Submitted:
05 July 2024
Posted:
08 July 2024
Read the latest preprint version here
Abstract
Recently, polyacetylene (PA) is receiving a renewed scientific attention due to its electrical properties potentially useful for energy applications (e.g., fabrication of electrodes for batteries and supercapacitors) and unique functional characteristics (e.g., flame retardant, oxygen scavenger, EMI-shielding, etc.). This chemical compound can be obtained in form of a polyacetylene-PVOH copolymers simply by chemical dehydration of poly(vinyl alcohol) (PVOH), which is a very common type of polymer, widely used in packaging and other technological areas. This very inexpensive chemical reaction for the large-scale synthesis of PA/polyvinylenes has been investigated by reacting PVOH with fuming sulfuric acid at room temperature. In this process, PVOH, shaped in form of film, is dipped in fuming sulfuric acid (i.e., H2SO4 at 95-97%) and, after complete chemical-dehydration, it has been mechanically removed from the liquid phase by using a nylon sieve. The reduction process leads to a substantial PVOH film conversion to PA as it has been proved by infrared spectroscopy (ATR-mode). Indeed, the ATR spectrum of the reaction product included all characteristic absorption bands of PA. The reaction product has been also characterized by UV-Vis spectroscopy in order to evidence the presence in the structure of conjugated carbon-carbon double bonds of various lengths. Differential scanning calorimetry (DSC) and thermogravimetric analysis have been used respectively to investigate the PA solid-state cis-trans isomerization and thermal stability in air and nitrogen, respectively.

Keywords:
Polyacetylene
; Polyenes
; Polyvinylenes
; Polyvinylalcohol
; Chemical-dehydration
; Fuming sulfuric acid
; Infrared spectroscopy
; Optical spectroscopy
; Isomerization
; Thermal analysis (DSC)
1. Introduction
Among the electrically-conductive organic materials, polyacetylene (i.e., -[CH=CH]n-) has been the first intrinsically conductive polymer to be developed. Initially, only the cis- and trans-isomers with undoped conductivity values of respectively 10-11 and 10-6 ohm-1·cm-1 were studied. In the most famous synthesis scheme, polyacetylene (PA) has been obtained by Zielgler-Natta polymerization of acetylene [1]. In particular, a polymeric film resulted on the surface of the glass reactor by exposing the acetylene gas to a titanium salt catalyst. Subsequently, several complex chain and step polymerization methods for the high-regular PA synthesis have been developed (e.g., ring-opening metathesis polymerization from precursor, reverse Diels-Alder reaction processes, modified-Ziegler homogeneous, radiation polymerization) [1].
Polyacetylene (PA) has very attractive electrical and non-electrical properties, potentially useful for different technological applications. In particular, the good electrical conductivity of doped PA can be advantageously exploited in the field of energy for fabricating electrodes for rechargeable batteries, current collectors, supercapacitors, etc. [2,3]. The most important drawback for a PA technological exploitation is represented by the very poor processability of this polymer type, which follows to its complete insolubility in organic solvents and failure to give a molten phase by heating. However, this problem could be overcome by generating PA by chemical transformation of an easily processable polymeric precursor. In this case, the polymeric precursor is first shaped in the required geometry (e.g., planar electrode) and then this piece is chemically converted to polyacetylene. With this purpose, there are a few little known approaches for the PA synthesis that use the conversion of a polymeric precursor to PA. These approaches are based on the dehydroalogenation of polymers [4] and on the polyvinyl alcohol (PVOH) thermal dehydration [5,6,7]. The dehydrochlorination of poly(vinyl chloride) (PVC) by treatment with a very strong base (e.g., t-BuO-K+) in a polar medium has been the first example of PA synthesis based on reducing a plastic precursor, however this elimination reaction never reaches the completion and hence it is unsuitable for producing a highly electrically-conductive material. Differently, the PVOH thermal-dehydration used for PA films synthesis [5,6] has shown great technological potentialities because it leads to a high conversion yield. Before the discovery of the PA Ziegler-Natta synthesis, it was found that extended polyenic groups, (-CH=CH-CH=CH-)n, at that time named: ‘polyvinylenes’, were generated in polyvinyl alcohol (PVOH) molecules simply by exposing the polymer to a very common chemical-dehydrating agent, known as fuming sulfuric acid (H2SO4) [8]. This chemical treatment leads to whole PA molecules by operating under H2SO4 refluxing conditions for a few days. A great potentiality of this very simple chemical approach is represented by the possibility to control the heterogeneous reaction conversion degree. For example, the process of solid PVOH reduction to PA can be end at a desired conversion degree by slightly diluting the sulfuric acid or by acting at room temperature for different time periods. The whole PVOH conversion to PA can result only under very drastic dehydration conditions (i.e., hot/boiling fuming sulfuric acid), while polyene-PVOH copolymers (i.e., -(CH=CH)n-(CH2-CH(OH))m-, with n>m) are usually achieved by polymer dehydration treatments based on H2SO4 aqueous solutions. Thus, depending on the H2SO4 concentration and reaction temperature, different polyene/PVOH ratios are possible in the final linear macromolecular product. The physical properties (color, fluorescence, isomeric composition, etc.) of these polyene-PVOH copolymers significantly differ and consequently the obtained polymeric product is suitable for numerous technological applications (color filters, fluorescent plastics, molecular memories, etc.).
In particular, at quite high concentrations (‘fuming conditions’), sulfuric acid contains a small percentage of sulfuric anhydride (SO3) and this in situ generated chemical specie results an extremely strong dehydrating agent. As well known, fuming sulfuric acid is capable to act on alcohols and polyols at 140-180°C for generating olefins and polyenes [9]; also carbohydrates are readily converted to carbon materials by dehydration with fuming sulfuric acid at room temperature [10]. Similarly, sulfuric acid can act on the hydroxyl groups present in the linear PVOH molecules for generating conjugated polyenic groups of different extensions in the molecular chain, up to the limit case of a whole PA molecule formation. In this case, an hydrocarbon (CnHn) is obtained instead of a carbon material because PVOH has an hydrogen content slightly higher than carbohydrates; indeed, one H2O molecule per each carbon atom is contained in carbohydrates (the carbohydrate formula can be written as: Cm(H2O)n, with m=n or very close) and therefore pure elemental carbon results from their dehydration. Concerning the exact type of chemical specie acting as dehydrating agent under such drastic reduction conditions (i.e., highly concentrated sulfuric acid), a compound named pyrosulfuric acid (H2SO4.xSO3) and also known as disulfuric acid (ySO3.H2O) should be present. However, when PVOH dehydration takes place under milder conditions like, for example, by treatment with H2SO4 aqueous solutions (e.g., 50% by volume of H2SO4) at room temperature, sulfuric anhydride (SO3) is not present and only protons/hydrogenions (H+, H3O+) are involved in the dehydration process. Consequently, acid solutions lead to a mild PVOH reduction to polyene-PVOH copolymers and during this reaction also a chemical cross-linking process with condensated sulfate bridge formation could take place [11].
