Submitted:
11 April 2024
Posted:
15 April 2024
You are already at the latest version
Abstract
Glycosylation, the most significant modification of proteins, plays an essential role in cellular processes. Congenital disorders of glycosylation (CDG) are multisystemic diseases resulting from mutations affecting glycosylation steps. Diagnosis of CDG relies on biochemical assays, such as isoelectric focusing. Despite its significance, CDG cases remain unreported in Cuba due to the lack of implemented diagnostic methods. This study aimed to evaluate transferrin glycoforms for diagnosing congenital N-glycosylation defects. We conducted a cross-sectional descriptive study using serum samples from 17 patients stored at the National Center for Medical Genetics of Cuba for suspected CDG. Results revealed abnormal glycosylation patterns in 7 patients, suggesting congenital N-glycosylation defects. We conclude that the implementation of transferrin glycoform analysis could pioneer a preliminary diagnosis of CDG in Cuba.
Keywords:
Congenital disorders of glycosylation
; CDG
; N-glycosylation
; serum transferrin isoelectric focusing
I. Introduction
Congenital disorders of glycosylation (CDG), genetic diseases determined by alterations in one or more stages of the protein or lipid glycosylation process, manifest with multisystemic clinical presentations of varying severity.
Jaeken et al. first described these disorders in 1980 [1], and subsequent cases have steadily accumulated. Orphanet’s database reported a collective prevalence at birth of 1.5/100000 live births in 2016 [2]. In 2014, a project conducted in the United States theoretically predicted CDG prevalence to be 1/1000 in Americans of European descent and 2/1000 in Americans of African descent [3]. The evident disparity in prevalence rates has prompted the CDG research community to recognize these conditions as both epidemiologically elusive and underdiagnosed [2,3,4,5,6].
CDG generally exhibit an autosomal recessive inheritance pattern, although defects with X-linked and autosomal dominant inheritance have been described [3].
CDG are characterized by a broad spectrum of symptoms, including mental retardation, severe growth and developmental delay, structural abnormalities in the central and peripheral nervous systems, cardiac defects, hormonal imbalances, coagulation problems, among others. The majority of CDG cases have an unfavorable prognosis [7].
II. Theoretical Framework
II.1. Protein Glycosylation
Glycosylation is defined as the synthesis of glycans (oligosaccharides) and their covalent attachment to proteins, mediated by enzymes, resulting in glycan-prosthetic group conjugated proteins: glycoproteins. These processes occur either co- or post-translationally [4,8].
With the exception of albumin, all plasma proteins are glycosylated. In fact, more than half of all proteins undergo glycosylation [9]. Glycoconjugates play vital roles in the human body, including molecular recognition, conformation-function of glycoproteins, cell migration, resistance to proteases, and antigenicity [10]. Hence, individuals with CDG manifest multisystemic diseases of variable severity.
Glycoproteins are classified based on the type of bond between the oligosaccharide and the involved amino acid residue. The bonds implicated in CDG are primarily of two types:
- Attachment through the nitrogen (N) of the amide group of the asparagine (Asn) amino acid (or less commonly, glutamine), resulting in N-glycoproteins (the mechanism known as N-glycosylation).
- Attachment through the oxygen (O) of the hydroxyl group of the serine (Ser) or threonine (Thr) amino acids, forming O-glycoproteins (the mechanism known as O-glycosylation).
The majority of glycoproteins exhibit the first type of bond, hence most CDG are due to alterations in N-glycosylation [8,11,12].
For the purposes of this work, only the N-glycosylation pathway is required. Therefore, we consider it prudent to simplify the theoretical content of the topic to almost exclusively focus on this pathway. This means that both the O-glycosylation process and other forms of glycosylation, such as lipid glycosylation resulting in glycosylphosphatidylinositol (GPI) anchors and glycosphingolipids [11,13], as well as a type of protein glycosylation resulting from the attachment of the glycan to the carbon 2 of the indole group of a tryptophan residue, known as C-glycosylation [14], are excluded.
II.1.1. N-Glycosylation
The biosynthesis of N-glycoproteins is primarily cotranslational, proceeding through successive steps that take place in the cytosol, endoplasmic reticulum (ER), and Golgi apparatus (GA). It consists of two stages:
- The first stage involves the synthesis of the standard oligosaccharide (or precursor oligosaccharide) and its transfer to the nascent protein.
- The second stage involves the final processing of the oligosaccharide attached to the protein.
The first stage begins on the cytoplasmic side and culminates on the luminal side of the ER membrane. Generally, it occurs through the transfer of monosaccharides onto the lipid carrier dolichol phosphate (Dol-PP) (an isoprenoid lipid) by a specific glycosyltransferase. Monosaccharides are sequentially added to form the standard oligosaccharide consisting of 14 sugar residues assembled as follows: 2 residues of N-acetylglucosamine (NAcGlc), 9 residues of mannose (Man), and 3 residues of glucose (Glc) (formulated as NAcGlc2Man9Glc3), attached to a molecule of Dol-PP [8].
To form NAcGlc2Man9Glc3, both the cytoplasmic and luminal portions of the ER membrane are required. In the cytoplasmic portion, the covalent attachment of the first seven monosaccharides is facilitated by the activated nucleotide sugars uridine diphosphate N-acetylglucosamine (UDP-NAcGlc) and guanosine diphosphate Mannose (GDP-Man), which serve as donor substrates for glycosyltransferases [8]. The resulting heptasaccharide is then composed of NAcGlc2Man5 and is ready to be translocated to the ER lumen through a flip-flop mechanism catalyzed by a flippase [17,18,19]. In the ER lumen, 4 mannose residues and 3 glucose residues are added; dolichol-phosphomannose (Dol-P-Man) and dolichol-phosphoglucose (Dol-P-Glc), respectively, serve as substrates for these reactions. Dol-P-Man is derived from the addition of a mannose residue to a dolichol phosphate (Dol-P) molecule, provided in the extraluminal space by GDP-Man, while dolichol-phosphoglucose results from the addition of a glucose residue to a Dol-P molecule, provided in the extraluminal space by UDP-Glc. In both cases, translocation occurs via a flip-flop mechanism. The substrate specificity of glycosyltransferases ensures a defined assembly route that culminates with the addition of a glucose residue (the last of the three) linked by an α 1-2 glycosidic bond, an important determinant in substrate recognition by oligosaccharyltransferase (OST), the enzyme responsible for transferring the standard oligosaccharide to the nascent glycosylatable protein [8]. It is important to note that sugar donors are nucleotide sugars in the cytosol and monosaccharides attached to a dolichol phosphate molecule in the ER lumen [17,18,19].
Proteins susceptible to glycosylation, typically lysosomal, membrane-bound, or destined for secretion, are characterized by having an N-terminal signal sequence that marks them for cotranslational translocation into the lumen of the ER. This process requires the signal recognition particle (SRP) to recognize and direct the free ribosome to SRP-bound receptors on the cytosolic face of the ER. The growing polypeptide chain is transferred into the ER lumen by a polypeptide translocation complex (translocon) that directly interacts with the ribosome. The signal sequence is removed by a signal peptidase, which hydrolyzes it at its carboxyl side [20]. Subsequently, the protein undergoing synthesis becomes susceptible to the incorporation of the standard oligosaccharide, provided that the process has occurred with the required fidelity.
The OST recognizes a consensus site for glycosylation on the nascent protein, denoted as Asn-X-Ser/Thr, where X is any amino acid except Proline (Pro). It then transfers the oligosaccharide en bloc to the Asn residue, forming a β N-glycosidic bond, leaving Dol-PP free. This glycosylated protein will then proceed to the second stage, meaning it is ready to be processed [21,22].
The second part of the pathway begins after the glycan is transferred. It starts with the removal of the first glucose residue, followed by the cleavage of the remaining 2 glucose residues along with the first mannose residue, in consecutive steps; this process takes place in the ER. Subsequently, the glycoprotein is transported to the GA via transport vesicles, where 5 mannose units are removed. Enzymes responsible for such removals are specific glycosidases. Then, several residues of N-acetylglucosamine (NAcGlc), galactose (Gal), sialic acid (N-acetylneuraminic acid, Neu5Ac), and fucose (Fuc) are added through glycosyltransferases. This occurs progressively from the cis to the trans face of the GA [8,17,18,19].
For each addition, the donors must be transported to the GA by activated nucleotide sugar transporters [23]. The order of addition is controlled by the substrate specificities of the enzymes involved but is not strict because cells exhibit variations in the expression and activity of these enzymes, resulting in a wide spectrum of N-glycan structures [8]. Additionally, some enzymes compete for the same protein-bound glycan at certain points in the pathway. Depending on abundance, affinity, and localization, glycosyltransferases may or may not favor the synthesis of specific glycans [8,17,18]. Essentially, three types of N-glycoproteins are obtained: high-mannose, complex, and hybrid, depending on whether they have a high mannose content, are highly variable in composition, or fall somewhere in between, respectively. Nonetheless, all of them retain a common pentasaccharide core with the formula NAcGlc2Man3 [24].
II.2. CDG Where N-Glycosylation Is Affected. Current Nomenclature
CDG where the N-glycosylation pathway is affected have been divided into two groups for study, depending on which part of the glycosylation process is altered. Thus, Group I include defects in the first stage of the process, while Group II includes those in the final stage. These diseases were formerly designated as CDG, followed by the group to which they belong (I or II) with a hyphen (-) in between, and then a lowercase letter related to the enzymatic defect (a, b…). Currently, the recommended nomenclature begins with the name of the affected gene followed by the acronym CDG, with a hyphen in between. Cases identified where the molecular defect is unknown are designated as CDG-X [25,26].
II.3. Molecular Basis of CDG
The molecular bases of CDG where N-glycosylation is affected are rooted in the glycosylation process itself. Therefore, the molecular bases of CDG-I involve the impairment of some of the glycosyltransferases involved, a decrease in the synthesis of activated sugars, or their availability in the cytoplasmic face and lumen of the ER. Additionally, catalytic impairments of the flippase [27] and OST [21] have been implicated. CDG-II includes defects in vesicular trafficking of N-glycoproteins from the ER to the GA, and within the GA, from the cis to the trans portions. Moreover, there is a decrease in the availability of activated sugars in the GA due to problems in their synthesis or transportation, impairment of glycosidases and glycosyltransferases involved, and the involvement of proteins related to the proper functionality of the GA [8,28].
We consider it necessary to clarify that, henceforth, alterations in the first or second stage of glycosylation will be referred to as CDG types (CDG-I and CDG-II, from the former nomenclature), and specific defects in enzymes, transporters, among others (corresponding to the lowercase letters of the former nomenclature), will be termed CDG subtypes. Furthermore, for convenience, both the old and current nomenclatures will be used interchangeably.
Up to now, 15 subtypes of defects in the N-glycosylation pathway associated with the structure of some glycosyltransferases have been described: ALG6-CDG, ALG3-CDG, ALG12-CDG, ALG8-CDG, ALG2-CDG, DPAGT1-CDG, ALG1-CDG, ALG9-CDG, ALG11-CDG, DDOST-CDG, ALG13-CDG, STT3A-CDG, STT3B-CDG, MGAT2-CDG, and Β4GALT1-CDG. Additionally, 12 subtypes of defects in these diseases are caused by the impairment of the availability and biosynthesis of monosaccharides and their respective sugar-nucleotide diphosphate and sugar-Dol-P donors. These include: PMM2-CDG, MPI-CDG, DPM1-CDG, MPDU1-CDG, DPM3-CDG, PGM1-CDG, DPM2-CDG, GNE-CDG, GFPT1-CDG, GFPT2-CDG, SRD5A3-CDG, and DK1-CDG [29]. Furthermore, 2 subtypes result from deficiencies in enzymes that remove monosaccharide units: MOGS-CDG and MAN1B1-CDG; 4 are the product of dysfunctional monosaccharide transporter proteins: SLC35C1-CDG, SLC35A1-CDG, SLC35A2-CDG, and SLC35A3-CDG, while 1, RFT1-CDG, corresponds to a flippase alteration. Similarly, in 3 CDG subtypes, the structures of proteins involved in vesicular movement of glycoproteins from the ER to the GA are affected: SEC23A-CDG, SEC23B-CDG, and SSR4-CDG.
