Submitted:
03 May 2024
Posted:
07 May 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Chiroptera includes of over 1,400 bat species with at least 35 of these present in Italy. Due to their role as lyssavirus reservoirs, in Italy, dead bats and oral swabs are routinely submitted to the laboratory network of the Istituti Zooprofilattici Sperimentali within in the framework of the rabies national passive and active surveillance programme. Carcasses and biological samples collected from January to December 2021 in Latium and Tuscany Regions were further screened for the presence of Coronaviruses (CoVs) and Herpesviruses, using pan-family virus PCR tests and relative PCR products were Sanger sequenced. Subsequent to the viral investigations, genetic bat species was also carried out. Viral characterization detected AlphaCoVs in Miniopterus schreibersii and Beta- and Gamma-Herpesviruses in Tadarida Teniotis.
Keywords:
Chiroptera
; Miniopterus schreibersii
; Tadarida Teniotis
; Alphacoronavirus
; Betaherpesvirus
1. Introduction
Chiroptera include to date, more than 1,400 species, representing one-fifth of all mammalian species worldwide [1]. In Italy, at least thirty-five species [2] divided into eleven different genera and four families are documented [3].
During the second half of the 20th century, bat populations in Europe declined significantly, probably due to different factors, such as agricultural intensification, destruction of roosts, habitat loss and massive use of toxic chemicals.
Bats are considered the ancestral hosts of Lyssaviruses (LYSVs), and, to date, they serve as reservoirs to 15 of the currently known 17 [4] of these viruses.
In Europe, five different LYSVs, namely European bat 1 lyssavirus (EBLV-1), European bat 2 lyssavirus (EBLV-2), Bokeloh bat lyssavirus (BBLV), Lleida bat lyssavirus (LLEBV), Kotalahti bat lyssavirus (KBLV), and the newly described Divača bat lyssavirus [5,6], are reported in specific bat host species [7]. In the same geographical area, especially in the last two decades, some bat species were also found to be associated with other zoonotic pathogens such as Coronavirus, Herpesvirus, and others [8,9,10]. Bats host viruses from at least 28 different families, most of which are likely to be host-specific with limited zoonotic potential [11]. Correct host species identification is of crucial importance to also increase knowledge of the ecology and evolutionary pattern of bats and bat viruses.
The detection of microbes in some bat species, close to those that cause human diseases, may indicate only an evolutionary relationship and not that a pathogen currently circulating in bat populations will infect humans [12].
Chiroptera pathogen surveillance plays a crucial role in the management of public health safety, as the early detection of potentially newly emerging zoonotic pathogens is fundamental to prevent their diffusion, especially those with RNA genome, in view of their higher mutation rate [13].
As bats play an important role as reservoirs and in the circulation of LYSVs, even if their risk in both humans and bats is at present low [14], it is crucial to continue monitoring their presence, owing to the broad spectrum of affected bat species and the persistent zoonotic threat of infection [5]. Respectively, since 1980 and 2008, passive and active surveillance in bats are carried out to verify rabies-related LYSV circulation, based also on the analysis of dead bats, especially those that could have come in contact with humans or domestic animals. A report on active surveillance describes LYSV serological evidence in at least six bat colonies, present across the country, suggesting their circulation, even if no direct viral evidence was obtained that probably could be due to the low number of carcasses present or because the habitual LYSV host bat species are not present in great numbers in Italy [7].
Since 2021, lyssavirus active surveillance in Chiroptera is mandatory, in compliance with the Commission Implementing Regulation (EU) 2018/1882, and adopted in Italy with a national decree of the President of the Republic 357/97 [15].
In view of the potential role of bats as reservoirs of SARs CoV-2, during the Covid-19 pandemic, as indicated by the scientific literature and the sequences of the viruses available in the public databases [16], objective of the present study was to evaluate the presence of Coronavirus and also investigate the presence of another wide group of DNA viruses, represented by herpesviruses, that are extensively distributed in nature among several vertebrates [17].
2. Materials and Methods
Oral swabs of LYSVs susceptible bat species were collected during 2021 from a colony in Arezzo, Italy, within the national surveillance programme, set up subsequent to the detection of West Caucasian Bat Lyssavirus (WCBL) in 2020, in a cat living in the vicinity. During the same year, brain, lung and intestines were sampled from bat carcasses collected in Latium and Tuscany.
Nucleic acid extraction from biological tissues was performed using the automatic extractor QIAsymphony with the DSP Virus/Pathogen Kit, Qiagen, GmbH, Hilden, Germany, according to the manufacturer instructions. Briefly, 0.1 g of each sample was added to 0.9 ml of ATL buffer (QIAGEN, GmbH, Hilden, Germany) and a 5 mm stainless steel bead and homogenized by Tissue Lyser II (QIAGEN, GmbH, Hilden, Germany), followed by a centrifugation at 16,000 g. Oral swabs were suspended in 500 µl of a 1/1 phosphate saline buffer solution (PBS) and ATL buffer. A volume of 400 µl of supernatant from each sample was used for RNA/DNA extraction. The concentration and purity of the extracted nucleic acids was evaluated by spectrophotometric analysis, based on the absorbance values (A), respectively at 260 and 280 nm wavelengths. All individuals were genetically identified for species identification as described elsewhere [18].
LYSVs infection was first ruled out, by testing brain tissues and oral swabs using real-time RT-PCR [19,20]. Lungs, intestines and oral swabs were tested using a panCoV RT-PCR assay, targeting the RNA-dependent, RNA polymerase gene (RdRp) [21]. A subset of samples for which material was still available for analysis (n=77/129 lungs and n=64/111 oral swabs) were also tested using a pan-Herpes RT-PCR assay [22,23]. Positive amplicons were sequenced using an automated sequencer (3500 Genetic Analyzer, A. Biosystems, Foster City, CA, USA). The sequences were assembled and analysed using the Geneious Prime 2021.0.3 (Biomatters, Auckland, New Zealand) and compared with those present for the viruses, object of this study, in the National Center for Biotechnology Information (NCBI) GenBank database, using the BLAST online tool (Rockville Oike, USA).