Here, the possibility to prepare small PA pieces by chemical dehydration of conveniently shaped PVOH films with H2SO4 at room temperature has been investigated. Such a process can be usefully exploited for the preparation of electrode materials, Lithium anode coatings and separators for supercapacitors and rechargeable batteries. In order to prove the formation of cis-PA isomer and to establish its amount, the dehydration product has been characterized by spectroscopic techniques (ATR and UV-Vis) and thermal analysis (DSC and TGA).
2. Experimental Part
When the PVOH semi-crystalline powder (Aldrich, MW=89,000-98,000, 99+% hydrolyzed) was dispersed into pure H2SO4 (J.T. Baker, 95-97%) at room temperature, the dehydration reaction took place immediately, according to the observed color change of white powder to reddish-brown/black. In particular, during this process, H2SO4 is largely absorbed by the PVOH grains and a PVOH/H2SO4 gel is formed. This PVOH/H2SO4 gel rapidly evolves in a new gel system made of PVOH-PA copolymer and sulfuric acid and a very strong H2SO4 absorption is observed too. When dehydration is completed (full conversion of PVOH to PA), the H2SO4 can be quite effectively stripped by distilled water addition.
In order to verify the possibility to isolate the product (reduced polymer), opportunely shaped, from the very aggressive (i.e., strong acid and hygroscopic) liquid medium at dehydration end, this heterogeneous reaction was preferentially performed by reacting PVOH in form of films. Indeed, in this case, the reduced PVOH film can be mechanically separated from the liquid phase simply by using a plastic (nylon) sieve. In particular, PVOH films with the desired thickness (ca. 0.5mm) were prepared by solution-casting technique. The PVOH semi-crystalline powder was dissolved in hot distilled water (3g of PVOH in 30ml of H2O at 70°C) and the obtained stable aqueous solution was cast into a Petri dish. After water evaporation, the achieved films were cut in small pieces and successively dried in oven under vacuum (3h at 60°C) in order to achieve a completely dried reactant. During the heterogeneous reaction (polymeric films dipped in H2SO4 without stirring), the formation of extended polyenic groups in PVOH caused a slow darkening of the initially transparent and colorless films (a copolymer: -(CH=CH)n-(CH2-CH(OH))m- with n>>m resulted). In particular, the films were swollen by sulfuric acid and became gradually reddish-brown and then reddish-black colored. Such darkening process can be attributed to the formation of chromophoric groups in the PVOH films with nonselective absorption in visible spectral region. These chromophores consisted of both conjugated carbon-carbon double bonds and some isolated olefinic groups in the macromolecular backbone. When the darkened residual films were removed from the reaction medium, they had a gelatinous consistency because of the swelling phenomenon due to the adsorbed sulfuric acid.
In a typical preparation, the film was reacted for one week at room temperature, the reduced solid material (swollen by H2SO4) was mechanically separated from the polyene/H2SO4 liquid solution and, in order to eliminate residual sulfuric acid, it was repeatedly washed first by distilled water and then by ethanol in presence of ultrasounds. During the reduced film drying in air, a significant volume decrease occurred. Figure 1A shows a piece of dehydrated PVOH film after such washing/drying treatment. In addition, overtime a little amount of sulfuric acid was segregated at surface of the solid phase probably because of a progressive H2SO4 segregation from the solid material. In addition to the solid-state PVOH dehydration, a small fraction of this polymeric reactant (probably low-molecular-weight PVOH chains) dissolved in the viscous liquid (fuming H2SO4), thus leading to a progressively increasing coloration of the liquid phase. Like the solid phase, the liquid medium coloration changed to reddish-brown and then to reddish-black for the presence of reduced oligomers dissolved in it (see Figure 1B). This remaining part of produced PA can be separated from the reactive medium by precipitating the polymer with distilled water. In particular, the reactive medium was added dropwise to distilled water under magnetic stirring (typically, 80ml of liquid medium was dissolved in 300ml of distilled water) and a PA flocculation process followed. The flocculated PA was separated by vacuum filtration on a paper filter (Whatman 2 filter papers, hydrophilic membrane). In order to completely remove residual sulfuric acid from this product, it was washed repeatedly by distilled water and ethanol, then recovered from the filter by a spatula and dried in air.
Infrared analysis of the PA samples (ATR-mode) was performed by using a Fourier-Transform Infrared Spectrometer (FT-IR) (PerkinElmer, Fronties, with micro ATR). Absorption optical characterization has been performed by using a double-beam UV-Vis spectrophotometer (VWR, UV-6300PC, China). In particular, distilled water, pristine fuming H2SO4 and H2SO4 aqueous solution (50:50 by volume) were used as both solvents and references for pristine PVOH, PA, and polyvinylene, respectively. Quartz cuvettes were used for tests. The solid-state cis-trans isomerization of PA samples was investigated by Differential Scanning Calorimetry (DSC, PerkinElmer, Discovery) and the PA thermal stability was investigated by Thermo-gravimetric Analysis (TGA) (Q500, TA Instruments). Electrical properties of the PA samples were measured by a LCR-meter (UT612, Uni-Trend, China).
3. Results and Discussion
At beginning of the chemical dehydration process, the PVOH films resulted purple colored and also slightly fluorescent, with a white emission, when observed under the UV-light of a mercury-vapor lamp (TLC lamp, short-wave: 254nm) (see Figure 2A,B). Such visible coloration/fluorescence phenomenon, that reduced and then disappeared with progress of PVOH conversion to PA, has been already described in the literature for thermally-dehydrated PVOH films [12]. The visible coloration/fluorescence of partially reduced PVOH could be ascribed to the initial formation of short chains of conjugated carbon-carbon double bonds (i.e., dienes, trienes, etc.). The final polymeric sample was a soft black solid with a density higher than water (relative density: 1.2). The dry reaction product resulted electrically conductive and behaved like a highly resistive material (ca. 10 MΩ/square at 10kHz), as measured by a LCR-meter.
