Submitted:
13 May 2024
Posted:
14 May 2024
You are already at the latest version
Abstract
Bosentan, an endothelin-receptor antagonist (ERA), has potential anti-atherosclerotic properties. We investigated the complementary effects of bosentan and atorvastatin on the progression and composition of the atherosclerotic lesions in diabetic mice. Forty-eight male apoE-/- mice were fed high-fat diet (HFD) for 14 weeks. At week 8, diabetes was induced with streptozotocin and mice were randomized into 4 groups: 1) Control/COG: no intervention. 2) ΒΟG: bosentan 100 mg/kg/day per os. 3) ATG: atorvastatin 20mg/kg/day per os). 4) BO+ATG: Combined administration of bosentan and atorvastatin. The intra-plaque contents of collagen, elastin, monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor-a (TNF-a), matrix-metalloproteinases (MMP-2,-3,-9), and TIMP-1 concentrations were determined. The percentage of lumen stenosis significantly decreased across all treated groups: BOG: 19.5±2.2%, ATG: 12.8±4.8%, BO+ATG: 9.1±2.7% compared to controls (24.6±4.8%, p<0.001). Both atorvastatin and bosentan significantly increased collagen content and fibrous cap thickness versus COG (p<0.01). All intervention groups reduced the relative intra-plaque concentrations of MCP-1, MMP-3,-9, and increased TIMP-1 compared to COG (p<0.001). Importantly, bosentan showed modest but additive to atorvastatin effects on the latter parameters compared COG (p<0.05). Bosentan treatment in diabetic, atherosclerotic apoE-/- mice delayed the atherosclerosis progression and enhanced plaques’ stability, showing modest but complementary effects with atorvastatin, which are promising in atherosclerotic cardiovascular diseases.
Keywords:
endothelin-receptor antagonist (ERA)
; bosentan
; atorvastatin
; atherosclerosis
; plaque stability
; matrix metalloproteinases
1. Introduction
Τhe onset of endothelial dysfunction occurs prior to the formation of the atherosclerotic plaque. The latter process is multi-faceted including lipid core formation, macrophages, dendritic cells, and monocytes, as well as elements of the adaptive immune system, such as T cells, can be found [1,2]. Plaques which are prone to rupture are considered "vulnerable", leading to acute blockage of the artery, and clinically to either acute ischemic heart attack or stroke [3,4]. The co-existence of diabetes mellitus (DM) multiplies the risk for acute plaque destabilization and consequent adverse cardiovascular events [5,6]. On the other hand, statins have been long proved to exert “pleiotropic” actions and remain an essential part of the secondary prevention of atherosclerotic cardiovascular diseases (ASCVDs). Among their beneficial impact, they stabilize vulnerable atherosclerotic plaques by inhibiting the inflammatory response and attenuating lipid deposition [7,8,9].
The activation of the endothelin (ET) system is closely linked to various pathological conditions, encompassing a broad spectrum of cardiovascular diseases, like essential hypertension, atherosclerosis, arterial restenosis, heart failure, coronary vasospasm, myocardial infarction, and pulmonary hypertension [10]. Endothelin was first described by Yanagisawa in 1988 and is known for its potent vasoconstrictor properties [11]. There are three isoforms of endothelin, namely ET-1, ET-2, and ET-3, with ET-1 being the most abundant and relevant in the context of atherosclerosis [12]. ET-1 is a multisource derivative and exerts its effects by activating two distinct G protein-coupled receptors, ETA and ETB. Both receptors are present on vascular smooth muscle cells (VSMCs), leading to vasoconstriction. Additionally, the ETB receptor, which is also present on endothelial cells, mediates vasodilation by releasing nitric oxide (NO) or prostacyclin. ET-1 primarily acts in a paracrine manner, being secreted from endothelial cells towards the VSMCs of the arterial media [13]. In addition to myocardial and vascular fibrosis [14,15,16], ET further activates neutrophils, mast cells, and monocytes, influencing the production of a wide range of cytokines involved in the inflammatory cascade. Diabetic patients typically exhibit elevated levels of ET-1 [17] and endothelial dysfunction, regardless the presence of coronary artery disease (CAD) [18,19].
The pharmaceutical agent named bosentan acts as a dual endothelin receptor antagonist (ERA) against both ETA and ETB. It has gained a significant place in pulmonary arterial hypertension (PAH), to relax pulmonary vasculature and inhibit the proliferation of VSMCs, promoting a favorable remodeling of pulmonary arterioles. Bosentan alleviates the symptoms associated with PAH and improves the overall condition of those patients [20,21]. Up to now, there is a single experimental study documenting the potential anti-atherosclerotic effect of bosentan, using a similar to our study animal model of atherosclerosis [22]. The actions of bosentan on arterial wall and atherosclerosis unambiguously require more robust evidence [23].
In the present study, we aimed to evaluate the impact of bosentan on atherosclerotic plaque progression and vulnerability using a widely accepted animal model of diabetic atherosclerosis. Moreover, regarding the well-known atheroprotective effects of statins, we hypothesized the complementary to atorvastatin effects of bosentan on atherosclerosis progression.
2. Results
2.1. Body Weight and Biochemical Analysis
All mice appeared with weight gain throughout the study independent the group. Between groups comparison revealed no statistically significant difference in weight at the end of the study (p > 0.05). Similarly, there were non-significant differences in total triglycerides and glucose levels between all groups (p > 0.05). On the other hand, total cholesterol levels differed between groups at the end of the study. As expected, atorvastatin-treated mice (both ATG and BO+ATG) exhibited decreased total cholesterol levels compared to controls (p < 0.05), whereas BOG and COG showed equivalent levels of total cholesterol at the end of the study (758 ± 182 mg/dl vs 759 ± 131 mg/dl; p > 0.05) (Table 1).
2.2. Morphometry, Collagen, and Elastin
The effect of bosentan treatment on atherosclerotic lesion progression, alone or combined with atorvastatin, was evaluated by measuring luminal stenosis at the level of aortic valve. The degree of luminal stenosis was significantly less in all treatment groups in comparison to COG (p< 0.001). Although the atherosclerosis regression after bosentan administration was much less than atorvastatin, the combined treatment resulted in a further decrease in stenosis percentage than atorvastatin alone, with bosentan showing the weaker effect among others on (p=0.045). Measurements are depicted in Table 2 and Figure 1 includes representative images of the hematoxylin/eosin (H&E) staining.
