
Submitted:
10 April 2024
Posted:
11 April 2024
You are already at the latest version
Abstract
Enteroviruses (EV) are important pathogens causing human disease with various clinical manifestations. To date, treatment of enteroviral infections is mainly supportive since no vaccination or antiviral drugs are approved for their prevention or treatment. Here, we describe the antiviral properties and mechanisms of action of leucoverdazyls - novel heterocyclic compounds with antioxidant potential. The lead compound, 1a, demonstrated low cytotoxicity along with high antioxidant and virus-inhibiting activity. A viral strain resistant to 1a was selected, and the development of resistance was shown to be accompanied by mutation of virus-specific non-structural protein 2C. This resistant virus had lower fitness when grown in cell culture. Taken together, our results demonstrate high antiviral potential of leucoverdazyls as novel inhibitors of enterovirus replication and support previous evidence of an important role of 2C proteins in EV replication.
Keywords:
enteroviruses
; coxsackievirus
; leucoverdazyls
; antioxidant
; antiviral
; 2C protein
1. Introduction
Enteroviruses (EV) represent a diverse group of small icosahedral non-enveloped viruses with a single-stranded non-segmented positive RNA genome belonging to Picornaviridae family Enterovirus genus, encompassing EV A-L and rhinovirus A-C species [1]. EV are characterized by high resistance to harsh environments and the ability to cause both self-limiting infections as well life-threatening diseases and outbreaks, especially among newborns and children [2]. EV-A species members, such as coxsackieviruses (A6, A16) and enterovirus 71, are etiological agents of the largest outbreaks of hand, foot and mouth disease (HFMD), both in Asia and Western countries. Enterovirus D68 (EV-D68) infection is associated with respiratory and neurologic disease worldwide. Although EVs are mainly associated with acute infections, more evidence is emerging on the long persistence of EVs in target organs, such as heart and pancreas [3,4,5].
Among EV-induced diseases, HFMD is one of the most widely spread pathologies. It is mostly mild self-limiting disease that occurs in children under the age of five. Its symptoms include sores in the mouth, anorexia, mild fever, as well as ulcers on the hands, feet, and mouth [6]. In some cases, however, EV infection leads to fatal neurological or cardiopulmonary complications such as myocarditis, pulmonary edema and neuroinflammation causing meningoencephalitis and cognitive impairment. EV infection is also of high danger for children with immunodeficiencies or accompanying diseases. Therefore, HFMD is a significant concern for public health [7].
Vast majority of HFMD cases are caused by group A enteroviruses, mainly EV-A71, CV-A16, CV-A10, and CV-A6. EV’s of B-family also can cause sporadic cases of HFMD [8,9].
Enteroviruses are characterized by high genotypic and phenotypic diversity, which significantly complicates the development of broad-spectrum vaccines for the prevention of EVI. Currently, vaccination is available only for the prevention of poliomyelitis and for EV71-associated HFMD [10,11,12]. A large amount of research carried out in the field of the EV life cycle has paved the way for the development of antivirals for the treatment of EVI [13,14]. Among the previously studied enterovirus inhibitors, the following groups can be distinguished: inhibitors that bind to the viral capsid and prevent its penetration into the cell; capsid binders (pleconaril, pirodavir, vapendavir, pokapavir, disoxaril); inhibitors of viral proteases (rupintrivir and drug AG74/04); viral polymerase inhibitors (ribavirin, gemcitabine, amiloride); and viral ATPase inhibitors (dibucaine, fluoxetine) [15,16,17]. Despite numerous efforts of the scientific community and pharmaceutical companies, no direct acting antivirals were approved for the treatment of EVI. Therefore, there is an unmet need for new antivirals targeting EVs.
