Submitted:
11 April 2024
Posted:
12 April 2024
You are already at the latest version
Abstract
Accurate diagnosis for the 400 million people with rare diseases is critical for healthcare decisions, prognosis, understanding disease mechanisms, and identification of treatments. Despite advances in genome sequencing, barriers such as high interpretation costs, diagnostic expertise, throughput associated delays, and uncertain variant classifications persist, with demand exceeding capacity. Many variant classification methods focus narrowly on specific consequences, leading to the use of complex integrative pipelines that often overlook transcript variability and lack prediction transparency. To overcome these limitations, we introduce DITTO. This transparent, transcript-aware machine-learning method demonstrates superior overall performance in accuracy, recall, and precision when compared to existing tools. DITTO is publicly available at https://github.com/uab-cgds-worthey/DITTO
Keywords:
rare disease
; genomics
; explainable
; machine learning
; pathogenic
; variant consequence
; classification
; prioritization
1. Background
Rare diseases, generally defined as those affecting about 1 in 10 individuals, affect at least 400 million people worldwide [1]. More than 8,000 rare diseases have been described, with >80% believed to be genetic in nature [1]. In a rare disease setting, identification of causal and exclusion of benign variants is critical for medical and family decision-making. Identification of disease-causing variants can also generate new insights into the molecular basis of disease, support identification of novel therapeutic avenues, and generate prognostic information. Over the last decade, application of high-throughput sequencing has revolutionized the field of molecular diagnostics, facilitating reductions in cost and time to diagnosis [1,2,3,4,5]. Development of computational approaches for variant identification, classification, and interpretation has been critical in supporting these advances [1,6,7,8,9,10].
Even with these genomic and computational advances, challenges remain, and many patients face roadblocks [1,3]. Even in countries such as the USA and UK where the method is being applied somewhat routinely (although generally excluded from insurance coverage), the number of patients who might benefit dramatically outweighs the current possible throughput and biases based on patient geographic location and socioeconomic status exist [11,12,13]. When genomics-based testing is performed, roughly one third of tested individuals are returned a report containing at least one variant of uncertain significance (VUS) [14,15]. Since the clinical significance of these VUS has not been established, healthcare providers exercise caution in using these results, management and treatment plans are generally not based solely on VUS results [14,15]. Receiving a VUS result can be confusing and distressing for patients due to the uncertainty it presents.
Biases impact the likelihood of receiving a VUS, with individuals of or with ancestry from underrepresented backgrounds (e.g. Native American, Hispanic, South Asian, Greater Middle Eastern, or African) having fewer reported pathogenic variants and more VUSs [16,17]. This repository, like the field, is also biased towards identification and classification of variants within protein coding or splice regions (accounting for ~2% of the genome) and the methods for prediction of the pathogenicity of missense and other protein altering variants are much more mature than for other genomic locations [18,19].
Newly identified VUSs join an ever-increasing backlog of existing VUSs in need of periodic reanalysis and/or reevaluation [14,15]. Of the more than 2.1 million variants so far deposited in ClinVar; 50% are VUSs and another 5% have conflicting interpretations [15,20]. Collaborative efforts are underway, but the cost of human-mediated variant interpretation is high [21,22,23]. Addressing this bottleneck will require a multifaceted approach, combining wet lab and computational technological, methodological, and collaborative efforts.
For instance, a number of projects are underway with the goal of generating high-throughput scalable multiplexed assays capable of identifying molecular and cellular impact of variants. These encompass a variety of assays including cell growth and morphology, protein abundance and activity, and transcriptomic promoter and enhancer activity [15,24,25,26]. These projects will undoubtedly provide a wealth of data and improve variant classification. The timeline for these types of large-scale projects can be long and, in the meantime, patients are waiting today for definitive diagnoses that will change clinical and family planning decisions, suggest novel treatments, and/or guide therapeutic decision-making [27].
In the meantime, the development of more sophisticated computational tools to accelerate classification as part of the reinterpretation process is needed. Application of machine learning (ML) methods offer a potential solution for the cost, turnaround, and expertise limitations of more manual approaches. Conventional ML methods, such as VarSight, MetaLR and MetaSVM, improved impact prediction and classification [28,29]. Neural network-based predictors such as MetaRNN [30], DEOGEN2 [31] and EVE [32] have been developed and found to outperform the conventional methods. Many of these advances are being integrated into diagnostic lab pipelines. Despite these advances, challenges remain. No methods exist that classify across all transcripts, classes of small variants (e.g. SNVs and Indels) and consequences (e.g. splice, missense). Many methods focus on a limited set of consequences (e.g. MetaLR for missense or Splice AI for splice variants) resulting in poor prediction accuracy for others (e.g. intergenic, Untranslated regions (UTR) etc.) [33]. In diagnostic labs, this necessitates integration of multiple predictors, which can make pipeline development, maintenance, and validation more complex requiring additional computational expertise [34]. Most methods provide predictions for only a single transcript, despite the fact that numerous studies have identified deleterious effects limited to a single or subset of transcripts, influenced by alternative splicing, gene regulation, and expression [32,33,35]. This can make it challenging to interpret variants where the impact is seen only in a non-canonical transcript [36,37]. In addition, many of these methods remain “black boxes” providing no indication as to why a variant has been predicted to be deleterious [2,10,38]. This can limit or bias interpretation and reporting with variants being overly conservatively classified as VUSs [2,10,38]. Cumulatively these limitations reduce our ability to accurately predict variant impact and represent a challenge in molecular diagnostics and precision medicine [1,10,39,40,41,42].
To address these limitations, we developed DITTO, an XML (explainable machine learning) neural network-based variant impact predictor that generates interpretable and transparent explanations for all predictions. DITTO provides scores that accurately predict variant impact across all consequences taking into account known transcripts. By providing a single classification and explanation system for any class of small variant, including substitutions, insertions, indels etc., and across all consequences including missense, frameshifts, splice site, intronic, intergenic etc., this method streamlines variant interpretation. Most importantly, our DITTO variant classification method outperforms existing methods (even single use methods) across variant classes and consequences.
2. Methods
Development of a classifier capable of differentiating between benign and deleterious variants is a multi-step process. The variants to be used in training and testing of the model are gathered, annotated, and preprocessed. Next, feature engineering methods are applied to identify, and process annotations to features from these datasets. Finally, the model is trained using the processed dataset with stepwise tuning to avoid overfitting and to optimize performance.
2.1. Implementation and Statistical Analysis
DITTO is a tunable neural network implemented in Python (v3.10.11) [43] making use of the deep learning frameworks, Keras to build the neural network architecture and Optuna to tune the architecture [44,45]. Hyperopt [46] was used to optimize training steps and select the optimal parameters for the best fit of given data without over/under fitting. The program was executed on UAB high-performance computing cluster (Cheaha) to enable efficient training and evaluation of the neural network. The Python scikit-learn [47] package was used to evaluate DITTO’s performance and compare against other methods. Figures were generated using Matplotlib and Seaborn libraries [48,49]. The DITTO software, complete with data processing and tuning parameters used, is available on GitHub at https://github.com/uab-cgds-worthey/DITTO.
2.2. Dataset Extraction and Variant Annotation
We downloaded 2,177,684 variants from the ClinVar repository along with corresponding ACMG classes and other annotations [50] (ClinVar accessed on 2023/06/06). Variants were filtered to exclude those with less than a 2-star ClinVar review status retaining only those with supporting evidence: “assertion criteria, from multiple submitters, with no conflicts”, “reviewed and agreed by an expert panel”, or reaching the level of evidence of “practice guidelines”. After preprocessing (see next section), 329,136 variants remained and were annotated using OpenCRAVAT v2.4.1 [51], an open-source framework for variant analysis and interpretation that supports rapid quality-controlled annotation using data from repositories and prediction tools. We used 69 OpenCRAVAT annotators to annotate our variants with 117 distinct Ensemble transcript-level annotations, including but not limited to, conservation scores, allelic frequencies, functional impact testing, known or predicted disease association, damage predictions, and others [51] (see Table S1 for annotation categories by OpenCRAVAT).
2.3. Data Preprocessing
Several levels of filtering and preprocessing were applied to prepare the downloaded ClinVar variants for training. 39,970 variants were excluded as they had missing values for at least 30% of all features, leaving 2,137,714 variants extending to 11,153,639 variant - transcript pairings. Variants were assigned to a class based on their ClinVar extracted ACMG classification [50] collapsing as follows: Pathogenic, Likely Pathogenic and Pathogenic/Likely Pathogenic were assigned to the “High impact” category with 302,734 variant-transcript pairs; Benign, Likely Benign and Benign/Likely Benign were assigned “Low impact” category with 748,092 variant-transcript pairs. Low impact and High impact variants were then used as binary classes with an 80:20 random split for training and testing (Table 1). Since we are interested in the variant impact across transcripts influenced by alternative splicing, start codon usage etc., variants were considered based on variant-transcript pairings [36,37]. A total of 842,659 variant-transcript pairs were selected for training, and 208,167 for testing (see Table S2 for test-training breakdown by variant consequence). After the filtering step, only two variants from chrY and none from chrM remained. Both the chrY variants were included in training.
We performed correlation analysis on our training data split to determine whether any of the features significantly correlated with the binary class (High/Low impact) (Figure S1). We identified six features (aloft, MetaSVM, Polyphen2, varity_r, dbscsnv and gnomad3 allele frequency [16,52,53,54,55,56] that showed correlation (pearson r2 > 0.95) to one another but not to the binary class. Upon review, we opted not to drop these features; reasoning whilst correlated, they convey distinct information. For example, MetaSVM and dbscSNV both make use of conservation scores, but MetaSVM provides damage prediction scores for missense variants, whilst dbscSNV provides annotations for SNVs within splicing consensus regions [57]. This data was then used to train DITTO's neural network architecture.
2.4. Model Training and Architecture
Our DITTO variant deleteriousness classifier is based on a tunable neural network that can optimize itself for all hyperparameters (network shape, depth, optimizer, dropout rate and activation function). For architecture, we used Keras, an easy-to-use high level python framework for writing neural networks, and Optuna, which offers a flexible architecture for tuning machine learning algorithms [44,45,58]. For optimizing parameters, we used Hyperopt, which uses Tree of Parzen Estimator’s (TPE) [59] stochastic search algorithm for optimized parameters supporting quicker convergence and better results compared to grid or random search. 'Loss', calculated as the error between the predicted values and actual values, is used to minimize the error in making correct predictions.
As depicted in Figure 1, the input layer of the network consisted of the preprocessed features along with variant consequences (i.e., missense, synonymous, etc.). The subsequent layers of the network perform feature extraction and transformation to learn the complex relationships between the input features and pathogenicity. The final layer of the network is a fully connected layer that outputs the probability of deleteriousness of the variant. This structure facilitates the learning of complex relationships between the input features and the target variable, ultimately generating deleterious score predictions ranging from 0 to 1, with 0 representing those most likely to be benign.
