
Submitted:
10 April 2024
Posted:
11 April 2024
You are already at the latest version
Abstract
Bursera fagaroides popularly used in México possess bioactive lignans. These compounds are low in the bark, and its extraction endangers the life of the trees. The aim of the present investigation was to search for alternative sources of cytotoxic compounds from B. fagaroides as leaves and in vitro callus cultures. The friable callus of B. fagaroides were established using a combination of phytoregulators: 4 mgL-1 of 2,4-dichlorophenoxyacetic acid (2,4-D), 1 mgL-1 Naphthaleneacetic Acid (NAA) and 1 mgL-1 Zeatin. The maximum cell growth was at day 28 with a specific growth rate μ= 0.059 days-1 and duplication time td=11.8 days. Scopoletin with a concentration of 10.7 µg/g dry weight was the main compound inducible as a phytoalexin by the addition of high concentrations of 2,4-D, as well as by the absence of nutrients in the culture medium. The com-pounds γ-sitosterol, and stigmasterol were also present. Open column chromatography was used to separate and identify: yatein, acetyl podophyllotoxin and 7'8'-dehydropodophyllotoxin in the leaves of the wild plant. Cytotoxic activity on four cancer cell lines was tested, being PC-3 prostate carcinoma (IC50 of 12.6±4.6) the most sensitive to the wild-type plant extract and HeLa cervical carcinoma (IC50 of 72±5) to the callus culture extract.
Keywords:
Bursera fagaroide
; callus culture
; leaves
; scopoletin
1. Introduction
Bursera fagaroides (Kunth) Engl. belongs to the Burseraseae family and is commonly known as “aceitillo”, “copalillo”, “copal”, “cuajiote amarillo”, “palo del diablo” or “papelillo” [1,2,3]. Its bark is yellowish gray, it is used in traditional medicine to relieve inflammation, skin tumors and warts [4]. The chemical study of B. fagaroides over the years shows the characterization of 19 lignans that have been isolated from the bark, using solvents with different polarities and these compounds present cytotoxic activity, and some have been evaluated by their antitumor effect. It has been demonstrated that some of them promote mitotic arrest, delay cell migration, and disrupt microtubules [5,6,7,8,9,10,11]; however, their extraction and purification has been carried out from the bark and requires large amounts of plant biomass. Given the magnitude of tropical deforestation in recent decades, the fact that about 70% of tropical hardwood forests have been drastically lost [12,13], and that it is also one of the most threatened ecosystems in the country, it is necessary to seek alternatives for its extraction.
Plant cell culture is a biotechnological tool that allows the cultivation of plant explants in appropriate culture media [14,15]. Plant material can be used to obtain callus cultures that can be scaled up in liquid systems, allowing controlled production of secondary metabolites independent of environmental conditions. For several years, there has been a particular interest in the establishment of calluses from various plants for mass propagation or production of compounds of interest to be scaled up in suspension cultures [16,17,18]. However, research in the Burseraceae family is limited.
In vitro cultures of some species of the Burseraceae family have been established and compounds of biological interest have been obtained, such as guggulsterone (plant sterol that inhibits the growth of a wide variety of tumor cells and induces apoptosis). This compound was synthesized in the callus of Commiphora wightii grown in all treatments with 2, 4-D and kinetin [19]. On the other hand, callus cultures and cell suspensions of Commiphora gileadensis can be used to produce secondary metabolites (quercetin, 10-hydroxycamptothecin, floginax and puromycin) with antimicrobial and cytotoxic effects [20]. Another study was the production of boswellic acid (anti-inflammatory and anti-arthritic agent) in calluses obtained from root, stem, cotyledon, and leaf explants of Boswellia serrata [21].
Bursera fagaroides is a plant with medicinal, antitumor, and cytotoxic properties due to the different components it synthesizes. The traditional method of extraction or chemical synthesis has been done using the bark, but this practice is considered unsustainable due to high production costs, low yields, and severe damage to the plant. Therefore, it is necessary to look for attractive alternatives for its production. The aim of this work was to study the metabolism present in friable callus cultures of Bursera fagaroides and to compare their production with that of the leaves from the wild plant.
2. Results
2.1. Obtaining Leaves from the Cultivation by Cuttings
Cuttings grown in pots with coconut fiber and maintained under greenhouse conditions showed good development and growth as shown in Figure 1. At 12 days the first leaf buds of approximately 0.5 cm in length developed (1A) and at 25- and 70-days healthy leaves of between 2 and 3 cm in length developed (1B and 1C).
2.2. Callus Induction
The disinfection process allowed obtaining 90% of leaves free of contamination in the different treatments and conditions tested (Figure 2A). The MS medium and using a combination of 2 mgL-1 of 2,4-D and 2 mgL-1 of Kinetin had 86.67% of dedifferentiated cells with respect to the rest of the concentrations tested as summarized in Table 1. However, as shown in Figure 2B, there was a higher proliferation of cells in the center of the explant after 30 days, in addition, a more compact callus developed, of green coloration that, after the subcultures, oxidized and turned brown, so they were transferred to B5 medium using 4 mgL-1 of 2,4-D, 1 mgL-1 of NAA and 1 mgL-1 of Zeatin. As shown in Figure 2C, this process promoted a better callus formation and growth response with more friable characteristics, and individual cells were observed with a predominance of elongated morphology, but circular cells were also observed with 98% cell viability (Figure 2D).
2.3. Callus Culture Growth Kinetics
The growth curve of the callus culture with the weights of the fresh and dry biomass is shown in Figure 3A. The growth kinetics allowed to obtain a sigmoidal behavior during 63 days in which an adaptation phase is from days 1 to 14, followed by an exponential phase from days 14 to 42 reaching a specific growth rate of µ = 0.059 days-1 and a td =11.8 days, then a stationary phase between days 42 to 56 and finally a death phase that is seen from days 56 to 63. It should be noted that the highest growth rate (GI) was observed on day 28.
2.4. Appearance and Percentage of Viability of the Callus Culture
The cell appearance and viability of the callus culture is shown in Figure 3. The culture started with 81% cell viability that increased slightly to 91% during the adaptation phase (days 0 to 14), here, the cultures presented a friable consistency with a light yellow coloration, later in the exponential phase (days 21 to 42), the callus took a coloration that went from light green to dark green with an average of between 80 to 90% viability, then in the stationary and death phases the cultures turned dark brown until reaching 56% viability.
2.5. Chemical Profile of B. fagaroides Callus Culture
The CH2Cl2 extract of the dry plant biomass from each of the days of B. fagaroides growth kinetics was first analyzed qualitatively by TLC (Figure 4A). In the adaptation phase, a minimal production of secondary metabolites was observed compared to the stationary phase, where low polarity compounds associated with cell growth were observed. However, in the stationary phase, their production increased considerably, maintaining the maximum concentration from days 42 to 56. This analysis suggests that the callus culture of this species synthesizes compounds of lower polarity in the stationary phase, possibly in response to the absence of nutrients in the medium. It should be noted that, during the culture, one of the major compounds was the one that presented an Rf equal to 0.35, with a bright blue coloration observed at 360 nm, which was obtained constantly from day 14 onwards, being maintained until the end of the exponential phase, decreasing its presence considerably in the cell death phase. This molecule was compared with the scopoletin standard (TLC-S). To confirm its presence, first a commercial standard was used and analyzed by HPLC at 360 nm, which presented a RT= 10.27 minutes (Figure 4B), and then each of the spots of the callus culture were also analyzed. Their concentration expressed in μg of compound per g of dry biomass was quantified. As can be seen in Figure 4C, on 21 day corresponding to the beginning of the exponential phase, a first production peak of the metabolite (10.7 μg) is observed, which decreases as the cell is in growth and cell division; on the other hand, when the cells enter the stationary phase, its production increases again, observing that at the end of this phase (day 56), there was a second production peak (9.7 μg), possibly in response to the absence of nutrients.
Finally, the scopoletin production of the extract from the callus culture was compared against the extract from the leaves of the wild plant and it was observed that the accumulation in the wild plant is low with respect to the callus culture (Figure 4D).
After analyzing the production of scopoletin, open column fractionation of the callus culture allowed the identification of the following compounds by gas chromatography coupled to mass spectrometry (GC-MS): gamma-sitosterol (1), dehydrodiosgenin (2), stigmasterol (3), and scopoletin (4) (Figure 5).
2.6. Chemical Profile of wild B. fagaroides Leaves
Phytochemical analysis of the CH2Cl2 extract of the wild-type plant allowed the identification of three lignan-type compounds namely: yatein (5), 7‘8‘-dehydropodophyllotoxin (6) and acetyl podophyllotoxin (7) and two pentacyclic triterpenes; friedelin (8) and lupeol (9) as well as α-tocospiro-A (10) (Figure 6). Compounds 1-3 were isolated by column chromatography as described in section 4.7.2 and identified by 1H and 13C NMR analysis and compared with literature data [5,6]. Compounds 4-6 were identified by Gas Chromatography coupled to Mass Spectrometry (GC-MS) (NIST 1.7a).
