Submitted:
18 April 2024
Posted:
22 April 2024
You are already at the latest version
Abstract
A dioxomolybdenum catalyst [(MoO2Cl2-O-MoO2Cl2)(L)] (1) (with L (1S,2S)-N,N´-bis(2-pyridinecarboxamide)-1,2-cyclohexane) was prepared and used as catalyst for the desulfurization of a multicomponent model fuel containing the most refractory sulfur compounds in real fuels. This complex showed to have a high efficiency to oxidize the aromatic benzothiophene derivative compounds present in fuels, mainly using a biphasic 1:1 model fuel/MeOH system. This process conciliates catalytic oxidative and extractive desulfurization, occurring the oxidation of the sulfur compounds in the polar organic solvent. The oxidative catalytic performance of (1) showed to be influenced by the presence of water in the system. Using 50% aq. H2O2 it was possible to reuse the catalyst and the extraction solvent MeOH during ten consecutive cycles without loss of desulfurization efficiency.
Keywords:
Oxidative desulfurization
; Diesel
; Multicomponent Model Fuel
; Dioxomolybdenum complex
; Hydrogen Peroxide
1. Introduction
Fossil fuels still will be the main available natural resource to supply the impressive increment of the energy consumption expected until 2050, associated to the expansion of the planet population [1]. However, fossil fuels usage compromise the health of our planet and the humanity due to the toxic gases large emissions [3]. Sulfur oxide emission has been one of the most concerns since the heavy crudes and fuel oils present a high concentration and high variety of sulfur compounds. Desulfurization of fuel oil is of real importance to reduce environmental pollution due to the emission of sulfur oxides from fuel combustion. The environmental rules are very strict, in many countries the sulfur content in fuel oil must be decreased below 10 ppm [1]. In order to decrease sulfur emissions to the environment, it is important to remove the sulfur compounds from fuels before fuel combustion. The emissions of SOx are responsible for the formation of acid rain, causing irreparable damage to vegetation, buildings, and even human health [2]. Therefore, the desulfurization processes capable to produce low-sulfur or even sulfur-free fuels are of high importance, and this is an actual worldwide research topic.
The conventional industrial process to obtain ultra-low sulfur diesel (bellow 10 ppm of S), is the hydrodesulphurization (HDS). However, this technology requires high pressures and high temperatures leading to an increase in energy consumption and cost of the overall process [3]. Oxidative desulfurization (ODS) can be seen as a potential complementary or alternative technology since it requires milder reaction conditions (< 100°C and atmospheric pressure) and is more efficient in removing aromatic sulfur compounds. In the ODS process, the sulfur compounds are oxidized to sulfoxides in the presence of oxidants (H2O2, molecular oxygen, or organic oxidants) and then further oxidized to sulfones. The latter compounds have a strong polarity and can be easily removed by extraction, adsorption, precipitation, from the fuel oil [3]. Our research group has been combining the ODS with extractive processes (ECODS), and very good results have been achieved for model and real diesels [4].
Several cis-dioxomolybdenum(VI) complexes have been investigated as catalysts in ODS and ECODS of simulated or real diesel fuel. Specifically, some of us reported cis-dioxomolybdenum(VI) complexes based on oxygen donor ligands, using 1-butyl-3-methylimidazolium hexafluorophosphate ([BMIM]PF6) [5], or polyethylene glycol (PEG) based deep eutectic solvents (DES) [6] as extraction solvents, and more recently, bipyridine ligands with different substituents and using different extraction solvents (acetonitrile, [BMIM][PF6] and PEG based DES) [4a]. Ferella et al. investigated also the activity of several binuclear cis-dioxomolybdenum(VI) complexes bearing different α-aminoacids ligands, in the presence of imidazolium based ionic liquids [7]. All the referenced complexes proved to be stable and efficient for ECODS of simulated and real diesel with H2O2 as oxidant. [4a-f, 8]
Herein, a binuclear complex based on cis-dioxomolybdenum moieties and on the chiral ligand (1S,2S)-N,N´-bis(2-pyridinecarboxamide)-1,2-cyclohexane (S,S-L) with a Mo-O-Mo bridge was prepared ([(MoO2Cl2-O-MoO2Cl2)(S,S-L)]) (1), characterized and investigated as homogeneous catalyst for ODS. Bispyridylamides, with the general formula LH2, were firstly prepared by Ojima in 1967 and since then their coordination chemistry with a great variety of metal ions and their catalytic applications have been largely explored [9]. In particular, chiral bispyridylamides ligands are very efficient in Mo-catalyzed asymmetric alkylation reactions using [Mo(CO)3(EtCN)3] [10] and Mo(CO)6 precursors [11]. To the best of our knowledge, this type of ligand has not yet been explored for ODS.
In this work, the desulfurization of model fuel has been studied using dioxomolybdenum(VI) catalyst [(MoO2Cl2)2 (L)] (1) (with L as (1S,2S)-N,N´-bis(2-pyridinecarboxamide)-1,2-cyclohexane), in the presence of different extraction solvents and under a solvent-free system. Two different amount of catalyst and H2O2 as oxidant were used. After optimization of the catalytic system for the oxidative desulfurization of a multicomponent model fuel, the reusability of the catalyst was investigated.
2. Materials and Methods
2.1. Chemicals
(1S,2S)-N,N´-bis(2-pyridinecarboxamide)-1,2-cyclohexane (S,S-L) was kindly provided by ChiraTecnics. Molybdenum(VI) dichloride dioxide (Aldrich) was used as received. Air-sensitive procedures for the synthesis of the complex were performed using standard Schlenk techniques under nitrogen atmosphere.
The reagents for ECODS studies: 1-benzenothiophene (Fluka), dibenzothiophene (Aldrich), 4-methyldibenzothiophene (Aldrich), 4,6-dimethyldibenzothiophene (Alfa Aesar), decane (Carlo Erba), tetradecane (Aldrich), (1-butyl-3-methylimidazolium hexafluorophosphate (Sigma-Aldrich), acetonitrile (Fisher Chemical), methanol (Fisher Chemical), ethanol (Fisher Chemical), polyethylene glycol (Sigma ), 30 and 50 wt% hydrogen peroxide (Aldrich) were purchased from commercial sources and used without further purification.
2.2. Instrumentation
FT-IR spectra were recorded on a Perkin-Elmer FT-IR Spectrometer Spectrum Two equipped with an attenuated total reflection (ATR) cell in the range of 4000–400 cm−1.
Solution 1 H NMR spectra were obtained with a Bruker AMX400 at 400.13 MHz. Elemental analyses (EA) were carried out with a Thermofinnigan Flash EA 112 series.
FT-Raman spectra were collected on a RFS-100 Bruker FT-spectrometer equipped with a Nd:YAG laser with an ex- citation wavelength of 1064 nm.
Mass spectrometry (MS) was performed by laser desorption ionization time of flight (LDI-TOF-MS) in the absence of matrix compounds. Compound (1) was dissolved in acetonitrile to 1 mg/ml, and 1 µl of solution was directly applied to the MALDI target plate and let to air dry. Spectra were acquired in a Bruker ultrafleXtreme mass spectrometer operated in the positive-ion reflector mode, using delayed extraction in the range m/z 200–3500 with approximately 1500 laser shots. Full MS spectra were externally calibrated with a peptide mixture according to the manufacturer’s instructions.
Electrospray ionization (ESI) high resolution mass spectra of the same solutions were acquired on an LTQ-orbitrap XL mass spectrometer (Thermo Scientific). ESI was shown to promote dissociation of the metal complex and only the free ligand could be observed.
Catalytic reactions were periodically monitored by GC-FID analysis carried out in a Varian CP-3380-GC-FID chromatograph (Germany). Hydrogen was used as carrier gas (55 cm.s−1) and fused silica Supelco capillary columns SPB-5 (30 m × 0.25 mm i. d.; 25 μm film thickness) were used.
Gas chromatography coupled to mass spectrometry (GC-MS) was performed in a Thermo Scientific Trace 1300 chromatograph coupled to a Thermo Scientific ISQ Sin-gle Quadruplo MS device. In both cases, TG-5MS columns (30 m; 0.25 mm (i.d.); 0.25 µm) were used.
2.3. Catalyst Synthesis
Synthesis of [(MoO2Cl2)2(S,S-L)] (1)
The solvent adduct MoO2Cl2(THF)2 was prepared from the evaporation to dryness of a solution of MoO2Cl2 (0.340 g, 1.7 mmol) in THF (5 mL). This adduct was then dissolved in CH2Cl2 (5 mL) and added to a solution of (1S,2S)-N,N´-bis(2-pyridinecarboxamide)-1,2-cyclohexane (S,S-L) in CH2Cl2 (5 mL). The formation of a white precipitated was observed immediately. The mixture was stirred for 2h at room temperature, filtered under nitrogen and washed with n-hexane. The resultant white solid was dried under vacuum. Yield:82%. Anal. Found: C, 28.40; H, 2.67; N, 6.80; Mo, 22.8; C19H22Cl4Mo2N4O6.CH2Cl2 requires C, 28.28; H, 2.75; N, 6.94; Mo, 23.78;
Selected FT-IR (ATR, cm -1): ν = 3334 (m), 3058 (w), 2934 (m), 2858 (m),1651 (w), 1622 (s), 1598 (s), 1558 (m), 1518 (m), 1475 (m) 1350 (w), 1292 (w), 1263 (w) 1142 (m), 1086 (m), 1044 (m), 1022 (m), 998 (m), 948 (s, νasym (Mo=O)), 912 (vs, νsym (Mo=O)), 883 (m), 809 (m), 748 (s), 733 (s), 700 (m), 688 (s), 777 (vs), 763 (vs), 732 (m), 644 (m), 607 (s), 598 (m), 409 (m), 645 (s), 619 (s), 597 (m), 481 (m), 451 (m), 408 (m). Selected FT-Raman (cm –1 ): 1602 (m), 1575 (w), 1559 (w), 1381 (w), 1353 (m), 1141 (w), 1021 (m), 950 (s), 917 (m), 877 (w), 877 (w), 382 (w), 307 (m), 243 (m), 219 (m), 193 (m), 144 (m). 1H NMR (400 MHz, RT, CD3CN): δ = 9.24 (d, 2H, J = 5.1 Hz), 9.09 (br, 2H), 8.35 (d, 2H, J = 8.0 Hz), 8.27 (t, 2H, J = 7.9 Hz), 7.89 (t, 2H, J = 6.4 Hz), 4.58 (br, 2H), 1.85 (m, 4H), 1.59 (m, 4H). MS (LDI-TOF-MS): [MoO(S,S-L)]+ (m/z = 436.12), [MoOCl(S,S-L)H]+.
