Submitted:
12 April 2024
Posted:
15 April 2024
You are already at the latest version
Abstract
Hereditary Hemorrhagic Telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is a rare and inherited vascular disorder, characterized by the development of arteriovenous malformations (AVMs) in various organs and telangiectasia (small AVM) in the mucocutaneous. The majority of HHT patients have haploinsufficiency of genes involved in the transforming growth factor-beta (TGFβ) signaling pathway, including endoglin (ENG), activin receptor-like kinase 1 (ALK1, also known as ACVRL1), or SMAD4. Active angiogenesis is also required for telangiectasia and AVM development. Anti-angiogenic strategies have been tested in patients and animal models extensively. However, the exact mechanisms for telangiectasia and AVM development remain unclear. In this review, we discuss recent advances in identifying disease mechanisms, and potential therapeutic targets.
Keywords:
Hereditary Hemorrhagic Telangiectasia
; Arteriovenous malformations
; Pathogenesis
; Therapeutics.
1. Introduction
Hereditary hemorrhagic telangiectasia (HHT) is a rare disorder characterized by arteriovenous malformations (AVMs) in multiple organs telangiectasia (small AVM) in the mucocutaneous [1]. This condition affects about 1 in 5000-8000 worldwide [2]. Telangiectasias in mucocutaneous can cause repeated gastrointestinal and nasal bleeding. AVMs in major organs, like the lungs, liver, and brain, can cause life-threatening hemorrhage and heart failure. Approximately 90% of HHT cases carry a heterozygous mutation in either the endoglin gene (ENG, HHT-1) or the activin-like receptor kinase 1 (ALK1, also known as ACVRL1, HHT-2). These pathogenic variants follow an autosomal dominant inheritance pattern. A subset of patients with HHT (about 2% of cases) carry a mutation in the SMAD4 gene, leading to a condition known as Juvenile Polyposis Syndrome (JPS). In this syndrome, the predominant clinical features include juvenile polyps and anemia [3]. Alteration of growth differentiation factor 2 (GDF2, also known as BMP-9) has lately been identified as a cause of HHT [4]. This rare form of HHT is also known as HHT5 [5]. Loss of function variants in EPHB4 (encoding ephrin receptor B4) were reported in a few individuals exhibiting atypical HHT symptoms and HHT-like hepatic abnormalities [6]. It has been shown that endothelial-specific DROSHA, a key enzyme for microRNA (miRNA) biogenesis, regulates the TGF-β and BMP pathway through interaction with SMADs. Inactivation of Drosha induced vascular malformation similar to HHT in zebrafish and mice, indicating a link between Drosha dysfunction and HHT [7,8].
In addition to a few randomized trials, currently, there are no standard medical therapies for HHT patients [4,9]. Therefore, the development of effective drug therapy is urgently needed. In recent years, various teams have collaboratively demonstrated several new disease mechanisms and potential therapeutic targets. This review aims to discuss the new advances within this field.
2. Pathophysiology
The pathophysiology of HHT involves haploinsufficiency of ALK1, ENG and SMAD4 genes [10]. However, this theory cannot explain several aspects of the pathogenesis and phenotypes of HHT patients. Recent studies show that somatic second hit, environmental second hit, and alternation of BMP9 and BMP10 function all play important roles in HHT pathogenesis.
2.1. Second Hit Hypothesis
The prevailing theory for the progression of HHT is the haploinsufficiency model [10], which reduces the production of proteins from the altered genes. Over 900 pathogenic variants have been identified in the ENG and ALK1 genes, including deletions, insertions, missense, and splice site mutations [11]. The reduction of the protein of the affected gene impairs the normal signaling pathways mediated by ENG or Alk1. However, the haploinsufficiency model cannot explain why AVMs/telangiectasia only develop in a few organs or part of the mucocutaneous, even if all cells in the body carry the same mutation. The haploinsufficiency model also fails to explain why all members carry the same mutations in one family have symptomatic HHT. This enigma has led researchers to propose the existence of a "second hit" hypothesis (also known as the Knudson hypothesis), suggesting that additional factors interact with HHT gene heterozygosity to trigger the formation of vascular lesions [10].
2.1.1. Genetic Two-Hit
Recently, the second somatic mutations were identified in dermal telangiectasia of both HHT1 and HHT2 patients (Figure 1). The second somatic mutation was first discovered by Snellings et al. in 2019 in skin telangiectasia [12]. Hence, local somatic mutations are present and may play a role in initiating focal vascular malformation [13]. In these studies, the authors checked (1) if there's a mutation in the same gene as the one causing germline HHT; (2) if both the germline and somatic mutations occur in pairs (bi-allelic); and (3) if both the mutations lead to loss of function (LoF). Capture-based library preparations were used to sequence 19 telangiectasia samples, focusing on three genes associated with HHT (ENG, ALK1, and SMAD4), along with an additional 13 genes related to vascular malformations. They discovered that among 19 telangiectasia samples, nine have had somatic mutations in the same gene of the pathogenic germline mutation [12]. Importantly, these mutations are not present in the constitutional (germline) DNA. To determine the biallelic nature of the mutations, they investigated whether these mutations were arranged in cis or trans configuration by sequencing amplicons encompassing positions of both somatic and germline mutations in a single molecule. They found that all seven mutation pairs are arranged in a trans configuration corresponds to a p-value of 0.008, indicating a significant bias toward a trans arrangement. This observation supports the idea that both somatic and germline mutations tend to occur in pairs, exhibiting a bi-allelic pattern. It also confirms that all bi-allelic germline and somatic mutations are highly likely to result in LoF [12]. Collectively, these data suggest that the occurrence of bi-allelic loss of function of ENG, ALK1, or SMAD4 genes may be a crucial step in the development of telangiectasia.
Animal studies have also supported the two-hit model. There is a significant impact of LoF of the remaining wild-type allele on HHT phenotype development, regardless of how the LoF occurs (e.g loss of heterozygosity or loss of protein during inflammation) [14]. Elimination of one allele of genes implicated in HHT, like Eng or Alk1, mimics particular characteristics of the human disease in animal models, primarily in older animals [15,16]. However, loss of both alleles of any HHT-causative gene is embryonically lethal in mice [17,18]. Further, conditional homozygous deletion of Eng [14] or Alk1 [19,20] in mice results in striking vascular malformations, resembling the AVMs found in HHT patients. It is important to acknowledge that, while the genomic second-hit hypothesis is a reasonable explanation for the development of vascular malformation in HHT patients, further research is needed to fully understand how bi-allelic loss of function of ENG or ALK1 in a few cells lead to the development of telangiectasia or AVM lesions, [21].
2.1.2. Angiogenesis
Pathogenic variants in Alk1 or Eng can initiate the development of AVMs in fetuses and neonates. During embryonic stages in mice, Alk1 is prominently expressed in arterial ECs. However, in adult mice, Alk1 expression is consistently high in lung arterial ECs only and during processes like wound healing and tumor angiogenesis [22].
The important of angiogenesis in AVM formation has been shown by many studies. Conditional global or EC-specific Alk1 deletion is lethal in neonates and adults, resulting in AVMs and hemorrhage in various organs that have active angiogenesis [20,23,24]. However, homozygous loss of Alk1 alone does not trigger cutaneous arteriovenous (AV) shunt formation in adults. Dermal wounding in Alk1-deficient mice led to the formation of AVMs in the skin [20,25]. Local application of angiogenic or inflammatory stimulators, like VEGF or Lipopolysaccharide (LPS), triggers skin AVM formation, while VEGF neutralizing antibody reduces wound-induced AVMs. Additionally, angiogenic stimulation is required for the development of brain AVM in adult mice [26,27,28]. Deletion of Eng in neonates leads to brain AVM development spontaneously when brain angiogenesis is still active. However, in adults, skin and brain AVMs only developed in mice with global or EC-specific Eng deletion when subject to wound or angiogenic stimulation [27]. Together, these data indicate that angiogenesis is necessary for AVM development.
2.1.3. Inflammation
In addition to angiogenesis, inflammation is also a second-hit event in the development of vascular malformation in HHT patients. Down regulation of ALK1 in human umbilical ECs (HUVECs) or Eng in mouse ECs upregulated pro-inflammatory and innate immune signaling [29], which may facilitate leukocyte infiltration and extraction [30]. An abnormally high number of macrophages are present in and around vascular walls in human brain AVM lesions, with or without hemorrhage [31,32,33], a pattern also observed in brain AVMs in mice with Eng or Alk1 deletion [27,28,34]. A persistent infiltration and pro-inflammatory differentiation of monocytes have also been observed in mouse brains with Eng deletion, indicating the effect of inflammation on HHT pathophysiology [35].
