Submitted:
13 May 2024
Posted:
14 May 2024
You are already at the latest version
Abstract
Pulmonary hypertension (PH) is a serious condition linked to immune-system dysfunction. Myositis-specific/associated antibodies (MSA/MAA) play a role in idiopathic inflammatory myopathy (IIM) and interstitial lung disease (ILD), but their significance in PH remains unclear. We believe the presence of these antibodies, may be underestimated. This study analyzed adult PH patients without pre-existing IIM for MSA/MAA prevalence using a line-blot assay. We compared PH patients with and without ILD signs to a cohort clinically suspected of IIM/ILD (n=558). Our PH cohort (n=121) showed a significantly higher prevalence of overall weak positive MSA/MAAs and positive overlap syndrome-associated MAAs than the suspected IIM/ILD group (p
Keywords:
Pulmonary Hypertension
; Myositis specific antibodies
; Myositis associated antibodies
; Immunology
; auto-antibodies
1. Introduction
Pulmonary hypertension (PH), a severe condition with high morbidity and mortality, significantly impacts patients' quality of life [1]. Elevated pressure within the pulmonary vasculature leads to right-sided heart failure and potential death. Vascular remodeling within the pulmonary arterioles is a hallmark of pre-capillary PH, while post-capillary PH arises from changes in the venous vasculature often diagnosed in patients with left-sided heart disease. Right heart catheterization (RHC) distinguishes these subtypes. PH is a hemodynamic diagnosis and has diverse etiologies. This is illustrated by the fact that the World Health Organization (WHO) has classified PH into five different clinical subtypes, indicating similarities in pathobiology, although overlap between classes remains [2]. Immune dysregulation plays a critical role in the pulmonary vascular and interstitial changes underlying PH [3]. This is best exemplified by the increased risk of developing pulmonary arterial hypertension (PAH) among patients with systemic autoimmune diseases, like systemic sclerosis (SSc) and systemic lupus erythematosus (SLE), which are grouped as WHO type 1 PH and subclassified as connective tissue disease (CTD)-PAH [4]. Importantly, immune involvement extends to idiopathic PAH (IPAH). Different immunological pathways that initiate or maintain the vasculopathy and vascular remodeling of IPAH have been elucidated [3,5,6,7,8,9,10,11,12,13,14]. Polarization of innate immune cells into pro-fibrotic subtypes, peri-vascular inflammation with extensive lymphocyte involvement, and association with autoimmunity by the presence of auto-antibodies against endothelial cells (AECAs) that influence vascular stromal cells contribute to endothelial activation and may propagate vascular remodeling in PAH [3,8,9,10,11,12,15,16,17,18]. However, whether these immune-mediated vascular changes may precede or contribute to the development of interstitial lung disease (ILD) remains a topic of investigation [19].
Although PH is associated with connective tissue diseases (CTDs), it is rarely observed in idiopathic inflammatory myopathy (IIM), a condition characterized by myositis, ILD, and myositis-specific/associated antibodies (MSA/MAAs) [20,21,22,23,24,25]. The prevalence of MSA/MAAs in PH patients remains unclear, with reported rates varying widely. For instance, the prevalence of anti-synthetase antibodies can vary widely, ranging from 8% to as high as 29% [26,27]. However, it's important to note that methodological differences in studies can significantly impact this reported percentage. When PH occurs in patients with IIM, it is often accompanied by extensive end-stage ILD. The presence of PAH in patients with IIM without ILD is very rare. The involvement of immune-mediated vascular remodeling is likely to contribute to the pathogenesis, but much remains unknown due to its rarity. Vice versa, a large PAH cohort study screened 5223 patients for IIM, which yielded only 34 cases. Most of these had severe ILD, with only three patients having isolated PAH [28]. PH is currently not included in the diagnostic criteria of IIM, although mild PH may occur during more severe stages of ILD, classified under WHO type 3 [21,22,27,28,29,30,31].
In our expertise referral clinic, we questioned whether MSA/MAAs are prevalent in patients presenting with PH but who had not been recognized as patients with an underlying systemic autoimmune disease and who had not been treated as such. We analyzed a cohort of pre-capillary and combined pre- and post-capillary PH patients for the presence of MSA/MAAs. Because PH can develop in patients after or alongside pulmonary interstitial changes, often diagnosed as ILD, we also stratified our cohort based on the presence of pulmonary interstitial changes on radiological imaging suggestive of ILD. We hypothesize that MSA/MAAs are more prevalent in PH than currently suggested, especially in PH patients with ILD.
2. Materials and Methods
2.1. Patient Selection
A retrospective cohort study was performed on adult patients who were diagnosed with PH in the past 20 years at the Maastricht University Medical Center (MUMC+) according to the diagnostic criteria at the time, with a mPAP ≥ 25mmHg, a PVR ≥ 3 Wood Units assessed by RHC. Patients with combined pre- and post-capillary PH were also included. Patients with congenital heart disease, pulmonary veno-occlusive disease, and porto-pulmonary hypertension were excluded. Patients who were already diagnosed with an IIM were also excluded. PH patients underwent clinical evaluation by a clinical immunologist who performed immunological diagnostics accordingly to evaluate the presence of a systemic immunological disease. Clinical characteristics were assessed and analyzed at the time of diagnosis of PH, including WHO type of PH, New York Heart Association (NYHA) class, hemodynamics measured by RHC, pulmonary function tests, 6-minute walking test (6MWT) and laboratory parameters, like N-terminal-pro-brain natriuretic peptide (NT-pro-BNP) and C-reactive protein (CRP). To determine the underlying cause of pulmonary hypertension (PH), all patients underwent high-resolution computed tomography (HRCT) as part of the diagnostic workup. Patients were stratified into the PH with ILD group when signs of pulmonary interstitial changes were present. To compare the prevalence observed in our PH cohort, routine clinical data were collected from patients suspected to have IIM (n=558) and, hence, were tested for MSA/MAA. Obviously, in these patients, a diagnosis of IIM might be made or rejected. The clinical suspicion of IIM in these patients was established based on signs of myositis, like elevated creatine kinase (CK), proximal muscle weakness, muscle biopsy, electromyography, and/or typical skin lesions, with or without the presence of ILD. This study complies with the Declaration of Helsinki and was performed according to the local ethics committee’s approval.