2.7. Evaluation of Cytotoxic Activity
The results of the cytotoxic evaluation of the CH2Cl2 extracts of callus culture and wild plant are displayed in Table 2. It was generally observed that the wild-type plant extract showed important cytotoxicity over HepG2 (IC50 =20.3±3.38), HeLa (IC50 =18.6±5.2), and PC3 (IC50 = 12.6±4.6), this last being the most sensitive. On the other hand, this extract did not show activity against H1299 (IC50 = 69.1±6.2) and comparing with its effect against the non-cancerous cell line HFF-1 (IC50 = 79.6±5.2), selectivity for cancer cell lines was observed. Treatment of the five cell lines with the callus culture extract resulted in a negligible effect against all the cell lines tested: HepG2 (IC50 = 155.4±4.95), HeLa (IC50 = 72±5), PC3 (IC50 = 141V5.8), H1299 (IC50 = 210.4±5) and HFF-1 (IC50 = 95.4±6.2). The HeLa cell line was the most sensitive (IC50 = 72±5).
3. Discussion
The accumulation of secondary metabolites in plants is low and slow because it is spatially and temporally regulated. Indeed, it occurs in specific cells, organs, and tissues in specific phases of the plant life cycle, under seasonal or stress conditions [22,23]. Therefore, an alternative to produce these metabolites of interest is the use of in vitro callus culture systems. Callus induction involves a process of dedifferentiation and constant cell division, which depends on the type of explant, culture medium, type, concentration, and combination of plant growth regulators [24,25,26].
The leaves produced by the cultivation of cuttings of B. fagaroides were 90% free of pathogens, so this method was effective not only for obtaining plant material but also for the dedifferentiation of explants and obtention of calluses.
However, there are few studies on callus formation and development in the Burseraceae family because some species are arboreal, woody, with high content of endophytic fungi, some of them are endangered and highly recalcitrant, which leads to reduced germination [13,19,27]. In addition, the copales and cuajiotes of the Bursera genus present difficulties for germination and/or obtaining seedlings and calluses from seeds. These difficulties are because the fruit has relatively hard covers and passes through the digestive system of certain species of birds, where it softens and facilitates germination [28,29]. Another option is to use pregermination treatments such as hydrochloric acid immersion or mechanical scarification to obtain plants in vitro.
The cell wall that covers a plant cell prevents it from expanding. The wall structure loosens and is regulated by small molecules called auxins that allow the expansion of the intracellular vacuole by incorporating water, leading to its elongation [30].
It has been reported that the use of some auxins to obtain callus in the genus Bursera, such as indole butyric acid (IBA) and naphthalene acetic acid (NAA), allowed the formation of 25-87% of callus [27,31,32].
Among the different types of auxins, is 2,4-D, a synthetic auxin that causes rapid cell proliferation, formation, and optimal callus growth, besides being the most widely used growth regulator in cereal tissue culture, it has been used for callus induction by initiating dedifferentiation from day 5 in cultures [33,34,35].
Some studies suggest that when the concentration of 2,4-D is equal to or higher than 3 mgl-1, disorganized growth is observed which favors callus formation, but when increasing concentrations are used, they cause inhibition of cell division [33,34,36].
In this study, the best callus induction response was observed with concentrations of 4 mgL-1 of 2,4-D in combination with NAA and Zeatin 1:1 mgL-1 in B5 medium, producing a yellow-light colored callus, friable, without oxidation, with high percentages of cell viability, with circular and elongated morphologies in the different growth stages (Figure 2). These results partially coincide with those reported by Souza-Pádua and collaborators [37], who showed two types of callus morphologies in Coffea arabica. It should be noted that in this medium and with the combination of PGR previously described, the accumulation of biomass was favored, allowing the characterization of the culture.
The growth curve determined by the points analyzed, reported in fresh and dry biomass, presented a sigmoid curve with four distinct phases (adaptation, exponential, stationary and death) (Figure 3A).
In the exponential phase, the kinetic parameters μ =0.059 days-1, td = 11.76 days, with a growth rate greater than 28 days were obtained. This is the first report on the characterization of a callus culture on Bursera fagaroides. Mishra and Kumar [19] performed callus induction on Commiphora wightii (Burseraceae) using 5 mg-1 of 2,4-D and 0.5 mgL-1 of BAP, obtaining 5.78 g of maximum fresh biomass. On the other hand, Al-Abdallat and collaborators [20], obtained 32.41 g of fresh biomass and 0.89 g of dry biomass in a callus culture of Commiphora gileadensis (Burseraceae) using high concentrations of auxin with respect to cytokinin. In our study we obtain higher concentrations of dry biomass.
It is important to perform the growth kinetics to decide the replanting of the crop and to determine in which stage the highest amount of the secondary metabolite of interest is produced.
With the aim to identify the blue-fluorescent compound observed by TLC (Figure 4A) a GC-MS analysis of the extract from callus biomass at different culture stages was performed. This analysis showed the presence of a major compound, further identified as scopoletin (7-hydroxy-6-methoxy coumarin) 14 to 56 days, it is a simple coumarin derived from the C6-C3 carbon skeleton.
It should be noted that in the B. fagaroides callus culture, scopoletin had two points of production (Figure 4C), the first one being associated with the beginning of cell growth, with a maximum peak at day 21, where it decreased drastically thereafter. This may be because the culture medium is supplemented with 4 mgL-1 of 2,4-D. Previous studies have reported that when scopoletin plus 1 mgL-1 of 2,4-D is added endogenously to Nicotiana tabaccum callus cells, they absorb scopoletin from the medium and it is accumulate it in its glycoconjugate (scopolin) mainly in the vacuoles [38,39]. Likewise, some elicitors have been used for its synthesis. For example, it has been reported that the synthesis of scopoletin is associated with cell growth in callus and suspension cultures of Tilia americana after increasing the copper concentration up to 1.2 μM [40], and copper sulfate stimulates its production in suspension cultures of Angelica archangelica [41].
The second point of scopoletin production started on day 42 (Figure 4C) with a peak on day 56, i.e., it was associated with the lack of nutrients in the culture medium. It should be noted that this compound is one of the phytoalexins induced by different types of stress [40,41,42].
There are very few reports on the effect of natural coumarins on cell division and callogenesis in plant tissues cultured in vitro [43]. The culture of Ammi majus callus on Linsmaier-Skoog’s medium with the PGRs NAA and BAP (1:1 mgL-1) was the best for the accumulation of coumarins (furan coumarins) that favors the formation of embryogenic callus. The levels of these metabolites in the in vitro culture were several times higher than those found in the vegetative organs of the plants [44].
It should be noted that, this metabolite has been found to possess antibacterial, cytotoxic, and antifungal activities [45,46,47,48], and prevent weight gain and lipemia effect in mice [49]. It has been isolated from several plant families such as Apiaceae, Rutaceae, Asteraceae, Fabaceae to mention a few [50]. Also, it has also been found in the bark of some species of the genus Bursera such as B. grandifolia [49], B. serrata [47,48], B. fagaroides [51], as well as in B. simuraba leaves [52]. However, so far there are no reports of its presence in the leaves of B. fagaroides.
In the future, it is important to start liquid suspension cultures that can be scale-up into bioreactors.
4. Materials and Methods
4.1. Identification of Plant Material
A specimen of Bursera fagaroides (Kunth) Engl. was collected on July 2021 at the locality “El Mango”, in the municipality of Puente de Ixtla (14 Q 046300, UTM 2043732, DDQH9), altitude: 1382 m. by Biologist Fidel Ocampo Bautista, and was identified by Biologist Gabriel Flores Franco. Deposited in the HUMU Herbarium of the Universidad Autónoma del Estado de Morelos (UAEM), with voucher number 39796.
4.2. Obtaining of Plant Material
Fragments of approximately 20 cm in length were randomly cut from terminal branches of an adult tree and submerged in water for 12 hours. Subsequently, a rooting agent (Raizone Plus) was added to each tip and 10 of them were placed in a pot containing coconut fiber as substrate. The experiment was carried out in triplicate. The pots were placed in greenhouse conditions at 41±2° C, watered every third day by nebulization for 139 days. The plant material obtained was used for the establishment of the in vitro culture.