Notably, both ENG and ALK1 are expressed not only in ECs, but also in mononuclear cells (MNCs). They play essential roles in the maturation of MNCs within the bone marrow and their subsequent migration into the circulation [30,36,37,38,39]. Alterations in signaling through ENG and ALK1 in MNCs have been suggested for modifying immune responses in HHT patients, which present with heightened incidence of infections or leukopenia [40]. The observed alterations in the migration and release of various interleukins and mediators in HHT cases suggest an immunodeficiency in these patients [41]. Deletion of Eng in macrophages impaired mouse immune response [3,42]. However, deletion of Eng in macrophages did not cause bAVM development in mice, suggesting that inflammation could not cause AVM formation, but can act as a second-hit to promote AVM progression [3,27].
In addition, inflammation can alter the collagen I and III ratios and render brain AVMs prone to rupture. The collagen I/ collagen III ratios ratio was positively correlated with the number of CD68+ microglia/macrophages and GPNMB+ monocytes in Alk1 and Eng deficient mouse bAVMs. Microglia, one of the primary immune cells in the central nervous system, play a crucial role in neuroinflammation. Increased microglial accumulation was noted in bAVMs in both human and mouse models [31,43,44]. Studies revealed heightened macrophage infiltration in bAVMs with hemorrhage compared to those without [45]. However, the connection between microglial activation and hemorrhage remains unclear.
Several other environmental triggers have also been proposed as potential second-hits in HHT pathogenesis, including mechanical stress and sun exposure. A study on 103 HHT patients revealed a higher number of telangiectasias on the dominant hand and on the lower lip, which are expected to be more frequently exposed to mechanical stimuli, than the undominant hand and the upper lip [46].
Above evidence indicates that haploinsufficiency of HHT causative genes alone is not sufficient to induce AVM lesion formation. Additional insult (second hit), such as somatic mutation of the normal allele, angiogenesis and aforementioned factors are needed to trigger or promote AVM formation and development.
2.2. Role of BMP9 and BMP10
BMP 9 and 10, the principal ligands within the TGF-β family, are involved in the HHT development [47]. Mutations in BMP-9 have been reported to cause a vascular anomaly syndrome with phenotypic overlap with HHT [48]. BMP10 is the closest family member of BMP9, sharing many biochemical properties with BMP9, including regulating ALK1 downstream signals, such as SMAD1/5 phosphorylation and Id1 expression [49]. BMP10-null mice died at embryonic day 10.5 with phenotypes like Alk1 null embryos [50]. HHT-like phenotypes have also been shown in zebrafish with both bmp10 and bmp10-like genes knocked down, which phenocopied ALK1 mutants [51] and bmp10-null adult fish [52]. These ligands exert their impact through a heteromeric transmembrane receptor of serine/threonine type I (RI) and type II (RII), followed by the activation of the SMAD cascades [53]. It has been found that BMP9 induces the expression of VEGFR1, thus reduces downstream VEGF signaling [54]. In addition, BMP9 and BMP10 promote vascular maturation and quiescence [49].
3. Advancements in Developing New Therapies
The use of anti-angiogenic agents like bevacizumab (an anti-VEGF antibody) has shown promising in treating HHT patients. Anti-angiogenic approaches promise to modify the disease process and slow down the development of telangiectasias in addition to potentially causing regression of existing vascular malformations [61]. The available evidence thus far indicates a potential benefit of anti-angiogenic agents in treating small, mucosal telangiectasias, but not effective in treating larger visceral AVMs [62]. The limitations associated with the use of anti-angiogenic agents include limited efficacy and considerable adverse effects, which make them unsuitable for long-term utilization. Topical nasal spray of bevacizumab (Avastin®), a recombinant, humanized VEGF antibody, did not improve epistaxis severity in comparison with placebo [63,64]. An in vitro study showed that high bevacizumab concentration reduced VEGF expression but posed a higher risk of toxic effects on ECs [65]. A more recent systematic review revealed a significant gap in sociodemographic variables that identify the race and ethnicity of participants with established HHT in receiving intravenous delivery of bevacizumab treatment [66]. All current treatments for brain AVM, including surgery, radiation, or endovascular embolization, are associated with considerable risks of stroke or death. Treatment of patients with unruptured bAVMs has become increasingly controversial because the natural history of these patients may be less morbid than invasive therapies [67]. There is currently no FDA-approved treatment for HHT [68].
3.1. Enhancing ALK1 or ENG Expression
An alternative therapeutic approach for HHT patients is to enhance the expression of ENG and ALK1 genes in ECs. Kim et al. reported that overexpression of Alk1 in ECs can rescue HHT phenotype in mice with Alk1 or Eng gene deleted, without untoward effects [69]. This observation indicates that overexpression of Alk1 in ECs is well tolerated by mice. Similar finding has been shown in a zebrafish model in which alk1 is stably expressed in all ECs. [51] This zebrafish line has been continuously maintained for more than 12 years. The fact that overexpression of Alk1 rescues the HHT phenotypes in Eng mutant mice suggests that overexpression of Alk1 in ECs is a promising therapeutic strategy to treat both HHT2 (ALK1 mutation) and HHT1 (ENG mutation) patients, which together make up more than 80% of the HHT patient population.
The absence of hepatic endothelial Alk1 changes liver sinusoidal EC differentiation, hinders the differentiation of large vessels by stimulating angiogenesis and arterialization, and cause the formation of shunts within the hepatic vascular system. Therefore, hepatic endothelial ALK1 signaling protects from development of vascular malformations by preserving organ-specific endothelial differentiation and angiocrine signaling [70]. This evidence further supports the finding that an increase in ALK1 or ENG levels can reduce HHT phenotype severity.
Through screening FDA-approved drugs, Tacrolimus was found to be a potent activator of ALK1 signaling [71]. A case report demonstrated that oral administration of low-dose Tacrolimus improved HHT-related epistaxis without impacting pulmonary artery hypertension (PAH) progression in a patient with HHT2 and PAH [72]. Clinical trials using tacrolimus in HHT patients undergoing liver transplantation enhanced control of epistaxis. This medication upregulates the expression of ENG and ALK1, promotes tubulogenesis and cell migration [73]. In another study, oral administration of tacrolimus significantly increased hemoglobin levels and decreased epistaxis and/or gastrointestinal bleeding and the needs of blood transfusion in HHT patients [73]. Adverse effects however were common. At least one adverse effect occurred in 64% of patients during tacrolimus treatment. The most frequently observed adverse effects were headache, abdominal pain, diarrhea, and insomnia. 25% of patients still have adverse effects after the trial has finished [73]. Another study reported outcomes of 11 HHT patients treated with low tacrolimus doses (0.5–2 mg/day) on an off-label prescription basis and demonstrated that low doses of tacrolimus is a promising treatment for epistaxis and gastrointestinal bleeding in HHT [74]. Overall, treatment with tacrolimus significantly reduced severe bleeding, improved hemoglobin levels, and decreased transfusion needs in patients with HHT. Nevertheless, side effects of tacrolimus were prominent. Among 11 patients, two patients had to stop treatment due to gastric intolerance [74]. Despite the potential therapeutic benefit, its safety should be further investigated in a randomized controlled clinical trial. A recent study found that Neuropilin-1 interacts with ALK1 and ENG in vascular smooth muscle cells (SMCs). Neuropilin-1 deletion in vascular SMCs leads to reduced ALK1, ENG, and pSMAD1/5/8 signaling, and reduced cell death associated with HHT. Therefore, in addition to Tacrolimus, Neuropilin can be considered as a new therapeutic target for the treatment of HHT [75].
3.2. Anti-Angiogenic Gene Therapy
Anti-angiogenic approaches promise to reduce hemorrhage and modify the disease process [61]. Many agents can block VEGF signaling, e.g., anti-VEGF antibodies [26,61,76,77,78,79,80] and tyrosine kinase inhibitors (TKIs) [81,82,83]. However, these agents have considerable side effects [84,85,86,87]. In addition, TKIs showed a differential effect on skin and intestinal AVMs [88]. Genetic deletion of the angiogenic signal-transducing VEGF receptor-2 (VEGFR-2) prevented excessive angiogenesis but did not fully revert AVM formation [89]. This data suggests that blocking angiogenesis alone is not sufficient to treat AVMs. In addition, systemic therapies with antibodies and TKIs have many drawbacks and side effect [81,84,85,90,91]. Telangiectasia and brain AVMs are chronic lesionin which angiogenesis can be activated over time[87,92,93]. To control the disease progression, systemic delivery of antibodies or TKIs must be repeated for an extended period, which may be intolerable to some patients.
