Submitted:
27 April 2024
Posted:
30 April 2024
You are already at the latest version
Abstract
Aβ peptides are known to bind neural plasma membranes in a process leading to the deposit of Aβ-enriched plaques. These extracellular structures are characteristic of Alzheimer’s disease, the major cause of late-age dementia. The mechanisms of Aβ plaque formation and deposition are far from being understood. A vast amount of literature describes the efforts to analyze those mechanisms, using a variety of tools. The present review focuses on biophysical studies, mostly carried out with model membranes, or with computational tools. The review starts by describing basic physical aspects of lipid phases, and commonly used model membranes (monolayers and bilayers). This is followed by a discussion of biophysical techniques applied to these systems, mainly but not exclusively Langmuir monolayers, isothermal calorimetry, density-gradient ultracentrifugation, and molecular dynamics. The methodological section is followed by the core of the review, a summary of important results obtained with each technique. The last section is devoted to an overall reflection and an effort in understanding Aβ-bilayer binding. Concepts such as Aβ peptide membrane binding, adsorption, and insertion are defined and differentiated. The roles of membrane lipid order, nanodomain formation, and electrostatic forces in Aβ-membrane interaction are separately identified and discussed.
Keywords:
Aβ42
; β-amyloid
; Aβ membrane binding
; ganglioside
; sphingomyelin
; cholesterol
; isothermal calorimetry
; Langmuir balance
; Alzheimer’s disease
1. Introduction
The neurodegenerative disorder known as Alzheimer's disease (AD) is the most frequent cause of late-age dementia [1]. It is characterized by a progressive loss of memory and cognitive abilities. Plaques were detected on the central nervous system structures from the very early descriptions of the disease, but it was only in 1984 when Glenner and Wong [2] put forward the idea that the cause of AD disease could be the accumulation of the amyloidogenic peptide Aβ. These authors showed that this serum peptide, which they had isolated, would be the chief component of amyloid plaques. An amyloid precursor protein (APP) is sequentially processed by β- and γ-secretases [2,3], giving rise to the pathogenic Aβ peptide. Aβ contain 39–43 amino acid residues. The most common isoforms are Aβ40 and Aβ42, the latter considered to be particularly pathogenic [4]. Excessive APP cleavage, or insufficient Aβ clearance, would give rise to accumulation of self-aggregating Aβ peptides. The amyloid hypothesis is now the dominant model of AD pathogenesis, and it has given rise to an impressive amount of research work (see Selkoe and Hardy [3] for a review).
Plaque formation by an Aβ peptide appears to require conversion of the monomeric form to an aggregated fibrillar species. Specifically, Aβ peptide is released from cells in a soluble form, and progressively undergoes aggregation forming oligomers, multimers, and fibrils, ending with deposition of extracellular plaques. Several investigations converge on the notion that soluble Aβ aggregates, namely oligomers and protofibrils are the major players in the clinical syndrome of AD dementia causing synaptic impairment, neuronal stress, spreading of tau pathology and cell death. Oligomers and protofibrils can further aggregate forming insoluble β-pleated sheets that form mature insoluble amyloid fibrils and plaques. The formation of insoluble fibrils and plaques has been suggested to be a protective mechanism that mitigates and reduces oligomer toxicity [5]. Extensive investigations indicate that cell membranes play a significant catalytic role in increasing Aβ aggregation rates (see, e.g., [4,5,6,7] for reviews). A large part of the above studies, including investigations from this laboratory [8,9,10,11,12], examined the interaction of β-amyloid with model membranes, the consensus view being that Aβ, when interacting with lipid surfaces of various compositions, especially anionic lipids, would aggregate at a much faster rate than in solution.
The present contribution is aimed at reviewing the most relevant studies on the interaction of Aβ peptide with lipids in model systems, monolayers or bilayers. Some open questions to be dealt with are e.g. the conceptual and experimental distinction between peptide binding to, adsorption onto and insertion into membranes, the surface catalysis of peptide aggregation, the influence of the bilayer physical properties (physical state or phase, fluidity, molecular order, electrostatic charge) on peptide-lipid interaction, the suitability of the experimental methods used, and the pathophysiological relevance of the biophysical studies with model membranes. For simplicity, along the review, ‘binding’ will refer to any form of detectable peptide-lipid interaction, unless otherwise indicated. Aβ, Aβ peptide, or amyloidogenic peptide Aβ will be used as synonym.
2. Lipid Phases, and Their Significance
A short discussion of the meaning and implications of the lipid phase concept is essential for a proper understanding of the interaction of Aβ peptide with lipids. A phase is defined as a region of space throughout which all physical properties of a material are uniform. “Phase” is synonym of “state of matter”, e.g. water can exist in the solid, liquid or vapour phases, or states. Phases are thermodynamic concepts, i.e. ideal entities to which real objects resemble more or less. The condition of uniformity included in the definition must be understood macroscopically, at least at the micrometer scale in the context of membrane lipids. In the last century a number of phases were identified with properties intermediate between liquid and solid. They are collectively known as mesophases. A well known example is the liquid-crystalline phase, in which cell membranes appear mostly to exist, that is characterized by exhibiting a liquid-like fluidity, with its molecules being oriented in a crystal-like way. Lipids dispersed in water can adopt a rich variety of mesophases, depending on the lipid chemical structure, temperature, pressure, amount of water, and other variables. Lipids are said to be mesomorphic [13].
The best method for describing a lipid phase in aqueous environment is X-ray scattering [14,15,16]. In some instances 31P NMR can provide useful information [17]. Moreover in favorable cases a phase transition may be observed by increasing or decreasing temperature (thermotropic phase transitions). The method of choice for detecting the latter kind of transitions is differential scanning calorimetry [18,19]. The main phases (mesophases) adopted by pure membrane lipids when dispersed in water are the so-called lamellar (L), micellar (M), inverted hexagonal (HII) and inverted cubic (QII) phases [20].
The lamellar (L) phases, consisting of two lipid layers whose non-polar moieties are in contact and away from water constitute the disposition spontaneously adopted by most phospholipids and glycolipids. Several lamellar phases are known, that are relevant in the study of cell membranes [20]. Most saturated membrane lipids can give rise to a gel (or solid) Lβ lamellar phase at a given temperature, and to a fluid (or liquid crystalline) Lα phase at a higher temperature. In the Lα, but not in the Lβ phase, the lipids exhibit unhindered translational and rotational diffusion (alkyl chain disorder). For instance, dipalmitoyl phosphatidylcholine (DPPC) exists in the Lβ phase below 35 °C and in the Lα phase above 41 °C. Also, at lower temperatures, subgel lamellar phases have been observed.
A liquid ordered lamellar phase is also known to exist [21], formed in the presence of certain saturated phospholipids and cholesterol. In this phase the lipid molecules have free lateral diffusion, i.e. they are fluid, but rotation around the alkyl chain C-C bonds is restricted (fatty acyl chains are ordered). The nomenclature for the liquid ordered, or fluid ordered phase is unclear, Lo or lo being often used.
3. Model Membranes: Monolayers and Bilayers
A number of methodological and conceptual aspects, which are not always clear in the literature on Aβ peptide-lipid interactions, are discussed now. Lipid monolayers and bilayers, studied either in situ or after transfer to solid supports are an indispensable tool for understanding the behavior of lipids, and the lipid-protein interactions, in membranes.
3.1. Monolayers
Many physical phenomena displayed by lipids in biomembranes can also be observed and modelled in lipid monolayers, spread over an air/water interface [8]. These phenomena, which reflect the chemical and structural features of different lipids, include phase transitions, changes in lateral diffusion, alterations in lateral compressibility/elasticity, and mixing interactions that can result in critical points as well as immiscible or coexisting lateral phases (domains) [12,22,23,24,25].
Although early monolayer studies involving construction of lipid phase diagrams were limited to mixtures consisting of only two different lipid components [23,24,26], more recently direct insights into the monolayer phase behaviour of “raft” ternary mixtures of phosphatidyl choline/sphingomyelin/cholesterol (PC/SM/Chol) (Figure 1) have been stimulated by using epifluorescence microscopy to monitor the distribution patterns of trace amounts of lipids containing covalently attached reporter fluorophores [27,28,29,30]. Also, because this same combination of lipids can form stable giant unilamellar vesicles (GUVs, see Section 3), direct comparison of lipid mixing, domain formation, and construction of ternary phase diagrams has been possible in bilayers (GUVs) and in monolayers using epifluorescence microscopy. Not surprisingly, interesting similarities and differences have emerged.
Also yielding valuable insights into monolayer lipid mixing behaviour is a variation of the in situ approach involving transfer of the monolayers from the air/water interface to alkane-coated, solid supports (i.e. supported monolayers) for study by various techniques. While the basic strategy is well-established for mapping phase diagrams in mixed lipid monolayers (e.g., [22]), applications to ternary lipid combinations have emerged only recently in combination with epifluorescence microscopy [31,32], atomic force microscopy [33,34,35] and/or imaging mass spectrometry. The latter approach, used by Winograd and colleagues [36,37,38], provides an exciting means for direct imaging of the mixing behaviour of complex lipid combinations in the absence of fluorophore probes that can alter microdomain morphology. Moreover, the imaging mass spectrometry approach enables lipid identification and quantitative estimates of different lipid components within differing domains. Ternary mixtures of PC/SM/Chol, have been analyzed in detail, confirming formation of domains enriched in sphingomyelin and cholesterol as well as the importance of acyl composition in domain formation [36,37,38].
3.2. Bilayers
Lipids are organized in a double layer, or bilayer [20,39]. Membrane lipids are amphipathic, i.e. they possess both a hydrophobic and a hydrophilic moiety. This occurs in phospholipids, glycolipids, and sterols. Because of this amphiphatic character, in an aqueous medium they can organize themselves on both sides of an imaginary plane, with the hydrophobic portions facing each other, and the polar moieties oriented to the outer, aqueous space. In fact, when dry lipids are mixed with water, they spontaneously organize themselves in bilayers, e.g. during liposome formation. (Note however that certain lipids do not give rise spontaneously to bilayers, they are the so-called “non-lamellar lipids”, see below). The bilayer in aqueous medium provides a simple method for the thermodynamic stabilization of a population of molecules that are neither entirely hydrophobic nor entirely hydrophilic. As mentioned above, Singer and Nicolson recovered the bilayer concept from Danielli and Davson, after the idea had been severely criticized in the 60s.
Membrane proteins can be associated either to the lipid bilayer polar headgroups (peripheral proteins) or to the hydrophobic matrix (integral proteins). Protein binding to the bilayer outer region had been proposed by Danielli and Davson, but the idea of proteins embedded in a hydrophobic milieu, while supported by experimentation in the late 60s and early 70s, had never been proposed in a clear and explicit way before. In fact peripheral (or extrinsic) and integral (or intrinsic) proteins [40] were independently defined in a purely operational way: peripheral proteins would be those that could be released from membranes using relatively gentle methods, such as changes in buffer pH, or ionic strength, while integral proteins would be amphipathic molecules requiring the use of more drastic agents, e.g. detergents, or organic solvents. In practice, the correspondence between these two groups of proteins classified after their solubilization properties, and the two ways of protein association to bilayers in the Singer–Nicolson model have led to the almost always accurate identification of the two kinds of proteins in the model with the corresponding two groups of differently solubilized membrane proteins in the test tube.
4. Biophysical Methods in the Study of Aβ Peptide Interaction with Membranes
A large variety of experimental and numerical methods are available for the study of Aβ-lipid interaction. Of those, a selection of the most widely used ones follows.
4.1. The Langmuir Balance and Langmuir Monolayers
Surface pressure experiments are carried out with a Langmuir trough, under constant stirring. The aqueous phase consists of a buffered saline solution. The appropriate lipid, or lipid mixture, dissolved in chloroform/methanol (2:1), is gently spread over the surface until the desired initial surface pressure is attained. Aβ binding is assayed with lipid monolayers at an initial surface pressure πi above the maximum Δπ caused by adsorption of pure Aβ at an air-water interface [8]. The protein is injected with a micropipette through a hole connected to the subphase. The increment in surface pressure versus time is recorded until a stable signal is obtained.
Once a Langmuir monolayer is established at the air-water interface, several reflection techniques can be applied on it. Grazing incidence X-ray diffraction [41,42] reports on peptide-induced changes in the lipid monolayer. Infrared reflection absorption spectroscopy (IRRAS) allows the measurement of the conformation and orientation of the peptide adsorbed to the air–water interface and to a lipid monolayer [41,42]. Brewster angle microscopy allows detection of fibrils in the monolayers [43,44]. Furthermore, when the Langmuir monolayers are transferred into a solid, inert surface, e.g. mica, they can be examined under the atomic force microscope, from which manipulation novel aspects of Aβ-lipid interaction have been obtained [43,44]. Finally, fluorescence techniques have been combined with monolayer experiments, as discussed below (Section 4.6).