2.2. Determination of Antibodies
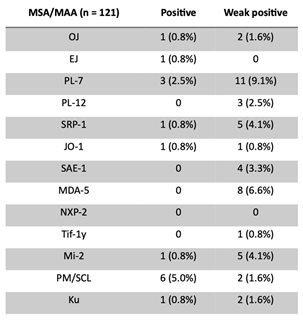
Antibodies were detected in serum using a line-blot assay (EUROLINE Autoimmune Inflammatory Myopathies, EUROIMMUN, Lubeck, Germany). The assay identified MSA antibodies against Jo-1, EJ, OJ, PL-7, PL-12 (anti-synthetase antibodies), Mi-2α, Mi-2β, TIF1-γ, MDA5, NXP2, and SAE1 (dermatomyositis antibodies) and MAA antibodies against Ku, PM/Scl-75 and PM/Scl-100 (overlap syndrome antibodies). A patient was considered positive for Mi-2 and PM/Scl antibodies if they tested positive for any subtype. In cases of both positive and weak positive results, the highest result was used to determine overall positivity. Data of Ro52 reactivity was excluded from our analysis. The immunoblot strips were analyzed with the EUROLINE Scan software (EUROIMMUN, Lubeck, Germany) according to the manufacturer’s recommendations for the EUROLINE Autoimmune Inflammatory Myopathies line blot assay. Strips were scored as negative, weak positive and positive, which corresponds with intensity levels of respectively 0–10, 11–25 and >25 [32]. Antibody reactivity on a positive and weak positive intensity level was separately evaluated. A patient is considered positive for MSA/MAAs if at least one antibody reveals a reactivity >25; or weak positive if at least one antibody reveals a reactivity of 11-25, but none >25; all other patients are considered negative. Prevalence of MSA/MAA in our comparative cohort, as well as in healthy cohorts from the literature, was used for comparison [23,33,34].
2.3. Statistical Analysis
Categorical characteristics were expressed as numbers and percentages. Numerical data were expressed as mean and standard deviation (SD) or median and interquartile range (IQR) based on normal distribution estimated by Shapiro/Wilk test and data visualization. Statistics were performed using the Chi-Square and Fisher’s Exact tests, respectively. The statistical analysis was performed by software IBM SPSS version 24.0. A p-value less than 0.05 was considered as statistically significant. Figures were made with R version 4.0 and GraphPad Prism version 9.5.0.
3. Results
3.1. Study Population
This study analyzed data from 269 patients with pulmonary hypertension (PH) registered in the MUMC+ PH database. Of these, 121 patients met the inclusion criteria and underwent myositis blot testing (Figure 1). The study cohort (n=121) had a median age of 68 years (61.5-73.0) and was 57.9% female. Patients were classified according to the World Health Organization (WHO) system: 55 (45.5%) as WHO type 1 PAH, 9 (7.4%) as WHO type 2 (underlying heart disease with significant pre-capillary PH component), 35 (28.9%) as WHO type 3 (underlying pulmonary disease), 18 (14.9%) as WHO type 4 CTEPH, and 4 (3.3%) as WHO type 5 (multifactorial etiologies). The median follow-up duration was 4.2 years (2.2-6.0). Baseline characteristics for this cohort are summarized in Table 1.
Figure 1.
Flow chart for inclusion and exclusion of PH patients. PAH, pulmonary arterial hypertension; LFU, lost to follow up; PVR, pulmonary vascular resistance; WU, wood units; IIM, idiopathic inflammatory myopathy; PPH, porto-pulmonary hypertension; CHD, congenital heart disease; MSA, myositis specific antibodies; MAA, myositis associated antibodies.
Figure 1.
Flow chart for inclusion and exclusion of PH patients. PAH, pulmonary arterial hypertension; LFU, lost to follow up; PVR, pulmonary vascular resistance; WU, wood units; IIM, idiopathic inflammatory myopathy; PPH, porto-pulmonary hypertension; CHD, congenital heart disease; MSA, myositis specific antibodies; MAA, myositis associated antibodies.
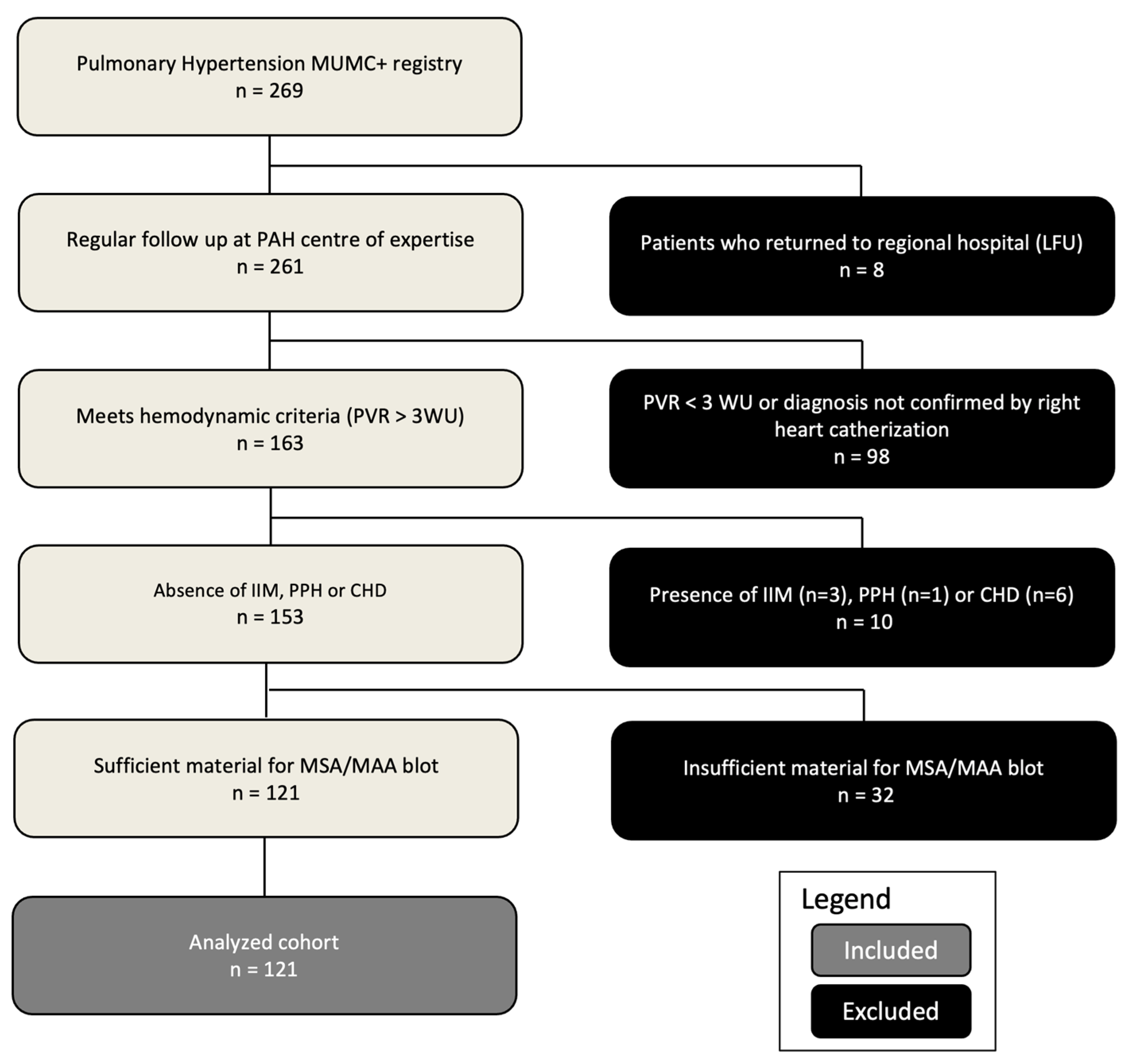

Table 1.
Demographic, clinical characteristics and laboratory features of the study population.