The soluble FMS-related tyrosine kinase 1 (sFLT1) contains the extracellular domain of VEGF receptor-1 and can bind all isoforms of VEGF, thereby inhibiting their downstream signaling. Thus, the effect of sFLT1 could be broader than VEGF antibodies that are specific to an individual VEGF isotope, and kinase inhibitors. Mice carrying two modified Flt1 alleles which lack the cytoplasmic tyrosine kinase signaling domain but retain the ability to express the sFlt1 isoform, are viable with mild adult phenotypes [94,95,96]. This suggests that signaling by FLT1 is not essential and supports a primary role for FLT1 and sFLT1 as decoy receptors, limiting the availability of VEGF to signal through its receptors.
It has been shown that in mice, intravenous (i.v.) injection of AAV9-sFLT1 (a recombinant AAV packaged in serotype 9 capsid) reversed bAVM phenotypes [97]. However, uncontrolled systemic sFLT1 expression caused minor liver inflammation and growth arrest of young mice [97]. New engineered AAV capsids and different delivery routes may minimize the side effects. AAV has many advantages over direct systemic delivery of anti-angiogenic protein because it can mediate long-term transgene expression [98,99,100,101,102,103], which makes a need for repeated dosing unlikely. In addition, many engineered AAV capsids make targeted delivery of genes possible. Therefore, AAV is a promising vector for developing specific gene therapy strategy for treating telangiectasia and AVMs.
3.3. Angiopoetin-2 (Angpt2) Antibodies
Alk1 germline deletion increased Angpt2 expression in rodent brain and spinal cord AVMs [19]. Angpt2 expression was found to increase in the postnatal retina of Smad4 mutant mice, as detected through RNA- and ChIP-sequencing on BMP9- stimulated ECs, as well as in mice with Smad4 knocked out specifically in ECs [104]. Increased Angpt2 expression was also detected in postnatal brain ECs of Alk1, Eng, and Smad4 mutant mice [104,105]. Administration of Angpt2 monoclonal antibodies prevented and resolved retinal AVMs of Smad4 mutant mice [104] and improved brain vascular morphology in other HHT models [105]. However, other studies showed that the blood ANGPT2 levels were decreased in HHT2 patients and unchanged in HHT1 patients [106,107]. Further, cultured blood outgrowth ECs from HHT1 and HHT2 patients, along with several tissues collected from Eng+/− mice exhibited substantial decreases in Angpt2 levels [108,109]. Studies have also indicated that ANGPT2 expression is depended on the type of HHT, and tissue/organ assessed [107,110,111]. Nevertheless, an inductive role for ANGPT2 in both brain and retinal AVMs has been suggested [105]. In clinical trials anti-ANGPT2 antibodies have shown potential in normalizing vessel caliber in HHT patients [104]. A Robust and ectopic Angpt2 expression in the brain ECs was evident in all the 3 mouse models that have Smad4, Alk1, or Eng knockout in ECs. Loss of HHT signaling in HUVECs, via ALK1-Fc, resulted in increased secretion of ANGPT2 into the media [105]. Thus, Angpt2-targeted therapies may represent a novel approach for treating vascular abnormalities in HHT patients.
3.4. PI3-Kinase Inhibitor and Other Agents
Because the PI3-kinase pathway operates downstream of both VEGF and ANGPT2, researchers have tested PI3-kinase inhibitors in HHT patients. Treatment with Buparlisib, a PI3-kinase inhibitor, reduced the frequency of epistaxis in HHT2 patient [112,113]. However, long-term utilization of this inhibitor could pose substantial challenges related to safety and tolerability. Adverse effects like depression, increased lipase, hyperglycemia, and anxiety of first-generation PI3 kinase inhibitors warrant careful consideration for prolonged use [114]. Among the AKT inhibitors, perifosine and uprosertib, VAD044 (a small molecule) exhibited superior efficacy in inhibiting retinal AVM formation by reducing phospho-S6 and AKT activity [115]. A phase I trial known as INSIGHT is currently underway to investigate the benefits and risks of VAD044 [116].
In addition to PI3-kinase inhibitors, other drugs with anti-angiogenic property have been tested. Non-selective beta-blockers, such as propranolol and timolol have been applied locally to reduce bleeding from telangiectasia [117]. Etamsylate (a nasal spray) has recently approved to treat HHT patients with high susceptibility to nosebleeds [118].
3.5. Modulation of BMP9-10/ENG/ALK1/SMAD Pathway as an Emerging Therapeutic Target
Based on the insights gained from genetic studies, it becomes feasible to develop tailored treatments or repurposing existing medications to modulate the BMP9-10/ENG /ALK1/SMAD pathway (Figure 2) [62]. Various therapeutic approaches targeting Alk1 are under investigation, including monoclonal antibodies, small molecule inhibitors, and gene therapy to modulate Alk1 expression and signaling [69,119]. Ruiz et al. found that Sirolimus and nintedanib in combination prevented vascular pathology in the oral mucosa, lungs and liver of the BMP9/BMP10-immunoblocked mice [120]. Sirolimus binds FKBP12 and inhibits mTOR downstream of PI3K and AKT. Post-liver transplantation, Sirolimus ameliorated HHT symptoms by correcting haploinsufficiency or upregulating protein levels of ENG and ALK1. HHT2 patients with hepatic AVMs and high-output cardiac failure experienced significant improvement in epistaxis after receiving treatment of immunosuppressive agent tacrolimus or sirolimus after liver transplantation [71,121].
Ruiz et al. found that Sirolimus and nintedanib in combination prevented vascular pathology in the oral mucosa, lungs and liver of the BMP9/BMP10-immunoblocked mice [120]. Tacrolimus is a strong activator of the BMP9/ALK1/Smad signaling cascade and it has been used for treatment of animal HHT and decreased the development of retinal AVMs [71]. A case study reported a substantial decrease in epistaxis in HHT2 patients following orally administration of a low dose of tacrolimus [72].
Together, these data shown that agents that can modulate gene expression in BMP9-10/ENG /ALK1/SMAD pathway have potential to reduce bleeding from telangiectasia and AVM.
4. Summary and Future Perspective
In this review, we summarized the advances in HHT pathophysiology and newly explode therapeutic strategies. The identification of second-somatic hit and enhanced ALK1 expression in ECs rescues the phenotype of Alk1 and Eng knockout mice, thus opening new revenues in our understanding of HHT pathogenesis and therapeutic targets. Furthermore, the exploration of drugs like Tacrolimus and Sirolimus, which have the potential to reactivate the BMP9-10/ ENG/ALK1/Smad1/5/9 signaling pathway, offers a novel direction in addressing the disrupted molecular mechanisms central to the HHT pathogenesis. ANGPT2 antibodies and AAV mediated gene therapies are also promising therapeutic strategy for reducing phenotype severity of HHT patients.
Further research should focus on exploring future potential treatments comprehensively utilizing existing data. Although the existence of somatic second hit has been proven, further investigations on how the few ECs with biallelic mutations can lead to the development of vascular malformations are needed. Although the above-mentioned therapeutic strategies are promising, they are still in the experimental and research stages. Clinical trials and further studies are necessary to determine their safety and effectiveness in treating HHT patients.