2.4. Oxidative Desulfurization Studies (ODS)
The ODS studies were performed using a multicomponent model fuel containing four sulfur compounds: 1-benzothiophene (1-BT), dibenzothiophene (DBT), 4-methyldibenzothiophene (4-MDBT) and 4,6-dimethyldibenzothiophene (4,6-DMDBT), in decane (with a total sulfur concentration of 2000 ppm, with approximately 500 ppm of sulfur from each compound). The experiments were carried out under air in a closed borosilicate 5 mL vessel, equipped with a magnetic stirrer and immersed in a thermostatically controlled liquid paraffin bath at 70 °C. In a typical experiment, 5 or 10 µmol of catalyst was added to the model fuel (750 µL, containing approximately 47 μmol of S) and to the immiscible extraction solvent (750 µL). The resulting mixture was stirred for 10 min, after which, the catalytic step was initiated by adding 30% w/v of the oxidant, i.e., hydrogen peroxide (75 µL, 1.2 μmol). The sulfur content in the model fuel was periodically quantified by GC analysis using tetradecane as the external standard. The reusability of the catalyst was evaluated by removing the desulfurized model fuel at the end of each ODS cycle and adding a new portion of model fuel and oxidant maintaining all the experimental condition.
3. Results and Discussion
3.1. Catalyst Characterization
The molybdenum(VI) complex [(MoO2Cl2)2(S,S-L)] (1) was obtained by reaction of 3 equivalents of the chiral ligand (1S,2S)-N,N´-bis(2-pyridinecarboxamide)-1,2-cyclohexane (S,S-L) with the adduct [MoO2Cl2(THF)2] in dichloromethane. The product was isolated as a white solid with a high yield (82%) and was characterized by elemental analysis, ICP, FTIR-ATR, FT-Raman and 1H NMR spectroscopies and LDI-TOF-MS. The obtained values of elemental analysis and ICP suggested the coordination of two molybdenum centers to the chiral ligand as the Mo/N ratio is 2.0 and C/N ratio is 4.9 (calculated value is 4.5). The FTIR spectrum (Figure 1) shows the vibration corresponding to ν(N-H) at 3333 cm-1 suggesting the coordination of the metal without the deprotonation of the amide. The ν(C=O) vibration of the amide was shifted to lower frequency (from 1651 cm-1 in the ligand to 1622 cm-1 in the complex) and the δ(N-H) and ν(C-N)) vibrations found was shifted to higher frequency (from 1505 cm-1 in the ligand to 1518 cm-1 in (1)). These changes are indicative of the coordination of the metal through the oxygen of the amide group [12]. The aromatic ring and skeletal vibrations found at 1590 and 1570 cm-1 in the free ligand are shifted to higher frequencies 1622 cm-1 and 1597 cm-1 in the complex 1 indicating the coordination to the pyridyl unities. The presence of cis-[MoO2]2+ moieties is confirmed by νasym(Mo=O) and νsym(Mo=O) at 912 and 948 cm-1, respectively [13]. FT-Raman spectrum (Figure 2) contains the vibrational modes νasym(Mo=O) and νsym(Mo=O) at 917 and 950 cm-1, respectively, νasym(Mo-Cl)/ νsym(Mo-Cl) at 307/243, 219 cm-1 and νsym(Mo-N)/νasym(Mo-N) at 219 or 193/144 cm-1, and the out- of-plane deformation mode γMoO2 at 382 cm-1 which are in agreement with binuclear dichlorodioxomolybdenum(VI) complexes [14]. The 1H NMR spectrum (Figure 3) of complex 1 contains the ligand peaks corresponding to the pyridyl rings shifted to the downfield region corroborating the coordination of the pyridyl nitrogen atom to the molybdenum center. The protons of the NH amide groups are still observed and shifted to downfield region, which confirms that the deprotonation does not occurs and the metal coordinates through the oxygen atom of the amide.
The molybdenum complex (1) proved to be labile under MS conditions. ESI was found to promote complex dissociation, and with the release of the metal ions, only the free ligand (S,S-L) could be observed from solutions of (1). Nevertheless, spectra demonstrating the coordination of Mo by S,S-L could be obtained by LDI-TOF-MS (Figure 4), despite similar observations of complex dissociation and ligand degradation under the harsh direct laser ionization. The binuclear complex (1) was also found to dissociate under LDI conditions and two m/z values were observed corresponding to the moiety [MoOCl(S,S-L)] whose ionisation occurred either by loss of the chloride anion to yield [MoO(S,S-L)]+ (m/z = 436.12) or by protonation to yield [MoOCl(S,S-L)H]+ (m/z = 472.08).
These results suggest the formation of the binuclear complex represented in Figure 5.
3.2. Desulfurization Studies
The catalytic efficiency of [(MoO2Cl2-O-MoO2Cl2)(L)] (1) (with L as (1S,2S)-N,N´-bis(2-pyridinecarboxamide)-1,2-cyclohexane) as catalysts was investigated for the desulfurization of a multicomponent model fuel containing four refractory sulfur compounds usually present in real diesel: 1- benzothiophene (1-BT), dibenzothiophene (DBT), 4-methyldibenzothiophene (4-MDBT) and 4,6-dimethyldibenzothiophene (4,6-DMDBT), with an individual concentration of approximately 500 ppm of sulfur for each compound, in decane. The catalytic ODS process was conciliated with the ECODS, using a biphasic liquid-liquid system, containing equal volume amount of model fuel and extraction solvent. The desulfurization process of the multicomponent model fuel was performed in two main steps: first an initial liquid-liquid extraction occurred in the presence of the catalyst by stirring the 1:1 model fuel/extraction solvent during 10 min at 70 °C (grey highlight region in Figure 6; and after this initial equilibrium, the desulfurization in only increased by the application of the catalytic oxidation stage. This last is initiated by adding the oxidant (promoting the oxidation of sulfur compounds to the corresponding sulfoxides and/or sulfones, mainly in the extraction solvent medium).
To maximize desulfurization efficiency of the process and minimize reactants and costs, the optimization of various parameters was performed. Because of the large volume of solvents used in industry, finding safer and more sustainable alternatives is a great challenge for green chemistry research. Firstly, a comparison of different extraction solvents was studied, using several polar organic solvents: acetonitrile, methanol, ethanol, and PEG. Furthermore, an ionic liquid was used ([BMIM]PF6). The initial conditions adopted were 0,75 mL of model fuel, 0,75 mL of extraction solvent and 75 µL of 30% aq. H2O2 at 70 °C.
Figure 5 displays the desulfurization profiles using various extraction solvents and when no solvent is used. The desulfurization efficiency is similar for the different solvents, with exception to PEG and ethanol, that resulted in lower desulfurization (73% and 80% of desulfurization after 5 h). For all the ECODS systems, the desulfurization of the multicomponent fuel is finished after 2 h of reaction. On the other hand, using the solvent-free system, an increase of oxidative desulfurization efficiency was observed during the 5 h, achieving 96% of conversion at this time (77% after 2 h). After the 3 h of reaction the desulfurization profile was similar to the most active ECODS systems, i.e., the ionic liquid [BMIM]PF6 (PF6), methanol (MeOH) and acetonitrile (MeCN). Using these last extraction solvents the desulfurization efficiency obtained after 2 and 5 h was between 93% and 92%. The initial extraction results obtained with the ECODS systems, shows that EtOH and MeOH presented slightly higher extractive desulfurization, followed by the ionic liquid, and lower for MeCN and PEG. This is due to the affinity of the sulfur compound with the extraction solvent. MeOH and EtOH are less polar solvents than the ionic liquid and PEG, but more polar than the decane model fuel.
However, the higher extractive desulfurization did not result in a higher oxidative desulfurization, i.e., the best extractive solvent is not the most suitable to promote the highest catalytic oxidation, since the most efficient ECODS systems after 5 h were the ionic-liquid, MeCN, and MeOH. This must be related to the stability of the binuclear dioxomolybdenum(VI) catalyst in different solvent media. In all the extraction solvents, the catalyst showed to dissolved easily, and the opposite was observed for the solvent-free system. In this case a partial dissolution was observed. This resulted in a slower desulfurization profile, achieving a high efficiency only after 5 h. Contrarily, the ECODS technology achieved their highest desulfurization after only 2 h.
