM may vary ranging from asymptomatic to life-threatening ventricular arrhythmias. Both of our patients were symptomatic but the clinical presentations were diametrically different. Syncope during exercise was an initial symptom in one adolescent patient diagnosed with Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC). SCD is referred to be the first clinical manifestation of the disease specifically in the young people [27] similar to one adolescent patient of our cohort. The diagnosis of ARVC was established by autopsy report.
Pediatric CMPs are highly heterogeneous in origin. A known or presumed cause of CMP patients enrolled in our study was identified in 40% of children which correlates with other studies [8,19]. Inborn errors of metabolism were diagnosed in 10 % of children (1 infant and 3 school aged children). Metabolic disease evaluation was positive mostly in children with HCM phenotype (23,5% of all HCM patients) but inborn error of metabolism was diagnosed also in 1 patient with DCM phenotype. Similar results were reported in The Pediatric Cardiomyopathy Registry of children diagnosed with HCM, where 8.7% had inborn errors of metabolism [21]. In the study of Kindel et al [28] significant rates of metabolic causes in children with CMP (19%) were identified confirming the importance of a broad differential for clinical evaluation of children.
The progress in understanding the genetic base of CMP has shown that the genetics of CMPs in children is extremely heterogeneous and complex [17]. Genetic variants causing CMP in children can be isolated (merely cardiac involvement) or they can have systemic features (multiorgan involvement). The RASopathies are the most well-known syndromic causes of pediatric cardiomyopathy [17,28]. In our cohort there was only one patient with genetically confirmed diagnosis of Noonan syndrome and HCM phenotype. The most common neuromuscular disorders with cardiac manifestations are the muscular dystrophies. The type and extent of cardiac manifestations are specific to the type of neuromuscular disorder. The most common cardiac findings include DCM or HCM phenotype [29]. One adolescent patient with diagnosis of Duchenne’s muscular dystrophy and DCM phenotype from the pediatric neuromuscular disorders cohort was enrolled. Acute myocarditis was the initial diagnosis in one child with DCM (the diagnosis was established by CMR imaging). Viral myocarditis is the most common cause of inflammatory DCM in children [2]. Results from the endomyocardial biopsy are still the reference standard for diagnose of myocardial inflammation. Given the difficulties of providing inflammation in the myocardium, the clinical diagnosis is based on non invasive diagnostic methods (CMR imaging) [2].
Genetic evaluation was performed in nearly one third of cases (32,5%) of all CMP patients. DNA analysis was positive in 22,5% of children. Genetic testing revealed pathogenic variant in 69% of evaluated children.45% of these pathogenic variants were variants in genes encoding sarcomeric proteins. These results of our patients DNA analysis are similar to previous literature. Pediatric CMPs are genetically heterogeneous with many different causative genes and multiple mutations in each gene. Mutations in genes encoding sarcomeric proteins are the most common abnormalities in children with isolated HCM [1], but they are also associated with other CMPs [30]. Desmosomal gene variants are associated with ACM [2,17].
The present study findings should be viewed talking into account the study limitations including retrospective study design, small sample size of the reference population, and limited diagnostic evaluation regarding underlying etiology (genetics) due to associated cost.
5. Conclusions
The incidence of pediatric CMP in Crete is higher than in other population-based studies manifesting also regional clusters mainly in the isolated rural and mountain western regions. However the relative and age distribution of pediatric CMP cases was similar to that previously described. An etiology was identified in 40%, being genetic in 22,5 % of cases. Further studies are needed to investigate whether Mediterranean populations are at increased risk of pediatric CMP and for the underlying genetic factors of any differences.
Author Contributions
A.B.: Conceptualization; investigation; methodology; writing – original draft; formal analysis; writing – review and editing; F.P.: Conceptualization;methodology; validation; writing – review and editing; formal analysis. G.C.: Conceptualization; methodology; validation; statistical analysis; A.A.: Conceptualization; methodology; validation;writing – review and editing; formal analysis; supervision. E.G.: Conceptualization; methodology; validation; writing – review and editing; supervision I.G.: Conceptualization; investigation; writing – original draft; methodology; validation; writing – review and editing; formal analysis; project administration; supervision.
Funding
This research received no external funding.