Author Contributions
A.Y.: conceptualization, writing, original draft preparation, review, and editing; Z.S.N.: writing-review-editing; A.S.: writing-review-editing, J.K.: writing-review-editing H.S.: Supervision, conceptualization, original draft, and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding Statement: This study was supported by the National Institutes of Health R01 HL122774 (H.S.) NS027713 (H.S.), NS112819 (H.S.), and by the Michael Ryan Zodda Foundation (H.S.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This is a review article. No new data was created.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Labeyrie P-E, Courthéoux P, Babin E, Bergot E, Touzé E, Pelage J-P (2016) Neurological involvement in hereditary hemorrhagic telangiectasia. Journal of Neuroradiology 43 (4):236-245. [CrossRef]
- de Gussem EM, Kroon S, Hosman AE, Kelder JC, Post MC, Snijder RJ, Mager JJ (2020) Hereditary Hemorrhagic Telangiectasia (HHT) and Survival: The Importance of Systematic Screening and Treatment in HHT Centers of Excellence. Journal of Clinical Medicine 9 (11). [CrossRef]
- Arthur HM, Roman BL (2022) An update on preclinical models of hereditary haemorrhagic telangiectasia: Insights into disease mechanisms. Frontiers in Medicine 9. [CrossRef]
- Balachandar S, Graves TJ, Shimonty A, Kerr K, Kilner J, Xiao S, Slade R, Sroya M, Alikian M, Curetean E et al. (2021) Identification and validation of a novel pathogenic variant in GDF2 (BMP9) responsible for hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations. American Journal of Medical Genetics Part A 188 (3):959-964. [CrossRef]
- Farhan A, Yuan F, Partan E, Weiss CR (2022) Clinical manifestations of patients with GDF2 mutations associated with hereditary hemorrhagic telangiectasia type 5. Am J Med Genet A 188 (1):199-209. [CrossRef]
- Guilhem A, Dupuis-Girod S, Espitia O, Rivière S, Seguier J, Kerjouan M, Lavigne C, Maillard H, Magro P, Alric L, et al. (2023) Seven cases of hereditary haemorrhagic telangiectasia-like hepatic vascular abnormalities associated withEPHB4pathogenic variants. Journal of Medical Genetics 60 (9):905-909. [CrossRef]
- Jiang X, Wooderchak-Donahue WL, McDonald J, Ghatpande P, Baalbaki M, Sandoval M, Hart D, Clay H, Coughlin S, Lagna G et al. (2018) Inactivating mutations in Drosha mediate vascular abnormalities similar to hereditary hemorrhagic telangiectasia. Sci Signal 11 (513). [CrossRef]
- Hata A, Lagna G (2019) Deregulation of Drosha in the pathogenesis of hereditary hemorrhagic telangiectasia. Current Opinion in Hematology 26 (3):161-169. [CrossRef]
- Kritharis A, Al-Samkari H, Kuter DJ (2018) Hereditary hemorrhagic telangiectasia: diagnosis and management from the hematologist’s perspective. Haematologica 103 (9):1433-1443. [CrossRef]
- Bernabeu C, Bayrak-Toydemir P, McDonald J, Letarte M (2020) Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. Journal of Clinical Medicine 9 (11). [CrossRef]
- Karlsson T, Cherif H (2018) Mutations in the ENG, ACVRL1, and SMAD4 genes and clinical manifestations of hereditary haemorrhagic telangiectasia: experience from the Center for Osler's Disease, Uppsala University Hospital. Ups J Med Sci 123 (3):153-157. [CrossRef]
- Snellings DA, Gallione CJ, Clark DS, Vozoris NT, Faughnan ME, Marchuk DA (2019) Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am J Hum Genet 105 (5):894-906. [CrossRef]
- Weakley SM, Jiang J, Kougias P, Lin PH, Yao Q, Brunicardi FC, Gibbs RA, Chen C (2010) Role of somatic mutations in vascular disease formation. Expert Rev Mol Diagn 10 (2):173-185. [CrossRef]
- Mahmoud M, Allinson KR, Zhai Z, Oakenfull R, Ghandi P, Adams RH, Fruttiger M, Arthur HM (2010) Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res 106 (8):1425-1433. [CrossRef]
- Srinivasan S, Hanes MA, Dickens T, Porteous ME, Oh SP, Hale LP, Marchuk DA (2003) A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum Mol Genet 12 (5):473-482. [CrossRef]
- Bourdeau A, Faughnan ME, Letarte M (2000) Endoglin-deficient mice, a unique model to study hereditary hemorrhagic telangiectasia. Trends Cardiovasc Med 10 (7):279-285.
- Urness LD, Sorensen LK, Li DY (2000) Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet 26 (3):328-331. [CrossRef]
- Sorensen LK, Brooke BS, Li DY, Urness LD (2003) Loss of distinct arterial and venous boundaries in mice lacking endoglin, a vascular-specific TGFbeta coreceptor. Dev Biol 261 (1):235-250.
- Milton I, Ouyang D, Allen CJ, Yanasak NE, Gossage JR, Alleyne CH, Jr., Seki T (2012) Age-dependent lethality in novel transgenic mouse models of central nervous system arteriovenous malformations. Stroke 43 (5):1432-1435. [CrossRef]
- Park SO, Wankhede M, Lee YJ, Choi EJ, Fliess N, Choe SW, Oh SH, Walter G, Raizada MK, Sorg BS et al. (2009) Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest 119 (11):3487-3496. [CrossRef]
- Shaligram SS, Zhang R, Zhu W, Ma L, Luo M, Li Q, Weiss M, Arnold T, Santander N, Liang R et al. (2022) Bone Marrow-Derived Alk1 Mutant Endothelial Cells and Clonally Expanded Somatic Alk1 Mutant Endothelial Cells Contribute to the Development of Brain Arteriovenous Malformations in Mice. Transl Stroke Res 13 (3):494-504. [CrossRef]
- Seki T, Yun J, Oh SP (2003) Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ Res 93 (7):682-689. [CrossRef]
- Morine KJ, Qiao X, Paruchuri V, Aronovitz MJ, Mackey EE, Buiten L, Levine J, Ughreja K, Nepali P, Blanton RM et al. (2017) Conditional knockout of activin like kinase-1 (ALK-1) leads to heart failure without maladaptive remodeling. Heart and Vessels 32 (5):628-636. [CrossRef]
- Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL et al. (2008) ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2 (HHT2). Blood 111 (2):633-642. [CrossRef]
- Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E (2000) Activin receptor-like kinase 1 modulates transforming growth factor- beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A 97 (6):2626-2631. [CrossRef]
- Walker EJ, Su H, Shen F, Degos V, Amend G, Jun K, Young WL (2012) Bevacizumab attenuates VEGF-induced angiogenesis and vascular malformations in the adult mouse brain. Stroke 43 (7):1925-1930. [CrossRef]
- Choi EJ, Chen W, Jun K, Arthur HM, Young WL, Su H (2014) Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS One 9 (2):e88511. [CrossRef]
- Chen W, Sun Z, Han Z, Jun K, Camus M, Wankhede M, Mao L, Arnold T, Young WL, Su H (2014) De novo cerebrovascular malformation in the adult mouse after endothelial Alk1 deletion and angiogenic stimulation. Stroke 45 (3):900-902. [CrossRef]
- Ma L, Zhu X, Tang C, Pan P, Yadav A, Liang R, Press K, Nelson J, Su H (2024) CNS resident macrophages enhance dysfunctional angiogenesis and circulating monocytes infiltration in brain arteriovenous malformation. Journal of Cerebral Blood Flow & Metabolism. [CrossRef]
- Rossi E, Sanz-Rodriguez F, Eleno N, Duwell A, Blanco FJ, Langa C, Botella LM, Cabanas C, Lopez-Novoa JM, Bernabeu C (2013) Endothelial endoglin is involved in inflammation: role in leukocyte adhesion and transmigration. Blood 121 (2):403-415. [CrossRef]
- Chen Y, Zhu W, Bollen AW, Lawton MT, Barbaro NM, Dowd CF, Hashimoto T, Yang GY, Young WL (2008) Evidence of inflammatory cell involvement in brain arteriovenous malformations. Neurosurgery 62 (6):1340-1349; discussion 1349-1350. [CrossRef]
- Guo Y, Tihan T, Kim H, Hess C, Lawton MT, Young WL, Zhao Y, Su H (2014) Distinctive distribution of lymphocytes in unruptured and previously untreated brain arteriovenous malformation. Neuroimmunol Neuroinflamm 1 (3):147-152. [CrossRef]
- Ma L, Guo Y, Zhao YL, Su H (2015) The Role of Macrophage in the Pathogenesis of Brain Arteriovenous Malformation. Int J Hematol Res 1 (2):52-56. [CrossRef]
- Chen W, Guo Y, Walker EJ, Shen F, Jun K, Oh SP, Degos V, Lawton MT, Tihan T, Davalos D et al. Akassoglou K, Nelson J, Pile-Spellman J, Su H, Young WL (2013) Reduced mural cell coverage and impaired vessel integrity after angiogenic stimulation in the Alk1-deficient brain. Arterioscler Thromb Vasc Biol 33 (2):305-310. [CrossRef]
- Zhang R, Han Z, Degos V, Shen F, Choi EJ, Sun Z, Kang S, Wong M, Zhu W, Zhan L et al. (2016) Persistent infiltration and pro-inflammatory differentiation of monocytes cause unresolved inflammation in brain arteriovenous malformation. Angiogenesis 19 (4):451-461. [CrossRef]
- van Laake LW, van den Driesche S, Post S, Feijen A, Jansen MA, Driessens MH, Mager JJ, Snijder RJ, Westermann CJ, Doevendans PA et al. (2006) Endoglin has a crucial role in blood cell-mediated vascular repair. Circulation 114 (21):2288-2297. [CrossRef]
- Post S, Smits AM, van den Broek AJ, Sluijter JP, Hoefer IE, Janssen BJ, Snijder RJ, Mager JJ, Pasterkamp G, Mummery CL et al. (2010) Impaired recruitment of HHT-1 mononuclear cells to the ischaemic heart is due to an altered CXCR4/CD26 balance. Cardiovasc Res 85 (3):494-502. [CrossRef]
- Shen F, Degos V, Chu PL, Han Z, Westbroek EM, Choi EJ, Marchuk D, Kim H, Lawton MT, Maze M et al. (2014) Endoglin deficiency impairs stroke recovery. Stroke 45 (7):2101-2106. [CrossRef]