4.2. Sucrose Gradient Ultracentrifugation
Liposomal flotation assays allow the direct measurement of bound and unbound protein to lipid vesicle membranes. Vesicles containing bound protein are denser, i.e. sediment more readily, than the protein-free ones, the latter tending to float to the top of the gradient. This technique is useful because the composition and curvature that cell membranes would adopt in vivo could be mimicked with liposomes. Ultracentrifugation was used initially by Masserini and co-workers [45] in the study of anti-Aβ MAb-decorated nanoliposomes to Aβ peptides.
More recently, Ahyayauch et al. have made extensive use of the technique [9] for assessing Aβ42 binding to liposomes containing gangliosides in their composition. A fixed concentration of pure Aβ (typically around 15 μM) was incubated with liposomes (1:200 protein:lipid mol ratio) and the mixture was subjected to an equilibrium sucrose gradient centrifugation, in which the protein-free vesicles have the capacity to float up, and the amount of protein interacting with these liposomes can be quantified. Rhodamine-stained liposomes are commonly used (0.5 mol % Rho-PE) to facilitate detection.
4.3. Calorimetric Methods
The main calorimetric method used in the study of Aβ-lipid interactions is isothermal calorimetry (ITC). Specifically, the enthalpy change upon partitioning of Aβ42 into liposomal membranes (usually large unilamellar vesicles, LUV) is often measured with high-sensitivity ITC. The calorimetric cell is filled with an Aβ solution, usually around 20 µM. Lipid vesicles at about 15 mM lipid concentration are injected into the cell. Typically, the injections are made at 10 min intervals and at 2 s/μL. Constant stirring is maintained during the experiment to ensure proper mixing after each injection. Dilution heats of lipid vesicles into the buffer are determined in separate experiments and subtracted from experimental heats of binding. At each lipid injection, free Aβ peptide monomers partition into the bilayer membrane and the corresponding heat of reaction is measured. The heat of reaction becomes progressively smaller as less peptide remains free in solution. The integration of each calorimetric peak yields a heat of reaction. These heats are plotted versus the lipid concentration. The obtained isotherm is used to determine the thermodynamic parameters of partitioning [7,8,46,47,48]. A model of partition equilibrium of the peptide molecules between aqueous phase and the membrane is assumed for the interaction, in which Henry's law applies, i.e., there is a linear relationship between the concentration of peptide free in solution and the concentration bound to the membrane [47].
A different calorimetric method is the so-called differential scanning calorimetry (DSC), generally used to detect thermotropic phase transitions in lipid-water systems, and also protein thermal denaturation. Martinez-Senac et al. [49] used DSC to demonstrate that the effect of Aβ25-35 on the gel to fluid transition was small at neutral pH for negatively charged phospholipids and practically nil for DMPC. Furthermore, these authors suggested, on the basis of the above studies, that the peptide-lipid interactions were basically electrostatic in origin, and that these interactions favored peptide aggregation. DSC was also applied, in the absence of peptides, to explore the phase behavior of sphingomyelin vesicles containing GM1 ganglioside and cholesterol [50].
4.4. Thioflavin T (ThT) Fluorescence Assays
Thioflavin T (ThT) fluorescence is an example of a facile, useful technique, frequently used because of its simplicity rather than for its heuristic possibilities. ThT fluorescence increases with β-sheet contents of peptides [51,52], and this property is used to study, in a semi-quantitative way, the monomer-oligomer, or in general the monomer-aggregate transitions of Aβ peptides in solvents, which are accompanied by a transition from helical (or random) to β-sheet conformation. Briefly, ThT is prepared in glycine (50 mM, pH 8.2) and filtered (0.22 µm). Stock Aβ is added to each vesicle solution to yield incubation mixtures containing typically 5 μM Aβ and a 1:200 peptide-to-lipid mole ratio at 37 °C. ThT is then added, and fluorescence is recorded in a spectrofluorometer (λex = 446 nm, λem = 485 nm). Control samples of pure Aβ and peptide-free vesicles are also prepared and measured as indicated. Among the abundant examples of ThT in the study of β-sheet formation in Aβ peptides, the following may be mentioned: [8,9,10,11,43,44,53].
4.5. Computational Methods
The methodology published by Ahyayauch et al. [8,10] can serve as an example for this kind of studies. Starting coordinates of the protein structure are obtained from a data bank, e.g. PDBid 1IYT for Aβ42 [54]. This three-dimensional structure, obtained with NMR, shows two helical regions encompassing residues 8–25 and 28–38, connected by a type I β-turn. Aβ42 is modeled as a zwitterionic structure, with ionized C- and N-termini. The first protomer of this PDB entry is typically placed at 1 nm from the surface of a bilayer constituted of 128 lipids, whose coordinates are taken from [55,56]. In each case, the systems are embedded into simulation boxes containing SPC water and 150 mM NaCl. The GROMOS53a64 force field is commonly used [57]. Energy minimization, isotropic equilibration in two steps (10 ns at 100 K and 298 K, respectively) and molecular dynamics production runs are performed using a GROMACS 4.6.7 package [58]. A combination of the force field parameters developed by Tieleman [59] and the GROMOS 96 [60] generic parameters can be used for the description of sphingomyelin and other phospholipids. For cholesterol, the parameters developed by Hoeltje et al. in conjunction with the Berger parameters are used [61]. Production runs are performed using two simulation schemes. First, all systems are simulated for 100 ns at 298 K. Subsequently, a dual resolution solvent approach is implemented as detailed in [62,63]. Molecules beyond 1 nm of the protein or membrane are substituted by coarse-grained (CG) water using the WatFour model [64]. The position of ions laying in the CG region is preserved and the interaction parameters are changed to those corresponding to CG electrolytes. Simulations using this dual solvation approach proceed up to 4 μs, achieving a 3-fold speed up in the calculations. All simulations are performed with a time step of 2 fs, updating the neighbor list every 10 steps and using the PME algorithm for long range electrostatics [65], with a direct cut-off of 1 nm. Temperature coupling is achieved with the V-rescale algorithm [66]. Pressure coupling to 1 atm is obtained using the Parrinello-Rahman algorithm [67]. Standard analysis utilities from GROMACS are commonly used. Trajectories can be visualized and analysed with an VMD viewer [68].
4.6. Miscellaneous Techniques
The techniques discussed above, and particularly Langmuir monolayers, isothermal calorimetry, and molecular dynamics, are by far the ones that have been more extensively used, and provided most detailed information, on the interactions of Aβ peptides with lipid molecules in membranes. However, several other methods, even if they are less informative, or less frequently used, deserve some discussion.
Circular dichroism (CD) provides a simple method to detect peptide/protein conformational changes in solution, although its quantitative information is limited to estimating the percent amino acid residues involved in α-helix conformation. CD is therefore more used in studies of Aβ in solution, rather than in analyses of peptide-lipid interaction. Still, it has provided useful information on Aβ oligomerization [7,69,70,71,72], and it may be very useful to complement the ThT fluorescence measurements.
Resonance techniques. Nuclear magnetic resonance (NMR) and electron spin resonance (ESR) (also called electron paramagnetic resonance, EPR) have been occasionally applied to the study of Aβ peptide-lipid interaction. In these techniques a nucleus, or an electron, are respectively oriented in a magnetic field. NMR requires little or no chemical modification of the lipids, but the sensitivity of the technique is rather low. The opposite happens with ESR, for which very small amounts of chemically modified lipids are required. Terzi et al. [69] used deuterium and phosphorus-31 solid-state NMR to study the lipid molecular structure in the presence of Aβ40. Phosphatidylcholine (PC) was selectively deuterated at the choline headgroup and at the cis-double bond of the oleic acyl chain, and mixed with phosphatidylglycerol (PG). Phosphorus-31 NMR showed that the lipid phase retained the bilayer structure at all lipid-to-protein ratios. Deuterium NMR revealed no change in the headgroup conformation of the choline moiety or in the flexibility and ordering of the hydrocarbon chains upon the addition of βAP (1−40). Chang et al. [73] have applied solid-state NMR to analyze Aβ oligomerization in reverse micelles, while Matsuzaki et al. have studied the conformational change of Aβ in the presence of ganglioside by NMR [74]. ESR was used by Vitiello et al. [75] to establish how omega-3 fatty acids regulate the interaction of Aβ25-35 peptide with lipid membranes. The latter technique requires usually the chemical modification of lipids with spin probes, e.g. TEMPO, and this may lead to artefactual results unless careful control experiments are performed.
Fluorescence techniques. Fluorescence is a phenomenon in which an atom or molecule absorbs radiation of a given wavelength and, after a time lapse of nanoseconds, emits radiation of a longer wavelength (smaller energy). Fluorescence offers innumerable applications in Biology. In the context of our studies, Chi et al. [76] used Texas Red 1, 2-dihexadecanoyl 3-phosphoethanolamine (TR-DHPE) to specifically stain certain domains in monolayers. Dark patches representing condensed domains, where bulky head-group-labeled TR-DHPE dye molecules were excluded, were seen together with bright regions representing the liquid expanded phase where the dye preferentially resided. At low GM1 concentrations, Aβ preferentially inserted into the disordered, liquid expanded phase. At higher GM1 concentrations, Aβ inserted more uniformly into the monolayer, resulting in no detectable preferences for either the expanded or condensed phase. Dynamic (time-resolved) fluorescence imaging of monolayers was applied by Lin et al. [77] to establish the interaction of Aβ40 with lipid monolayers containing negatively charged lipids and Chol. Hossain et al. [53] studied the intrinsic fluorescence of Tyr residues in the Aβ42 peptide and the effects of docosahexaenoic acid on amyloidogenesis. They found that this polyunsaturated ω-3 fatty acid decreased the levels of Tyr intrinsic fluorescence, suggesting a decreased fluidity in the microenvironment, and a certain level of anti-amyloidogenic protection.
5. A Review of Selected Results
5.1. Monolayer Studies
Perhaps the earliest study on the interaction of Aβ peptide with lipid monolayers was performed by Seelig and co-workers in 1997 [69]. Aβ40 was found to insert into acidic monolayers provided the lateral pressure was low (20 mN/m). The extent of incorporation increased distinctly with the content of acidic lipid in the monolayer. However, at a lipid packing density equivalent to that of a bilayer (lateral pressure ≥32 mN/m), no insertion of Aβ40 was observed. These studies were later confirmed and extended by Maltseva et al. using monolayers composed of phosphatidyl ethanolamine [41] or of several zwitterionic or negatively charged phospholipids [42]. Thakur et al. [78] described the complex effects of cholesterol, in mixtures with several phospholipids, on the binding of Aβ40.
Ahyayauch et al. [8] explored the initial stages of Aβ42 deposition on membranes. Aβ42 is considered to be more pathogenic than Aβ40. In the absence of lipids, injection of Aβ42 into the aqueous phase caused an increase in surface pressure, the latter reaching equilibrium after ≈1 h. This showed that Aβ42 was surface active, like many other peptides [79]. The increase in surface pressure was dose-dependent and reached a plateau at ∼10 mN/m, for Aβ concentrations slightly above 1 μM. Thus, at these and higher concentrations the interface was saturated with adsorbed peptide and the peptide partitioned between the interface and the bulk water [79]. Previous studies had found plateau values in the 12–17 mN/m range with Aβ42 or Aβ40 [62,63,64,66], the origin of the variability being unclear.
Aβ insertion into lipid monolayers was assayed in a different set of experiments, in which a lipid monolayer was extended at the air-water interface and the peptide was injected into the aqueous subphase. The initial surface pressure of the lipid monolayer was fixed as desired, at values >10 mN/m to avoid simultaneous peptide insertion and peptide adsorption. Aβ insertion into the lipid monolayer at the interface caused a further increase in surface pressure Δπ (Figure 2A). As the initial pressure increased, Δπ decreased (Figure 2B) until a point was reached, at 30–33 mN/m for this system, at which peptide insertion was no longer possible. When the negatively charged dimyristoyl phosphatidic acid (DMPA) was included in the monolayer lipid composition, and insertion was assayed in the 10–30 mN/m range, peptide insertion became easier in the order: SM/Chol (1:1) < SM/Chol/DMPA (40:40:20) < SM/Chol/DMPA (47.5:47.5:5), all figures given as mole ratios. However, the limiting initial pressure for all three lipid compositions was the same, close to 30 mN/m, supposed to be (with large fluctuations) the average surface pressure in the cell membranes. The same limiting initial pressure of 30 mN/m was found by Terzi et al. [69] and by Maltseva et al. [41,42] for Aβ40. This may mean that, for a hypothetical cell membrane domain in the liquid-ordered state, Aβ would exist in equilibrium between the free and membrane-bound forms. The fact that insertion was facilitated by the presence of negatively charged lipids in the monolayer supports the role of electrostatic interactions in stabilizing Aβ42 insertion into lipid monolayers, in agreement with previous suggestions [41,78]. A compression isotherm of the SM/Chol/DMPA (40/40/20) mixture indicated an average area/molecule of ≈ 0.4 nm2, in agreement with MD calculations (see Section 5.4 below).