Plaque stability was assessed by measuring collagen and elastin concentrations within the atherosclerotic plaques and fibrous cap thickness (Table 2). Compared to COG, sirius red staining revealed a statistically significant higher collagen content along intervention groups: BOG (p=0.023), ATG (p<0.001) and BO+ATG (p<0.001). Notably, atorvastatin yielded even greater increase in collagen content than bosentan (p<0.01). A similar pattern was observed for the fibrous cap, where all interventions significantly increased its thickness than controls and the atorvastatin-treated groups had even thicker fibrous cap over the bosentan-treated mice (ATG or BO+ATG vs BOG; p < 0.05). In case of elastin, atorvastatin treatment had the determinant role. In particular, bosentan did not significantly affect elastin levels compared to controls (p > 0.05). On the other hand, ATG and BO+ATG had considerably higher elastin levels than both BOG and COG (p<0.001) implicating that in the combined treatment atorvastatin administration drove the increase in elastin content. All morphometric data are presented in Table 2.
2.3. Immunohistochemistry
In comparison to COG all treated groups showed higher a-actin concentrations but only BOG and BO+ATG achieved statistically significant difference from controls (p < 0.05). The positive-stained area for macrophages decreased along BOG (p=0.015), ATG (p<0.001) and BO+ATG (p<0.001) groups compared to COG, respectively. Atorvastatin was more effective than bosentan to reduce macrophages contents within plaques. Overall, bosentan and atorvastatin showed additive effects on the reduction in macrophages concentration.
Atorvastatin treatment, led to a considerable decrease in MMP-2 concentrations compared to COG (ATG: 9.56 ± 1.21%, BO+ATG: 10.02 ± 2.17% vs COG: 18.12 ± 4.62%, p<0.001). Regarding that the reduction in MMP-2 after bosentan treatment was not significant than controls (15.24±3.54%, p=0.212), it seems that atorvastatin entirely affected MMP-2 in the group of combined treatment.
All treatment groups were associated with statistically significant lower concentrations of MMP-3 and MMP-9, while TIMP-1 was statistically increased compared to COG (p < 0.05). Bosentan showed complementary effects to atorvastatin in the above parameters, since its impact was significant but to a lesser extent than atorvastatin (p<0.05), while the changes after combined treatment were greater than each single therapy with either bosentan or atorvastatin.
Finally, the pro-inflammatory agents, TNF-a and MCP-1, presented a similar trend, with all interventions leading to significantly lower concentrations of these markers compared to COG (p<0.05). Both bosentan and atorvastatin single therapies considerably decreased TNF-a contents compared to controls (p=0.023, p<0.001, respectively), but atorvastatin to a greater extent explaining the absence of significant difference between BO+ATG and ATG. On the other hand, MCP-1 concentrations were equivalently lower after both bosentan and atorvastatin single therapies than control mice. Notably, their combination had complementary effects with further significant lowering of MCP-1 levels than all other groups (p<0.05) (see Figure 2 and Figure 3).
3. Discussion
To our knowledge this is the first experimental study demonstrating the beneficial effects of bosentan on diabetic atherosclerosis progression and vulnerability in a valid animal model. Moreover, this is the first time to combine an ERA with the established anti-atherosclerotic treatment of atorvastatin. Although the magnitude of atherosclerosis regression, fibrous cap thickening and the increase of collagen content within plaque was modest after bosentan administration, it achieved statistically significant level compared to the untreated mice. Bosentan significantly ameliorated MMP-3, MMP-9, TIMP-1, TNF-a and MCP-1 concentrations. Moreover, the addition of bosentan to atorvastatin administration significantly further reduced the intraplaque levels of MMP-3, MMP-9, TIMP-1 and MCP-1, implicating a complementary to statins favorable impact of bosentan on intraplaque inflammation and stability (p<0.05).
ET-1 represents the predominant isoform of endothelins, it exerts a paracrine and autocrine role and is associated with deleterious effects in the cardiovascular system [24]. It triggers vasoconstriction through the activation of ETA-receptors and has been involved in the development of atherosclerosis. Both ET-1 and its receptors appear to be overexpressed in atheromatous lesions found in animal models as well as in humans [25]. In animals fed a HFD, ET-1 and its receptors have been also located in endothelial cells, VSMCs and macrophages along different stages of atherosclerotic plaque progression [26,27]. A recent study using a valid animal model of atherosclerosis (ApoE-/- mice fed HFD) demonstrated reduced atherosclerosis burden after administration of an identical dose of bosentan [22]. Compared to that study, we confirmed the regression of atherosclerotic lesions in our animal model of diabetic atherosclerosis. Moreover, we comparatively evaluated the anti-atherosclerotic effect of bosentan and a statin member. The former had modest effect than atorvastatin in most examined parameters, which seemed unrelated to lipids homeostasis, since bosentan therapy left unaltered blood lipids in our study. The interplay of ET-1 with hyperlipidaemia has been mentioned in patients with dyslipidemia and higher levels of ET-1 [28]. However, there is no clear evidence supporting the hypolipidemic effects of ERA [29]. Both lipid-lowering and “pleiotropic” actions of statins have been associated with atherosclerosis regression in animal studies [30]. The induction of atherosclerosis regression after bosentan beyond lipid modification is resounding, but the extrapolation in clinical ASCVDs needs further and meticulous investigation.
In addition to atherosclerosis burden, we also assessed the complementary effects of bosentan to atorvastatin, on atherosclerosis composition in ApoE−/− mice with diabetic atherosclerosis. We confirmed that after 6 weeks of atorvastatin administration all connective tissue components of plaque stability were improved (collagen, elastin and fibrous cap thickness). To our knowledge, this is the first study demonstrating plaque stabilization after bosentan administration either as a single therapy or on the top of atorvastatin therapy. We concluded that bosentan contributed to a more resilient plaque structure based on collagen, fibrous cap thickness and VSMC content measurements. However, that effect was significantly smaller compared to the exaggerated amelioration of the same parameters culminated in plaque vulnerability in atorvastatin receivers. Nevertheless, the beneficial effect of bosentan on plaque stability in an additive manner may encourage its use not as a stand-alone therapy but as a complementary regimen to established anti-atherosclerotic therapies, like statins. The use of pharmaceutical combinations has become a popular therapeutic approach in clinical practice of ASCVDs, since it promotes pharmaceutical synergy, inhibits a wider spectrum of atherosclerotic mechanisms and magnifies their effectiveness [31,32].