- Han Z, Shaligram S, Faughnan ME, Clark D, Sun Z, Su H (2020) Reduction of endoglin receptor impairs mononuclear cell-migration. Explor Med 1:136-148. [CrossRef]
- Dingenouts CK, Goumans MJ, Bakker W (2015) Mononuclear cells and vascular repair in HHT. Front Genet 6:114. [CrossRef]
- Meurer SK, Weiskirchen R (2020) Endoglin: An 'Accessory' Receptor Regulating Blood Cell Development and Inflammation. Int J Mol Sci 21 (23). [CrossRef]
- Ojeda-Fernandez L, Recio-Poveda L, Aristorena M, Lastres P, Blanco FJ, Sanz-Rodriguez F, Gallardo-Vara E, de las Casas-Engel M, Corbi A, Arthur HM et al. (2016) Mice lacking endoglin in macrophages show an impaired immune response. PLoS Genet 12 (3):e1005935. [CrossRef]
- Park ES, Kim S, Yao DC, Savarraj JPJ, Choi HA, Chen PR, Kim E (2022) Soluble Endoglin Stimulates Inflammatory and Angiogenic Responses in Microglia That Are Associated with Endothelial Dysfunction. Int J Mol Sci 23 (3). [CrossRef]
- Germans MR, Sun W, Sebök M, Keller A, Regli L (2022) Molecular Signature of Brain Arteriovenous Malformation Hemorrhage: A Systematic Review. World Neurosurgery 157:143-151. [CrossRef]
- Nakamura Y, Sugita Y, Nakashima S, Okada Y, Yoshitomi M, Kimura Y, Miyoshi H, Morioka M, Ohshima K (2016) Alternatively Activated Macrophages Play an Important Role in Vascular Remodeling and Hemorrhaging in Patients with Brain Arteriovenous Malformation. Journal of Stroke and Cerebrovascular Diseases 25 (3):600-609. [CrossRef]
- Geisthoff U, Nguyen H-L, Lefering R, Maune S, Thangavelu K, Droege F (2020) Trauma Can Induce Telangiectases in Hereditary Hemorrhagic Telangiectasia. Journal of Clinical Medicine 9 (5). [CrossRef]
- Tillet E, Bailly S (2015) Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front Genet 5:456. [CrossRef]
- Wooderchak-Donahue WL, McDonald J, O'Fallon B, Upton PD, Li W, Roman BL, Young S, Plant P, Fülöp GT, Langa C et al. (2013) BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am J Hum Genet 93 (3):530-537. [CrossRef]
- David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S (2007) Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109 (5):1953-1961. [CrossRef]
- Chen D, Zhao M, Mundy GR (2004) Bone morphogenetic proteins. Growth Factors 22 (4):233-241. [CrossRef]
- Laux DW, Young S, Donovan JP, Mansfield CJ, Upton PD, Roman BL (2013) Circulating Bmp10 acts through endothelial Alk1 to mediate flow-dependent arterial quiescence. Development 140 (16):3403-3412. [CrossRef]
- Capasso TL, Li B, Volek HJ, Khalid W, Rochon ER, Anbalagan A, Herdman C, Yost HJ, Villanueva FS, Kim K et al. (2020) BMP10-mediated ALK1 signaling is continuously required for vascular development and maintenance. Angiogenesis 23 (2):203-220. [CrossRef]
- Hata A, Chen Y-G (2016) TGF-β Signaling from Receptors to Smads. Cold Spring Harbor Perspectives in Biology 8 (9). [CrossRef]
- Larrivee B, Prahst C, Gordon E, Del Toro R, Mathivet T, Duarte A, Simons M, Eichmann A (2012) ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell 22 (3):489-500. [CrossRef]
- Choi H, Kim B-G, Kim YH, Lee S-J, Lee YJ, Oh SP (2022) BMP10 functions independently from BMP9 for the development of a proper arteriovenous network. Angiogenesis 26 (1):167-186. [CrossRef]
- Bouvard C, Tu L, Rossi M, Desroches-Castan A, Berrebeh N, Helfer E, Roelants C, Liu H, Ouarné M, Chaumontel N et al. (2022) Different cardiovascular and pulmonary phenotypes for single- and double-knock-out mice deficient in BMP9 and BMP10. Cardiovascular Research 118 (7):1805-1820. [CrossRef]
- Chen H, Brady Ridgway J, Sai T, Lai J, Warming S, Chen H, Roose-Girma M, Zhang G, Shou W, Yan M (2013) Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc Natl Acad Sci U S A 110 (29):11887-11892. [CrossRef]
- Ricard N, Ciais D, Levet S, Subileau M, Mallet C, Zimmers TA, Lee SJ, Bidart M, Feige JJ, Bailly S (2012) BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 119 (25):6162-6171. [CrossRef]
- Ruiz S, Zhao H, Chandakkar P, Chatterjee PK, Papoin J, Blanc L, Metz CN, Campagne F, Marambaud P (2016) A mouse model of hereditary hemorrhagic telangiectasia generated by transmammary-delivered immunoblocking of BMP9 and BMP10. Sci Rep 5:37366. [CrossRef]
- Al Tabosh T, Liu H, Koça D, Al Tarrass M, Tu L, Giraud S, Delagrange L, Beaudoin M, Rivière S, Grobost V et al. (2024) Impact of heterozygous ALK1 mutations on the transcriptomic response to BMP9 and BMP10 in endothelial cells from hereditary hemorrhagic telangiectasia and pulmonary arterial hypertension donors. Angiogenesis. [CrossRef]
- Al-Samkari H, Kasthuri RS, Parambil JG, Albitar HA, Almodallal YA, Vazquez C, Serra MM, Dupuis-Girod S, Wilsen CB, McWilliams JP et al. (2021) An international, multicenter study of intravenous bevacizumab for bleeding in hereditary hemorrhagic telangiectasia: the InHIBIT-Bleed study. Haematologica 106 (8):2161-2169. [CrossRef]
- Al Tabosh T, Al Tarrass M, Tourvieilhe L, Guilhem A, Dupuis-Girod S, Bailly S (2024) Hereditary hemorrhagic telangiectasia: from signaling insights to therapeutic advances. Journal of Clinical Investigation 134 (4). [CrossRef]
- Whitehead KJ, Sautter NB, McWilliams JP, Chakinala MM, Merlo CA, Johnson MH, James M, Everett EM, Clancy MS, Faughnan ME et al. (2016) Effect of topical intranasal therapy on epistaxis frequency in patients with Hereditary Hemorrhagic Telangiectasia: a randomized clinical trial. JAMA 316 (9):943-951. [CrossRef]
- Dupuis-Girod S, Ambrun A, Decullier E, Fargeton AE, Roux A, Breant V, Colombet B, Riviere S, Cartier C, Lacombe P et al. (2016) Effect of bevacizumab nasal spray on epistaxis duration in Hereditary Hemorrhagic Telangectasia: a randomized clinical trial. JAMA 316 (9):934-942. [CrossRef]
- Sadick H, Schäfer E, Weiss C, Rotter N, Müller C, Birk R, Sadick M, Häussler D (2022) An in vitro study on the effect of bevacizumab on endothelial cell proliferation and VEGF concentration level in patients with hereditary hemorrhagic telangiectasia. Experimental and Therapeutic Medicine 24 (3). [CrossRef]
- Galiatsatos P, Wilson C, O’Brien J, Gong AJ, Angiolillo D, Johnson J, Myers C, Strout S, Mathai S, Robinson G et al. (2022) A lack of race and ethnicity data in the treatment of hereditary hemorrhagic telangiectasia: a systematic review of intravenous bevacizumab efficacy. Orphanet Journal of Rare Diseases 17 (1). [CrossRef]
- Mohr JP, Parides MK, Stapf C, Moquete E, Moy CS, Overbey JR, Al-Shahi Salman R, Vicaut E, Young WL, Houdart E et al. (2014) Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet 383 (9917):614-621. [CrossRef]
- Snodgrass RO, Chico TJA, Arthur HM (2021) Hereditary Haemorrhagic Telangiectasia, an Inherited Vascular Disorder in Need of Improved Evidence-Based Pharmaceutical Interventions. Genes (Basel) 12 (2). [CrossRef]
- Kim YH, Phuong NV, Choe SW, Jeon CJ, Arthur HM, Vary CP, Lee YJ, Oh SP (2020) Overexpression of Activin Receptor-Like Kinase 1 in Endothelial Cells Suppresses Development of Arteriovenous Malformations in Mouse Models Of Hereditary Hemorrhagic Telangiectasia. Circ Res. [CrossRef]
- Schmid CD, Olsavszky V, Reinhart M, Weyer V, Trogisch FA, Sticht C, Winkler M, Kürschner SW, Hoffmann J, Ola R et al. (2023) ALK1 controls hepatic vessel formation, angiodiversity, and angiocrine functions in hereditary hemorrhagic telangiectasia of the liver. Hepatology 77 (4):1211-1227. [CrossRef]
- Ruiz S, Chandakkar P, Zhao H, Papoin J, Chatterjee PK, Christen E, Metz CN, Blanc L, Campagne F, Marambaud P (2017) Tacrolimus rescues the signaling and gene expression signature of endothelial ALK1 loss-of-function and improves HHT vascular pathology. Human Molecular Genetics 26 (24):4786-4798. [CrossRef]
- Sommer N, Droege F, Gamen KE, Geisthoff U, Gall H, Tello K, Richter MJ, Deubner LM, Schmiedel R, Hecker M et al. (2018) Treatment with low-dose tacrolimus inhibits bleeding complications in a patient with hereditary hemorrhagic telangiectasia and pulmonary arterial hypertension. Pulmonary Circulation 9 (2):1-3. [CrossRef]
- Hessels J, Kroon S, Boerman S, Nelissen RC, Grutters JC, Snijder RJ, Lebrin F, Post MC, Mummery CL, Mager JJ (2022) Efficacy and Safety of Tacrolimus as Treatment for Bleeding Caused by Hereditary Hemorrhagic Telangiectasia: An Open-Label, Pilot Study. J Clin Med 11 (18). [CrossRef]
- Álvarez-Hernández P, Patier JL, Marcos S, Gómez del Olmo V, Lorente-Herraiz L, Recio-Poveda L, Botella LM, Viteri-Noël A, Albiñana V (2023) Tacrolimus as a Promising Drug for Epistaxis and Gastrointestinal Bleeding in HHT. Journal of Clinical Medicine 12 (23). [CrossRef]
- Kilari S, Wang Y, Singh A, Graham RP, Iyer V, Thompson SM, Torbenson MS, Mukhopadhyay D, Misra S (2022) Neuropilin-1 deficiency in vascular smooth muscle cells is associated with hereditary hemorrhagic telangiectasia arteriovenous malformations. JCI Insight 7 (9). [CrossRef]
- Sullivan LA, Carbon JG, Roland CL, Toombs JE, Nyquist-Andersen M, Kavlie A, Schlunegger K, Richardson JA, Brekken RA (2010) r84, a novel therapeutic antibody against mouse and human VEGF with potent anti-tumor activity and limited toxicity induction. PLoS One 5 (8):e12031. doi:ARTN e1203110.1371/journal.pone.0012031.