2.5. Model Evaluation and Comparison
Our trained model was evaluated on our independent test dataset to assess performance. The evaluation metrics used included accuracy, sensitivity, specificity, and area under the receiver operating characteristic curve (AUC-ROC). ROC curves are graphical representations of the sensitivity (true positive rate) versus the 1-specificity (false positive rate) of a binary classifier system [60]. The area under the ROC curve (AUC) provides a measure of the overall performance of the classifier, with higher AUC values indicating better performance. Precision-Recall (PR) curves are graphical representations of the precision (true positives / all positives) versus the recall (true positives / all actual positives) of a binary classifier system [60,61]. The precision-recall AUC (PR-AUC) is a measure of the classifier's ability to identify true positives while minimizing false positives, focused on performance with respect to the minority class for imbalanced datasets [61]. The F1 score measures model performance by calculating the harmonic mean of precision and recall for the minority positive class [62]. It is calculated as:
The calculation is based on precision (ratio of correctly predicted positive observations to the total predicted positives) and recall (or sensitivity; the ratio of correctly predicted positive observations to all observations in the actual class) for identification of pathogenic variants.
To further evaluate model performance, we compared DITTO’s performance against a set of widely used and highly regarded variant classification tools (CADD [63], ClinPred [64], MetaSVM [28], GERP [65], SpliceAI [33] and Revel [66]). We performed comparison based on: overall performance, by chromosome, and by variant consequence category. For testing purposes, we considered only the transcript with highest DITTO score for each variant since all but MetaSVM and Revel are not transcript aware. We normalized comparator tool scores to a (0,1) range, and calculated performance metrics comparing to our previously defined ground truth (binary class: High/Low impact). In order to control for biases and provide as fair a comparison as possible, we normalized using the minimum and maximum scores for each comparison performed i.e. min/max scores for chr1 will be different from min/max scores for chr2, and min/max scores for missense variants will be different from min/max scores for splice variants.
Each variant can have multiple consequences. For example, a missense variant within two bases of a canonical splice site can be annotated as both “missense” and “splice site” [67]. Since we wished to perform a fair and unbiased comparison, we segmented comparisons based on variant consequence, categorizing each variant in to just one category for use in comparisons, choosing the one considered most likely to be deleterious. For instance, a variant that is both splice site and missense would be analyzed in the splice category. The hierarchy of these consequence categories is detailed in Table S3. Model performance was then determined based on comparisons of F1-scores across each of these methods. The F1 score was used for comparisons based on our binary classification, somewhat uneven class distribution, and equal interest in correctly identifying both deleterious and benign variants for our diagnostic use case. F1 scores range from 0 (worst) to 1 (perfect precision and recall).
2.6. Model Interpretability
Feature importance scores were derived using SHapley Additive exPlanations (SHAP) calculations [68]. SHAP calculations facilitate quantification of each feature's contribution to the model, defining their significance both in terms of overall classification accuracy and individual prediction performance. Visual representations in the form of Shapley plots were created to depict incorporation of features within the model. These plots illustrate the trajectory of the model's output from a baseline prediction to the final outcome, offering an easily explainable transparent view of the influence each feature had on the prediction [68,69].
2.7. Web Application
A web application was developed using Streamlit [70] to allow users to easily submit small variants, specified in the GRCh38 HGNC nomenclature, for DITTO score prediction and SHAP-based explanation without requiring personal computational resources for model construction or hosting. The application is available at https://cgds-ditto.streamlit.app/. It features an automatic validation system for the input variant information, leveraging the UCSC genome browser's API [71]. Upon submission, the app retrieves and displays the DITTO score with pathogenicity classification and annotations. These annotations include information on overlapping/corresponding gene(s), transcript(s), allele frequencies from population repositories, functional consequences, among others, sourced from the OpenCRAVAT database via its API. Additionally, the application provides SHAP values and visualizations to illustrate the influence of each feature on the DITTO score, offering users a deeper understanding of the underlying prediction logic.
3. Results
3.1. DITTO Tuning and Training
DITTO can adaptively tune its parameters based on the training and validation dataset, ensuring it neither overfits nor underfits the data (Figure 1a). Table 2 summarizes the hyperparameters available for tuning. In order to determine optimal hyperparameters for our model, we conducted two-phase tuning with 500 trials, each utilizing a distinct combination of hyperparameters. Initially, we employed a random search strategy with 200 trials, assigning random hyperparameter combinations to each.
This approach facilitated exploration of a diverse pool of configurations offering preliminary insights into performance. For each trial, we used the “accuracy score” as the performance metric for the next trial to improve upon. Leveraging the insights gained from the initial 200 trials, we advanced to a more refined tuning strategy using Bayesian optimization via the TPESampler [59] from Hyperopt. This method optimizes the selection process by leveraging outcomes from previous trials, considering both the hyperparameter configurations and corresponding accuracy scores. By adopting the TPESampler proposed hyperparameter combinations for the remaining 300 trials, we optimized the search space to find the highest performant hyperparameter combination.
To improve accuracy and mitigate over/under fitting, we structured each trial by splitting the preprocessed training data into training (80%) and validation (20%) subsets. This approach allowed the model to train on a substantial dataset while its performance was evaluated independently on the validation set. Each trial ran for 500 epochs (iterations through the training data).
To prevent stagnation, enhance efficiency, conserve resources, and address potential over/under fitting, an early stopping mechanism was implemented that halted the trial if no improvement in the model's validation accuracy occurred for 10 consecutive epochs. Out of the total 500 trials, 325 successfully completed. 175 were pruned due to poor performance or terminated by the early stopping mechanism (Figure S2). This tuning process yielded the optimal hyperparameter configuration used for DITTO (Figure 1c). The optimal set of hyper parameters was [# layers: 1, activation (input and hidden layers): elu, # neurons: 161, kernel initializer (hidden layer): he_normal, dropout_l0: 0.808, kernel initializer (output layer): glorot_normal, optimizer: Adamax, batch_size: 267]
The neural network optimized with the best parameters underwent training on the full training dataset with the model saved for testing and benchmarking purposes. To streamline analysis from VCF files, a nextflow [72] pipeline was developed using OpenCRAVAT’s annotation service. The pipeline computes DITTO scores using the pre-trained DITTO model and generates deleteriousness predictions for variants alongside relevant annotations (Figure 1b).
3.2. DITTO Has High Precision, Recall, and Accuracy in Predicting Variant Deleteriousness
The DITTO neural network achieved extremely high accuracy in distinguishing between deleterious and benign variants, achieving an overall accuracy of 99.9% on the held back test dataset (Table 3). The model demonstrated exceptional performance across Precision, Recall, Loss, and F1-scores (0.99, 0.99, 0.07, and 0.99, respectively). These results indicate that the model reliably predicts both positive and negative outcomes, shows excellent discriminative ability between positive and negative classes, and performs well even in scenarios with imbalanced data.
3.3. Feature Contribution
SHAP plots provide a visual explanation of the importance of specific features to model predictions (Figure 2). The application of SHAP plots can help us understand the features most important for model predictions, and in doing so can also help identify potential biases within the model. Figure 2b shows the extent to which each feature is contributing to model prediction across 10,000 randomly selected variants. SHAP analyses can be used to delineate the top contributing features at the individual variant level. For instance, Figure 2c, shows an example in which gnomAD3, SpliceAI, and CADD emerge as top features influencing the model towards classifying a variant as deleterious with a DITTO score of 1.
3.4. Performance Comparison against Existing Methods
For benchmarking of DITTO’s performance, we conducted analysis on a set of 37,027 variants split from the test dataset before training. This evaluation compared DITTO against several established and industry standard pathogenicity prediction methods, including CADD, ClinPred, MetaSVM, GERP [77], SpliceAI and Revel [66]. Of note, a number of these methods are themselves annotations used as features in the training of DITTO. Comparisons were based on the normalized scores for each tool as described in the Methods. The comparative performance of these tools was assessed with Receiver Operating Characteristic (ROC) and Precision Recall (PR) curves. DITTO outperformed all other evaluated classifiers, achieving an ROC-AUC of 0.99 and a PR-AUC of 0.99 (Figure 3a,b). In contrast, the other methods achieved ROC-AUC scores ranging from 0.59 to 0.88 and PR-AUC scores from 0.39 to 0.81, with CADD and ClinPred emerging as the next most effective classifiers (Figure 3a,b).
We reasoned that potential chromosome-specific biases stemming from variations in GC content, gene, count, size, or density, or evolutionary history might impact predictions, and so compared DITTO predictions against the other tools, examining F1 scores on a chromosome-by-chromosome basis. As shown in Figure 3c, DITTO consistently outperformed other evaluated tools across all chromosomes. It is important to note that variants from Chromosome Y (chrY) and the Mitochondrial Chromosome (chrMT) were excluded from this analysis due to low numbers and failure to meet the predefined filtering criteria. Of note, a marked variation in the performance of several tools was observed across different chromosomes, pointing to the potential of chromosome-specific biases within the training datasets or the algorithms themselves.
3.5. Model Performance by Variant Consequence
To ensure fair comparison, we segmented our analysis based on variant consequence providing a fairer evaluation of each tool's effectiveness taking into account specializations. Based on this, we noted that DITTO demonstrated superior performance compared to the other tools evaluated. It generated accurate predictions across all chromosomes and variant consequences (Figure 4 and Tables S4 and S5).
DITTO was able to accurately classify 98% of missense variants. It achieved the highest overall F1 score (0.99), greater than that of the meta predictors ClinPred, MetaSVM, and Revel (Figure 4b). DITTO also showed superior performance in accurately classifying variant consequences often overlooked by other tools (Figure 4c–e). For example, intergenic variants are more challenging to classify and interpret. They are scarce in variant repositories (partly due to limited consideration or exclusion from molecular lab workflows) and are less likely to be identified as deleterious (partly due to limited availability of annotated feature data as compared to other variant classes). Despite these challenges, DITTO was able to accurately classify 98.5% of intergenic variants achieving the highest F1 score of 0.92 among all classifiers tested in this category (Figure 4c). ClinPred was the next most accurate, with a 72% accuracy rate. Many consequence agnostic prediction tools also perform poorly on splice variants, prompting development of specialized classifiers such as SpliceAI. For splice site variants, DITTO achieved an F1 score of 0.99, surpassing SpliceAI, as well as CADD and GERP (scoring 0.97, 0.98, and 0.99, respectively; Figure 4d). Similarly, intronic variants outwith the canonical splice sites have also posed a challenge for variant impact interpretation and classification again often leading to exclusion from consideration. DITTO was again able to accurately predict the majority of these variants with an F1 score of 0.88, outperforming CADD and SpliceAI (0.59 and 0.63, respectively; Figure 4e).
SHAP-based feature importance analysis was performed to explore feature importance for each set of predictions across each variant consequence (Figure 5). ClinPred, gnomAD and CADD emerge as the top features contributing to DITTO predictions overall regardless of the specific variant consequence (Figure 5). GnomAD, funseq2, and CADD were the top contributing features for missense variants (Figure 5b). GnomAD, CADD and SpliceAI were found to be the top contributing features for splice impacting variants (Figure 5c). GnomAD, funseq2, and CADD were the top contributing features for intronic variants (Figure 5d).