Count (percentage), mean ± standard deviation, or median (interquartile range) were given, as appropriate. P-values were calculated using Chi-square / Fisher’s exact test, analysis of variance, or Kruskal-Wallis test, respectively. ILD, interstitial lung disease (stratification based on radiological imaging); BMI, body mass index; NYHA, New-York Heart Association; 6MWT, six-minute walking test; NT-pro BNP, N-terminal pro brain natriuretic peptide; CRP, C-reactive protein; mPAP, mean pulmonary arterial pressure; PVR, pulmonary vascular resistance; FEV%, forced expiratory volume %; FVC%, forced vital capacity %; TLC%, total lung capacity %;KCO, corrected carbon mono-oxide transfer coefficient; WHO, world health organization; MCTD, mixed connective tissue disease.
3.2. Cardiopulmonary Characteristics
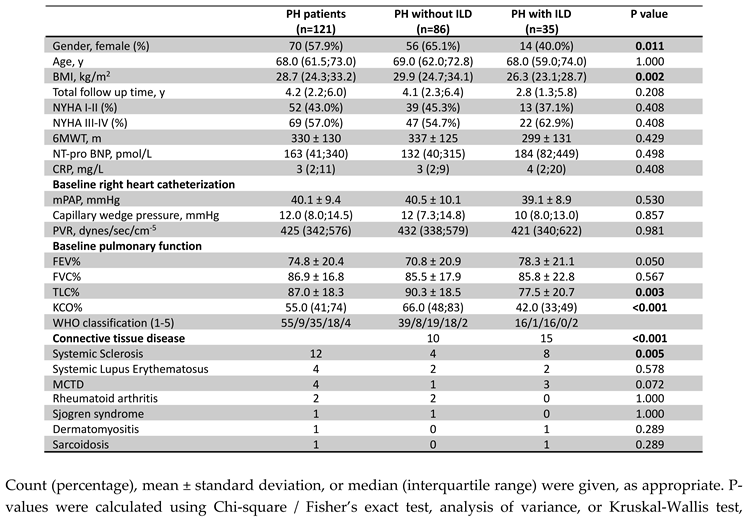
Baseline hemodynamics confirmed PH, with a mean mPAP of 40.1 ± 9.4 mmHg. Median PAWP was 12 mmHg (8-14.5 mmHg), and median PVR was 425 dynes/sec/cm-5 (342-576 dynes/sec/cm-5). Exercise capacity was impaired, with a mean 6-minute walk distance (6MWD) of 330 ± 130 m. Most patients (57%) were in NYHA functional class III/IV. Table 1 provides additional cardiac and hemodynamic data. Pulmonary function testing revealed a mean forced expiratory volume in one second (FEV1) of 74.8% ± 20.4% predicted, a mean forced vital capacity (FVC) of 86.9% ± 16.8% predicted, and a mean total lung capacity (TLC) of 87.0% ± 18.3% predicted. The median diffusing capacity of the lungs for carbon monoxide (KCO) was reduced at 55% predicted (41-74%). Significant differences in TLC and KCO were observed between PH patients with and without ILD on imaging (p=0.003, p<0.001, Table 1). Radiological imaging revealed interstitial changes in 35 patients (38.9%). The distribution of HRCT ILD patterns is shown in Table 2. Systemic sclerosis was five times more prevalent in the PH with ILD group (p=0.005).
Table 2.
ILD patterns on HRCT in PH patients (n=35).
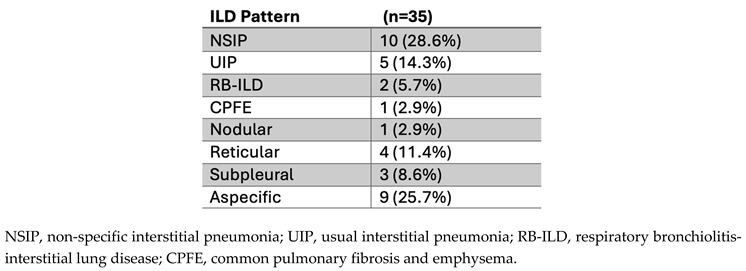
3.3. Prevalence of Myositis-Specific Antibodies (MSA) and Myositis-Associated Antibodies (MAA) in PH Patients
In the PH cohort (n=121), 16 patients (13.2%) had at least one positive MSA/MAA, while 26 patients (21.5%) had no positive but at least one weak positive MSA/MAA (Table 3). In this cohort in total 17 positive and 44 weak positive reactivities were observed (Table 4). Distribution of single and multiple MSA/MAA (weak)positivity is depicted in Figure 2.
We compared total MSA/MAA prevalence, grouped by associated clinical myositis syndromes, to a cohort of suspected IIM/ILD patients. This revealed a significantly higher prevalence of total weak positive (36.4%, n/N=44/121) MSA/MAAs in the PH cohort when compared with weak positive MSA/MAAs (19.2%, n/N=107/558, p<0.001) in the suspected IIM/ILD cohort (Figure 2a). No significant difference was observed when the prevalence of positive MSA/MAAs in the PH cohort (14.0%, n/N=17/121) was compared with the suspected IIM/ILD cohort (9.3%, n/N=52/558, p=0.222) (Figure 2a).
Specifically, the PH cohort did not demonstrate a significant difference in the prevalence of positive anti-synthetase syndrome-associated MSAs (5.0%, n/N=6/121 vs. 2.9%, n/N=16/558, p=0.25) in suspected IIM/ILD). However, weak positive anti-synthetase syndrome-associated MSAs were significantly more prevalent in the PH cohort (14.0%, n/N=17/121 vs. 6.8%, n/N=38/558 in suspected IIM/ILD, p=0.008) (Figure 2b, c). Positive dermatomyositis-associated MSAs were less prevalent in the PH cohort (0.8%, n/N=1/121) compared to the suspected IIM/ILD cohort (4.5%, n/N=25/558, p=0.058). However, weak positive dermatomyositis-associated MSA/MAAs were more prevalent in the PH cohort (14.9%, n/N=18/121 vs. 6.8%, n/N=38/558 in suspected IIM/ILD, p=0.011) (Figure 2b, c). The prevalence of positive overlap syndrome-associated MSA/MAAs was significantly higher in the PH cohort (7.4%, n/N=9/121) in comparison with the suspected IIM/ILD cohort (1.8%, n/N=10/558, p<0.001). No significant difference was observed in the prevalence of weak positive overlap syndrome-associated MSA/MAAs (3.3%, n/N=4/121 in PH vs. 3.6%, n/N= 20/558 in suspected IIM/ILD, p=0.783) (Figure 2b, c).
Figure 2.
Distribution of single and multiple (weak) positivity of MSA and MAA antibodies in PH patients (n=121).
Figure 2.
Distribution of single and multiple (weak) positivity of MSA and MAA antibodies in PH patients (n=121).
Table 3.
Distribution of single and multiple (weak) positivity of MSA and MAA antibodies in PH patients with or without ILD.
Table 3.
Distribution of single and multiple (weak) positivity of MSA and MAA antibodies in PH patients with or without ILD.