Phylogenetic analysis was performed for our positive amplicons. Similar sequences were retrieved aligning newly submitted protein sequences with BLASTp program from NCBI, against sequences obtained from the DBatVir [16] database and the NCBI Non-redundant (nr) Database. In case of Coronavirus amplicons, a taxonomic subgenera classification analysis was carried out using the R library MyCoV [24]. Identity and similarity were calculated by global pairwise alignments using the EMBOSS Needle program (version 6.6.0), based on the Needleman-Wunsch algorithm and available on the European Bioinformatics Institute webpage [25].
Multiple sequence alignment were obtained using MUSCLE program [26] applying standard parameters. Prottest version 3.4.2 program [27] and jModelTest version 2.1.10 program [28] took as input each multiple alignment to select each best model among all distributions, considering smallest Bayesian Information Criterion (BIC) scores. Phylogenetic relationships were inferred by using the Maximum Likelihood method with the PhyML version 3.3.20211231 program [29]. Tree images were plotted with R-studio program 30 using ape, ggtree, tidytree libraries [31,32,33].
3. Results
In 2021, tissue organs of 128 bat carcasses were analysed, of which 36 were from Tuscany and 92 were from Latium. Moreover, 111 oral swabs collected from the aforementioned Arezzo colony, for which presence of Miniopterus schreibersii was confirmed, were also examined 20. Bats examined belonged mostly to Tadarida teniotis (n = 71, 29.7%), that were all collected from an urban colony which since 2008 had registered cyclical mass mortality 7, followed by Pipistrellus kuhlii (n = 23, 9.6%), Hypsugo savii (n = 18, 7.5%), Pipistrellus sp. (n=10, 4.2%), Myotis daubentonii and Plecotus austriacus (both with n = 1, 0.4%) (Table 1). A small percentage of bats (n = 4, 1.7%) were not identified, probably due to the storage conditions before submission to the laboratory which could have caused a nucleic acid degradation; probably for the same reason it was not possible to arrive to species identification for some of the subjects.
All samples were negative for the presence of LYSVs. The date relative to CoV is including that derived from a previous research project in which we participated [34]. In the present studied, CoV was detected in three oral swabs. Herpesvirus were detected in nine lung samples (n=9/77, 11.68%) and due to the poor quality of some of the amplicon bands when visualized on electrophoresis gel, only two were confirmed by Sanger sequencing.
Using Sanger sequencing, we obtained three sequences for CoVs respectively identified as M. schreibersii Italy 1/2021, M. schreibersii Italy 2/2021 and M. schreibersii Italy 3/2021, while for Betaherpesviruses and Gammaherpesviruses we respectively obtained the following sequences: WJK71402.1 (OQ807168.1 nucleic) and WJK71403.1 (OQ807169.1 nucleic. The sequences obtained in this study will be referred to as novel sequences. Relative to CoV, only two of three novel sequences are considered as their length was sufficient for determining sub-genera taxonomic classification and an informative phylogenetic analysis. M. schreibersii Italy 1/2021 (OP776451.1 nucleotide and UZC49592.1 protein accession numbers) has a nucleotide length of 333 bp and a protein of 110 amino acids. While, M. schreibersii Italy 2/2021 (ON834690.1 nucleotide and UUG60948.1 protein accession numbers) and M. schreibersii Italy 3/2021 (OP627105.1 nucleotide and UYI58598.1 protein accession numbers) are identical and are 441 bp long, which correspond to 147 amino acids. Comparing these novel isolates with blastn program, OP776451.1 M. schreibersii Italy 1/2021 exhibits a 78.08% percentage identity with ON834690.1 M. schreibersii Italy 2/2021 and OP627105.1 M. schreibersii Italy 3/2021, aligning with a word size parameter of 15. Using blastp and needle emboss, UZC49592.1 M. schreibersii Italy 1/2021 aligned with UUG60948.1 M. schreibersii Italy 2/2021 and UYI58598.1 M. schreibersii Italy 3/2021 showing an 88.18% percentage identity and a similarity score of 102/110. Table 2 reports all the information on the comparison among the three novel CoVs, using as query M. schreibersii Italy 2/2021.
The phylogenetic analysis was carried out using multiple sequence alignments of 129 amino acid and 397 nucleotide sequence lengths, obtained by including 29 sequences from bat hosts. The nucleotide Alpha-CoVs tree (log likelihood -3518.55687) illustrated in Figure 1, which is mainly built from Miniopterus Alpha-CoVs, indicates a group belonging to the Minunacovirus subgenera and composed of HKU8, HKU7, Bat corona virus 1A and 1B taxonomy classifications, that separates from the HKU2, HKU6 and M.sch/A/Spain/2004 (HQ184049.1) isolates. Considering all the Minunacovirus subgenera, the uppermost part represents the Miniopterus bat coronavirus 1 taxonomy, which consists of bat coronavirus 1A and 1B and other unclassified bat coronaviruses, whereas the lowermost part includes the Miniopterus bat coronavirus HKU8 classification. The other reference taxonomy classification, which is bat coronavirus HKU7, stands between the two parts, but nearer to the HKU8 classification. The Minunacovirus subgenera tree topology is also confirmed by past studies, where the HKU7 and HKU8 classifications are close to each other [35,36], and more separated from the Miniopterus bat coronavirus 1 (Bat coronavirus 1A and 1B classifications (cit)). The two samples considered, which are ON834690.1 M. schreibersii Italy 2/2021 and OP627105.1 M. schreibersii Italy 3/2021, fall next to the group of the Miniopterus bat coronavirus HK8. Furthermore, they share the same clade with the French (KY423482.1 France 2014) and the Algerian (MN701038.1 Algeria 2017) isolates. M. schreibersii Italy 2/2021 and M. schreibersii Italy 3/2021 are identical to KY423482.1 France 2014 while they have a 98% percentage identity with MN701038.1 Algeria 2017. The branch of the Algerian isolate, that describes a short distance from M. schreibersii Italy 2/2021, M. schreibersii Italy 3/2021 and France 2014, is also caused by an insertion of an adenine in the seventh position of the pairwise alignment between it and one of the novel Italian isolates.