Institutional Review Board Statement
This study was performed in line with the principles of the Declaration of Helsinki. University Hospital Heraklion, Ethics Committee approval (1007, 33/2.12.2020) was obtained for collection and processing of data from medical records of each study patient.
Informed Consent Statement
Informed consent was obtained from all parents of children involved in the study.
Data Availability Statement
The data presented in this study are available upon request from the corresponding authors. The data are not publicly available due to restrictions eg privacy or ethical.
Acknowledgments
None
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Elliott, P.; Andersson, B.; Arbustini, E.; et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; et al. Cardiomyopathy in children: Classification and diagnosis. Circulation 2019, 140, e9–e68. [Google Scholar] [CrossRef] [PubMed]
- Tsatsopoulou, A.; Protonotarios, I.; Xylouri, Z.; et al. Cardiomyopathies in children: An overview. Hellenic J Cardiol 2023, 72, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Sleeper, L.A.; Towbin, J.A.; et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 2003, 348, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.W.; Piers, B.S.; Daubeney, M.B.; et al. The epidemiology of childhood cardiomyopathy in Australia. N Eng J Med 2003, 348, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Arola, A.; Jokinen, E.; Ruuskanen, O.; et al. Epidemiology of idiopathic Cardiomyopathies in Children and Adolescents. Am J Epidemiol 1997, 146, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimero, J.R.; et al. ESC guidelines for the management of cardiomyopathies. European Heart Journal 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.D.; Sleeper, L.A.; Alvarez, J.A.; Bublik, N.; Lipshultz, S.E. The Pediatric Cardiomyopathy Registry: 1995-2007. Prog Pediatr Cardiol 2008, 25, 31–36. [Google Scholar] [CrossRef] [PubMed]
- The Boston Children’s Hospital z-score system. https://zscore.chboston.org/.
- Hellenic Statistical Authority. https://www.statistics.gr/el/statistics/-/publication/SPO18.
- De Sanctis, V.; Kattamis, C.; Canatan, D.; et al. Beta-thalassemia distribution in the Old World: an ancient disease seen from a historical standpoint. Mediterr J Hematol Infect Dis 2017, 9, e2017018. [Google Scholar] [CrossRef]
- Lancieri, M.; Bustaffa, M.; Palmeri, S.; et al. An Update on Familial Mediterranean Fever. Int. J. Mol. Sci. 2023, 24, 9584. [Google Scholar] [CrossRef]
- Charoute, H.; Bakhchane, A.; Benrahma, H.; et al. Mediterranean Founder Mutation Database (MFMD):Taking Advantage from Founder Mutations in Genetics Diagnosis, Genetic Diversity and Migration History of the Mediterranean Population. HUMAN MUTATION Database in Brief 2015, 36, E2441–E2453. [Google Scholar] [CrossRef] [PubMed]
- Tzagournissakis, M.; Foukarakis, E.; Samonakis, D.; et al. High Hereditary Transthyretin-Related Amyloidosis Prevalence in Crete. Genetic Heterogeneity and Distinct Phenotypes. Neurol Genet 2022, 8, e200013. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Charron, P.; Blanes, J.R.G.; on behalf of the EORP Cardiomyopathy Registry Pilot Investigators; et al. European Cardiomyopathy Pilot Registry: EURObservational Research Programme of the European Society of Cardiology. European Heart Journal 2016, 37, 164–173. [Google Scholar] [CrossRef]