Ahyayauch et al. [9,10,11,12] have further developed the possibilities of Langmuir monolayers to analyze the lipid interactions of Aβ42. The binding of A42 peptide monomers to sphingomyelin/cholesterol (1:1 mol ratio) monolayers containing 5 mol% gangliosides (either GM1, or GT1b, or a mixture of brain gangliosides) was assayed [10]. In general, gangliosides facilitated monolayer-peptide binding. In the 10–30 mN/m range, peptide insertion became easier in the following order: SM/Chol < SM/Chol/total gangliosides ≈ SM/Chol/GT1b < SM/Chol/GM1. However, the limiting initial pressure for all four lipid compositions was the same, close to 32–34 mN/m. The monolayer data were independent of geometric fluctuations that could be produced by gangliosides in bilayers, and they confirmed that gangliosides facilitate the binding of Aβ42 to membrane lipids. In a different series of experiments [10], four different lipid compositions were tested in monolayer form, namely POPC with 0, 1, 3, and 5 mol% GM1. In the 12–22 mN/m range, peptide binding became easier the higher the ganglioside concentration. However, the limiting initial pressure for all three lipid compositions was the same, close to 22 mN/m. Considering that 30 mN/m is supposed to be the average surface pressure in the (mostly fluid) cell membranes, the data indicate that, in fluid disordered bilayers, Aβ42 would adsorb onto the lipid surface, without being able of becoming inserted in the bilayer.
The state of aggregation of Aβ42 (monomer, oligomer or fibril) and its influence on the interaction with sphingomyelin/cholesterol (with or without gangliosides) in lipid monolayers has also been examined by Ahyayauch et al. [11,12]. The Langmuir balance demonstrated the capacity of all three peptide preparations to become inserted in lipid monolayers of any composition and initial π in the range 10–30 mN/m, although fibrils were less capable to do so than oligomers or monomers, their maximum initial π being ≈25 mN/m. When the various lipid compositions were compared, monolayers lacking gangliosides were most resistant to peptide insertion, while GM1 ganglioside allowed an easier insertion along a wide range of πi. The effect of ganglioside concentration in the monolayers is as follows: For total ganglioside and for GM1, ganglioside concentrations below 5% did not clearly favor peptide insertion in the monolayer, as compared to the ganglioside-free SM/Chol mixture. A more gradual facilitation was observed, however, for GT1b. Moreover, when the monolayer electric charge was varied, through addition of electronegative phosphatidic acid, cardiolipin or gangliosides, the data showed that: (i) Aβ42 aggregation hindered peptide insertion into the monolayer, insertion decreasing in the order monomer > oligomer > fibril; (ii) lipid composition did not cause large differences in insertion, apart from a slight facilitation of monomer and oligomer insertion by gangliosides; (iii) SM/Chol constituted an exception to the above rule, in that it exhibited a particularly low binding to fibrils [12].
Meanwhile, the group of Fidelio and co-workers have also published interesting results on the interaction of Aβ40 fibrils with lipid monolayers [43,44,80,81]. Alvarez et al. [43] examined the surface properties of Aβ40 amyloid peptides mixed with 1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC) (liquid crystalline state) or 1,2-distearoyl-phosphatidylcholine (DSPC) (gel state) phospholipids in the form of Langmuir monolayers. They observed, at a low amyloid peptide proportion (2.5-10% of total area), formation of a fibril-like structure when mixed with POPC, but not with DSPC. Fibrils were evidenced directly from the monolayers by using Brewster angle microscopy, as thioflavin T-positive structures when observed by fluorescence microscopy, and as an amyloid network by atomic force microscopy when the films were transferred onto a mica support. The authors concluded that amyloid fibrillogenesis on the membrane is regulated at least by the physical state of the water-lipid interface and by the relative content of amyloid protein at the interface. Later, the same authors described a “dynamical smelting”, or dissolution, process when pre-formed fibrils in amyloid/phospholipid monolayers was laterally mixed with gangliosides [44]. They postulated that the ganglioside-dependent smelting of amyloid fibrils on the membrane surface resulted from the new interfacial environment imposed by the complex glycosphingolipids.
More recently Alvarez et al. [80] described how Aβ40 fibrils alter the topography and mechanical properties of lipid membranes. When Aβ was assembled with DMPC in a binary film, the resulting mixed monolayers were heterogeneous, with a peptide-enriched phase distributed in a network-like pattern. The fibers covered most of the interface area and remained stable at higher pressures (20-30 mN·m-1, depending on Aβ content) than pure peptide films (17 mN·m-1). Furthermore, the fibers induced a compressional hysteresis in the film, similar to that of pure peptide films, which is nonexistent in the pure lipid monolayer, even at low peptide proportions. Thus, due to peptide intermolecular interactions, Aβ may have drastic effects on the molecular arrangement and mechanical properties of membranes. Conversely, the molecular lipid order can modulate interfacial Aβ fibril formation [81]. These authors studied the surface properties of DPPC and Aβ40 mixed monolayers at different temperatures (from 15 to 40 °C). In this temperature range the surface properties of pure Aβ40 remain unchanged, whereas DPPC undergoes its characteristic liquid-expanded → liquid-condensed two-dimensional phase transition. As the initially expanded mixed film was isothermally compressed, the fibril-like structure of Aβ40 was triggered specifically in the liquid-expanded region, and excluded from the DPPC liquid-condensed domains. As the monolayers were compressed (from 11 mN/m to 20 mN/m) the condensed domains became irregular, perhaps because the fibrils were imposing additional lateral stress sequestering lipid molecules from the surrounding liquid-expanded phase to self-organize into amyloid.
5.2. Isothermal Calorimetric Studies
The earliest calorimetric studies of Alzheimer beta-amyloid fragments were also carried out by Seelig and co-workers, in 1994. They initially examined the random coil – β-sheet transition of a short fragment (residues 25-35) [70]. Using ITC, they estimated an enthalpy of association ΔH ≈ -3 kcal/mol. This was followed by an investigation of the binding of Aβ peptides to lipid vesicles containing negatively charged lipids [71]. The presence of lipids shifted the random coil – β-sheet equilibrium almost completely toward β-sheet structure. The Aβ(25-35)OH peptide exhibited an exothermic binding enthalpy ΔH ≈ -2 kcal/mol, with an intrinsic binding constant, after correction for electrostatic effects, K ≈ 2 M-1. The same authors went on to examine the self-association of the much longer Aβ(1-40) in solution and binding to lipid membranes, using calorimetry together with other techniques [7]. The binding isotherm, as measured with high sensitivity titration calorimetry, was approximately linear in the initial binding phase and exhibited an apparent saturation behavior. The apparent binding constant decreased with concentration from Kapp ≈ 2100 M-1 at low concentration to 700 M-1 at the highest concentration measured. These authors were the first to propose peptide penetration into the lipid membrane and peptide aggregation at the membrane surface as possible mechanisms to explain the lipid-induced random coil – β-sheet transition [7], the latter considered nowadays as the earliest step in pathogenic plaque formation [3,6,8].
In more recent years, Ahyayauch et al. have carried out extensive and systematic calorimetric measurements of the interaction of Aβ42 peptide with lipids, in the form of liposomes (large unilamellar vesicles, LUV) [8,10,11,12]. When LUVs were composed of SM and Chol only, in the absence of negatively charged lipids, no measurable heats of interaction with monomeric Aβ were observed [8]. This may be related to the relatively poor insertion of Aβ into SM/Chol monolayers, described above (Section 5.1). However, reliable measurements were obtained with LUV containing the negatively charged lipid DMPA. The peptide association constant (Ka) decreased regularly (ΔG was made less negative) with increasing DMPA concentrations, in agreement with the surface pressure measurements of peptide insertion (Figure 2B). A different negatively charged phospholipid, cardiolipin, favored Aβ42 interaction with bilayers even more than DMPA.
The SM/Chol mixtures used in the above experiments give rise to bilayers in the Lo state. With the aim of testing the influence of membrane order on Aβ42 binding, a number of experiments were performed in parallel with Ld bilayers, based on either pure eggPC, or on eggPC/DMPA mixtures. The more disordered membranes bound Aβ42 monomers with higher affinity than those in the Lo phase, even pure PC bilayers bound the peptide [8].
In a different series of experiments, Ahyayauch et al. applied isothermal calorimetry to study the influence of gangliosides on Aβ42 interaction with either Ld or Lo bilayers. In the preparation of Ld bilayers [10], POPC, either pure or in mixtures with 1, 3, or 5 mol% GM1 ganglioside was used. The peptide association constant (Ka) increased regularly (and correspondingly ΔG became more negative) when GM1 concentration increased from 0 to 3 mol%, and it decreased slightly (ΔG was made slightly less negative) for 5 mol% GM1. Aβ42 binding to fluid lipid bilayers appeared to be favored by GM1 at concentrations up to 3 mol%, while higher concentrations did not exhibit any clear effect [10]. With bilayers in the Lo state, based on SM/Chol (1:1), mixed with 5 mol% ganglioside (GM1, GT1b, or a porcine brain total ganglioside extract) [11], monomer binding could be reliably measured, unlike the situation in the absence of ganglioside. The peptide association constant (Ka) was of the same order of magnitude (≈105 M−1) irrespective of the nature of the ganglioside. Correspondingly ΔG was in all three cases negative and very similar (−7 to −8 kcal/mol). Similar studies were carried out with varying ganglioside concentrations, namely 1, 3, and 5 mol%. The values for Ka and ΔG were very similar irrespective of ganglioside structure or concentration [11]. Note however that the rather constant ΔG along the various conditions was the result of mutually compensating changes in enthalpy and entropy, the two latter functions of state varying widely with the ganglioside species.
All of the above calorimetric studies have been performed with Aβ42 in monomeric form. However, it was important to explore the effect of the peptide state of aggregation (monomer, oligomer, fibril) on its membrane binding capacity. The calorimetric results of such studies have been summarized in Ahyayauch et al. [11,12]. Aβ peptide oligomers were able to bind bilayers composed of SM and Chol only, in the absence of gangliosides, under conditions when measurable amounts of heat were released [11]. The association constant Ka was of the order of 105 M−1 with no gangliosides, or with 5 mol% total gangliosides, or with 5 mol% GT1b, corresponding to ΔG of about −7 kcal/mol. However, when GM1 ganglioside was present, Ka was lower, of the order of 103 M−1, thus ΔG was of about −4 kcal/mol. As described for the case of Aβ monomers, similar ΔG values occur through mutually compensating changes of ΔH and ΔS. The low ΔG in the presence of GM1 was due to a low ΔH, not compensated by a high ΔS. When the Aβ peptide was in the form of fibrils [11], a measurable amount of heat was also released as a result of peptide-vesicle interaction even in the absence of ganglioside. The heat exchange (absolute value) decreased in the order: total ganglioside > > GM1 > GT1b ≈ no ganglioside, and ΔS increased accordingly, so that ΔG was in all cases of about −7 kcal/mol, corresponding to Ka of the order of 105 M−1 [11].
Moreover, the SM/Chol bilayers were doped with the negatively charged phospholipids DMPA or CL, instead of gangliosides, under conditions similar to those used in [8] for monomers, but with the Aβ42 peptide in the form of oligomers or fibrils [12]. Studies with soluble oligomers have the additional interest that those appear to be most active from the pathogenic point of view [82]. One major difference with monomers is that oligomers were able to interact with SM/Chol bilayers even in the absence of added negatively charged lipids (Table 2). They did so with a rather robust ΔG = −7.88 kcal/mol, in which an important entropic component (T · ΔS = −5.77 kcal/mol) occurred. Mixtures containing negatively charged lipids bound Aβ42 oligomers with ΔG rather similar to the case of the monomers, in the 5 to 7 kcal/mol range [12]. An additional important difference between oligomers and monomers is the smaller ΔG (in absolute value) of the CL-containing mixtures with oligomers, as compared with those involving monomers. In general, the thermodynamic parameters describing binding equilibria appeared to be less dependent on bilayer composition for oligomers than for monomers. The remarkable positive values of ΔS observed with oligomers irrespective of lipid composition appear to speak in favor of a large disordering effect imposed by the oligomeric structures.
The interaction of Aβ(1-42) fibrils with LUV bilayers was assessed in the same way; the results showed that the association constants (Ka), related to the standard variation of the Gibbs’ free energy (ΔGº) and the actual changes in ΔG under our experimental conditions, were remarkably independent of the bilayer lipid composition [12]. As discussed above for some examples of monomer binding, the constancy of ΔG was the result of compensating ΔH and ΔS values. ΔS was largest (most positive) for fibril interaction with SM/Chol than in any other system under study; this could be interpreted considering that the binary SM/Chol bilayer exhibited the largest degree of lipid order; thus, it was more perturbed than others by the fibril insertion. Conversely, in the samples containing the total ganglioside mixture, which is rich in trisialic gangliosides, the insertion of fibrils would cause a marked reorganization of the water molecules solvating the ganglioside sugar moieties, with the consequence of a decrease in entropy, compensating a large, exothermic (ΔH < 0) enthalpy change [12].