Reactive oxygen species (ROS) generated by cells as a byproduct of oxidative metabolism are able to inactivate DNA, proteins and lipids, thereby inducing cell death and providing a general defense against many pathogens. Surprisingly, some viruses, including influenza viruses, coronaviruses, herpes viruses and enteroviruses use oxidative stress induction via different mechanisms for effective reproduction and enhanced pathogenesis [18,19,20,21]. ROS can be an attractive target for antiviral therapy design [22]. The use of antioxidants (including but not limited to resveratrol, N-acetyl cysteine, quercetin and their derivatives) for the prevention and treatment of viral diseases has been actively studied [23,24,25]. However, currently there are no approved drugs inhibiting enterovirus replication through antioxidant mechanisms.
Previously, we described leucoverdazyls as a promising group of substances with antioxidant properties that potently reduce the replication of group B enteroviruses in vitro [26]. Thereafter, we extended the library of leucoverdazyls through directed modifications and investigated virus inhibiting properties of novel leucoverdazyls against enteroviruses A, B, C in vitro. According to the results of mechanistic studies, the lead compound targets the 2C protein of Coxsackieviruses, with inhibition of viral genome replication. The results obtained in this study can be used to develop therapeutic agents to combat EVI.
2. Materials and Methods
2.1. Compounds
Leucoverdazyls (2-(1-aryl-3-phenyl-5,6-dihydro-4H-1,2,4,5-tetrazin-1-yl)-1,3-benzothiazoles 1-4) were synthesized by alkylation of 1-aryl-5-(1,3-benzothiazol-2-yl)-3-phenylformazans with haloalkanes in alcoholic alkali, followed by cyclization of N-alkyl derivatives, according to the procedure described previously [27]. The structures of leucoverdazyls synthesized are given at the Figure 1 below.
2.2. Viruses and Cell Lines
Influenza A virus (IAV, strain A/Puerto Rico/8/1934 (H1N1)), influenza B virus (IBV, strain B/Florida/04/06, Yamagata lineage), Coxsackievirus B3 (CVB3, strain Nancy), Coxsackievirus B4 (CVB4, strain Powers), herpes simplex virus type 1 (HSV1), human adenovirus type 5 (Ad5), and SARS-CoV2 virus (Wuhan strain) were obtained from the viral collection of the Pasteur Institute (St. Petersburg, Russia). Clinical isolates of ECHO30 (specimen number 4972), Сoxsackievirus В5 (GenBank: OQ939946), Coxsackievirus A16 (specimen number 10120), Coxsackievirus A24 (specimen number 68427) were obtained from The Subnational polio laboratory (St. Petersburg, Russia). Virus isolation from stool specimens and identification with enterovirus typing antisera were performed according to the WHO manual [28]. Identification of Сoxsackievirus В5 strain was performed by partial sequencing of the VP1 genomic region using EV-specific primers [29]. Influenza A virus (IAV, strain A/Puerto Rico/8/1934 (H1N1)), influenza B virus (IBV, strain B/Florida/04/06, Yamagata lineage), Coxsackievirus B3 (CVB3, strain Nancy), Coxsackievirus B4 (CVB4, strain Powers), herpes simplex virus type 1 (HSV1), human adenovirus type 5 (Ad5), and SARS-CoV2 virus were obtained from the viral collection of the Pasteur Institute (St. Petersburg, Russia). Clinical isolates of ECHO30 and Сoxsackievirus В5 were obtained from Regional Centre for Epidemiological Surveillance of Poliomyelitis (St. Petersburg, Russia). The following permissive cell lines obtained from ATCC were used in the studies: MDCK (ATCC # CCL-34), Vero (ATCC #CCL-81), RD (ATCC #CCL-136), and A549 (ATCC# CCL-185). Infectious titers (in 50%-tissue culture infection dose, TCID50) were determined in MDCK for IAV, in Vero cells for CVB3, CVB4, CVB5, HSV1, SARS-CoV-2, in RD cells for ECHO30, CVA16 and CVA24 viruses, and in A549 cells for Ad5. The endpoint titration assay was performed using the following procedure. Permissive cells were seeded into 96-wells plates in Eagles minimal essential medium (MEM) supplemented with 5% fetal bovine serum (FBS). After 24 h, the media was aspirated, the wells were washed with saline, fresh MEM without FBS was added to the wells, and the cells were infected with ten-fold serial dilutions of viral stocks (100 µL per well, 4 wells for each dilution). Trypsin TPCK was added to the titration media for influenza viruses only. The plates were incubated at +37°C in 5% CO2 and observed daily for cytopathic effect (CPE). Viral titer was calculated in TCID50 using the Spearmen-Carber method.