The products of 8 genes are related to the structure and functionality of the GA. Their structural and functional modifications have been linked to 8 mixed-type glycosylation disorders, where both N- and O-glycosylation are affected: COG7-CDG, COG1-CDG, COG8-CDG, COG4-CDG, COG4-CDG, TMEM165-CDG, COG6-CDG, and ATP6V0A2-CDG [30].
II.4. Diagnosis of Congenital Disorders of N-Glycosylation
Hundreds of different types of proteins susceptible to damage are involved in the glycosylation process, leading to the belief that new subtypes will be diagnosed in the coming years [39]. In fact, new defects in both N- and O-glycosylation pathways are identified each year, and since it is estimated that there are around 500 genes involved in the synthesis or function of glycoproteins, the currently known set may only be “the tip of the iceberg”. This concerns the international scientific community due to the high morbidity and mortality associated with CDG [40].
The organic alterations of CDG can manifest at different stages of life, involving any system. However, the broad clinical spectrum mainly affects the central and peripheral nervous systems, digestive system, musculoskeletal system, hematological system, immune system, and integumentary system. Among the associated symptoms and signs, those most commonly highlighted include psychomotor retardation, failure to thrive, seizures, hypotonia, abnormal fat accumulations, inverted nipples, and various clinical manifestations associated with hepatopathies, enteropathies, and coagulopathies [39,40,41,42,43,44,45].
The diagnosis of CDG is complicated because they involve nearly all clinical specialties, and also due to the limited knowledge of such defects among healthcare providers. Moreover, only a small number of centers screen for CDG due to technical complexities and result analysis. Additionally, CDG are not clinically diagnosable as their symptoms and signs are nonspecific. Furthermore, they mimic some diseases that are better understood, for instance, PMM2-CDG, the subtype with the highest number of confirmed cases, mimics mitochondrial diseases such as MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) and NARP (neuropathy, ataxia, and retinitis pigmentosa) [46,47]. Therefore, it is not surprising that there is a significant underdiagnosis of these conditions.
The positive diagnosis is purely biochemical, and the primary biological marker is serum transferrin (Tf), a glycoprotein responsible for transporting iron (Fe3+) in plasma [7,8,27,39,40].
Tf is synthesized and metabolized primarily in hepatocytes and is present in the blood at high concentrations. For instance, in children under 1 year, the range is from 125 to 270 mg/dl, and in those over 1 year, it is from 200 to 350 mg/dl. It is composed of a polypeptide chain of 679 amino acids with a molecular weight of approximately 80 kDa and an isoelectric point (pI) that can vary between 5.4 and 5.9. Each transferrin molecule consists of two lobes of similar internal and independent structure for the binding of Fe3+; the lobe disposed toward the N-terminal contains residues 1-336, and the other (C-terminal) contains residues 337-679. The glycosidic prosthetic group is found in this latter lobe and is constituted by two complex N-glycan chains linked to residues Asn 413 and Asn 611. These chains vary in their degree of branching, each may present up to 4 antennas [48,49,50]. If the N-glycosylation process involved here proceeds without defects, each antenna will terminate in a Neu5Ac residue (mononegatively charged).
Tf presents various isoforms, each with a pI and a molecular mass that represents it; thus, they can be identified based on these characteristics.
The Tf isoforms arise from:
- Differential composition of the amino acid sequence in the primary structure, resulting from genetic polymorphisms. Among the allelic variants, the most prevalent is Tf C; of these, 16 subtypes have been described, with Tf C1 being present in over 95% of cases and having a pI of 5.4. Variant Tf B has a pI of 5.2 and Tf D, 5.7. This comparison of pI is made considering they have the same Fe3+ content and carbohydrate composition [51,52,53].
- Differential composition of carbohydrate chains (glycoforms). The major isoform, known as tetrasialotransferrin (tetrasialoTf) (pI 5.4), presents two biantennary N-glycan chains, corresponding to four terminations in Neu5Ac residues. However, isoforms can vary from asialotransferrin (asialoTf) to octasialotransferrin (octasialoTf), meaning from no chains to two tetraantennary ones, respectively. Nonetheless, the isoforms following tetrasialoTf in concentration are pentasialotransferrin (pentasialoTf) and trisialotransferrin (trisialoTf). Additionally, very small amounts (less than 2.5%) of isoforms with fewer than three Neu5Ac residues are determined; these isoforms are generally termed carbohydrate deficient transferrin (CDT), corresponding to asialoTf (pI 5.9), monosialotransferrin (monosialoTf) (pI 5.8), and disialotransferrin (disialoTf) (pI 5.7). From a more concrete perspective, the Neu5Ac content can range from 0-8 and determines the microheterogeneity of the transferrin molecule. The variations in the pIs of these isoforms are 0.1 units for each Neu5Ac residue attached [11,40,53].
- Differential Fe3+ content. Each Tf molecule can contain a maximum of two Fe3+ depending on the iron supply to the body. The pI of the Tf molecule decreases by approximately 0.2 units for each Fe3+ bound [53].
In patients with defects in N-glycosylation, as expected, there is an increase in the concentration of CDT and trisialoTf, which forms the theoretical basis for the diagnosis of CDG of the N-glycosylation pathway through isoelectric focusing (IEF) with immunofixation of serum transferrin, provided that the isoforms of genetic polymorphisms and Fe3+ content remain constant [11,40,53].
In polyacrylamide gel IEF, each type of protein can be “focused” into a narrow band based on its pI. Small amounts of the solution containing the protein in question are applied onto a polyacrylamide gel support [11,40,53].
The gel is formed by polymerization of the acrylamide monomer [CH2=CH-C(=O)-NH2], which generates linear chains that are crosslinked together by the crosslinking comonomer N,N’-methylenebisacrylamide (or simply bisacrylamide) [CH2=CH-C(=O)-NH-CH2-NH-C(=O)-CH=CH2]. The concentration of acrylamide used determines the average length of the polymer chain, while the concentration of bisacrylamide determines the degree of crosslinking. Therefore, the proportion in which both are present determines the size of the pore. When the electrokinetic separation aims to exclude the steric effect induced by the protein’s molecular mass, the best resolution is obtained under acrylamide concentrations less than 5% and bisacrylamide concentrations less than 3%, which avoids excessive sieving. Thus, it is carried out in highly porous dispersion media [54].
Polymerization is carried out using redox catalytic systems, employing chemical and/or photochemical catalysts. In the former case, the most commonly used are: ammonium persulfate, (NH4)2S2O8 or APS, as an oxidizing agent, causing the formation of free radicals from the monomer, which are produced by the action of oxygen free radicals (due to the action of persulfate ions), and N,N,N’,N’-tetramethylethylenediamine (TEMED) as a reducing agent, which produces the formation of persulfate free radicals. This reaction is strongly inhibited by high levels of oxygen, so the solution must be degassed to achieve reproducible gel formation. In photochemical catalysis, primarily riboflavin mononucleotide (FMN) and TEMED are used. This polymerization is induced by the free radicals that appear in the chemical decomposition of APS (S2O82- to 2SO4-) or the photodecomposition of riboflavin to leucoflavin in the presence of O2. In either case, TEMED, a free radical stabilizer, is commonly added to the gel mixture. Photopolymerization with riboflavin is not inhibited by oxygen; however, in both cases, TEMED acts as a mild reducing agent and accelerates polymerization [54].
The APS has a high tendency to spontaneously decompose, and undergoes homolytic cleavage, forming sulfate radicals that are unstable and tend to recombine. Although this initiates the polymerization reaction, the addition of TEMED as a propagator is required. TEMED forms stable free radicals in the presence of sulfate radicals, which contributes to the propagation of the polymerization reaction and prevents its extinction. By adjusting the amounts of persulfate and TEMED, the polymerization rate can be controlled. Additionally, polymerization can also be achieved with FMN instead of APS. When riboflavin is illuminated with ultraviolet radiation, its photodecomposition occurs, generating free radicals that initiate polymerization. In this case, TEMED is not essential, but it can be added as it facilitates the process [54].
IEF is considered as a stable gradient electrophoresis of H+ ion concentration, measured as pH. Therefore, macromolecules will migrate through the pH gradient as long as they maintain their net positive or negative charge until they reach the point in the gradient that corresponds to their pI value. At this point, since the net charge is zero, migration ceases. The pH gradient is established through the use of low molecular weight amphoteric substances, called ampholytes, oligomers with a low molecular mass (300-600 D) that carry amino and carboxyl groups with a range of pI values. The gradient involves a decrease in pH from the cathode to the anode. Under the influence of an electric field, in a solution, different classes of ampholytes will separate according to their pI values, so that the most basic ones gradually step towards the cathode while the most acidic one’s step from the anode. These molecules have buffering capacity, so the pH of that zone will be that of the corresponding pI. When equilibrium is reached, a pH gradient is formed along the gel. The pH gradient is generated and maintained by establishing an electric field [54].
The technique of serum Tf IEF is the most commonly used diagnostic test for detecting N-glycosylation defects. In patients with CDG, Tf IEF shows a cathodic shifting profile, indicative of a defect in negative charge(s), namely, in Neu5Ac(s) [7,8,39,40,55].
In individuals with CDG-I, an IEF pattern is observed that suggests a relative deficiency of tetrasialoTf (not fully manifested) with an increase in the isoforms asialoTf, disialoTf, and eventually trisialoTf, referred to as pattern-I. On the other hand, in patients with CDG-II, patterns are presented where the remaining low-sialic acid isoforms are added. The monosialoTf isoform, as can be inferred, is excluded from pattern-I and appears in pattern-II; therefore, it is considered a good differential indicator of patterns [8,55].
Some types of CDG cannot be identified through IEF analysis because in special cases sialylation is not altered, for example: MOGS-CDG, SLC35C1-CDG, in alterations of one of the two GDP-fucose transporters of AG, and in TUSC3-CDG. This, coupled with the laboriousness and time required by the IEF technique, has led to the need for costly techniques such as high-performance liquid chromatography (HPLC), capillary zone electrophoresis (CZE), and mass spectrometry (MS) in the last two decades [56,57,58,59,60,61].
Tf polymorphisms and variations in Fe3+ content can also produce anomalous patterns in IEF. These situations can be respectively ruled out by performing a preincubation of the sample with neuraminidase and saturating it with Fe3+. In cases with polymorphism, it is useful to perform IEF of other glycoproteins, for example: haptoglobin, hexosaminidase, and α1-antitrypsin, to search for a generalized glycosylation defect. Since different polymorphic variants of Tf C1 are infrequent, neuraminidase treatment can be reserved for cases where alterations in IEF bands are observed compared to internationally reported standards in the scientific literature. Saturation should precede all serum Tf IEF for standardization purposes [53,63,64].
It is valid to clarify that there are other conditions in which the IEF analysis of Tf may appear normal, in addition to those previously mentioned. For example, fucose defects cannot be detected since they do not interfere with the addition of Neu5Ac to the glycoprotein. Therefore, in patients suspected of having a fucose defect, blood groups should be determined, as they are all of the Bombay blood group [65].
A normal IEF pattern, in the case of MOGS-CDG, has only been described in one case; however, it has been observed in several adolescent and adult patients with PMM2-CDG, the most frequent subtype. This may be due to the mild-to-moderate severity of these cases, which, combined with a very slight alteration in the Tf profile, may not be detected with conventional procedures. In fact, in CDG, the degree of alteration in the serum Tf profile seems to correlate with the severity of the clinical picture.
Alterations in the Tf IEF profile may also be observed secondary to diseases other than CDG, for example: fructosemia and galactosemia at the time of diagnosis or with poorly controlled disease, as they cause altered glycosylation. Similarly, this altered Tf pattern is observed in chronic alcohol consumption. This implies that these alterations (fructosemia, galactosemia, and chronic alcohol consumption) should be ruled out before confirming a positive diagnosis of CDG through IEF [68,69,70,71,72].
Therefore, for accurate interpretation, it is necessary to use both a positive and a negative control. A serum treated with neuraminidase serves as a positive control, or more precisely, positive cases diagnosed using the combined HPLC-MS method. As a negative control, serum from a healthy individual, confirmed likewise by HPLC-MS, should be used [53,73].