From the ensemble of their isothermal calorimetry studies, Ahayayauch et al. [8,10,11,12] concluded that: (i) the binding of Aβ42 fibrils, oligomers, and monomers was spontaneous (ΔG < 0) for all six lipid bilayer compositions tested, except that monomers could not interact with SM/Chol binary bilayers; (ii) Aβ42 fibrils, oligomers, and monomers could bind bilayers in the liquid-ordered state, with the said exception for monomers and SM/Chol bilayers; (iii) both ΔH and ΔS were very sensitive to lipid composition, even if, in most cases, the composition was changed by only 5 mol%, and (iv) very similar values of ΔG were often attained through marked compensatory changes of ΔH and ΔS.
5.3. Molecular Dynamics (MD) Studies
To our knowledge, Yechun Xu et al. [83] in 2005, were the first to apply molecular dynamics to examine Aβ peptides, specifically Aβ40, in an explicit bilayer environment. These authors first studied the conformational transitions of monomeric Aβ40 by a series of long time MD simulations. They found that conformational transitions from α-helix to coil occur through helix/β-sheet mixed conformations, in agreement with previous experimental work [71]. This was interpreted as suggestive of helix/β-sheet mixed conformations as putative intermediates in Aβ oligomerization. Four glycines (G25, G29, G33, and G37) were found to be important for Aβ40 to form β-sheet in aqueous solution. In a DPPC bilayer, the major secondary structure of Aβ40 was a helix; moreover, the peptide exhibited a tendency to exit the membrane environment and lie down on the bilayer surface. Lys-28 was assigned to the interface region between the bilayer and the aqueous environment [83].
A similar Aβ40/DPPC system was examined by Lemkul and Bevan [84]. Unlike the previous study, they found that a portion of the peptide remained embedded in the bilayer in all cases. When deeper insertion occurred, Aβ40 adopted a near-transmembrane orientation, drawing water molecules into the bilayer to associate with its charged amino acids. In the more frequent case of shallower insertion, the peptide associated strongly with the membrane–water interface and the phosphatidylcholine headgroups of the bilayer. These authors concluded that Aβ-membrane association was highly dynamic and capable of adopting a number of conformations. To explain the fact that the peptide was not seen to leave the bilayer, Lemkul and Bevan suggested that maybe the length of time of the simulations was not long enough to observe such behavior. More importantly, these authors implied that a nucleation site was needed to initiate the aggregation process.
Davis and Berkowicz [85] introduced a new factor in these MD studies, namely the DPPC lamellae were compared with dioleoyl phosphatidylserine (DOPS) bilayers, the latter lipid bearing unsaturated chains, and a net negative charge. In this case the peptide was Aβ42. In the light of their results, the authors suggest that the Aβ peptide, cleaved from the APP, would be brought close to the bilayer surface. With a mostly zwitterionic bilayer, the peptide would strongly interact with the hydrophobic core of the bilayer, this binding precluding any secondary structure change, thus extensive interactions with other nearby peptides. In turn, an anionic lipid membrane appeared to promote aggregation by locally increasing Aβ concentration on the bilayer surface, due to the highly favorable free energy of binding, and by decreasing the local pH on the bilayer surface to promote an Aβ configuration that would drive oligomerization [85]. These authors did not take into account, in their interpretation of the data, that DPPC and DOPS bilayers would exist in different states at room temperature, respectively the gel and fluid disordered states.
Ahyayauch et al. [8] considered the interactions of Aβ42 with mixed bilayers, composed of SM and Chol at equimolar amounts plus different proportions (5 or 20 mol%) of the saturated, negatively charged DMPA. Apart from the mixed composition, an important difference with respect to the previous MD studies was that, because of the high concentrations of SM and Chol, these bilayers were in the liquid-ordered state. It is reasonable to expect some liquid-ordered micro- or nanodomains in the cell membranes, while the presence of gel domains would be, unless under exceptional circumstances, disallowed [20]. MD pictures were different for the low- and high-charge bilayers (respectively 5 and 20 mol% DMPA), in the former case the peptide was bound through many contact points to the bilayer, whereas for the latter case only a small fragment of the peptide appeared to be bound. Moreover, the MD methods indicated the development of a β-sheet structure by the peptide in bilayers containing 5% DMPA, whereas in the presence of 20 mol % DMPA the peptide would retain a partially helical conformation. This would lead, under equilibrium conditions, to the situation of maximum β-sheet at low DMPA concentrations experimentally observed by ITC, Langmuir balance, and spectroscopic measurements [8]. The MD results indicated that the binding and fibril formation on the membrane surface depended on the composition of the bilayer, and was the result of a subtle balance of many inter- and intramolecular interactions between Aβ42 and membrane. The prevailing role of Lys-28 in bilayer binding was also underlined in this study, in agreement with previous data [83].
Gangliosides had been shown to promote the structural conversion of Aβ and increase the rate of peptide aggregation, but the exact nature of the interactions driving these processes was not understood [74,76]. Manna and Mukhopadhyay [86] investigated the behavior of Aβ42 monomers and dimers in GM1 ganglioside-containing liquid-ordered membranes, composed basically of 1-pamitoyl-2-oleoyl phosphatidylcholine (POPC) and Chol at a 3:1 mol ratio. The oligosaccharide head-group of GM1 was observed to act as scaffold for Aβ-binding through sugar-specific interactions. Starting from a monomer in a helical peptide conformation, a β-hairpin motif was formed at the C-terminus of the GM1-bound Aβ peptide, which did not appear in the absence of ganglioside. In the case of Aβ dimers, the β-structure was further enhanced by peptide-peptide interactions with the GM1-containing surfaces, which might influence the propensity of Aβ to aggregate into higher-ordered structures. Ganglioside effects were also considered in the MD studies by Ahyayauch et al. [10], performed with POPC lipid bilayers, to which small amounts of GM1 ganglioside (1–5 mol%) were incorporated. These bilayers were, thus, in the liquid-disorderd state (Ld). MD measurements concurred with surface pressure (Langmuir monolayer) and thioflavin T data in showing that, under those conditions, the Aβ42 peptide bound (adsorbed onto) the bilayer surface, but did not become inserted into it at surface pressures compatible with the cell membrane conditions (πi ≈ 30 mN/m). Moreover, those authors detected a very low degree of peptide oligomerization/aggregation under their conditions [10]. All these data supported the notion that Aβ42 binding to lipid membranes was facilitated by lipid chain disorder. GM1 tends to increase lipid order [50], and this property may also be responsible for the dual effect of GM1 on Aβ42 binding, its ordering properties compensating the pro-binding effects of the negative charge and H-bonding network.
The subject of Aβ42 oligomerization, or, more specifically, of tetramer formation in bilayers, was considered in at least three contributions [82,83,84]. Poojari et al. [87] used atomistic MD simulations to investigate the behavior of Aβ42 in zwitterionic and anionic lipid bilayers. These authors simulated transmembrane β-sheets (monomer and tetramer), as well as a helical structure, the latter obtained from an NMR study. In all cases Aβ42 remained embedded in the bilayer. It was found that zwitterionic surfaces and unsaturated lipids promoted Aβ42 transmembrane stability, and that the β-sheet tetramer was most stable as a result of inter-peptide interactions. A novel, interesting aspect of this study was that the translocation of water in the Aβ42-bilayer systems was analyzed. The process was slower in the presence than in the absence of peptide. The rate-limiting step was identified as the permeation through the hydrophobic core, where interactions between Aβ42 and permeating H2O molecules slowed the translocation process. The β-sheet tetramer allowed more water molecules to pass through the bilayer than the monomeric Aβ, and the authors concluded that the experimentally observed permeabilization of membranes must be due to membrane-bound Aβ oligomers, and not monomers. In turn, Brown and Bevan [88] utilized MD simulations to show the formation of a tetramer unit by four separate Aβ42 peptides, in the absence of lipid. Aβ42 tetramers showed a significant increase in β-strand formation relative to the monomers, suggesting that tetramerization could be a step in fibril formation. The previously formed Aβ42 tetramer was used in subsequent MD simulations in the presence of pure POPC or of a POPC/SM/Chol (1:1:1 mol ratio) membrane. The tetramer was elongated in the presence of the bilayers. With bilayers containing SM and Chol, formation of a more rod-like structure was observed, which could lead to generation of fibril-seeding aggregates.
The presence of tetramer led to more ordered, rigid membranes, with the pure POPC being affected to a greater extent than the 3-component membrane. In a complementary study on Aβ oligomerization, Tachi et al. [89] found that Aβ40 formed an α-helix, then a β-hairpin structure at the air-water interface containing GM1. The β-hairpin promotes the formation of oligomers with intermolecular β-sheets. The results suggest that helix formation, which is the first step in the conformational changes toward pathological aggregation, is initiated at the GM1-glycan moieties rather than at the lipid-ceramide moieties.
Brains of Alzheimer’s disease patients exhibit a substantial decrease in unsaturated lipid contents, particularly lipids containing (𝜔 – 3) docosahexaenoic (22:6cis) fatty acid chains in the frontal gray matter [90,91]. Hossain et al. [53], using spectroscopic and microscopic methods, demonstrated that docosahexaenoic acid inhibits Aβ42 fibril formation, suggesting that membranes rich in polyunsaturated lipids would hamper formation of Aβ aggregates on their surface. Moreover, electronic spin resonance experiments [75] showed that docosahexaenoic-containing lipids enhanced Aβ25−35 peptide interaction with lipid membrane, favoring deep peptide internalization, and inhibiting peptide release and subsequent fibrillization. Then, Ntarakas et al. [92] carried out MD simulations of Aβ1-28 and Aβ26-40 peptides in four different lipid bilayers: DMPC, 1-stearoyl-2-docosahexaenoyl PC (SDPC), and mixtures of distearoyl phosphatidyl ethanolamine (DSPE), didocosahexaenoyl PE (DDPE), DPPC, and dioleoyl PC (DOPC), mimicking neuronal membranes in "healthy" and "Alzheimer’s" brains. The simulations showed that the presence of polyunsaturated lipids caused stronger adsorption of Aβ peptides to the membrane and led to weaker binding between peptides when the latter form aggregates, thus confirming the previous experimental results [53,75].
More recently, Matthes and de Groot [93] have considered the role of Aβ42 oligomer conformations in membrane permeabilization. They used Aβ42 oligomer structures, previously determined in a membrane-mimicking environment, as model systems to study the pore formation process in phospholipid bilayers with all-atom molecular dynamics simulations. These authors found that pore formation and ion permeation occurred in the presence of β-sandwich structures with exposed side-by-side β-strand pairs formed by residues 9 to 21 of Aβ42. Water or ions were only in contact with the outermost, exposed β-sheet hydrophilic edges, along which water and ion conductivity took place. The extent of pore formation and ion permeation depended on the insertion depth of hydrophilic residues 13 to 16 (HHQK domain). These results suggest that membrane-inserted, layered β-sheet edges are a key structural motif in pore-forming Aβ42 oligomers and aggregate-induced membrane permeabilization.
6. An Effort in Understanding
This closing section summarizes a number of data and hypotheses that should be taken in mind for a proper understanding of Aβ peptide binding to cell membranes. General considerations will be discussed, together with more specific examples.
6.1. The Validity of Models
Virtually all the data in this review have been obtained either from model membranes (monolayers, vesicles, supported bilayers) or from simulation studies. The use of model membranes hardly needs to be justified, so vast is the amount of our knowledge on cell membranes that was born in the form of a liposome or other model study. Model membranes should not be identified with cell membranes, since, as their own name implies, they are intended to be simplified models of the biological structures. Yet simplification is at the core of any science, cell biology being not an exception.
Computer simulations can be seen as an extreme form of simplification, but here comes to mind the aphorism, attributed to A. Einstein, “Everything should be made as simple as possible, but not simpler”. Lipid bilayer simulations have been in use for the last four decades, and their explanatory and predictive powers have increased in parallel with the availability of more powerful computers. MD, which in fact predates computers, has been used for analyzing the physical movements of atoms and molecules in membranes. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. The time steps in an MD simulation must be short, typically only a few femtoseconds (10–15 s) each. Most of the events of biophysical interest, for example, structural changes in lipid chains, take place on timescales of nanoseconds, microseconds, or longer. In consequence, a typical simulation requires millions or billions of time steps. In these days, advances in computing allow reaching simulations in the millisecond time scale, with reasonable computation times [94]. Not infrequently, experimental and computational techniques have been applied in parallel in the study of Aβ-membrane interactions, of which some examples are given above. The good correlation of both kinds of results have improved the acceptability of MD simulations in biophysical studies in general.
6.2. Aβ Peptide Generation, Membrane Binding, Adsorption, and Insertion
One of the difficulties in assessing the literature in this field is that Aβ peptide generation, membrane binding, and membrane insertion are often confused, being as they are structurally and functionally very different, even if some of them may occur almost simultaneously.
Aβ production occurs at the membrane level, as a result of the action of membrane proteases, whose outcome is the release of a peptide in the extracellular aqueous medium. Aβ arises from the processing of the APP precursor protein, a transmembrane glycoprotein that influences neuron growth. APP is hydrolyzed through the sequential activity of β- and γ-secretases, leading mainly to Aβ40 and Aβ42 peptides, the latter being more hydrophobic and fibrillogenic [95].