2.3. Antioxidant Activity Testing
Antioxidant activity was evaluated by spectrophotometric monitoring of the hydrogen transfer reaction with a stable chromogen radical, 2,2-diphenyl-1-picrylhydrazyl (DPPH), using vitamin C (Vc) as described before [27]. A solution of DPPH in methanol with a concentration of 200 μM was added to a solution of dihydrotetrazines in the same solvent (concentration 5 to 50 μM). The reaction vessel (test tube) was wrapped in foil and kept for 30 min at 30°C, and the optical density was measured at λ 517 nm (DPPH absorption maximum). The antioxidant activity (AO) was calculated by the formula
where Atest is the optical density of a solution containing a compound to be tested and DPPH, and Acontr is the optical density of a solution containing DPPH alone. The half inhibitory concentration (IC50) corresponding to the reduction of the initial DPPH concentration by 50% was determined from the DPPH inhibition percentage plotted against concentrations of compounds 2 through 5 using the OriginPro 8.5 program (Model DoseResp).
AO = (1 – Atest/Acontr)×100%,
2.4. Cytotoxicity Assay
The microtetrazolium test (MTT) was used to study the cytotoxicity of the compounds [30]. Permissive cells were seeded in 96-well plates in Eagles minimal essential medium (MEM) supplemented with 10% FBS. After 24 h, the media was removed, and the wells were washed with saline. Compounds were dissolved in DMSO, and a series of two-fold dilutions of each compound (1000-4 µg/mL) in MEM without FBS were added to the cells in triplicates (200 µL per well). The maximal concentration of DMSO was 0.5%; MEM with 0.5% DMSO was added to cell control wells. Cells were incubated for 24 h or 48h at 37°C in 5% CO2 and thereafter the MTT was performed. The optical density of cells was then measured on Multiskan multifunctional reader (ThermoFisher Scientific) at a wavelength of 540 nm and plotted against the concentration of the compounds to generate the dose–response curve. The 50% cytotoxic dose (CC50) of each compound (i.e., the compound concentration that causes the death of 50% of cells in a culture, or decreases the optical density twice as compared to the control wells) was calculated using a four-parameter logistic nonlinear regression model (GraphPad Prism 6). CC50 values in μg/mL were then converted into micromoles.
2.5. Antiviral Activity Determination
Viral yield reduction assay was used for antiviral activity evaluation. The respective permissive cell lines were seeded in MEM supplemented with 5% FBS in 24-well plates. The next day, compounds tested were dissolved in DMSO, and a series of three-fold dilutions of each compound (final concentrations 100–1 µg/mL) in MEM without FBS was added to the cells (500 µL per well), followed by incubation (37°C, 5% CO2). After 1 h, the media was discarded, and equal volumes of fresh serial dilutions of each compound (200-2 µg/mL) and viral suspension in MEM without FBS at MOI 0.01 were added to all the wells of the plate (the final volumes were 500 µl per well). In cell control wells, only MEM without FBS was added. In virus control wells, no compounds were added. The plates were incubated at 4°C for 1 h. Thereafter, unbound virus was washed away, and again three-fold dilutions of each compound (final concentrations 100–1 µg/mL) in MEM without FBS was added to the wells (1 mL). After 24 h (for enteroviruses and SARS-CoV-2) or 48 h (for HSV1, Ad5 and influenza viruses) of incubation at 37°C in 5% CO2, the infectious titer of viral progeny (in TCID50) for each compound concentration, cell control, and virus control wells were determined in permissive cell lines by endpoint dilution assay (described above).