The protein Alpha-CoVs tree (-1143.82593) shown in Figure 2, where only the major differences among the isolates are observed: in the Minunacovirus isolates the separation between the Miniopterus bat coronavirus 1 and Miniopterus bat coronavirus HKU8 is clear. This topology confirms that UUG60948.1 M. schreibersii Italy 2/2021 and UYI58598.1 M. schreibersii Italy 3/2021 are closer to the Miniopterus bat coronavirus HKU8 classification, and in fact the percentage of identity and similarity score between the two novel Italian strains and Miniopterus bat coronavirus HKU8 isolates are high. For example, considering ABB77042.1, they are 98.4% and 128/129. UUG60948.1 M. schreibersii Italy 2/2021 and UYI58598.1 M. schreibersii Italy 3/2021, when compared to Miniopterus bat coronavirus 1 isolates, that show slightly lower percentage of identities and similarity score than to Miniopterus bat coronavirus HKU8 isolates. In fact, they exhibit percentages of identities and similarity scores respect to YP_001718603.1 Miniopterus bat coronavirus 1/Bat coronavirus 1A and ABG75573.1 Miniopterus bat coronavirus 1/Bat coronavirus 1B, that are for both equal to 93.8% and 124/129. The QNJ45063.1 Algeria 2017, which in Figure 1 is in the same group of our novel strains, shows a longer branch than UUG60948.1 M. schreibersii Italy 2/2021 and UYI58598.1 M. schreibersii Italy 3/2021, that is caused by the presence of a WL instead of LP at position 1 and 2 of the pairwise alignment of the region selected in this study, and also by D5I R7M. The percentage of identity and the similarity score between UUG60948.1 M. schreibersii Italy 2/2021 and UYI58598.1 M. schreibersii Italy 3/2021 and QNJ45063.1 Algeria 2017 are for both 96.8% and 125/129, respectively.
For Betaherpesviruses, the phylogenetic inference was achieved from a 155 amino acid length multiple sequence alignment with 15 protein sequences, including different Betaherpesviruses and a Gammaherpesvirus, hosted by bats.
As reported in Figure 3, the phylogenetic tree (log likelihood -2527.03558) shows the submitted sequence with accession number WJK71402.1, mainly grouping with other three European Betaherpesvirus sequences, sampled from Tadarida teniotis in 2008, having a percentage of identity and a similarity score of 100 % and 143/143 with AMY98784.1, 89.5%, and 136/143 with AMY98785.1, 78.3% and 125/143 with AMY98783.1. QJX19381.1, whose host is Molossus molossus from Martinique, presenting a long branch considering those of the isolates from Tadarida teniotis; in addition when aligned to WJK71402.1, the percentage of identity, similarity score and number of gaps are 70.7%, 119/147 and 4. The last gap is related to the last lacking amino acid of the partial sequence QJX19381.1 An interesting evidence, even if only partial DNA polymerase B is considered, is that, the isolates belonging to the same host taxonomic family are grouped in the same node, especially for Molossidae, Miniopteridae and Vespertilionidae, as already seen in a previous study [17].
As reported in Table 3, the main differences between our novel sequence WJK71402.1 and other European DNA polymerases, with the exception for Tadarida teniotis hosts, constantly appear at the starting and ending positions.
In Table 4, the Italian Betaherpesvirus strain protein sequence was also compared to several Betaherpesvirus sequences hosted by different animals and all classified within the Betherpesvirinae taxon family (Taxonomy ID: 10357).
Phylogenetic analysis of Gammaherpesvirus was made with a multiple sequence alignment of 71 amino acid sequence length (from position 2 to 72 of the protein accession number WJK71403.1) using 23 protein sequences. A limitation was a short sequence length that affected the phylogenetic analysis. Table 5 reports identity percentages and similarity scores of the submitted sequence against other bat-hosted virus sequences.
4. Discussion
In view of the results obtained, virus circulation in the limited number of bat species examined and identified is sporadic and could be due to the difficulty in obtaining samples representative of the different species present in Italy, that could also be due to their having different behavior for roosting and migration [37].
Species identification is based on molecular methods, both because of the advanced state of decomposition in which some of the carcasses were found and because some species have similar morphological features that does not allow an accurate species identification [1]. This is essential to monitor especially in case of an upsurge in their death rate, considering that most of our activity is based on passive surveillance, as also the confirmation of the presence of bat species, that are more susceptible to viral infections, such as LYSVs, as in the case of Miniopterus schreibersii and Myotis daubentonii, that are reported in our study, and other bat species as Eptesicus serotinus, Myotis brandtii, Myotis dasycneme and Myotis nattereri that were not detected [38].
Relative to the viral characterization, it would be interesting to correlate the migratory behavior of bats to the phylogenetic data obtained as the viral strains detected are related to those identified in geographically close regions [39]. In fact, in the phylogenetic reconstructions, significative differences reveal that M. schreibersii Italy 2/2021 and M. schreibersii Italy 3/2021, which are identical to each other, are located in a group composed by two strains detected in France and Algeria. Moreover, they were classified with R library MyCoV [24] as Minunacovirus taxonomic subgenus of Alphacoronavirus genus that are more related to the Miniopterus bat coronavirus HKU8. On the other hand, M. schreibersii Italy 1/2021, even if the amplicon obtained was too short to be analysed phylogenetically, the software used still classified it as Minunacovirus taxonomic subgenus by MyCoV [24]. In addition, even if it presents differences with M. schreibersii Italy 2/2021 and M. schreibersii Italy 3/2021, the three CoV positive swab samples were collected at the same moment, within the same colony. From the data obtained it can be noted that different Minunacovirus taxonomic subgenus viruses can circulate within the same colony as previously reported [40] that could be due to a high genetic variability of these viruses.
Betaherpesviruses phylogenetic relationships demonstrate a strict association between novel strain and virus species hosted by European Tadarida teniotis bats, especially with the Tadarida teniotis betaherpesvirus 2. Considering the importance of the DNA pol B region, when our novel strain was aligned with other Betaherpevirinae taxon family strains, hosted by other species (Table 4), the majority of similar and matching amino acids were located in the central part of its protein sequence, with the 5’ and 3’ ends being the most dissimilar parts. Indeed, the DNA-directed, DNA polymerase family B multifunctional domain (InterPro IPR006134) in our novel strain starts from 18 to 119 aa position. The inference of phylogenetic relationships of novel Gammaherpesvirus is very limited as the sequence length is too short for a reliable reconstruction. Comparing its short sequence with other deposited sequences, as shown in Table 5, it does not show huge differences with the exception of a vespertilionid gammaherpesvirus 3 (ATA58242.1).