Since the synthesized PA is insoluble in all types of organic solvents (e.g., chloroform, acetone, ethanol), its characterization was carried out at solid state. In particular, the degree of PVOH conversion to polyene molecules was established by ATR analysis of the solid reaction product. Spectroscopic analysis is very sensible to the presence of adsorbed water molecules, which produces a strong and broad signal at 3400cm-1 (OH stretching vibration) and a medium-intensity signal at 1650cm-1 (HOH bending vibration) [13] and these two bands could obscure important sample information within the mid-IR spectrum. Therefore, the reaction product was accurately dried before ATR-mode analysis by mildly heating the sample under vacuum first at 40°C for 8h and then at 60°C for 3h. The ATR spectrum of the pure dehydrated product is shown in Figure 3A. The incomplete reduction of the polyalcohol to an unsaturated hydrocarbon is readily noticed for the presence of residual hydroxyl groups (OH) absorption in the spectrum. Indeed, these groups produce the characteristic intense and broad absorption band due to the O-H stretching vibration, which is centred at 3398cm-1. Such absorption is accompanied by the small intensity absorption band due to C-O stretching vibration at 1041cm-1 and a less intensive OH bending vibration band located at 1250cm-1. The small IR resonance appearing at 2934cm-1 is generated mainly by the terminal methyl groups, that are present also in the PA molecules but also the stretching vibration of methylene/methine groups in residual PVOH may contribute. Such aliphatic groups also generate bending vibrations appearing at 1039cm-1. As for the case of -OH stretching, these signals have strongly reduced intensities compared to that in pristine PVOH spectrum (see spectral comparison in Figure 2B). However, the IR spectrum of reduction product also shows clear evidences of conjugated olefinic groups formation. Indeed, the IR spectrum of the dehydration product contains the four main characteristic absorption bands generated by conjugated olefinic groups: conjugated carbon-carbon double bond (C=C) stretching vibration, conjugated C-C single bond stretching vibration, in-plane =C-H olefinic (vinyl) =C-H bending vibrations, and out-of-plane olefinic =C-H bending vibrations [14,15,16,17,18,19]. In particular, the C=C stretching vibration band appears at a wavenumber of ca. 1632cm-1 and it is characterized by medium intensity absorption. This band extents over a wide spectral region (from ca. 1602cm-1 to ca. 1705cm-1) because the conjugation phenomenon variously extends in the linear polymer chains and even isolated C=C could be present in these molecules. In particular, the wavenumber of the carbon-carbon double bond absorption band decreases with the extent of conjugation since the bond order (i.e., force) reduces. On the other hand, owing to the same conjugation phenomenon, the C-C single bond stretching absorption appears at higher wavenumber (i.e., 1387cm-1), with an absorption band of quite high intensity. Very intensive absorption bands are also generated by the in-plane =C-H bending resonance, that is visible at 1192 cm-1, and by the out-of-plane (oop) =C-H bending absorptions, that are located at 851cm-1 for the cis-isomer and at 902cm-1 for the trans-isomer. In particular, the comparison between the intensities of these =C-H oop bending vibration bands allows to easily distinguish the product between cis and trans isomers [17]. In the present case, both isomers seems to be contained in the dehydrated product; however, according to the band intensities, conformation mostly corresponded to the cis-type. The generated molecular structure should be largely consisting of cis-PA, also because cis-alkenes have a non symmetric structure, and therefore they are capable to absorb more strongly than trans-alkenes at 1632cm-1, as found for our sample. It must be pointed out that polyenes should show also vinyl =C-H bond stretching absorption, appearing at ca. 3080cm-1, but in our PA sample this band is probably obscured by the broader residual hydroxyls absorption centered at ca. 3000cm-1 (stretching vibration). Yet carbon-carbon double bond (C=C) bending vibration is located outside the explored infrared spectral region (i.e., below 400cm-1 [14]). Finally, comparison between the ATR spectra of pristine and dehydrated PVOH, shown in Figure 3B, clearly evidences as the most intensive absorptions bands of the two compounds do not correspond. The fuming sulfuric acid treatment has caused variations in the intensity and position of bands and the appearance of completely new absorptions corresponding mostly to that of a cis-rich PA sample.
Similarly, the process of PVOH dehydration caused a radical modification of the polymeric material optical properties. Indeed, the perfectly transparent and colorless PVOH film, whose optical absorption spectrum, measured on PVOH aqueous solution by a double-beam UV-Vis spectrophotometer, showed no absorption peaks above 400nm (see the red-curve in Figure 4A). This type of spectrum indicates the absence of conjugated carbon-carbon double bonds as defects in the original polymeric sample. Differently, the spectrum of the dehydrated fraction recovered from the liquid phase showed a strong absorption band in the visible spectral region whose exact profile strictly depended on the dehydration extent. In particular, as visible in Figure 4A, this electronic absorption spectrum of PA was characterized by a very broad absorption band, which extended over the 250-600nm spectral range and was generated by the convolution of five main elementary absorptions with maxima located at 272, 324, 433, 489, and 589nm, that are generated by π→π* electronic transitions. These elementary bands correspond to the optical absorption of the generated linear polyenes, that are characterized by a variable number of conjugated carbon-carbon double bonds as predicted by the Fieser-Kuhn law [20]. In particular, the positioning of some bands up to a quite high wavelength value indicates the presence in the product of polyvinylene unities with very extended conjugation (higher than 14 carbon-carbon double bonds). The observed UV-Vis absorption bands corresponded exactly to the optical behavior usually ascribed to PA [21] (these spectra were obtained by using sulfuric acid as both dispersing medium for the PA molecules/nanoparticles and reference).
Polyenes obtained by reacting PVOH with H2SO4 aqueous solutions (e.g., 50% by volume of H2SO4) show a quite similar UV-Vis spectrum (see Figure 4B). The temporal evolution of the liquid phase optical spectrum during the chemical dehydration reaction by fuming H2SO4 at room temperature is shown in Figure 4C; these spectra have been acquired at time intervals of 15 minutes. According to the distribution of peak intensities in the spectra shown in Figure 4A–C, highly conjugated carbon-carbon double bonds are much more abundant in PVOH films treated by fuming H2SO4 respect to the case of dehydration by concentrated H2SO4 aqueous solution (50% by volume of H2SO4). The optical spectrum of the purified PA solid film is shown in Figure 4D. As visible, the absorbance spectrum showed a continuum absorption band, covering half of the visible spectral region and it was characterized by a slightly visible fine structure.
Solutions of PVOH-PA copolymer in fuming H2SO4 optically behave according to the Fieser-Kuhn equation, written with numerical coefficients adequate for this molecular structure. In particular, the mathematical expression to predict the spectral properties of the PVOH-PA copolymers assumes the following parabolic form:
where λmax is the wavelength of the electronic band maximum absorption and n is the number of conjugated carbon-carbon double bonds in the copolymer. Indeed, the PVOH blocks present along the copolymer molecules do not contribute to the electronic spectrum, because they have only n→σ* and σ→σ* transitions that fall outside the experimentally measured ultraviolet spectral region (i.e., below 190nm). Only the various polyene families in the copolymer chains, that are characterized by different lengths, may contribute to the optical spectrum. In particular, the above mathematical law predicts an increase of the wavelengths of absorption maxima of polyene bands up to 14 conjugated carbon-carbon double bonds and then a decrease of the band wavelength. In particular, with increasing of polyene block extension (n) from one to 14, the band moves from the ultraviolet spectral region (UV-A) to the middle of the visible spectral region, while it comes back to the blue region and then to the ultraviolet spectral region with further increasing of polyene block extension (i.e., n>14). A graphical representation of the absorption band wavelength vs. the number of carbon-carbon double bonds present in each polyene unit is given in Figure 5.
λmax = -1.7·n2+48·n+124
According to the differential scanning calorimetry (DSC) thermogram (1st run, 10°C/min) shown in Figure 6, the obtained chemical product undergoes an exothermic thermal transition in the 100-150°C temperature rage. Such exothermic transition should correspond to a solid-state cis-trans isomerization [17], which consists in the conversion of the as synthesized cis-isomer to the more thermodynamically stable trans-isomer. However, this thermally-activated solid-state transition shows a complex behavior; indeed, the exothermic signal is superimposed to an endothermic signal. The phenomenon starts as an exothermic process (I step of thermal activation in the range 100-150°C), but it can be accomplished only by a simultaneous endothermic collapse of the previously formed cis-PA crystalline structure (II step from 150°C to ca. 220°C). However, the melting peak seems constituted by the overlapping of three different endothermic signals that are probably produced by different phenomena like the evaporation of contaminants (sulfuric acid, water and other solvents trapped in the PA crystalline structure) and/or the thermal dehydration of residual PVOH blocks.