The titer of viral progeny was plotted against log concentration of the compounds tested to generate the dose–response curve. The 50% inhibition concentration (IC50) of each compound tested (i.e., the compound concentration that decreases the infectious viral progeny titer twice as compared to the control wells) was calculated using four-parameter logistic nonlinear regression model (GraphPad Prism 6). IC50 values in μg/mL were then converted into micromoles. Selectivity index (SI) was calculated for each compound tested as a ratio of CC50 to IC50 values.
2.6. Thermostability Assay
Thermostability assay was performed as described previously [31]. Briefly, CVB4 Powers (104 TCID50) was pre-incubated with the compound (10 µg/ml), pleconaril (10 µg /ml), or an equal volume of MEM (virus control) for 30 min at +37 °C in sterile thin-walled 200 ul PCR-tubes (5 tubes per each treatment condition) in a BioRad CFX PCR-machine. Then, a thermal gradient of 37–55°C for 2 min, followed by rapid cooling to 4°C, was applied. Subsequently, the infectious viral activities of the samples were quantified by end-point dilution assay.
2.7. Time-of-Addition Assay
Time-of-addition assay was performed according to the recommendation described earlier [32]. Vero cells were seeded in 24-well plates for 24 hours before the beginning of the assay in order to reach 90% confluence. The leader compound was sequentially added in the following time points to the respective individual wells in the plate: (-2), (-1), 0, 2, 4, 6, where (-2) means 1 hour before addition of the virus, (-1) – addition of the virus, 0 – completion of viral sorption on the cell surface, and 2, 4, 6 – in 2, 4 or 6 hours after virus sorption. At timepoint (-1), a suspension of 106 TCID50 of CVB4 was added to all the wells (except cell control), and the plate was incubated at +4°C for 1 hour in order to synchronize the infection in all conditions. Afterwards, unbound virus was washed off, and the plate was returned to +37°C. The capsid binder pleconaril was used as a reference compound. Eight hours after the completion of virus sorption, the experiment was stopped, and the infectious viral titer was measured in each well using end-point titration in Vero cells.
2.8. Selection and Analysis of the Drug-Resistant Strain
In order to study the development of resistance to the lead compound, CVB3 virus (Nancy) was serially passaged in Vero cells in the presence of increased concentrations of the compound. Cells were infected with the virus and incubated for 2-3 days (37°C, 5% CO2) until a cytopathic effect was observed. The culture supernatants were used for sequential selection. In total, nine passages were performed using the following 1a concentrations: 0.5, 1, 2, 3, 4, 5, and 6 µg/mL, following by two passages at 7 µg/ml to obtain the resistant (R) variant. The wild type (WT) virus was passaged in Vero cells in the absence of 1a. The values of IC50 for original, WT and R viruses were further determined by viral yield reduction assay. Three viral variants (original, WT, R) were plaque-purified, and full genomes of three clones from each virus were sequenced. Viral RNA was extracted using the kit Ribo-prep (Amplisense, Russia). After reverse transcription using MMLV RT kit (Evrogen, Russia) and amplification of cDNA using the highfidelity polymerase Tersus plus (Evrogen, Russia) and CVB3-specific primers, purified PCR-products were analyzed on ABI-3500 XL Genetic Analyser (Applied Biosystems) using BigDye® Terminator v3.1 Chemistry and POP-7™ polymer (Thermo Fisher Scientific). CVB3 specific primers used for cDNA amplification and sequencing are listed in Supplementary materials, Table S1 (primer sequences were adapted from the publication by Liu et al. [33]. The chromatograms were converted into contigs using Unipro Ugene free software (version 45.1) [34]. The sequences were aligned to a reference CVB3 sequence (GenBank: M16572.1). The nucleotide sequences were translated into amino acids by free online software (https://web.expasy.org/translate/) (accessed on 12 February 2022) [35].
2.9. Growth Kinetics
The in vitro growth kinetics of CVB3-WT and -R variants were determined in Vero A cells (96-well tissue culture plates, 5 × 104 cells/well). At the zero timepoint, cells were infected with the respective viral variants (MOI 0.001). After 2 h of virus absorption, cells were washed three times with medium to remove non-adsorbed virus, and 200 μL of medium was added to each well. At 8, 24, 36, 48, 60, and 72 h post-infection, culture supernatants were collected to quantify the number of infectious viral particles at each time point by end-point titration.