MMPs and its inhibitor (TIMP-1) regulate the extracellular matrix (ECM) remodeling and inflammatory cells infiltration, determining atherosclerosis progression in diabetic condition [33]. The balance of proteolytic and anti-proteolytic enzymes (MMPs/TIMPs) has been long studied as a surrogate marker of plaque vulnerability [34]. To our knowledge, this is the first study demonstrating the favorable changes in MMP members, like MMP-3, MMP-9 and their inhibitor TIMP-1 within atherosclerotic lesions after bosentan therapy. Most importantly the combined therapy with atorvastatin exerted a complementary effect of those two medications on MMPs/TIMP-1 homeostasis. In particular, the addition of bosentan to atorvastatin-treated mice further ameliorated intra-plaques contents of MMP-3, MMP-9 and TIMP-1 and those changes paralleled the beneficial changes in collagen and fibrous cap thickness after combined therapy. Perhaps the downregulation of the proteolytic activity of MMPs/TIMP-1 in an additive pattern led to remarkably less degradation of collagen and elastin, enhancing the connective tissue scaffold within the atherosclerotic plaques. In addition to this, bosentan administration rather than atorvastatin triggered plaque infiltration by VSMCs, the predominant cellular sources of the ECM [35]. Our findings are of clinical relevance, since the alteration in MMP/TIMP balance has been associated with atherosclerotic plaque vulnerability and adverse cardiovascular events in clinical setting [36,37]. Hence, bosentan seems to possess the potential of reducing plaque vulnerability, even at a lower extent than atorvastatin, by shifting the balance between production and degradation of ECM towards increased intra-plaque collagen and elastin within the atherosclerotic lesions [38]. The proposed combination could be clinically implemented concerning statins as an integral part of ASCVDs and bosentan as a novel add-on therapy.
A previous study supported the anti-atherosclerotic effect of bosentan demonstrating its association with suppressed expression of pro-apoptotic agents and increased miRNA-21 expression in the aortic arch endothelium [22]. Our study provides a novel anti-atherosclerotic mechanism of bosentan regarding its anti-inflammatory effects. We and other investigators have demonstrated the strong contribution of inflammation to atherosclerosis vulnerability [8,39,40]. We also noticed a complementary pattern after bosentan and atorvastatin in the modulation of inflammatory mediators, focusing on TNF-a and MCP-1. Experimental and clinical data have supported the relationship of inflammation with atherosclerosis development and progression [41]. Previous studies have reported reduction of two multifunctional inflammatory agents, TNF-a and MCP-1 after statin administration [42,43]. A number of studies from a wide spectrum of ASCVDs have implicated the interaction between ET-1 and inflammation [44]. An increased expression of ET-1 receptors has been found in inflammatory cells (macrophages, T lymphocytes) and in the smooth muscle fibers of the vessels. It has been suggested that foam cells and T-lymphocytes regulate a switch in expression from ETA to ETB-receptors in vascular endothelial smooth muscle fibers [45]. However, only in vitro data have reported that bosentan has the potential of suppressing inflammation through the modification of inflammatory agents like TNF-a and MCP-1 in models simulating atherosclerosis [46,47]. In our study, single therapy with either bosentan or atorvastatin inhibited macrophages infiltration and paralleled the decreased macrophage-derived atherogenic cytokines, TNF-a and MCP-1, within plaques. The combination therapy seemed to precipitate further the suppression of TNF-a and MCP-1 and the consequent plaque stabilization with the highest, elastin, collagen and VSMCs contents and the lowest macrophages concentration among groups. Many pharmaceutical interventions have targeted plaque stabilization, but the promising supplementary clinical relevance of bosentan remains to be proved.
The present study had several limitations. Firstly, we used histochemistry-based measurements which provide a semi-quantified molecular concentration, but do not depict the absolute protein activity and its gene expression. Secondly, apoE-/- mice fed HFD is a valid animal model of atherosclerosis, but plaque rupture is rarely identified and thereby the estimation of plaque vulnerability is indirect. For this purpose, we and other investigators we used parameters associated with plaque texture Thirdly, we could not examine the interaction between bosentan and atorvastatin unless their combined effects on studied parameters. Finally, we did not measure ET-1 levels and their receptors within plaques which could directly depict the impact of bosentan on ET-1 homeostasis within plaques.
In conclusion, the combined treatment of bosentan and atorvastatin in diabetic atherosclerotic apoE-/- mice induced additive beneficial effects on plaque texture, favoring its stabilization. The amelioration of ECM content within plaques yielded to a more stable plaque phenotype, which was associated with a beneficial modification of MMP-3, -9, TIMP-1 and MCP-1. After comparative evaluation bosentan showed a modest effect but it could act as a supplement of atorvastatin. Our findings outline the potential clinical importance of pharmaceutical combinations in the management of established atherosclerosis. More studies are required to clarify the anti-atherosclerotic effects of bosentan.
4. Materials and Methods
4.1. Animal Model and Experimental Design
Forty-eight male C57BL/6J ApoE double knockout (ApoE-/- KO) mice, were hosted in the Institute of Biomedical Research, Academy of Athens. At the age of 8 weeks, all mice started feeding with a western-type high-fat diet (HFD - Harlan, Teklad; 88137) for 14 weeks, in order to develop atherosclerotic lesions. Diabetes was induced after 8 weeks from the beginning of the study by intra-peritoneal injections of streptozotocin (STZ) for 5 consecutive days (0.05 mg/g body weight in 0.05 mol/L citrate buffer, pH 4.5). After STZ injections, mice maintaining fasting glucose levels >200 mg/dL were considered diabetic and were included in the study analysis. This is a valid animal model for diabetic atherosclerosis development which we have previously used in pharmaceutical studies [48,49]. After diabetes induction (8th week of experiment), mice continued HFD and were randomized into the following four groups (n=12) for the last 6 weeks of the study period:
(1) Control group (COG): Normal saline was administered every day by esophageal gavage to make all interventions comparable between groups.
(2) Bosentan group (BOG): Bosentan was administered by esophageal gavage (100 mg/kg/day).
(3) Atorvastatin group (ATG): Mice were treated with atorvastatin (20mg/kg/day) administered by esophageal gavage. The detailed protocol has been described in previous publication8.
(4) Bosentan and atorvastatin (BO+ATG): Concomitant bosentan and atorvastatin administration by esophageal gavage, as described previously, for 6 weeks.
On the last day of the experimental period (14th week of experiments) body weights were measured after overnight fasting. All mice were euthanized under isoflurane deep anesthesia. After euthanasia, the whole blood was collected through heart puncture and fasting glucose, triglycerides and total cholesterol plasma levels were immediately assayed in an automatic enzymatic analyser (Olympus AU560, Hamburg, Germany). Afterwards, the heart along the aorta were excised.