III. Scientific Problem and Our Hypothesis
In Cuba, there are patients who present with multisystemic clinical manifestations compatible with some undiagnosed congenital error of metabolism because all diagnostic tests implemented by the National Center for Medical Genetics (NCMG) have resulted negative. These tests cover a large part of these metabolic defects, such as galactosemia, mucopolysaccharidoses, aminoacidopathies, defects in mitochondrial β-oxidation, lysosomal and peroxisomal diseases, etc.; however, in terms of CDG, they are not useful. In summary, Cuba does not have an algorithm for the diagnosis of CDG.
III.1. Scientific Problem
There is a need to implement a method to diagnose CDG and thereby determine their presence or absence in Cuba.
III.2. Hypothesis
The implementation of serum transferrin isoelectric focusing contributes to the genetic-biochemical diagnosis of CDG in patients with multisystemic clinical manifestations who have been ruled out for other inborn errors of metabolism.
IV. Objectives
IV.1. General Objective
- To propose a method that allows the evaluation of transferrin glycoforms for the diagnosis of congenital defects of N-glycosylation in Cuba.
IV.2. Specific Objectives
- To implement the procedure of serum transferrin glycoform isoelectric focusing for the biochemical diagnosis of congenital defects of N-glycosylation.
- To evaluate the isoelectric focusing patterns of serum transferrin glycoforms in stored patient samples who presented with clinical manifestations suggestive of CDG.
V. Materials and Methods
We conducted a descriptive cross-sectional prevalence study. Our population included patients seen in Clinical Genetics consultations in Cuba, suspected of having undiagnosed inborn errors of metabolism, utilizing standardized methods at the NCMG; whose serum samples were preserved at the institution. Our study sample comprised 17 patients from the population. The patients in our sample met the inclusion criteria and were exempt from exclusion criteria.
V.1. Inclusion and Exclusion Criteria
V.1.1. Inclusion Criteria
Patient serum samples stored at the NCMG, which were documented in the institution's records as having multisystem manifestations and clinical evidence of suffering from a genetically unclassified biochemical disease.
V.1.2. Exclusion Criteria
Samples from patients who had a history of ethyl alcohol consumption were excluded; additionally, samples from patients documented to have basic clinical manifestations of hereditary fructose intolerance were also excluded. (The clinical manifestations considered basic for hereditary fructose intolerance are: hypersomnia, seizures, vomiting, or irritability after consuming foods rich in fructose or sucrose (fruits, commercial beverages, honey), and clinical improvement after removing these foods.)
Additionally, we considered a withdrawal criterion from the study: we considered for removal from the study those serum samples that did not meet the quality parameters for processing in the laboratory according to ISO 15189 Standard and WHO standards L/31, L/32, L/33, and L/34 in the Quality Manual related to pre-analytical procedures.
V.2. Methodological Design
The serum samples were obtained from the Biological Sample Warehouse of the NCMG, where they are coded (without personal patient data). In the warehouse, they were stored at -20°C.
In addition to the previously mentioned samples, control samples were included (one negative control, one positive control for pattern-I (from a CDG-Ia patient), and another positive control for pattern-II (from a CDG-X patient)) tested by HPLC-MS, provided and verified by the Glycobiology Laboratory of the Faculty of Sciences at the Autonomous University of Morelos, Mexico.
The pretreatment of the serum samples involved saturating the serum Tf with iron under alkaline conditions. For this purpose, sodium bicarbonate (NaHCO3) and iron (III) chloride (FeCl3) were added [47,76].
The IEF technique employed required the preparation of polyacrylamide gel as a support, using ampholytes with a pH range of 5-7. For the immunofixation to the Tf gel, a polyclonal antibody against human Tf (anti-Tf Ab) was used. Subsequently, the revelation was carried out by staining with Coomassie blue, which required prior removal of the remaining serum proteins through washing with sodium chloride (NaCl) [77,78,79].
The IEF technique was in accordance with the methods described by van Eijk, et al. in 1983 [80], and additionally, with the procedure described in the instruction manual of the BIORAD model 111 Mini IEF Cell.
V.2.1. Variables
-
Variable: Suspected CDG (Qualitative nominal dichotomous).
- -
- CDG Phenotype: Clinical presentation indicative of suspected CDG, characterized by the exhaustion of the diagnostic resources protocolized at the NCMG, along with one or more of the following alterations: mental retardation, severe delay in growth and development, structural and functional abnormalities of the central and peripheral nervous systems (hypotonia, seizures, etc.), cardiac defects, hormonal imbalances, abnormal fat accumulations, inverted nipples, hepatopathy, enteropathies, and coagulopathies.
- -
- Unknown Phenotype: Phenotypic presentation characterized by any symptom and/or sign unrelated to the CDG Phenotype.
-
Variable: Biochemical Diagnosis of CDG (Qualitative nominal dichotomous).
- -
- Positive: Identification of Pattern I or II in IEF of Tf.
- -
- Negative: Absence of abnormal bands characteristic of hyposialylation patterns in the IEF of Tf, indicating a normal result and the absence of CDG.
V.2.2. Algorithm for Standardizing the Conditions of the Manual Polyacrylamide Gel IEF Method for Serum Transferrin Glycoforms
For the standardization of the IEF method, the following conditions were evaluated:
- Saturation and dilution of serum samples,
- Manual preparation of polyacrylamide gel and sample loading,
- Electrophoretic run-in terms of times and voltages,
- Quality of resolution in terms of pH gradients generated by ampholytes,
- Amount of anti-Tf antibody for immunofixation,
- Washing technique,
- Fixation of content to the gel,
- Staining and destaining.
V.2.3. Methods of Data Processing and Analysis
The result of the IEF technique was qualitatively analyzed by visualizing patterns suggestive of CDG and comparing them with international standards. For this analysis, a negative control and two positive controls (for both patterns) were used, provided and certified by the Glycobiology Laboratory of the Faculty of Sciences at the Autonomous University of Morelos, Mexico, using HPLC-MS.
The obtained IEF patterns underwent validation, repeatability, and reproducibility assays.
For method validation, the aforementioned controls were used and compared with reports from the scientific literature.
To evaluate repeatability, the assay was conducted by mounting duplicate samples, in this case from patients with CDG Phenotype. The procedure was then repeated with the same working conditions, by a single analyst, sequentially, and within a short interval of time between techniques. For the first assay, three gels were prepared, and each pair of samples was mounted on separate gels. The same procedure was followed for the second assay. Due to the time required for the procedure, it was not possible to perform more than two experiments; the time intervals between the first assay and a third one would deviate from the requirements for evaluating repeatability.
Reproducibility was estimated by performing the IEF procedure on patient samples in duplicate, but this time, processed by different analysts, in different laboratories, with a 7-day interval between assays. Three reproducibility tests were conducted, and for each one, gels were prepared as described for repeatability assessment.
The results were archived in the database of the Department of Genetics-Biochemistry of the NCMG.
The analytical procedures used in the research are standardized and documented in the Standard Operating Procedures (known internationally by the acronym SOP) of the Genetics-Biochemistry Laboratory.
V.3. Ethical Aspects
We conducted a purely descriptive, non-experimental research. We utilized stored samples from patients who had previously remained undiagnosed with an inborn error of metabolism at the NCMG, where surplus serum was reserved at the institution. We did not have access to personal patient data; only clinical data associated with a coded sample was accessible. At the NCMG, biological samples stored from patients are consistently utilized for biomedical research purposes. Consequently, both patients and their legal guardians are duly informed, aligning with the principles outlined in the 2014 Declaration of Helsinki by the World Medical Association [81,82,83] (A model of Informed Consent for the treatment of stored biological samples is shown in Appendix A). Laboratory test results were collected in a password-protected database.
VI. Results
The following are the conditions under which the best resolutions in the isoelectric bands of Tf were achieved.
VI.1. Saturation and Dilution of Samples
The following components were placed in a vial:
- 100 μl of serum,
- 5 μl of FeCl3 at 20 mM, and
- 5 μl of NaHCO3.
It was incubated for 1 hour at 25°C, followed by 15 minutes at 4°C (to stop the saturation reaction). Subsequently, the serum was diluted in distilled water at a ratio of 1/10. The sample could be used immediately, although in the case of the present investigation, it was stored at -20°C until further use [80].
To begin the preparation, the working material, which could be cleaned, was cleaned with 70% ethanol. Then, the following reagents were added in order to a test tube:
- 2.75 ml of distilled water,
- 1 ml of 24.25% acrylamide solution / 0.75% bisacrylamide solution,
- 1 ml of 25% glycerol,
- 250 μl of pH 5-7 ampholyte,
- 7.5 μl of 10% APS,
- 25 μl of 0.1% FMN, and
- 2 μl of TEMED.
After gently stirring on a vortex mixer for 30 s, the prepared solution was placed in contact with the support plate, according to the recommendations of the IEF equipment. It was polymerized directly under white light from the visible spectrum for 1 hour, with the light source placed 30 cm away from the plate. Finally, the comb (with 0.5 cm wells) was placed, and 2 μl of each sample was allocated to each well. The sample was allowed to absorb into the gel for 15 minutes, and then the comb was removed [80].
The electrodes were placed according to the recommendations of the IEF equipment. The run proceeded as follows:
- 15 minutes at 100 V (for the migration of the ampholytes),
- 15 minutes at 200 V (to align the sample proteins, including Tf), and
- 60 minutes at 450 V (to separate the transferrins according to their isoelectric points).
At the end of the run, the gel (along with the support) was removed, and immunofixation was performed by applying 100 μl of anti-Tf antibody uniformly. Then, it was incubated at 4 ºC for 30 minutes.
Washing, to select Tf, was performed with 0.9% NaCl and left on a horizontal shaker for 20 hours. Subsequently, a second wash was carried out with distilled water for 10 minutes to remove excess NaCl [80].
To fix the contents to the gel, a solution prepared with 30 ml of pure methanol, 12.5 g of trichloroacetic acid, and 4 g of sulfosalicylic acid was added to the gel, topped up to 100 ml with distilled water. This mixture was kept on a horizontal shaker for 30 minutes.
After fixing, another wash was performed on a horizontal shaker for 2 minutes with 100 ml of a solution called A plus 100 ml of another solution, B.
For staining, 100 ml of solution A plus 100 ml of solution C were added, and moderate agitation was performed on a shaker for 15 minutes. At this point, it was possible to obtain a good visual appreciation of the bands.
|
Solution A was prepared by mixing 1 g of copper sulphate (CuSO4) and 100 ml of acetic acid, and then making up to 500 ml with distilled water. Solution B was prepared by mixing 600 ml of pure methanol with 400 ml of distilled water. Solution C was prepared by mixing 0.6 g of Coomassie blue, 180 ml of pure methanol, to make up to 300 ml with distilled water. |
Decolorization was carried out for 30 minutes using 100 ml of solution A plus 100 ml of solution B.
Finally, the gel was allowed to dry for 20 hours.
There was a correlation between the isoform patterns of serum Tf using the standardized method in the scientific literature [40,55,84,85] and the controls analyzed on the model 111 Mini IEF Cell equipment (Figure 1).
Figure 1.
Validation of the qualitative IEF method by comparing the negative control and the positive controls used in the assay of this study with standardized reports from the scientific literature. The image corresponds to the IEF result obtained from the controls performed at the Laboratory of Glycobiology, Faculty of Sciences, Autonomous University of Morelos, Mexico. The numbers arranged horizontally correspond to the control, pattern-I (CDG-Ia), and pattern-II (CDG-X), respectively, from left to right. The vertically arranged numbers correspond to the bands associated with different forms of Tf, including asialoTf, monosialoTf, disialoTf, trisialoTf, tetrasialoTf, and pentasialoTf.
Figure 1.
Validation of the qualitative IEF method by comparing the negative control and the positive controls used in the assay of this study with standardized reports from the scientific literature. The image corresponds to the IEF result obtained from the controls performed at the Laboratory of Glycobiology, Faculty of Sciences, Autonomous University of Morelos, Mexico. The numbers arranged horizontally correspond to the control, pattern-I (CDG-Ia), and pattern-II (CDG-X), respectively, from left to right. The vertically arranged numbers correspond to the bands associated with different forms of Tf, including asialoTf, monosialoTf, disialoTf, trisialoTf, tetrasialoTf, and pentasialoTf.