2.10. Transmission Electron Microscopy (TEM)
Vero cells in 6-well plates were incubated with CVB3 virus, MOI= 100, at 4°C for 1 hour. Unbound virions were removed by washing the cells twice with cold MEM, medium containing 100 µM of 1a was added, and cells were incubated for either 3 or 6 hours at 36°C in 5% CO2. In wells with control virus, MEM without 1a was added. After incubation, cells were collected from the wells, transferred into tubes, and centrifuged at 2000×g for 15 min. Cell pellets were fixed with 1.5% glutaraldehyde in PBS overnight, followed by post-fixation with 1.5% OsO4 for 1 h and uranyl acetate for 45 min at room temperature. They were then dehydrated in graded acetone and embedded in Epon/Araldit resin (Serva Feinbiochemica, Heidelberg, Germany). Thin sections (90 nm) were stained with lead citrate and examined in a JEM-100S electron microscope (JEOL, Tokyo, Japan).
2.11. Computer Modeling
The model of 2C protein were prepared using AlphaFold software. Molecular docking for modeling the interaction between 1a and 2C was done by Hex online server (http://hexserver.loria.fr/) [36].
2.12. Statistics
All in vitro experiments were repeated three times. The results are represented as M±SE. Viral titers were plotted against the logarithm of concentration, and the IC50 values for each virus were calculated using GraphPad Prism (v.8.0) software using four-parameter logistic curves.
3. Results
3.1. Leucoverdazyls Are Potent Inhibitors of EVs at Low Micromolar Concentrations
At the beginning of the study, the virus-inhibition activity of leucoverdazyls in viral yield reduction assay was determined. Cytotoxicity of the compounds for permissive cell lines used was evaluated in MTT assay. The results are summarized in Table 1 below. Pleconaril was used for comparison as a reference compound due to the fact that CVB3 Nancy is a pleconaril-resistant strain [37].
As can be seen from the data presented in the table, the least toxic compounds were the following: 1b, 2b, 3b, 3d, and 4d. Two of them (1b, 2b) contain a methoxy group in the aromatic fragment. Halogen containing compounds 2d and 2e showed the greatest toxicity. In viral yield reduction assay, a large majority (57 %) of leucoverdazyls from the library (11 out of 19 tested) demonstrated remarkable anti-enteroviral activity in vitro against pleconaril resistant CVB3 strain in comparison to pleconaril. SI values for these compounds exceeded 10, which is indicative of high anti-enteroviral potential. Compounds 1a-1c exhibited the most pronounced antiviral activity, with IC50 values much lower than that of pleconaril. It should be noted that the structure of these compounds lacks a substituent at position 6 of the tetrazine ring. In addition, the best values of the selectivity index were also noticed for compounds that do not contain a substituent in this position of the tetrazine ring (compounds 1a-1d), as well as for compounds containing the least bulky substituent (Me) – compounds 2a, 2b. Compound 1a with the lowest IC50 value (2.7 µM) and the highest SI (230) was selected as the leader for further study.
In order to trace the effect of the substituent at the position 6 of the tetrazine ring on the antioxidant activity of dihydrotetrazines, a DPPH test was performed in the series of 1a,b, 2a,b, 3a,b and 4a,b. The results are presented in Figure 2.
As can be seen from the data obtained, the most pronounced antioxidant activity was detected for compounds 1a and 1b, and their antioxidant potentials were even superior to vitamin C used as the reference compound. Therefore, we suppose that addition of substituent in the sixth position of the tetrazine ring has a negative impact on the antioxidant activity of dihydrotetrazines in vitro.
The data on antioxidant activity indicate that compounds without substituent in the sixth position of the tetrazine ring, as well as compounds with a methoxy group in the aromatic fragment at N1, turned out to possess the highest antioxidant activity.