Obviously, such thermal behavior is an irreversible phenomenon; indeed, as shown in Figure 7A,B, there are not signals in the subsequent DSC cooling/heating runs performed on the same DSC specimen (a thermal decomposition is visible in Figure 7B, at temperatures above 300°C). Indeed, the generated trans-PA isomer can melt (and completely decompose) only above the investigated temperature range (i.e., at ca. 467°C [22]). Owing to these unique thermal properties of the synthesized cis-PA, the material could be technologically exploited for developing new types of thermally activated molecular memories [23].
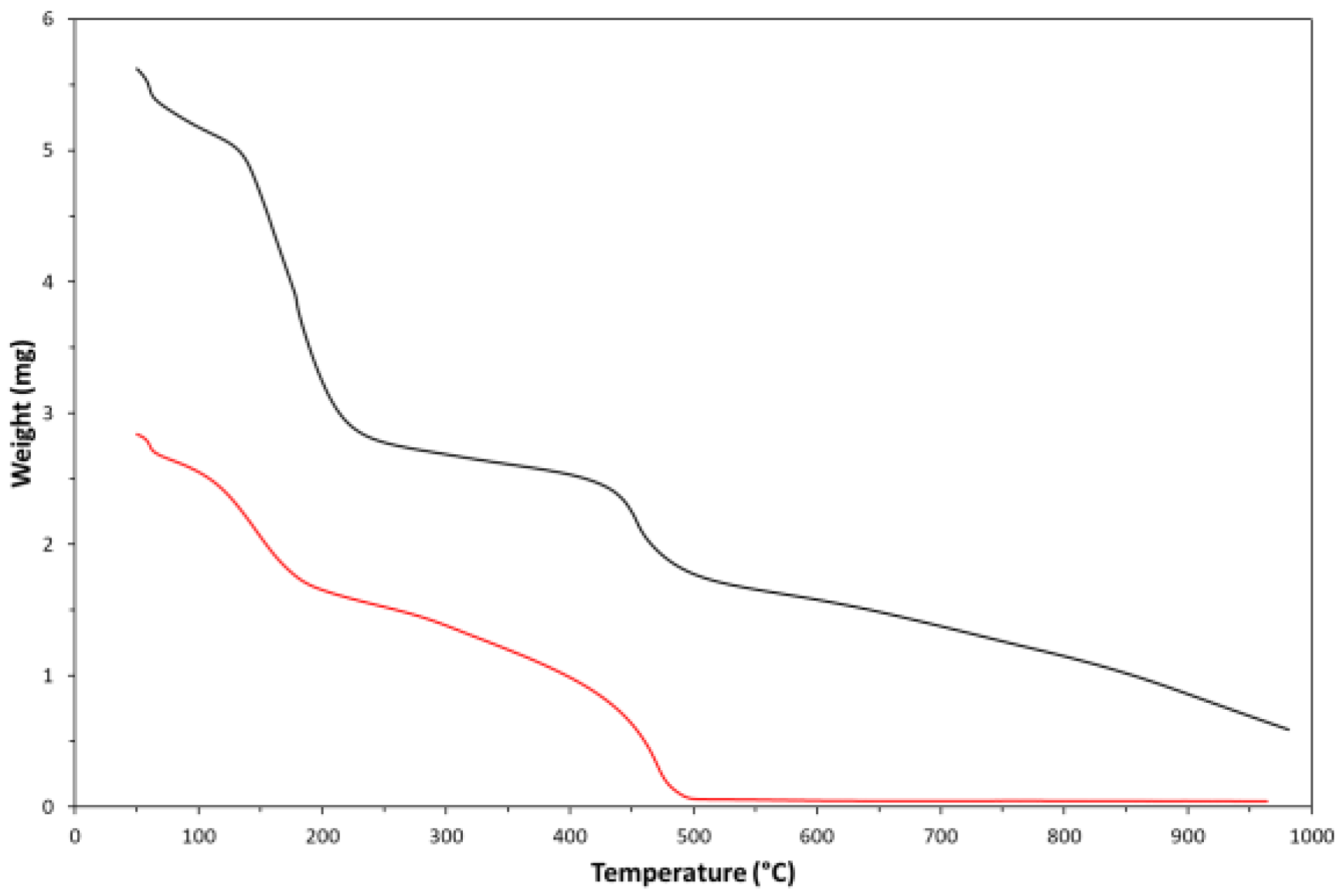
The synthesized cis-PA was investigated by thermogravimetric analysis (TGA), both in fluxing nitrogen and air atmospheres, to evaluate its thermal stability. As visible in Figure 8, both TGA-thermograms showed a quite similar behaviour, which is substantially characterized by two weight-losses, occurring at quite different temperature values. The first weight-loss is centered at ca. 150°C and it is probably due to different phenomena like for example the thermal dehydration of residual PVOH blocks present in the copolymer chain, the elimination by evaporation of H2SO4 molecules trapped in the copolymer, etc. The second weight-loss is centered at ca. 450°C and it could be related to molecular hydrogen elimination from the PA molecules [24]. Therefore, the mechanism of thermal decomposition should be the same, independently from the type of used atmosphere. The ratio between these two weight variations depends on the conversion degree of the chemical dehydration process. The residual weights in the two thermograms have different values (0.05 mg in air at 964°C) and according to these values the synthesized PA sample was characterized by an exceptional thermal stability in nitrogen atmosphere. Indeed, 30-40% of the polymer endured the temperature of ca. 1000°C under nitrogen (and up to ca. 500°C in air). Such a behaviour could be explained on the basis of a ‘quasi-carbon’ nature of this type of polymer.
4. Conclusion
In conclusion, a substantial PVOH chemical transformation has occurred during the polymeric precursor room temperature treatment by fuming sulphuric acid. The IR spectral assignments have shown as the chemically synthesized compound mostly corresponded to a cis-rich PA sample with some residual alcoholic chain portions (PVOH) in its molecule. Such conformational analysis based on the ATR data has been confirmed by DSC; and TGA has confirmed the presence of residual PVOH blocks in the copolymer chain. The UV-Vis investigation of the synthesized product has shown the presence of variously extended conjugated carbon-carbon double bonds along the polymer chain.