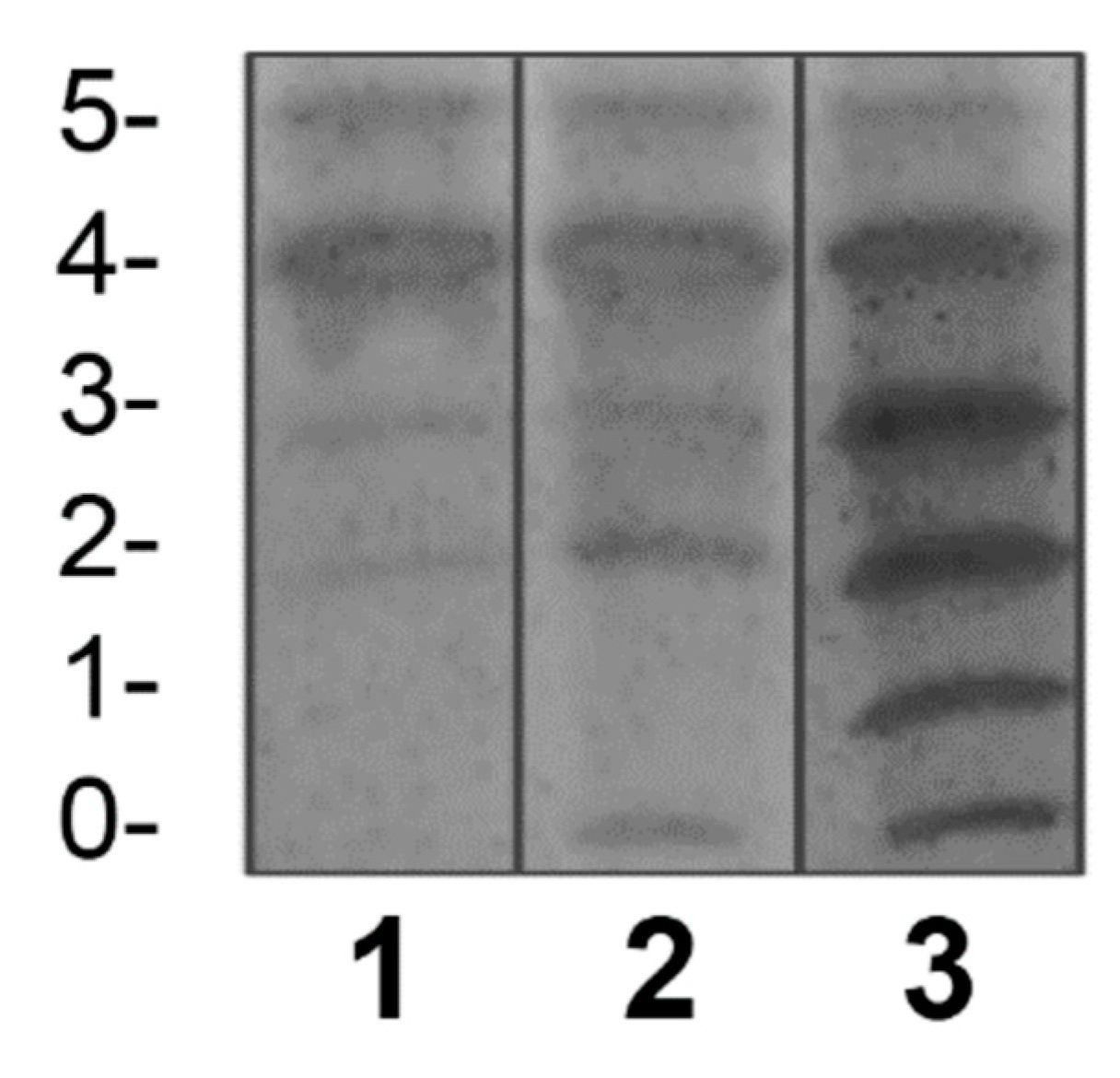
Figure 2.
Validation of the qualitative IEF method by comparing the negative control and the positive controls used in the assay of this study with standardized reports from the scientific literature. The left image is the same as the image in Figure 1 (from the Laboratory of Glycobiology in Mexico). The image on the right corresponds to the result obtained by us using the conditions of the NCMG in Cuba. (The numbers arranged horizontally and vertically are analogous to those in Figure 1.).
Figure 2.
Validation of the qualitative IEF method by comparing the negative control and the positive controls used in the assay of this study with standardized reports from the scientific literature. The left image is the same as the image in Figure 1 (from the Laboratory of Glycobiology in Mexico). The image on the right corresponds to the result obtained by us using the conditions of the NCMG in Cuba. (The numbers arranged horizontally and vertically are analogous to those in Figure 1.).
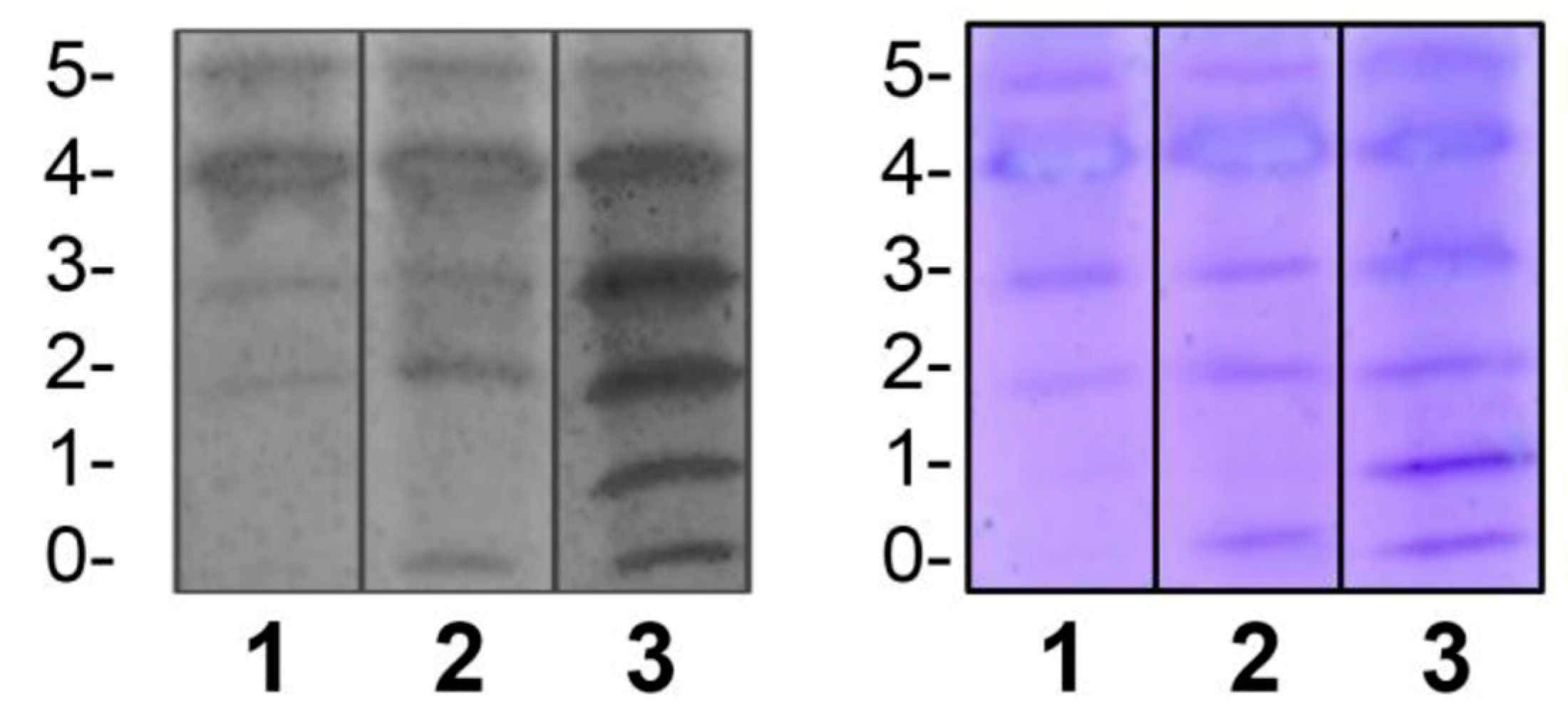
Regarding the assessment of repeatability, we found that intra-assay precision was feasible in terms of standardization (Figure 3).
Inter-laboratory precision, as indicated by the reproducibility of the method, was satisfactory when comparing the results provided by the different analysts involved (Figure 4).
When the analysis of serum Tf glycoforms was performed using IEF on the 17 patients with CDG Phenotype, both types of IEF patterns were observed in some samples. Additionally, negative and doubtful cases were also observed (Figure 5).