3.6. DITTO Can Predict Deleterious Score by Transcript
The impact of a variant can vary depending on transcript-specific effects. A variant might lead to alternative splicing and resultant frameshift in one transcript but be deep intronic with no impact in another. Transcripts can also exhibit tissue-specific expression patterns, such that a variant’s impact is seen in one tissue and not another [78,79]. DITTO’s ability to predict impacts based on transcripts can be helpful in exploring these occurrences. As shown in Table S7, we have identified numerous instances where a variant has notably differing DITTO scores across transcripts. In some cases, the presence of overlapping genes is a critical factor. This is the case for the chr5:g.176629656C>A variant spanned by MIR4281, SNCB, and EIF4E1B where DITTO scores of 0.121, 0.332, and 0.997 are seen for the different genes, respectively. This is also the case for the chr1:g.43384485C>G variant present in both MED8 and its antisense RNA MED8-AS1 (0.99 and 0.31, respectively). In other instances, DITTO scores are disparate for variants spanned by transcripts belonging to only a single gene, such as the case for a CHDR2 variant that scores 0.560, 0.791, and 0.971 in different transcripts with the same consequence with transcript length, gene expression, amino acid position, and underlying predicted features (e.g., chasmplus and VEST) contributing to the scores. In other cases, the variant is present at a position with different gene structure in different transcripts. This is the case of a variant affecting the PPT1 gene. In one transcript, the variant affects the 3’ UTR receiving a DITTO score of zero. In another, it is located in an alternative exon leading to a missense change and DITTO score of 0.914. It is also the case for a variant affecting the TEKT2 gene, which is deeper intronic in one transcript and exonic resulting in a stop gain in another with DITTO scores of 0.0 and 1.0, respectively. Not considering transcript context in such cases might well lead to inaccuracies in classification.
3.7. Dissemination and Access to DITTO Predictions
As shown in Figure 6, our pre-trained DITTO model is accessible via a user-friendly streamlit app allowing users to generate DITTO scores for their variants of interest (https://cgds-ditto.streamlit.app/). The app can be used to explore variant annotations and feature importance for all extracted variant-transcript mappings, presenting SHAP values for each feature to better explain DITTO predictions.
We also generated a Nextflow pipeline to be used to facilitate the generation of DITTO scores for genome sequencing VCF files (Figure 1b). This pipeline is hosted and publicly accessible on GitHub: https://github.com/uab-cgds-worthey/DITTO. Leveraging this pipeline, we computed DITTO scores for all possible SNVs and for all known Insertions/Deletions/Indels catalogued in the gnomADv3.0 database. These scores have been compiled in to tabix indexed format [80] and are available for download via this link: https://s3.lts.rc.uab.edu/cgds-public/dittodb/dittodb.html. Completion of these additional resource intensive tasks ensures that these predictions are made widely available especially for those who may lack the necessary infrastructure to complete the task themselves.
3.8. Validation on Previously Unseen NF1 Dataset
To evaluate DITTO's applicability on an additional test dataset, we used it to analyze variants previously identified in the Neurofibromin (NF1) gene, the causal gene for autosomal dominant Neurofibromatosis type-1 disease (NF1). We selected NF1 as the gene is of interest for ongoing projects in our lab. And also, because the NF1 gene is large, spanning 60 exons, many transcripts, and ~350kb. It functions as a tumor suppressor gene by negatively regulating Ras [81,82]. NF1 can present with café au lait spots, disfiguring cutaneous and/or plexiform neurofibromas, optic nerve gliomas, malignant peripheral nerve sheath tumors, skeletal defects, attention and learning deficits, and other cognitive disabilities [83,84]. As de novo variants are a common cause of NF1, NF1 locus has a higher spontaneous mutation rate than most gene loci. The phenotypic diversity in NF1 is partly attributed to hypomorphic variants that maintain partial function. Variant classification in NF1 is a complex task. By applying DITTO in the case of NF1, we sought to determine the tool's effectiveness in classification and prioritization in this type of complex situation.
The Leiden Open Variation Database (LOVD) is an open access, flexible web-based platform designed to support compilation and display of DNA variants linked to genetic disorders https://databases.lovd.nl/shared/variants/NF1/) [85,86,87]. Generation of this database was supported by the European Community's Seventh Framework Programme (FP7/2007-2013) as part of the GEN2PHEN project [88]. LOVD is a manually curated dataset providing not only variant data, but also curated patient-centered data where available [85,86,87]. We extracted 3,759 NF1 variants from LOVD (accessed on 2/16/2023) and associated ACMG classes from ClinVar for the following analysis. From this set, we excluded the 497 NF1 variants previously used as part of the DITTO training set leaving 3,262. We also excluded 207 variants with conflicting interpretations. 937 of the variants remaining had pathogenic/likely pathogenic (733) or benign/likely benign (204) classifications and were used for testing.
In this analysis, we refined our DITTO score deleterious cutoff to 0.91 (mean of all P/LP variants -1SD) and benign cutoff to 0.11 (mean of all B/LB variants +1SD). Using this cutoff, DITTO classified 927/937 (99%) of these variants in agreement with ClinVar with precision, recall, and F1-scores of 0.99, 0.99, and 0.99, respectively. This constituted 725/733 of ClinVar pathogenic/likely pathogenic variants and 202/204 of the benign/likely benign variants. Clearly, we can significantly reduce the number of variants to review using a refined gene specific cutoff. Figure 7a shows the distribution of these variants across the gene with ClinVar classifications. Across the entire NF1 test set, DITTO accuracy outperformed the other methods tested with CADD and GERP scoring 0.97 and 0.84 respectively (Figure S3). SpliceAI’s performance was equivalent to DITTO’s (scoring 0.99) when tested exclusively on splice variants.
Out of 937 variants with known classifications, five were discordant between DITTO and ClinVar. These were reviewed using the canonical NF1 transcript ENST00000356175 as the basis for all annotations (Table 4). Variant 1 was classified as likely benign in ClinVar yet had a DITTO score of 1. Inspection of the region in the UCSC genome browser [71] identified four similar pathogenic splice variants within three nucleotides of this position supporting the accuracy of the DITTO deleterious prediction. We hypothesis that this variant is misclassified in ClinVar.
Variant 2, although synonymous, has been experimentally confirmed to generate an alternate splice donor site with exclusion of normally exonic sequence [89,90]. This rarer consequence is presumably not well accounted for in the DITTO model due to limited numbers of these types of events in the training dataset. Variants 3, 4, and 5 are all identified as likely or known pathogenic in ClinVar, but deeper intronic and complex variants in the case of 3 and 5 and as such also presumably not well accounted in the training dataset [89,91,92]. DITTO performed exceptionally well in this case study accurately classifying 99.57% of variants. This is despite NF1 being a complex disorder characterized by presence of many de novo and hypomorphic alleles, a wide range of causal variant consequences including many non-protein coding variants, and complex genotype to phenotype relationships.
We next used DITTO to generate likely pathogenic or likely benign classifications for the 665 NF1 variants currently identified as VUSs. We were able to classify 333 as Likely deleterious and 242 as Likely benign (90 remained as VUSs). The ClinVar classified VUS p.Gly309Ile (Variation ID: 1766337) that we reclassified to Likely deleterious with DITTO. There is good supporting evidence for this variant. It results in substitution of glycine for isoleucine, an amino acid with dissimilar properties. This position is highly conserved, the variant is not seen in population databases, and two individuals (one with NF1 and the other with Hereditary cancer-predisposing syndrome) were previously identified. A number of additional pathogenic or likely pathogenic variants have been identified in the region.
Finally, we classified the 1,950 variants not currently submitted to ClinVar. DITTO classified 294 as Likely benign and 1,589 as Likely pathogenic, with 67 remaining as VUSs. Many classes of variants including premature stops, splice variants, and frameshifts were predominantly classified as Likely pathogenic, whilst missense variants were also classified as Likely benign. We have shown through this study that this type of ML classification can be used to classify variants, prioritize variants for curation/experimental validation, and identify misclassified variants.
4. Discussion
Our study demonstrates that our classifier DITTO can accurately classify any class and consequence of small variant with an overall accuracy of 99.6%. DITTO demonstrated superior performance compared to the other tools evaluated, generating accurate predictions spanning all chromosomes and variant consequences. The performance of our tool is on par with specialized tools for specific variant consequences. DITTO also showed superior performance in accurately classifying variant consequences that are often overlooked by other tools, including those occurring in deep intronic and intergenic regions. These improvements can be attributed to several factors. DITTO’s neural network was trained on a larger dataset of ClinVar curated variants comprehensively annotated using OpenCRAVAT. A focus on training data preparation to minimize potential biases. DITTO is based on a tunable neural network and as such can optimize itself for all hyperparameters, tuning itself for number of layers, neurons per layer, activation method for each layer, dropout layers and batch size, thereby reducing bias and over/under-fitting.
The most significant limitation relates to biases present in the available training data: For historical and societal reasons, particular diseases (e.g. cancer, neurologic disorders), organs or tissues (e.g. brain, heart), or genes (e.g. BRCA1, TP53, CFTR) tend to have gathered more attention and are overrepresented. There are also enrichment biases for variants that have an early or developmental effect (birth defects versus aging related disorders). Similarly, certain genic (splice sites and exons versus untranslated regions) and protein (binding pockets, channel walls versus disordered regions) locations have received greater focus. Some molecular products or their functions (e.g. metabolic enzymes) are easier to experimentally validate than others. Omic based molecular diagnostics has also focused on rare disorders as opposed to more common disorders and repositories contain fewer variants associated with more common diseases. Finally, biases exist related to the preponderance of data from individuals of European ancestries and lesser contribution of variants from underrepresented groups. As a result, variants available for use in model training tend to skew towards having low population frequencies, high conservation, and Mendelian inheritance patterns and towards certain subsets of genes, regions, tissues, ancestries, and functions. These biases in variant databases undoubtedly impact the models making use of them. Improvements addressing these biases are identified as vital next steps.
5. Conclusions
Despite advances in technology allowing for genome sequencing as part of diagnostic testing, many rare disease families still lack a definitive molecular diagnosis. Whilst the cost of the sequencing has dropped dramatically, the cost for interpretation has remained static. Even in wealthy countries applying these methods, the number of patients who might benefit dramatically outweighs current throughput. The sequencing is not the bottleneck, but rather the interpretation. DITTO represents a significant advance in the field of variant classification that can be used to address these challenges. Its methods can be implemented easily and used to classify across all types of small variants and transcripts with exceptional accuracy, recall, and precision. It provides explanations as to its predictions. The precision, recall, and F1-score of DITTO were high, indicating that the tool can be used effectively for clinical applications. With careful application of cutoffs, it can be used to dramatically reduce the overall numbers of variants to be reviewed without sacrificing accuracy and precision.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Tables and Figures are available and attached to this manuscript.
Author Contributions
E.A.W. conceived the project and obtained funding and supervised the study. T.K.K.M., and E.A..W. developed and refined the experimental design. T.K.K.M. processed the data and trained the DITTO model. T.K.K.M., M.G., B.M.W., and E.A.W.. devised the data analysis methodology. T.K.K.M. and E.A.W. performed variant, genes, and cohort level analyses. T.K.K.M., M.G., and B.M.W. built the DITTO tool website. M.G. and B.M.W. performed code review for DITTO GitHub repo. T.K.K.M. and E.A.W. wrote the initial draft of the manuscript and refined it along with M.G. and B.M.W.. All authors read and approved the final manuscript.