About the future perspectives of this study, PA has been considered potentially useful as polymer principally for its very good electrical conductivity after doping, that can be exploited in the energy field for fabricating electrodes for rechargeable batteries, supercapacitors, etc. PA films have been used also as coatings for anodes in Li-batteries in order to prevent dendrite formation, which may cause rechargeable battery explosion [25]. In addition, PA could be very useful also in other technological fields for producing coatings, films or filaments to be used as electrical wires, sensors, electrical collectors, etc. Owing to the PA infusibility and insolubility in all organic solvents, the PVOH precursor polymer should be used to fabricate electrodes with the desired shape and then this component should be converted to the electrically conductive PA by the H2SO4 treatment. As described in the literature [26], PA can be produced also as nano-powder to make possible its processing in form of colloidal suspension. However, PA could have also a number of non-electrical applications [27,28,29,30,31], where it is used in form of powder or granular material. For example, owing to the presence of a very large number of carbon-carbon double bonds, PA can be exploited for EMI shielding, gas-trap, antioxidant, oxygen scavenger, adsorbent for organic substances, hydrogen storage medium (in a transition-metal decorated form), etc. The here described very simple chemical reaction scheme allows to produce high quality cis-PA films or powders to be used for both electrical and non-electrical applications. In addition, this approach could be used also for reusing a PVOH plastic waste; indeed, PVOH is an appealing target for recycling because it is a high-production-volume polymer.
Acknowledgments
The authors are grateful to Maria Rosaria Marcedula of IPCB-CNR for the technical support and useful discussion.
References
- Olabisi, Olagoke, and Kolapo, Adewale, eds. Handbook of thermoplastics. Vol. 41. CRC press, 2016. Cap. 34.
- G.C. Farrington, R. Huq, “Polyacetylene electrodes for non-aqueous lithium batteries”, Journal of Power Sources 14(1-3)(1985)3-9. [CrossRef]
- T. Nagatomo, C. Ichikawa, O. Omoto, “All-plastic batteries with polyacetylene electrodes”, J. Electrochem. Soc. 134(2)(1987)305-308.
- S.E. Eusyukov, Yu P. Kudryavtsev, Yu V. Korshak, “Chemical dehydroalogenation of halogen-containing polymers”, Russian Chemical Reviews 60(4)(1991)373-390.
- I. Yu. Prosanov, N.F. Uvarov, “Electrical properties of dehydrated polyvinyl alcohol”, Physics of the Solid State 54(2)(2012)421-424. [CrossRef]
- I. Yu. Prosanov, A.A. Matvienko, “Study of PVA thermal destruction by means of IR and Raman spectroscopy”, Physics of the Solid State 52(10)(2010)2203-2206. [CrossRef]
- I. Yu. Prosanov, A.A. Matvienko, B.B. Bokhonov, “Influence of urea on polyvinyl alcohol molecular superstructure formation”, Physics of the Solid State 53(6)(2011)1302-1306. [CrossRef]
- S.B. Mainthia, P.L. Kronick, M.M. Labes, “Electrical measurements on polyvinylene and polyphenylene”, J. Chem. Phys. 37(1962)2509-2510. [CrossRef]
- D.J. Ward, D.J. Saccomando, G. Walker, S.M. Mansell, “Sustainable routes to alkenes: applications of homogeneous catalysis to the dehydration of alcohols to alkenes”, Catal. Sci. Technol. 13(9)(2023)2638-2647. [CrossRef]
- D.A. Dolson, R. Battino, T.M. Letcher, K.H. Pegel, N. Revaprasadu, “Carbohydrate dehydration demonstration”, J. Chem. Edu. 72(10)(1995)927. [CrossRef]
- B.S. Minhas, D.G. Peiffer, J.L. Soto, D.L. Stern, “Process for the recovery of sulfuric acid using polymeric membranes, PTC patent, WO 2004/074811 A2 (2 September 2004).
- K. Iwahana, P. Knoll, H. Kuzmany, M. Riegler, B. Hubmann, “Luminescence from trans-polyacetylene degraded by laser irradiation”. In: Kuzmany; H., Mehring, M.; Roth, S. (eds.) Electronic Properties of Polymers and Related Compounds. Springer Series in Solid-State Sciences, Vol. 63, Springer, Berlin, Heidelberg (1985).
- C. Hill, M. Altgen, P. Penttilä, L. Rautkari, “Review: interaction of water vapour with wood and other hygro-responsive materials”, J. Mater. Sci. 59(2024)7595-7635. [CrossRef]
- H. Shirakawa, S. Ikeda, “Infrared spectra of poly(acetylene)”, Polymer Journal 2(2)(1971)231-244. [CrossRef]
- F. Cataldo, “A spectroscopic study of polyacetylene prepared by using Rh(I) catalysts”, Polymer 35(24)(1994)5235-5240. [CrossRef]
- J.-Y. Kim, J.-T. Kim, M.-H. Kwon, D.-K. Han, S.-J. Kwon, “ATR-infrared spectroscopic study of n-doped polyacetylene films”, Macromolecular Research 15(1)(2007)5-9. [CrossRef]
- H.W. Gibson, S. Kaplan, R.A. Mosher, W.M. Prest, R.J. Weagley, “Isomerization of polyacetylene films of the Shirakawa type-spectroscopy and kinetics”, Journal of the American Chemical Society 108(22)(1986)6843-6851. [CrossRef]
- B. Alvarez, A. Sarmiento-Santos, E. Vera-Lopez, “Acetylene polymerization in plasma of direct current”, Journal of physics: Conference Series 1386(2019)012044. [CrossRef]
- Huan Li, G. Chen, P.N. Duchesne, P. Zhang, Y. Dai, H. Yang, B. Wu, S. Liu, C. Xu, N. Zheng, “A nanoparticulate polyacetylene-supported Pd(II) catalyst combining the advantages of homogeneous and heterogeneous catalysts”, Chinese Journal of Catalysis 36(2015)1560-1572. [CrossRef]
- D.C. Nandgaye, A.N. Daf, U.B. Lade, D.W. Moharkar, “Woodward Fisher regulation for calculating absorption maxima”, International Journal of Pharmaceutical and Bio-Medical Science 3(7)(2023)340-344.
- W. Zhao, Y. Yamamoto, S. Seki, S. Tagawa, “Formation of conjugated double bonds in poly(vinyl alcohol) film under irradiation with γ-rays at elevated temperature”, Chemistry Letters (1997)183184. [CrossRef]
- F.D. Kleist, N.R. Byrd, “Preparation and properties of polyacetylene”, J. Polym. Sci.: Part A-1 7(1969)3419-3425. [CrossRef]
- H.-J. Yen, C. Sham, L. Wang, P. Xu, M. Zhou, H.-L. Wang, “Development of conjugated polymers for memory device applications”, Polymers 9(1)(2017)25. [CrossRef]
- T. Luo, X. Xu, M. Jiang, Y.-z. Lu, H. Meng, C.-x. Li, “Polyacetylene carbon materials: facile preparation using AlCl3 catalyst and excellent electrochemical performance for supercapacitors”, RSC Adv. 9(2019)11986. [CrossRef]
- D.G. Belov, O.V. Yarmolenko, A. Peng, O.N. Efimov, “Lithium surface protection by polyacetylene in situ polymerization”, Synthetic Metals 156(2006)745-751. [CrossRef]
- V.M. Kobryanskii, “Nanopolyacetylene: optical properties and practical application”, Proceedings of SPIE, Volume 4277, Integrated Optics Devices V, Symposium on Integrated Optics, 2001, San Jose, CA, United States.