3.2. 1a Possesses Wide-Range Activity against Group A, B and C Enteroviruses
To assess the prospects for further development of the most potent compound (1a), the spectrum of its anti-enterovirus activity was assessed against a panel of group A, B and C enteroviruses including both Coxsakievirus B4 (strain Powers) and patient isolates in viral yield reduction assay. The following patients isolates of viruses were used: CVA16, CVB5, ECHO30, CVA24. Among them CVA16 is common agent of HFMD, while CVB5 and ECHO30 has been reported to be associated with HFMD also [38,39,40]. The results are presented in the Table 2 below. Guanidine hydrochloride targeting initiation step of viral RNA synthesis was used as reference drug [41].
Compound 1a showed high activity towards other strains of group A, B and C enteroviruses superior to reference compound guanidine hydrochloride, though guanidine hydrochloride was less toxic.
We further investigated whether 1a is capable of inhibiting the life cycle of other phylogenetically distinct RNA or DNA viruses. Compound 1a was tested against influenza (ss RNA-negative enveloped virus), HSV1 (dsDNA enveloped virus), Ad5 (dsDNA non-enveloped virus), and SARS-CoV-2 (ssRNA-positive enveloped virus) in viral yield reduction assay (Table 3).
According to the results, compound 1a showed only modest activity against influenza viruses, Ad5, and HSV1. Surprisingly, however, it inhibited replication of another ssRNA-positive enveloped virus, namely SARS-CoV-2. This leads us suppose that 1a targets some biological entity (protein or process) important to both the enterovirus and coronavirus life cycle.
3.3. 1a Does Not Increase Virion Thermostability, and Inhibits Late Stages of the Enterovirus Life Cycle
The plausible mechanism of action for 1a was studied using in vitro assays. We addressed whether 1a has capsid binding properties using a thermal stability assay. It is known that capsid binders (pleconaril and its derivatives) directly interact with capsid of enteroviruses and stabilize its structure, thereby preventing the virus from entering the host cell. This interaction increases the resistance of the viral capsid to a short-term temperature increase, and the heated virus retains its ability to infect a permissive cell line. The results are presented in the Figure 3 below.
In the virus control, as well as when test drug 1a was mixed with the virus, no infectious particles were detected when heated above 45 degrees. As expected, the reference capsid binding drug pleconaril had a thermostabilizing effect on Coxsackie B4 virus, ensuring the presence of infectious particles even when heated to 55 degrees (the highest temperature used). Thus, it was concluded that 1a does not belong to the capsid-binding group of inhibitors.
Next, we focused on the stage in the viral cycle when 1a demonstrated the highest inhibitory activity in time-of-addition assay. CVB4 was propagated in Vero cells with addition and removal of 1a at distinct time points before or after the zero point when virus was added. After one cycle of replication (8 hpi), the infectious titer of viral progeny was determined in end-point dilution assay. The titer of viral progeny versus interval of 1a presence in the media is presented in Figure 4.
The most pronounced inhibitory effect of 1a was demonstrated if it was present in the culture medium starting from -2 to 4 h post-infection (hpi). Inhibition of viral replication was not observed if the substance was added later than 6 hours after the adsorption of the virus. Pleconaril used as a reference compound demonstrated the highest activity between (-2) and 0 hours, as expected for early-stage inhibitors. The results obtained suggest that the substance acts on steps involved in viral replication.
The Coxsackievirus lifecycle is relatively short (6–8 h) and has been extensively studied previously. Briefly, after receptor mediated endocytosis, viral genomic RNA is translated into a polyprotein, which in turn is proteolytically processed by viral 2Apro and 3Cpro to release viral proteins, and viral RNA replication begins. Viral RNA replication is performed via a dsRNA intermediate in specialized replication organelles. Nascent viral (+)RNA is encapsidated by structural proteins to form new virions, which are released either lytically or non-lytically (in autophagic vesicles).
According to previously obtained results, viral RNA replication is initiated 2–3 h after infection, and translation of viral capsid proteins is detectable as early as after 4 hpi [42]. Therefore, the inhibitory effect of 1a spans the following stages of the viral life cycle: cell attachment, penetration, genomic RNA transcription, proteolytic processing, and RNA replication.