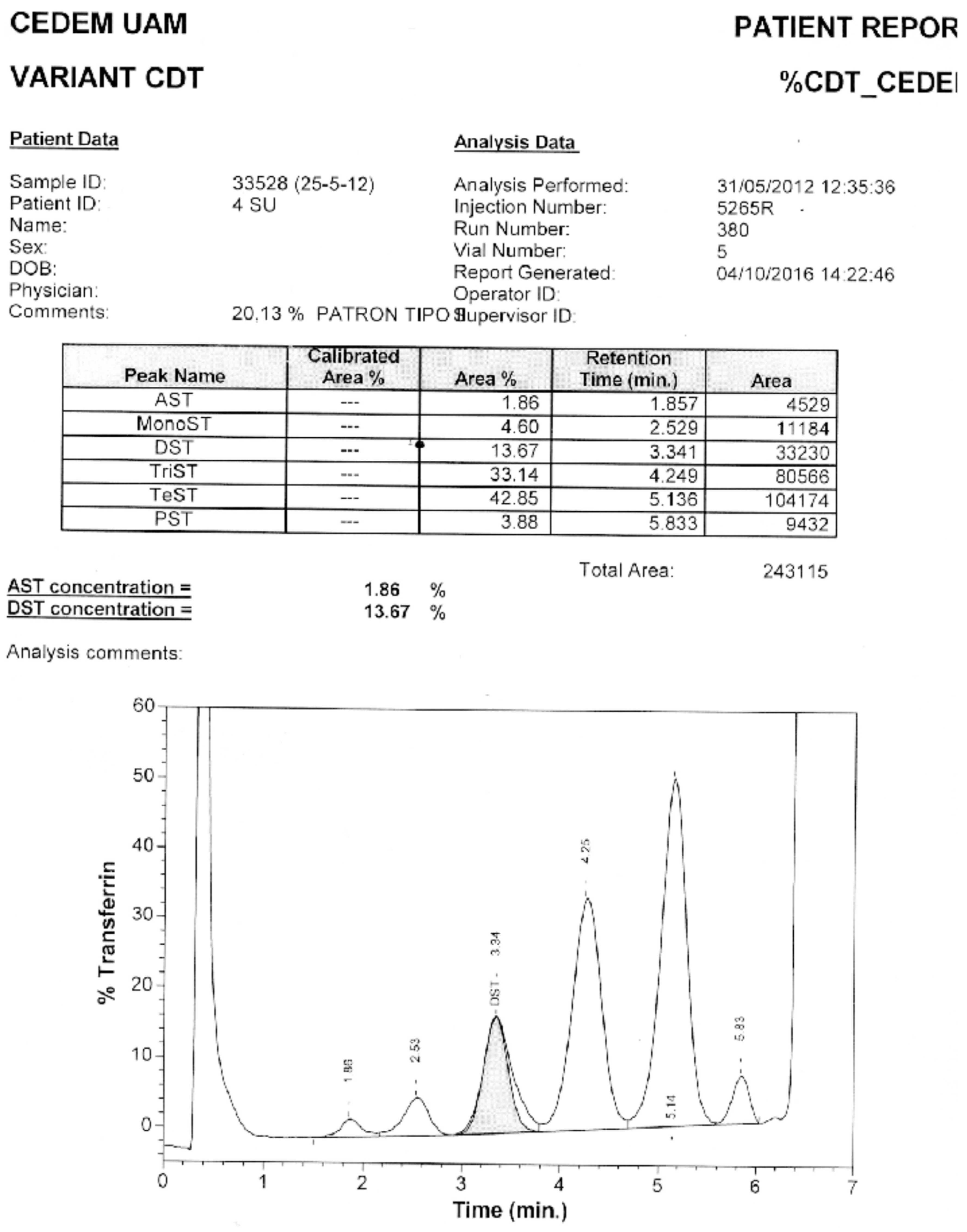
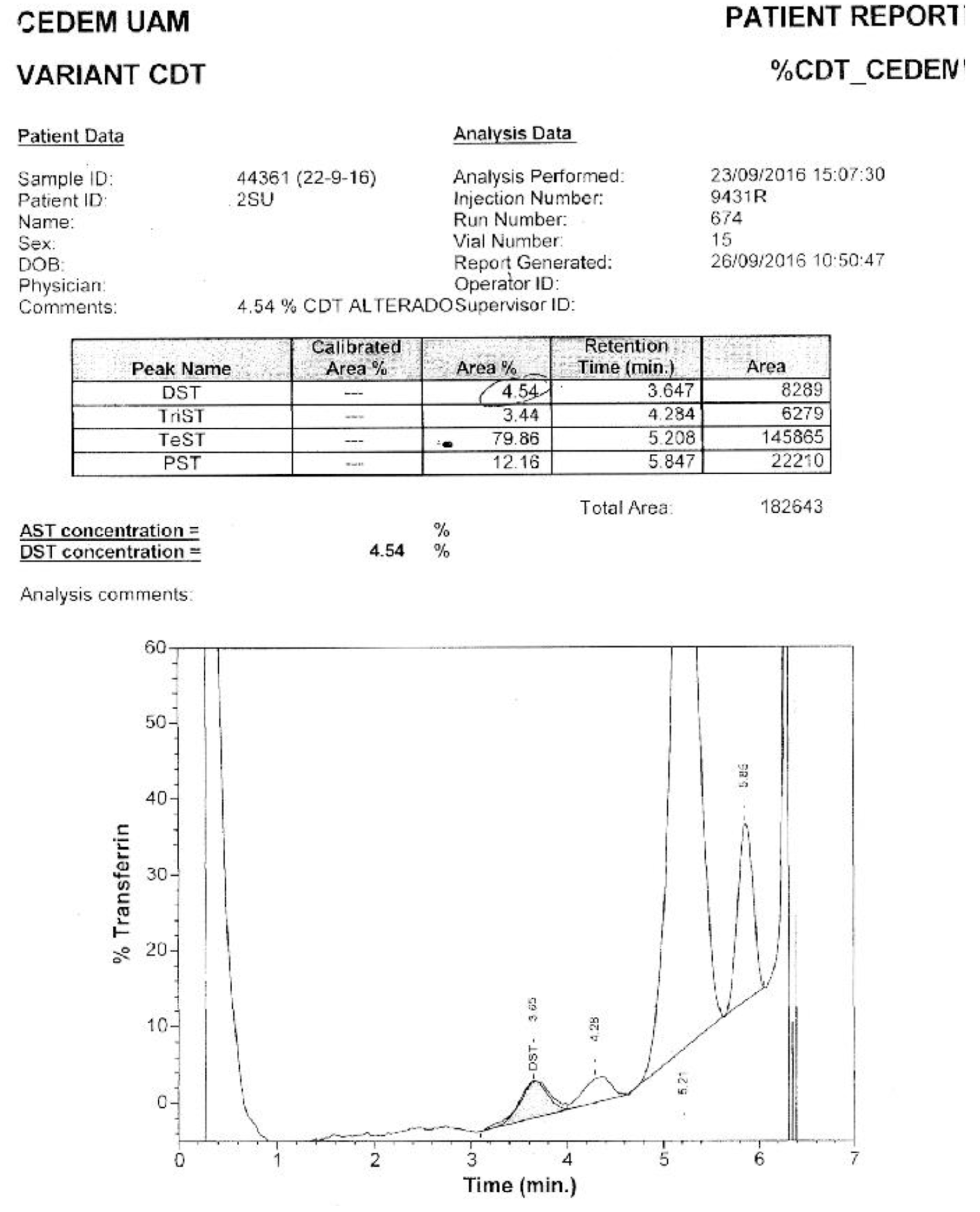
Of the samples with positive patterns suggestive of CDG, those numbered as 6 and 10 (Figure 5) were confirmed by HPLC at the Molecular Disease Diagnosis Center of the Autonomous University of Madrid, Spain (Appendix B and Appendix C). At the aforementioned center, the samples were also processed by HPLC, and samples 9 and 12, which were considered doubtful; sample number 12 turned out to be negative, and sample 9 remained doubtful.
When the analysis of serum Tf glycoforms was performed via IEF on the 17 patients with CDG Phenotype, both types of isoelectric patterns were observed in some samples. Additionally, negative and dubious cases were also observed.
In general, the analysis of isoelectric patterns in the previous assays and the confirmation of some samples at the Center for Molecular Disease Diagnosis of the Autonomous University of Madrid yielded the following results:
- Pattern-I: samples 10, 13, and 17.
- Pattern-II: samples 1, 5, 6, and 14.
- Dubious case: sample 9.
- Possibly negative cases: samples 2, 3, 4, 7, 8, 11, 12 (previously dubious), 15, and 16.
VII. Discussion
In the last three decades, biomedical scientists have collaborated in the identification and characterization of each subtype of CDG. The N-glycosylation pathway is one of the most studied. Congenital defects in N-glycosylation present a wide spectrum of symptoms and signs, including mental retardation, severe growth and developmental delay, hypotonia, seizures, abnormal fat accumulations, inverted nipples, and other symptoms and signs associated with hepatopathies, endocrinopathies, enteropathies, and coagulopathies. This heterogeneity of clinical manifestations makes clinical diagnosis extremely complex, thus biochemical techniques are required for accurate diagnosis.
We conducted the standardization of the IEF method for Tf, which allowed the analysis of stored samples from 17 patients with CDG Phenotype and the positive diagnosis of some of them. This demonstrates the presence of CDG in Cuba. Our goal has been to expand knowledge about these defects and to “take the first step” in the implementation of Tf IEF as a protocol in the arsenal of diagnostic methods for congenital metabolic errors in Cuba.
Various assays were conducted in the standardization process.
Through saturation and dilution tests of the samples, saturation homogeneity in Fe3+ of transferrins was achieved, thereby keeping constant the possible modifications of the pI of each one due to this. The 1/10 dilution proved optimal for proper band resolution.
The tests for the manual preparation of polyacrylamide gel and for sample loading allowed for the preparation of an appropriate gel with the required conditions of porosity, hydration, and pH gradient. Additionally, a convenient separation between intra-sample and inter-sample bands was achieved. Through electrophoretic run assays, precise voltages and times for the migration of ampholytes, alignment of sample proteins, and subsequent separation based on their isoelectric points were determined.
In experiments for immunofixation, the quantity of anti-Tf antibodies necessary for selecting Tf within the set of sample proteins was established. The incubation time required for sufficient antigen-antibody complexes to develop, which would select Tf, was also obtained. Purging of the remaining proteins was best achieved with a 20-hour wash time (with NaCl). Furthermore, the time to remove excess NaCl resulting from the mentioned wash was normalized. Regarding the fixation of content to the gel, staining, and destaining of the sample, sufficient quantities were determined to obtain the best possible visualization of the bands relative to their surroundings.
For the validation of the IEF method for Tf on manually prepared polyacrylamide gels, the technique described by van Eijk et al. in 1983 [80] was used. This technique, employed to detect the microheterogeneity of serum Tf, was applied to different systems (immobilized pH gradients and PhastSystem) by the same author (van Eijk) in collaboration with de Jong in 1988 [55] and van Noort in 1992 [86]. In all cases, it was concluded that the resolution achieved by IEF with manually prepared gels is sufficient and efficient for the analysis of Tf glycoforms [86]. Therefore, the experimental use of IEF for the evaluation of serum Tf glycoforms in the present study is justified. To validate the technique and its interpretation, the standardized isoelectric patterns given by Pérez- Cerdá et al. in 2006 [40], de Jong and van Eijk in 1988 [55], Lefeber et al. in 2011 [84], and Denecke in 2009 [85] with controls confirmed by HPLC-MS. The bands, characteristic of each Tf glycoform, corresponded to international standards; hence, the validation of the IEF method for serum Tf on manually prepared gels was assumed.
Repeatability, assessed by immediate replication of the technique under constant working conditions, proved feasible, meaning there was no variation in results when measurements were taken iteratively.
Reproducibility, evaluated under variable working conditions, analysts, and days, was equally satisfactory in the tests conducted, meaning measurable results were obtained. Thus, the technique employed in this study can be reproduced by other analysts and/or in other laboratories.
Once the technique was standardized, we evaluated serum samples from patients with CDG phenotype to detect whether they indeed had N-glycosylation defects. The finding that 7 of the cases exhibited characteristic isoelectric patterns confirmed the suspicion.
However, it is known that the most frequent defect is PMM-CDG (CDG-Ia), resulting in a pattern-I [40,67]. Nevertheless, in this study, 4 samples with pattern-II were found. This does not contradict the literature; determining the frequency of such defects in Cuba would require a different type of investigation with strict statistical analysis, which is beyond the scope of this study.
In the Orphanet Report Series in 2016, the most prevalent subtypes of CDG-I were found to be, after PMM-CDG (which reports hundreds of cases and continues to increase): ALG6-CDG or CDG-Ic (58 cases), MPI-CDG or CDG-Ib (20 cases), ALG1-CDG or CDG-Ik (15 cases), DPM1-CDG or CDGIe (14 cases), and ALG12-CDG or CDG-Ig (11 cases), just to mention a few of the most prevalent ones [2]. At the same site, regarding CDG-II subtypes, the most prevalent ones were published as: COG5-CDG or CDG-IIi (9 cases), COG7-CDG or CDG-IIe (9 cases), MAN1B1-CDG (7 cases), TMEM165-CDG or CDG-IIk (5 cases), SLC35A2-CDG or CDG-IIm (4 cases), and MGAT2-CDG or CDGIIa (4 cases); the remaining subtypes have varying numbers of cases reported, ranging from 3 to 1 [2]. Therefore, it is probable that the patients studied in this research belong to some of the aforementioned subtypes. However, we do not intend to classify these patients into subtypes as this is not justified in this study. Deschamps et al. in 2003, mention that for practical purposes, it is important to conduct an initial study to identify protein hypoglycosylation for the initial biochemical diagnosis, which justifies the conduct of more complex molecular studies [53].
Grunewald, et al., 2002 propose that experience with serum Tf IEF has demonstrated that Tf is the biomarker par excellence for CDG, and furthermore, a good differential indicator between CDG-I and CDG-II [87]. Specifically, the pattern-I found refers to various possibilities of alterations in the first stage of the N-glycosylation process. These include defects in glycosyltransferases, decreased synthesis of activated sugars, or their availability in the cytoplasmic face and lumen of the ER, as well as affecting flipase [27] and OST [21]. However, the pattern-II found may be due to defects in vesicular trafficking of N-glycoproteins from the ER to the GA, and within the GA, from the cis to the trans portions; decreased availability of activated sugars in the GA due to problems in their synthesis or transportation; involvement of glycosidases and glycosyltransferases participating in this stage, and involvement of proteins related to the functionality of the GA [8,28].
In total, 7 cases with characteristic isoelectric patterns pointing towards a positive diagnosis of a congenital N-glycosylation defect were found. Among them, 3 corresponded to the involvement of the first stage of the process (pattern-I) and 4 to the second stage.
The Molecular Disease Diagnosis Center of the Autonomous University of Madrid supported this research by confirming samples 6 and 10 using HPLC, which turned out to be CDG-II and CDG-I, respectively. Additionally, it allowed for the confirmation of negativity of a doubtful sample, number 12.
The IEF method applied to samples from patients with CDG Phenotype yielded diverse band patterns. Thus, through the particularized analysis of these bands in each sample, each case was discussed as follows:
- Sample 1, with pattern-II (bands of asialoTf, monosialoTf, disialoTf, and trisialoTf, homogeneous and with intensity related to control II): patient with a defect in the second stage of N-glycosylation.