5. Conclusions
Although the risk of disease transmission is due to a close interrelationship between humans and animals, it is crucial to follow correct procedures to prevent such events. In relation to the recent Covid-19 pandemic, monitoring the presence of other pathogens, such as CoVs, of which bats are known to be reservoirs, is fundamental due to their ability for genetic reassortments and possibly be responsible for interspecies transmission [41]. The zoonotic potential of Viruses discovered in bats has not yet fully been demonstrated, such as Lloviu virus (LLOV), a filovirus closely related to Ebola virus. Despite this, bats are often in contact with humans and domestic animals and they host a number of pathogens that have ecological opportunities to emerge in humans [42].
Due to the reduction of natural habitats and the increase in urbanization, many species have become anthropophilic with a greater probability of contact and interaction with humans [43]. These flying mammals are able to host many pathogens without showing clinical signs of disease, probably because they are capable to develop antiviral immune responses that control virus replication, limiting self-damaging inflammatory responses [13]. Monitoring the presence of viruses and potentially susceptible species is useful in early detection of virus introduction and circulation for timely adoption of sanitary suitable measures of containment and prevention of spread, as proven by the detection in Italy of a LYSVs 7 reported only once in bats, in the Caucasian region many years earlier [44].
This study provides useful data about virus circulation and bat species identification of those present in Latium and Tuscany, central Regions of Italy.
As our country is part of the agreement on the Act for the Conservation of Population of European Bats (UNEP/EUROBATS), which aims to protect all 51 European bat species through specific legislation, education, conservation measures and international co-operation, species identification using genetic sequencing is useful for monitoring species present in a specific area especially whenever the morphological identification could be complicated. Submission of dead bats for laboratory analysis is of crucial importance to detect and identify pathogenic agents, as well as the bat species involved, to be proactive in the prevention of the transmission of pathogens within and between different host species and prevent spillover of microorganisms.
Ethics
Bat carcasses were collected by the local veterinary services and the wildlife animal rescue and rehabilitation centres (Associazione Tutela Pipistrelli and LIPU-Lega italiana protezione uccelli). No animal was killed for diagnostic purposes. Oral swabs were collected in the context of active surveillance against lyssaviruses carried out by the CRN for rabies located at IZSVe, approved by the ministry of health and subjected to authorization for handling by the animal upon positive opinion from ISPRA, in derogation of Presidential Decree 357/97 implementing 92/43/EEC habitat.
Author Contributions
“Conceptualization, S.T., I.R. and M.T.S.; methodology, S.T., G.M., R.C., G.P. and E.T.; software, G.M., R.C. and D.L.R.; validation, G.M.; formal analysis, S.T. and G.P.; investigation, A.L., R.C., G.P., M.S., R.G. and A.D; resources, A.L. and M.S.; data curation, S.T., I.R. and G.M..; writing—original draft preparation, S.T., I.R.,G.M., E.T. and M.Sc.; writing—review and editing, S.T., I.R., G.M., A.C., R.N. and M.T.S.; visualization, S.T., I.R.,G.M. and M.Sc.; supervision, M.T.S..; project administration, M.T.S.; funding acquisition, M.T.S.”. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially supported by the Italian Ministry of Health, Project IZSVE 01/20 RCS, “Suscettibilità dei mammiferi a SARS-COV-2: rischi di zoonosi inversa e possibilità in medicina traslazionale”.
Data Availability Statement
“Not applicable”.
Acknowledgments
we warmly acknowledge Alessandra Tomassini (Associazione Tutela Pipistrelli), Francesca Manzia and the whole personnel from CRFS Lipu of Rome, to CRUMA (Centro Recupero Uccelli Marini e Acquatici, Livorno) and other Wildlife Rehabilitation Centres in the territory for having submitted bat carcasses. Calogero Terregino, Paola De Benedictis, Petra Drzewniokova, Stefania Leopardi and Francesca Festa (Istituto Zooprofilattico Sperimentale delle Venezie) are also acknowledge for continuous support, protocol sharing and results’ harmonization. Stefania Leopardi, Francesca Festa and Dino Scaravelli (Studi Ecologici Ricerca Natura Ambiente - ST.E.R.N.A., Forlì) are finally acknowledged for performing active surveillance on the Miniopterus schreibersii in Tuscany region.
Conflicts of Interest
“The authors declare no conflict of interest.”.
References
- Arnaout, Y.; Djelouadji, Z.; Robardet, E.; Cappelle, J.; Cliquet, F.; Touzalin, F.; Jimenez, G.; Hurstel, S.; Borel, C.; Picard-Meyer, E. Genetic Identification of Bat Species for Pathogen Surveillance across France. PLoS One 2022, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schede mammiferi https://www.mammiferi.org/specie/.
- Ruffo S., S. F. Checklist e Distribuzione Della Fauna Italiana. 10.000 Specie Terrestri e Delle Acque Interne; Agnoli G.L. & Rosa P., Ed.; Verona, 2022.