3.4. Transmission Electron Microscopy
In order to visualize the effect of 1a on viral morphogenesis, we performed an analysis of the ultrastructure of infected cells in the presence of 1a versus non-infected cells and infected cells without treatment (Figure 5). In infected cells without treatment, typical cytoplasmic membranous vesicles were observed, representing replication organelles typical for enterovirus infection [43]. Treatment of cells with 1a abrogated these changes in the cytoplasm. As can be seen, compound 1a fully eliminates signs of viral replication in infected cells (no replication organelles are visible).
3.5. 1a-Resistant Strain Selection and Its Genomic and Phenotypic Characteristics
In order to assess the genetic barrier to resistance development to 1a, we further passaged CVB3 (Nancy strain) in Vero cells at increasing concentrations of 1a, evaluated the emerging resistance level, and identified amino acid substitutions. Three viral strains were analyzed: o’riginal’ CVB3 from the bank, which was used to generate w’ild-type’ virus (CVB3 WT, passaged without 1a in Vero cells), and r’esistant’ strain (CVB3 R, passaged at increasing concentrations of 1a). After nine subsequent passages of the virus in cell culture, the IC50 of 1a was determined to be 12.9 μM for the resistant strain, which was 7-fold higher than that of the original virus (IC50=1.8 µM), and higher than wild type virus (IC50=0.48 µM). Therefore, 1a stimulates the selection of resistance of CVB3 virus, suggesting its direct antiviral activity and a virus-specific target (Figure 6). We also investigated the growth characteristics of resistant virus in comparison to the wild type one in vitro (Figure 7). In the presence of 1a, the resistant strain was able to effectively propagate in contrast to the wild type virus. Nevertheless, without 1a, the growth speed of the resistant strain was significantly lower than that of the wild-type virus during the first 48 hours (p<0.05 by Mann–Whitney U-test).
After the resistant viral variant was obtained, viruses were plaque purified, and full genomes of three clones from each viral type (initial, wild-type, 1a-resistant) were sequenced. Their nucleotide sequences were translated to localize amino acid substitutions. After a comparison of the amino acid sequences, substitution S1209I was identified. Since position 1209 of viral polyprotein corresponds to 2C protein, here and further we use amino acid numeration corresponding to this specific protein (p‘osition 109’, instead of 1209). AlphaFold software was used to generate the complete structural model of 2C protein. Based on the model, the position 109 in 2C was mapped (Figure 8).
Therefore, with sequential passage of the Coxsackievirus in the presence of 1a, a decrease in the sensitivity of the virus to 1a occurs. This is accompanied by a deterioration in the growth characteristics of the resistant virus and the appearance of mutation in the 2C protein. This indicates an influence of 1a on processes associated with replication of the viral genome.
3.6. Molecular Modeling
Further, we compared the localization of the S109I amino acid substitution in 2C protein of 1a-resistant virus with the localization of the probable binding site of 1a. As shown in Figure 9, the binding site for 1a appeared to be located in close proximity to the S109I substitution, thus corresponding to the target for 1a among virus-specific proteins.
4. Discussion
Enteroviruses represent a clinically important group of human pathogens with neither vaccine nor direct antiviral available for enteroviral infection management. In the present study, we showed the high antiviral potential of a novel class of compounds, leucoverdazyls, against enteroviruses A, B and C, including both laboratory strains and patient isolates and described its mechanism of action. We also assessed the possibility of selection of a viral variant resistant to lead compound 1a and characterized its properties including fitness, susceptibility to the inhibitor, and genomic composition. Our results suggest that the compounds of this class exert their virus-inhibiting activity at early stages of the viral cycle (before 4 hpi). The drug resistant Coxsackie B3 viral variant featured an IC50 value seven-fold higher than that of the wild-type virus. Amino acid substitution S109I in the 2C viral protein was detected in the resistant virus. Molecular docking of 1a to 2C protein showed that the ligand and substituted amino acid are localized at the same domain of 2C.