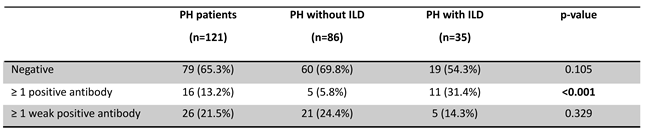
Table 4.
Prevalence of MMA/MSA antibodies in PH patients (n=121).
- Prevalence of myositis specific antibodies (MSA) and myositis associated antibodies (MAA) in PH patients with or without radiological signs of ILD
We stratified the PH cohort based on the presence or absence of interstitial changes on imaging and MSA/MAA findings (Table 3). The PH with ILD group had a significantly higher prevalence of patients with at least one positive MSA/MAA (n/N=11/35; 31.4%) compared to the PH without ILD group (n/N=5/86; 5.8%, p<0.001). The prevalence of patients with >1 weak positive, but no positive, MSA/MAA did not differ significantly between the two groups. See Table 4 and Appendix for multi-antibody positivity, distribution, antigen specificity, and antibody prevalence per patient (Table A1).
The total prevalence of positive MSA/MAAs was significantly higher in the PH with ILD cohort (34.3%, n/N=12/35) compared to the PH without ILD cohort (5.8%, n/N=5/86, p<0.001) (Figure 3d). This was also significantly higher when compared to the suspected IIM/ILD cohort (9.3%, n/N=52/558, p<0.001). The total prevalence of weak positive MSA/MAAs was significantly higher in the PH without ILD cohort (43.0%, n/N=37/86) compared to the PH with ILD cohort (20.0%, n/N=7/35, p=0.005) (Figure 3d).
Figure 3.
Percentages of positive and weak positive antibodies with MSA/MAA grouped by association with disease in the PH with and without ILD and the suspected IIM/ILD cohort. a) Total positive and weak positive MSA/MAA in the PH cohort (n=121) and suspected IIM/ILD patients (n=558). b) positive MSA/MAA in PH and suspected IIM/ILD patients. c) weak positive MSA/MAA in PH and suspected IIM/ILD patients. d) Total positive and weak positive MSA/MAA in PH with (n=81) / without (n=35) ILD patients. e) positive MSA/MAA in PH with/without ILD patients f) weak positive MSA/MAA in PH with/without IL patients. MSA/MAA were grouped as follows: anti-synthetase (Jo1, OJ, PL7, PL12, EJ), dermatomyositis (Mi2, NXP2, SAE1, MDA5, Tif1y), CTD overlap syndrome (Ku, Pm/Scl). Significance is depicted with * (<0.05), ** (<0.02) and ***(<0.001).
Figure 3.
Percentages of positive and weak positive antibodies with MSA/MAA grouped by association with disease in the PH with and without ILD and the suspected IIM/ILD cohort. a) Total positive and weak positive MSA/MAA in the PH cohort (n=121) and suspected IIM/ILD patients (n=558). b) positive MSA/MAA in PH and suspected IIM/ILD patients. c) weak positive MSA/MAA in PH and suspected IIM/ILD patients. d) Total positive and weak positive MSA/MAA in PH with (n=81) / without (n=35) ILD patients. e) positive MSA/MAA in PH with/without ILD patients f) weak positive MSA/MAA in PH with/without IL patients. MSA/MAA were grouped as follows: anti-synthetase (Jo1, OJ, PL7, PL12, EJ), dermatomyositis (Mi2, NXP2, SAE1, MDA5, Tif1y), CTD overlap syndrome (Ku, Pm/Scl). Significance is depicted with * (<0.05), ** (<0.02) and ***(<0.001).
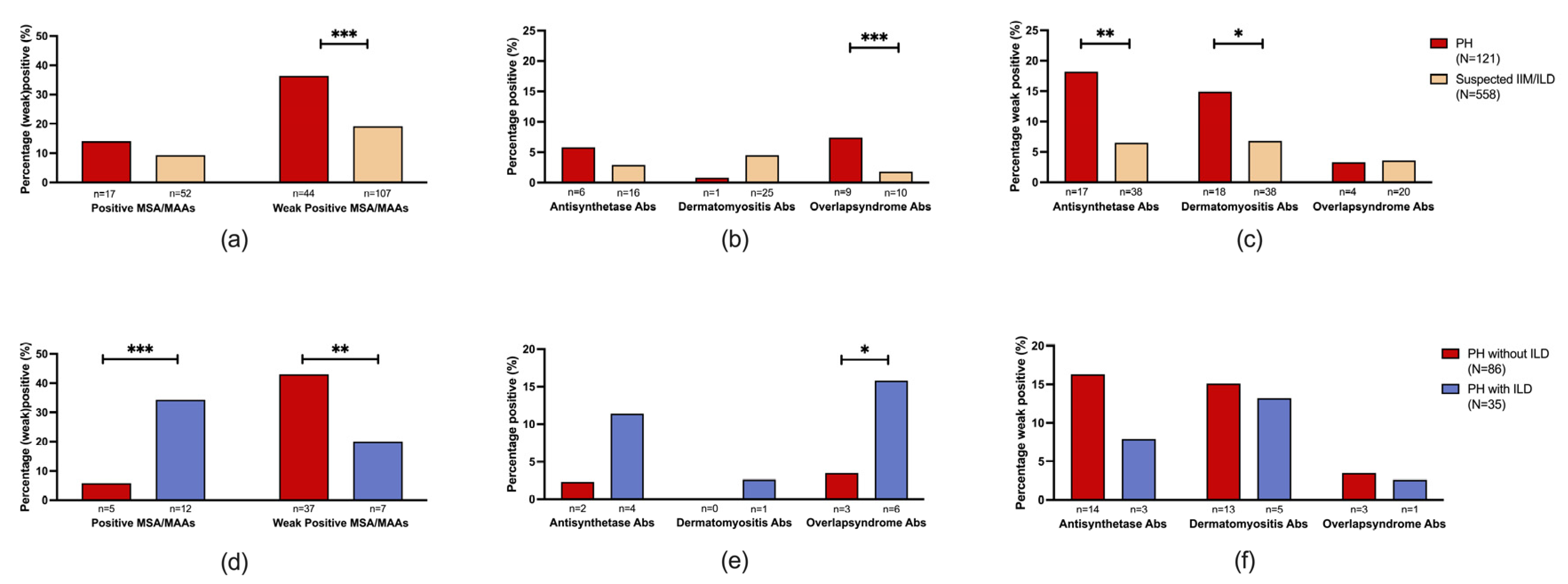
No significant difference was observed when the prevalence of positive anti-synthetase syndrome-associated MSAs was compared between the PH with and without ILD patients. Positive overlap syndrome-associated MAAs were significantly more prevalent in the PH with ILD cohort (17.1%, n/N=6/35) compared to the PH without ILD cohort (3.5%, n/N=3/86, p=0.017) (Figure 3e). No significant difference was observed when the prevalence of weak positive anti-synthetase syndrome-associated, dermatomyositis-associated, and overlap syndrome-associated antibodies were compared between PH patients with or without ILD.