Extracellularly produced Aβ interacts with the cell membrane lipid bilayer [96]. The newly released peptide is hydrophobic enough to partition into the neighboring cell membrane. This is often called “membrane binding”, although the name includes two different processes, if not two stages of the same process, specifically peptide adsorption onto the membrane/bilayer, and peptide insertion into the membrane/bilayer. The term deposition is sometimes used, mostly meaning adsorption, but its use is not recommended.
Adsorption depends chiefly on H-bonding and electrostatic interactions (although hydrophobic bonds should not be excluded at this stage). Adsorption is rather easily reversible, by e.g. changing ionic strength of the media, or submitting the membrane-peptide system to gradient centrifugation. The adsorption phenomenon, in the absence of other events, is rather conveniently observed in the Langmuir balance (Section 4.1 and Section 5.1). Ahayayauch et al. [10], using a combination of MD, isothermal calorimetry and Langmuir balance measurements, observed that Aβ42 peptide adsorbed but did not insert into ganglioside-containing phospholipid membranes in the liquid-disordered state.
Insertion is an essentially irreversible process, dominated by hydrophobic interactions, in which the previously adsorbed peptide penetrates the non-polar matrix to interact with the lipid fatty acyl chains [7]. Insertion can be confidently assessed through density gradient centrifugation assays (Section 4.2). A good example of peptide insertion was presented by Ahyayauch et al. [9], using a combination of Langmuir balance, ultracentrifugation and thioflavin T fluorescence, to show the insertion of Aβ42 peptide monomers into sphingomyelin/cholesterol/ganglioside bilayers.
One key consequence of such interactions is that Aβ conformation changes markedly, with an overall increase in β-structure and a tendency towards oligomer and fibril formation. The latter changes depend on whether the peptide has been inserted into, or merely adsorbed to the membrane. β-sheet formation is usually presumed to occur in adsorbed peptides [95]. However, the post-binding effects on Aβ conformation are beyond the scope of this review.
6.2. Ordered and Disordered Bilayers
The plasma membrane is laterally heterogeneous (Section 2). The supporting lipid matrix is mostly organized as a continuous fluid-disordered (or liquid-crystalline) Lα phase, in which discontinuous domains, either Lα or Lo, co-exist. Lo, or liquid-ordered domains, are rich in Chol. In the liquid-ordered domains, the lateral diffusion of the lipid molecules is fast, as in the case of Lα, while the C-C rotational frequency of the acyl lipid chains is low, i.e. lipid molecular order is high. Note that the size of the discontinuous domains may be very small, of the order of nanometers (nanodomains) [CPL].
The fluidity and molecular order of a membrane, or membrane domain, largely resulting from their lipid composition, greatly influence Aβ binding and subsequent folding/oligomerization. Lα bilayers bind Aβ42 peptides with higher affinity than those in the Lo phase, but this does not mean that Aβ42 fibrils, oligomers, and monomers cannot, to a smaller extent, bind bilayers in the Lo state (Section 5). A previous study [8] intended to clarify a discussion on the influence of lipid order on Aβ42-bilayer interactions. In summary, Aβ accumulation had been related to the presence of cholesterol [97] and lipid rafts [98]. However, other authors [99,100] had challenged the significance of the previous experiments. In our opinion, some previous studies [97,98] were in fact measuring the joint result of two different processes, namely APP hydrolysis by β- and γ-secretases, yielding Aβ42, and Aβ deposition and aggregation. β-secretase appears to be located preferentially in Lo domains [97,101], thus Aβ is probably generated in those domains, however, adsorption/insertion and aggregation may occur in different domains. In several of our previous studies [8,9,11,12] Aβ binding to fluid-ordered bilayers was studied in particular detail, because of the association of β-secretase to that sort of domains. Interestingly, Ahyayauch et al. [8] found that the Aβ42 affinity, measured e.g. by isothermal calorimetry, for Lα membranes was higher than that for the Lo environment in which it is originally generated. Moreover, particular care should be taken to define the aggregation state of Aβ when initially added to the membrane suspension, monomers should be used when the initial stages of the process are to be described.
6.3. Electrostatic Forces
Phospholipid headgroups are electrostatically charged in general, even if in some of them no net electric charge is detected, because of internal salt (zwitterion) formation between positive (usually NR4+) and negative (PO3-) groups, e.g. PC, PE, SM. The partial electric charges are enough to direct the binding of some amino acid residues toward the bilayer surface, thus starting adsorption and further putative insertion [49]. Thus, electrostatic interactions influence the attachment of Aβ to the cell membrane. The hydrophilic N-terminus of Aβ contains 6 positively charged amino acids (lysine, arginine, histidine) [102]. The latter may constitute the initial anchor for Aβ peptide adsorption. In particular, MD studies have pointed at Lys-28 as a plausible candidate for the initial anchoring.
Apart from the zwitterionic phospholipids, some lipids bearing a net negative charge exist in the plasma membrane that could attract the basic amino acid residues in Aβ. Phosphatidic acid and cardiolipin are acidic phospholipids that have been used in experimental studies of Aβ binding [12]. The most abundant acidic phospholipid in the plasma membrane is probably phosphatidyl serine (PS), but it is almost in its entirety oriented towards the cytosolic side, thus with little influence on extracellular Aβ binding. Glycosphingolipids (GSL), in turn, are phosphate-free lipids, some of them bearing one or more net negative charges in their sialic acid moieties, as is the case of gangliosides. GSL exist in the plasma membrane with the large polar headgroup oriented towards the extracellular medium, thus being optimally located for interaction with Aβ. GSL occur at low concentrations in the membrane, around 5 mol% of all lipids, and not all GSL bear a net negative charge, but, as seen in many model membrane studies, even a low proportion should be enough to facilitate electrostatic Aβ anchoring.
In relation with the above mentioned nanodomains (Section 6.3), it should be kept in mind that heterogeneity of lipid composition may mean heterogeneity of electric charge. It is thus possible that certain domains are enriched in gangliosides, hence in net negative charges, which make them good targets for Aβ anchoring. It should be remembered, in this context, the proposed existence of the nanodomains called “lipid rafts”, enriched in cholesterol and GSL, thus in a physical state close to the Lo phase [98,103], for which several bilayer compositions used in this kind of studies constitute a good model.
6.4. The Role of Specific Lipids
Even if much of this has been already discussed, it is perhaps suitable to review, in the briefest way, the role of specific lipids in the process of Aβ binding.
Phosphatidylcholine (PC). The most abundant phospholipid in mammalian cell membranes, and perhaps the most commonly used one in biophysical studies of Aβ-bilayer binding. The polar group is doubly ionized, forming a zwitterion. PC exhibits a marked tendency to self-organize in the form of bilayers (L phases). PC of natural origin (egg, liver) gives rise usually to fluid disordered Lα phases, and the same is true of the synthetic 1-palmitoyl-2-oleoyl and 1,2-dioleoyl derivatives, respectively POPC and DOPC. Aqueous dispersions of POPC, in the form of unilamellar vesicles, constitute a simple, satisfactory model for cell membranes. In turn 1,2-dipalmitoyl PC (DPPC) exists in the gel Lβ phase at room temperature, and is often employed when, for experimental reasons, this non-physiological condition must be mimicked. Equimolar DPPC/Chol mixtures form good examples of bilayers in the liquid-ordered Lo state.
Sphingomyelin (SM). A structural analog of PC, but a sphingolipid, in which the phosphorylcholine group is esterified to sphingosine C1 (OH), instead of to glycerol C3 (OH). SM is the most abundant sphingolipid in membranes [104]. Its biological activity is rather low, its main biological role appears to be to act as a sphingolipid depot, from which bioactive lipids, e.g. ceramides or gangliosides, can be readily synthesized in response to a given need. SM of natural origin are often in the gel state, or close to the gel-fluid interface, at the physiological temperatures of mammals. Equimolar palmitoyl SM/Chol mixtures give rise to robust bilayers in the Lo state.
Gangliosides. A complex group of glycosphingolipids (no phosphorus in their structure) containing one or more sialic acid residues in their glycosyl moieties. In turn, sialic acids, or N-acetylneuraminic acids, are a diverse group of 9-carbon carboxylated monosaccharides, thus endowing gangliosides with one or more net negative charges (Section 6.4). Gangliosides are typically located, albeit at low concentrations, on the outer surface of nerve cells, and are presumed to provide a primary anchor site for Aβ binding. It is possible that Aβ, or at least a portion of it, binds the protruding saccharide portion of gangliosides, enriched in domains, just where it is generated, with immediate effects on peptide aggregation, and therefore that ganglioside-containing Lo domains play a pivotal role in the paradigm of Aβ aggregation.
Cholesterol (Chol). The only lipid, in this short catalog, that is not biosynthetically related to fatty acids, and in many senses a most mysterious molecule [105]. Unlike the above lipids, it is a rigid, flat molecule, its basic structure being constituted by a tetracycle (cyclopentane perhydrophenanthrene). Chol, or close relatives, exist in all eukaryotic membranes. From the biophysical point of view, Chol appears to act as a fluidity buffer, decreasing fluidity of Lα phases and increasing it when added to lipids in Lβ phases. In fact, when Chol is added to DPPC or palmitoyl SM in the gel phase, a particular fluid phase, the Lo liquid-ordered phase, is obtained. Even in the absence of other lipids, palmitoyl SM/Chol mixtures can bind Aβ oligomers or fibrils, while pure SM cannot.
6.5. Aggregation State
The consensus view of Aβ binding to lipid bilayers is that the process is initiated by an Aβ molecule in monomeric form, probably freshly hydrolyzed from APP at the extracellular side of the plasma membrane. After re-joining the membrane, the peptide (either adsorbed or inserted) will oligomerize and eventually give rise to a fibril, and to a plaque. In what is probably a sound strategy, most biophysical studies are focused on the initial stages of the process, consequently the vast majority of available data refer to monomeric Aβ interaction with lipids. But this should not mask the smaller, but significant amount of investigations in which pre-formed oligomers or fibrils have been added to lipid bilayers. Note that oligomers appear to be particularly neurotoxic [82,92]. Sparing the details of these sometimes complex data, it should be stressed at this point that oligomers and fibrils form and remain membrane-bound when interacting with bilayers [88,89]. According to equilibrium studies (calorimetry, Langmuir balance) binding appears to be entropically driven in most cases, due to the peptide disordering the lipid phase upon binding [11]. A naïve interpretation of this observation would be that, if Aβ aggregates could not become/remain associated to the membranes, the system at equilibrium would be displaced towards oligomers/fibrils being expelled from the bilayers, a situation far different from what we know to be the case. In cell membranes of HEK293 of neuronal origin, oligomeric Aβ42, but not monomers or fibers, have been shown to form voltage-independent, non-selective ion channels, suggestive of peptide insertion into the cell membrane [106]. In contrast, Aβ40 oligomers, fibers, or monomers failed to form channels. A final note of caution on this aspect of Aβ studies: it should not be forgotten that the experimental results reported as obtained with “monomers” cannot be always guaranteed to arise from oligomer-free samples, due to the intrinsic experimental difficulty to obtain “pure monomer” preparations.
6.6. Data Integration
Combining the different available data into a single table or figure is a simple but effective way to obtain reliable conclusions. As an example, ultracentrifugation, calorimetric and Langmuir balance data for Aβ42 in monomer or fibril form, interacting with SM/Chol monolayers/bilayers, composed of SM/Chol ± gangliosides (12 independent parameters, from various publications) have been collected in Table 1. The table could have been made much more extensive, but it is useful in its present form as a methodological example. Several conclusions, already discussed above, are seen more clearly here, e.g. (i) gangliosides facilitate Aβ42-lipid interaction, (ii) fibrils interact with lipids less favorably than monomers, (iii) similar association constants Ka are often the result of very different, mutually compensating enthalpic (ΔH) and entropic (ΔS) components, (iv) monomers, but not fibrils, become inserted in lipid monolayers at surface pressures compatible with the values found in cell membranes, and (v) the fact that Aβ42 monomer interaction with SM/Chol bilayers did not elicit measurable heat exchanges, while ultracentrifugation or Langmuir balance demonstrated a clear interaction, deserves a more detailed investigation, but it may be related to the fact that these bilayers are in the liquid-ordered state. Aβ42 monomer adsorption onto fluid bilayers gives off measurable amounts of heat [10].
Figure 3.
ITC calorimetric studies. (A) A representative titration calorimetry curve of unilamellar vesicles composed of SM/Chol/GT1b ganglioside (47.5/47.5/5 mol ratio) with Aβ42 peptide fibrils. The calorimetric trace was observed upon successive injections of lipid vesicles into a Aβ42 solution contained in the reaction cell. (B) Cumulative heats of reaction obtained from the integration of the peaks displayed in the top plot. The solid line represents the fitting of the experimental data to a partitioning model. The calorimetric cell was filled with a 28 μM Aβ42 solution. Lipid vesicles at 35 mM lipid concentration were injected into the cell (1.43 mL) in 10- μL steps, i.e., leading to a 143-fold dilution of lipid vesicles. The titration experiments were performed at 37 °C. (Taken from [11].
Figure 3.