All experiments were performed in the Biomedical Research Foundation of the Academy of Athens (BRFAA) and the protocol was evaluated and approved by the Veterinary Service of the Prefecture of Athens (permit number 2697/26-04-2013), as required by the Greek legal requirements for animal experimentation. Bosentan pure substance was supplied by Actelion Pharmaceuticals LTD, Allschwil, Switzerland.
4.2. Histological Parameters
After euthanasia, the heart along with the aortic root was perfused with normal saline via heart puncture. The procedure of aortic valve sectioning with microtome and paraffin blockage for histomorphometric analysis is valid and has been previously described [8]. We used 3 nonconsecutive aortic slices (at equal intervals of 20μm) for each staining per mouse. Aortic plaque area, collagen and elastin contents were quantitatively assessed, by staining cross-sections with hematoxylin/eosin (H&E), sirius red and orcein, respectively, according to a standardized protocol [49]. Finally, by means of immunohistochemistry, we used antibodies for the staining of the corresponding antigens: MCP-1, TNF-a, MMP-2, MMP-3, MMP-9 (MBL, International Corporation, Woburn, MA, USA), Mac-3 antigen of murine macrophages (BD Pharmigen, Franklin Lakes, NJ, USA), alpha-smooth muscle isoform of actin (Biocare Medical, LLC, Concord, CA, USA) and TIMP-1 (Acris Antibodies GmbH, Herford, Germany).
4.3. Histomorphometry
Using a bright-field microscope (Leica DM LS2, Wetzlar, Germany), we observed all sections from the aortic valves and digital pictures were acquired and analyzed using an “Altra 20 Soft Imaging System” computer software. Total plaque area and total lumen area (in μm2) circumscribed by the internal elastic lamina (IEL) were measured in H&E-stained sections. The percentage of luminal stenosis was calculated as the proportion of the total lumen area occupied by all atherosclerotic plaques in each section, and we then averaged luminal stenoses.
In Sirius-red and Orcein-stained sections, we measured the relative concentrations of collagen and elastin, respectively, and the fibrous cap thickness of each atherosclerotic plaque. For each parameter, we then averaged all values of all sections per mouse. For the measurement of the relative concentrations of the stained molecules by immunohistochemistry, the segmental stained plaque area was expressed as the percentage of the whole atherosclerotic plaque area [39]. We then averaged the values of all plaques per animal and then for each group.
4.4. Statistical Analysis
The results of the study are presented as mean values ± standard deviation. Normality of distribution was assessed using the Kolmogorov-Smirnov test. For continuous variables, the comparisons between groups were made using one-way ANOVA and post-hoc Tukey test, respectively. A two-tailed p value <0.05 was considered statistically significant. All statistical analyses were based on SPSS v26.0 (IBM, Armonk, NY, USA).
Author Contributions
N.P.E. K.: conceptualization, methodology, experiments, writing—review and editing; M.S.: writing—original draft preparation, data curation; N.K.: technical support, resources. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
The authors acknowledge the personnel of the Laboratory Animal Core Facility of the Biomedical Research Foundation of the Academy of Athens for their contribution during the experimental process.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Libby, P.; Nahrendorf, M.; Swirski, F.K. Monocyte Heterogeneity in Cardiovascular Disease. Semin Immunopathol 2013, 35, 553–562. [Google Scholar] [CrossRef]
- Hansson, G.K.; Libby, P.; Schönbeck, U.; Yan, Z.Q. Innate and Adaptive Immunity in the Pathogenesis of Atherosclerosis. Circ Res 2002, 91, 281–91. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis--an Inflammatory Disease. N Engl J Med 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of Atherosclerosis Plaque Progression. Heart Lung Circ 2013, 22, 399–411. [Google Scholar] [CrossRef]
- Eckel, R.H.; Bornfeldt, K.E.; Goldberg, I.J. Cardiovascular Disease in Diabetes, beyond Glucose. Cell Metab 2021, 33, 1519–1545. [Google Scholar] [CrossRef]
- Pleus, S.; Tytko, A.; Landgraf, R.; Heinemann, L.; Werner, C.; Müller-Wieland, D.; Ziegler, A.G.; Müller, U.A.; Freckmann, G.; Kleinwechter, H.; et al. Definition, Classification, Diagnosis and Differential Diagnosis of Diabetes Mellitus: Update 2023. Exp Clin Endocrinol Diabetes 2024, 132, 112–124. [Google Scholar] [CrossRef]
- Howard-Alpe, G.; Foëx, P.; Biccard, B. Cardiovascular Protection by Anti-Inflammatory Statin Therapy. Best Pr. Res Clin Anaesthesiol 2008, 22, 111–133. [Google Scholar] [CrossRef]
- Stasinopoulou, M.; Kadoglou, N.P.E.; Christodoulou, E.; Paronis, E.; Kostomitsopoulos, N.G.; Valsami, G.; Liapis, C.D.; Kakisis, J. Statins’ Withdrawal Induces Atherosclerotic Plaque Destabilization in Animal Model-A “Rebound” Stimulation of Inflammation. J Cardiovasc Pharmacol Ther 2019, 24, 377–386. [Google Scholar] [CrossRef]
- Wierzbicki, A.S.; Poston, R.; Ferro, A. The Lipid and Non-Lipid Effects of Statins. Pharmacol Ther 2003, 99, 95–112. [Google Scholar] [CrossRef]
- Gallo, G.; Savoia, C. New Insights into Endothelial Dysfunction in Cardiometabolic Diseases: Potential Mechanisms and Clinical Implications. Int J Mol Sci 2024, 25, 2973. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A Novel Potent Vasoconstrictor Peptide Produced by Vascular Endothelial Cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef]