4. Discussion
This is the first study to systematically assess MSA/MAA prevalence in a large PH cohort. We observed a significantly higher prevalence of positive MSA/MAAs in PH patients with radiological signs of ILD and weak positive MSA/MAAs in PH patients without radiological signs of ILD compared to a cohort with suspected IIM/ILD, who were specifically tested for the presence of these autoantibodies. Notably, MSA/MAAs were detected in PH patients both with and without interstitial abnormalities on HRCT, although positivity tended to be weaker in the absence of radiographic ILD. We employed established nomenclature for clinical associations, with anti-synthetase and anti-overlap syndrome antibodies being the most frequently detected [20]. Importantly, patients in this study were referred to our PH clinic without prior diagnosis or treatment for immune-mediated inflammatory myopathies or ILD and lacked signs of myositis at presentation. These findings suggest that PH should be considered within the diagnostic criteria for MSA/MAA-associated diseases.
Previous studies report varying MSA/MAA prevalence in healthy controls. Ghirardello et al. showed a prevalence of 28.6% of MSA/MAA in healthy controls [23]. However, Ro52 was included and was positive in 73.4% of the MSA/MAA positive cases. Besides, assay results were positive ≥11 AU. Vulsteke et al. showed MSA/MAA positivity of 3/40 (7.5%) in blood donors; however, cut-off values were not mentioned [33]. Espinosa et al. showed MSA/MAA positivity of 3/60 (5%) in healthy controls; however also, cut-off values of ≥11AU were used [34]. Prior studies in IIM or ILD patients demonstrate a variable MSA/MAA prevalence. One study showed a prevalence of 27% MSA/MAA positivity in ILD patients. However, 36% of these patients were anti-Ro52 positive [24]. Others showed a prevalence of MSA/MAAs in IIM of around 50% [33]. Remarkably, the cut-off value for the positivity of MSA/MAAs was ≥11 AU in that study. Our study employed stricter criteria in which we excluded Ro52 and used a higher cut-off of ≥26 AU, resulting in a potentially more specific assessment of MSA/MAA positivity. This lack of standardization complicates direct comparisons with our findings. To address this, we compared our PH cohort to MSA/MAA line blot results from patients suspected of IIM (n=558) tested at our hospital. This internal control group allows for meaningful comparisons, as the same assay and criteria were used. The significantly higher prevalence of MSA/MAAs in our PH cohort suggests an association between these antibodies and PH.
The association of idiopathic inflammatory myopathies, anti-synthetase syndrome and PH has been established in the literature before [22,27,28,29,35]. However, in most cases, a diagnosis of active myositis and often severe ILD was found. Patients in our cohort had no signs of active myositis or severe end-stage ILD. Although the prevalence of MSA/MAAs was highest in the PH with ILD group, most patients in our cohort positive for MSA/MAAs showed, to some extent, interstitial pulmonary disease on radiographical imaging. However, these patients often did not present with a classified ILD diagnosis prior to PH diagnosis. Interestingly, even the patients without interstitial pulmonary involvement did also show, however weak, positivity of MSA/MAAs. If the cut-off values used in other studies were applied, these patients would have been classified as positive. When we compared pulmonary voluminal measurements between patients with positive MSA/MAAs and patients with weak antibody positivity, no significant difference was observed (Figure A1). Moreover, since the ILD observed was classified as only moderately mild based on pulmonary functional testing, the elevated pulmonary arterial pressure could not be explained directly by interstitial changes alone [36]. This is also known as out-of-proportion PAH, which could suggest that PH in these patients might have developed due to immune-mediated vascular remodeling. Notably, our findings suggest that in the PH patients with radiological signs of ILD who tested positive for MSA/MAA, a retrospective diagnosis of IIM should be considered even without overt clinical myositis. This highlights the importance of screening for MSA/MAA in PH, especially in the presence of mild radiological signs of ILD, as IIM prevalence may be underestimated in this population. This study has limitations inherent to its retrospective design, including potential selection bias and confounding factors. The single-center nature limits generalizability. This study opted for a disease-control group rather than a healthy-control group due to concerns that MSA/MAA levels detected in healthy individuals with no apparent illness might partially reflect assay limitations or subclinical immune processes. ILD diagnosis in this study relied on HRCT evaluation by different radiologists. This approach may introduce bias since HRCT interpretation can be subjective, particularly for subtle or early-stage ILD.
Until now, PH associated with MSA/MAAs has been merely interpreted as a complication of severe or end-stage ILD in IIM patients [29]. However, this study provides new immunological insights into pre-capillary and combined pre-post capillary PH associated with MSA/MAAs, showing a high prevalence of (weak) positive MSA/MAAs with and without interstitial pulmonary disease. The progressive nature of immune-mediated diseases, with their shifting pathophysiological mechanisms, underscores the importance of early recognition of autoimmunity in PH. PH patients with lower MSA/MAA titers without overt interstitial changes may represent an earlier disease stage. We propose a case series with longitudinal follow-up of pulmonary function and imaging in patients presenting with (weak) MSA/MAA positivity and PH. This study could elucidate disease progression and further investigate the role of immunosuppressive treatment in PH. Based on our findings, we strongly advocate for routine MSA/MAA screening in newly diagnosed PH patients, especially in those with (mild) radiological signs of ILD. Integrating MSA/MAA positivity into screening tools, such as the one validated by Parikh et al. [37,38] for PH in ILD, could enhance early detection. Future research should also focus on elucidating the fundamental immunological mechanisms associated with MSA/MAAs and their role in vascular remodeling in pre-capillary and combined pre- and post-capillary PH patients, to substantiate immunological targeted therapy that could help prevent further progression of vascular remodeling into end-stage PH.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Cardiopulmonary hemodynamic and pulmonary functional measurements and 5-year survival in PH patients with positive, weak positive and negative MSA/MAAs.; Table S1: Clinical characteristics and MSA/MAA reactivity per patient.
Author Contributions
Conceptualization, R.T. and J.D.; methodology, R.T and J.D.; formal analysis, R.T.and D.vD.; investigation, R.T.; data curation, R.T. and J.P.; writing—original draft preparation, R.T.; writing—review and editing, R.T., J.P., J.D., V.vE. and P.vP.; visualization, R.T.; supervision, J.D and P.vP.; project administration, P.vP.; funding acquisition, P.vP. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of AzM/UM protocol code 2021-3010, approval date 7 November 2021 and NL57351.068.17/METC 172021 approval date 1 November 2017.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study
Data Availability Statement
All data that has been used for this article has been provided within this article. Additional data can be downloaded as a supplementary file.
Acknowledgments
We like to thank Caroline Bijnens (Central Diagnostic Laboratory, Maastricht University Medical Center, Maastricht, The Netherlands) for her laboratory assistance and performance of the line-blot assay. We also like to thank Lisa Kalisvaart for the data acquisition and the development of the suspected IIM/ILD cohort.
Conflicts of Interest
All authors declare no conflict of interest.
Appendix A
Figure A1.