- Maurizi, N.; Passantino, S.; Spaziani, G.; et al. Long-term Outcomes of Pediatric-Onset Hypertrophic Cardiomyopathy and Age-Specific Risk Factors for Lethal Arrhythmic Events. JAMA Cardiol 2018, 3, 520–525. [Google Scholar] [CrossRef]
- Lodato, V.; Parlapiano, G.; Cali, F.; et al. Cardiomyopathies in children and systemic disorders when is it useful to look beyond the heart? J. Cardiovasc. Dev Dis. 2022, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Drineas, P.; Tsetsos, F.; Plantinga, A.; et al. Genetic history of the population of Crete. Ann Hum Gen 2019, 83, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.E.; Fenton, M.J.; Ridout, D.A.; Burch, M. New-onset heart failure due to Heart muscle disease. A prospective study in the United Kingdom and Ireland. Circulation 2008, 117, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; et al. Incidence, causes and outcomes of dilated cardiomyopathy in children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Colan, S.D.; Lipshultz, S.E.; Lowe, A.M.; et al. Epidemiology and cause specific outcome of hypertrophic cardiomyopathy in children. Circulation 2007, 115, 773–781. [Google Scholar] [CrossRef]
- Marston, N.A.; Han, L.; Olivotto, I.; et al. Clinical characteristics and outcomes in childhood- onset hypertrophic cardiomyopathy. European Heart Journal 2021, 42, 1988–1996. [Google Scholar] [CrossRef]
- Alexander, P.M.A.; Nugent, A.W.; Daubeney, P.E.F.; et al. Long-Term Outcomes of Hypertrophic Cardiomyopathy Diagnosed During Childhood Results From a National Population-Based Study. Circulation 2018, 138, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.; Weintraub, R. Overview of cardiomyopathies in childhood. Frontiers in pediatrics 2021, 9, 708732. [Google Scholar] [CrossRef] [PubMed]
- Kaski, J.P.; Syrris, P.; Burch, M.; et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008, 94, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic cardiomyopathy. Circ. Res. 2017, 121, 785–802. [Google Scholar] [CrossRef]
- Anastasakis, A.; Papatheodorou, E.; Ritsatos, K.; et al. Sudden unexplained death in the young: epidemiology, aetiology and value of the clinically guided genetic. Europace 2018, 20, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Kindel, J.S.; Miller, E.M.; Gupta, R.; et al. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail 2012, 18, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Baban, A.; Lodato, V.; Parlapiano, G.; et al. Myocardial and Arrhythmic Spectrum of Neuromuscular Disorders in Children. Biomolecules 2021, 11, 1578. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Singer, E.S.; Wacker, J.; et al. Genetic Basis of Childhood Cardiomyopathy. Circ Genom Precis Med 2022, 15, e003686. [Google Scholar] [CrossRef]
Figure 1.
Prevalence and annual incidence of pediatric CMP 2002-2022 in Crete.
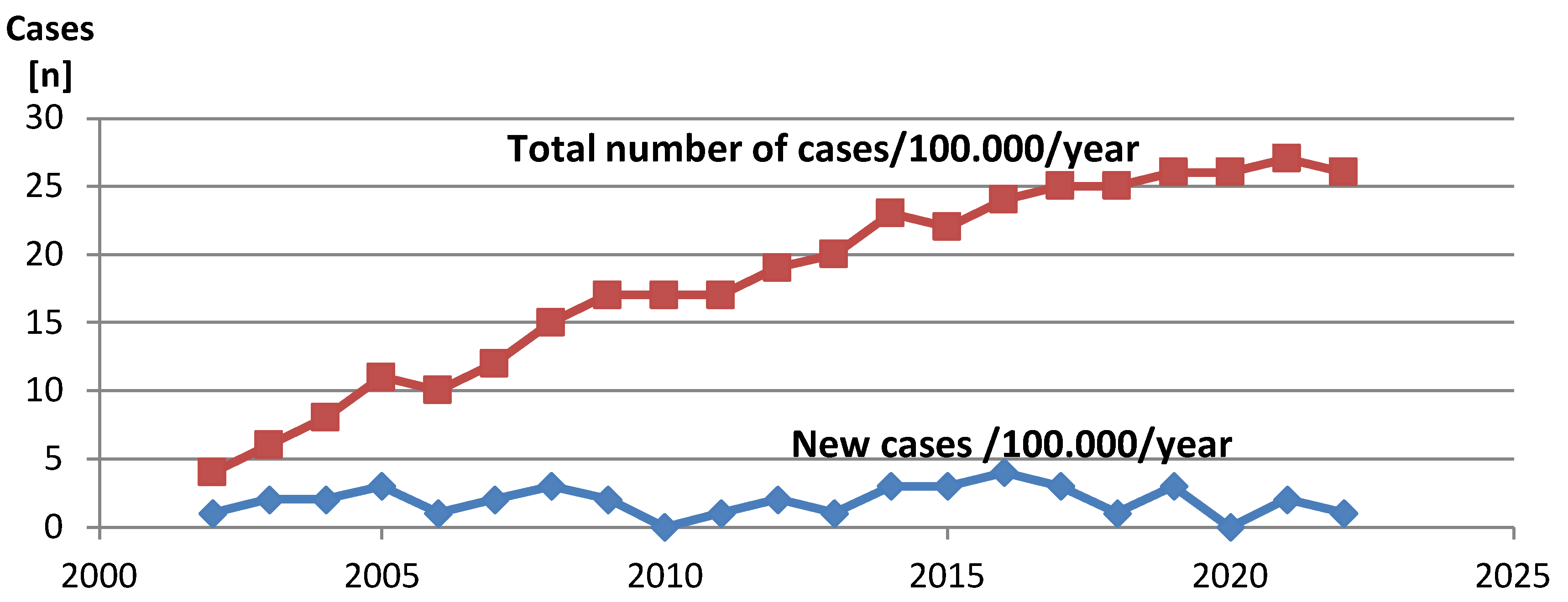
Table 1.