4.3. Callus Induction
Young leaves of between 2 and 3 cm in length developed from the stake culture were used as explants for callus induction. They were washed with sterile miliQ water and neutral soap (Hycel) for 4 min, then an antifungal was added, followed by successive washes with 40% and 10% sodium hypochlorite for 3 min each. Subsequently, inside the laminar flow hood, they were immersed with a broad-spectrum antibiotic (Curamycin Agricola 500) for 4 minutes, rinsed with sterile miliQ water, dried with filter paper, and finally cuts were made with a scalpel and placed in 9 cm diameter Petri dishes containing 25 mL of Murashige and Skoog (1962) culture medium, using as carbon source 30 gL-1 of sucrose and supplemented with 6 gL-1 of Polyvinylpyrrolidone (PVP), 10 mgL-1 of gentamicin, 1. 5 gL-1 of phytagel, adjusting the pH to 5.7 plus phytoregulators. The culture medium was previously sterilized at 121 °C, 15 psi, for 15 minutes. Ten explants were seeded per Petri dish with three replicates and placed in photoperiod (16/8 light-dark) at 25±2 °C for 30 days. To determine which combination induced better callus formation, ten combinations with MS medium, plus B5 medium (Gamborg, 1968), ten combinations with of 2,4-D plus Kinetin (2,0; 2,1; 2,2; 3,1; 3,2; 3,3; 4,1; 4,2; 4,3 y 4,4 mgL-1) were used. Callus obtained from the 2,4-D: Kinetin combination (2:2 mgL-1), which showed better appearance, were transferred to B5 medium containing 4 mgL-1 of 2,4-D, 1mg/L-1 of NAA and 1 mgL-1 of Zeatin, using as carbon source sucrose (30 gL-1) and supplemented with PVP (6 gL-1), phytagel (1. 5 gL-1) and adjusting the pH to 5.7. Friable calluses were obtained and reseeded every 28 days for 6 months. The cultures were incubated at 25±2 °C in a photoperiod with white-fluorescent light (50 µmol m-2s-1).
4.4. Growth Kinetics of Callus Culture
Growth kinetics was performed by triplicate by placing an inoculum with 1 g of fresh biomass per flask with the previously mentioned culture conditions and incubating in a photoperiod of 16 h light / 8 hours dark at 25±2 °C. Fresh and dry biomass was determined at seven-days intervals after inoculation up to 63 days. During the assay, the weight of fresh biomass was recorded by placing it on a previously weighed filter paper, subsequently to determine the dry biomass, the plant material was lyophilized at -50 °C and 0.1-0.3 mBar until a constant weight was reached. The callus growth curve was plotted with the culture time against the weights of the fresh and dry biomasses to know the phases of cell growth (adaptation, exponential, stationary and death). Kinetic parameters were calculated: the specific growth rate µ (time -1), the cell doubling time (td) using the following formula td=ln(2)/μ [15,16,17,18,19,20,21,22,23] and the growth index (GI) given by the final dry biomass minus the initial dry biomass divided by the initial dry biomass.
4.5. Cell Morphology and Viability
Cell viability was measured with the fluorescein diacetate method (FAD) [53]. The percentage viability of each sample was determined based on the number of cells that stained green divided by the total number of cells counted. For both cell morphology and viability, observations were made using an epifluorescence microscope [54]. Objectives with 40X and 10X magnification were used.
4.6. Phytochemical Analysis of the Callus Culture
4.6.1. Obtaining the Extracts
The dry plant biomass of the callus collected from the different growth stages was used to determine the production of scopoletin with respect to time of growth. For this purpose, the samples were ground to a fine powder. The pulverized material was macerated in triplicate with dichloromethane (CH2Cl2) for 24 h with a ratio of 1 g of plant material per 10 mL of solvent, the samples were concentrated by separating the solvent by distillation at reduced pressure using a rotary evaporator (Büchi R-100) [5,6]. The product obtained was brought to complete dryness at room temperature and the procedure was repeated three times. The yield expressed in percentage was determined for each of the extracts and stored until further use.
To separate, obtain and identify the majority compounds, a sample of 530 g of fresh callus biomass from 15 to 35 days of culture was lyophilized obtaining 23 g of dry biomass, crushed until pulverized, then macerated with CH2Cl2 following the procedure previously described, obtaining 3.3 g of extract (0.6% yield).
4.6.2. Chemical Profile and Identification of Major Compounds
Once the extracts from each day of callus culture were obtained, the chemical profile was analyzed.
First, Thin Layer Chromatography (TLC, ALUGRAM® SIL G/UV 254 silica gel plates) was performed, using hexane-ethyl acetate (5:5) as the elution system. The plates without developer were exposed to irradiation with shortwave UV light at 254 nm, longwave at 360 nm, and then ammoniated with ammoniated ceric sulfate [(NH4)4Ce(SO4)4*2H2O] as a chemical developer at 1% in H2SO4 2N. The plates without chemical treatment and exposed to irradiation with ultraviolet light at 360 nm allowed the identification of scopoletin (bright blue spot with Rf=0.32) as the major compound.
Subsequently, to confirm the presence of scopoletin in the callus culture, a 10 µL aliquot of the extract was injected with CH2Cl2 from the culture at day 49 (2mg/mL), using a high-performance liquid chromatograph (Waters 996), with binary pumps (2695), coupled to a diode array detector (2996) with a UV detection range from 190 to 600 nm and operated by the Millenium System Manager Software (Empower 1) [49], using an elution system of water/acetonitrile mixture (70:30), and maintaining a constant flow rate of 1 mL/min for 30 min. The column used was a Supelco RP-18 (5μm, 4.6 mm x 25cm).
For the quantification of scopoletin a calibration curve was performed. A commercial standard was used as reference (Sigma Aldrich). Five different concentrations were used: 50, 25, 12.5, 6.25 and 3.125 µg/mL of the compound dissolved in HPLC grade acetonitrile. The chromatogram was read at a wavelength of 350 nm.
Finally, the production of scopoletin was quantified for each point of the growth kinetics by plotting the dry biomass against the production of this metabolite. The analysis was performed in triplicate and the results were shown as µg/mg dry biomass. Finally, the results obtained were compared against the extract of the leaves of the wild plant.
After performing the identification and quantification analysis of scopoletin on the different days of callus culture, we proceeded to separate and identify the majority compounds. The extract with CH2Cl2 (3.3 g) obtained from the callus culture from days 15 to 35 was fractionated by Open Column Chromatography, previously packed with silica gel (6 g, 70-230 mesh; Merck). Fractions of 150 mL were collected, obtaining 182 fractions monitored by TLC. The fractions that showed similarity in TLC were grouped obtaining 6 groups: NPM 1 (1-25) eluted with hexane-acetate (95:5), NPM 2 (26) eluted with hexane-acetate (90:10), NPM 3 (27-53) eluted with hexane-acetate (85:15), NPM 4 (107-114), NPM 5 (115-151) eluted with hexane-acetate (75:25) and NPM 6 (152-182) eluted with hexane-acetate (70:30).
NPM 2 group (white crystals, 12 mg) showed by TLC to contain a single compound, and the GC/MS analysis indicated that it was constituted by gamma sitosterol (1), NPM 4 group (light yellow wax, 4.9 mg) presented a mixture of the compounds dehydrodiosgenin (2) and stigmaesterol (3), finally NPM 6 group (light yellow solid, 28.1 mg) indicated the presence of a mixture of several compounds, the main was scopoletin (4).
4.7. Phytochemical Analysis of Wild Plant Leaves
4.7.1. Obtaining the Extracts
To obtain the extracts from the wild plant, fresh leaves were first collected and dried in the open air and under the shade for 20 days, obtaining 1.2 kg of dry plant material. Subsequently, it was ground to a particle size of 2-5 mm, a maceration was performed in triplicate with CH2Cl2, following the same protocol. A 15.3 g extract (1.25% yield) was obtained.
4.7.2. Chemical Profile and Identification of Major Compounds
The isolation and purification of compounds from the CH2Cl2 extracts was carried out by a successive fractionation by Column Chromatography using various organics solvents. From the last open column fractionation on silica gel 60 (15 g, 70-230 mesh; Merck) eluted with hexane-ethyl acetate. Five groups were obtained., from these fractions: NPM-40-2, NPM-40-3, and NPM-40-4 were passed through activated carbon to remove the color.
The final separation of the compounds was performed by HPLC analysis as reported by Rojas-Sepúlveda in 2012 [5], because the three samples were found to contain a mixture of several compounds. For this purpose, a semi-preparative X-Terra RP-18 reversed-phase column (7.8 mm × 50 mm; 5 µm particle size) was used; the mobile phase was 25:75 acetonitrile-water at a flow rate of 1.0 mL min-1 with a separation module and a diode array detector with detection at 215, 250, 300 and 350 nm and a flow rate of 1 mL/min, with an isocratic profile for 30 min. Each injection was 10 μL and the solvents used were HPLC grade.
Fraction NPM-40-2 was purified by HPLC to afford 2.9 mg of a compound with tR = 13.57 and identify as yatein (5, tR = 13.57 min). Similarly, the purification of fraction NPM-40-3 allowed the obtaining of 1.6 mg 7‘8‘-dehydropodophyllophyllotoxin (6, tR = 11.76 min). Finally, HPLC fractionation of NPM-40-4 afforded 1.5 mg of acetyl podophyllotoxin (7, tR = 16.24 min). It should be noted that in addition to the fractionation, the volatile compounds from the total extract were separated and analyzed by GC/MS, showing friedelin (8), lupeol (9) and α-tocospiro-A (10) as the main compounds.