Funding
This work was supported by UAB Worthey Lab Start-Up funds (EAW), the UAB Pilot Center for Precision Animal Modeling (NIH 1U54OD030167; EAW), Children’s Tumor Foundation Hack4NF award (CTF; TKKM and EAW), The UAB Carmichael Scholar award for academic achievement (TKKM). The funders had no role in the design of the study or in collection, analysis, and interpretation of data or writing of the manuscript.
Data Availability Statement
Code used for this project has been made available at https://github.com/uab-cgds-worthey/DITTO. DITTO score for all possible SNVs and gnomAD indels are available at https://s3.lts.rc.uab.edu/cgds-public/dittodb/dittodb.html. DITTO webapp is available for public use - https://cgds-ditto.streamlit.app/. NF1 variants are available to download from LOVD here - https://databases.lovd.nl/shared/variants/NF1/. Databases to annotate variants are downloaded from OpenCravat. Please find the instructions in our project repo - https://github.com/uab-cgds-worthey/DITTO/blob/main/docs/install_openCravat.md.
Acknowledgments
The authors gratefully acknowledge the resources provided by the University of Alabama at Birmingham IT-Research Computing group for high performance computing (HPC) support and CPU time on the Cheaha compute cluster. The authors also wish to specifically thank and acknowledge William Warriner from UAB HPC, who greatly assisted us with architecting the necessary resources to run pipelines on Cheaha compute cluster. Finally, we wish to thank the additional members of the UAB Center for Computational Genomics and Data Sciences (CGDS) and the UAB Biological Data Sciences Core (U-BDS) for helpful discussions during this project's development.
Conflicts of Interest
The authors declare that they have no competing interests.
Ethics Approval and Consent to Participate: Not applicable.
Consent for Publication: Not applicable.
Abbreviations
ROC - receiver operating characteristic
PR – Precision Recall
HGMD - Human Gene Mutation Database
ACMG - American College of Medical Genetics and Genomics
P/LP – Pathogenic/Likely pathogenic
B/LB – Benign/Likely benign
VUS – Variant of Uncertain Significance
NMD – Nonsense mediated decay
NF1 – Neurofibromin 1
LOVD - Leiden Open Variation Database
XML – eXplainable Machine Learning
SNV – Single Nucleotide Variation
HGNC - HUGO Gene Nomenclature Committee
SHAP - SHapley Additive exPlanations
VCF – Variant Call Format
References
- Splinter K, Adams DR, Bacino CA, Bellen HJ, Bernstein JA, Cheatle-Jarvela AM, et al. Effect of Genetic Diagnosis on Patients with Previously Undiagnosed Disease. New Engl J Med [Internet]. 2018;379:2131–9. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30304647.
- Marshall CR, Chowdhury S, Taft RJ, Lebo MS, Buchan JG, Harrison SM, et al. Best practices for the analytical validation of clinical whole-genome sequencing intended for the diagnosis of germline disease. npj Genom Med. 2020;5:47.
- Zastrow DB, Kohler JN, Bonner D, Reuter CM, Fernandez L, Grove ME, et al. A toolkit for genetics providers in follow-up of patients with non-diagnostic exome sequencing. J Genet Couns. 2019;28:213–28.
- Bick D, Fraser PC, Gutzeit MF, Harris JM, Hambuch TM, Helbling DC, et al. Successful Application of Whole Genome Sequencing in a Medical Genetics Clinic. J Pediatric Genetics [Internet]. 2017;6:61–76. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28496993.
- Prokop JW, May T, Strong K, Bilinovich SM, Bupp C, Rajasekaran S, et al. Genome sequencing in the clinic: the past, present, and future of genomic medicine. Physiol Genomics [Internet]. 2018;50:563–79. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29727589.
- Ramoni RB, Mulvihill JJ, Adams DR, Allard P, Ashley EA, Bernstein JA, et al. The Undiagnosed Diseases Network: Accelerating Discovery about Health and Disease. Am J Hum Genetics. 2017;100:185–92.
- Wojcik MH, Reuter CM, Marwaha S, Mahmoud M, Duyzend MH, Barseghyan H, et al. Beyond the exome: What’s next in diagnostic testing for Mendelian conditions. Am J Hum Genet. 2023;110:1229–48.
- Holt JM, Wilk B, Birch CL, Brown DM, Gajapathy M, Moss AC, et al. VarSight: prioritizing clinically reported variants with binary classification algorithms. Bmc Bioinformatics. 2019;20:496.
- Worthey, EA. Analysis and annotation of whole-genome or whole-exome sequencing-derived variants for clinical diagnosis. Curr Protoc Hum Genetics Éditor Board Jonathan L Haines Et Al [Internet]. 2013;79:Unit 9 24. Available from: https://www.ncbi.nlm.nih.gov/pubmed/24510652.
- Stenton SL, O’Leary M, Lemire G, VanNoy GE, DiTroia S, Ganesh VS, et al. Critical assessment of variant prioritization methods for rare disease diagnosis within the Rare Genomes Project. medRxiv. 2023;2023.08.02.23293212.
- Angelis A, Tordrup D, Kanavos P. Socio-economic burden of rare diseases: A systematic review of cost of illness evidence. Heal Polic. 2015;119:964–79.
- Marshall DA, Gerber B, Lorenzetti DL, MacDonald KV, Bohach RJ, Currie GR. Are We Capturing the Socioeconomic Burden of Rare Genetic Disease? A Scoping Review of Economic Evaluations and Cost-of-Illness Studies. PharmacoEconomics. 2023;41:1563–88.
- Currie GR, Gerber B, Lorenzetti D, MacDonald K, Benseler SM, Bernier FP, et al. Developing a Framework of Cost Elements of Socioeconomic Burden of Rare Disease: A Scoping Review. PharmacoEconomics. 2023;41:803–18.
- Rehm HL, Alaimo JT, Aradhya S, Bayrak-Toydemir P, Best H, Brandon R, et al. The landscape of reported VUS in multi-gene panel and genomic testing: Time for a change. Genet Med. 2023;25:100947.
- Fowler DM, Rehm HL. Will variants of uncertain significance still exist in 2030? Am J Hum Genet. 2024;111:5–10.
- Gudmundsson S, Singer-Berk M, Watts NA, Phu W, Goodrich JK, Solomonson M, et al. Variant interpretation using population databases: Lessons from gnomAD. Hum Mutat. 2022;43:1012–30.
- Chen E, Facio FM, Aradhya KW, Rojahn S, Hatchell KE, Aguilar S, et al. Rates and Classification of Variants of Uncertain Significance in Hereditary Disease Genetic Testing. JAMA Netw Open. 2023;6:e2339571.
- Aguirre J, Padilla N, Özkan S, Riera C, Feliubadaló L, Cruz X de la. Choosing Variant Interpretation Tools for Clinical Applications: Context Matters. Int J Mol Sci. 2023;24:11872.
- Shendure J, Findlay GM, Snyder MW. Genomic Medicine–Progress, Pitfalls, and Promise. Cell. 2019;177:45–57.
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8.
- Sciascia S, Roccatello D, Salvatore M, Carta C, Cellai LL, Ferrari G, et al. Unmet needs in countries participating in the undiagnosed diseases network international: an international survey considering national health care and economic indicators. Front Public Heal. 2023;11:1248260.
- Taruscio D, Baynam G, Cederroth H, Groft SC, Klee EW, Kosaki K, et al. The Undiagnosed Diseases Network International: Five years and more! Mol Genet Metab. 2020;129:243–54.
- Taruscio D, Salvatore M, Lumaka A, Carta C, Cellai LL, Ferrari G, et al. Undiagnosed diseases: Needs and opportunities in 20 countries participating in the Undiagnosed Diseases Network International. Front Public Heal. 2023;11:1079601.
- Esposito D, Weile J, Shendure J, Starita LM, Papenfuss AT, Roth FP, et al. MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol. 2019;20:223.
- Gasperini M, Starita L, Shendure J. The power of multiplexed functional analysis of genetic variants. Nat Protoc. 2016;11:1782–7.
- Weile J, Roth FP. Multiplexed assays of variant effects contribute to a growing genotype–phenotype atlas. Hum Genet. 2018;137:665–78.
- Bauskis A, Strange C, Molster C, Fisher C. The diagnostic odyssey: insights from parents of children living with an undiagnosed condition. Orphanet J Rare Dis. 2022;17:233.
- Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet [Internet]. 2015;24:2125–37. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25552646.
- Holt JM, Wilk B, Birch CL, Brown DM, Gajapathy M, Moss AC, et al. VarSight: prioritizing clinically reported variants with binary classification algorithms. Bmc Bioinformatics. 2019;20:496.
- Li C, Zhi D, Wang K, Liu X. MetaRNN: differentiating rare pathogenic and rare benign missense SNVs and InDels using deep learning. Genome Med. 2022;14:115.
- Raimondi D, Tanyalcin I, Ferté J, Gazzo A, Orlando G, Lenaerts T, et al. DEOGEN2: prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Res. 2017;45:gkx390-.
- Frazer J, Notin P, Dias M, Gomez A, Min JK, Brock K, et al. Disease variant prediction with deep generative models of evolutionary data. Nature. 2021;599:91–5.
- Jaganathan K, Panagiotopoulou SK, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176:535-548.e24.
- Spielmann M, Kircher M. Computational and experimental methods for classifying variants of unknown clinical significance. Cold Spring Harb Mol Case Stud. 2022;8:a006196.
- Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020;12:103.
- Bhuiyan SA, Ly S, Phan M, Huntington B, Hogan E, Liu CC, et al. Systematic evaluation of isoform function in literature reports of alternative splicing. BMC Genom. 2018;19:637.
- Chen H, Shaw D, Zeng J, Bu D, Jiang T. DIFFUSE: predicting isoform functions from sequences and expression profiles via deep learning. Bioinformatics. 2019;35:i284–94.
- Gargis AS, Kalman L, Bick DP, Silva C da, Dimmock DP, Funke BH, et al. Good laboratory practice for clinical next-generation sequencing informatics pipelines. Nat Biotechnol [Internet]. 2015;33:689–93. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26154004.
- Pena LDM, Jiang YH, Schoch K, Spillmann RC, Walley N, Stong N, et al. Looking beyond the exome: a phenotype-first approach to molecular diagnostic resolution in rare and undiagnosed diseases. Genet Med [Internet]. 2018;20:464–9. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28914269.
- Shashi V, Schoch K, Spillmann R, Cope H, Tan QK, Walley N, et al. A comprehensive iterative approach is highly effective in diagnosing individuals who are exome negative. Genet Med [Internet]. 2019;21:161–72. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29907797.
- Samani A, English KG, Lopez MA, Birch CL, Brown DM, Kaur G, et al. DOCKopathies: A systematic review of the clinical pathologies associated with human DOCK pathogenic variants. Hum Mutat. 2022;43:1149–61.