The influence of the amount of binuclear dioxomolybdenum catalyst was investigated. Two different amounts of catalyst (5 and 10 µmol) were employed, maintaining all the experimental conditions, and the results obtained are displayed in Figure 7. After 5 h of reaction, the desulfurization obtained using 5 and 10 µmol of catalyst was similar (between 92 and 96% of desulfurization) using MeOH and MeCN extraction solvents. An influence of catalyst amount was found using the ionic liquid as extraction solvent, and also under a solvent-free system. Using the ionic liquid [BMIM]PF6 as solvent extraction, higher desulfurization was achieved when 5 µmol of catalyst was used. The difference of catalyst activity was even more pronounced under a solvent-free system. In these last cases, higher amount of catalyst decreased the conversion of sulfur oxidation. This must be related to the higher amount of insoluble catalyst in the reaction medium that can difficult the oxidation of sulfur in the interface between the model fuel phase and the ionic liquid phase, and also to an increase of not effective H2O2 oxidant usage by the presence of solid particles.
The mechanism involved in the sulfoxidation reaction catalysed by dioxomolybdenum (VI) catalyst, was deeply studied recently by Bullock et al. [15]. Initially the activation of the H2O2 oxidant occurs by a proton transfer to the Mo=O moiety from the complex (1), forming a five-coordinate molybdenum (VI) intermediary specie. Then, it is suggested that the oxidation of sulfide occurs by a nucleophilic attack of this with the -Mo(OOH) hydroxoperoxyl group previously formed, and a consequent regeneration of the pre-catalyst dioxomolybdenum (VI) occurs (Figure 8). Second cycle proceeds for the oxidation of the sulfoxide to the sulfone.
3.3. Reusing Tests
The recyclability of the binuclear dioxomolybdenum complex was studied for the solvent-free system and using the ECODS system, using MeOH as extraction solvent, since this is cheaper than ionic liquid, more sustainable than MeCN, and most easily recovered from reaction by distillation than PEG. The reusability of the ECODS system was evaluated for consecutive cycles, by recovering the desulfurized model fuel phase after each cycle and starting a new cycle by adding fresh portions of model fuel and oxidant to the extraction solvent phase (in the case of ECODS studies), containing the entrapped catalyst.
The reusing capacity of the binuclear dioxomolybdenum catalyst was investigated for the solvent-free system. In this case, the model fuel was removed after each catalytic cycle and the solid was left to the reactor without any cleaning and activation process. The results are presented in Figure 9, and it is possible to observe a large deactivation of the catalyst after the 1st cycle. This must the due to the loss of catalyst with the model fuel after 1st cycle and/or a large adsorption of the sulfones formed in together with the solid catalyst. Other possibility is the transformation of the catalyst into less active complex with the interaction of the oxidant H2O2, for example into peroxo-compounds [16].
To investigate the reusing capacity under an ECODS system, the catalysis was reused for consecutive cycles using MeOH as extraction solvent. Figure 10 displays the results obtained for the seven consecutive cycles using the ECODS process with MeOH, where it is possible to observe a slightly loss of activity during the reusing process (total of 10% of desulfurization from the 1st to the 7th cycle). The small increase noticed for the 5th cycle must be related to the experimental error. This was calculated to be of 5%. During the seven ECODS cycles, the oxidized sulfur compounds (sulfones) precipitated as a white solid (image inserted in Figure 10). The amount of sulfones increased over the sequence of the seven reusing cycles. This has been observed previously for comparable ECODS systems using multicomponent model diesels with high concentrations of S.[4f, 6] Therefore, the decrease of desulfurization efficiency during the reusing process must be due to the saturation of the extraction solvent with the oxidized sulfur compounds, which decreases the capacity of MeOH to receive more sulfur compounds from model fuel, retarding the effect of total desulfurization capacity of the system.
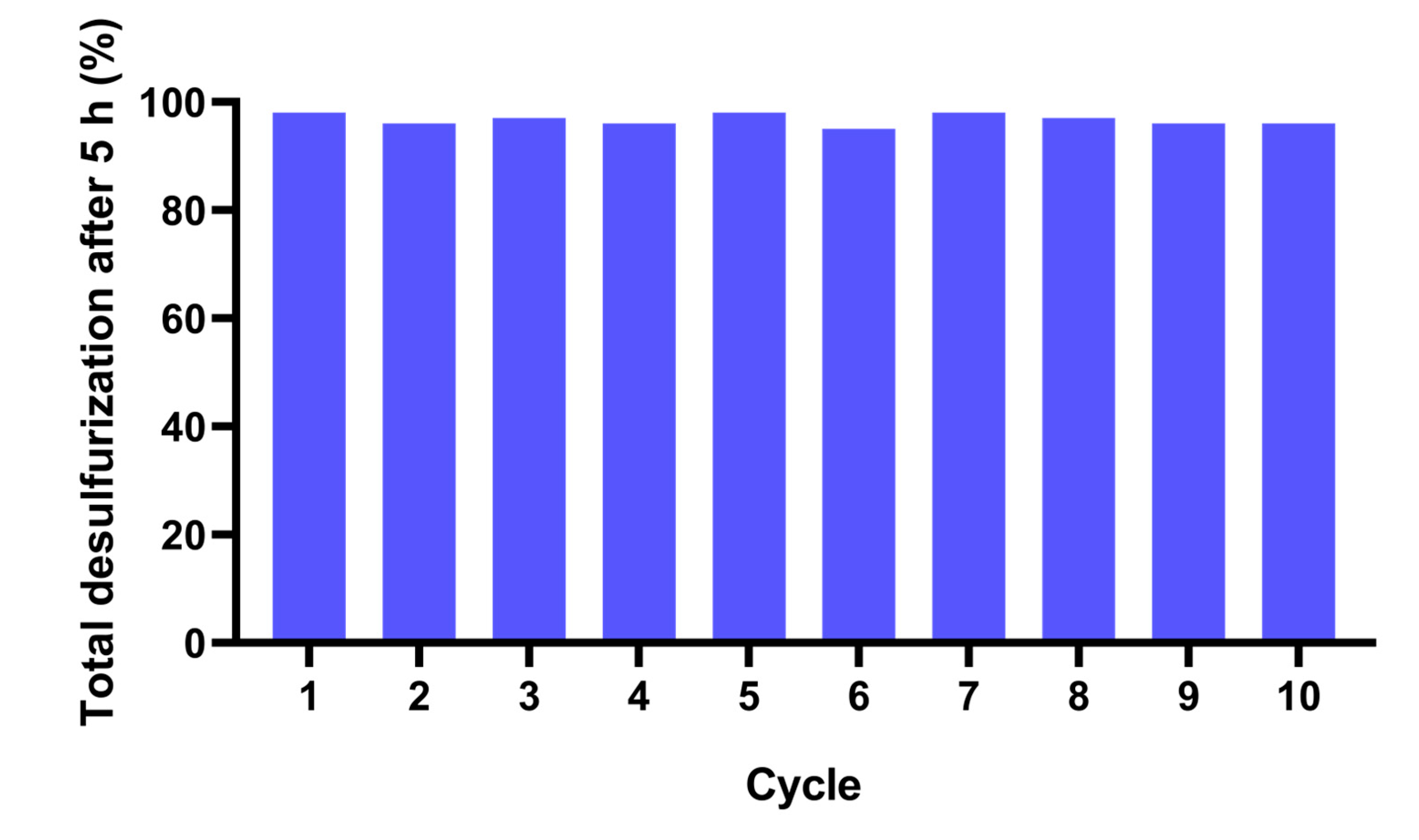
Furthermore, the same system was investigated using an oxidant with less water content, i.e., using 50% aq. H2O2, instead of 30% aq. H2O2. Results obtained are displayed in Figure 11 and it is possible to observe that for ten consecutive cycles practically no loss of activity was observed. The difference of total desulfurization observed between 98% and 95% are in the same order as the experimental error (5%). These are promising results surpass the above reported data using the same system with higher amount of water present in the H2O2 oxidant. These demonstrates that the content of water present in the system has a strong influence in the catalyst performance. This must be associated to the low stability of the complex (1) in the presence of water.
4. Conclusions
The [(MoO2Cl2)2(L)] complex (with L as (1S,2S)-N,N´-bis(2-pyridinecarboxamide)-1,2-cyclohexane) was prepared, characterized and tested as catalyst for the desulfurization of a multicomponent model fuel containing the most refractory sulfur compounds present in read diesel, using hydrogen peroxide as oxidant. Two different desulfurization systems were investigated: a solvent-free system, that forces the catalytic oxidation of sulfur compounds to occur in the model diesel phase; and a biphasic liquid-liquid system, where a polar immiscible organic extraction solvent was used with equal volume to the multicomponent model fuel. Under these different systems, the maximum of desulfurization efficiency was achieved faster using the biphasic system with MeOH, MeCN and ionic liquid [BMIM]PF6 as extraction solvents (92-93% of total desulfurization after 2 h). The advantage of this system is to conciliate the oxidative catalytic desulfurization and extractive desulfurization. On the other hand, using the solvent-free system was possible to achieve slightly higher desulfurization after 5 h (98%). But this system showed do not be favorable for the catalyst reusing in consecutive reaction cycles, probably due to the catalyst deactivation in the presence of aqueous oxidant. The low stability of the dioxomolybdenum catalyst in the presence of water was verified by its reutilization in the biphasic system. Using MeOH as extraction solvent, the catalyst and the extraction phase could be reused for ten consecutive cycles, in the presence of 50% aq. H2O2. Some loss of desulfurization efficiency (10% during seven reaction cycles) was achieved using less concentrated 30% aq. H2O2.