4.8. Analysis of Callus and Leaf Extracts by Gas Chromatography Coupled to Mass Spectrometry (GC/MS)
The dichloromethane extracts of the callus and wild-type plant were analyzed by GC-MS Agilent GC 6890, MSD 5973N (Agilent Technologies, USA) to determine the chemical composition of the major compounds. The analysis was performed using the HP-5MS column (30 mm × 0.25 mm × 0.25 µm). The carrier gas was helium with a gas flow rate of 1 mL/min and a linear velocity of 37 cm/s. The injector temperature was set to 250 ◦C. Splitless. The initial oven temperature was 40 ◦C and was increased to 250 ◦C for 5 min and 10 ◦C/min, and the final temperature was maintained at 285 ◦C for 20 min. The mass spectrometer was operated in the electron ionization mode at 70 eV and the electron multiplier voltage at 1859 V. Compounds were identified by comparison of retention times and fragmentation patterns of reference compounds from the NIST database version 1.7a [55].
4.9. Cytotoxicity Assay
The cytotoxic activity of the CH2Cl2 extracts from the leaves of the wild plant and the callus culture was evaluated using the MTS method [56]. Different human cancer cell lines were used such as HepG2 (hepatocellular carcinoma), HeLa (cervical carcinoma), PC3 (prostate carcinoma) and H1299 (lung carcinoma). The cell lines were obtained from ATCC. (American Type Culture Collection USA). We also included an immortalized human fibroblast cell line (HFF-1) as a control of non-cancerous cells. The PC3 and H1299 cell lines were cultured in RPMI-1640 medium (Sigma Aldrich, St. Louis, MO, USA), while HepG2, HeLa, MCF7, and HFF-1 were cultured in DMEM medium (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (SFB, Invitrogen) and 2 mM glutamine. All cultures were incubated at 37°C in a 5% CO2 atmosphere.
Cells (5 × 103 cells/well) were seeded in 96-well plates to initiate cytotoxic evaluation. The compounds were solubilized in DMSO, the concentrations used for the evaluation of the compounds in the cancer cell lines were 200, 100, 50, 25 and 12.5 µg/mL, while in HFF-1 cells they were 400, 200, 100, 50, 25 µg/mL, after treatment the cells were incubated at 37°C in 5% CO2 atmosphere for 48 h. Paclitaxel was used as a positive control. For determining the number of viable cells in proliferation we used a CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI, USA), following the manufacturer’s instructions. Cell viability was determined by absorbance at 450 nm using an automated ELISA reader (Promega, Madison, WI, USA). The experiments were conducted in triplicate in three independent experiments. Data were analyzed using the Prism 8.0 statistical program (Graphpad Software Inc., La Jolla, CA, USA) and the half-maximal inhibitory concentrations (IC50) were determined by regression analysis.
4.10. Statistical Analysis
All experiments were conducted with three replicates for each treatment and were repeated three times and are presented as means ± SDs. Statistical analyses were performed by one-way analysis of variance (ANOVA), followed by a multiple comparison of means (Tukey). GraphPad Prism 9.4.1 (681) software was used, performing a P value ˂0.05 were considered to indicate statistical significance.
5. Conclusions
For the first time, conditions were established to obtain an in vitro culture of friable callus from B. fagaroides using a combination of PGR: 4 mgL-1 2,4-D, 1 mgL-1 NAA and 1 mgL-1 Zeatin. Four phases of cell growth were clearly identified in this culture. The maximum cell growth point was identified at day 28 with a μ= 0.059 days-1 and td=11.8 days. It was observed that in this culture scopoletin was synthesized as the main compound (inducible as a phytoalexin by the addition of high concentrations of 2,4-D, as well as by the absence of nutrients in the culture medium), as well as γ-sitosterol, dehydrodiosgenin and stigmasterol.
Open column chromatography was used to separate and identify three abundant compounds: yatein, acetyl podophyllotoxin and 7‘8‘-dehydropodophyllotoxin in the extract of the leaves of the wild plant; it should be noted that yatein is the first time that has been described in leaves.
The cytotoxic activity of four cell lines was tested, the most sensitive being PC3 (IC50 of 12.6±4.6) in the wild-type plant extract and HeLa (IC50 of 72±5) in the callus culture extract.
Author Contributions
Conceptualization, M.L.V, A.O.-C., and L.A.; methodology, J.N.S.-C., L.G.-M., A.O.-C., M.G.-C., and L.A.; validation, M.L.V., and L.A.; formal analysis, N.P.-M. and J.N.S.-C.; investigation, N.P.-M. and J.N.S.-C.; resources, J.N.S.-C., L.G.-M., M.G.-C., A.O.-C., and L.A.; writing—original draft preparation, N.P.-M.; writing—review and editing, L.A., M.L.V., J.N.S.-C., and A.O.-C.; supervision, L.A., M.L.V., and A.O.-C; funding acquisition, L.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
This research was supported in part by a scholarship grant to N.P-M. (fellowship 288473) and grant CB-240801. The authors thank Laboratorio Nacional de Estructura de Macromoléculas, UAEM (LN Conhacyt 321131) for the use of NMR and gas chromatography coupled with mass spectrometry. Maria Gregoria Medina Pintor (Centro de Investigaciones Químicas, UAEM) for skillful technical assistance in the analysis and GC/MS experiments. Biol. Ixchel Gómez Palacios (Centro de Investigación Biomédica del Sur-CIBIS) for advice, assistance and use of HPLC. Biol. Isaac Rodríguez Sánchez for its support in the greenhouse cultivation of the cuttings and Dra. Mariana Sánchez Ramos for providing materials for the realization of growth kinetics.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Noge, K. and Becerra, J.X. Germacrene D, a common sesquiterpene in the genus Bursera (Burseraceae). Molecules 2009, 14, 5289–5297. [Google Scholar] [CrossRef] [PubMed]
- Rzedowski, J.; Medina, L.R.; and Calderón, de R. G. Las especies de Bursera (Burseraceae) en la cuenca superior del río Papaloapan (México). Acta Bot. Mex. 2004, 66, 23–151. [Google Scholar] [CrossRef]
- Rzedowski, J.; Medina, L.R.; and Calderón, de R. G. Inventario del conocimiento taxonómico, así como de la diversidad y del endemismo regionales de las especies mexicanas de Bursera (Burseraceae). Acta Bot Mex. 2005, 70, 85–111. [Google Scholar] [CrossRef]
- Marcotullio, M.C.; Curini, M.; and Becerra, J.X. An Ethnopharmacological, Phytochemical and Pharmacological Review on Lignans from Mexican Bursera spp. Molecules. 2018, 23, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Sepúlveda, A.M.; Mendieta-Serrano, M.; Antúnez, M.M. Y.; Salas-Vidal, E.; Marquina, S.; Villarreal, M.L.; Puebla, A.M.; Delgado, J.; and Alvarez, L. Cytotoxic podophyllotoxin type-lignans from the steam bark of Bursera fagaroides var. fagaroides. Molecules. 2012, 17, 9506–9519. [Google Scholar] [CrossRef] [PubMed]
- Antúnez, M.M.; León, A.; Rojas-Sepúlveda, A.M.; Marquina, S.; Mendieta-Serrano, M.A.; Salas-Vidal, E.; Villarreal, M.L.; and Alvarez, L. Aryldihydronaphthalene-type lignans from Bursera fagaroides var. fagaroides and their antimitotic mechanism of action. RSC. 2016, 6, 4950–4959. [Google Scholar] [CrossRef]
- Antúnez-Mojica, M.; Romero-Estrada, A.; Hurtado-Díaz, I.; Miranda-Molina, A.; and Alvarez, L. Lignans from Bursera fagaroides: Chemistry, Pharmacological Effects and Molecular Mechanism. A Current Review. Life. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Bianchi, E.; Sheth, K.; and Cole, J.R. Antitumor agents from Bursera fagaroides (Burseraceae). (β -peltatin-a-methylether and 5′-desmethoxy-β-peltatin-a-methylether). Tetrahedron Lett. 1969, 10, 2759–2762. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Jiménez, R. , Torres-Valencia, J.M.; Cerda-García-Rojas, C.M.; Hernández-Hernández, J.D.; Román-Marín, L.U.; Manríquez-Torres, J.J.; Gómez-Hurtado, M.A.; Valdez-Calderón, A.; Motilva, V.; García-Mauriño, S.; Telero, E.; Ávila, J. and Joseph-Nathan, P. Absolute configuration of podophyllotoxin related lignans from Bursera fagaroides using vibrational circular dichroism. Phytochem. 2011, 72, 2237–2243. [Google Scholar] [CrossRef]
- Acevedo, M.; Nuñez, P.; González-Maya, L.; Cardoso, T.A. and Villarreal, M.L. Cytotoxic and Anti-inflamatory activities of Bursera species from Mexico. J. Clin. Toxicol. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- Antúnez-Mojica, M.; Rojas-Sepúlveda, A.M.; Mendieta-Serrano, M.A.; Gonzalez-Maya, L; Marquina, S. ; Salas-Vidal, E. and Alvarez, L. Lignans from Bursera fagaroides affect in vivo cell behavior by disturbing the tubulin cytoskeleton in zebrafish embryos. Molecules. 2019, 24, 1–13. [Google Scholar] [CrossRef]
- Ceballos, G.; Martínez, L., García, A., Espinoza, E.; Bezaury, C.J. and Dirzo, R. Diversidad, amenazas y áreas prioritarias para la conservación de las selvas secas del Pacífico de México. 1st ed. Fondo de Cultura Económica, CONABIO, México D.F. 2010. 9-11.