- Colombi, D.; Serra-Cobo, J.; Métras, R.; Apolloni, A.; Poletto, C.; López-Roig, M.; Bourhy, H.; Colizza, V. Mechanisms for Lyssavirus Persistence in Non-Synanthropic Bats in Europe: Insights from a Modeling Study. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Banyard, A. C.; Evans, J. S.; Luo, T. R.; Fooks, A. R. Lyssaviruses and Bats: Emergence and Zoonotic Threat. Viruses 2014, 6, 2974–2990. [Google Scholar] [CrossRef] [PubMed]
- Černe, D.; Hostnik, P.; Toplak, I.; Presetnik, P.; Maurer-Wernig, J.; Kuhar, U. Discovery of a Novel Bat Lyssavirus in a Long-Fingered Bat (Myotis Capaccinii) from Slovenia. PLoS Negl. Trop. Dis. 2023, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Leopardi, S.; Priori, P.; Zecchin, B.; Poglayen, G.; Trevisiol, K.; Lelli, D.; Zoppi, S.; Scicluna, M. T.; D’Avino, N.; Schiavon, E.; et al. Active and Passive Surveillance for Bat Lyssaviruses in Italy Revealed Serological Evidence for Their Circulation in Three Bat Species. Epidemiol. Infect. 2019, 147. [Google Scholar] [CrossRef] [PubMed]
- Shipley, R.; Wright, E.; Selden, D.; Wu, G.; Aegerter, J.; Fooks, A. R.; Banyard, A. C. Bats and Viruses: Emergence of Novel Lyssaviruses and Association of Bats with Viral Zoonoses in the EU. Trop. Med. Infect. Dis. 2019, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Schatz, J.; Fooks, A. R.; Mcelhinney, L.; Horton, D.; Echevarria, J.; Vázquez-Moron, S.; Kooi, E. A.; Rasmussen, T. B.; Müller, T.; Freuling, C. M. Bat Rabies Surveillance in Europe. Zoonoses Public Health 2013, 60, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Mühldorfer, K.; Speck, S.; Kurth, A.; Lesnik, R.; Freuling, C.; Müller, T.; Kramer-Schadt, S.; Wibbelt, G. Diseases and Causes of Death in European Bats: Dynamics in Disease Susceptibility and Infection Rates. PLoS One 2011, 6(12). [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Seifert, S. N.; Olival, K. J.; Plowright, R. K.; Munster, V. J. Bat-Borne Virus Diversity, Spillover and Emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X. Lou; Wang, X. G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H. R.; Zhu, Y.; Li, B.; Huang, C. L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Baker, M. L.; Kulcsar, K.; Misra, V.; Plowright, R.; Mossman, K. Novel Insights Into Immune Systems of Bats. Front. Immunol. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Festival, C. B.; Vora, N. M.; Osinubi, M. O. V; Davis, L.; Abdurrahman, M.; Adedire, E. B.; Akpan, H.; Aman-oloniyo, A. F.; Audu, S. W.; Blau, D.; et al. Bat and Lyssavirus Exposure among Humans in Area That. Emerg. Infect. Dis. 2020, 26, 1399–1408. [Google Scholar]
- D.P.R. 8 Settembre 1997, n. 357. 1997.
- Jian Yang. DATABASE OF BAT-ASSOCIATED VIRUSES http://www.mgc.ac.cn/cgi-bin/DBatVir/main.cgi?func=update&update=2023-03.
- Pozo, F.; Juste, J.; Vázquez-Morón, S.; Aznar-López, C.; Ibáñez, C.; Garin, I.; Aihartza, J.; Casas, I.; Tenorio, A.; Echevarría, J. E. Identification of Novel Betaherpesviruses in Iberian Bats Reveals Parallel Evolution. PLoS One 2016, 11, 1–17. [Google Scholar] [CrossRef]
- Baldwin, C. C.; Mounts, J. H.; Smith, D. G.; Weigt, L. A. Genetic Identification and Color Descriptions of Early Life-History Stages of Belizean Phaeoptyx and Astrapogon (Teleostei: Apogonidae) with Comments on Identification of Adult Phaeoptyx. Zootaxa 2009, 22, 1–22. [Google Scholar] [CrossRef]
- Wadhwa, A.; Wilkins, K.; Gao, J.; Condori Condori, R. E.; Gigante, C. M.; Zhao, H.; Ma, X.; Ellison, J. A.; Greenberg, L.; Velasco-Villa, A.; Orciari, L.; Li, Y. A Pan-Lyssavirus Taqman Real-Time RT-PCR Assay for the Detection of Highly Variable Rabies Virus and Other Lyssaviruses. PLoS Negl. Trop. Dis. 2017, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Leopardi, S.; Barneschi, E.; Manna, G.; Zecchin, B.; Priori, P.; Drzewnioková, P.; Festa, F.; Lombardo, A.; Parca, F.; Scaravelli, D.; Ponti, A. M.; De Benedictis, P. Spillover of West Caucasian Bat Lyssavirus (Wcbv) in a Domestic Cat and Westward Expansion in the Palearctic Region. Viruses 2021, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- De Benedictis, P.; Leopardi, S.; Markotter, W.; Velasco-Villa, A. The Importance of Accurate Host Species Identification in the Framework of Rabies Surveillance, Control and Elimination. Viruses 2022, 14, 1–11. [Google Scholar] [CrossRef]
- Vandevanter, D. R.; Warrener, P.; Bennett, L.; Schultz, E. R.; Coulter, S.; Garber, R. L.; Rose, T. M. Detection and Analysis of Diverse Herpesviral Species by Consensus Primer PCR. J. Clin. Microbiol. 1996, 34, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, B.; Borchers, K.; Grund, C.; Frölich, K.; Ludwig, H.; Buhk, H. J. Detection of New DNA Polymerase Genes of Known and Potentially Novel Herpesviruses by PCR with Degenerate and Deoxyinosine-Substituted Primers. Virus Genes 1999, 18, 211–220. [Google Scholar] [CrossRef]
- Wilkinson, D. A.; Joffrin, L.; Lebarbenchon, C.; Mavingui, P. Analysis of Partial Sequences of the RNA-Dependent RNA Polymerase Gene as a Tool for Genus and Subgenus Classification of Coronaviruses. J. Gen. Virol. 2021, 101, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Pearce, M.; Tivey, A. R. N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and Sequence Analysis Tools Services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R. C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G. L.; Doallo, R.; Posada, D. ProtTest 3: Fast Selection of Best-Fit Models of Protein Evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G. L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9(8), 772. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52(5), 696–704. [Google Scholar] [CrossRef]
- 3.6.3, R. D. C. T. A Language and Environment for Statistical Computing. R Found. Stat. Comput. 2020, 1, https://www.R-project.org.
- Guangchuang, Yu. Data Integration, Manipulation and Visualization of Phylogenetic Trees, 1st ed.; Chapman and Hall/CRC, Ed.; Taylor & Francis: New York, 2022. [Google Scholar] [CrossRef]
- Yu, G. Using Ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinforma. 2020, 69, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35(3), 526–528. [Google Scholar] [CrossRef] [PubMed]
- Leopardi, S.; Desiato, R.; Mazzucato, M.; Orusa, R.; Obber, F.; Averaimo, D.; Berjaoui, S.; Canziani, S.; Capucchio, M. T.; Conti, R.; Di Bella, S.; Festa, F.; Garofalo, L.; Lelli, D.; Madrau, M. P.; Mandola, M. L.; Martin, A. M. M.; Peletto, S.; Pirani, S.; Robetto, S.; Torresi, C.; Varotto, M.; Citterio, C.; Terregino, C. Erratum: One Health Surveillance Strategy for Coronaviruses in Italian Wildlife (Epidemiology & Infection 151 (E96) DOI: 10.1017/S095026882300081X). Epidemiol. Infect. 2023, 151. https://doi.org/10.1017/S0950268823001000.