Cardiopulmonary hemodynamic and pulmonary functional measurements and 5 year survival in PH patients with positive, weak positive and negative MSA/MAAs. A) mean pulmonary arterial pressure in mmHg, B) pulmonary vascular resistance in dynes/sec/cm-5, C) forced expiratory volume as percentage of predicted, D) total lung capacity as percentage of predicted, E) forced vital capacity as percentage of predicted, F) carbon oxide transfer coefficient as percentage of predicted, G) C-reactive protein in mg/L, H) 5 year survival of pulmonary hypertension patients stratified according to MSA/MAA positive, weak positive or negative results. Horizontal bars represent either median values with interquartile range or mean with standard deviation according to the normality test. Statistical analysis was performed using the Kruskal-Wallis and Mann-Whitney test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Survival was analyzed using the Gehan-Breslow-Wilcoxon test.
Figure A1.
Cardiopulmonary hemodynamic and pulmonary functional measurements and 5 year survival in PH patients with positive, weak positive and negative MSA/MAAs. A) mean pulmonary arterial pressure in mmHg, B) pulmonary vascular resistance in dynes/sec/cm-5, C) forced expiratory volume as percentage of predicted, D) total lung capacity as percentage of predicted, E) forced vital capacity as percentage of predicted, F) carbon oxide transfer coefficient as percentage of predicted, G) C-reactive protein in mg/L, H) 5 year survival of pulmonary hypertension patients stratified according to MSA/MAA positive, weak positive or negative results. Horizontal bars represent either median values with interquartile range or mean with standard deviation according to the normality test. Statistical analysis was performed using the Kruskal-Wallis and Mann-Whitney test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Survival was analyzed using the Gehan-Breslow-Wilcoxon test.
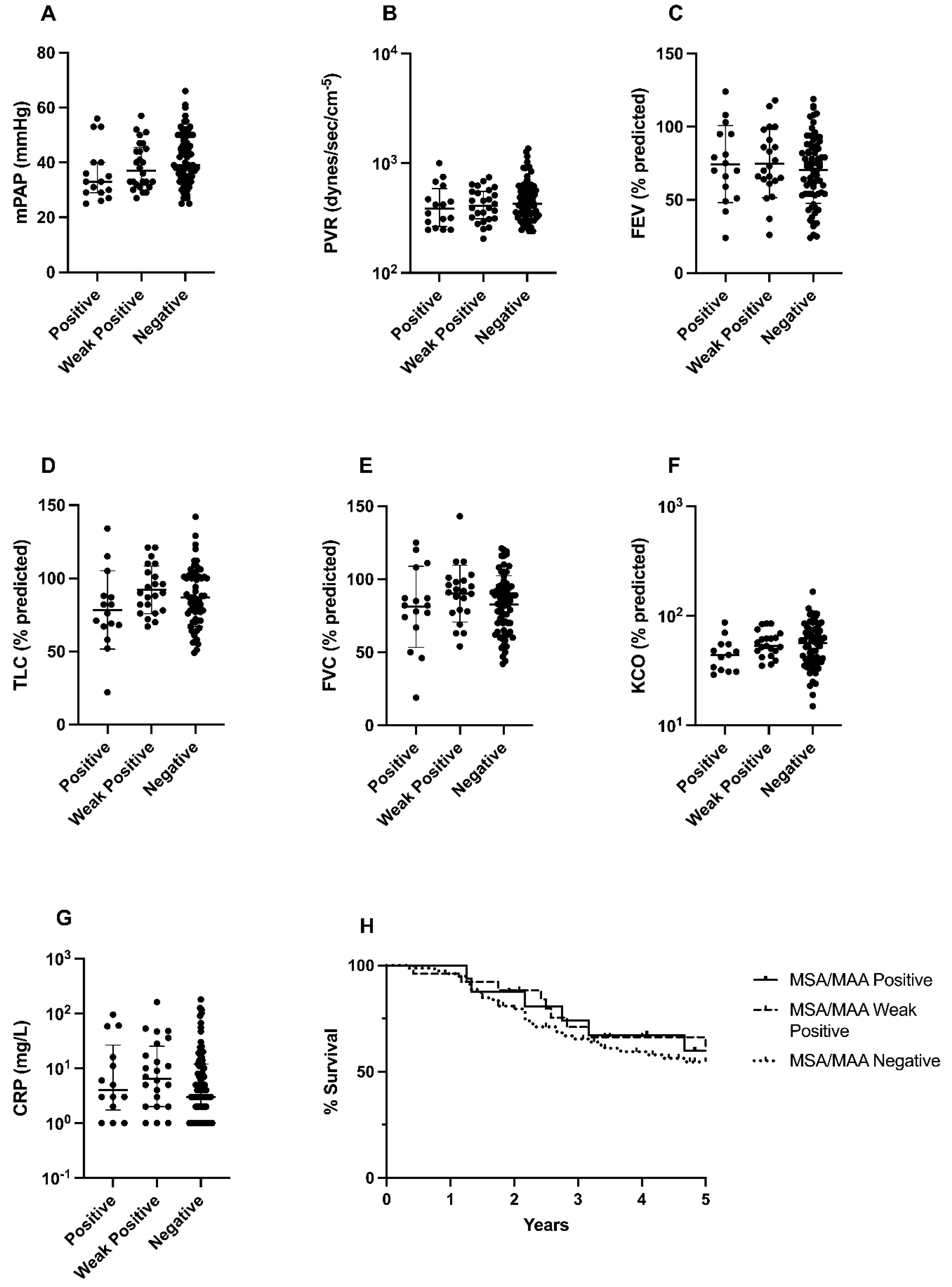
Table A1.
Clinical characteristics and MSA/MAA positivity per patient.