- Böhm, F.; Pernow, J. The Importance of Endothelin-1 for Vascular Dysfunction in Cardiovascular Disease. Cardiovasc Res 2007, 76, 8–18. [Google Scholar] [CrossRef]
- Masaki, T.; Miwa, S.; Sawamura, T.; Ninomiya, H.; Okamoto, Y. Subcellular Mechanisms of Endothelin Action in Vascular System. Eur J Pharmacol 1999, 375, 133–138. [Google Scholar] [CrossRef]
- Peacock, A.J.; Dawes, K.E.; Shock, A.; Gray, A.J.; Reeves, J.T.; Laurent, G.J. Endothelin-1 and Endothelin-3 Induce Chemotaxis and Replication of Pulmonary Artery Fibroblasts. Am J Respir Cell Mol Biol 1992, 7, 492–499. [Google Scholar] [CrossRef]
- Ito, H.; Hirata, Y.; Hiroe, M.; Tsujino, M.; Adachi, S.; Takamoto, T.; Nitta, M.; Taniguchi, K.; Marumo, F. Endothelin-1 Induces Hypertrophy with Enhanced Expression of Muscle-Specific Genes in Cultured Neonatal Rat Cardiomyocytes. Circ Res 1991, 69, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, L.; Gray, M.; Honbo, N.Y.; Chentoufi, J.; Bergman, M.; Karliner, J.S. Endothelin-1 Stimulates Cardiac Fibroblast Proliferation through Activation of Protein Kinase C. J Mol Cell Cardiol 2000, 32, 565–576. [Google Scholar] [CrossRef]
- Takahashi, K.; Ghatei, M.A.; Lam, H.C.; O’Halloran, D.J.; Bloom, S.R. Elevated Plasma Endothelin in Patients with Diabetes Mellitus. Diabetologia 1990, 33, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Settergren, M.; Pernow, J.; Brismar, K.; Jörneskog, G.; Kalani, M. Endothelin-A Receptor Blockade Increases Nutritive Skin Capillary Circulation in Patients with Type 2 Diabetes and Microangiopathy. J Vasc Res 2008, 45, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Mather, K.J.; Mirzamohammadi, B.; Lteif, A.; Steinberg, H.O.; Baron, A.D. Endothelin Contributes to Basal Vascular Tone and Endothelial Dysfunction in Human Obesity and Type 2 Diabetes. Diabetes 2002, 51, 3517–3523. [Google Scholar] [CrossRef]
- Galiè, N.; Beghetti, M.; Gatzoulis, M.A.; Granton, J.; Berger, R.M.; Lauer, A.; Chiossi, E.; Landzberg, B. Bosentan Randomized Trial of Endothelin Antagonist Therapy-5 (BREATHE-5) Investigators Bosentan Therapy in Patients with Eisenmenger Syndrome: A Multicenter, Double-Blind, Randomized, Placebo-Controlled Study. Circulation 2006, 114, 48–54. [Google Scholar] [CrossRef]
- Gatzoulis, M.A.; Beghetti, M.; Galiè, N.; Granton, J.; Berger, R.M.; Lauer, A.; Chiossi, E.; Landzberg, M. BREATHE-5 Investigators Longer-Term Bosentan Therapy Improves Functional Capacity in Eisenmenger Syndrome: Results of the BREATHE-5 Open-Label Extension Study. Int J Cardiol 2008, 127, 27–32. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, Z.; Li, G. The Protective Effect of Bosentan against Atherosclerosis in Apolipoprotein E-Deficient Mice Is Mediated by miRNA-21. Biomed Res Int 2019, 2019, 8348430. [Google Scholar] [CrossRef]
- Mulder, P.; Richard, V.; Derumeaux, G.; Hogie, M.; Henry, J.P.; Lallemand, F.; Compagnon, P.; Macé, B.; Comoy, E.; Letac, B.; et al. Role of Endogenous Endothelin in Chronic Heart Failure: Effect of Long-Term Treatment with an Endothelin Antagonist on Survival, Hemodynamics, and Cardiac Remodeling. Circulation 1997, 96, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, R.M.; Yanagisawa, M. Endothelin System: The Double-Edged Sword in Health and Disease. Annu Rev Pharmacol Toxicol 2001, 41, 851–876. [Google Scholar] [CrossRef] [PubMed]
- Ihling, C.; Szombathy, T.; Bohrmann, B.; Brockhaus, M.; Schaefer, H.E.; Loeffler, B.M. Coexpression of Endothelin-Converting Enzyme-1 and Endothelin-1 in Different Stages of Human Atherosclerosis. Circulation 2001, 104, 864–869. [Google Scholar] [CrossRef]
- Kowala, M.C.; Rose, P.M.; Stein, P.D.; Goller, N.; Recce, R.; Beyer, S.; Valentine, M.; Barton, D.; Durham, S.K. Selective Blockade of the Endothelin Subtype A Receptor Decreases Early Atherosclerosis in Hamsters Fed Cholesterol. Am J Pathol 1995, 146, 819–826. [Google Scholar] [PubMed]
- Best, P.J.; Lerman, A. Endothelin in Cardiovascular Disease: From Atherosclerosis to Heart Failure. J Cardiovasc Pharmacol Ther 2000, 35, S61–63. [Google Scholar] [CrossRef] [PubMed]
- Jabarpour, M.; Rashtchizadeh, N.; Argani, H.; Ghorbanihaghjo, A.; Ranjbarzadhag, M.; Sanajou, D.; Panah, F.; Alirezaei, A. The Impact of Dyslipidemia and Oxidative Stress on Vasoactive Mediators in Patients with Renal Dysfunction. Int Urol Nephrol 2019, 51, 2235–2242. [Google Scholar] [CrossRef]
- Rivera-Gonzalez, O.; Wilson, N.A.; Coats, L.E.; Taylor, E.B.; Speed, J.S. Endothelin Receptor Antagonism Improves Glucose Handling, Dyslipidemia, and Adipose Tissue Inflammation in Obese Mice. Clin. Sci. 2021, 135, 1773–1789. [Google Scholar] [CrossRef]
- Moustardas, P.; Kadoglou, N.P.; Katsimpoulas, M.; Kapelouzou, A.; Kostomitsopoulos, N.; Karayannacos, P.E.; Kostakis, A.; Liapis, C.D. The Complementary Effects of Atorvastatin and Exercise Treatment on the Composition and Stability of the Atherosclerotic Plaques in ApoE Knockout Mice. PLoS One 2014, 9, e108240. [Google Scholar] [CrossRef]
- Lee, S.G.; Lee, S.J.; Thuy, N.V.P.; Kim, J.S.; Lee, J.J.; Lee, O.H.; Kim, C.K.; Oh, J.; Park, S.; Lee, O.H.; et al. Synergistic Protective Effects of a Statin and an Angiotensin Receptor Blocker for Initiation and Progression of Atherosclerosis. PLoS One 2019, 14, e0215604. [Google Scholar] [CrossRef]