- Bonfil-Sanders, C.; Mendoza-Hernández-P. E., and Ulloa-Nieto, J.A. Root and callus development in cuttings of seven species of the genus Bursera. Agrociencia. 2007, 41, 103–109. [Google Scholar]
- Satyanarayana, B.N. and Varghese, D.B. Plant tissue culture: practices and new experimental protocols. I. K. International Pub. House. Pvt. Ltd., New Delhi. 2007; 9-54. ISBN: 978-81-89866-11-2.
- Verpoorte, R. and Alfermann, A. W. Metabolic Engineering of Plant Secondary Metabolism. Kluger Academic Publishers. Springer Netherlands, 2013, 1-46. ISBN: 978-94-015-9423-3.
- Rodríguez, B.M.M.; Latsague, V.M.I.; Chacón, F.M.A. and Astorga, B.P.K. In vitro induction of callogenesis and indirect organogenesis from explants of cotyledon, hypocotyl and leaf in Ugni molinae. Bosque. 2014, 35, 21–22. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Sugimoto, K. and Iwase, A. Plant Callus: Mechanisms of Induction and Repression. Plant Cell. 2013, 25, 3159–3173. [Google Scholar] [CrossRef] [PubMed]
- Robert, M.R.; Reyes, J. and Loyola, V.M. Biosíntes y bioconversión de metabolitos secundarios por células cultivadas in vitro. Cultivo de tejidos en la agricultura. 9th Chapter, 212-231.
- Mishra, S.K. and Kumar, A. Biosynthesis of guggulsterone in the callus culture of Commiphora wightii. Arnott. bhandari (Burseraceae). RJPBCS. 2010, 1, 35–41. [Google Scholar]
- Al-Abdallat, A.M.; Adayileh, B.K.; Sawwan, J.S.; Shibli, R.; Al-Qudah, T.S.; Abu-Irmaileh, B.; Albdaiwi, R.N.; Almaliti, J. and Bustanji, Y. Secondary Metabolites Profiling, Antimicrobial and Cytotoxic Properties of Commiphora gileadensis L. Leaves, Seeds, Callus, and Cell Suspension Extracts. Metabolites. 2023, 13, 1–14. [Google Scholar] [CrossRef]
- Nikam, T.D.; Ghorpade, R.P. Nitnaware K.M.; Ahire, M.L.; Lokhande, V.H. and Chopra, A. Micropropagation and non-steroidal anti-inflammatory and anti-arthritic agent boswellic acid production in callus cultures of Boswellia serrata Roxb. Physiol Mol Biol Plants. 2013, 19, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Ozyigit, I.I.; Dogan, I.; Hocaoglu-Ozyigit, A.; Yalcin, B.; Erdogan, A.; Yalcin, I.E. , Cabi, E. and Kaya, Y. Production of secondary metabolites using tissue culture-based biotechnological applications. Front Plant Sci. 2023, 14, 1–28. [Google Scholar] [CrossRef]
- Trejo-Tapia, G.; Rodríguez-Monroy, M. La agregación celular en la producción de metabolitos secundarios en cultivos vegetales in vitro. Interciencia. 2007, 32, 669–674. [Google Scholar]
- Larson, C.G.; Gómez, C.; Sánchez-Olate, M. and Ríos, D. Induction of indirect callogenesis in Eucalyptus globulus. Bosque. 2006, 27, 250–257. [Google Scholar] [CrossRef]
- Feeney, M.; Bhagwat, B.; Mitchell, J.S. and Lane, W.D. Shoot regeneration from organogenic callus of sweet cherry (Prunus avium L.). Plant Cell Tissue Organ Cult. 2007, 90, 201–214. [Google Scholar] [CrossRef]
- Rashmi, R. and Trivedi, M.P. Effect of Various Growth Hormone Concentration and Combination on Callus Induction, Nature of Callus and Callogenic Response of Nerium odorum. Appl Biochem Biotechnol. 2014, 172, 2562–2570. [Google Scholar] [CrossRef] [PubMed]
- Bonfil-Sanders, C.; Cajero-Lázaro, I. and Evans, R. Y. Seed germination of six Bursera species from central México. Agrociencia. 2008, 42, 827–834. [Google Scholar]
- Cultid-Medina, C.A. and Rico, Y. Los aliados emplumados de los Copales y Cuajiotes de México: aves y la dispersión de semillas de Bursera. RDU. 2020, 21, 1–9. [Google Scholar] [CrossRef]
- Rzedowski, J. y Guevara-Férer, F. Burseraceae. Flora del Bajío y de regiones adyacentes, fascículo 3. 1992, 1-46.
- Lodish, H.; Berk, A.; Kaiser, C.A.; Krieger, M.; Bretscher, A.; Ploegh, H.; Amon, A. and Scott, M.P. Biología celular y molecular. 7th. Buenos Aires: Panamericana, 2016; Volume 20, pp.967-71.
- Mc-Caughey-Espinoza, D.; Ayala-Astorga, G.; García-Baldenegro, C.; Buitimea-Cantúa, N.; Buitimea-Cantúa, G. and Ochoa-Meza, A. In vitro germination and induction of callus and root in Bursera laxiflora S. Watson. Abanico Agrofor. 2020, 2, 1–14. [Google Scholar]
- Pavón-Reyes, L.; Evangelista-Lozano, S.; Sepúlveda-Jiménez, G.; Chávez, A.V. and Rodríguez-Monroy, M. Cell Culture of Bursera linanoe in a Stirred Tank Bioreactor for Production of Linalool and Linalyl Acetate. Nat Prod Commun. 2017, 12, 319–322. [Google Scholar] [PubMed]
- Malik, S.I.; Rashid, H.; Yasmin, T. and Minhas, N. M. Effect of 2,4-dichlorophenoxyacetic Acid on Callus Induction from Mature Wheat (Triticum aestivum L.) Seeds. Int J Agric Biol. 2003, 6, 156–159. [Google Scholar]
- Zheng, M.Y. and Konzak, C.F. Effect of 2,4-dichlorophenoxyacetic acid on callus induction and plant regeneration in anther culture of wheat ( Triticum aestivum L.). Plant Cell Rep. 1999, 19, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Carsono, N.; Juwendah, E.; Liberty, L.; Sari, S.; Damayanti, F. and Rachmadi, M. Optimize 2,4-D concentration and callus induction time enhance callus proliferation and plant regeneration of three rice genotypes. Biodiversitas. 2021, 22, 2555–2560. [Google Scholar] [CrossRef]
- Ozias-Akins, P. and Vasil, I.K. Callus induction and growth from the mature embryo of Triticum aestivum (Wheat). Protoplasma. 1983, 115, 104–113. [Google Scholar] [CrossRef]
- Pádua, M.S.; Paiva, L.V.; Silva, L.C.D.; Livramento, K.G.D.; Alves, E. and Castro, A.H.F. Morphological characteristics and cell viability of coffee plants calli. Cienc. Rural. 2014, 44, 660–665. [Google Scholar] [CrossRef]