- Han C, Choe SW, Kim YH, Acharya AP, Keselowsky BG, Sorg BS, Lee YJ, Oh SP (2014) VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis 17 (4):823-830. [CrossRef]
- Bose P, Holter JL, Selby GB (2009) Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med 360 (20):2143-2144. [CrossRef]
- Oosting S, Nagengast W, de Vries E (2009) More on bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med 361 (9):931; author reply 931-932. [CrossRef]
- Brinkerhoff BT, Poetker DM, Choong NW (2011) Long-term therapy with bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med 364 (7):688-689. [CrossRef]
- Simons M, Eichmann A (2012) "On-target" cardiac effects of anticancer drugs: lessons from new biology. J Am Coll Cardiol 60 (7):626-627. [CrossRef]
- Levitzki A (2013) Tyrosine kinase inhibitors: views of selectivity, sensitivity, and clinical performance. Annu Rev Pharmacol Toxicol 53:161-185. [CrossRef]
- Faughnan ME, Gossage JR, Chakinala MM, Oh SP, Kasthuri R, Hughes CCW, McWilliams JP, Parambil JG, Vozoris N, Donaldson J et al. (2019) Pazopanib may reduce bleeding in hereditary hemorrhagic telangiectasia. Angiogenesis 22 (1):145-155. [CrossRef]
- Maestraggi Q, Bouattour M, Toquet S, Jaussaud R, Kianmanesh R, Durand F, Servettaz A (2015) Bevacizumab to treat cholangiopathy in hereditary hemorrhagic telangiectasia: Be cautious: a case report. Medicine (Baltimore) 94 (46):e1966. [CrossRef]
- Dupuis-Girod S, Ginon I, Saurin JC, Marion D, Guillot E, Decullier E, Roux A, Carette MF, Gilbert-Dussardier B, Hatron PY et al. (2012) Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA 307 (9):948-955. [CrossRef]
- Drabkin HA (2010) Pazopanib and anti-VEGF therapy. Open Access J Urol 2:35-40.
- Orphanos GS, Ioannidis GN, Ardavanis AG (2009) Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol 48 (7):964-970. [CrossRef]
- Kim YH, Kim MJ, Choe SW, Sprecher D, Lee YJ, Oh SP (2017) Selective effects of oral anti-angiogenic tyrosine kinase inhibitors on an animal model of hereditary hemorrhagic telangiectasia. J Thromb Haemost 15 (6):1095-1102. [CrossRef]
- Ola R, Dubrac A, Han J, Zhang F, Fang JS, Larrivee B, Lee M, Urarte AA, Kraehling JR, Genet G et al. (2016) PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat Commun 7:13650. [CrossRef]
- Tanvetyanon T, Murtagh R, Bepler G (2009) Rupture of a cerebral arteriovenous malformation in a patient treated with bevacizumab. J Thorac Oncol 4 (2):268-269. [CrossRef]
- Tabouret T, Gregory T, Dhooge M, Brezault C, Mir O, Dreanic J, Chaussade S, Coriat R (2015) Long term exposure to antiangiogenic therapy, bevacizumab, induces osteonecrosis. Invest New Drugs 33 (5):1144-1147. [CrossRef]
- Kim H, Marchuk DA, Pawlikowska L, Chen Y, Su H, Yang GY, Young WL (2008) Genetic considerations relevant to intracranial hemorrhage and brain arteriovenous malformations. Acta Neurochir Suppl 105:199-206. [CrossRef]
- Chen W, Choi EJ, McDougall CM, Su H (2014) Brain arteriovenous malformation modeling, pathogenesis, and novel therapeutic targets. Transl Stroke Res 5 (3):316-329. [CrossRef]
- Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M (1998) Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A 95 (16):9349-9354. [CrossRef]
- Sawano A, Iwai S, Sakurai Y, Ito M, Shitara K, Nakahata T, Shibuya M (2001) Flt-1, vascular endothelial growth factor receptor 1, is a novel cell surface marker for the lineage of monocyte-macrophages in humans. Blood 97 (3):785-791. [CrossRef]
- Niida S, Kondo T, Hiratsuka S, Hayashi S, Amizuka N, Noda T, Ikeda K, Shibuya M (2005) VEGF receptor 1 signaling is essential for osteoclast development and bone marrow formation in colony-stimulating factor 1-deficient mice. Proc Natl Acad Sci U S A 102 (39):14016-14021. [CrossRef]
- Zhu W, Shen F, Mao L, Zhan L, Kang S, Sun Z, Nelson J, Zhang R, Zou D, McDougall CM et al. (2017) Soluble FLT1 Gene Therapy Alleviates Brain Arteriovenous Malformation Severity. Stroke 48 (5):1420-1423. [CrossRef]
- Hadaczek P, Eberling JL, Pivirotto P, Bringas J, Forsayeth J, Bankiewicz KS (2010) Eight years of clinical improvement in MPTP-lesioned primates after gene therapy with AAV2-hAADC. Mol Ther 18 (8):1458-1461. [CrossRef]
- Kaplitt MG, Leone P, Samulski RJ, Xiao X, Pfaff DW, O'Malley KL, During MJ (1994) Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet 8 (2):148-154. [CrossRef]
- Riviere C, Danos O, Douar AM (2006) Long-term expression and repeated administration of AAV type 1, 2 and 5 vectors in skeletal muscle of immunocompetent adult mice. Gene Ther 13 (17):1300-1308. [CrossRef]
- Rivera VM, Gao GP, Grant RL, Schnell MA, Zoltick PW, Rozamus LW, Clackson T, Wilson JM (2005) Long-term pharmacologically regulated expression of erythropoietin in primates following AAV-mediated gene transfer. Blood 105 (4):1424-1430. [CrossRef]
- Rivera VM, Ye X, Courage NL, Sachar J, Cerasoli F, Jr., Wilson JM, Gilman M (1999) Long-term regulated expression of growth hormone in mice after intramuscular gene transfer. Proc Natl Acad Sci U S A 96 (15):8657-8662. [CrossRef]
- Koo T, Okada T, Athanasopoulos T, Foster H, Takeda S, Dickson G (2011) Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J Gene Med 13 (9):497-506. [CrossRef]