- Li, D.Q.; Lv, F.F.; Li, Z.C.; Dai, Z.Y.; Wang, H.X.; Han, Y. Anti-Atherosclerotic Effects between a Combined Treatment with Simvastatin plus Hirudin and Single Simvastatin Therapy in Patients with Early Type 2 Diabetes Mellitus. Ann Transl Med 2019, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- van der Vorst, E.P.C.; Weber, C.; Donners, M.M.P.C. A Disintegrin and Metalloproteases (ADAMs) in Cardiovascular, Metabolic and Inflammatory Diseases: Aspects for Theranostic Approaches. Thromb Haemost 2018, 118, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.A.; Wierer, M.; Dang, T.A.; Milic, J.; Moggio, A.; Sachs, N.; von Scheidt, M.; Hinterdobler, J.; Müller, P.; Werner, J.; et al. ADAMTS-7 Modulates Atherosclerotic Plaque Formation by Degradation of TIMP-1. Circ Res 2023, 133, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Lorentzen, L.G.; Yeung, K.; Eldrup, N.; Eiberg, J.P.; Sillesen, H.H.; Davies, M.J. Proteomic Analysis of the Extracellular Matrix of Human Atherosclerotic Plaques Shows Marked Changes between Plaque Types. Matrix Biol Plus 2024, 21, 100141. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.; Daskalopoulou, S.S.; Perrea, D.; Liapis, C.D. Matrix Metalloproteinases and Diabetic Vascular Complications. Angiology 2005, 56, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Zamilpa, R. Temporal and Spatial Expression of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases Following Myocardial Infarction. Cardiovasc Ther 2012, 30, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Johnson, J.A.; Fulp, A.; Sutton, M.A.; Lessner, S.M. Adhesive Strength of Atherosclerotic Plaque in a Mouse Model Depends on Local Collagen Content and Elastin Fragmentation. J Biomech 2013, 46, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P. The Beneficial Effects of a Direct Thrombin Inhibitor, Dabigatran Etexilate, on the Development and Stability of Atherosclerotic Lesions in Apolipoprotein E-Deficient Mice : Dabigatran Etexilate and Atherosclerosis. Cardiovasc Drugs Ther 2012, 26, 367–374. [Google Scholar] [CrossRef]
- Basiak, M.; Hachula, M.; Kosowski, M.; Machnik, G.; Maliglowka, M.; Dziubinska-Basiak, M.; Krysiak, R.; Okopien, B. The Effect of PCSK9 Inhibition on the Stabilization of Atherosclerotic Plaque Determined by Biochemical and Diagnostic Imaging Methods. Molecules 2023, 28, 5928. [Google Scholar] [CrossRef]
- Basiak, M. Impact of PCSK9 Inhibition on Proinflammatory Cytokines and Matrix Metalloproteinases Release in Patients with Mixed Hyperlipidemia and Vulnerable Atherosclerotic Plaque. Pharm. Basel 2022, 15, 802. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.E.; Velidakis, N.; Khattab, E.; Kassimis, G.; Patsourakos, N. The Interplay between Statins and Adipokines. Is This Another Explanation of Statins’ “pleiotropic” Effects? Cytokine 2021, 148, 155698. [Google Scholar] [CrossRef] [PubMed]
- Umebashi, K.; Yamamoto, M.; Tokito, A.; Sudou, K.; Takenoshita, Y.; Jougasaki, M. Inhibitory Effects of Simvastatin on IL-33-Induced MCP-1 via the Suppression of the JNK Pathway in Human Vascular Endothelial Cells. Int J Mol Sci 2023, 24, 13015. [Google Scholar] [CrossRef] [PubMed]
- Vieceli Dalla Sega, F.; Cimaglia, P.; Manfrini, M.; Fortini, F.; Marracino, L.; Bernucci, D.; Pompei, G.; Scala, A.; Trichilo, M.; De Carolis, B.; et al. Circulating Biomarkers of Endothelial Dysfunction and Inflammation in Predicting Clinical Outcomes in Diabetic Patients with Critical Limb Ischemia. Int J Mol Sci 2022, 23, 10641. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Pan, S.; Yu, H.; Hu, H.; Sun, Y.U.; Yang, Z.; Hoffman, R.M.; Yuan, H. Anti-Inflammatory and Anti-Thrombotic Efficacy of Targeted Ultrasound Microbubbles on LPS-Induced HUVEC Cells. Anticancer Res 2021, 41, 4761–4769. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Li, S.H.; Badiwala, M.V.; Weisel, R.D.; Fedak, P.W.; Li, R.K.; Dhillon, B.; Mickle, D.A. Endothelin Antagonism and Interleukin-6 Inhibition Attenuate the Proatherogenic Effects of C-Reactive Protein. Circulation 2002, 105, 1890–1896. [Google Scholar] [CrossRef]
- Yao, E.H.; Wang, H.J.; Xu, C.S. Effects of Tongxinluo on the Neointima Formation and Expression of Inflammatory Cytokines in Rats after Carotid Artery Balloon Injury. Indian J Pharmacol 2014, 46, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Dede, E.; Liapis, D.; Davos, C.; Katsimpoulas, M.; Varela, A.; Mpotis, I.; Kostomitsopoulos, N.; Kadoglou, N.P.E. The Effects of Exercise Training on Cardiac Matrix Metalloproteinases Activity and Cardiac Function in Mice with Diabetic Cardiomyopathy. Biochem Biophys Res Commun 2022, 586, 8–13. [Google Scholar] [CrossRef]
- Kadoglou, N.P. The Anti-Inflammatory Effects of Exercise Training Promote Atherosclerotic Plaque Stabilization in Apolipoprotein E Knockout Mice with Diabetic Atherosclerosis. Eur J Histochem 2013, 57, e3. [Google Scholar] [CrossRef]
Figure 1.
All active groups (BOG, ATG and BO+ATG) significantly reduced plaque formation compared to controls in ApoE−/− mice. Representative images and quantifications of aortic valve sections stained with hematoxylin/eosin, across all groups.
Figure 1.
All active groups (BOG, ATG and BO+ATG) significantly reduced plaque formation compared to controls in ApoE−/− mice. Representative images and quantifications of aortic valve sections stained with hematoxylin/eosin, across all groups.