- Sample 2, negative for patterns suggestive of N-glycosylation defect (No bands were observed with intensity comparable to positive controls, suggestive of marked hypoglycosylation. These bands are: asialoTf and monosialoTf): presumably, patient negative for N-glycosylation defect.
- Sample 3, negative for patterns suggestive of N-glycosylation defect: presumably, patient negative for N-glycosylation defect.
- Sample 4, negative for patterns suggestive of N-glycosylation defect: presumably, patient negative for N-glycosylation defect. Possible Tf polymorphism was observed (indicated by potential variation in the pI of glycoforms due to allelic variants).
- Sample 5, with pattern-II: patient with a defect in the second stage of N-glycosylation.
- Sample 6, with pattern-II: patient with a defect in the second stage of N-glycosylation and possible Tf polymorphism.
- Sample 7, negative for patterns suggestive of N-glycosylation defect: presumably, patient negative for N-glycosylation defect.
- Sample 8, negative for patterns suggestive of N-glycosylation defect: presumably, patient negative for N-glycosylation defect.
- Sample 9, doubtful pattern: remains doubtful because although bands of marked hypoglycosylation were visualized, they were not homogeneous in intensity (asialoTf and monosialoTf bands were less intense than the others), and HPLC analysis was inconclusive as well.
- Sample 10, pattern-I (asialoTf band evident and monosialoTf band slightly visible; in intensities comparable to control I): patient with a defect in the first stage of N-glycosylation.
- Sample 11, negative for patterns suggestive of N-glycosylation defect: presumably, patient negative for N-glycosylation defect. Possible Tf polymorphism was observed.
- Sample 12, previously doubtful for the same reasons as case 9, its negativity regarding patterns was confirmed: presumably, patient negative for N-glycosylation defect.
- Sample 13, pattern-I: patient with a defect in the first stage of N-glycosylation.
- Sample 14, with pattern-II: patient with a defect in the second stage of N-glycosylation.
- Sample 15, negative for patterns suggestive of N-glycosylation defect: presumably, patient negative for N-glycosylation defect. Possible Tf polymorphism was observed.
- Sample 16, negative for patterns suggestive of N-glycosylation defect: presumably, patient negative for N-glycosylation defect.
- Sample 17, pattern-I: patient with a defect in the first stage of N-glycosylation.
VIII. Conclusions
We have developed a process to standardize a manually prepared gel isoelectric focusing method aimed at evaluating serum transferrin glycoforms in stored samples from patients suspected of having a congenital glycosylation disorder. This approach is not only technically and methodologically rigorous but also holds profound significance in the context of public health.
Through this method, we have successfully confirmed, for the first time in Cuba, the presence of transferrin patterns I and II in the analyzed samples. This finding is pivotal as it suggests transferrin hypoglycosylation, indicative of potential defects in the N-glycosylation process. The identification of these biochemical abnormalities represents a significant milestone in Cuban medical research, shedding light on the nature and prevalence of congenital metabolic disorders in our population.
The early and accurate detection of N-glycosylation defects can have profound implications for the diagnosis, treatment, and management of affected patients. This research signifies a significant step forward in understanding and addressing congenital metabolic disorders in Cuba, with the potential to enhance medical care and improve the quality of life for our patients.
Acknowledgments
Acknowledgments to Tatiana Acosta Sánchez (Head of the Department of Genetics-Biochemistry, National Center of Medical Genetics of Cuba), who served as Alejandro Bermejo’s mentor during his specialization in Clinical Biochemistry and was the scientific supervisor of his thesis for this specialization, precisely in this aspect. Much of this study owes its guidance, dedication, knowledge, and advice to her invaluable mentorship.
Appendix A. Informed Consent
|
Informed Consent I, ________________________________________[Patient’s Name/Legal Guardian], voluntarily consent to the treatment of stored biological samples at the National Center for Medical Genetics for the detection of congenital metabolic disorders. I acknowledge that these samples will be used solely for diagnostic and screening purposes, and that all information will be treated confidentially. I agree that the samples may be used for anonymous scientific research and I have the right to withdraw this consent at any time. I understand and grant this consent willingly. |
| Signature |
| _____________________________________ |
Appendix B. Confirmation through HPLC. Sample 6. Confirmation of Pattern-II.
Appendix C. Confirmation via HPLC. Sample 10. Confirmation of Pattern-I.
References
- Jaeken, J.; Vanderschueren-Lodewyckx, M.; Caeaer, P.; Snoeck, L.; Corbeel, L.; Eggermont, E. Familial psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG deficiency, increased serum arylsulphatase A and increased CSF protein: a new syndrome? Pediatr Res. 1980, 14, 179. https://www.semanticscholar.org/paper/Familial-psychomotor-retardation-with-markedly-FSH-Jaeken-Vanderschueren%E2%80%90Lodeweyckx/080c31d7b117a869c7c545a8ef077ee7f268e25b. [CrossRef]
- Orphanet Report Series [database on the Internet]. Prevalence of rare diseases: Bibliographic data. Number 1. Available from: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf.
- Freeze, H.H.; Chong, J.X.; Bamshad, M.J.; Ng, B.G. Solving Glycosylation Disorders: Fundamental Approaches Reveal Complicated Pathways. Am J Hum Genet [serial on the Internet]. 2014, 94, 161–175. Available from: Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/. [CrossRef] [PubMed]
- Jaeken, J. Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! J Inherit Metab Dis. 2011, 34, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Freeze, H.H. Understanding human glycosylation disorders: biochemistry leads the charge. J Biol Chem. 2013, 288, 6936–6945. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.; Gadomski, T.; Kozicz, T.; Morava, E. Congenital disorders of glycosylation: new defects and still counting. J Inherit Metab Dis. 2014, 37, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Higuera, N.; Vázquez, S.; Palencia, R. Diagnóstico y tratamiento de los trastornos congénitos de la glicosilación de las proteínas. Bol Pediatr. 2011, 51, 188–193. [Google Scholar]
- Freeze, H.H.; Aebi, M. Altered glycan structures: the molecular basis of congenital disorders of glycosylation. Curr Opin Struct Biol. 2005, 15, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999, 1473, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Yarema, K.J.; Bertozzi, C.R. Characterizing glycosylation pathways. Genome Biol. 2001, 2, 0004.1-.10. https://genomebiology.biomedcentral.com/articles/10.1186/gb-2001-2-5-reviews0004. [CrossRef] [PubMed]
- Supraha, S.; Dabelic, S.; Dumic, J. Insights into complexity of congenital disorders of glycosylation. Biochemia Medica. 2012, 22, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J. Congenital disorders of glycosylation. Ann NY ACAD Sci. 2010, 1214, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Kinoshita, T. GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim Biophys Acta. 2012, 1821, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Furmanek, A.; Hofsteenge, J. Protein C-mannosylation: Facts and questions. ABP. 2000, 47, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Burda, P.; Aebi, M. The dolichol pathway of N-linked glycosylation. Biochim Biophys Acta. 1999, 1426, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Spiro, R.G. Glucose residues as key determinants in the biosynthesis and quality control of glycoproteins with N-linked oligosaccharides. J Biol Chem. 2000, 275, 35657–35660. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, T.; Denecke, J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr. 2003, 162, 359–379. [Google Scholar] [CrossRef]
- Mills, P.; Mills, K.; Clayton, P.; Johnson, A.; Whitehouse, D.; Winchester, B. Congenital disorders of glycosylation type I leads to altered processing of N-linked glycans, as well as under glycosylation. Biochem J. 2001, 359, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef]
- Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry, 5th ed.; W.H. Freeman and company New York: New York, 2011; https://search.worldcat.org/es/title/Lehninger-principles-of-biochemistry/oclc/191854286.
- Mohorko, E.; Glockshuber, R.; Aebi, M. Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. J Inherit Metab Dis. 2011, 34, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Gavel, Y.; von Heijne, G. Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 1990, 3, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Martínez, D.I.; Mollicone, R.; Codogno, P.; Oriol, R. The nucleotide sugar transporter family: a phylogenetic approach. Biochimie. 2003, 85, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, R.; Kornfeld, S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985, 54, 631–664. [Google Scholar] [CrossRef] [PubMed]
- Jaeken J, Hennet T, Matthijs G, Freeze HH. CDG nomenclature: time for a change! Biochim Biophys Acta. 2009, 1792, 825–826. [PubMed]
- Jaeken, J.; Hennet, T.; Freeze, H.H.; Matthijs, G. On the nomenclature of congenital disorders of glycosylation (CDG). J Inherit Metab Dis. 2008, 31, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Cylwik, B.; Naklicki, M.; Chrostek, L.; Gruszewska, E. Congenital disorders of glycosylation. Part I. Defects of protein N-glycosylation. ABP. 2013, 60, 151–161. [Google Scholar] [PubMed]
- Freeze, H.H. Genetic defects in the human glycome. Nature Rev Genet. 2006, 7, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Cantagrel, V.; Lefeber, D.J. From glycosylation disorders to dolichol biosynthesis defects: a new class of metabolic diseases. J Inherit Metab Dis. 2011, 34, 859–867. [Google Scholar] [CrossRef] [PubMed]
- OMIM Online Mendelian Inheritance in Man [database on the Internet]. An Online Catalog of Human Genes and Genetic Disorders. Available from: http://www.omim.org/.
- Oka, T.; Krieger, M. Multi-component protein complexes and Golgi membrane trafficking. J Biochem (Tokyo). 2005, 137, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Whyte, J.R.; Munro, S. The Sec34/35 Golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev Cell. 2001, 1, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Fotso, P.; Koryakina, Y.; Pavliv, O.; Tsiomenko, A.; Lupashin, V. Cog1p plays a central role in the organization of the yeast conserved oligomeric Golgi (COG) complex. J Biol Chem. 2005, 280, 27613–27623. [Google Scholar] [CrossRef] [PubMed]
- Ungar, D.; Oka, T.; Brittle, E.E.; Vasile, E.; Lupashin, V.V.; Chatterton, J.E.; et al. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J Cell Biol. 2002, 157, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, F.; Amyere, M.; Jaeken, J.; Zeevaert, R.; Schollen, E.; Race, V.; et al. TMEM165 deficiency causes a congenital disorder of glycosylation. Am J Hum Genet. 2012, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Brenner, V.; Nyakatura, G.; Rosenthal, A.; Platzer, M. Genomic organization of two novel genes on human Xq28: compact head to head arrangement of IDH-gamma and TRAP-delta is conserved in rat and mouse. Genomics. 1997, 44, 8–14. https://genome.fli-leibniz.de/publications/download/free/Brenner_1997.pdf. [CrossRef] [PubMed]
- Smith, A.N.; Lovering, R.C.; Futai, M.; Takeda, J.; Brown, D.; Karet, F.E. Revised nomenclature for mammalian vacuolar-type H(+)-ATPase subunit genes. Molec Cell. 2003, 12, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.; Dimopoulou, A.; Egerer, J.; Gardeitchik, T.; Kidd, A.; Jost, D.; et al. Further characterization of ATP6V0A2-related autosomal recessive cutis laxa. Hum Genet. 2012, 131, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- Martínez, D.I.; Palomares, A.L.; Sánchez, F.D.; Mollicone, R.; Ibarra, G.I. Trastornos congénitos de la glicosilación: abordaje clínico y de laboratorio. Acta Pediatr Mex. 2008, 29, 78–88. [Google Scholar]
- Pérez-Cerdá, C.; Ugarte, M. Congenital disorders of glycosylation. Their diagnosis and treatment. Rev Neurol. 2006, 43, 145–156. [Google Scholar]
- Funke, S.; Gardeitchik, T.; Kouwenberg, D.; Mohamed, M.; Wortmann, S.B.; Korsch, E.; Adamowicz, M.; Al-Gazali, L.; Wevers, R.A.; Horvath, A.; Lefeber, D.J.; Morava, E. Perinatal and early infantile symptoms in congenital disorders of glycosylation. Am J Med Genet A. 2013, 161, 578–584, Epub 2013 Feb 7. Erratum in: Am J Med Genet A. 2014 Jun;164A(6):1618. [Google Scholar] [CrossRef] [PubMed]
- Kouwenberg, D.; Gardeitchik, T.; Mohamed, M.; Lefeber, D.; Morava, E. Wrinkled skin and fat pads in patients with ALG8-CDG: Revisiting skin manifestations in congenital disorders of glycosylation. Pediatr Dermatol. 2014, 31, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wolthuis, D.F.G.J.; Janssen, M.C.; Cassiman, D.; Lefeber, D.J.; Morava-Kozicz, E. Defining the phenotype and diagnostic considerations in adults with congenital disorders of N-linked glycosylation. Expert Rev Mol Diagn. 2014, 14, 217–224. [Google Scholar] [CrossRef]
- Tegtmeyer, L.C.; Rust, S.; van Scherpenzeel, M.; Ng, B.C.; Losfeld, M.E.; Timal, S.; et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. New Engl J Med. 2014, 370, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Freeze, H.H.; Eklund, E.A.; Ng, B.G.; Patterson, M.C. Neurological aspects of human glycosylation disorders. Ann Rev Neurosci. 2015, 38, 105–125. [Google Scholar] [CrossRef]
- Briones, P.; Vilaseca, M.A.; García-Silva, M.T.; Pineda, M.; Colomer, J.; Ferrer, I.; et al. Congenital disorders of glycosylation (CDG) may be underdiagnosed when mimicking mitochondrial disease. Eur J Paediatr Neurol. 2001, 5, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Vilaseca, M.A.; Artuch, R.; Briones, P. Defectos congénitos de la glucosilación: últimos avances y experiencia española. Med Clin (Barc). 2004, 122, 707–716. https://dialnet.unirioja.es/servlet/articulo?codigo=876578. [CrossRef]
- Baker, E.N. Structure and reactivity of transferrins. Adv Inorg Chem. 1994, 41, 389–463. https://www.semanticscholar.org/paper/Structure-and-Reactivity-of-Transferrins-Baker/760f0c009919650618da410e4a04d71ccad067bd.