- H. Lee, W. Ih Choi, J. Jhm, “On hydrogen storage in metal-decorated trans-polyacetylene”, Journal of Alloys and Compounds 446-447(2007)373-375. [CrossRef]
- J. Liang, C. Song, J. Deng, “Optically active microspheres constructed by helical substituted polyacetylene and used for absorption of organic compounds in aqueous systems”, ACS Appl. Mater. Interfaces 6(21)(2014)19041-19049.
- M. Chen, G. Hu, T. Shen, H. Zhang, J.Z. Sun, B.Z. Tang, “Applications of polyacetylene derivatives in gas and liquid separation”, Molecules 28(2023)2748. [CrossRef]
- H. Li, G. Chen, P.N. Duchesne, P. Zhang, Y. Dai, H. Yang, B. Wu, S. Liu, C. Xu, N. Zheng, “A nanoparticulate polyacetylene-supported Pd(II) catalyst combining the advantages of homogeneous and heterogeneous catalysts”, Chinese Journal of Catalysis 36(2015)1560-1572. [CrossRef]
- A. Sebastian, A. Raghavan, “Advanced polymer composite with graphene content for EMI shielding”, International Research Journal on Advanced Engineering Hub 2(5)(2024)1334-1340.
Figure 1.
– Piece of synthesized PA as isolated from the reaction medium (A) and fuming sulfuric acid containing the dehydrated PVOH oligomers (B).
Figure 1.
– Piece of synthesized PA as isolated from the reaction medium (A) and fuming sulfuric acid containing the dehydrated PVOH oligomers (B).

Figure 2.
- A drop of fuming sulfuric acid dyes of purple a PVOH film (A) and this colored spot results also fluorescent (white emitting) under UV-light (254nm) exposure (B).
Figure 2.
- A drop of fuming sulfuric acid dyes of purple a PVOH film (A) and this colored spot results also fluorescent (white emitting) under UV-light (254nm) exposure (B).

Figure 3.
- ATR spectrum of dehydrated PVOH film (A) and overlapping of PVOH and dehydrated-PVOH spectra (B).
Figure 3.
- ATR spectrum of dehydrated PVOH film (A) and overlapping of PVOH and dehydrated-PVOH spectra (B).
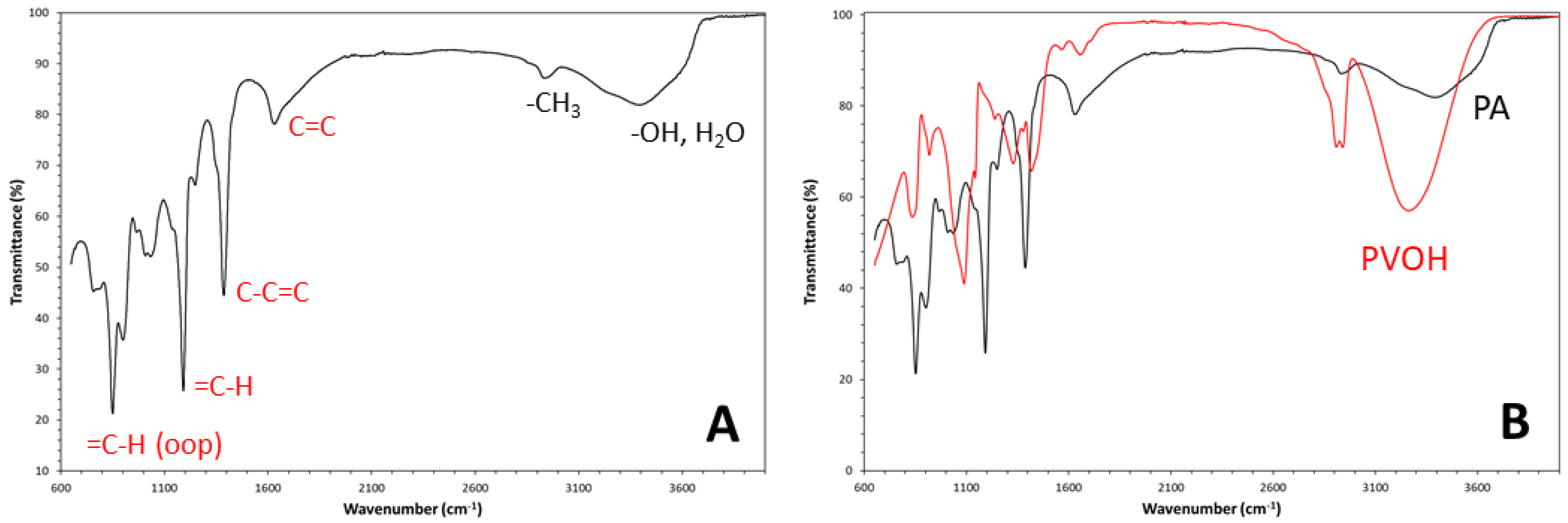
Figure 4.
– UV-Vis spectra of the starting PVOH film (H2O was used as solvent/reference) and reaction product in liquid phase (H2SO4 was used as solvent/reference) (A); similar comparison between PVOH and polyene UV-Vis spectra (H2SO4 aqueous solution 50:50 by volume was used as solvent/reference) (B); temporal evolution of UV-Vis spectrum of the reactive liquid phase (spectra were recorded at time intervals of 15min, using H2SO4 as reference) (C); and UV-Vis spectrum of the solid phase (D).
Figure 4.
– UV-Vis spectra of the starting PVOH film (H2O was used as solvent/reference) and reaction product in liquid phase (H2SO4 was used as solvent/reference) (A); similar comparison between PVOH and polyene UV-Vis spectra (H2SO4 aqueous solution 50:50 by volume was used as solvent/reference) (B); temporal evolution of UV-Vis spectrum of the reactive liquid phase (spectra were recorded at time intervals of 15min, using H2SO4 as reference) (C); and UV-Vis spectrum of the solid phase (D).