Author Contributions
conceptualization, F.M., A.S. and S.G.; methodology, S.S.B. and S.G.; validation, S.S.B, S.G.; formal analysis, F.M. and A.S.; investigation, F.M., A.S., C.N.D. and S.S.B.; resources, S.S.B and A.S.; data curation, S.G. and S.S.B.; writing—original draft preparation, F.M., A.S., C.N.D. and S.G.; writing—review and editing, S.S.B.; visualization, X.X.; supervision, S.S.B. and S.G.; project administration, S.S.B.; funding acquisition, S.S.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work received financial support from Fundação para a Cinência e a Tecnologia / Ministério da Ciência, Tecnologia e Ensino Superior (FCT/MCTES) by national funds, through LAQV / REQUIMTE (Ref. UIDP/50006/2020 DOI 10.54499/UIDP/50006/2020; LA/P/0008/2020 DOI 10.54499/LA/P/0008/2020; UIDB/50006/2020 DOI 10.54499/UIDB/50006/2020).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
S.S.B. thanks FCT/MCTES for supporting her contract position via the Individual Call to Scientific Employment Stimulus (Ref. CEECIND/03877/2018). The position held by I.S.-V. (Ref. 197_97_ARH-2018) was supported by national funds (OE). The authors acknowledge ChiraTecnics (www.chiratecnics.com) for the supply of the chiral diamine ligand used to generate the Mo catalyst. Authors thank Anthony Burke (University of Coimbra) and Isabel S. Gonçalves (CICECO, University of Aveiro) for valuable discussions on catalyst formation and behaviourConflicts of Interest: The authors declare no conflicts of interests.
References
- aY. Zhang, R. Wang, Applied Catalysis B: Environmental 2018, 234, 247-259; bY. Gu, W. Xu, Y. Sun, Catalysis Today 2020.
- Tugrul Albayrak, A. Tavman, Ultrasonics Sonochemistry 2022, 83, 105845.
- . aY. Tu, T. Li, G. Yu, L. Wei, L. Ta, Z. Zhou, Z. Ren, Energy & Fuels 2019, 33, 8503-8510; bN. S. El-Gendy, J. G. Speight, Handbook of refinery desulfurization, Vol. 140, CRC Press, 2015.
- aD. Juliao, A. C. Gomes, L. Cunha-Silva, M. Pillinger, I. S. Gonçalves, S. S. Balula, Journal of Organometallic Chemistry 2022, 967; bD. Juliao, A. C. Gomes, L. Cunha-Silva, M. Pillinger, A. D. Lopes, R. Valença, J. C. Ribeiro, I. S. Gonçalves, S. S. Balula, Catalysis Communications 2019, 128; cD. Juliao, A. C. Gomes, M. Pillinger, I. S. Gonçalves, S. S. Balula, Chemical Engineering & Technology 2020, 43, 1774-1783; dD. Juliao, A. C. Gomes, M. Pillinger, A. D. Lopes, R. Valença, J. C. Ribeiro, I. S. Gonçalves, S. S. Balula, Journal of Molecular Liquids 2020, 309; eD. Juliao, A. C. Gomes, M. Pillinger, R. Valenca, J. C. Ribeiro, I. S. Goncalves, S. S. Balula, Applied Organometallic Chemistry 2020, 34; fD. Juliao, A. C. Gomes, M. Pillinger, R. Valença, J. C. Ribeiro, I. S. Gonçalves, S. S. Balula, Dalton Transactions 2016, 45, 15242-15248; gR. G. Faria, D. Silva, F. Mirante, S. Gago, L. Cunha-Silva, S. S. Balula, Catalysts 2024, 14; hY. Gao, C. M. Granadeiro, L. Cunha-Silva, J. S. Zhao, S. S. Balula, Catalysis Science & Technology 2023, 13, 4785-4801; iD. F. Silva, R. G. Faria, I. Santos-Vieira, L. Cunha-Silva, C. M. Granadeiro, S. S. Balula, Catalysis Today 2023, 423.
- D. Julião, A. C. Gomes, L. Cunha-Silva, M. Pillinger, A. D. Lopes, R. Valença, J. C. Ribeiro, I. S. Gonçalves, S. S. Balula, Catalysis Communications 2019, 128, 105704.
- D. Julião, A. C. Gomes, M. Pillinger, A. D. Lopes, R. Valença, J. C. Ribeiro, I. S. Gonçalves, S. S. Balula, Journal of Molecular Liquids 2020, 309, 113093.
- F. Ferella, L. Biancalana, F. Marchetti, M. Crucianelli, Catalysis Today 2020, 357, 646-654.
- D. Juliao, A. C. Gomes, M. Pillinger, R. Valença, J. C. Ribeiro, I. S. Gonçalves, S. S. Balula, Applied Catalysis B-Environmental 2018, 230, 177-183.
- Belda, C. Moberg, Coordination Chemistry Reviews 2005, 249, 727-740.
- M. Palucki, J. Um, N. Yasuda, D. Conlon, F.-R. Tsay, F. Hartner, Y. Hsiao, B. Marcune, S. Karady, D. Hughes, P. Dormer, P. Reider, The Journal of organic chemistry 2002, 67, 5508-5516.
- S. W. Krska, D. L. Hughes, R. A. Reamer, D. J. Mathre, Y. Sun, B. M. Trost, Journal of the American Chemical Society 2002, 124, 12656-12657.
- aR. L. Chapman, R. S. Vagg, Inorganica Chimica Acta 1979, 33, 227-234; bT. F. Zafiropoulos, S. P. Perlepes, P. V. Ioannou, J. M. Tsangaris, A. G. Galinos, Zeitschrift für Naturforschung B 1981, 36, 87-93; cT. F. Zafiropoulos, S. P. Perlepes, P. V. Ioannou, J. M. Tsangaris, A. G. Galinos, 1981, 36, 87-93.
- aS. Gago, J. E. Rodríguez-Borges, C. Teixeira, A. M. Santos, J. Zhao, M. Pillinger, C. D. Nunes, Ž. Petrovski, T. M. Santos, F. E. Kühn, C. C. Romão, I. S. Gonçalves, Journal of Molecular Catalysis A: Chemical 2005, 236, 1-6; bF. E. Kühn*, A. M. Santos, A. D. Lopes, I. S. Gonçalves, J. E. Rodrı́guez-Borges, M. Pillinger, C. C. Romão*, Journal of Organometallic Chemistry 2001, 621, 207-217.
- C. Coelho, M. Nolasco, S. S. Balula, M. M. Antunes, C. C. L. Pereira, F. A. Almeida Paz, A. A. Valente, M. Pillinger, P. Ribeiro-Claro, J. Klinowski, I. S. Gonçalves, Inorganic Chemistry 2011, 50, 525-538.
- X. Bullock, C. S. Jamieson, P. Moënne-Loccoz, B. Taylor, J. A. M. Gonzalez, E. A. Draves, L. Y. Kuo, Inorganic Chemistry 2021, 60, 7762-7772.
- aR. Wang, G. Zhang, H. Zhao, Catalysis Today 2010, 149, 117-121; bY. Chen, Q. Tian, Y. Tian, J. Cui, G. Wang, Applied Sciences 2021, 11; cI. Kozhevnikov, Catalysts for fine chemical synthesis, catalysis by polyoxometalates, Vol. 2, Wiley, 2002.
Figure 1.
FT-IR spectra in the region 400-2000 cm-1 of the free ligand and of the complex 1.