- Taguchi, G.; Fujikawa, S.; Yazawa, T.; Kodaira, R.; Hayashida, N.; Shimosaka, M. and Okazaki, M. Scopoletin uptake from culture medium and accumulation in the vacuoles after conversion to scopolin in 2,4-D-treated tobacco cells. Plant Sci. 2000, 151, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, M.; Hino, F.; Kominami, K. and Miura, Y. Effects of Plant Hormones on Formation of Scopoletin and Scopolin in Tobacco Tissue Cultures. Agric Biol Chem. 1982, 46, 609–614. [Google Scholar] [CrossRef]
- Cisneros-Torres, D.; Cruz-Sosa, F.; González-Cortazar, M.; Martínez-Trujillo, A. and Nicasio-Torres, P. Enhancing the production of scopoletin and quercetin 3-O-β-d-glucoside from cell suspension cultures of Tilia americana var. mexicana by modulating the copper and nitrate concentrations. Plant Cell Tiss Organ Cult. 2019, 139, 305–316. [Google Scholar] [CrossRef]
- Siatka, T.; Chlebekb, J. and Hoštálková, A. Copper (II) Sulfate Stimulates Scopoletin Production in Cell Suspension Cultures of Angelica archangelica. Nat Prod Commun. 2017, 12, 1779–1780. [Google Scholar] [CrossRef]
- Gnonlonfin, G.J.B.; Sanni, A. and Brimer, L. Review Scopoletin – A Coumarin Phytoalexin with Medicinal Properties. Critical Reviews in Plant Sciences. 2012, 31, 47–56. [Google Scholar] [CrossRef]
- Bagni, N. and Fracassini, D.S. The effect of coumarin derivatives on organogenesis and callus growth of Cichorium Intybus roots and Helianthus tuberosus tubers in vitro. Experientia. 1971, 27, 1239–1241. [Google Scholar] [CrossRef]
- Ekiert, H. and Gomółka, E. Coumarin compounds in Ammi majus L. callus cultures. Die Pharmazie. 2000, 55, 684–687. [Google Scholar] [PubMed]
- Fritig, B.; Hirth, L. and Ourisson, G. Biosynthesis of the coumarins: Scopoletin formation in tobacco tissue cultures. Phytochem. 1970, 9, 1963–1975. [Google Scholar] [CrossRef]
- Modafar, C.E.; Clerivet, A.; Fleuriet, A. and Macheix, J.J. Inoculation of Platanus acerifolia with Ceratocystis fimbriata F. Sp. Platani induces scopoletin and umbelliferone accumulation. Phytochem. 1993, 34, 1271–1276. [Google Scholar]
- Habibullah, M.; Al-Mansur, M.A.; Mahboob, M.; Siddiqi, A.; Chowdhury, A.M.S.; Sohrab, M. and Hasan, C.M. Scopoletin Isolated from Stem Bark of Bursera serrata Wall. With Antimicrobial and Cytotoxic Activities of the Crude Extract. Dhaka Univ. J. Sci. 2010, 58, 287–289. [Google Scholar]
- Ara, K.; Rahman, M.S.; Rahman, A.H.M.; Hasan, C.M. and Rashid, M.A. Terpenoids and Coumarin from Bursera serrata Wall. Dhaka Univ J Pharm Sci. 2009, 8, 107–110. [Google Scholar] [CrossRef]
- Aguilar, S.L.; Romero, C.O.; González, C.M. and Tortoriello, J. Efecto de Bursera grandiflora sobre el peso corporal y lipemia en ratones obesos. BLACPMA. 2012, 11, 138–146. [Google Scholar]
- Matos, M.J.; Santana, L.; Uriarte, E.; Abreu, O.A.; Molina, E. and Yordi, E.G. Coumarins — An Important Class of Phytochemicals. Phytochemicals - Isolation, Characterization. in Human Health, Rao, A.V., Rao, L.G., Eds.; InTech: 2015; Volume 20, pp. 113–40. [CrossRef]
- Rojas-Sepúlveda, A.M. Búsqueda de metabolitos con actividad citotóxica y antitumoral en Bursera fagaroides var. fagaroides y Bursera morelensis, y evaluación de su efecto como inhibidores del ciclo celular en el modelo de pez cebra. Doctorado, Universidad Autónoma del Estado de Morelos, Cuernavaca, Morelos, junio 2012.
- Infante-Rodríguez, D.A.; Landa-Cansigno, C.; Gutiérrez-Sánchez, A.; Murrieta-León, D.L.; Reyes-López, C.; Castillejos-Pérez, A.B.; Pucheta-Fiscal, J.E.; Velázquez-Narváez, A.C.; Monribot-Villanueva, J.L. and Guerrero-Analco, J.A. Análisis fitoquímico y actividad antidiabética, antibacteriana y antifúngica de hojas de Bursera simaruba (Burseraceae). Acta Bot Mex. 2022, 129, 1–15. [Google Scholar] [CrossRef]
- Barry, L.N. Use of Fluorescein Diacetate in Citrus Tissue cultures for the determination of cell viability and the selection of mutants. Sci. Hortic. 1989, 39, 15–21. [Google Scholar]
- Jones, K.H. and Senft, J.A. An improved method to determine cell viability by viability by simultaneous staining with fluorescein diacetate-propidium Iodide. JHC. 1985, 33, 77–79. [Google Scholar]
- Ocampo-Bautista, F.; Mussali-Galante, P.; Alvarez, L.; Marquina-Bahena, S.; Valencia-Cuevas, L.; Valencia-A, S. and Tovar Sánchez, E. Natural Hybridization between Bursera bicolor × B. glabrifolia (Burseraceae). Forests. 2023, 14, 1–16. [Google Scholar] [CrossRef]
- Bork, P.M. , Schmitz, M.L.; Kuhnt, M.; Escher, C. and Heinrich, M. Sesquiterpene lactone containing Mexican Indian medicinal plants and pure sesquiterpene lactones as potent inhibitors of transcription factor NF-kappaB. FEBS Lett. 1997, 402, 85–90. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cultivation of B. fagaroides cuttings at A) 12, B) 25 and C) 70 days. Bar = 0.5 cm.

Figure 2.
Callus induction and development from B. fagaroides leaf explants in photoperiod. A) disinfection of explants, B) Dedifferentiated callus after 30 days in MS medium supplemented with 2,4-D: Kinetin 2:2 mgL-1, C) and D) culture and callus morphology at 25 days in B5 medium with 2,4-D: NAA: Zeatin 4:1:1 mgL-1 respectively at 30 days of culture. Bar = 0.5 cm and 40X objective.
Figure 2.
Callus induction and development from B. fagaroides leaf explants in photoperiod. A) disinfection of explants, B) Dedifferentiated callus after 30 days in MS medium supplemented with 2,4-D: Kinetin 2:2 mgL-1, C) and D) culture and callus morphology at 25 days in B5 medium with 2,4-D: NAA: Zeatin 4:1:1 mgL-1 respectively at 30 days of culture. Bar = 0.5 cm and 40X objective.

Figure 3.
A) Growth curve of callus from B. fagaroides and B) Percentage viability of callus from B. fagaroides.
Figure 3.
A) Growth curve of callus from B. fagaroides and B) Percentage viability of callus from B. fagaroides.
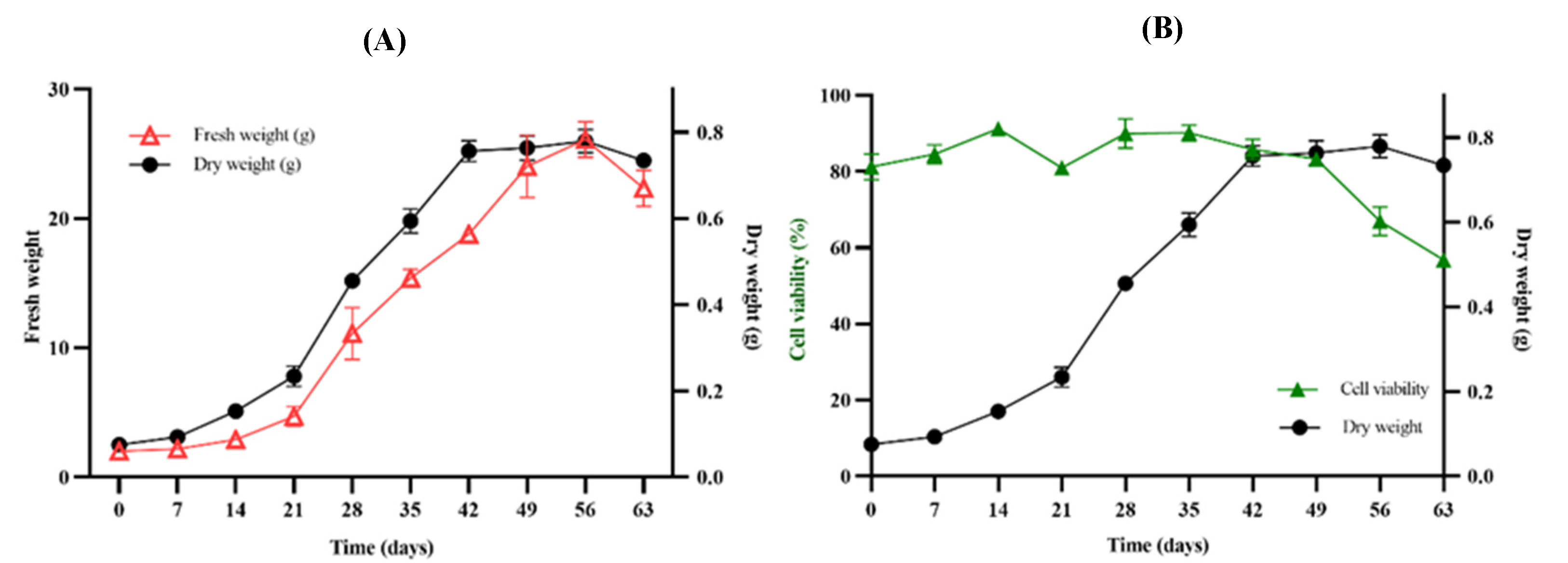
Figure 4.