- Jeffrey, P.D.; Bewley, M.C.; MacGillivray, R.T.; Mason, A.B.; Wood-worth, R.C.; Baker, E.N. Ligand-induced conformational change in transferrins: crystal structure of the open form of the N-terminal half-molecule of human transferrin. Biochemistry. 1998, 37, 13978–13986. [Google Scholar] [CrossRef] [PubMed]
- Ching-Ming Chung, M. Structure and function of transferrin. Biochemical education. 1984, 12, 146–154. [Google Scholar] [CrossRef]
- Fujita, M.; Satoh, C.; Asakawa, J.; Nagahata, Y.; Tanaka, Y.; Hazama, R.; et al. Electrophoretic variants of blood proteins in Japanese, VI. Transferrin. Jinrui Idengaku Zasshi. 1985, 30, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Kamboh, M.I.; Ferrell, R.E. Human transferrin polymorphism. Hum Hered. 1987, 37, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, E.M.; Miña, A.; Ciegues, M.A. Isoformas de la transferrina: Utilidad clínica de su determinación. Rev Digan Biol [artículo en Internet]. 2003 Mar [consulta: 2016 Oct];52(1). Disponible en: Http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S003479732003000100005&lng=es&nrm=iso&tlng=es.
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; John Wiley & Sons, Inc: United States of America, 2011; https://www.wiley.com/en-us/Biochemistry,+4th+Edition-p-9780470570951.
- de Jong, G.; van Eijk, H.G. Microheterogeneity of human serum transferrin: a biological phenomenon studied by isoelectric focusing in immobilized pH gradients. Electrophoresis. 1988, 9, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Helander, A.; Eriksson, G.; Stibler, H.; Jeppsson, J.O. Interference of transferrin isoform types with carbohydrate-deficient transferrin quantification in the identification of alcohol abuse. Clin Chem. 2001, 47, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Carchon, H.A.; Chevigne, R.; Falmagne, J.B.; Jaeken, J. Diagnosis of congenital disorders of glycosylation by capillary zone electrophoresis of serum transferrin. Clin Chem. 2004, 50, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Quintana, E.; Montero, R.; Casado, M.; Navarro-Sastre, A.; Vilaseca, M.A.; Briones, P.; et al. Comparison between high performance liquid chromatography and capillary zone electrophoresis for the diagnosis of congenital disorders of glycosylation. J Chromatogr B Analyt Technol Biomed Life Sci. 2009, 877, 2513–2518. [Google Scholar] [CrossRef] [PubMed]
- Sturiale, L.; Barone, R.; Garozzo, D. The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J Inherit Metab Dis. 2011, 34, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Sturiale, L.; Garozzo, D. Mass spectrometry in the characterization of human genetic N-glycosylation defects. Mass Spectrom Rev. 2009, 28, 517–542. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Nishikawa, A.; Okamoto, N.; Inui, K.; Tsukamoto, H.; Okada, S.; et al. Structure of serum transferrin in carbohydrate-deficient glycoprotein syndrome. Biochem Biophys Res Commun. 1992, 189, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Knopf, C.; Rod, R.; Jaeken, J.; Berant, M.; van Schaftingen, E.; Fryns, J.P.; et al. Transferrin protein variant mimicking carbohydrate-deficient glycoprotein syndrome in trisomy 7 mosaicism. J Inherit Metab Dis. 2000, 23, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Artuch, R.; Ferrer, I.; Pineda, J.; Moreno, J.; Busquets, C.; Briones, P.; et al. Western blotting with diaminobenzidine detection for the diagnosis of congenital disorders of glycosylation. J Neurosci Methods. 2003, 125, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Peters, V.; Korner, C.; Hoffmann, G.F. Improvement of CDG diagnosis by combined examination of several glycoproteins. J Inherit Metab Dis. 2004, 27, 581–590. [Google Scholar] [CrossRef]
- Körner, C.; Linnebank, M.; Koch, H.G.; Harms, E.; Von Figura, K.; Marquardt, T. Decreased availability of GDP-L-fucose in a patient with LAD II with normal GDP-D-mannose dehydratase and FX protein activities. J Leukoc Biol. 1999, 66, 95–98. [Google Scholar] [CrossRef] [PubMed]
- De Praeter, C.M.; Gerwig, G.J.; Bause, E.; Nuytinck, L.K.; Vliegenthart, J.F.; Breuer, W.; et al. A novel disorder caused by defective biosynthesis of Nlinked oligosaccharides due to glucosidase I deficiency. Am J Hum Genet. 2000, 66, 1744–1756. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, M.; Matthijs, G.; Jaeken, J.; van Schaftingen, E.; Nelson, P.V. Carbohydrate- deficient glycoprotein syndrome: beyond the screen. J Inherit Metab Dis. 2000, 23, 396–398. [Google Scholar] [CrossRef]
- Stibler, H.; Von Dobeln, U.; Kristiansson, B.; Guthenberg, C. Carbohydratedeficient transferrin in galactosaemia. Acta Paediatr. 1997, 86, 1377–1378. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Pirard, M.; Adamowicz, M.; Pronicka, E.; van Schaftingen, E. Inhibition of phosphomannose isomerase by fructose 1-phosphate: an explanation for defective N-glycosylation in hereditary fructose intolerance. Pediatr Res. 1996, 40, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Adamowicz, M.; Pronicka, E. Carbohydrate deficient glycoprotein syndrome-like transferrin isoelectric focusing pattern in untreated fructosaemia. Eur J Pediatr. 1996, 155, 347–348. [Google Scholar] [PubMed]
- Charlwood, J.; Clayton, P.; Keir, G.; Mian, N.; Winchester, B. Defective galactosylation of serum transferrin in galactosemia. Glycobiology. 1998, 8, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Landberg, E.; Pahlsson, P.; Lundblad, A.; Arnetorp, A.; Jeppsson, J.O. Carbohydrate composition of serum transferrin isoforms from patients with high alcohol consumption. Biochem Biophys Res Comm. 1995, 210, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Ideo, H.; Ohkura, T.; Fukushima, K.; Yuasa, I.; Ohno, K.; et al. Sugar chains of serum. J Biol Chem. 1993, 268, 5783–5789. [Google Scholar] [CrossRef] [PubMed]
- Norma ISO 15189:2007. Laboratorios clínicos. Requisitos particulares relativos a la calidad y competencia. https://www.une.org/encuentra-tu-norma/busca-tu-norma/norma?
- Quality Management Systems in the Medical Laboratory, D.M. Quality Management Systems in the Medical Laboratory, D.M. Browning. Mar 2004. [PubMed]
- Briones, P.; Vilaseca, M.A.; Schollen, E.; Ferrer, I.; Maties, M.; Busquets, C.; et al. Biochemical and molecular studies in 26 Spanish patients with congenital disorder of glycosylation type Ia. J Inherit Metab Dis. 2002, 25, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Andrews, A.T. Electrophoresis: theory, techniques, and biochemical and clinical applications, 2th ed.; Oxford University Press: Oxford, 1986; https://books.google.es/books/about/Electrophoresis.html?id=eZMRAQAAIAAJ&redir_esc=y.
- Westermeier, R. Electrophoresis in practice: a guide to methods and applications of DNA and protein separations, 2th ed.; VCH Press: Weinheim, 1997; https://beckassets.blob.core.windows.net/product/readingsample/16142615/3527338802_c01.pdf.
- Righetti, P.G. Isoelectric focusing: theory, methodology, and applications; Elsevier: Amsterdam, 1983; https://shop.elsevier.com/books/isoelectric-focusing-theory-methodology-and-application/righetti/978-0-444-80498-3.
- van Eijk, H.G.; van Noort, W.L.; Dubelaar, M.L.; van der Heul, C. The microheterogeneity of human transferrins in biological fluids. Clin Chim Acta. 1983, 132, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Cantín, M. Declaración de Helsinki de la Asociación Médica Mundial: Principios éticos para las investigaciones médicas en seres humanos. Revisando su última versión. Int J Med Surg Sci. 2014, 1, 339–346. https://www.wma.net/es/policies-post/declaracion-de-helsinki-de-la-amm-principios-eticos-para-las-investigaciones-medicas-en-seres-humanos/. [CrossRef]
- Estrella, L.; Castañeda, C.; Sánchez, J.; Zaharia, M. New version of the Declaration of Helsinki: shortcomings to resolve. Rev Peru Med Exp Salud Publica. 2014, 31, 803–804. [Google Scholar] [PubMed]
- World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013, 310, 2191–2194. https://jamanetwork.com/journals/jama/fullarticle/1760318.
- Lefeber, D.J.; Morava, E.; Jaeken, J. How to find and diagnose a CDG due to defective N-glycosylation. J Inherit Metab Dis. 2011, 34, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Denecke, J. Biomarkers and diagnosis of congenital disorders of glycosilation. Expert Opin Med Diagn. 2009, 3, 395–409. [Google Scholar] [CrossRef] [PubMed]
- van Eijk, H.G.; van Noort, W.L. The analysis of human serum transferrins with the PhastSystem: Quantitation of microheterogeneity. Electrophoresis 1992, 13, 354–358. [Google Scholar]
- Grunewald, S.; Matthijs, G.; Jaeken, J. Congenital disorders of glycosylation: a review. Pediatr Res. 2002, 52, 618–24. [Google Scholar] [CrossRef] [PubMed]
Figure 3.
Repeatability of the IEF method. The upper and lower sections show duplicated results of the repetitions. In this case, samples from patients with CDG Phenotype were used.
Figure 3.
Repeatability of the IEF method. The upper and lower sections show duplicated results of the repetitions. In this case, samples from patients with CDG Phenotype were used.