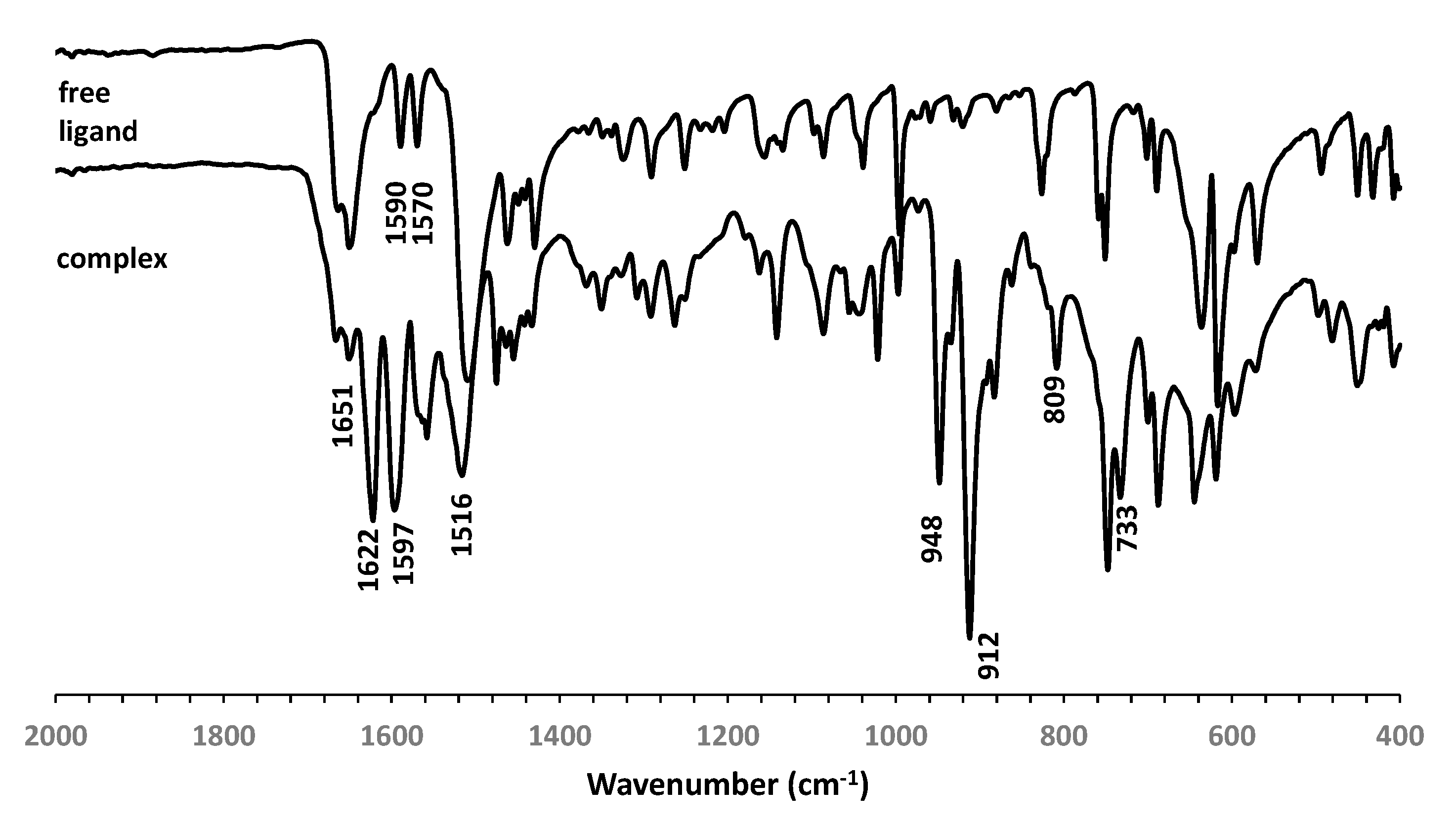
Figure 2.
Raman spectra in the region 150-2000 cm-1 of the free ligand and of the complex 1.
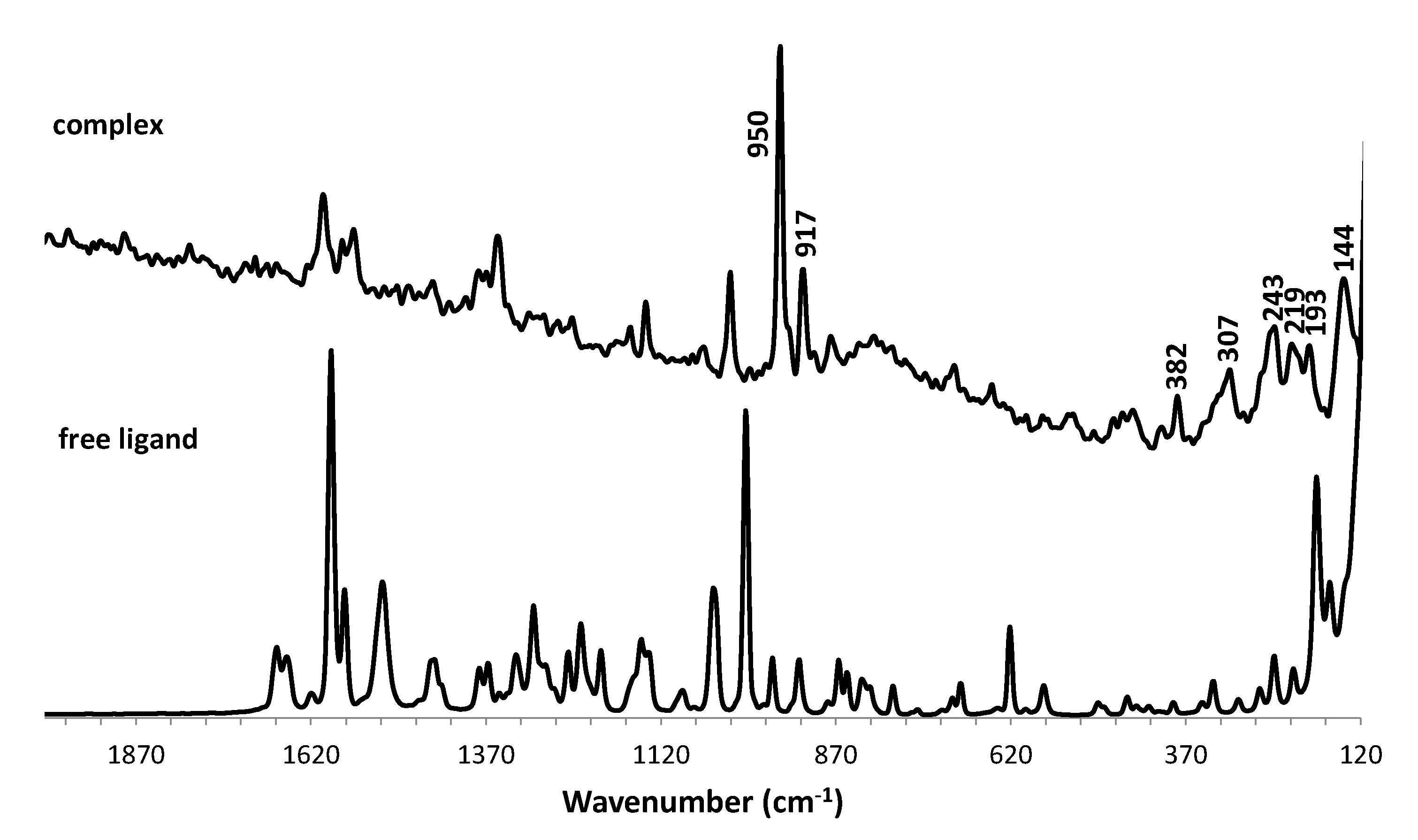
Figure 3.
Solution 1H NMR of the free ligand and of the complex 1 in acetonitrile in the range 7-10 ppm.
Figure 3.
Solution 1H NMR of the free ligand and of the complex 1 in acetonitrile in the range 7-10 ppm.
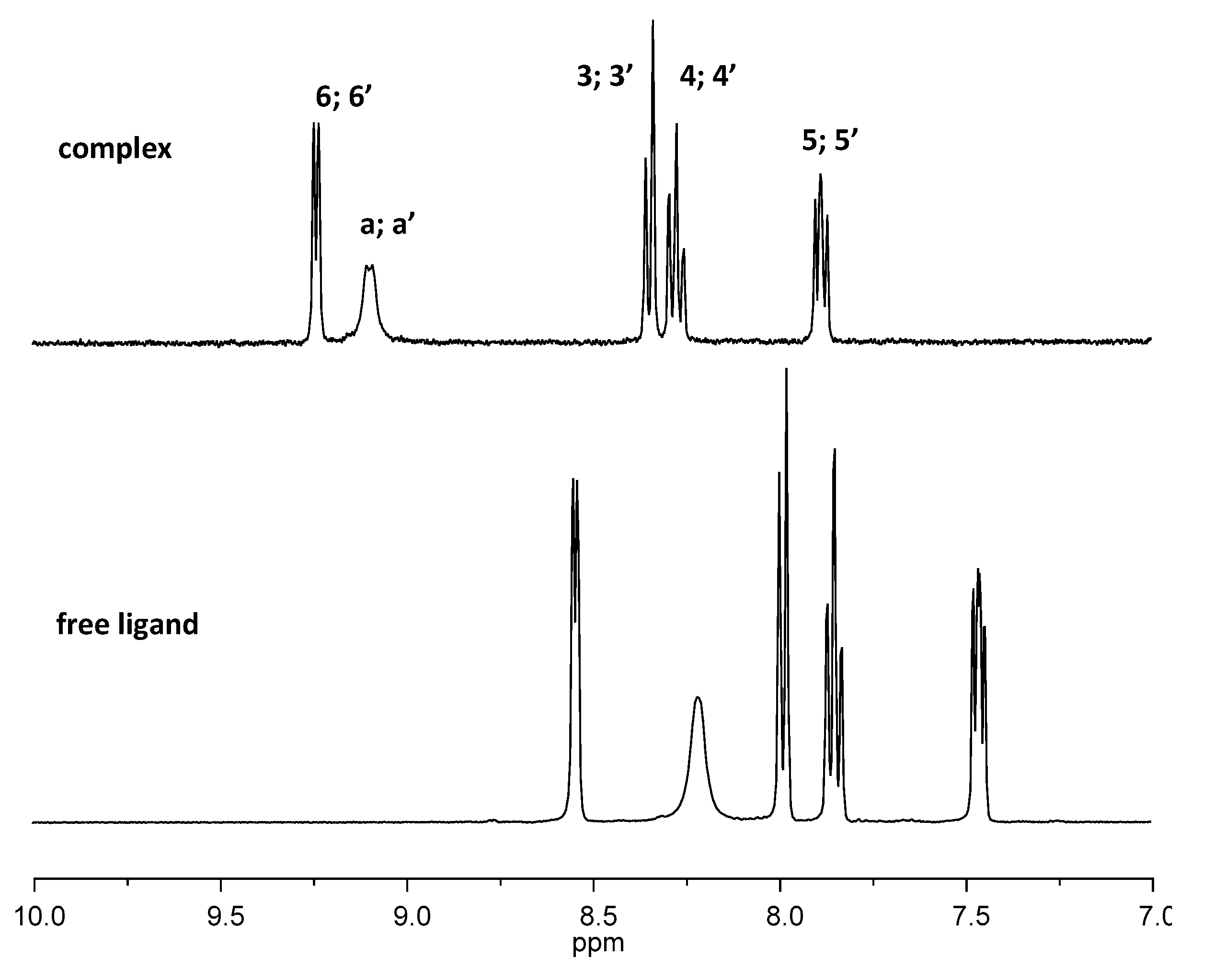
Figure 4.