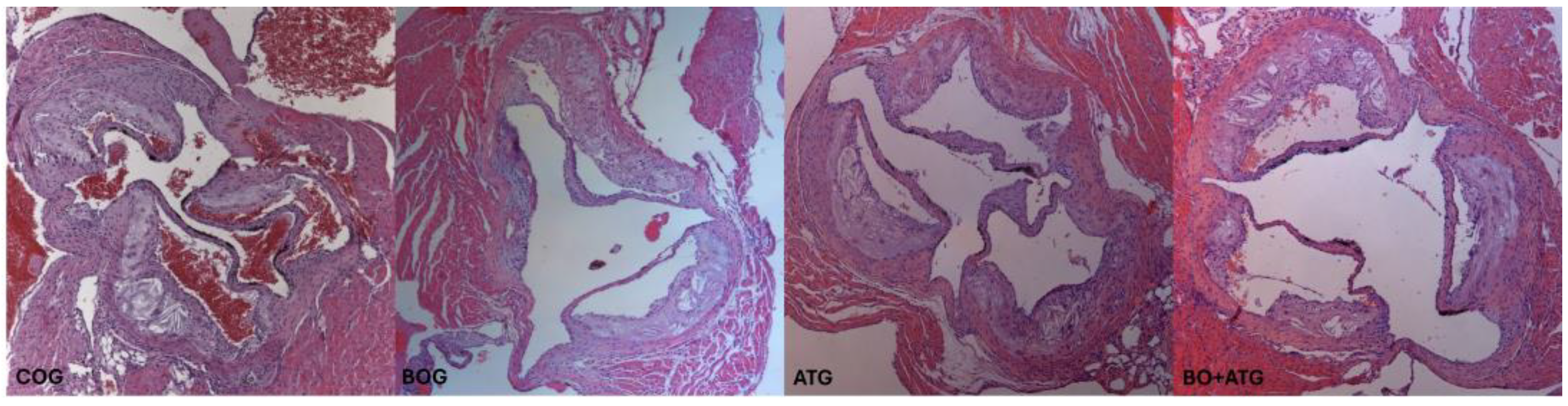
Figure 2.
Quantification of immunohistochemical staining with antibodies against MMP-2, MMP-3, MMP-9, TIMP-1, TNF-a and MCP-1. *p<0.05 vs COG, #p<0.05 vs BOG, ||p<0.05 vs AΤG.
Figure 2.
Quantification of immunohistochemical staining with antibodies against MMP-2, MMP-3, MMP-9, TIMP-1, TNF-a and MCP-1. *p<0.05 vs COG, #p<0.05 vs BOG, ||p<0.05 vs AΤG.
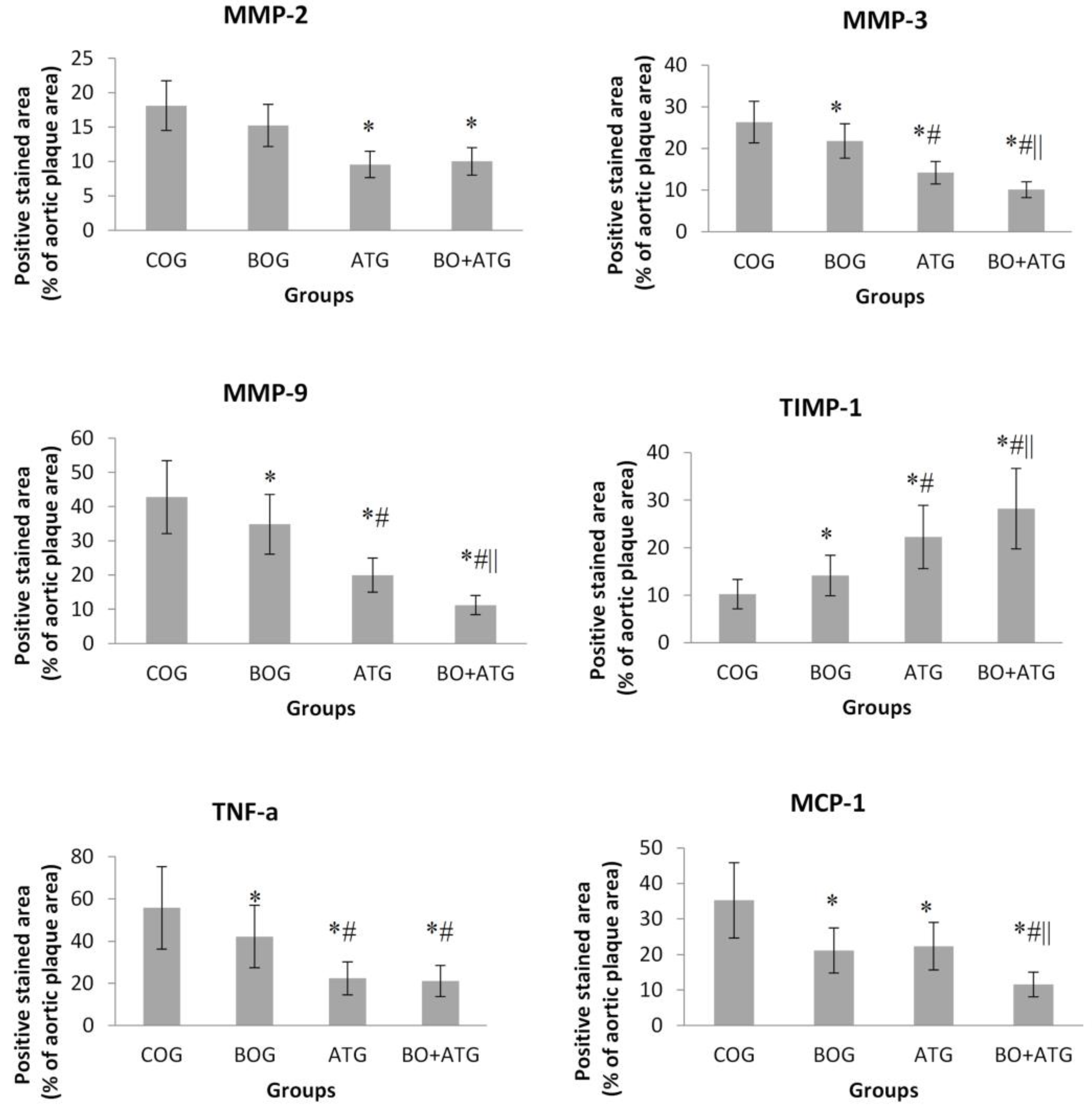
Figure 3.
Representative images of immunohistochemical staining across all groups with antibodies against (A) MMP-2 (upper panel), (B) MMP-3 (middle panel) and (C) TIMP-1 (lower panel). Sections were counterstained with H&E.
Figure 3.
Representative images of immunohistochemical staining across all groups with antibodies against (A) MMP-2 (upper panel), (B) MMP-3 (middle panel) and (C) TIMP-1 (lower panel). Sections were counterstained with H&E.