ITC calorimetric studies. (A) A representative titration calorimetry curve of unilamellar vesicles composed of SM/Chol/GT1b ganglioside (47.5/47.5/5 mol ratio) with Aβ42 peptide fibrils. The calorimetric trace was observed upon successive injections of lipid vesicles into a Aβ42 solution contained in the reaction cell. (B) Cumulative heats of reaction obtained from the integration of the peaks displayed in the top plot. The solid line represents the fitting of the experimental data to a partitioning model. The calorimetric cell was filled with a 28 μM Aβ42 solution. Lipid vesicles at 35 mM lipid concentration were injected into the cell (1.43 mL) in 10- μL steps, i.e., leading to a 143-fold dilution of lipid vesicles. The titration experiments were performed at 37 °C. (Taken from [11].
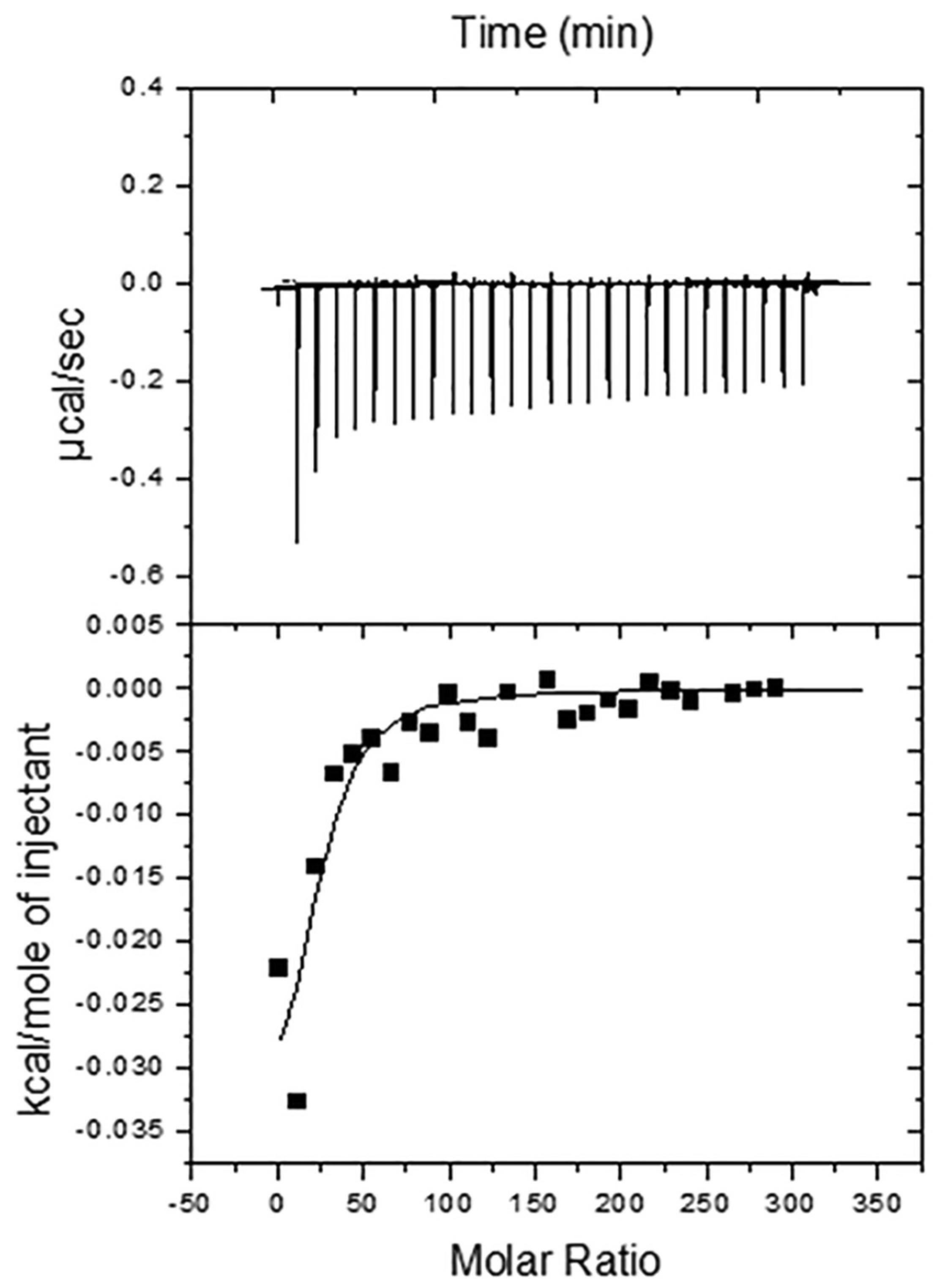
Figure 4.
Final state of the 500 ns MD simulation of the Aβ42 β-sheet tetramer in a POPC bilayer. The peptide is shown in cartoon and colored based on the physicochemical properties of the residues: blue, basic; red, acidic; white, hydrophobic; and green, polar. The bilayer phosphorus atoms are shown as Van der Waals spheres in tan color. Lipid tails and water molecules are not shown for clarity. (Taken from [87]).
Figure 4.
Final state of the 500 ns MD simulation of the Aβ42 β-sheet tetramer in a POPC bilayer. The peptide is shown in cartoon and colored based on the physicochemical properties of the residues: blue, basic; red, acidic; white, hydrophobic; and green, polar. The bilayer phosphorus atoms are shown as Van der Waals spheres in tan color. Lipid tails and water molecules are not shown for clarity. (Taken from [87]).
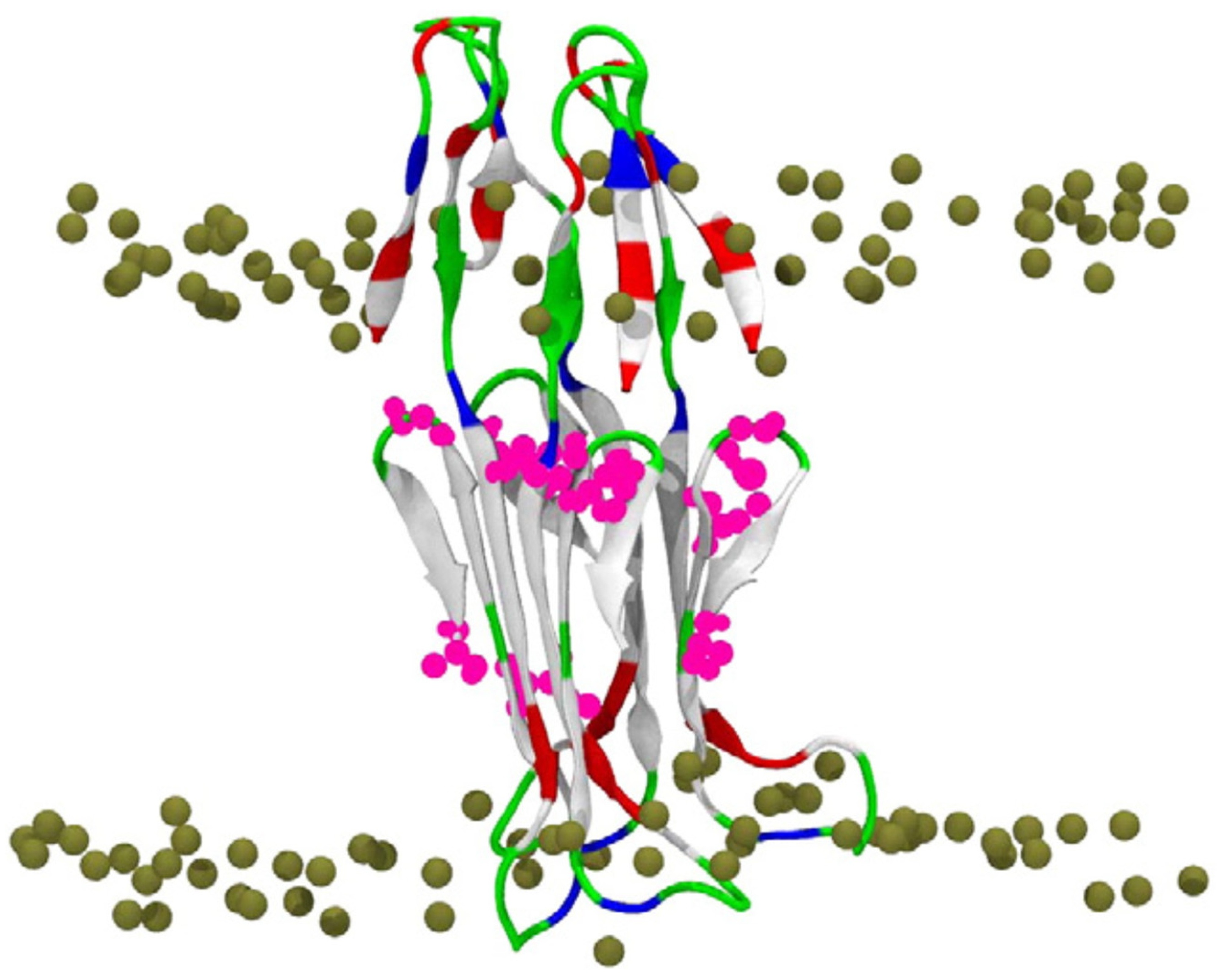
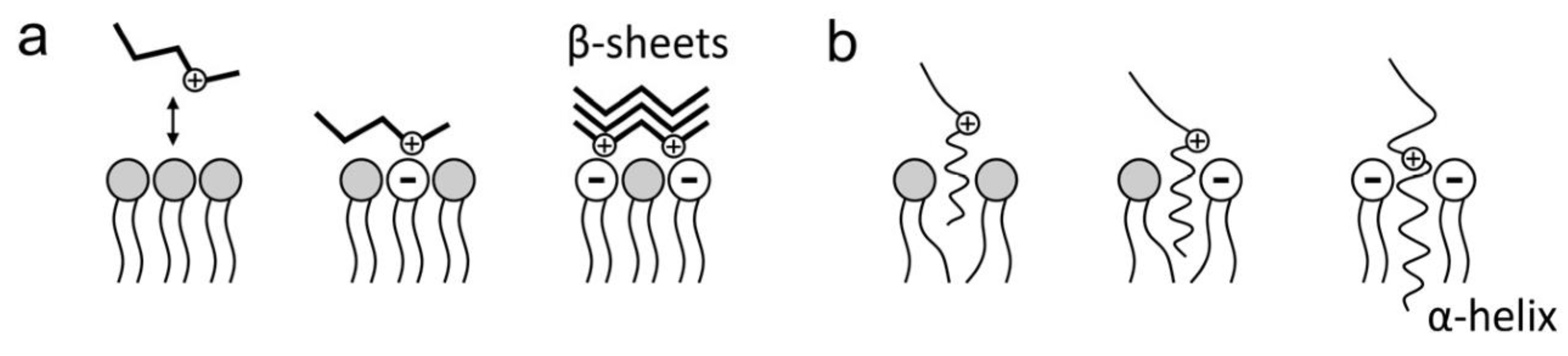
Figure 5.
Electrostatic interactions of amyloid peptides with cell membrane lipids. (a) Aβ adsorption on the membrane surface; (b) Aβ insertion in the membrane. (Taken from [94]).
Figure 5.
Electrostatic interactions of amyloid peptides with cell membrane lipids. (a) Aβ adsorption on the membrane surface; (b) Aβ insertion in the membrane. (Taken from [94]).

Author Contributions
All authors have contributed to a similar extent to the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded in part by the Spanish Ministry of Science, Innovation, and Universities (MCIU), Agencia Estatal de Investigación (AEI), Fondo Europeo de Desarrollo Regional (FEDER) (grant No. PID2021-124461NB-I00), the Basque Government (grant No. IT1625-22), Fundación Ramón Areces (CIVP20A6619), Fundación Biofísica Bizkaia, and the Basque Excellence Research Centre (BERC) program of the Basque Government.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations: Chol, cholesterol; CL, cardiolipin; GSL, glycosphingolipid(s); ITC, isothermal calorimetry; PA, phosphatidic acid; SM, sphingomyelin.