Laser desorption ionization time of flight mass spectra (LDI-TOF-MS) of compound (1). The molybdenum complex may be observed at m/z values 436.12 and 489.08, the two MS peaks corresponding to the [MoOCl(S,S-L)] complex which ionizes by two different mechanisms: respectively, loss of the chloride counter-ion [MoO(S,S-L)]+ or protonation [MoOCl(S,S-L)H]+. The isotopic patterns of these two peaks are characteristic of Mo complexes. Direct laser ionization of (1) was also found to promote complex dissociation and ligand degradation, with free ligand being observed at m/z = 325.22 and a degradation product found at m/z = 203.17.
Figure 4.
Laser desorption ionization time of flight mass spectra (LDI-TOF-MS) of compound (1). The molybdenum complex may be observed at m/z values 436.12 and 489.08, the two MS peaks corresponding to the [MoOCl(S,S-L)] complex which ionizes by two different mechanisms: respectively, loss of the chloride counter-ion [MoO(S,S-L)]+ or protonation [MoOCl(S,S-L)H]+. The isotopic patterns of these two peaks are characteristic of Mo complexes. Direct laser ionization of (1) was also found to promote complex dissociation and ligand degradation, with free ligand being observed at m/z = 325.22 and a degradation product found at m/z = 203.17.
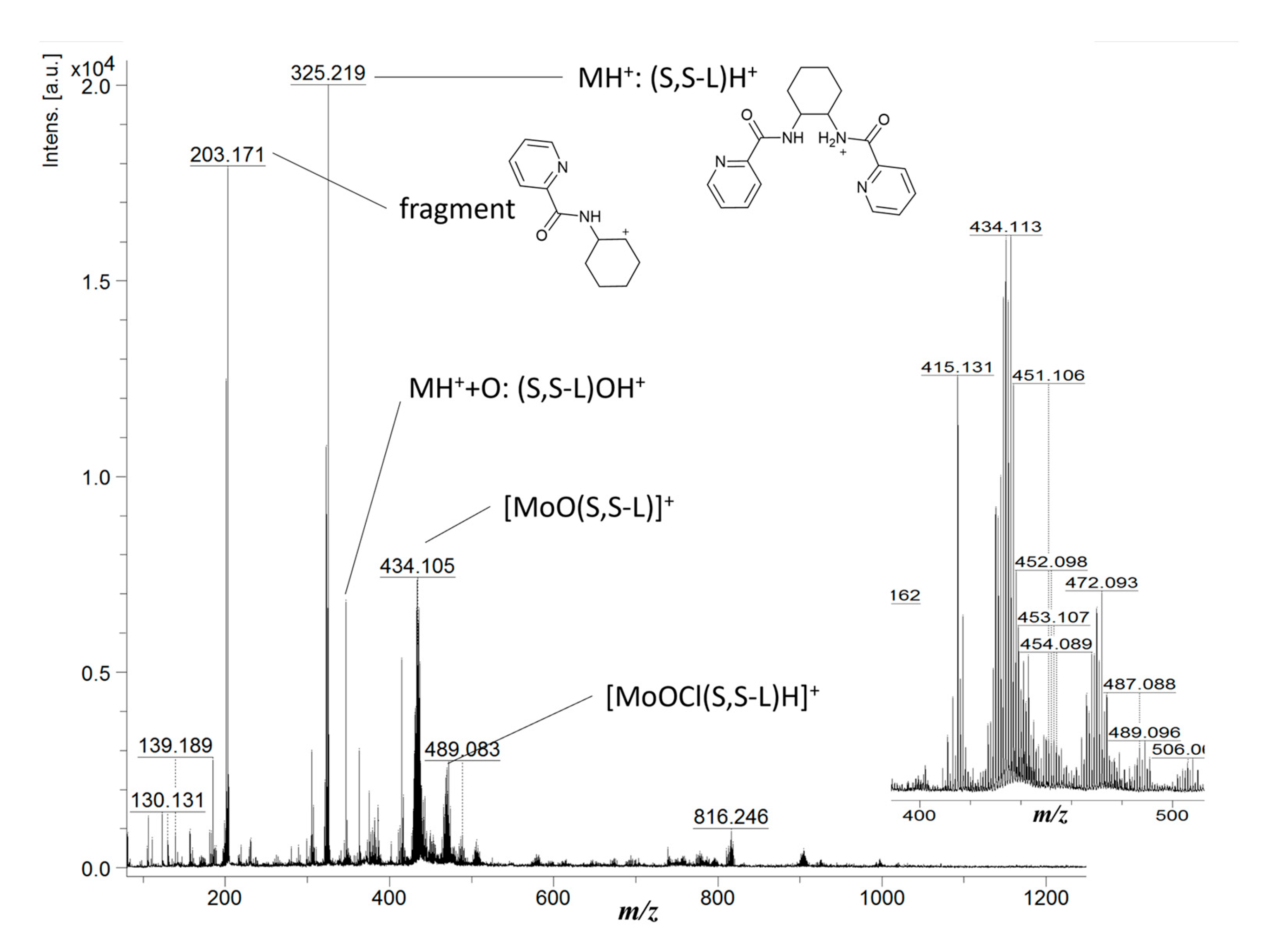
Figure 5.
Proposed structure for the obtained binuclear complex (1) and numbering assignment for 1H RMN spectrum.
Figure 5.
Proposed structure for the obtained binuclear complex (1) and numbering assignment for 1H RMN spectrum.
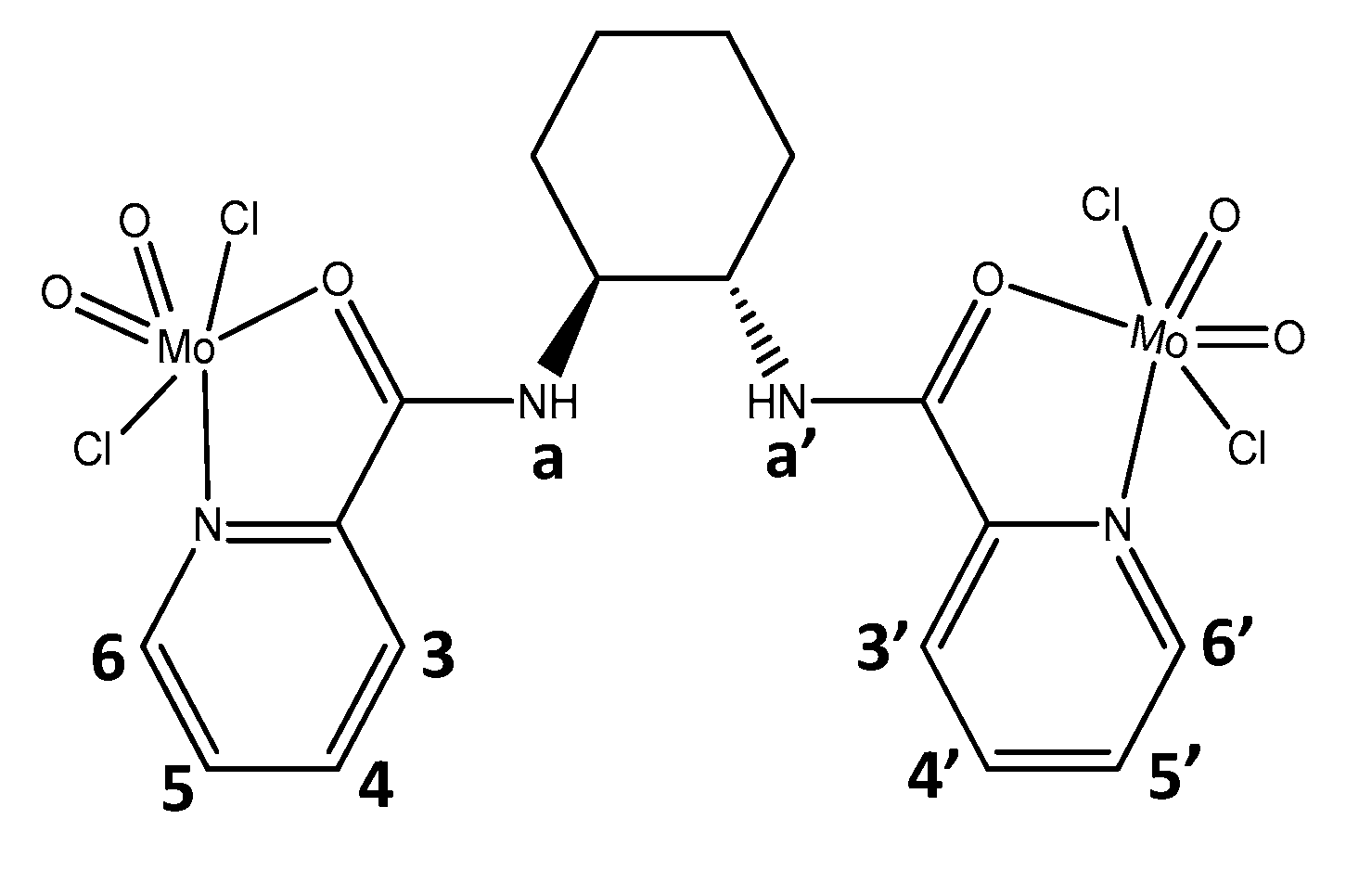
Figure 6.