- Crist AM, Zhou X, Garai J, Lee AR, Thoele J, Ullmer C, Klein C, Zabaleta J, Meadows SM (2019) Angiopoietin-2 Inhibition Rescues Arteriovenous Malformation in a Smad4 Hereditary Hemorrhagic Telangiectasia Mouse Model. Circulation 139 (17):2049-2063. [CrossRef]
- Zhou X, Pucel JC, Nomura-Kitabayashi A, Chandakkar P, Guidroz AP, Jhangiani NL, Bao D, Fan J, Arthur HM, Ullmer C, Klein C, Marambaud P, Meadows SM (2023) ANG2 Blockade Diminishes Proangiogenic Cerebrovascular Defects Associated With Models of Hereditary Hemorrhagic Telangiectasia. Arteriosclerosis, Thrombosis, and Vascular Biology 43 (8):1384-1403. [CrossRef]
- Ojeda-Fernandez L, Barrios L, Rodriguez-Barbero A, Recio-Poveda L, Bernabeu C, Botella LM (2010) Reduced plasma levels of Ang-2 and sEng as novel biomarkers in hereditary hemorrhagic telangiectasia (HHT). Clin Chim Acta 411 (7-9):494-499. [CrossRef]
- Wetzel-Strong SE, Weinsheimer S, Nelson J, Pawlikowska L, Clark D, Starr MD, Liu Y, Kim H, Faughnan ME, Nixon AB et al. (2021) Pilot investigation of circulating angiogenic and inflammatory biomarkers associated with vascular malformations. Orphanet Journal of Rare Diseases 16 (1). [CrossRef]
- Ardelean DS, Jerkic M, Yin M, Peter M, Ngan B, Kerbel RS, Foster FS, Letarte M (2014) Endoglin and activin receptor-like kinase 1 heterozygous mice have a distinct pulmonary and hepatic angiogenic profile and response to anti-VEGF treatment. Angiogenesis 17 (1):129-146. [CrossRef]
- Fernandez-Lopez A, Garrido-Martin EM, Sanz-Rodriguez F, Pericacho M, Rodriguez-Barbero A, Eleno N, Lopez-Novoa JM, Duwell A, Vega MA, Bernabeu C et al. (2007) Gene expression fingerprinting for human hereditary hemorrhagic telangiectasia. Hum Mol Genet 16 (13):1515-1533. [CrossRef]
- Winkler EA, Kim CN, Ross JM, Garcia JH, Gil E, Oh I, Chen LQ, Wu D, Catapano JS, Raygor K et al. (2022) A single-cell atlas of the normal and malformed human brain vasculature. Science:eabi7377. [CrossRef]
- Wälchli T, Ghobrial M, Schwab M, Takada S, Zhong H, Suntharalingham S, Vetiska S, Rodrigues Rodrigues D, Rehrauer H, Wu R et al. (2021). [CrossRef]
- Geisthoff UW, Nguyen H-LP, Hess D (2013) Improvement in hereditary hemorrhagic telangiectasia after treatment with the phosphoinositide 3-kinase inhibitor BKM120. Annals of Hematology 93 (4):703-704. [CrossRef]
- Robert F, Desroches-Castan A, Bailly S, Dupuis-Girod S, Feige J-J (2020) Future treatments for hereditary hemorrhagic telangiectasia. Orphanet Journal of Rare Diseases 15 (1). [CrossRef]
- Piha-Paul SA, Taylor MH, Spitz D, Schwartzberg L, Beck JT, Bauer TM, Meric-Bernstam F, Purkayastha D, Karpiak L, Szpakowski S et al. (2019) Efficacy and safety of buparlisib, a PI3K inhibitor, in patients with malignancies harboring a PI3K pathway activation: a phase 2, open-label, single-arm study. Oncotarget 10 (60):6526-6535. [CrossRef]
- Ola R, Hessels J, Hammill A, Friday C, Clancy M, Al-Samkari H, Meadows S, Iyer V, Akhurst R (2023) Executive summary of the 14th HHT international scientific conference. Angiogenesis 26 (S1):27-37. [CrossRef]
- Viteri-Noël A, González-García A, Patier JL, Fabregate M, Bara-Ledesma N, López-Rodríguez M, Gómez del Olmo V, Manzano L (2022) Hereditary Hemorrhagic Telangiectasia: Genetics, Pathophysiology, Diagnosis, and Management. Journal of Clinical Medicine 11 (17). [CrossRef]
- Mei-Zahav M, Gendler Y, Bruckheimer E, Prais D, Birk E, Watad M, Goldschmidt N, Soudry E (2020) Topical Propranolol Improves Epistaxis Control in Hereditary Hemorrhagic Telangiectasia (HHT): A Randomized Double-Blind Placebo-Controlled Trial. Journal of Clinical Medicine 9 (10). [CrossRef]
- Albiñana V, Giménez-Gallego G, García-Mato A, Palacios P, Recio-Poveda L, Cuesta A-M, Patier J-L, Botella L-M (2019) Topically Applied Etamsylate: A New Orphan Drug for HHT-Derived Epistaxis (Antiangiogenesis through FGF Pathway Inhibition). TH Open 03 (03):e230-e243. [CrossRef]
- Cunha SI, Pietras K (2011) ALK1 as an emerging target for anti-angiogenic therapy of cancer. Blood 117 (26):6999-7006. [CrossRef]
- Ruiz S, Zhao H, Chandakkar P, Papoin J, Choi H, Nomura-Kitabayashi A, Patel R, Gillen M, Diao L, Chatterjee PK et al. (2019) Sirolimus plus nintedanib treatments vascular pathology in HHT mouse models. [CrossRef]
- Dupuis-Girod S, Bailly S, Plauchu H (2010) Hereditary hemorrhagic telangiectasia (HHT): from molecular biology to patient care. J Thromb Haemost 8 (7):1447-1456. [CrossRef]
Figure 1.
Schematic representation of Hypothetical Two-hit model in HHT: The germline heterozygous mutation in the HHT gene cause haploinsufficiency (First hit). The second hits including inflammation, hypoxia, neo- angiogenesis, vascular injury, and somatic mutations in the previously unaffected normal allele contribute to HHT phenotype development and progression.
Figure 1.
Schematic representation of Hypothetical Two-hit model in HHT: The germline heterozygous mutation in the HHT gene cause haploinsufficiency (First hit). The second hits including inflammation, hypoxia, neo- angiogenesis, vascular injury, and somatic mutations in the previously unaffected normal allele contribute to HHT phenotype development and progression.
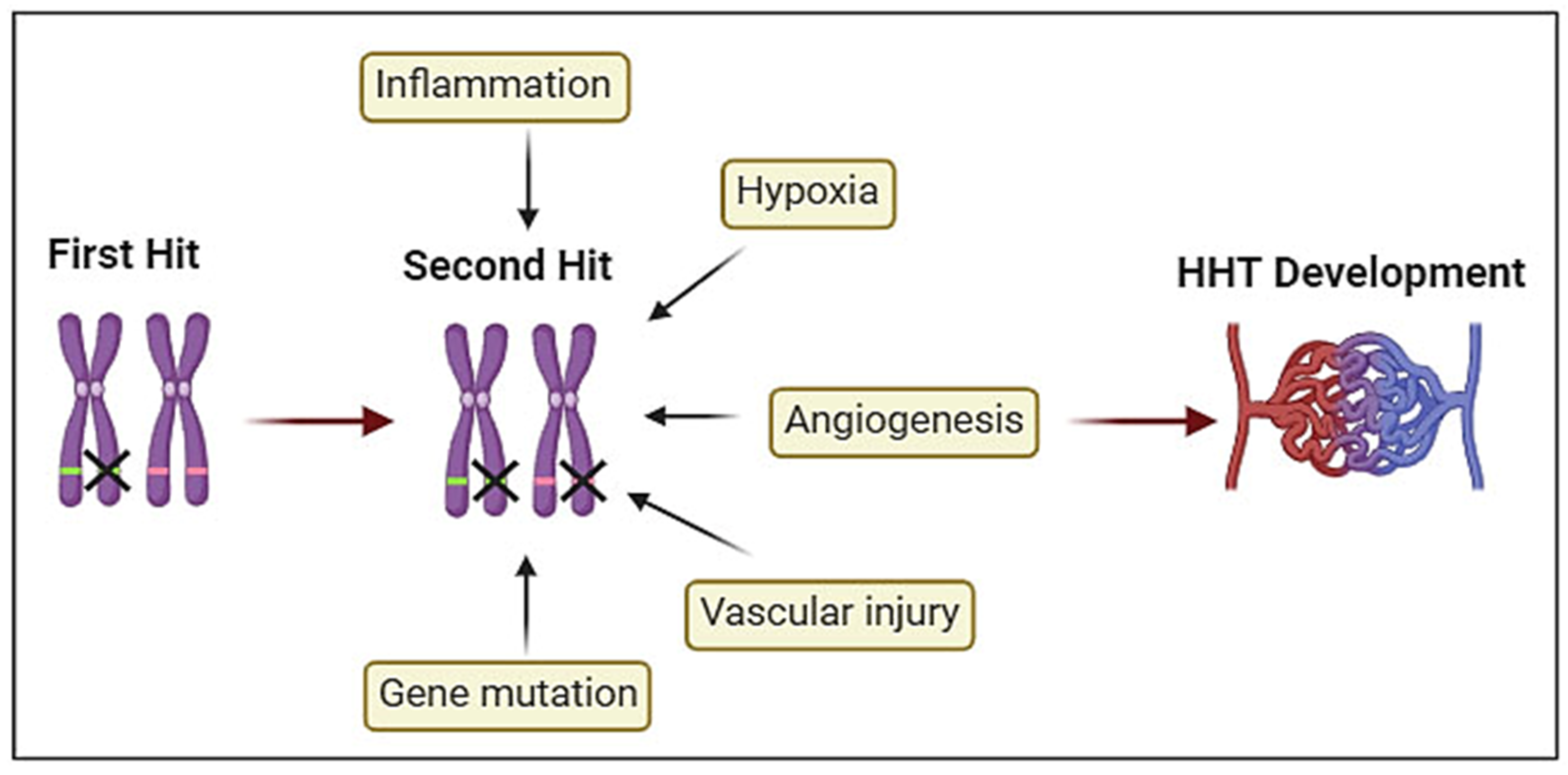
Figure 2.