References
- Cummings, J.L. Alzheimer disease, JAMA 2002, 287, 2335-2338. [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [CrossRef]
- Small, D.H.; Mok, S.S.; Bornstein, J.C. Alzheimer's disease and Abeta toxicity: from top to bottom, Nat. Rev. Neurosci. 2001, 2, 595–598. [CrossRef]
- Yin, Y.I.; Bassit, B.; Zhu, L.; Yang, X.; Wang, C.; Li, Y.M.J. {gamma}-Secretase substrate concentration modulates the Abeta42/Abeta40 ratio: implications for Alzheimer disease, Biol. Chem. 2007, 282, 23639–23644. [CrossRef]
- Karisetti, B.C.; Bhatnagar, A.; Armour, E.M.; Beaver, M.; Zhang, H.; Elefant, F. Amyloid-β peptide impact on synaptic function and neuroepigenetic gene control reveal new therapeutic strategies for Alzheimer’s disease, Front. Mol. Neurosci. 2020, 13, 577622. [CrossRef]
- Matsuzaki, K. Physicochemical interactions of amyloid beta-peptide with lipid bilayers, Biochim. Biophys. Acta 2007, 1768, 1935–1942. [CrossRef]
- Terzi, E.; Hölzemann, G.; Seelig, J. Self-association of beta-amyloid peptide (1-40) in solution and binding to lipid membranes, J. Mol. Biol. 1995, 252, 633–642. [CrossRef]
- Ahyayauch, H.; Raab, M.; J.V. Busto, J.V.; Andraka, N.; Arrondo, J.L.; Masserini, M.; Tvaroska, I.; Goñi, F.M. Binding of β-amyloid (1-42) peptide to negatively charged phospholipid membranes in the liquid-ordered state: modeling and experimental studies, Biophys. J. 2012, 103, 453–463. [CrossRef]
- Ahyayauch, H.; de la Arada, I.; Masserini, M.E.; Arrondo, J.L.R.; Goñi, F.M.; Alonso, A. The binding of aβ42 peptide monomers to sphingomyelin/ cholesterol/ ganglioside bilayers assayed by density gradient ultracentrifugation. Int. J. Mol. Sci. 2020, 21, 1674. [CrossRef]
- Ahyayauch, H.; García-Arribas, A.B.; Masserini, M.E.; Pantano, S.; Goñi, F.M.; Alonso, A. β-Amyloid (1-42) peptide adsorbs but does not insert into ganglioside-containing phospholipid membranes in the liquid-disordered state: modelling and experimental studies. Int. J. Biol. Macromol. 2020, 164, 2651-2658. [CrossRef]
- Ahyayauch, H.; Masserini, M.; Goñi, F.M.; Alonso, A. The interaction of Aβ42 peptide in monomer, oligomer or fibril forms with sphingomyelin/cholesterol/ ganglioside bilayers. Int. J. Biol. Macromol. 2021, 168, 611-619. [CrossRef]
- Ahyayauch, H.; Masserini, M.E.; Goñi, F.M.; Alonso, A. The influence of lipid electric charge on the binding of Aβ-amyloid (1–42) amyloid peptide to bilayers in the liquid-ordered state. Biomolecules, 2024, 14, 298. [CrossRef]
- Small, D.M. The Physical Chemistry of Lipids. From Alkanes to Phospholipids, Plenum Press, New York, 1986, 43–87.
- Luzzati, V.; Tardieu, A.; Taupin, D. A pattern-recognition approach to the phase problem: application to the X-ray diffraction study of biological membranes and model systems, J. Mol. Biol. 1972, 64, 269–286. [CrossRef]
- Quinn, P.J.; Wolf, C. An X-ray diffraction study of model membrane raft structures, FEBS J. 2010, 277, 4685–4698. [CrossRef]
- Caffrey, M. The study of lipid phase transition kinetics by time-resolved X-ray diffraction, Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 159–186. [CrossRef]
- Cullis, P.R.; de Kruijff, B. Lipid polymorphism and the functional roles of lipids in biological membranes, Biochim. Biophys. Acta 1979, 559, 399–420. [CrossRef]
- Ladbrooke, B.D.; Williams, R.M.; Chapman, D. Studies on lecithin–cholesterol–water interactions by differential scanning calorimetry and X-ray diffraction, Biochim. Biophys. Acta 1968, 150, 333–340. [CrossRef]
- McMullen, T.P.; Wong, B.C.; Tham, E.L.; Lewis, R.N.; McElhaney, R.N. Differential scanning calorimetric study of the interaction of cholesterol with the major lipids of the Acholeplasma laidlawii B membrane, Biochemistry 1996, 35, 16789–16798. [CrossRef]
- Goñi, F.M. The basic structure and dynamics of cell membranes: An update of the Singer–Nicolson model. Biochim. Biophys. Acta, 2014, 1838, 1467-1476. [CrossRef]
- Ipsen, J.H.; Karlström, G.; Mouritsen, O.G.; Wennerström, H.; Zuckermann, M.J. Phase equilibria in the phosphatidylcholine–cholesterol system, Biochim. Biophys. Acta 1987, 905, 162–172. [CrossRef]
- Sackmann, E. A charge-decoration technique for studying the heterogeneity of coexistent monolayer phases by electron microscopy. Nature, 1985, 313, 299–301. [CrossRef]
- McConnell, H.M. Structures and transitions in lipid monolayers at the air-water interface. Annu. Rev. Phys. Chem 1991, 42, 171–195. [CrossRef]
- McConnell, H.M.; Vrljic M. Liquid-liquid immiscibility in membranes. Ann. Rev. Biophys. Biomol. Struct 2003, 32, 469–492. [CrossRef]
- Brown, R.E.; Brockman, H.L. Using monomolecular films to characterize lipid lateral interactions. In: McIntosh, T.J., editor. Methods Molecular Biology. 398. Humana Press Inc.; Totowa, NJ, 2007. pp. 41-58. [CrossRef]
- Slotte, J.P. Lateral domain formation in mixed monolayers containing cholesterol and dipalmitoylphosphatidylcholine or N-palmitoylsphingomyelin. Biochim. Biophys. Acta 1995, 1235, 419–427. [CrossRef]
- Nag, K.; Pao, J.S.; Harbottle, R.R.; Possmayer, F.; Petersen, N.O.; Bagatolli, L.A. Segregation of saturated chain lipids in pulmonary surfactant films and bilayers. Biophys. J. 2002, 82, 2041–2051. [CrossRef]
- Veatch, S.L.; Keller, S.L. Organization in lipid membranes containing cholesterol. Phys. Rev. Lett. 2002, 8 9, 268101–268104. [CrossRef]
- Stottrup, B.L.; Keller, S.L. Phase behaviour of lipid monolayers containing DPPC and cholesterol analogs. Biophys. J. 2006, 90, 3176–3183. [CrossRef]
- Phillips, M.C. The physical state of phospholipids and cholesterol in monolayers, bilayers, and membranes. Prog. Surf. Membr. Sci. 1972, 5, 139–221. [CrossRef]
- Dietrich, C.; Bagatolli, L.A.; Volovyk, Z.N.; Thompson, N.L.; Levi, M.; Jacobson, K.; Gratton, E. Lipid rafts reconstituted in model membranes. Biophys. J. 2001, 80, 1417–1428. [CrossRef]
- Dietrich, C.; Volovyk, Z.N.; Levi, M.; Thompson, N.L.; Jacobson, K. Partitioning of Thy-1, GM1, and cross-linked phospholipid analogs into lipid rafts reconstituted in supported model membrane monolayers. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 10642–10647. [CrossRef]
- Yuan, C.; Johnston, L.J. Atomic force microscopy studies of ganglioside GM1 domains in phosphatidylcholine and phosphatidylcholine/cholesterol bilayers. Biophys. J. 2001, 81, 1059–1069. [CrossRef]
- Yuan, C.; Furlong, J.; Burgos, P.; Johnston, L.J. The size of lipid rafts: an atomic force microscopy study of ganglioside GM1 domains in sphingomyelin/DOPC/cholesterol membranes. Biophys. J. 2002, 82, 2526–2535. [CrossRef]
- Lawrence, J.C.; Saslowsky, D.E.; Edwardson, J.M.; Henderson, R.M. Real-time analysis of the effects of cholesterol on lipid raft behavior using atomic force microscopy. Biophys. J. 2003, 84, 1827–1832. [CrossRef]
- McQuaw, C.M.; Zheng, L.; Ewing, A.G.; Winograd, N. Localization of sphingomyelin in cholesterol domains by imaging mass spectrometry. Langmuir 2007, 23, 5645–5650. [CrossRef]
- McQuaw, C.M.; Sostarecz, A.G.; Zheng, L.; Ewing, A.G.; Winograd, N. Lateral heterogeneity of dipalmitoylphosphatidylethanolamine-cholesterol Langmuir-Blodgett films investigated with imaging time-of-flight secondary ion mass spectrometry and atomic force microscopy. Langmuir 2005, 21, 807–813. [CrossRef]
- Zheng, L.; McQuaw, C.M.; Ewing, A.G.; Winograd, N. Sphingomyelin/phosphatidylcholine and cholesterol interactions studied by imaging mass spectrometry. J. Am. Chem. Soc. 2007, 129,15730–15731. [CrossRef]
- Nagle, J.F.; Tristram-Nagle, S. Structure of lipid bilayers. Biochim. Biophys. Acta 2000, 1469, 159–195. [CrossRef]
- Chapman, D.; Gómez-Fernández, J.C.; Goñi, F.M. Intrinsic protein–lipid interactions. Physical and biochemical evidence. FEBS Lett. 1979, 98, 211–223. [CrossRef]
- Maltseva, E.; Brezesinski, G. Adsorption of amyloid b (1–40) peptide to phosphatidylethanolamine monolayers. ChemPhysChem. 2004, 5, 1185–1190. [CrossRef]
- Maltseva, E.; Kerth, A.; Brezesinski, G. Adsorption of amyloid b (1–40) peptide at phospholipid monolayers. ChemBioChem. 2005, 6, 1817–1824. [CrossRef]
- Alvarez, A.B.; Caruso, B.; Rodríguez, P.E.A.; Petersen, S.B.; Fidelio, G.D. Aβ-amyloid fibrils are self-triggered by the interfacial lipid environment and low peptide content. Langmuir. 2020, 36, 8056-8065. [CrossRef]
- Alvarez, A.B.; Rodríguez, P.E.A.; Fidelio, G.D. Gangliosides smelt nanostructured amyloid Aβ(1-40) fibrils in a membrane lipid environment. Biochim. Biophys. Acta Biomembr. 2022,1864,183749. [CrossRef]
- Gobbi, M.; Re, F.; Canovi, M.; Beeg, M.; Gregori, M.; Sesana, S.; Sonnino, S.; Brogioli, D.; Musicanti, C.; Gasco, P.; et al. Lipid-based nanoparticles with high binding affinity for amyloid-beta1-42 peptide. Biomaterials. 2010, 31, 6519–6529. [CrossRef]
- Arnulphi, C.; Sot, J.; García-Pacios, M.; Arrondo, J.L.; Alonso, A.; Goñi, F.M. Triton X-100 partitioning into sphingomyelin bilayers at subsolubilizing detergent concentrations: effect of lipid phase and a comparison with dipalmitoylphosphatidylcholine. Biophys. J. 2007, 93, 3504–3514. [CrossRef]
- Seelig, J. Titration calorimetry of lipid-peptide interactions, Biochim. Biophys. Acta 1997, 1331, 103–116. [CrossRef]
- Heerklotz, H.; Lantzsch, G.; Binder, H.; Klose, G.; Blume, A. Thermodynamic characterization of dilute aqueous lipid/detergent mixtures of POPC and C12EO8 by means of isothermal titration calorimetry. J. Phys. Chem. 1996, 100, 6764–6774. [CrossRef]
- Martinez-Senac, M.M.; Villalain, J.; Gomez-Fernandez, J.C. Structure of the Alzheimer b-amyloid peptide (25-35) and its interaction with negatively charged phospholipid vesicles. Eur. J. Biochem. 1999, 265, 744-753. [CrossRef]