- Fresard L, Smail C, Ferraro NM, Teran NA, Li X, Smith KS, et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat Med. 2019;25:911–9.
- Rossum GV, Drake FL. Python 3 Reference Manual. Scotts Valley, CA: CreateSpace;
- Chollet F, others. Keras. GitHub;
- Akiba T, Sano S, Yanase T, Ohta T, Koyama M. Optuna: A Next-generation Hyperparameter Optimization Framework. arXiv. 2019.
- Bergstra J, Yamins D, Cox D. Hyperopt: A Python Library for Optimizing the Hyperparameters of Machine Learning Algorithms. Proc 12th Python Sci Conf. 2013;13–9.
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research [Internet]. 2011;12:2825–30. Available from: http://jmlr.org/papers/v12/pedregosa11a.
- Hunter, J. D. Matplotlib: A 2D graphics environment. Computing in Science & Engineering. 2007;9:90–5.
- Waskom, M. seaborn: statistical data visualization. J Open Source Softw. 2021;6:3021.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med [Internet]. 2015;17:405–24. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25741868.
- Pagel KA, Kim R, Moad K, Busby B, Zheng L, Tokheim C, et al. Integrated Informatics Analysis of Cancer-Related Variants. JCO Clin Cancer Inform. 2020;4:CCI.19.00132.
- Balasubramanian S, Fu Y, Pawashe M, McGillivray P, Jin M, Liu J, et al. Using ALoFT to determine the impact of putative loss-of-function variants in protein-coding genes. Nat Commun. 2017;8:382.
- Kim S, Jhong J-H, Lee J, Koo J-Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017;10:2.
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet. 2013;76:7.20.1-7.20.41.
- Wu Y, Liu H, Li R, Sun S, Weile J, Roth FP. Improved pathogenicity prediction for rare human missense variants. Am J Hum Genet. 2021;108:1891–906.
- Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014;42:13534–44.
- Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum Mutat. 2016;37:235–41.
- Teredesai A, Kumar V, Li Y, Rosales R, Terzi E, Karypis G, et al. Optuna. Proc 25th ACM SIGKDD Int Conf Knowl Discov Data Min. 2019;2623–31.
- Watanabe, S. Tree-Structured Parzen Estimator: Understanding Its Algorithm Components and Their Roles for Better Empirical Performance. arXiv. 2023.
- Hanley, JA. Wiley StatsRef: Statistics Reference Online. 2017. [Google Scholar]
- Keilwagen J, Grosse I, Grau J. Area under Precision-Recall Curves for Weighted and Unweighted Data. PLoS ONE. 2014;9:e92209.
- Hand DJ, Christen P, Kirielle N. F*: an interpretable transformation of the F-measure. Mach Learn. 2021;110:451–6.
- Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018;47:D886–94.
- Alirezaie N, Kernohan KD, Hartley T, Majewski J, Hocking TD. ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants. Am J Hum Genet. 2018;103:474–83.
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++. PLoS Comput Biol. 2010;6:e1001025.
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99:877–85.
- Eilbeck K, Lewis SE. Sequence Ontology annotation guide. Comp Funct Genom. 2004;5:642–7.
- Lundberg S, Lee S-I. A Unified Approach to Interpreting Model Predictions. arXiv. 2017.
- Saarela M, Jauhiainen S. Comparison of feature importance measures as explanations for classification models. SN Appl Sci. 2021;3:272.
- streamlit/streamlit: Streamlit — A faster way to build and share data apps. [Internet]. [cited 2024 Feb 14]. Available from: https://github.com/streamlit/streamlit.
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006.
- Tommaso PD, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017;35:316–9.
- Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, et al. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 2014;15:480.
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res [Internet]. 2015;43:D805-11. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25355519.
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018;47:gky1015-.
- Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–61.
- Cooper GM, Stone EA, Asimenos G, Program NCS, Green ED, Batzoglou S, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res [Internet]. 2005;15:901–13. Available from: https://www.ncbi.nlm.nih.gov/pubmed/15965027.
- Cummings BB, Karczewski KJ, Kosmicki JA, Seaby EG, Watts NA, Singer-Berk M, et al. Transcript expression-aware annotation improves rare variant interpretation. Nature. 2020;581:452–8.
- Consortium TGte. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–30.
- Li, H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics. 2011;27:718–9.
- Trovó-Marqui A, Tajara E. Neurofibromin: a general outlook. Clin Genet. 2006;70:1–13.
- Scheffzek K, Welti S. Neurofibromatosis Type 1, Molecular and Cellular Biology. 2012;305–26.
- Rosenbaum T, Wimmer K. Neurofibromatosis type 1 (NF1) and associated tumors. Klinische P Diatrie [Internet]. 2014;226:309–15. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25062113.
- Peltonen S, Kallionpää RA, Peltonen J. Neurofibromatosis type 1 (NF1) gene: Beyond café au lait spots and dermal neurofibromas. Exp Dermatol. 2017;26:645–8.
- Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, Dunnen JT den. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–63.
- Fokkema IFAC, Kroon M, Hernández JAL, Asscheman D, Lugtenburg I, Hoogenboom J, et al. The LOVD3 platform: efficient genome-wide sharing of genetic variants. Eur J Hum Genet. 2021;29:1796–803.
- Minkelen R van, Bever Y van, Kromosoeto JNR, Withagen-Hermans CJ, Nieuwlaat A, Halley DJJ, et al. A clinical and genetic overview of 18 years neurofibromatosis type 1 molecular diagnostics in the Netherlands. Clin Genet. 2014;85:318–27.
- Thorisson GA, Muilu J, Brookes AJ. Genotype–phenotype databases: challenges and solutions for the post-genomic era. Nat Rev Genet. 2009;10:9–18.
- Wimmer K, Schamschula E, Wernstedt A, Traunfellner P, Amberger A, Zschocke J, et al. AG-exclusion zone revisited: Lessons to learn from 91 intronic NF1 3′ splice site mutations outside the canonical AG-dinucleotides. Hum Mutat. 2020;41:1145–56.
- Brinckmann A, Mischung C, Bässmann I, Kühnisch J, Schuelke M, Tinschert S, et al. Detection of novel NF1 mutations and rapid mutation prescreening with Pyrosequencing. ELECTROPHORESIS. 2007;28:4295–301.
- Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Roy NV, et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000;15:541–55.
- Evans DG, Bowers N, Burkitt-Wright E, Miles E, Garg S, Scott-Kitching V, et al. Comprehensive RNA Analysis of the NF1 Gene in Classically Affected NF1 Affected Individuals Meeting NIH Criteria has High Sensitivity and Mutation Negative Testing is Reassuring in Isolated Cases With Pigmentary Features Only. EBioMedicine. 2016;7:212–20.
Figure 1.
An overview of the training and testing methodology, and the architecture of DITTO model. a) 2,177,684 variants were extracted from ClinVar, filtered, and annotated for 117 distinct features using OpenCRAVAT. Following preprocessing and featuring engineering, a train:test variant split was performed. Training data was used to tune and train a neural network classifier that generates a deleteriousness score (0,1) for variant damage prediction. b) A Nextflow pipeline was developed to read VCF files, annotate variants using OpenCRAVAT, and run DITTO. c) The DITTO architecture is a single hidden layer neural network with 239 neurons in the input layer, 161 neurons in the hidden layer, and 1 output layer with 96,162 parameters.
Figure 1.
An overview of the training and testing methodology, and the architecture of DITTO model. a) 2,177,684 variants were extracted from ClinVar, filtered, and annotated for 117 distinct features using OpenCRAVAT. Following preprocessing and featuring engineering, a train:test variant split was performed. Training data was used to tune and train a neural network classifier that generates a deleteriousness score (0,1) for variant damage prediction. b) A Nextflow pipeline was developed to read VCF files, annotate variants using OpenCRAVAT, and run DITTO. c) The DITTO architecture is a single hidden layer neural network with 239 neurons in the input layer, 161 neurons in the hidden layer, and 1 output layer with 96,162 parameters.
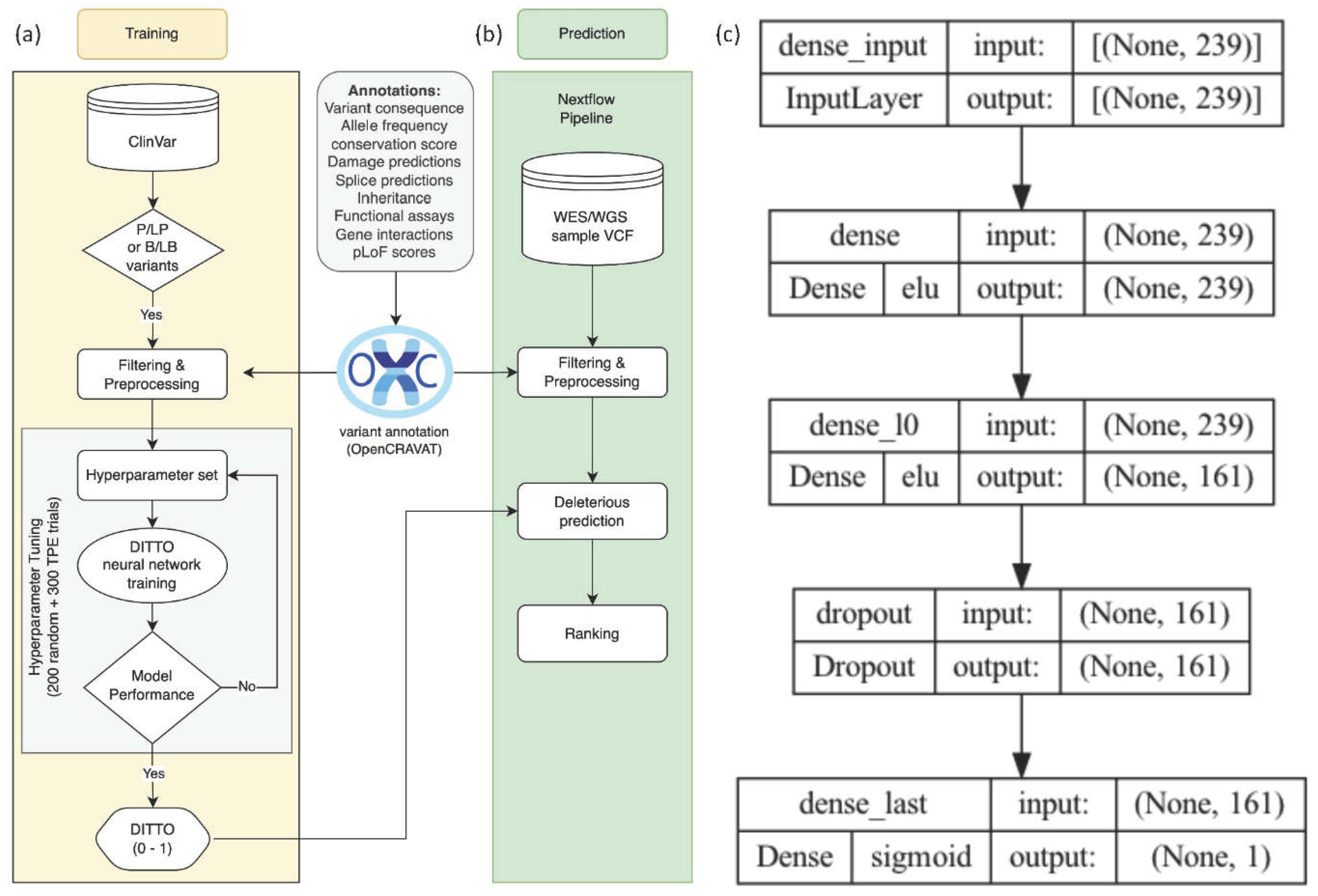
Figure 2.