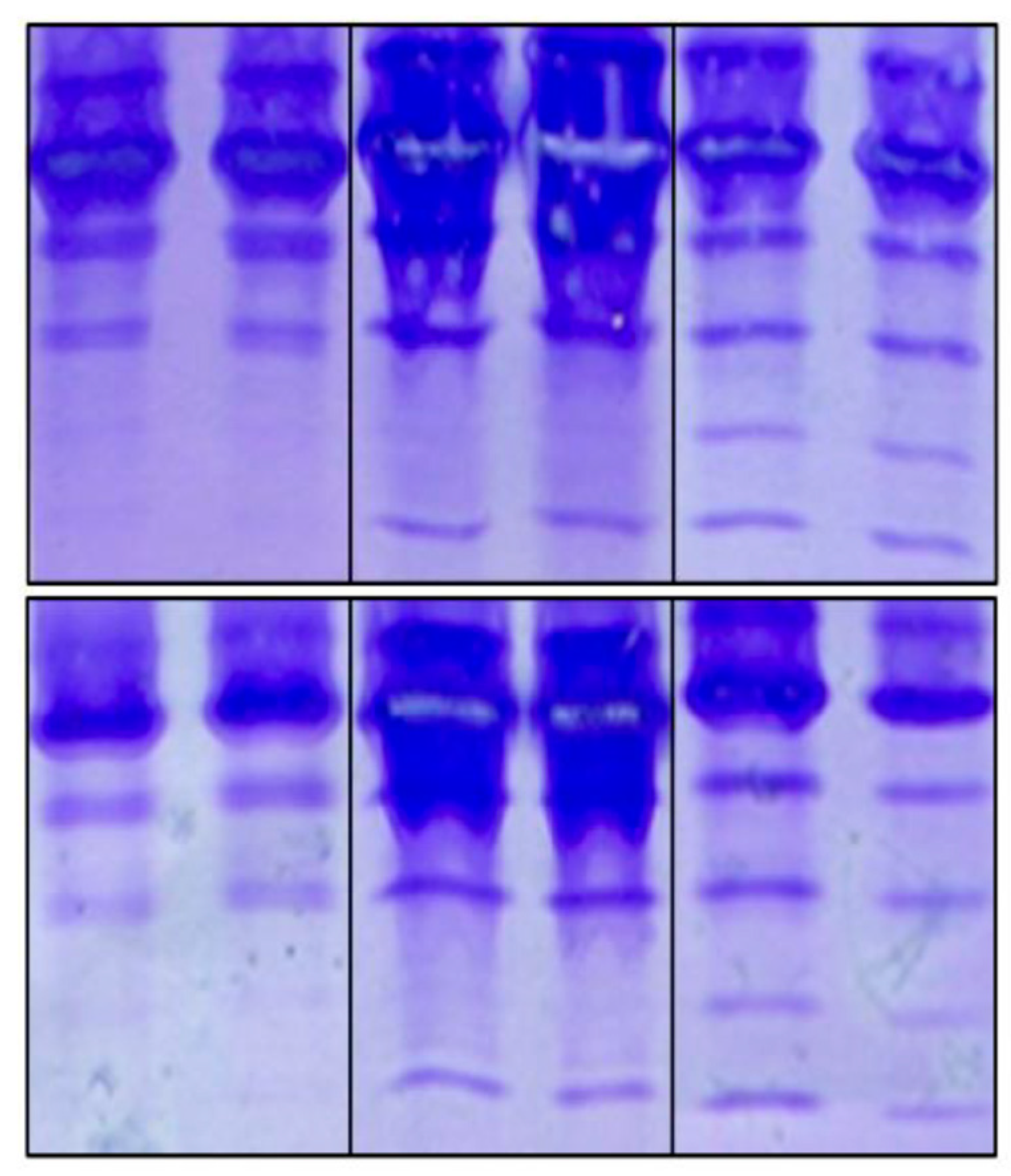
Figure 4.
Reproducibility of the IEF method. The duplicated results from different analysts are numbered. Number 1 corresponds to the test carried out by us, while numbers 2, 3 and 4 correspond to the other analysts. The latter case corresponds to the Laboratory of Glycobiology at the Faculty of Sciences, Autonomous University of Morelos, Mexico.
Figure 4.
Reproducibility of the IEF method. The duplicated results from different analysts are numbered. Number 1 corresponds to the test carried out by us, while numbers 2, 3 and 4 correspond to the other analysts. The latter case corresponds to the Laboratory of Glycobiology at the Faculty of Sciences, Autonomous University of Morelos, Mexico.
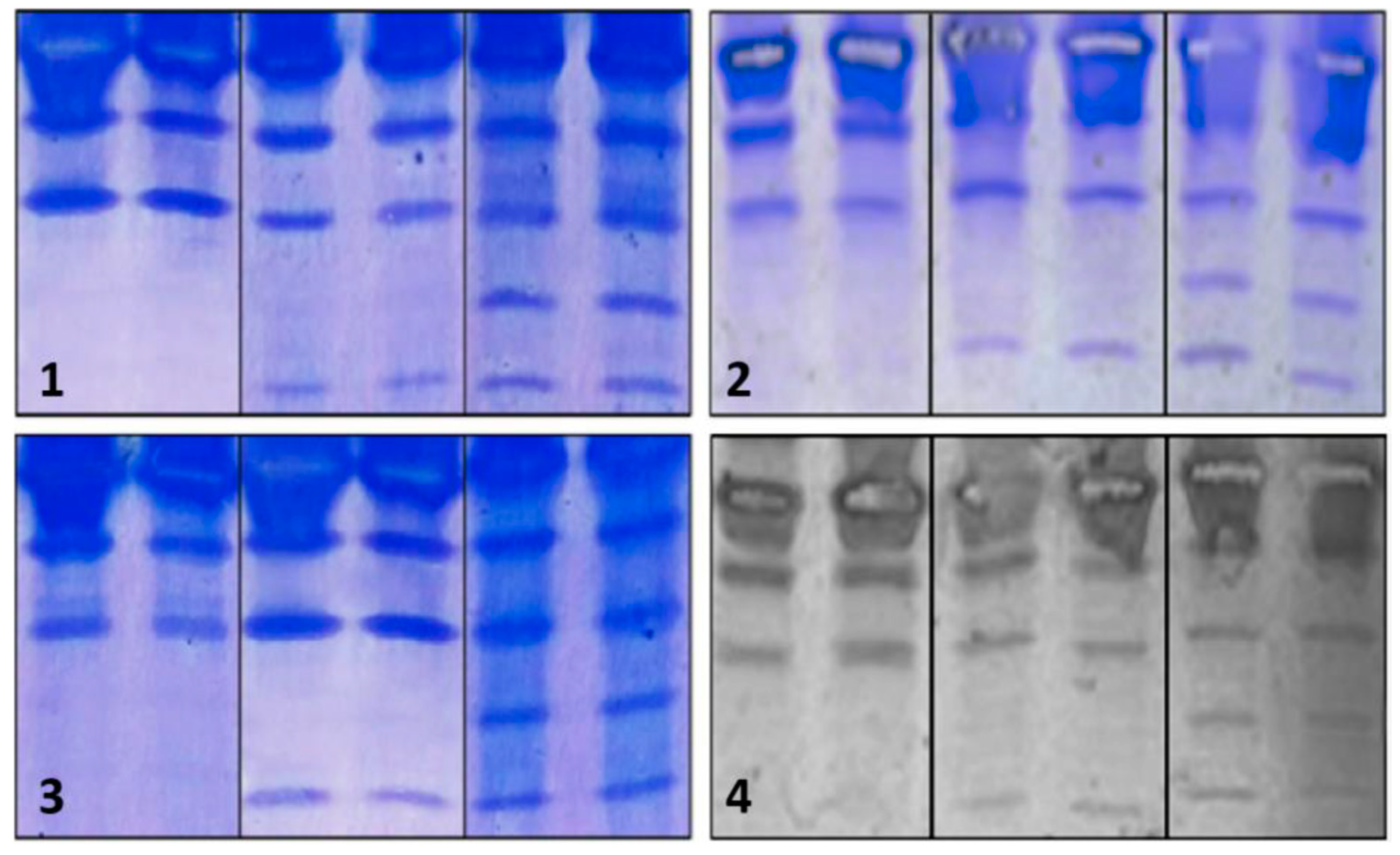
Figure 5.
IEF of Tf from patients with CDG Phenotype. C- designates the negative control; I and II designate the positive controls for patterns I and II, respectively, while Arabic numerals denote patient samples. A color code was utilized to differentiate various outcomes: red was associated with Pattern-I, light blue with Pattern-II, orange with ambiguous cases, black with negative cases, and green with repeatability assays.
Figure 5.
IEF of Tf from patients with CDG Phenotype. C- designates the negative control; I and II designate the positive controls for patterns I and II, respectively, while Arabic numerals denote patient samples. A color code was utilized to differentiate various outcomes: red was associated with Pattern-I, light blue with Pattern-II, orange with ambiguous cases, black with negative cases, and green with repeatability assays.
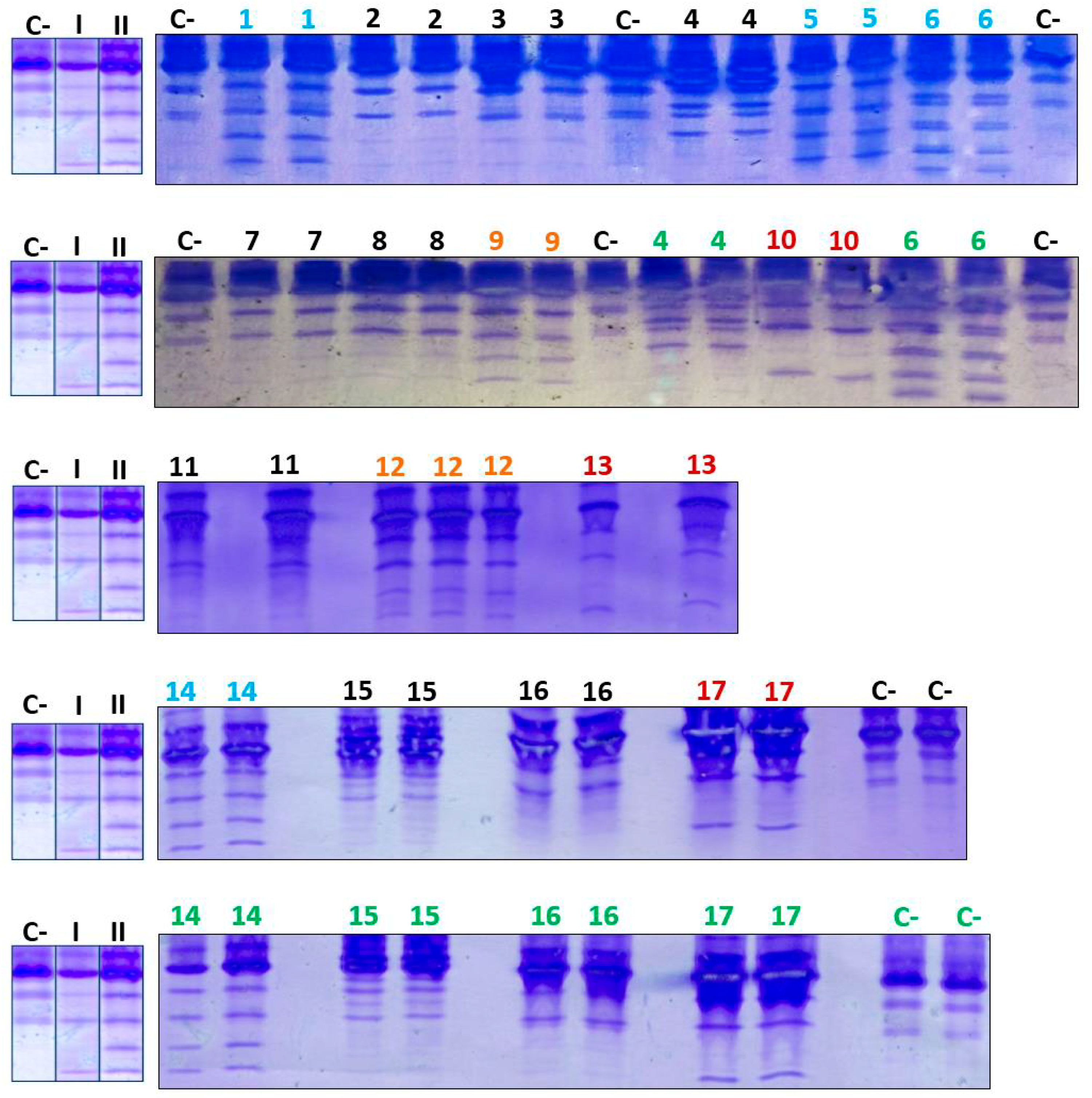
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