A) TLC of qualitative analysis of callus extracts on different days of culture, B) Chromatogram of scopoletin standard at 350 nm (50 mg/mL), C) Production curve of scopoletin and D) Production of scopoletin in callus extract and wild plant leaf of B. fagaroides.
Figure 4.
A) TLC of qualitative analysis of callus extracts on different days of culture, B) Chromatogram of scopoletin standard at 350 nm (50 mg/mL), C) Production curve of scopoletin and D) Production of scopoletin in callus extract and wild plant leaf of B. fagaroides.
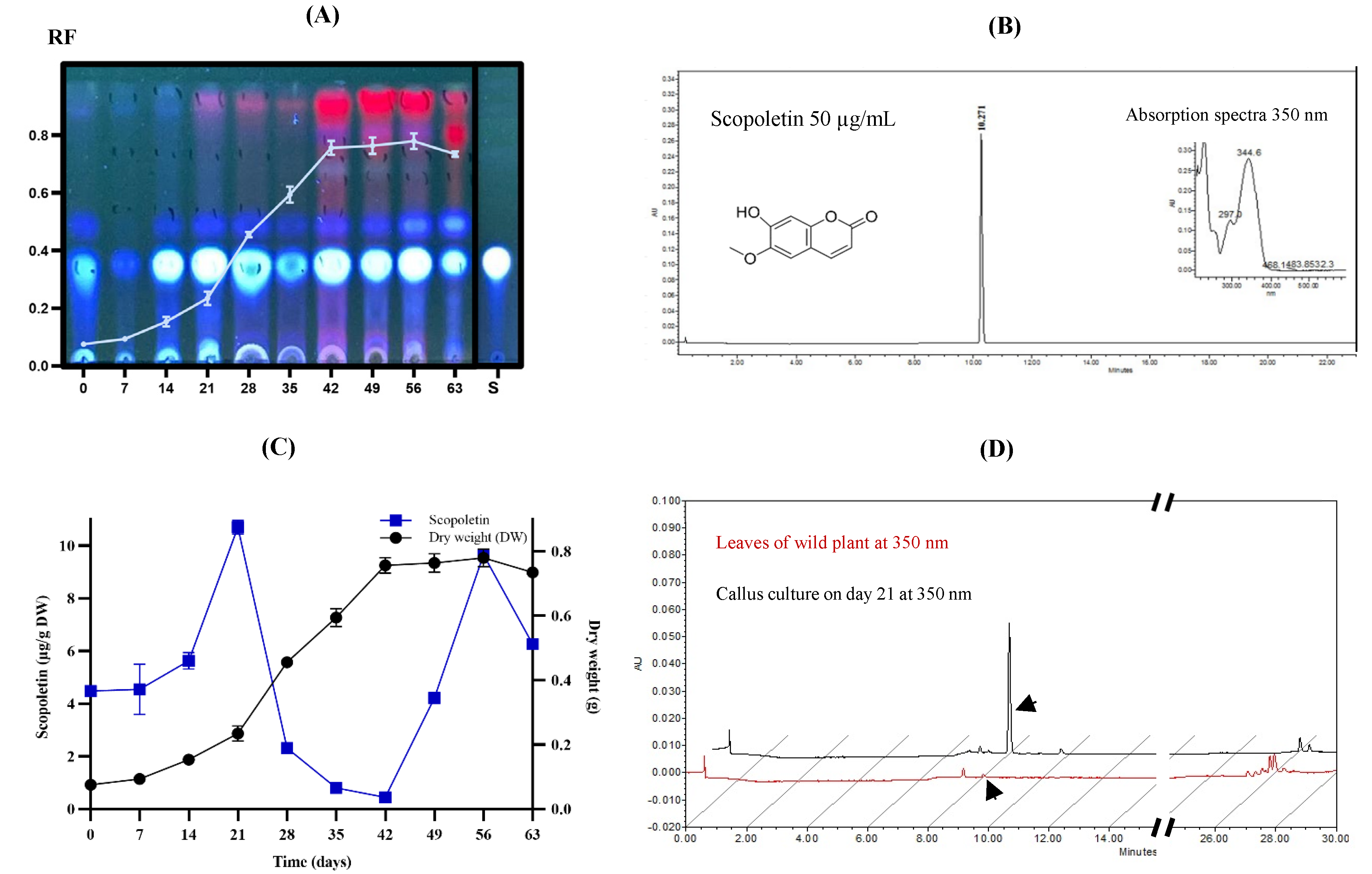
Figure 5.
Chemical structures of compounds identified and isolated from B. fagaroides callus.
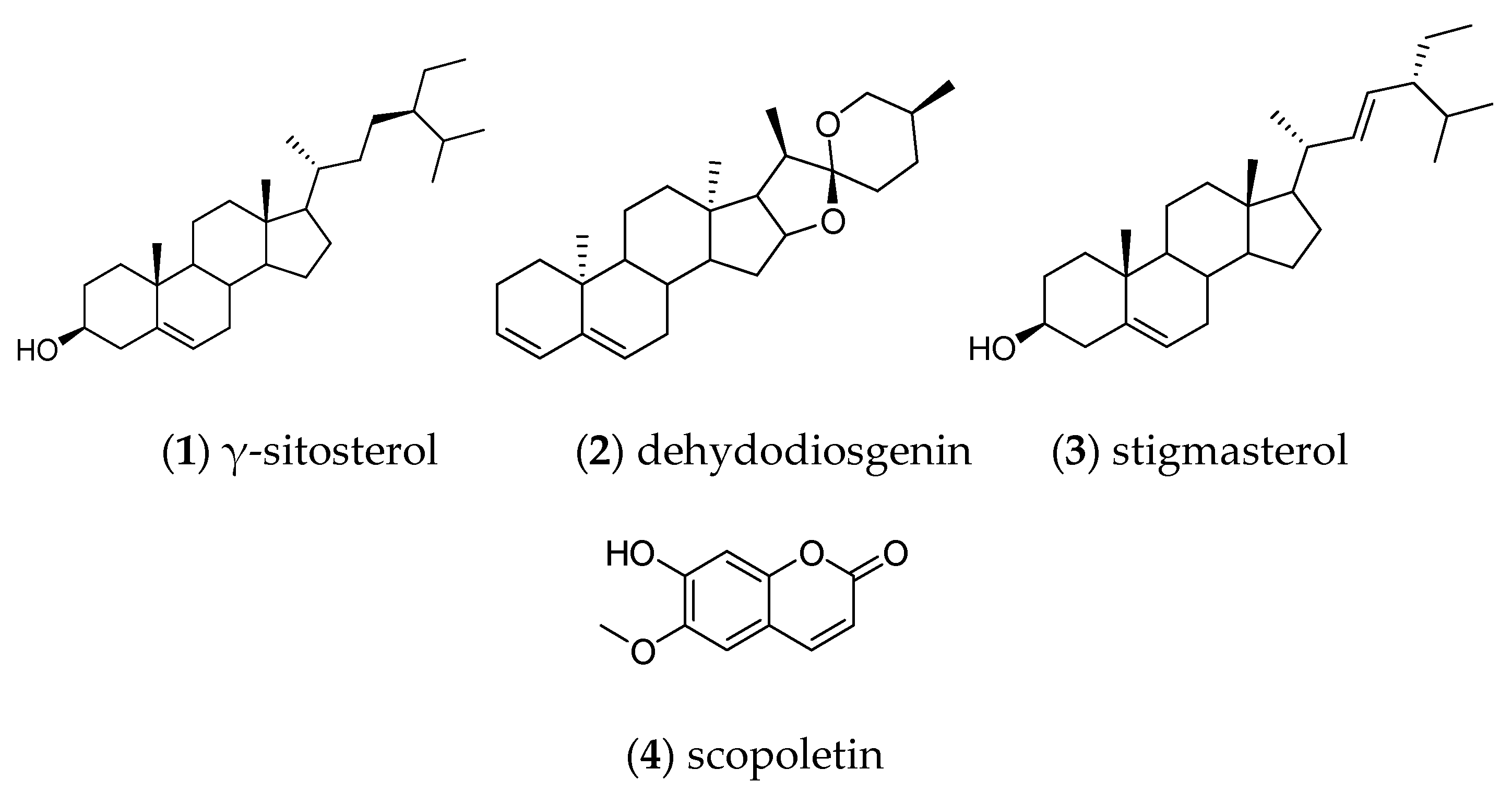
Figure 6.
Chemical structures of compounds identified and isolated from the leaves of wild B. fagaroides.
Figure 6.
Chemical structures of compounds identified and isolated from the leaves of wild B. fagaroides.