Explainability in DITTO predictions: a) Application of SHAP plots aids in explainability of DITTO predictions. b) The cumulative SHAP derived feature importance plot for the overall model derived from 10,000 random variants from training. The top features are GnomAD (gnomad and gnomad3 allele frequencies), cadd.phred (CADD score) [63], funseq2.score (FunSeq ranking of non-coding regulatory cancer variants) [73], clinpred.score (ClinPred missense variant predictions) [64], synonymous_variant (variant consequence), cosmic_gene.occurences (count of number of times the gene appears in Cosmic database) [74,75], coding_Yes (likelihood of variant altering the coding sequence), Lrt.omega (evolutionary conservation of the amino acid residue) [76] and spliceai.ds_dl (SpliceAI score) [33]. c) The SHAP plot provides an indication of specific feature contributions to the model's prediction for an example variant with a DITTO score of 1.
Figure 2.
Explainability in DITTO predictions: a) Application of SHAP plots aids in explainability of DITTO predictions. b) The cumulative SHAP derived feature importance plot for the overall model derived from 10,000 random variants from training. The top features are GnomAD (gnomad and gnomad3 allele frequencies), cadd.phred (CADD score) [63], funseq2.score (FunSeq ranking of non-coding regulatory cancer variants) [73], clinpred.score (ClinPred missense variant predictions) [64], synonymous_variant (variant consequence), cosmic_gene.occurences (count of number of times the gene appears in Cosmic database) [74,75], coding_Yes (likelihood of variant altering the coding sequence), Lrt.omega (evolutionary conservation of the amino acid residue) [76] and spliceai.ds_dl (SpliceAI score) [33]. c) The SHAP plot provides an indication of specific feature contributions to the model's prediction for an example variant with a DITTO score of 1.
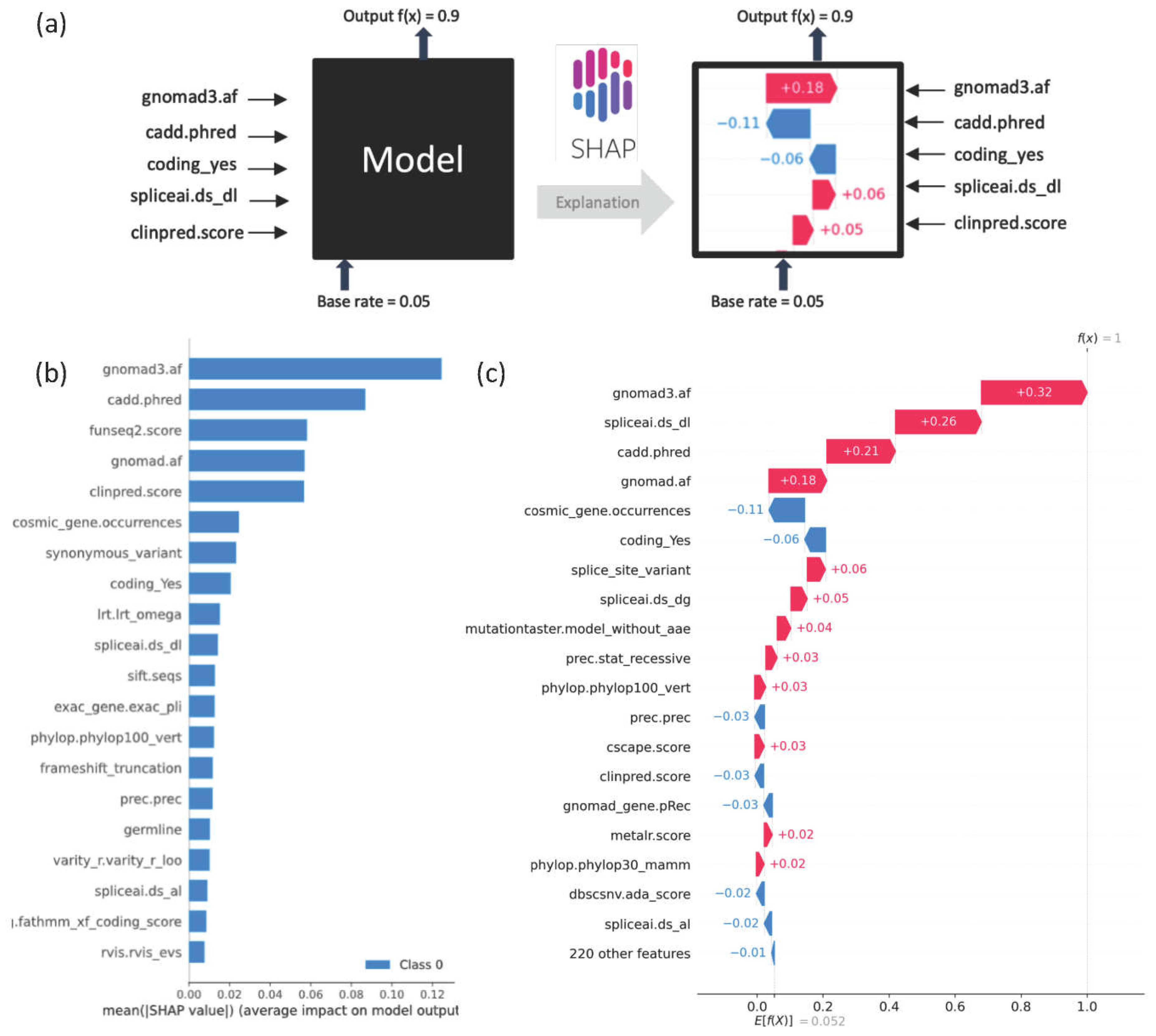
Figure 3.
Comparison of the performance of DITTO, CADD, ClinPred, MetaSVM, GERP, Revel, and SpliceAI. DITTO was noted to perform better overall than each of the other methods for variant scoring and/or classification tested. a) Receiver Operating Characteristic (ROC) and b) Precision Recall (PR) curves for each classifier are shown. c) As measured by F1 score DITTO outperformed other methods across all chromosomes. Underlying data for the plot is provided in Table S5.
Figure 3.
Comparison of the performance of DITTO, CADD, ClinPred, MetaSVM, GERP, Revel, and SpliceAI. DITTO was noted to perform better overall than each of the other methods for variant scoring and/or classification tested. a) Receiver Operating Characteristic (ROC) and b) Precision Recall (PR) curves for each classifier are shown. c) As measured by F1 score DITTO outperformed other methods across all chromosomes. Underlying data for the plot is provided in Table S5.
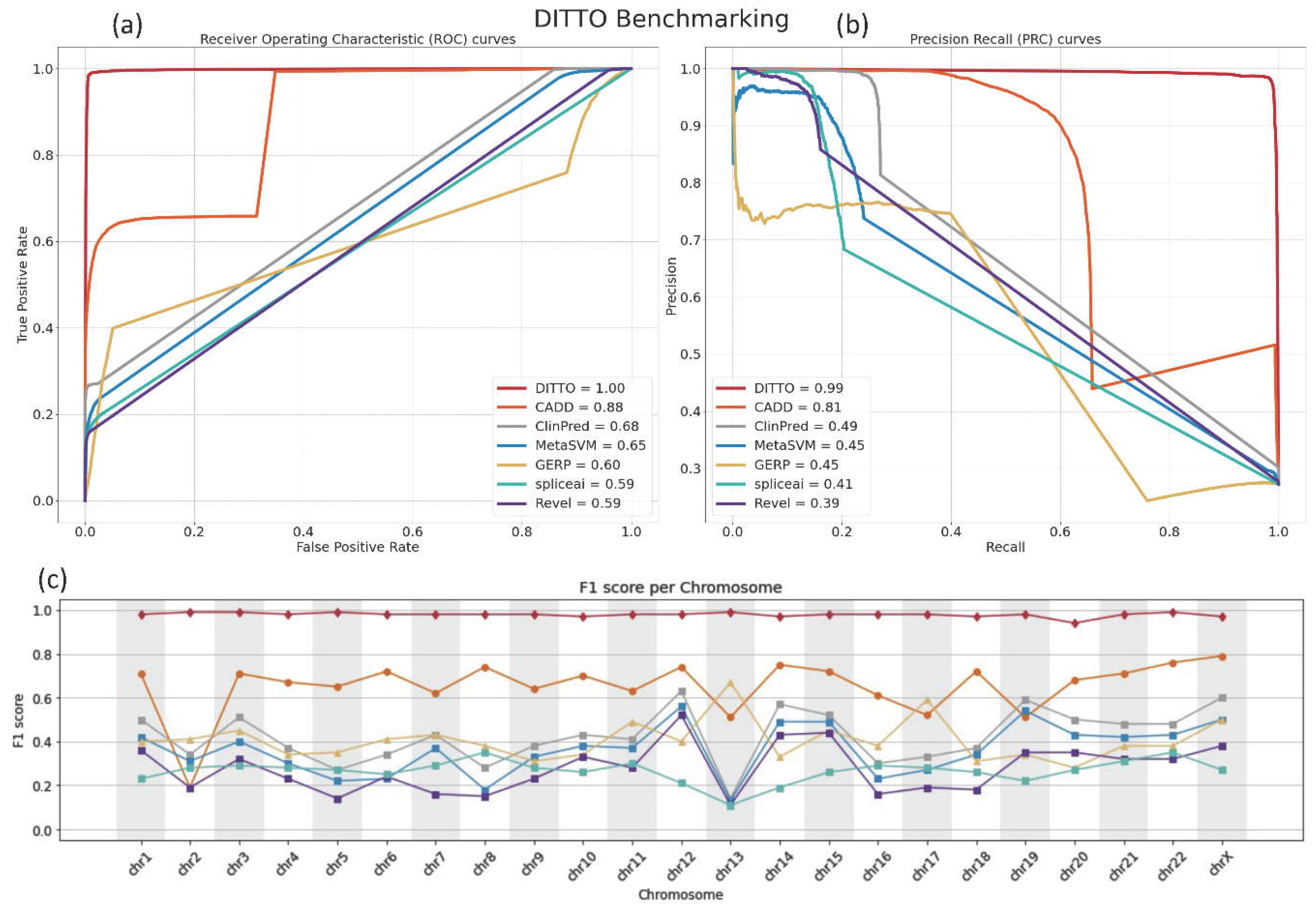
Figure 4.
F1 scores shown across variant consequences and per chromosome. The x-axis represents the chromosome, and the y-axis represents the F1 score. a) Performance across different methods tested for all variants. b-e) Scores for the fraction of variants identified as b) missense, c) intergenic, d) splice site, and e) intronic. F1 scores used to generate these plots, and for other variant consequence types not shown in this Figure due to space constraints, are provided in Table S5.