Epidemiology of pediatric CMP in Crete 2002 – 2022.
|
DCM N (%) |
HCM N (%) |
ACM N (%) |
RCM N (%) |
CMP N |
CMP % |
|
| Patients | 20 (50) | 17 (42,5) | 2 (5) | 1 (2,5) | 40 | 100 |
| Male | 16 (80) | 9 (53) | 1 (50) | 1 (100) | 27 | 67,5 |
| Female | 4 (20) | 8 (47) | 1 (50) | 0 | 13 | 32,5 |
| Age at diagnosis | ||||||
| < 1 year | 13 (65) | 5 (29,5) | 0 | 0 | 18 | 45 |
| 2-11 years | 7 | 4 | 0 | 0 | 11 | 27,5 |
| 12-18 years | 0 | 8 (47) | 2 (100) | 1 (100) | 11 | 27,5 |
| Clinical presentation at diagnosis | ||||||
| Clinical symptoms | ||||||
| Heart failure symptoms | 13 (65) | 0 | 0 | 0 | 13 | 32,5 |
| Murmur | 2 (10) | 6 (35) | 0 | 0 | 8 | 20 |
| Congenital malformations | 0 | 5 (29) | 0 | 0 | 5 | 12,5 |
| Exercise intolerance | 2 (10) | 2 (12) | 0 | 0 | 4 | 10 |
| Syncope/Seizures episodes | 0 | 2 (12) | 1 (50) | 1 (100) | 4 | 10 |
| Myocarditis | 1 (5) | 0 | 0 | 0 | 1 | 2,5 |
| Sudden cardiac death | 0 | 0 | 1 (50) | 0 | 1 | 2,5 |
| Asymptomatic | ||||||
| Cascade family screening | 1 (5) | 3 (17) | 0 | 0 | 4 | 20 |
| Preparticipation sports screening | 1 (5) | 1 (6) | 0 | 0 | 2 | 5 |
| Specific primary causes of CMP | ||||||
| Genetic causes | ||||||
| Pathogenic sarcomeric variant | 0 | 4 (23) | 0 | 0 | 4 | 10 |
| Pathogenic non-sarcomeric variant | 3 (15) | 2 (12) | 0 | 0 | 4 | 10 |
| Inborn errors of metabolism | ||||||
| Glycogen storage disease type 3 | 0 | 1 (6) | 0 | 0 | 1 | 2,5 |
| Primary carnitine deficiency | ||||||
| (disorders of fatty-acid metabolism) | 0 | 2 (12) | 0 | 0 | 2 | 5 |
| LCAD (disorders of fatty-acid metabolism) | 1 (5) | 0 | 0 | 0 | 1 | 2,5 |
| Neuromuscular disorders | ||||||
| Duchenne’s muscular dystrophy | 1 (5) | 0 | 0 | 0 | 1 | 2,5 |
| Inflammatory causes | ||||||
| Acute myocarditis | 1 (5) | 0 | 0 | 0 | 1 | 2,5 |
| Malformation syndromes | ||||||
| Noonan syndrome | 0 | 1 (6) | 0 | 0 | 1 | 2,5 |
Abbreviations: CMP – cardiomyopathy, DCM – Dilated cardiomyopthy, HCM – Hypertrophic cardiomyopathy, ACM – Arrhythmogenic cardiomyopathy, RCM – restrictive cardiomyopathy, LCAD – Long chain Acyl-CoA Dehydrogenase deficiency.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