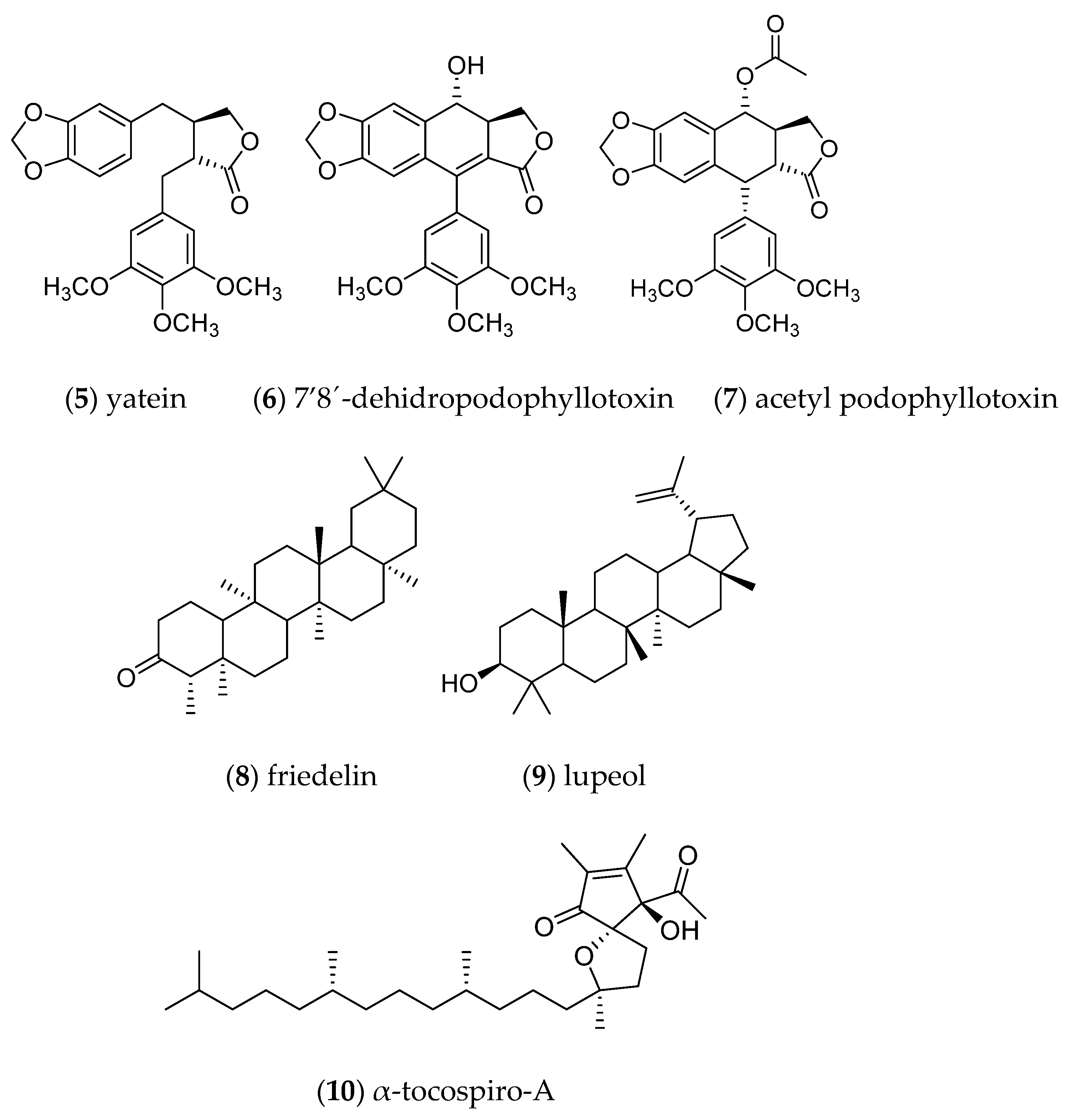

Table 1.
Effect of plant growth regulators and culture media on callus induction of Bursera fagaroides leaf explants.
Table 1.
Effect of plant growth regulators and culture media on callus induction of Bursera fagaroides leaf explants.
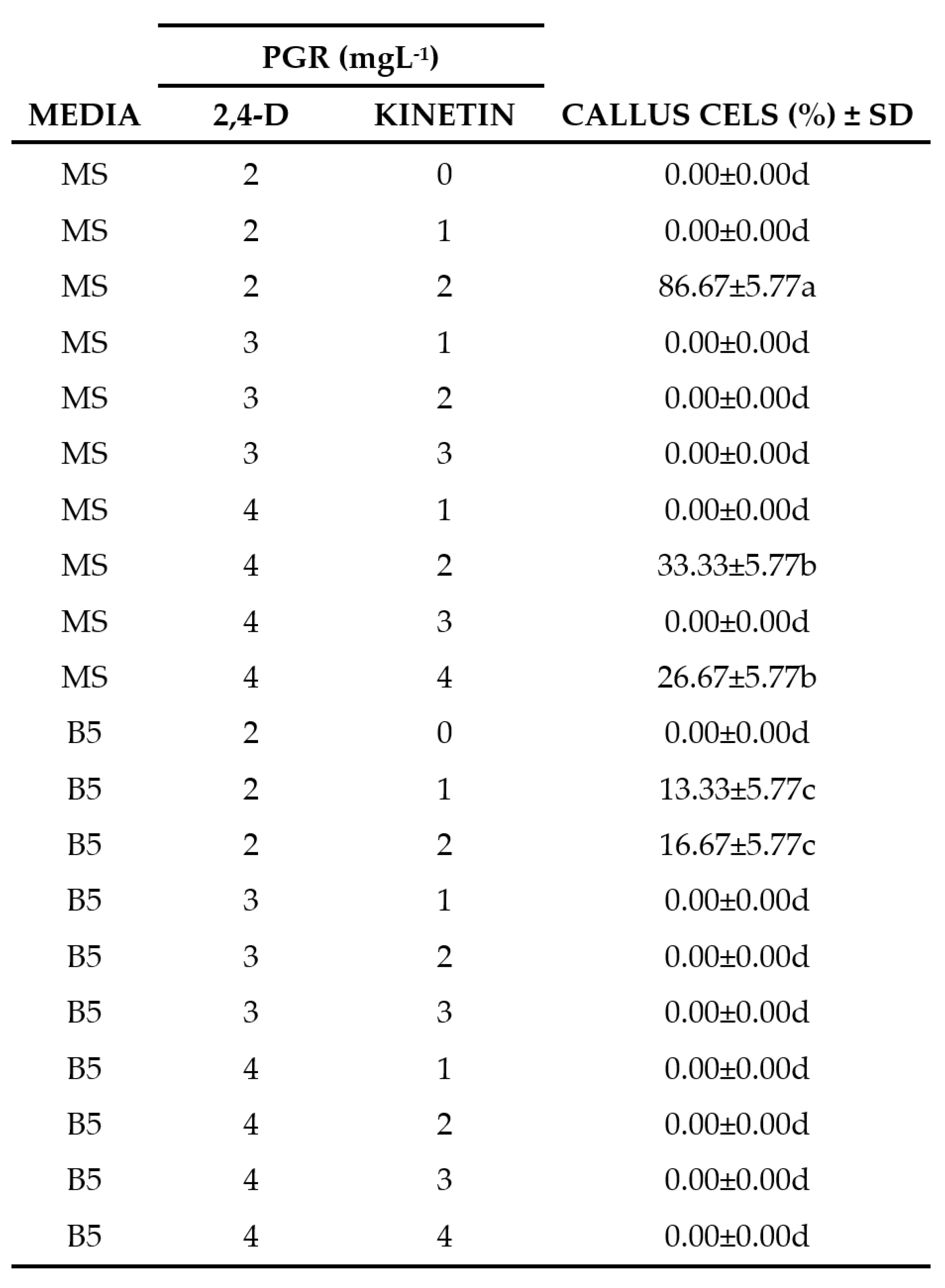
PGR = Plant Growth Regulators. Data represents the mean ± standard deviation. Values with the same letters in the columns are not statistically different according to the multiple comparison of means test (Tukey) P˂0.05.
Table 2.
(IC50) of CH2Cl2 extracts of wild-type plant and callus culture of B. fagaroides.
| Extract | HepG2 | HeLa | PC3 | H1299 | HFF-1 |
|---|---|---|---|---|---|
| Leavesb Callusb Paclitaxela |
20.3±3.38 155.4±4.95 10.12±2.5 |
18.6±5.2 72±5 40.23 ±7.2 |
12.6±4.6 141±5.8 17.5 ±2.8 |
69.1±6.2 210.4±5 45.25 ±7 |
79.6±5.2 95.4±6.2 989 ±21 |
aPaclitaxel values are in nM. bThe extracts of leaves and callus are in µg/mL.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