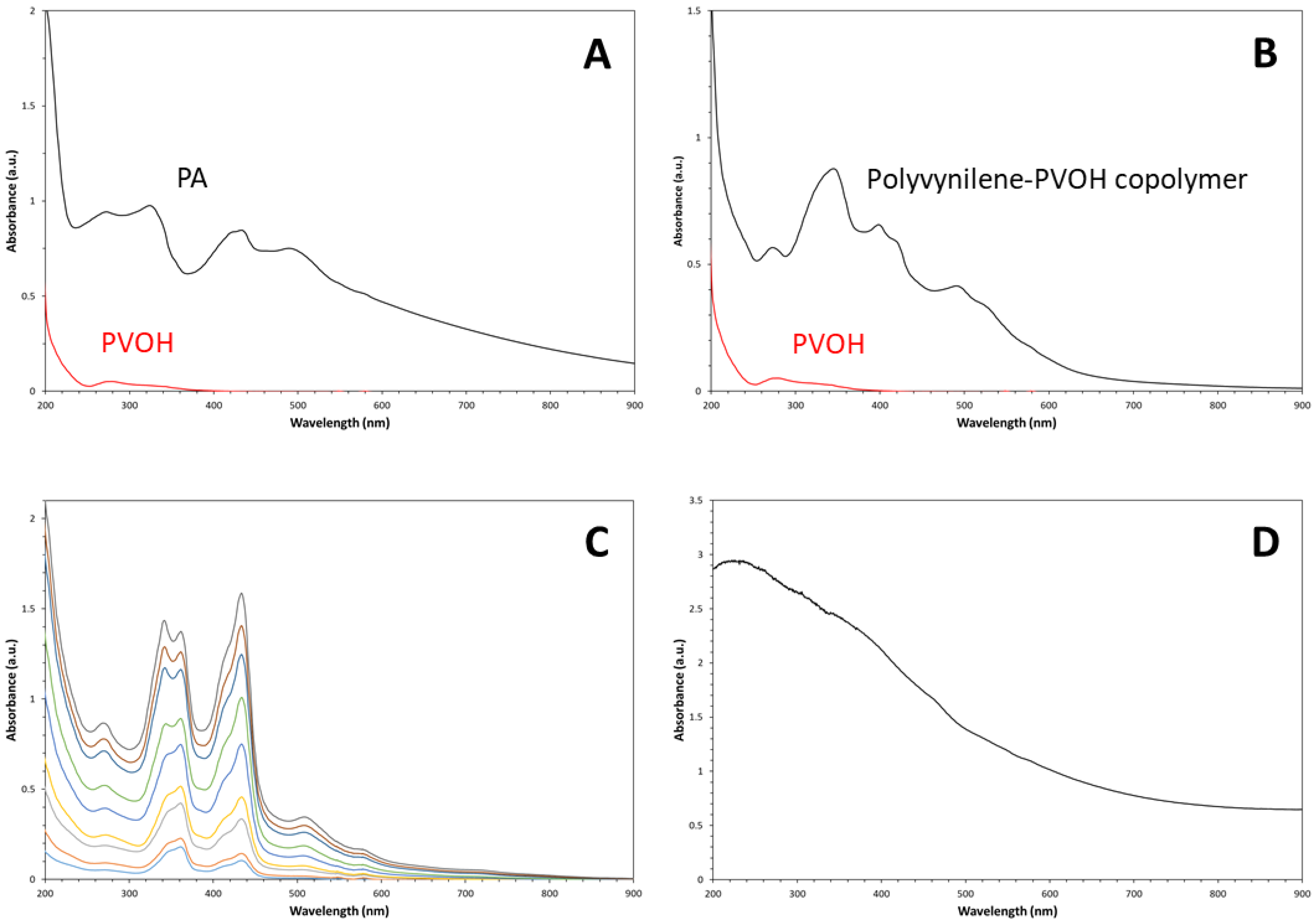
Figure 5.
–Behavior of the polyene block absorption wavelength (electronic band peak) with the number of carbon-carbon double bonds in the block.
Figure 5.
–Behavior of the polyene block absorption wavelength (electronic band peak) with the number of carbon-carbon double bonds in the block.
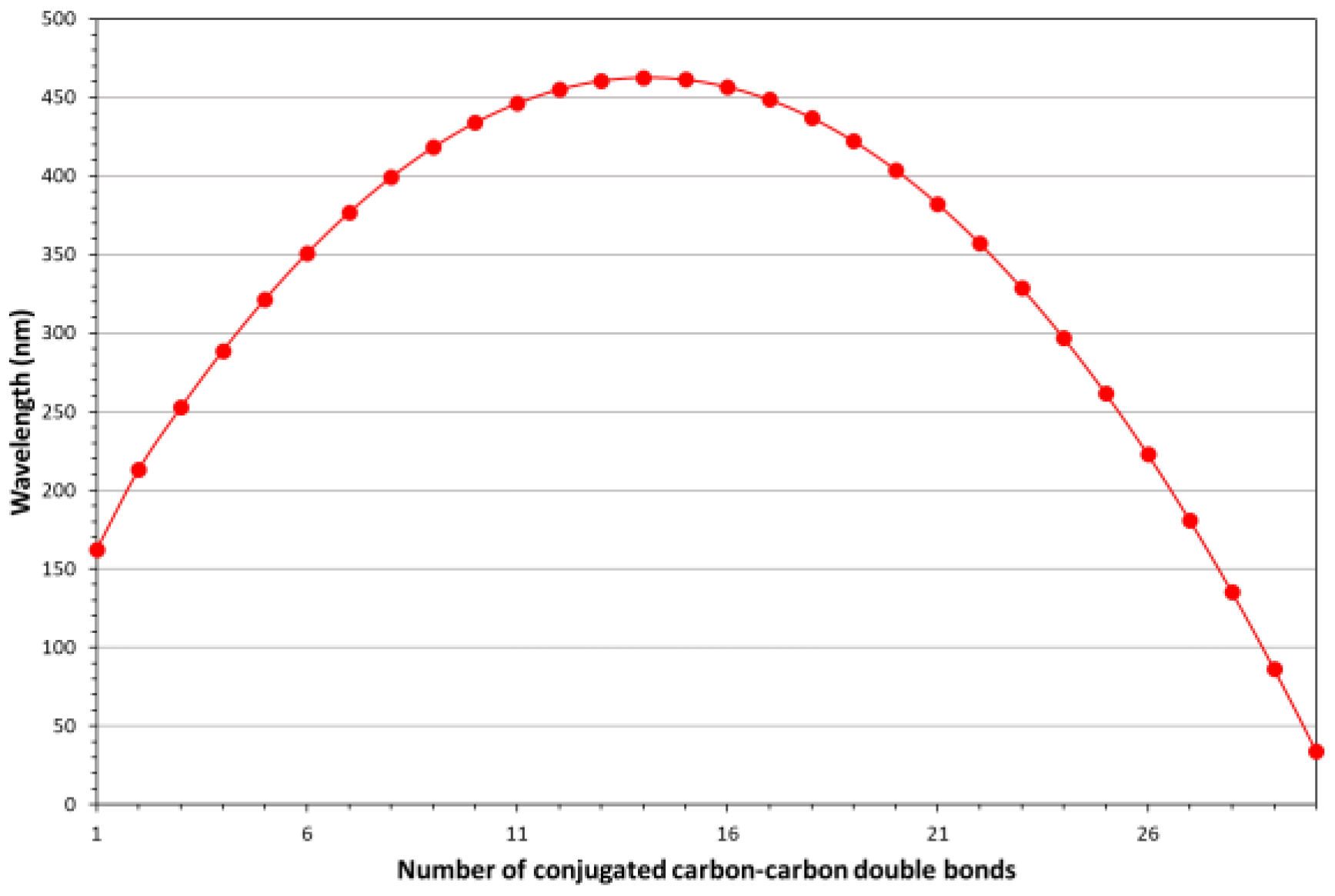
Figure 6.
- DSC-thermogram (1st heating run) showing the solid-state cis-trans isomerization of the synthesized PA.
Figure 6.
- DSC-thermogram (1st heating run) showing the solid-state cis-trans isomerization of the synthesized PA.