Desulfurization of a multicomponent model fuel (2000 ppm S) catalyzed by complex (1) (5 µmol), using different extraction solvents ([BMIM]PF6, MeCN, PEG, MeOH and EtOH)), or under a solvent-free system, H2O2 (75 µL, 1.2 μmol 30% aq.) as oxidant at 70 °C. Grey highlight region corresponds to extractive desulfurization, before oxidant addition.
Figure 6.
Desulfurization of a multicomponent model fuel (2000 ppm S) catalyzed by complex (1) (5 µmol), using different extraction solvents ([BMIM]PF6, MeCN, PEG, MeOH and EtOH)), or under a solvent-free system, H2O2 (75 µL, 1.2 μmol 30% aq.) as oxidant at 70 °C. Grey highlight region corresponds to extractive desulfurization, before oxidant addition.
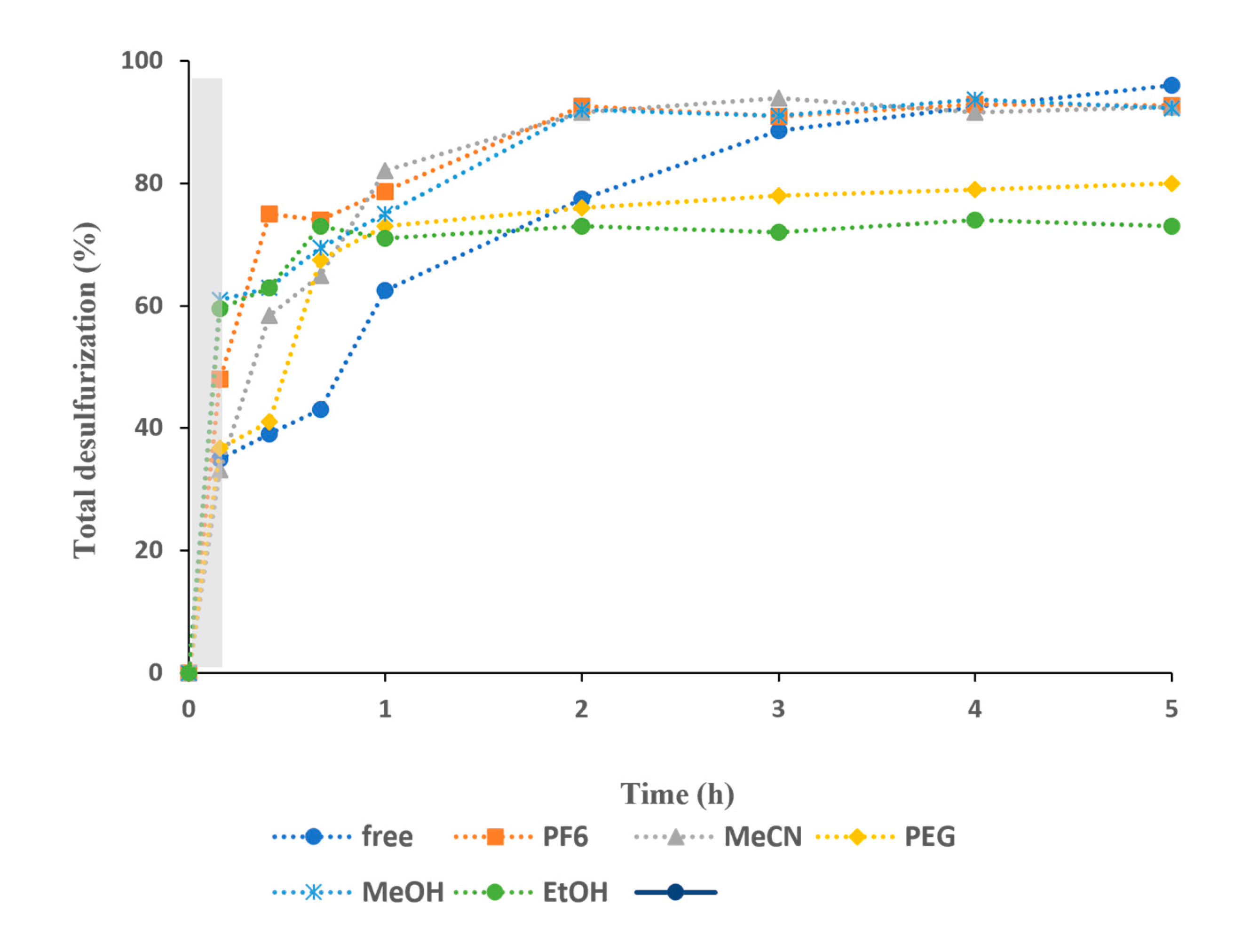
Figure 7.
Desulfurization of a multicomponent model fuel (2000 ppm S) catalyzed by complex (1), using two different amount (5 and 10 µmol), 75 µL (1.2 μmol) 30% aq. H2O2, and different extraction solvent, at 70 °C.
Figure 7.
Desulfurization of a multicomponent model fuel (2000 ppm S) catalyzed by complex (1), using two different amount (5 and 10 µmol), 75 µL (1.2 μmol) 30% aq. H2O2, and different extraction solvent, at 70 °C.
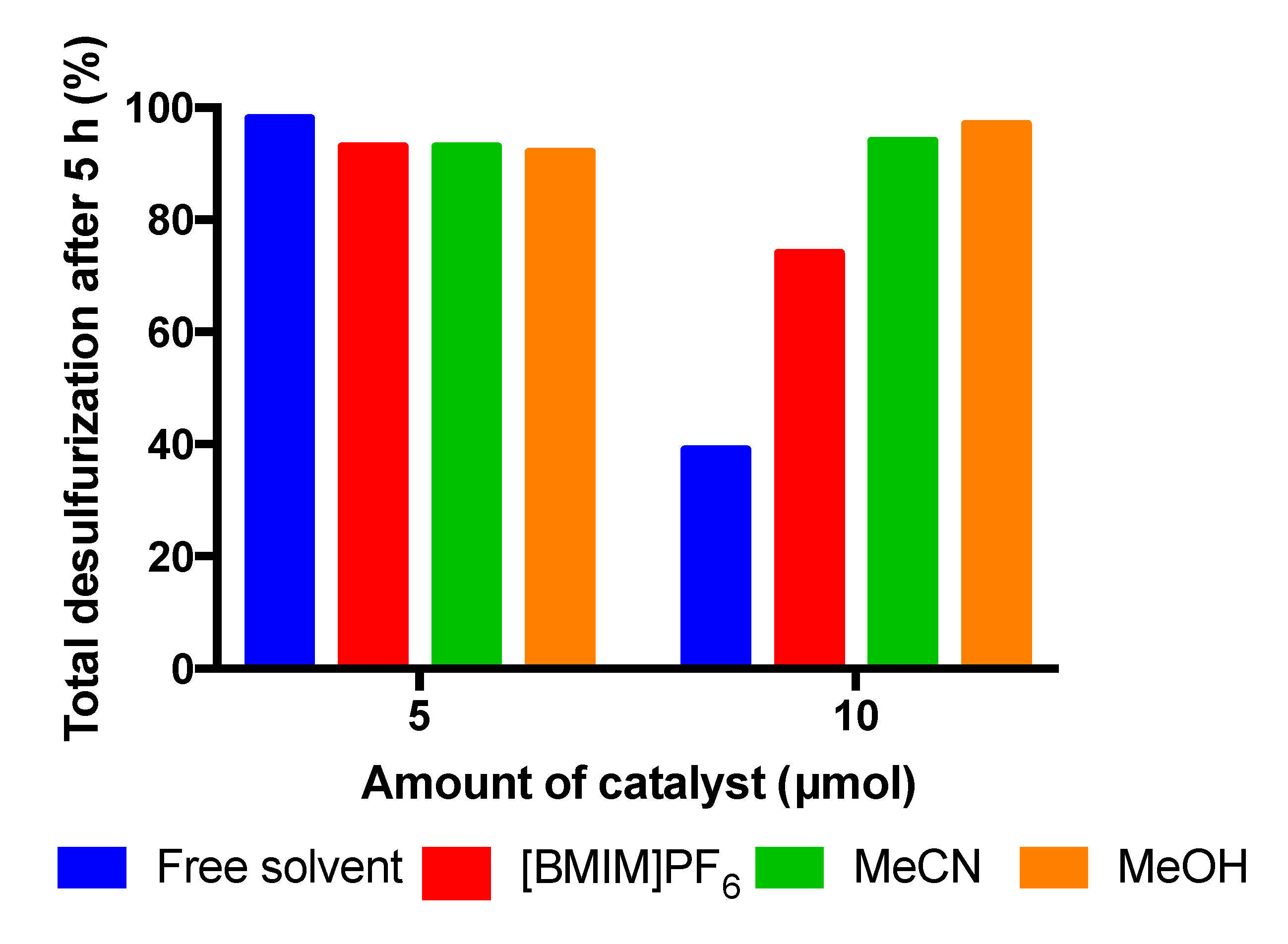
Figure 8.
Proposed mechanism for the oxidation of 1-benzothiophene to sulfoxide, catalyzed by with complex dioxomolybdenum (1).