Figure 4.
F1 scores shown across variant consequences and per chromosome. The x-axis represents the chromosome, and the y-axis represents the F1 score. a) Performance across different methods tested for all variants. b-e) Scores for the fraction of variants identified as b) missense, c) intergenic, d) splice site, and e) intronic. F1 scores used to generate these plots, and for other variant consequence types not shown in this Figure due to space constraints, are provided in Table S5.
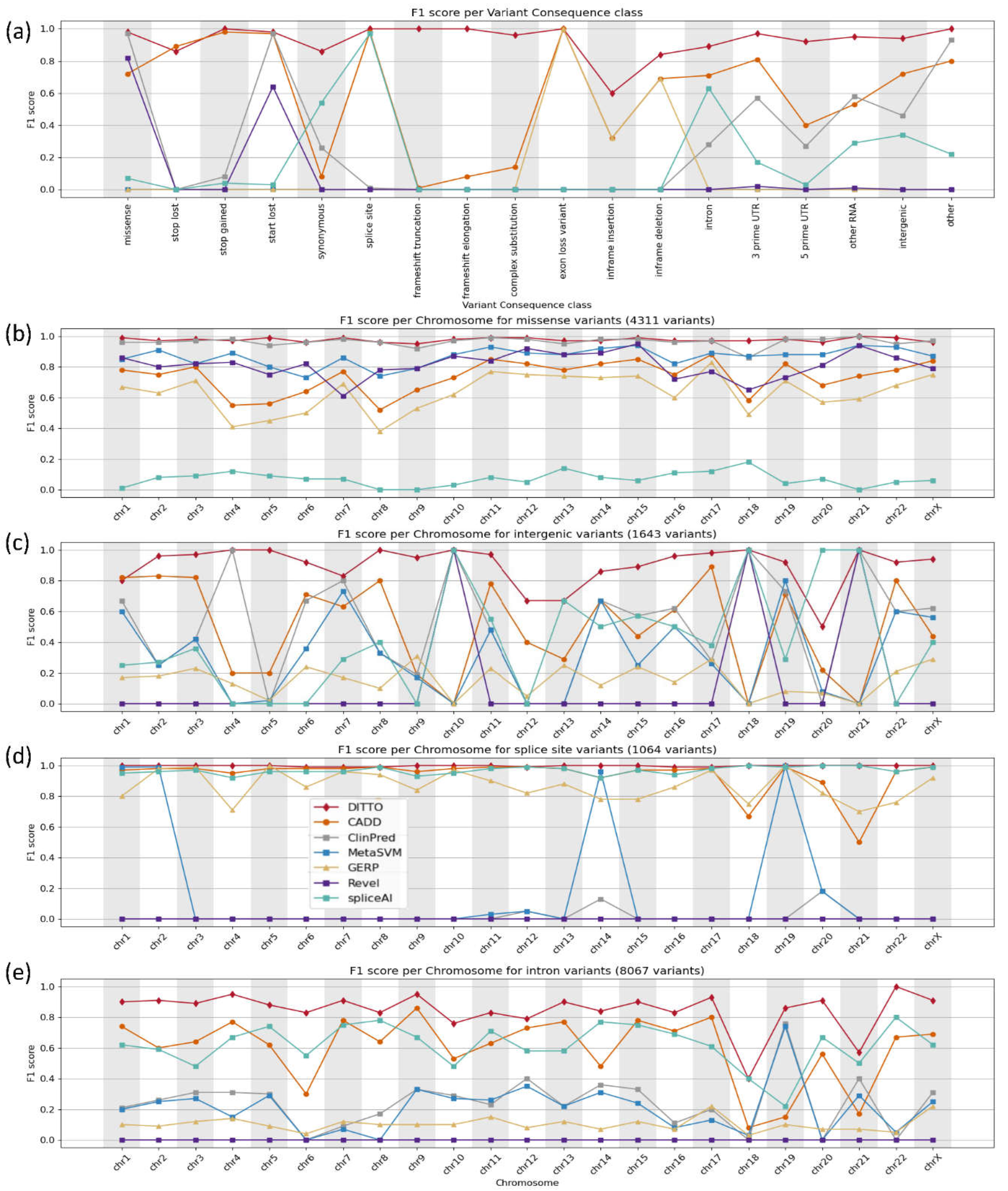
Figure 5.
SHAP plots showing top contributing features for DITTO predictions across each variant consequence. An overall and variant consequence specific plot is shown for: a) missense b) intergenic c) splice site, and d) intronic variants. Plots are limited to display only the top 15 features. The importance of features can be seen to differ amongst variant consequence although some trends, such as importance of allele frequencies, are shared.
Figure 5.
SHAP plots showing top contributing features for DITTO predictions across each variant consequence. An overall and variant consequence specific plot is shown for: a) missense b) intergenic c) splice site, and d) intronic variants. Plots are limited to display only the top 15 features. The importance of features can be seen to differ amongst variant consequence although some trends, such as importance of allele frequencies, are shared.
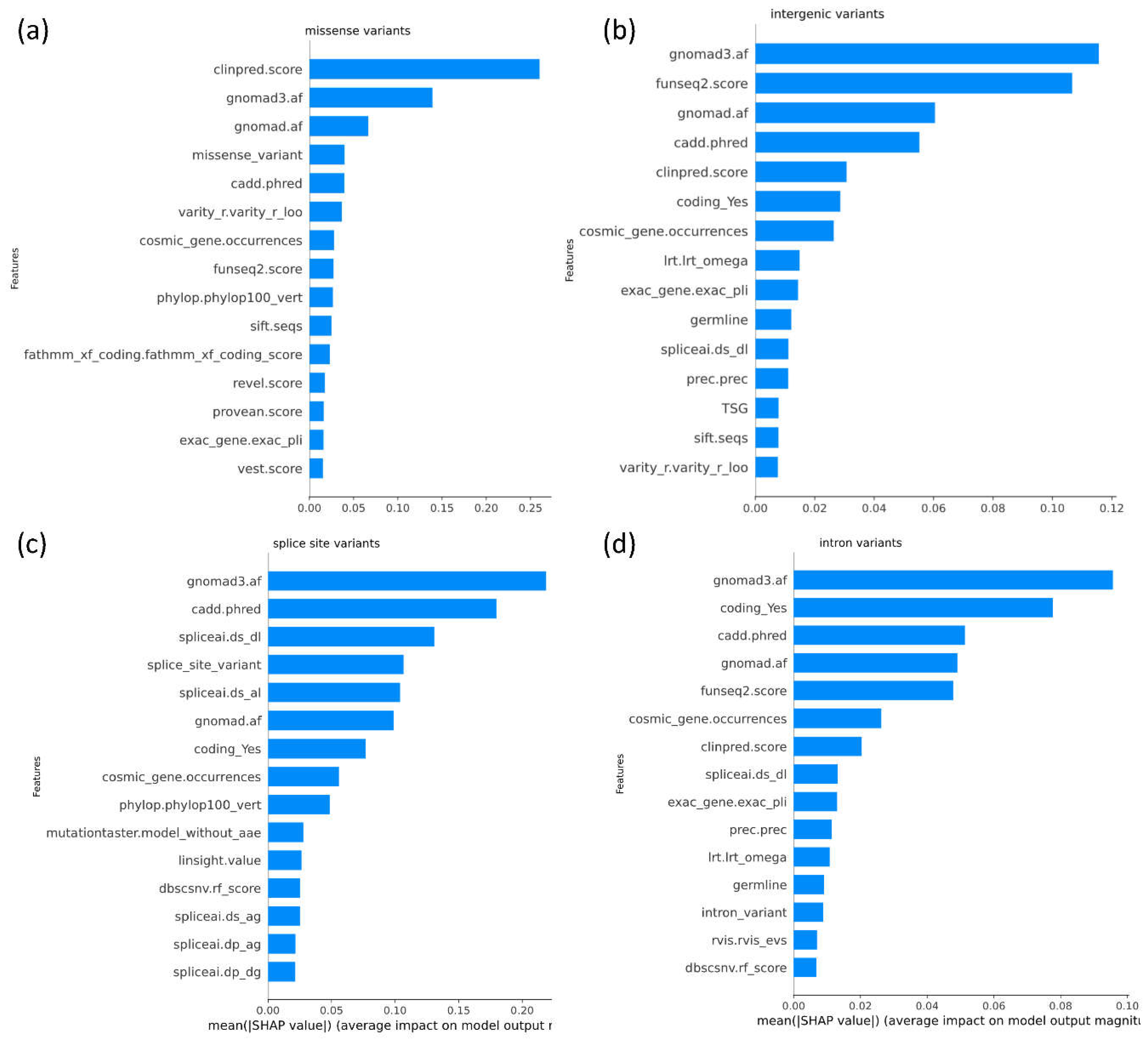
Figure 6.
DITTO webapp. In the illustrated example, a variant on Chromosome 5 is seen to be associated with nine transcripts overlapping 3 genes (EIF4E1B, MIR481 and SNCB). DITTO assigns a score of 1 to transcript “ENST00000318682.11 (EIF4E1B)”, indicating a high probability of pathogenicity. For transcript “ENST00000580852.1 (MIR481)”, DITTO assigns a score of 0.15, which leans towards a benign interpretation. Transcript-aware analysis offers valuable insights into the variable effects of a variant, highlighting potential impact across different biological contexts.
Figure 6.
DITTO webapp. In the illustrated example, a variant on Chromosome 5 is seen to be associated with nine transcripts overlapping 3 genes (EIF4E1B, MIR481 and SNCB). DITTO assigns a score of 1 to transcript “ENST00000318682.11 (EIF4E1B)”, indicating a high probability of pathogenicity. For transcript “ENST00000580852.1 (MIR481)”, DITTO assigns a score of 0.15, which leans towards a benign interpretation. Transcript-aware analysis offers valuable insights into the variable effects of a variant, highlighting potential impact across different biological contexts.