- Woo, P. C. Y.; Lau, S. K. P.; Li, K. S. M.; Poon, R. W. S.; Wong, B. H. L.; Tsoi, H. wah; Yip, B. C. K.; Huang, Y.; Chan, K. hung; Yuen, K. yung. Molecular Diversity of Coronaviruses in Bats. Virology 2006, 351, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Chu, D. K. W.; Peiris, J. S. M.; Chen, H.; Guan, Y.; Poon, L. L. M. Genomic Characterizations of Bat Coronaviruses (1A, 1B and HKU8) and Evidence for Co-Infections in Miniopterus Bats. J. Gen. Virol. 2008, 89(5), 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Kohl, C.; Nitsche, A.; Kurth, A. Update on Potentially Zoonotic Viruses of European Bats. Vaccines 2021, 9(7). [Google Scholar] [CrossRef] [PubMed]
- Drexler, J. F.; Gloza-Rausch, F.; Glende, J.; Corman, V. M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; Zhelyazkov, L.; Hermanns, U.; Vallo, P.; Lukashev, A.; Müller, M. A.; Deng, H.; Herrler, G.; Drosten, C. Genomic Characterization of Severe Acute Respiratory Syndrome-Related Coronavirus in European Bats and Classification of Coronaviruses Based on Partial RNA-Dependent RNA Polymerase Gene Sequences. J. Virol. 2010, 84(21), 11336–11349. [Google Scholar] [CrossRef] [PubMed]
- Vasenkov, D.; Desmet, J. F.; Popov, I.; Sidorchuk, N. Bats Can Migrate Farther than It Was Previously Known: A New Longest Migration Record by Nathusius’ Pipistrelle Pipistrellus Nathusii (Chiroptera: Vespertilionidae). Mammalia 2022, 86(5), 524–526. [Google Scholar] [CrossRef]
- Cerri, A.; Bolatti, E. M.; Zorec, T. M.; Montani, M. E.; Rimondi, A.; Hosnjak, L.; Casal, P. E.; Domenica, V. Di; Barquez, R. M.; Poljak, M.; Giri, A. A. Coronavirus Cross-Species Transmission. 2023, 11 (5).
- Chan, J. F. W.; To, K. K. W.; Tse, H.; Jin, D. Y.; Yuen, K. Y. Interspecies Transmission and Emergence of Novel Viruses: Lessons from Bats and Birds. Trends Microbiol. 2013, 21, 544–555. [Google Scholar] [CrossRef]
- Bergner, L. M.; Mollentze, N.; Orton, R. J.; Tello, C.; Broos, A.; Biek, R.; Streicker, D. G. Characterizing and Evaluating the Zoonotic Potential of Novel Viruses Discovered in Vampire Bats. Viruses 2021, 13(2). [Google Scholar] [CrossRef] [PubMed]
- Printz, L.; Jung, K. Urban Areas in Rural Landscapes – the Importance of Green Space and Local Architecture for Bat Conservation. Front. Ecol. Evol. 2023, 11, 1–11. [Google Scholar] [CrossRef]
- Botvinkin, A. D.; Poleschuk, E. M.; Kuzmin, I. V.; Borisova, T. I.; Gazaryan, S. V.; Yager, P.; Rupprecht, C. E. Novel Lyssaviruses Isolated from Bats in Russia. Emerg. Infect. Dis. 2003, 9(12), 1623–1625. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Phylogenetic tree of the RNA-dependent, RNA polymerase gene, partial cds, of coronaviruses. For each strain, the Accession Number, the year and the place of isolation, and known taxonomy classifications, are reported. Colors of the round tips refer to the Coronavirus subgenera. The sequences submitted and analyzed by us are in blue bold. The tree was performed using the HKY+G nucleotide substitution model (BIC 7402.134173), a 1000 bootstrapping value for resampling procedure (Nodes with bootstrapping supporting values greater than 600 are shown and are reported near nodes), and as outgroup a BetaCoVs HM211100.1. The outgroup branch length was manually reduced from 2.15 (original value) to 1.0 to perform the rescaling geom_treescale() function.
Figure 1.
Phylogenetic tree of the RNA-dependent, RNA polymerase gene, partial cds, of coronaviruses. For each strain, the Accession Number, the year and the place of isolation, and known taxonomy classifications, are reported. Colors of the round tips refer to the Coronavirus subgenera. The sequences submitted and analyzed by us are in blue bold. The tree was performed using the HKY+G nucleotide substitution model (BIC 7402.134173), a 1000 bootstrapping value for resampling procedure (Nodes with bootstrapping supporting values greater than 600 are shown and are reported near nodes), and as outgroup a BetaCoVs HM211100.1. The outgroup branch length was manually reduced from 2.15 (original value) to 1.0 to perform the rescaling geom_treescale() function.

Figure 2.
Phylogenetic tree of the RNA-dependent, RNA polymerase partial protein of coronaviruses. For each strain the Accession Number, the year and the place of isolation, and known taxonomy classifications, are reported. Colors of the round tips refer to the Coronavirus subgenera. The sequences submitted and analyzed by us are in blue bold. The tree was performed using the LG+G amino acid substitution model (BIC 2559.40), a 1000 bootstrapping value for resampling procedure (Nodes with bootstrapping supporting values greater than 600 are shown and are reported near nodes), and as outgroup a BetaCoVs ADM33573.1.
Figure 2.
Phylogenetic tree of the RNA-dependent, RNA polymerase partial protein of coronaviruses. For each strain the Accession Number, the year and the place of isolation, and known taxonomy classifications, are reported. Colors of the round tips refer to the Coronavirus subgenera. The sequences submitted and analyzed by us are in blue bold. The tree was performed using the LG+G amino acid substitution model (BIC 2559.40), a 1000 bootstrapping value for resampling procedure (Nodes with bootstrapping supporting values greater than 600 are shown and are reported near nodes), and as outgroup a BetaCoVs ADM33573.1.