Figure 8.
Proposed mechanism for the oxidation of 1-benzothiophene to sulfoxide, catalyzed by with complex dioxomolybdenum (1).
Figure 9.
Total desulfurization data of the model fuel catalyzed by complex (1) (using 5 μmol), for three ODS cycles under a solvent-free system, and 30% aq. H2O2 as oxidant (1.2 μmol), at 70°C.
Figure 9.
Total desulfurization data of the model fuel catalyzed by complex (1) (using 5 μmol), for three ODS cycles under a solvent-free system, and 30% aq. H2O2 as oxidant (1.2 μmol), at 70°C.
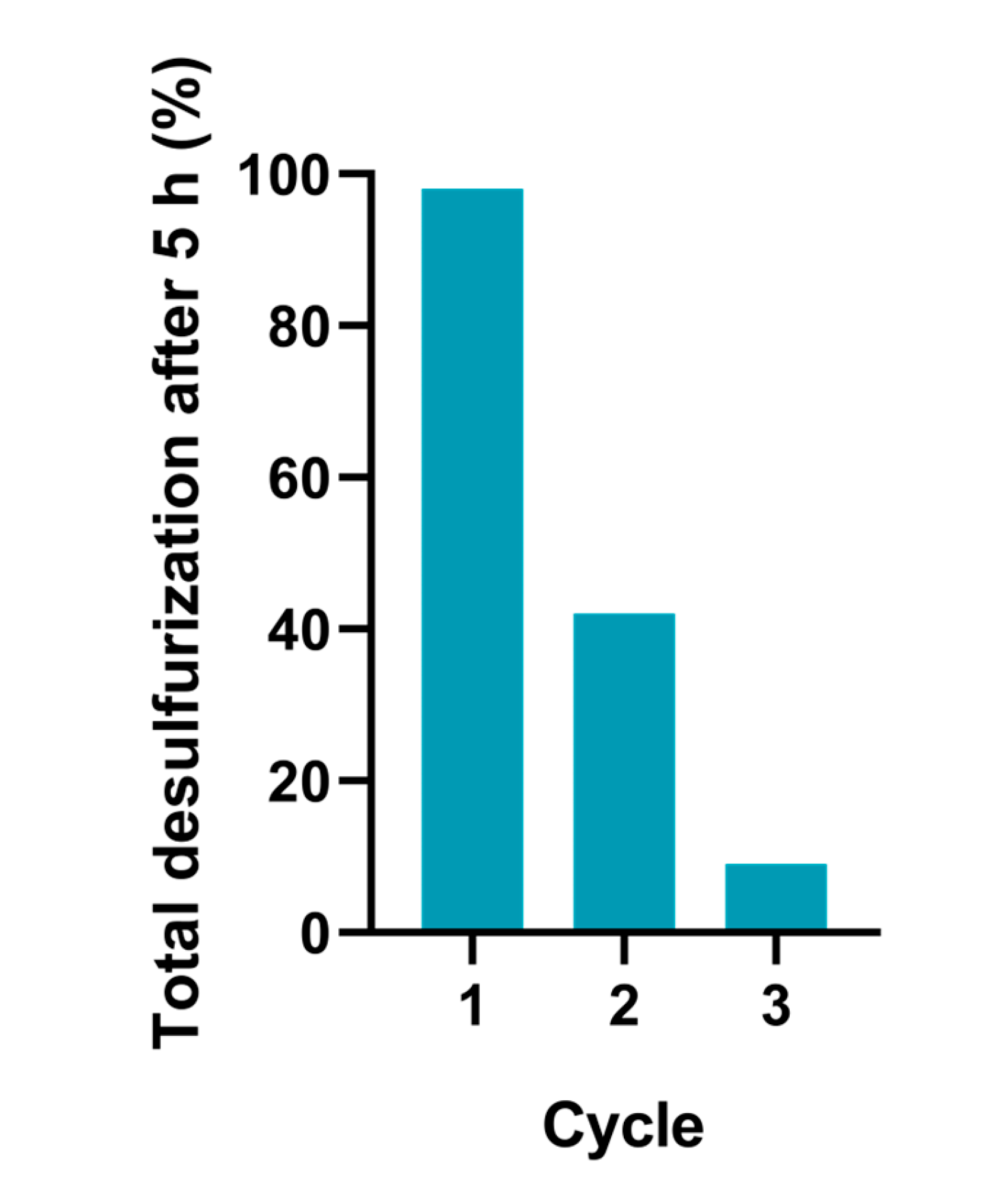
Figure 10.
Total desulfurization data of the model fuel catalyzed by complex (1) (using 5 μmol), for seven ECODS cycles using MeOH as the extraction solvent and 30% aq. H2O2 as oxidant, (1.2 μmol) at 70 °C. Image of reactor after the 7th cycle of desulfurization.
Figure 10.
Total desulfurization data of the model fuel catalyzed by complex (1) (using 5 μmol), for seven ECODS cycles using MeOH as the extraction solvent and 30% aq. H2O2 as oxidant, (1.2 μmol) at 70 °C. Image of reactor after the 7th cycle of desulfurization.
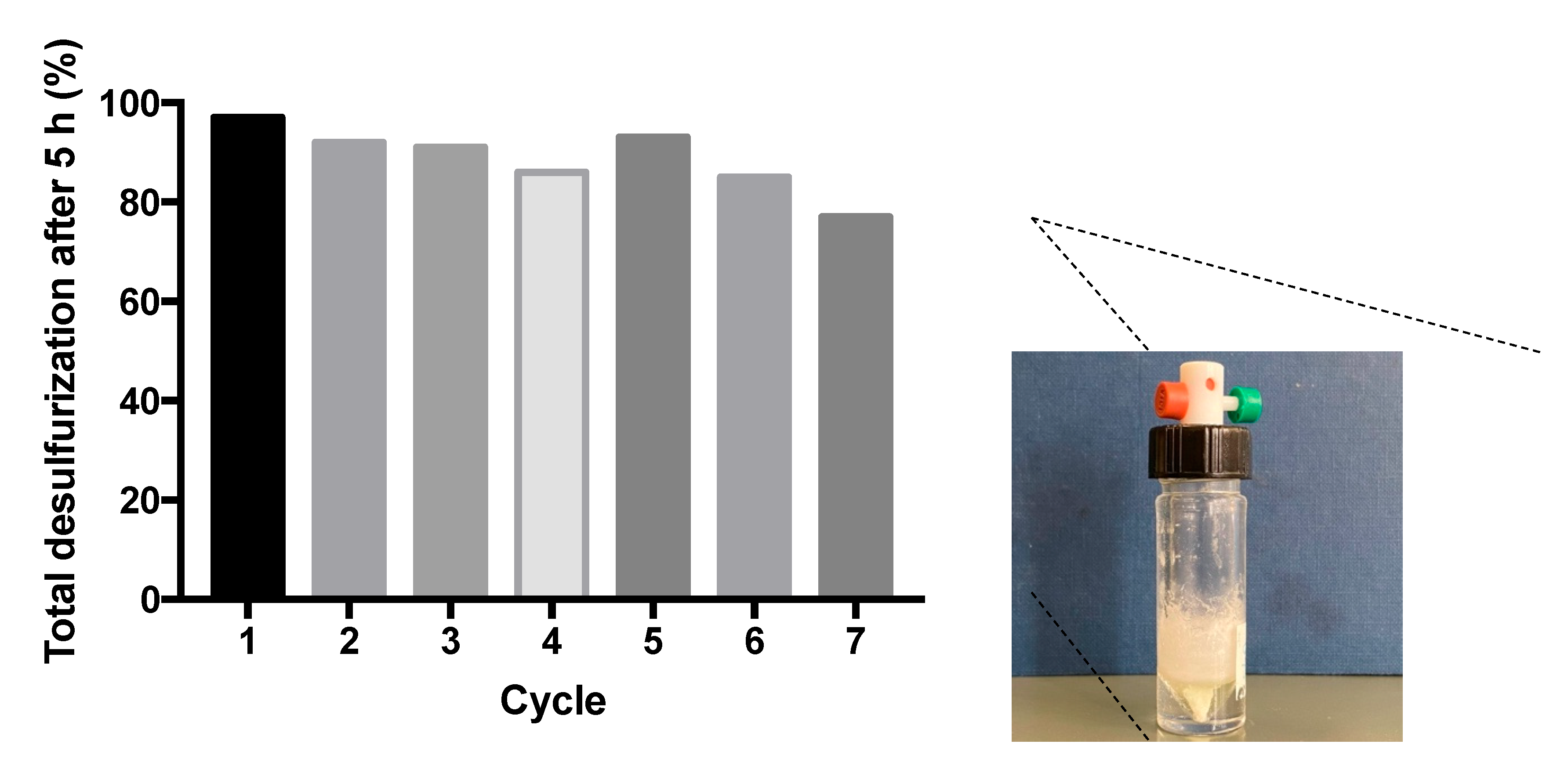
Figure 11.
Total desulfurization data of the model fuel catalyzed by complex (1) (using 5 μmol), for ten ECODS cycles using MeOH as the extraction solvent and 50% aq. H2O2 as oxidant (1.2 μmol), at 70 °C.
Figure 11.
Total desulfurization data of the model fuel catalyzed by complex (1) (using 5 μmol), for ten ECODS cycles using MeOH as the extraction solvent and 50% aq. H2O2 as oxidant (1.2 μmol), at 70 °C.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