Targets of therapeutic agents. Anti-VEGF antibodies bind VEGF in tissue prevent it interact with its receptors and thus reduce angiogenesis and AVM severity. ANGPT2 monoclonal antibodies bind with ANGPT2 in tissue prevent it interact with its receptors and thus reduce its downstream signaling and AVM severity. Sirolimus and Tacrolimus inhibit BMP9/BMP10 downstream signaling, which resulting in reduction of AVM severity. Tacrolimus can also reduce AVM severity through inhibit PI3K signaling pathway.
Figure 2.
Targets of therapeutic agents. Anti-VEGF antibodies bind VEGF in tissue prevent it interact with its receptors and thus reduce angiogenesis and AVM severity. ANGPT2 monoclonal antibodies bind with ANGPT2 in tissue prevent it interact with its receptors and thus reduce its downstream signaling and AVM severity. Sirolimus and Tacrolimus inhibit BMP9/BMP10 downstream signaling, which resulting in reduction of AVM severity. Tacrolimus can also reduce AVM severity through inhibit PI3K signaling pathway.
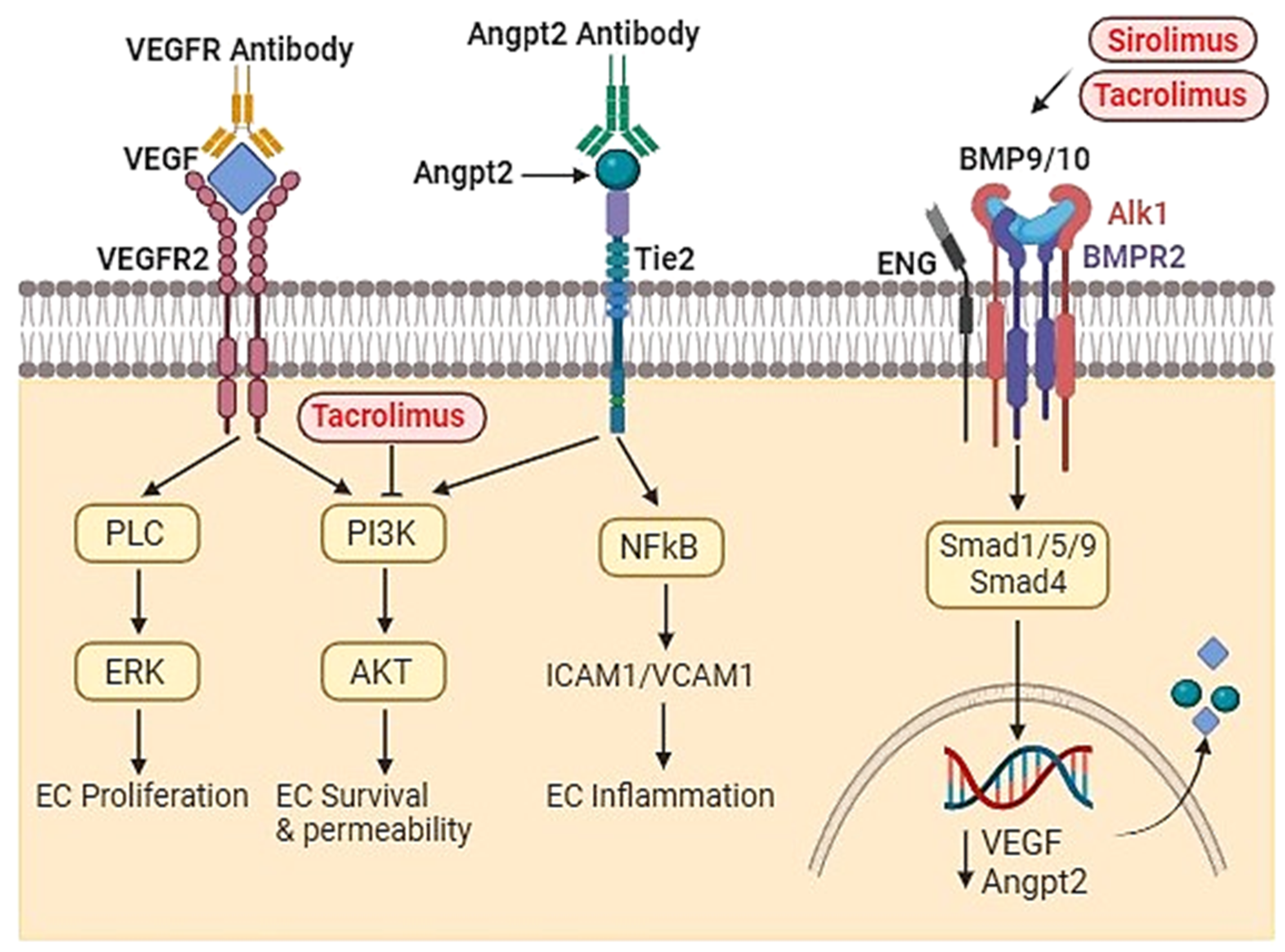

Table 1.
Summary of recent investigations of BMP9-10/ ENG/Alk1/SMAD signaling pathways.
| Subject | Title | Description | Ref. |
|---|---|---|---|
| Zebrafish | BMP10-mediated ALK1 signaling is continuously required for vascular development and maintenance. | The authors have found that loss of both bmp10 and bmp10-like genes leads to embryonic lethal cranial AVMs, which are distinguishable from alk1 mutants and concluded that BMP10 is vital for the maintenance of post-embryonic vascular development as a non-reductant ligand of Alk1. | [52] |
| Mice | Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. |
The study examined BMP9 and BMP10 in embryonic and postnatal development. The authors found that BMP9 is indispensable for postnatal vascular development in mice. BMP9 and BMP10 are ALK1's natural ligands. | [57] |
| BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. |
This study showed that administration of a neutralizing anti-BMP10 antibody to juvenile Bmp9-KO mice reduced retinal vascular expansion and vascular density. The data indicate that BMP9 and BMP10 are important in postnatal vascular remodeling of the retina and BMP10 can be a substitute for BMP9. | [58] | |
| A mouse model of HHT generated by trans-mammary-delivery of anti-BMP9/10 antibodies . | This study induced AVMs in postnatal retina through trans-mammary delivery of anti-BMP9/10 antibodies. This could be a practical and non-invasive method for the induction of HHT vascular pathology in the retina of postnatal mice. | [59] | |
| Human cell lines | Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in ECs . | The authors demonstrated that BMP9/BMP10 activate SMAD1/5/8 pathways and concluded that BMP9 and BMP10 serve as distinct ALK1 ligands and potentially elicit ALK1-mediated angiogenic effects. | [49] |
| Human | Impact of Heterozygous ALK1 mutations on the transcriptomic response to BMP9 and BMP10 in ECs from hereditary hemorrhagic telangiectasia and pulmonary arterial hypertension donors | Endothelial colony-forming cells (ECFCs) and microvascular ECs (HMVECs) were isolated from new-born HHT and adult PAH donors, and the impact of ALK1 mutations on BMP9 and BMP19 transcriptomic responses in ECs was consequently observed. RNA-sequencing was performed on these cells following an 18h stimulation with BMP9 or BMP10. The data showed that ALK1 heterozygosity modified a few of the BMP9/BMP19 regulated genes which are comparable to the controls. | [60] |
Abbreviations: BMP9-KO=Bone Morphogenetic Protein 9 Knockout, BMP10-KO= Bone Morphogenetic Protein 10 Knockout, PAH=pulmonary arterial hypertension.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