Figure 3.
Phylogenetic tree of the DNA polymerase B partial protein, of betaherpesviruses. For each strain the Accession Number, the host and the year of isolation are indicated. Blank values correspond to when information is not available. The sequence deposited and analyzed by us is in blue bold. The tree was performed using the LG+G amino acid substitution model (BIC 5223.60), a 1000 bootstrapping value for resampling procedure (Nodes with bootstrapping supporting values greater than 600 are shown and are reported near nodes), and as outgroup a Sturnira angeli Gammaherpesvirus (QJX19395.1). The tree was rescaled with geom_treescale() function.
Figure 3.
Phylogenetic tree of the DNA polymerase B partial protein, of betaherpesviruses. For each strain the Accession Number, the host and the year of isolation are indicated. Blank values correspond to when information is not available. The sequence deposited and analyzed by us is in blue bold. The tree was performed using the LG+G amino acid substitution model (BIC 5223.60), a 1000 bootstrapping value for resampling procedure (Nodes with bootstrapping supporting values greater than 600 are shown and are reported near nodes), and as outgroup a Sturnira angeli Gammaherpesvirus (QJX19395.1). The tree was rescaled with geom_treescale() function.
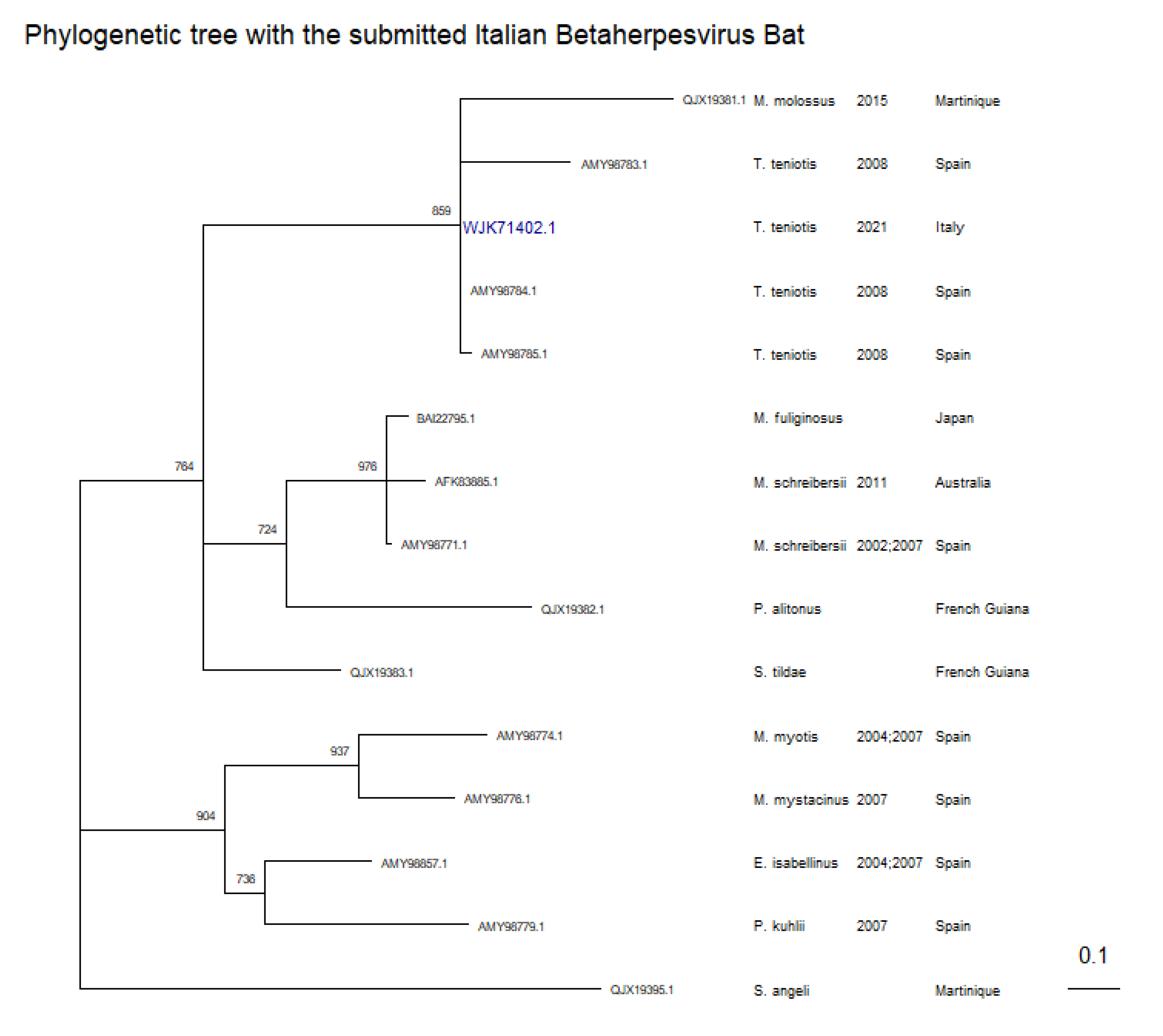

Table 1.
Distribution of bat species per region (*111 samples deriving from oral swabs; + samples analysed during passive surveillance for Herpesvirus were only from Latium: three were from Hypsugo savii, three were from Pipistrellus kuhlii, 71 were from Tadarida teniotis) and PCR test performed.
Table 1.
Distribution of bat species per region (*111 samples deriving from oral swabs; + samples analysed during passive surveillance for Herpesvirus were only from Latium: three were from Hypsugo savii, three were from Pipistrellus kuhlii, 71 were from Tadarida teniotis) and PCR test performed.
| Type of surveillance | Bat species | Total number per species (%) |
Bat species distribution per Region |
Virus family investigated |
||
|---|---|---|---|---|---|---|
| Latium | Tuscany | pan-CoV 21 | pan-Herpes 22,23 | |||
| Passive | Hypsugo savii | 18 (7.5) | 11 | 7 | 18 | 3+ |
| Pipistrellus kuhlii | 23 (9.6) | 9 | 14 | 23 | 3+ | |
| Pipistrellus Sp | 10 (4.2) | 1 | 9 | 10 | 0 | |
| Myotis daubentonii | 1 (0.4) | 0 | 1 | 1 | 0 | |
| Plecotus austriacus | 1 (0.4) | 0 | 1 | 1 | 0 | |
| Not identified | 4 (1.7) | 0 | 4 | 4 | 0 | |
| Tadarida teniotis | 71 (29.7) | 71 | 0 | 71 | 71+ | |
| Active | Miniopterus schreibersii | 111 (46.6) | 0 | 111* | 111 | 64 |
| Total | 239 (100) | 92 | 147 | 239 | 141 | |
Table 2.