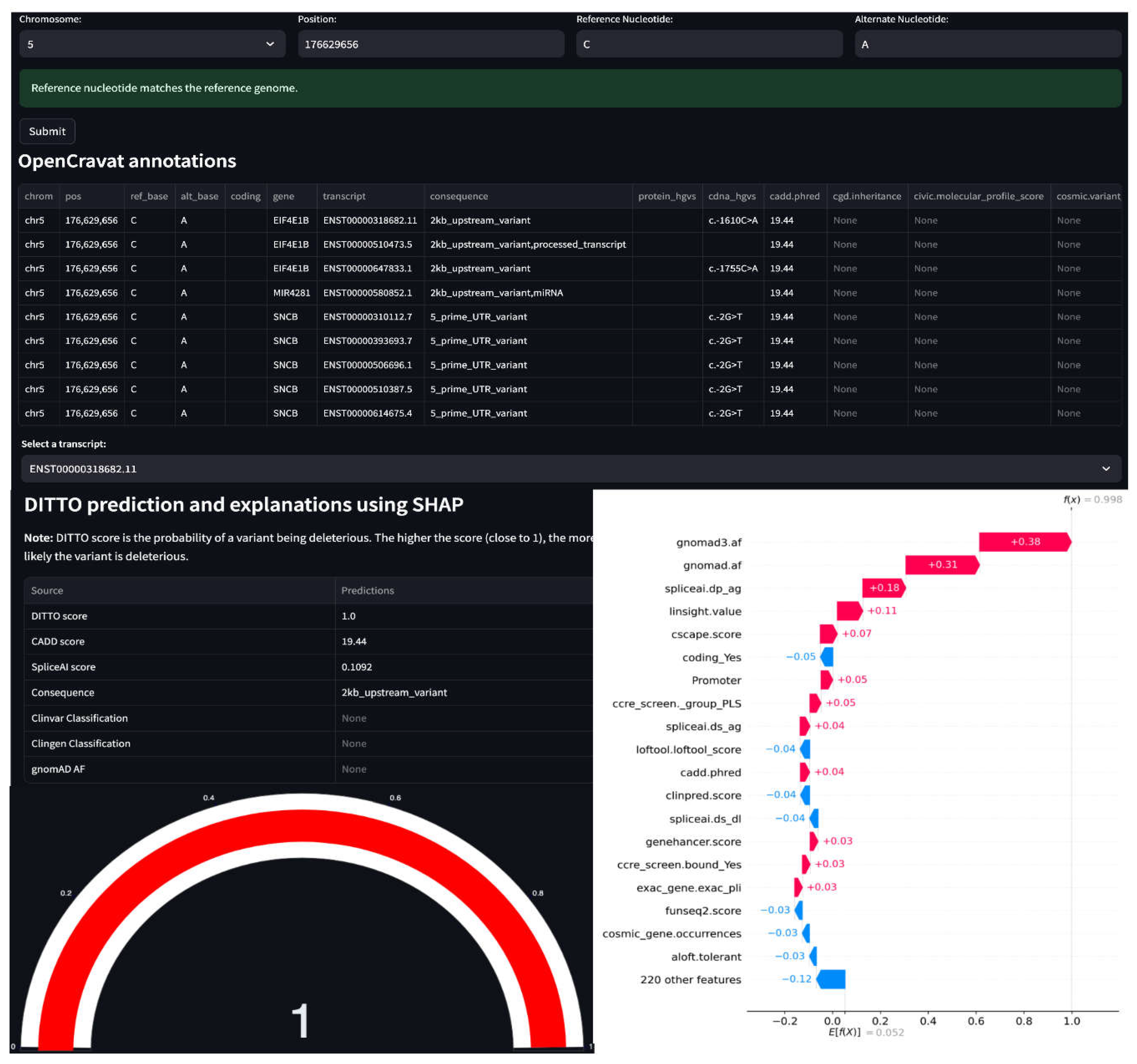
Figure 7.
Results from testing of DITTO on the NF1 LOVD derived dataset. a) A scatterplot showing the distribution of variants across the NF1 gene (x-axis) by DITTO score (y-axis), colored by ClinVar classification (blue for benign/likely benign, green for uncertain significance, red for pathogenic/likely pathogenic, and conflicting interpretations in gold). Intron-exon structure is shown by blue bar on top. The red dotted line represents a 0.91 cutoff for classification of a variant as deleterious with high confidence. b) Violin plots comparing DITTO scores against ClinVar class. c) A Sankey diagram showing the number of variants mapping from ClinVar classification to DITTO classification (see Table S6). Colors represent the same class across all plots. d) Breakdown of DITTO derived classifications for variants from the LOVD dataset not currently classified in ClinVar split by variant consequence. .
Figure 7.
Results from testing of DITTO on the NF1 LOVD derived dataset. a) A scatterplot showing the distribution of variants across the NF1 gene (x-axis) by DITTO score (y-axis), colored by ClinVar classification (blue for benign/likely benign, green for uncertain significance, red for pathogenic/likely pathogenic, and conflicting interpretations in gold). Intron-exon structure is shown by blue bar on top. The red dotted line represents a 0.91 cutoff for classification of a variant as deleterious with high confidence. b) Violin plots comparing DITTO scores against ClinVar class. c) A Sankey diagram showing the number of variants mapping from ClinVar classification to DITTO classification (see Table S6). Colors represent the same class across all plots. d) Breakdown of DITTO derived classifications for variants from the LOVD dataset not currently classified in ClinVar split by variant consequence. .
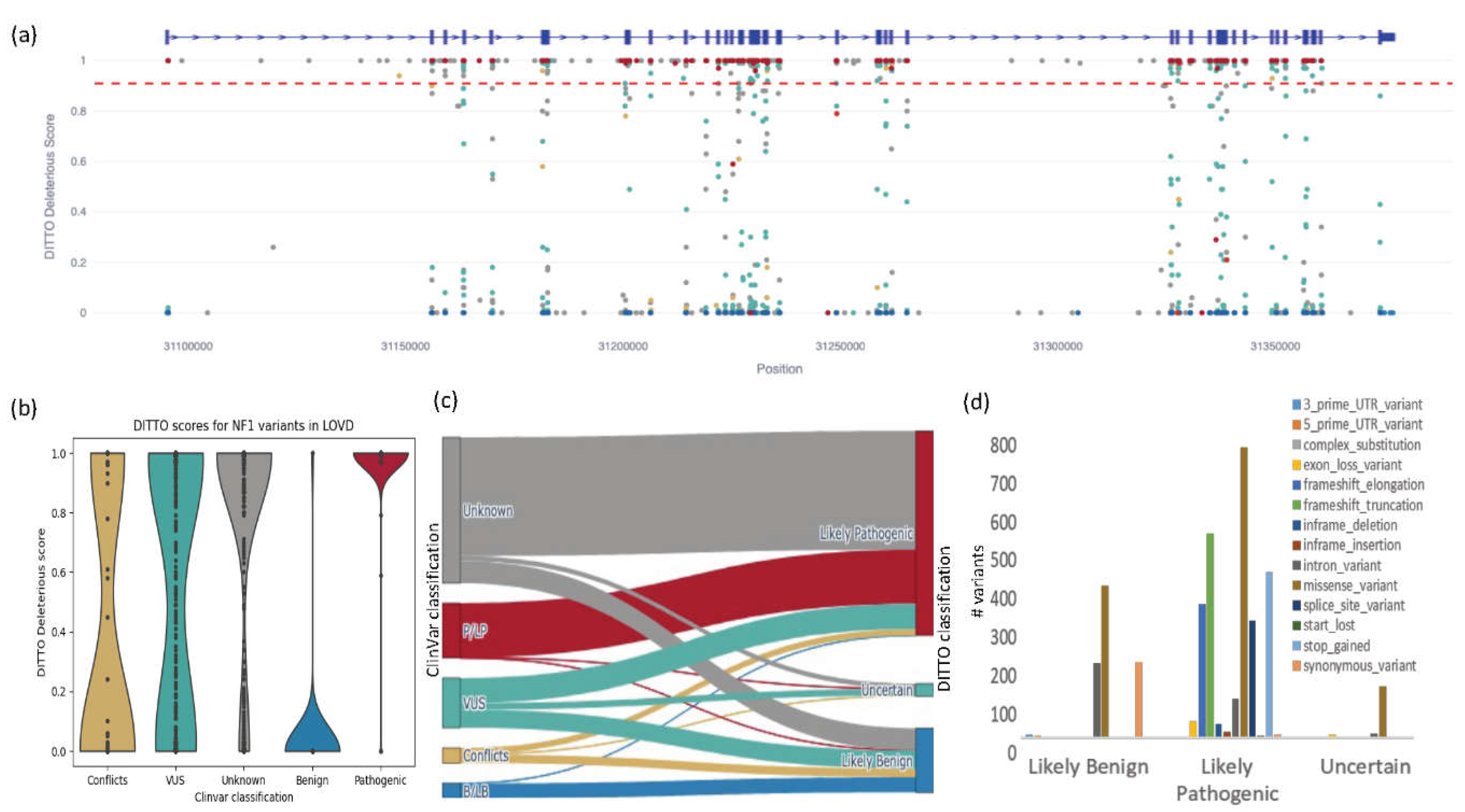

Table 1.
Breakdown of high and low impact categorized variants used for training and testing. The variant and variant-transcript test-training split is shown. The model learns from the training set and is evaluated on the separate, unseen test dataset in order to assess overfitting and model generalizability.
Table 1.
Breakdown of high and low impact categorized variants used for training and testing. The variant and variant-transcript test-training split is shown. The model learns from the training set and is evaluated on the separate, unseen test dataset in order to assess overfitting and model generalizability.
| High impact | Low impact | ||
|---|---|---|---|
| Training | variants | 40318 | 107790 |
| variant-transcript pairs | 242376 | 600283 | |
| Testing | variants | 10080 | 26947 |
| variant-transcript pairs | 60358 | 147809 | |
Table 2.
Overview of hyperparameter space for tuning of our DITTO model.
| # layers | Activation function | # neurons per layer | Kernel initializer | Dropout rate | Optimizer | Batch size |
|---|---|---|---|---|---|---|
| 1 – 30 | tanh | 1 - 200 | uniform | 0.0 – 0.9 | SGD | 10 - 1000 |
| softmax | lecun_uniform | RMSprop | ||||
| elu | normal | Adagrad | ||||
| softplus | zero | Adadelta | ||||
| softsign | glorot_normal | Adam | ||||
| relu | glorot_uniform | Adamax | ||||
| sigmoid | he_normal | Nadam | ||||
| hard_sigmoid | he_uniform | |||||
| linear |
Table 3.
DITTO performance metrics. Assessment of the DITTO model's predictive capabilities are provided across standard metrics.
Table 3.
DITTO performance metrics. Assessment of the DITTO model's predictive capabilities are provided across standard metrics.
| Accuracy | F1 | Loss | ROC AUC | PRC AUC | Recall | Precision |
|---|---|---|---|---|---|---|
| 0.99 | 0.99 | 0.07 | 0.99 | 0.98 | 0.99 | 0.99 |
Table 4.
Findings for variants that had discordant classifications. Annotations are shown for each of the five NF1 variants with discordant classification between ClinVar and DITTO. Annotations are all based on the ENST00000356175 NF1 transcript.
Table 4.
Findings for variants that had discordant classifications. Annotations are shown for each of the five NF1 variants with discordant classification between ClinVar and DITTO. Annotations are all based on the ENST00000356175 NF1 transcript.
| Consequence | cDNA (HGVS notation) | ClinVar class | ClinVar review status | DITTO | |
|---|---|---|---|---|---|
| 1 | Splice site | c.3198-3_3199del | Likely benign | no assertion criteria provided | 1 |
| 2 | synonymous | c.2709G>A (p.Val903=) | Pathogenic | criteria provided, multiple submitters, no conflicts | 0 |
| 3 | Exon loss, intronic | c.3975-1922_4111-2448delinsTTTACTTAGGT | Pathogenic | no assertion criteria provided | 0 |
| 4 | intronic | c.5206-38A>G | Likely pathogenic | criteria provided, single submitter | 0 |
| 5 | intronic | c.5750-1748_6184delinsCTA | Pathogenic | no assertion criteria provided | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