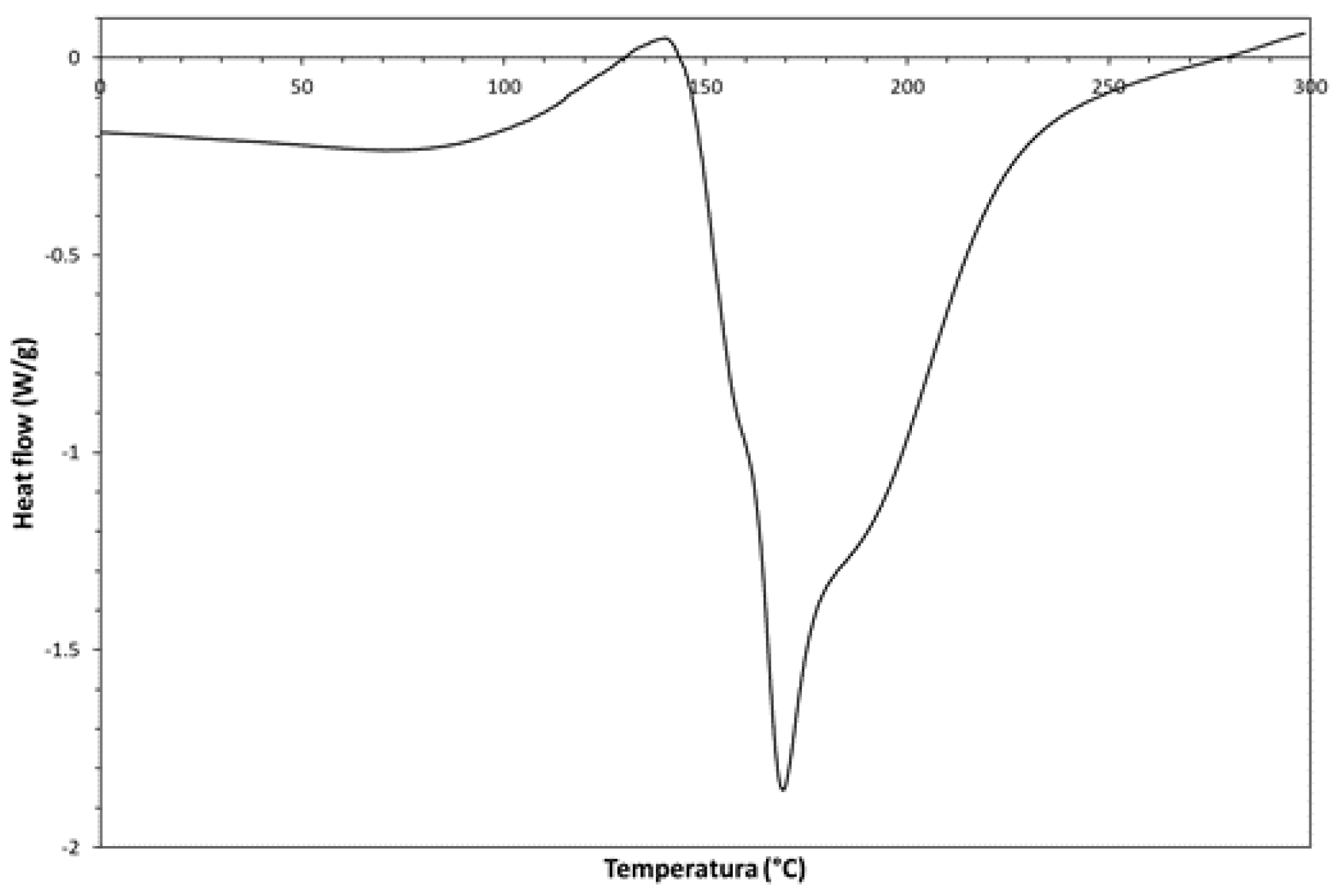
Figure 7.
– DSC thermograms (second and third runs): sample cooling thermogram from 300°C to -80°C (10°C/min) (A) and sample heating thermogram from -80°C to 400°C (10°C/min) (B).
Figure 7.
– DSC thermograms (second and third runs): sample cooling thermogram from 300°C to -80°C (10°C/min) (A) and sample heating thermogram from -80°C to 400°C (10°C/min) (B).
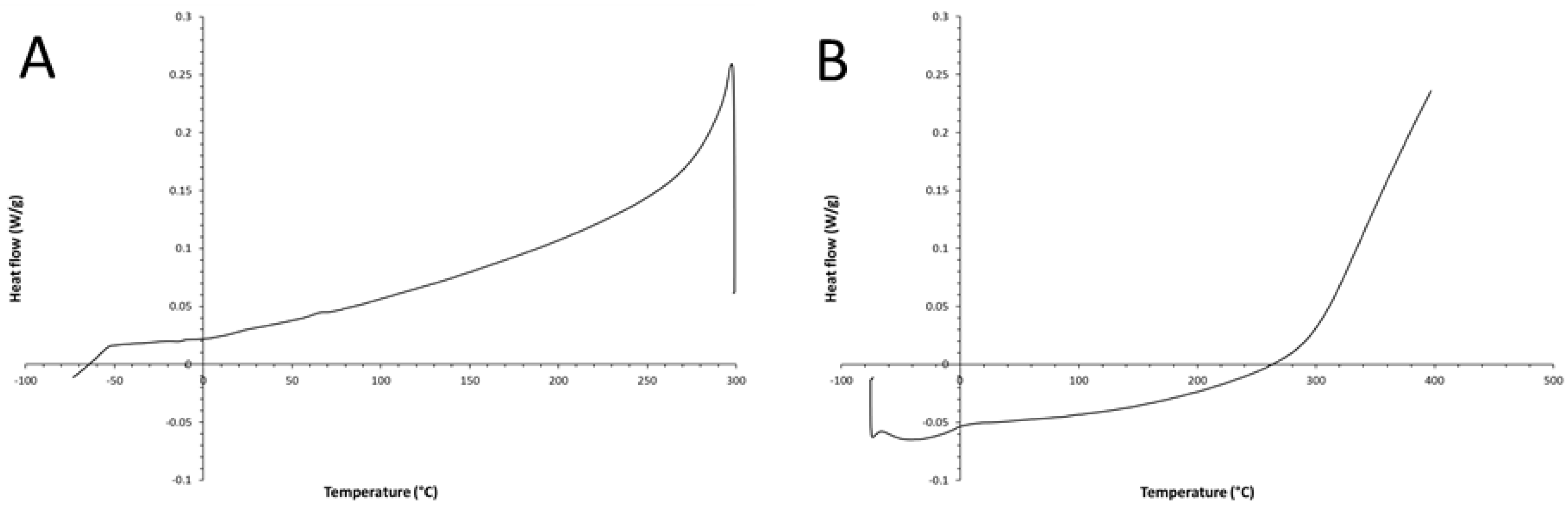
Figure 8.
- TGA thermogram of the synthesized cis-polyacetylene sample in fluxing air (red curve) and nitrogen (black curve).
Figure 8.
- TGA thermogram of the synthesized cis-polyacetylene sample in fluxing air (red curve) and nitrogen (black curve).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