| Patient ID | Sex m/f | Age at inclusion | PH WHO type | Radiological signs of ILD y/n | MMA/MSA |
| 1 | 0 | 64 | 1 | y | neg |
| 2 | 0 | 73 | 4 | n | wpos Jo1 |
| 3 | 1 | 56 | 5 | y | pos PM-Scl |
| 4 | 0 | 54 | 1 | y | pos PM-Scl |
| 5 | 0 | 59 | 3 | y | neg |
| 6 | 0 | 77 | 2 | n | pos EJ |
| 7 | 0 | 65 | 1 | y | neg |
| 8 | 0 | 59 | 1 | y | pos Jo1 |
| 9 | 1 | 82 | 2 | n | neg |
| 10 | 1 | 78 | 3 | y | pos PM-Scl |
| 12 | 0 | 75 | 3 | y | neg |
| 13 | 1 | 56 | 1 | y | neg |
| 15 | 1 | 77 | 3 | y | neg |
| 17 | 1 | 67 | 1 | n | neg |
| 18 | 1 | 53 | 1 | n | neg |
| 20 | 1 | 60 | 5 | y | wpos OJ, wpos Mi2 |
| 21 | 0 | 44 | 1 | n | neg |
| 23 | 0 | 79 | 1 | n | neg |
| 24 | 0 | 61 | 1 | n | neg |
| 26 | 0 | 66 | 4 | n | neg |
| 27 | 0 | 71 | 1 | n | wpos PL7 wpos MDA5 |
| 29 | 0 | 66 | 2 | n | wpos PL7 |
| 30 | 0 | 69 | 4 | n | wpos MDA5 |
| 31 | 0 | 63 | 1 | y | neg |
| 32 | 1 | 70 | 3 | n | neg |
| 33 | 0 | 75 | 3 | n | neg |
| 37 | 1 | 73 | 4 | n | neg |
| 38 | 1 | 65 | 3 | y | neg |
| 39 | 0 | 75 | 2 | n | wpos SRP1 wpos PL12 wpos Mi2 wpos MDA5 wpos Ku |
| 40 | 0 | 69 | 4 | n | pos PM-Scl wpos Mi2 |
| 41 | 1 | 75 | 4 | n | wpos PL7 |
| 43 | 0 | 68 | 1 | y | pos Ku wpos SAE1 wpos MDA5 |
| 46 | 0 | 66 | 1 | n | neg |
| 47 | 0 | 60 | 1 | n | neg |
| 50 | 0 | 59 | 3 | n | wpos SRP1 |
| 52 | 0 | 74 | 1 | n | neg |
| 53 | 0 | 69 | 1 | n | neg |
| 54 | 1 | 57 | 5 | n | wpos MDA5 |
| 56 | 1 | 67 | 1 | y | neg |
| 57 | 0 | 74 | 1 | y | pos PM-Scl |
| 64 | 0 | 59 | 1 | n | neg |
| 66 | 0 | 73 | 3 | y | pos OJ, |
| 67 | 0 | 80 | 2 | y | wpos Mi2 |
| 69 | 1 | 66 | 3 | n | neg |
| 70 | 0 | 62 | 5 | n | neg |
| 73 | 0 | 72 | 1 | n | wpos MDA5 |
| 78 | 1 | 40 | 1 | n | neg |
| 81 | 1 | 75 | 3 | y | neg |
| 83 | 0 | 64 | 4 | n | neg |
| 85 | 1 | 80 | 4 | n | neg |
| 86 | 0 | 65 | 3 | n | neg |
| 87 | 1 | 60 | 1 | y | neg |
| 88 | 0 | 75 | 1 | n | neg |
| 91 | 0 | 63 | 3 | n | neg |
| 93 | 1 | 68 | 3 | y | pos SRP1 |
| 94 | 0 | 75 | 2 | n | wpos SRP1 wpos PL7 |
| 95 | 1 | 77 | 3 | n | wpos OJ |
| 103 | 0 | 79 | 1 | n | wpos PL7 |
| 104 | 0 | 56 | 4 | n | neg |
| 106 | 0 | 64 | 1 | n | neg |
| 107 | 1 | 79 | 3 | n | pos Ku wpos PL7 |
| 108 | 1 | 50 | 1 | y | wpos Ku |
| 111 | 0 | 47 | 1 | n | neg |
| 119 | 0 | 70 | 4 | n | wpos PL7 wpos PM-Scl |
| 120 | 1 | 46 | 1 | n | wpos PL7 |
| 124 | 1 | 72 | 2 | n | neg |
| 126 | 1 | 39 | 1 | y | neg |
| 127 | 0 | 87 | 2 | n | pos PL7 wpos PM-Scl |
| 134 | 0 | 66 | 1 | n | wpos MDA5 |
| 141 | 0 | 66 | 4 | n | neg |
| 162 | 1 | 89 | 3 | n | neg |
| 164 | 1 | 23 | 4 | n | neg |
| 165 | 0 | 71 | 1 | n | pos PM-Scl |
| 168 | 1 | 76 | 3 | y | neg |
| 175 | 1 | 68 | 3 | y | neg |
| 181 | 0 | 81 | 4 | n | neg |
| 186 | 1 | 71 | 3 | y | neg |
| 191 | 0 | 73 | 1 | n | neg |
| 194 | 0 | 63 | 1 | n | neg |
| 195 | 1 | 72 | 1 | y | neg |
| 197 | 0 | 46 | 1 | n | neg |
| 198 | 0 | 66 | 1 | n | neg |
| 202 | 0 | 27 | 1 | n | neg |
| 205 | 0 | 71 | 1 | n | neg |
| 208 | 0 | 59 | 3 | y | neg |
| 213 | 0 | 63 | 1 | n | neg |
| 217 | 0 | 70 | 3 | n | neg |
| 220 | 1 | 75 | 3 | y | neg |
| 223 | 0 | 71 | 3 | n | neg |
| 228 | 0 | 70 | 3 | n | neg |
| 233 | 0 | 70 | 3 | n | neg |
| 246 | 0 | 64 | 1 | y | pos PL7 pos Ku |
| 250 | 1 | 72 | 1 | n | neg |
| 254 | 1 | 71 | 4 | n | neg |
| 271 | 1 | 74 | 3 | n | neg |
| 273 | 0 | 46 | 1 | n | wpos SAE1 |
| 279 | 1 | 57 | 1 | n | neg |
| 285 | 0 | 74 | 3 | n | neg |
| 293 | 1 | 78 | 3 | n | neg |
| 301 | 1 | 72 | 3 | y | wpos PL7 |
| 306 | 1 | 66 | 4 | n | neg |
| 309 | 1 | 55 | 2 | n | neg |
| 310 | 1 | 73 | 1 | y | pos Mi2, wpos PL12 |
| 311 | 0 | 71 | 1 | n | neg |
| 312 | 0 | 66 | 4 | n | wpos PL7 |
| 313 | 1 | 74 | 1 | n | neg |
| 314 | 1 | 75 | 3 | y | neg |
| 315 | 0 | 49 | 3 | n | neg |
| 316 | 1 | 57 | 1 | n | wpos Tif1y |
| 317 | 1 | 68 | 3 | y | wpos SAE1 wpos MDA5 |
| 318 | 0 | 69 | 4 | n | wpos SAE1 |
| 319 | 1 | 72 | 1 | n | wSAE1 |
| 320 | 0 | 63 | 1 | n | neg |
| 321 | 0 | 50 | 1 | y | pos PL7 |
| 322 | 1 | 73 | 1 | n | neg |
| 323 | 1 | 57 | 1 | n | neg |
| 324 | 0 | 64 | 3 | n | neg |
| 325 | 0 | 53 | 1 | n | neg |
| 326 | 1 | 65 | 4 | n | neg |
|
327 |
0 | 62 | 1 | n | wpos SRP1 wpos PL7 wpos PL12 wpos Mi-2 |
| 328 | 1 | 69 | 3 | n | neg |
References
- Chang KY, Duval S, et al. Mortality in Pulmonary Arterial Hypertension in the Modern Era: Early Insights From the Pulmonary Hypertension Association Registry. J Am Heart Assoc. 2022;11(9):e024969. [CrossRef]
- Humbert M, Kovacs G, et al. [2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension]. G Ital Cardiol (Rome). 2023;24(4):1e-116e. [CrossRef]
- Tobal R, Potjewijd J, et al. Vascular Remodeling in Pulmonary Arterial Hypertension: The Potential Involvement of Innate and Adaptive Immunity. Front Med (Lausanne). 2021;8:806899. [CrossRef]
- Thoreau B, Mouthon L. Pulmonary arterial hypertension associated with connective tissue diseases (CTD-PAH): Recent and advanced data. Autoimmun Rev. 2023:103506. [CrossRef]
- Dhala A. Pulmonary arterial hypertension in systemic lupus erythematosus: current status and future direction. Clin Dev Immunol. 2012;2012:854941. [CrossRef]
- Parperis K, Velidakis N, Khattab E, Gkougkoudi E, Kadoglou NPE. Systemic Lupus Erythematosus and Pulmonary Hypertension. Int J Mol Sci. 2023;24(6). [CrossRef]
- Naranjo M, Hassoun PM. Systemic Sclerosis-Associated Pulmonary Hypertension: Spectrum and Impact. Diagnostics (Basel). 2021;11(5). [CrossRef]
- Arends SJ, Damoiseaux JG, et al. Functional implications of IgG anti-endothelial cell antibodies in pulmonary arterial hypertension. Autoimmunity. 2013;46(7):463-70. [CrossRef]
- Koudstaal T, Boomars KA. Inflammatory biomarkers in pulmonary arterial hypertension: ready for clinical implementation? Eur Respir J. 2023;61(3).