- Ferraretto, A.; Pitto, M.; Palestini, P.; Masserini, M. Lipid domains in the membrane: thermotropic properties. Biochemistry 1997, 36, 9232–9236. [CrossRef]
- Nilsson, M. R. Techniques to study amyloid fibril formation in vitro. Methods 2004, 34, 151–160. [CrossRef]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta. 2010, 1804, 1405–1412. [CrossRef]
- Hossain, S.; Hashimoto, M.; Katakura, M.; Miwa, K.; Shimada, T.; Shido, O. Mechanism of docosahexaenoic acid-induced inhibition of in vitro Abeta1-42 fibrillation and Abeta1-42-induced toxicity in SH-S5Y5 cells. J. Neurochem. 2009, 111, 568-579. [CrossRef]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 2002, 269, 5642–5648. [CrossRef]
- Herrera, F.E.; Pantano, S. Salt induced asymmetry in membrane simulations by partial restriction of ionic motion. J. Chem. Phys. 2009, 130, 195105. [CrossRef]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. E. J. Chem. Theory Comput. 2008, 4, 435–447. [CrossRef]
- Tieleman, D. P.; Berendsen, H.J.C. Molecular dynamics simulations of fully hydrated dipalmitoylphosphatidylcholine bilayer with different macroscopic boundary conditions and parameters. J. Chem. Phys. 1996, 105, 4871–4880. [CrossRef]
- Scott, W.; Hunenberger, P.; van Gunsteren, W. The GROMOS biomolecular simulation program package. J. Phys. Chem. 1999, 103, 3596–3607. [CrossRef]
- Berger, O.; Edholm, O.; Jähnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. [CrossRef]
- Lemkul, J. A.; Bevan, D.R. A comparative molecular dynamics analysis of the amyloid b-peptide in a lipid bilayer. Arch. Biochem. Biophys. 2008, 470, 54–63. [CrossRef]
- Lemkul, J. A.; Bevan, D.R. Perturbation of membranes by the amyloid b-peptide—a molecular dynamics study. FEBS J. 2009, 276, 3060–3075. [CrossRef]
- Darré, L.; Machado, M.R.; Dans, P.D.; Herrera, F.E.; Pantano, S. Another coarse grain model for aqueous solvation: what for? J. Chem. Theory Comput. 2010, 6, 3793–3807. [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [CrossRef]
- Terzi, E.; Hölzemann, G.; Seelig, J. Interaction of Alzheimer beta-amyloid peptide(1-40) with lipid membranes. Biochemistry 1997,36, 14845-14852. [CrossRef]
- Terzi, E.; Hölzemann, G.; Seelig, J. Reversible random coil-beta-sheet transition of the Alzheimer beta-amyloid fragment (25-35). Biochemistry. 1994, 33, 1345-1350. [CrossRef]
- Terzi, E.; Hölzemann, G.; Seelig, J. Alzheimer beta-amyloid peptide 25-35: electrostatic interactions with phospholipid membranes. Biochemistry. 1994, 33, 7434-7441. [CrossRef]
- Kirkitadze, M.D.; Condron, M.M.; Teplow, D.B. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J. Mol. Biol. 2001, 312,1103-1119. [CrossRef]
- Chang, H-W.; Ma, H-I.; Wu, Y.-S.; … ; Chan, J.C.C. Site specific NMR characterization of abeta-40 oligomers cross seeded by abeta-42 oligomers. Chem Sci. 2022, 13, 8526–8535. [CrossRef]
- Matsuzaki, K.; Kato, K.; Yanagisawa, K. Abeta polymerization through interaction with membrane gangliosides. Biochim. Biophys. Acta 2010 1801, 868-877. [CrossRef]
- Vitiello, G.; Di Marino, S.; D’Ursi, A.M.; D’Errico, G. Omega-3 fatty acids regulate the interaction of the Alzheimer’s A(25–35) peptide with lipid membranes. Langmuir 2013, 29, 14239–14245. [CrossRef]
- Chi, E.Y.; Frey, S.L.; Lee, K.Y.C. Ganglioside GM1-mediated amyloid-beta fibrillogenesis and membrane disruption. Biochemistry 2007, 46, 1913–1924. [CrossRef]
- Lin, M. S.; Chen, X.B.; …; Chen, W.Y. Dynamic fluorescence imaging analysis to investigate the cholesterol recruitment in lipid monolayer during the interaction between b-amyloid (1-40) and lipid monolayers. Colloids Surf. B Biointerfaces. 2009, 74, 59–66. [CrossRef]
- Thakur, G.; Pao, C.; Leblanc, R.M. Surface chemistry of lipid raft and amyloid Ab (1-40) Langmuir monolayer. Colloids Surf. B Biointerfaces. 2011, 87, 369–377. [CrossRef]
- Sánchez-Magraner, L.; Cortajarena, A.L.; …; Ostolaza, H. Membrane insertion of Escherichia coli alpha-hemolysin is independent from membrane lysis. J. Biol. Chem. 2006, 281, 5461–5467. [CrossRef]
- Alvarez, A.B.; Rodríguez, P.E.A.; Fidelio, G.D.; Caruso, B. Aβ amyloid fibers drastically alter the topography and mechanical properties of lipid membranes. Langmuir. 2023, 39, 18923-18934. [CrossRef]
- Alvarez, A.B.; Rodríguez, P.E.A.; Fidelio, G.D. Interfacial Aβ fibril formation is modulated by the disorder-order state of the lipids: The concept of the physical environment as amyloid inductor in biomembranes. Biochim. Biophys. Acta Biomembr. 2024, 1866, 184234. [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The amyloid-beta oligomer hypothesis: beginning of the third decade. J. Alzheimers Dis. 2018, 64, S567-S610. [CrossRef]
- Xu, Y.; Shen, J.; Luo, X.; …; Jiang, H. Conformational transition of amyloid β-peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 5403–5407. [CrossRef]
- Lemkul, J.A.; Bevan, D.R. A comparative molecular dynamics analysis of the amyloid beta-peptide in a lipid bilayer. Arch. Biochem. Biophys. 2008, 470, 54-63. [CrossRef]
- Davis, C.H.; Berkowitz, M.L. Interaction between amyloid-β (1–42) peptide and phospholipid bilayers: a molecular dynamics study. Biophys. J. 2009, 96, 785-797. [CrossRef]
- Manna M.; Mukhopadhyay, C. Binding, conformational transition and dimerization of amyloid-β peptide on GM1-containing ternary membrane: insights from molecular dynamics simulation. PLoS ONE 2013, 8, e71308. [CrossRef]
- Poojari, C.; Kukol, A.; Strodel, B. How the amyloid-β peptide and membranes affect each other: an extensive simulation study. Biochim. Biophys. Acta 2013, 1828, 327-339. [CrossRef]
- Brown, A.M.; Bevan, D.R. Molecular dynamics simulations of amyloid β-peptide (1-42): tetramer formation and membrane interactions. Biophys. J. 2016, 111, 937–949. [CrossRef]
- Tachi, Y.; Itoh, S.G.; Okumura, H. Molecular dynamics simulations of amyloid-β peptides in heterogeneous environments. Biophys. Physicobiol. 2022, 19, e190010. [CrossRef]
- Söderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids 1991, 26, 3156–3168. [CrossRef]
- Torres, M.; Price, S.L.; …; Escribá, P.V. Membrane lipid modifications and therapeutic effects mediated by hydroxydocosahexaenoic acid on Alzheimer's disease. Biochim. Biophys. Acta 2014, 1838, 1680-1692. [CrossRef]
- Ntarakas, N.; Ermilova, I.; Lyubartsev, AP. Effect of lipid saturation on amyloid-beta peptide partitioning and aggregation in neuronal membranes: molecular dynamics simulations. Eur. Biophys. J. 2019, 48, 813–824. [CrossRef]
- Matthes, D.; de Groot, B.L. Molecular dynamics simulations reveal the importance of amyloid-beta oligomer β-sheet edge conformations in membrane permeabilization. J. Biol. Chem. 2023, 299, 103034. [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron. 2018, 99, 1129-1143. [CrossRef]
- Wiatrak, B.; Piasny, J.; Kuźniarski, A.; Gąsiorowski, K. Interactions of amyloid-β with membrane proteins. Int. J. Mol. Sci. 2021, 22, 6075. [CrossRef]
- Chang, C.C.; Edwald, E.; Veatch, S.; Steel, D.G.; Gafni, A. Interactions of amyloid-β peptides on lipid bilayer studied by single molecule imaging and tracking. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1616-1624. [CrossRef]
- Kalvodova, L.; Kahya, N.; Simons, K. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 2005, 280, 36815–36823. [CrossRef]
- Ehehalt, R.; Keller, P.; Simons, K. Amyloidogenic processing of the Alzheimer b-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [CrossRef]
- Park, I. H.; Hwang, E.M.; Mook-Jung, I. Lovastatin enhances Ab production and senile plaque deposition in female Tg2576 mice. Neurobiol. Aging 2003, 24, 637–643. [CrossRef]
- Abad-Rodriguez, J.; Ledesma, M.D.; Dotti, C.G. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol. 2004, 167, 953–960. [CrossRef]
- Riddell, D. R.; Christie, G.; Dingwall, C. Compartmentalization of b-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [CrossRef]
- Chauhan, A.; Ray, I.; Chauhan, V.P. Interaction of amyloid beta-protein with anionic phospholipids: possible involvement of Lys28 and C-terminus aliphatic amino acids. Neurochem. Res. 2000, 25, 423-9. [CrossRef]
- Goñi, F.M. "Rafts": A nickname for putative transient nanodomains. Chem. Phys. Lipids 2019, 218, 34-39. [CrossRef]
- Goñi, F.M. Sphingomyelin: What is it good for? Biochem. Biophys. Res. Commun. 2022, 633, 23-25. [CrossRef]
- Bukiya, A.N.; Dopico, A.M. (eds.) Cholesterol. From chemistry and biophysics to the clinic. Academic Press, London, 2022.
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion channel formation by amyloid-β42 oligomers but not amyloid-β40 in cellular membranes. J. Biol. Chem. 2017, 292, 1404 –1413. [CrossRef]
Figure 1.
Lipid structures.
Figure 2.
Changes in surface pressure of lipid monolayers, upon insertion of Aβ42 monomers at varying initial pressures. (A) A representative experiment, obtained with SM/Chol/DMPA (40/40/20) at 15 mN/m. (B) Equilibrium values. (●) SM/Chol (1:1); (▲) SM/Chol/DMPA (47.5/47.5/5); (■)SM/Chol/DMPA (40/40/20). Average values ± SE (n = 3). Aβ42 stock solution was 50 mM. Aβ42 final concentration in the trough was 1.22 mM. (Taken from [8]).
Figure 2.
Changes in surface pressure of lipid monolayers, upon insertion of Aβ42 monomers at varying initial pressures. (A) A representative experiment, obtained with SM/Chol/DMPA (40/40/20) at 15 mN/m. (B) Equilibrium values. (●) SM/Chol (1:1); (▲) SM/Chol/DMPA (47.5/47.5/5); (■)SM/Chol/DMPA (40/40/20). Average values ± SE (n = 3). Aβ42 stock solution was 50 mM. Aβ42 final concentration in the trough was 1.22 mM. (Taken from [8]).
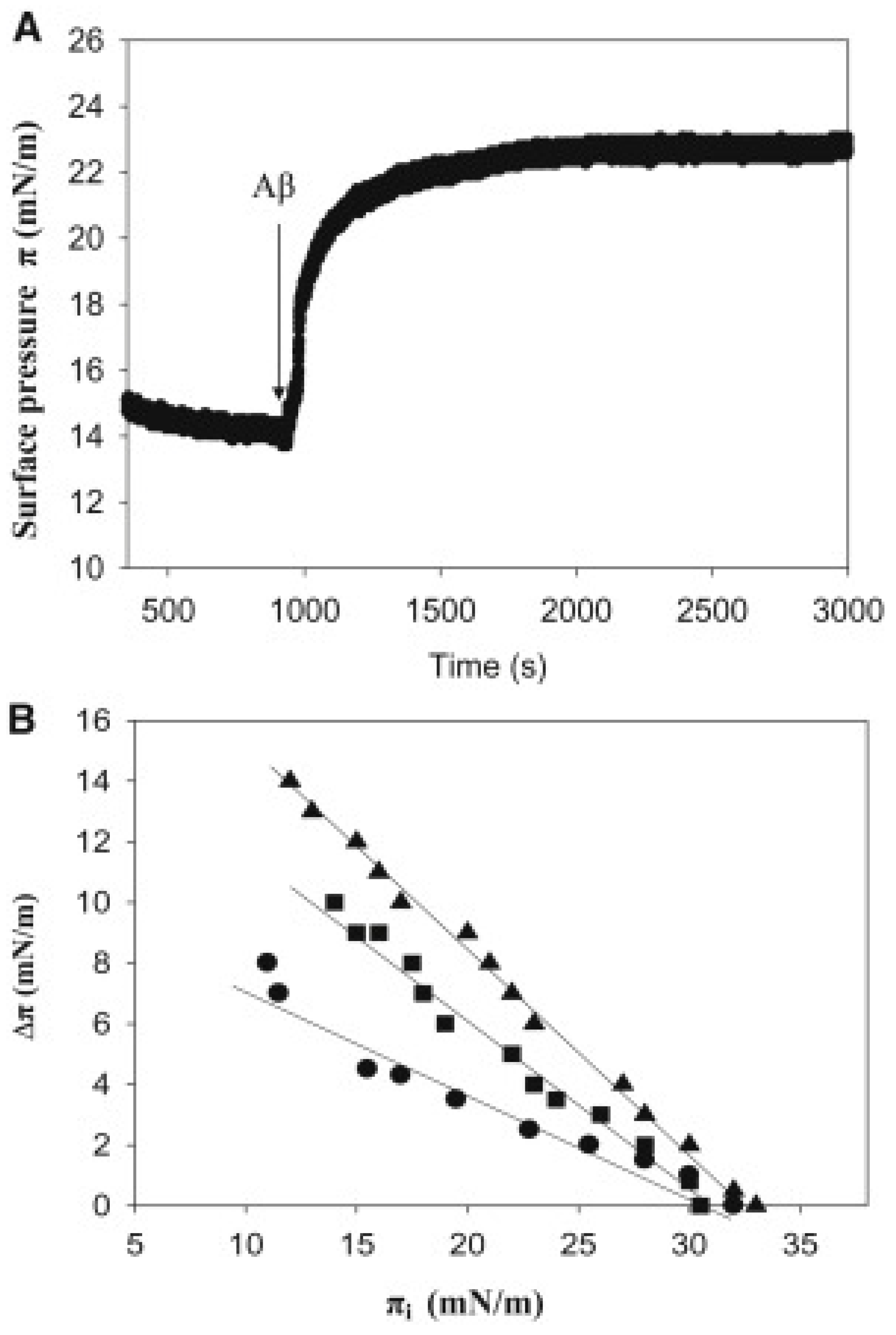
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