Results of nucleotide and protein alignments of M. schreibersii Italy 2/2021 (ON834690.1 nucleotide and UUG60948.1 protein accession numbers) as query against M. schreibersii Italy 1/2021 (OP776451.1 nucleotide and UZC49592.1 protein accession numbers) and M. schreibersii Italy 3/20212021 (OP627105.1 nucleotide and UYI58598.1 protein accession numbers).
Table 2.
Results of nucleotide and protein alignments of M. schreibersii Italy 2/2021 (ON834690.1 nucleotide and UUG60948.1 protein accession numbers) as query against M. schreibersii Italy 1/2021 (OP776451.1 nucleotide and UZC49592.1 protein accession numbers) and M. schreibersii Italy 3/20212021 (OP627105.1 nucleotide and UYI58598.1 protein accession numbers).
| Nucleotide characteristics | Protein characteristics | ||||
|---|---|---|---|---|---|
| Accession number | identity | Querycov | Accession number | identity | Querycov |
| OP776451.1 | 78.08% | 75% | UZC49592.1 | 88.2% | 102/147 |
| OP627105.1 | 100% | 100% | UYI58598.1 | 100% | 147/147 |
Table 3.
Results of pairwise global alignments between the novel strain with accession number WJK71402.1 aligned with other European DNA polymerases sequences. *AN: accession number.
Table 3.
Results of pairwise global alignments between the novel strain with accession number WJK71402.1 aligned with other European DNA polymerases sequences. *AN: accession number.
| AN* | % identity | Similarity score | Starting position | Amino Acids |
|---|---|---|---|---|
| AMY98779.1 | 48 | 97/150 | INS 13 | VRET |
| INS 145 | VI | |||
| AMY98857.1 | 54.0 |
100/150 |
INS 13 | LRDS |
| INS 137 | VN | |||
| AMY98776.1 | 48.1 |
100/160 |
INS 13 | LREE |
| INS 22 | DD | |||
| INS 118 | HAHFVDPEFR | |||
| INS 131 | FGERELSR | |||
| AMY98771.1 | 58.0 |
111/150 |
INS 13 | VATD |
| INS 139 | ER | |||
| AMY98783.1 | 78.5 | 126/144 | / | / |
| AMY98784.1 | 100 | 144/144 | / | / |
| AMY98785.1 | 89.6 | 137/144 | / | / |
| AMY98774.1 | 47.8 | 101/159 | INS 5 | FPENVDM |
| DEL 10 | GPG | |||
| INS 22 | AD | |||
| DEL 121 | PR | |||
| DEL 126 | LS | |||
| INS 137 | VEYLPG |
Table 4.
Results of pairwise global alignment from the alignment of WJK71402.1 (nucleic accession number OQ807168.1) with other members of the taxonomic Betaherpesvirinae family of other animal species. *AN: accession number.
Table 4.
Results of pairwise global alignment from the alignment of WJK71402.1 (nucleic accession number OQ807168.1) with other members of the taxonomic Betaherpesvirinae family of other animal species. *AN: accession number.
| Protein AN* | Virus Name | %Identity | Similarity score | Starting position | Amino Acids | Corresponding nucleic AN* |
|---|---|---|---|---|---|---|
| CCE57225.1:640-788 | Murid betaherpesvirus 1 | 52.7 | 106/150 | INS 7 | EG | HE610455.1 |
| INS 138 | PEA | |||||
| AAK71288.1:10-157 | Baboon cytomegalovirus | 58.1 | 106/148 | INS 9 | G | AF387664.1 |
| INS 16 | QV | |||||
| INS138 | P | |||||
| AAW57296.1:328-477 | Murid betaherpesvirus 2 | 50.7 | 103/150 | DEL 7 | ND | AY728086.1 |
| INS 11 | PEVSR | |||||
| INS 128 | KVI | |||||
| YP_008492977.1:583-733 | Suid betaherpesvirus 2 | 53.0 | 100/151 | INS 10 | DVTGI | NC_022233.1 |
| INS 132 | LS | |||||
| YP_007969814.1:627-764 | Elephantid betaherpesvirus 1 | 43.2 | 88/148 | INS 13 | LREE | NC_020474.2 |
| DEL 117 | QNVLPRSDVL | |||||
| AAP57912.1:454-594 | Human betaherpesvirus 5 | 52.0 | 98/148 | INS 7 | PGGE | AY304055.1 |
| DEL 138 | DVALKVI |
Table 5.
Results of pairwise global alignments between every Gammaherpesvirus bat sequences aligned with the complete novel strain with protein accession number WJK71403.1 (nucleic accession number OQ807169.1). The asterisk indicates that the deletion/insertion are at position 1 or 72 t of the sequence because the aligned sequence has the same length of WJK71403.1 but starts one amino acid after ours. *AN: accession number.
Table 5.
Results of pairwise global alignments between every Gammaherpesvirus bat sequences aligned with the complete novel strain with protein accession number WJK71403.1 (nucleic accession number OQ807169.1). The asterisk indicates that the deletion/insertion are at position 1 or 72 t of the sequence because the aligned sequence has the same length of WJK71403.1 but starts one amino acid after ours. *AN: accession number.
| AN* | % identity | Similarity score | Starting position | Amino Acids |
|---|---|---|---|---|
| AFM85234.1 | 66.7 | 57/72 | / | / |
| AFM85236.1 | 69.4 | 63/72 | / | / |
| AMA67369.1 | 68.1 | 61/72 | / | / |
| ATA58242.1 | 59.0 | 59/78 | DEL 51 | KK |
| INS 60 | PHAPAA | |||
| ATU31554.1 | 57.5 | 52/73 | * | * |
| BBB06458.1 | 70.8 | 63/72 | / | / |
| ATU31556.1 | 64.4 | 61/73 | * | * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