- Koudstaal T, Boomars KA, Kool M. Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective. J Clin Med. 2020;9(2). [CrossRef]
- Koudstaal T, van Uden D, et al. Plasma markers in pulmonary hypertension subgroups correlate with patient survival. Respir Res. 2021;22(1):137. [CrossRef]
- El Kasmi KC, Pugliese SC, et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J Immunol. 2014;193(2):597-609. [CrossRef]
- Heukels P, Corneth OBJ, et al. Loss of immune homeostasis in patients with idiopathic pulmonary arterial hypertension. Thorax. 2021;76(12):1209-18. [CrossRef]
- Price LC, Wort SJ, et al. Inflammation in pulmonary arterial hypertension. Chest. 2012;141(1):210-21. [CrossRef]
- Arends SJ, Damoiseaux JG, et al. Immunoglobulin G anti-endothelial cell antibodies: inducers of endothelial cell apoptosis in pulmonary arterial hypertension? Clin Exp Immunol. 2013;174(3):433-40.
- Arends SJ, Damoiseaux J, et al. Prevalence of anti-endothelial cell antibodies in idiopathic pulmonary arterial hypertension. Eur Respir J. 2010;35(4):923-5. [CrossRef]
- Stenmark KR, Nozik-Grayck E, et al. The adventitia: Essential role in pulmonary vascular remodeling. Compr Physiol. 2011;1(1):141-61.
- Marsh LM, Jandl K, et al. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2018;51(1). [CrossRef]
- Johnson SR, Granton JT. Pulmonary hypertension in systemic sclerosis and systemic lupus erythematosus. Eur Respir Rev. 2011;20(122):277-86.
- Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med. 2016;280(1):8-23. [CrossRef]
- Damoiseaux J, Mammen AL, Piette Y, Benveniste O, Allenbach Y, Group EtWS. 256(th) ENMC international workshop: Myositis specific and associated autoantibodies (MSA-ab): Amsterdam, The Netherlands, 8-10 October 2021. Neuromuscul Disord. 2022;32(7):594-608.
- Gasparotto M, Gatto M, Saccon F, Ghirardello A, Iaccarino L, Doria A. Pulmonary involvement in antisynthetase syndrome. Curr Opin Rheumatol. 2019;31(6):603-10. [CrossRef]
- Ghirardello A, Gatto M, et al. Detection of Myositis Autoantibodies by Multi-Analytic Immunoassays in a Large Multicenter Cohort of Patients with Definite Idiopathic Inflammatory Myopathies. Diagnostics (Basel). 2023;13(19). [CrossRef]
- Moll SA, Platenburg M, et al. Prevalence and clinical associations of myositis antibodies in a large cohort of interstitial lung diseases. PLoS One. 2022;17(11):e0277007. [CrossRef]
- Satoh M, Tanaka S, Ceribelli A, Calise SJ, Chan EK. A Comprehensive Overview on Myositis-Specific Antibodies: New and Old Biomarkers in Idiopathic Inflammatory Myopathy. Clin Rev Allergy Immunol. 2017;52(1):1-19. [CrossRef]
- . Foris V, Kovacs G, Matucci-Cerinic M, Olschewski H. PL-7 positive antisynthetase syndrome and pulmonary hypertension. J Rheumatol. 2013;40(10):1777-9. [CrossRef]
- Hervier B, Meyer A, et al. Pulmonary hypertension in antisynthetase syndrome: prevalence, aetiology and survival. Eur Respir J. 2013;42(5):1271-82. [CrossRef]
- Sanges S, Yelnik CM, et al. Pulmonary arterial hypertension in idiopathic inflammatory myopathies: Data from the French pulmonary hypertension registry and review of the literature. Medicine (Baltimore). 2016;95(39):e4911. [CrossRef]
- Lega JC, Reynaud Q, Belot A, Fabien N, Durieu I, Cottin V. Idiopathic inflammatory myopathies and the lung. Eur Respir Rev. 2015;24(136):216-38. [CrossRef]
- Ghigna MR, Mooi WJ, Grunberg K. Pulmonary hypertensive vasculopathy in parenchymal lung diseases and/or hypoxia: Number 1 in the Series "Pathology for the clinician" Edited by Peter Dorfmuller and Alberto Cavazza. Eur Respir Rev. 2017;26(144).
- Dhont S, Zwaenepoel B, Vandecasteele E, Brusselle G, De Pauw M. Pulmonary hypertension in interstitial lung disease: an area of unmet clinical need. ERJ Open Res. 2022;8(4). [CrossRef]
- Platteel ACM, Wevers BA, et al. Frequencies and clinical associations of myositis-related antibodies in The Netherlands: A one-year survey of all Dutch patients. J Transl Autoimmun. 2019;2:100013. [CrossRef]
- Vulsteke JB, De Langhe E, et al. Detection of myositis-specific antibodies. Ann Rheum Dis. 2019;78(1):e7.
- Espinosa-Ortega F, Holmqvist M, et al. Comparison of autoantibody specificities tested by a line blot assay and immunoprecipitation-based algorithm in patients with idiopathic inflammatory myopathies. Ann Rheum Dis. 2019;78(6):858-60. [CrossRef]
- Garcia-Fernandez A, Quezada-Loaiza CA, de la Puente-Bujidos C. Antisynthetase syndrome and pulmonary hypertension: report of two cases and review of the literature. Mod Rheumatol Case Rep. 2021;5(1):152-5. [CrossRef]
- Lopes AJ, Capone D, Mogami R, Lanzillotti RS, Melo PL, Jansen JM. Severity classification for idiopathic pulmonary fibrosis by using fuzzy logic. Clinics (Sao Paulo). 2011;66(6):1015-9. [CrossRef]
- Parikh R, Konstantinidis I, O'Sullivan DM, Farber HW. Pulmonary hypertension in patients with interstitial lung disease: a tool for early detection. Pulm Circ. 2022;12(4):e12141. [CrossRef]
- Parikh R, O'Sullivan DM, Farber HW. The PH-ILD Detection tool: External validation and use in patients with ILD. Pulm Circ. 2023;13(3):e12273. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