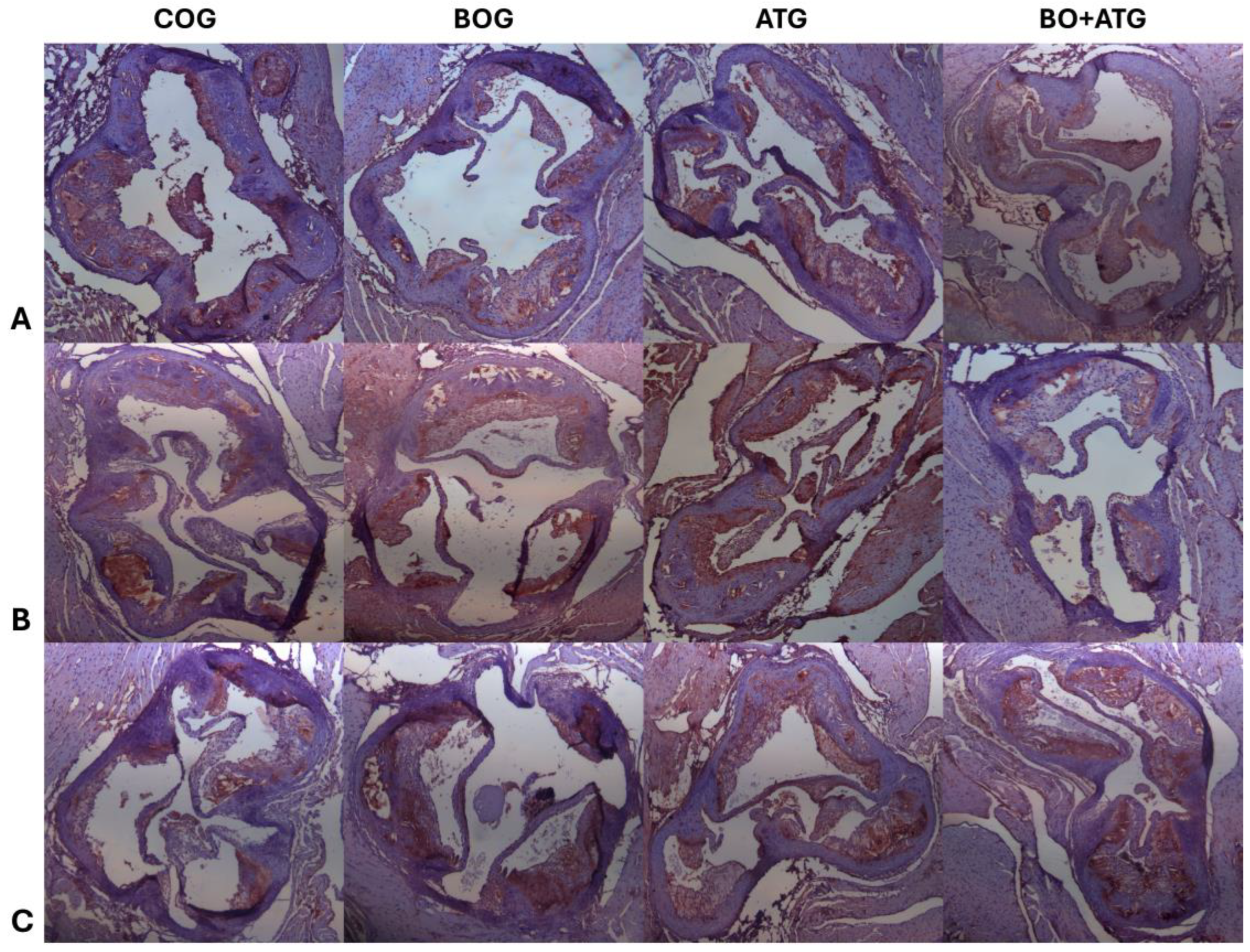

Table 1.
Body weight, lipid parameters and glucose at the end of the study.
| COG (n=12) |
BOG (n=12) |
ATG (n=12) |
BO+ATG (n=12) |
p | |
|---|---|---|---|---|---|
| Weight (g) | 34.4±4.2 | 33.3±4.8 | 32.9±3.9 | 33.5±4.2 | 0.512 |
| FPG (mg/dl) | 281±29 | 289±55 | 295±45 | 309±49 | 0.794 |
| TC (mg/dl) | 759±131 | 758±182 | 511±131* | 502±191* | <0.001 |
| TG (mg/dl) | 155±41 | 145±34 | 129±30 | 131±33 | 0.400 |
CG, control group; BOG, bosentan group; ATG, atorvastatin group; BO+ATG, bosentan + atorvastatin group. FPG, fasting plasma glucose; TC, total cholesterol; TG, triglycerides; P, one-way ANOVA. *Tuckey test, p<0.05, vs COG.
Table 2.
Atherosclerotic lumen stenosis (percentage of plaques are/lumen area) (H&E staining), intra-plaque contents of collagen (sirius red staining), elastin (orcein staining), vascular smooth muscle cells (VSMCs – a-actin) and macrophages (Mac-3).
Table 2.
Atherosclerotic lumen stenosis (percentage of plaques are/lumen area) (H&E staining), intra-plaque contents of collagen (sirius red staining), elastin (orcein staining), vascular smooth muscle cells (VSMCs – a-actin) and macrophages (Mac-3).
| COG (n=12) |
BOG (n=12) |
ATG (n=12) |
BO+ATG (n=12) |
p | |
|---|---|---|---|---|---|
| Lumen Stenosis (%) | 24.6±4.8 | 19.5±2.2 a,c,d | 12.8±4.8a,b,d | 9.1±2.7a,b,c | <0.001 |
| Elastin (%) plaque | 8.12±2.10 | 10.62±6.52c,d | 25.17±6.91a | 31.02±5.23a,b | <0.001 |
| Collagen (%) plaque | 14.21±4.21 | 22.83±4.79 a,c,d | 31.88±5.97 a,b | 40.33±8.72 a,b,c | <0.001 |
| Fibrous cap thickness (μm) | 9.12±3.10 | 13.12±3.23 a,c,d | 23.12±5.44 a,b,d | 48.12±6.21a,b,c | <0.001 |
| a-actin (VSMCs) (%) plaque | 17.13±3.21 | 26.88±6.06a | 20.53±6.97 | 28.10±6.82a | 0.005 |
| Mac-3 (macrophages) (%) plaque | 34.56±10.25 | 26.46±6.82a,c,d | 15.09±3.22a,b,c | 10.12±3.78a,b,c | <0.001 |
CG, control group; BOG, bosentan group; ATG, atorvastatin group; BO+ATG, bosentan + atorvastatin group. P, one-way ANOVA p value. *Tuckey test, significant differences of each intervention group (p<0.05) compared to other groups based on post-hoc one-way ANOVA analysis: a COG, b BOG, c ATG, d BO+ATG .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