The life cycle of enteroviruses has been described. Following cell entry, capsid disassembly and exposure of viral RNA to the cellular translational system, the viral genome is translated into a single polyprotein which is further processed by viral protease into structural and non-structural virus-specific proteins [44]. Double-stranded RNA is an essential intermediate in the process of viral genome replication in cellular cytoplasm wherein host pathogen recognition receptors (PRRs), in particular dsRNA sensors, provide protection from invading viral pathogens [45]. dsRNA is one of the most important pathogen-associated molecular patterns (PAMPs). Therefore, in order to avoid contact with PRRs and further progression of innate antiviral immune reactions, enteroviruses induce formation of single- and double membrane vesicles called replication organelles (RO). There, formation of replication and transcription complexes, as well as processes of RNA replication and virion assembly, take place being protected from antiviral host cell defense mechanisms [46,47].
To provide large amounts of membrane required for RO formation, enteroviruses developed numerous ways to manipulate host cell pathways for biogenesis and functionality of the membranous structures in infected cells [48,49]. Some viral proteins must be therefore associated with membranes to properly execute their function and realize the viral life cycle. The 2C protein is one of the most conserved proteins within the Picornaviridae family with multiple functions [50,51]. This non-structural protein of 322-330 amino acids is involved in virus uncoating, host cell membrane binding and rearrangement, formation of the viral cytoplasmic replication vesicles, RNA binding and RNA synthesis, and possibly encapsidation [52,53,54]. However, in infected cells, it is localized in Golgi-related membranes [44]. It possesses ATP-dependent RNA helicase and ATP-independent chaperoning activities [53]. Recently, 2C protein was demonstrated to possess ATPase-independent nuclease activity with a preference for polyU ssRNAs [55]. The amino acids essential for RNAse activity have been mapped to the central pore of the hexameric ring. In addition, enterovirus 2C proteins affect the NF-κB signaling pathway, one of the most important mechanisms of innate antiviral immunity, by recruitment of protein phosphatase (PP1) for suppression of IKKβ phosphorylation [56] and by direct binding to IKKb, as well as with p65 and MDA5 [57,58]. In addition, EV71 2C induces degradation of another innate immunity protein, APOBEC3G by its ubiquitination utilizing the autophagy lysosome pathway [59].
For proper action, 2C has to be oligomerized into a ring-shaped hexamer [60,61,62]. However, detailed structural and functional characterization of 2C is impeded by the presence of an N-terminal amphipathic helix which makes the protein insoluble and impossible to be crystallized [63,64,65]. Most of the structural studies, therefore, have been done with a soluble fragment of 2C covering the ATPase domain, a cysteine-rich zinc finger, and a C-terminal helical domain. In our study, we took advantage of the predictive ability of AlphaFold software to build a complete model of CVB3 2C protein based on its amino acid sequence.
The 1a resistance-associated mutation S109I was shown to be located within the interface between the head of 2C and its tail containing amphilin, i.e., the possible domain where 2C is inserted in cytoplasmic membranes. To the best of our knowledge, no 2C mutations that confer viral resistance to clinical or experimental compounds were detected in the domain we described in our study. No specific function of 2C, therefore, can be hypothesized to be altered by 1a. In 2000, Klein et al. demonstrated that due to 2C mutations in or near the NTP binding domain, resistance to 2-(α-hydroxybenzyl)-benzimidazole (HBB) and guanidine was achieved [66]. Viruses with lowered sensitivity to other benzimidazole derivatives, TBZE-029 and MRL-1237, guanidine-HCl and hydantoine, were obtained bearing mutations in positions 64, 65, 120, 125, 133, 142, 143 and several others located at even longer distance from the position 109 we found [67,68]. Based on the docking results, none of these mutations could alter binding of leucoverdazyl derivative to 2C (Figure 9). The mechanism of action of 1a is, therefore, distinct from that of all described compounds including guanidine-HCl and benzimidazole derivatives.
As indicated above, the N-terminus of 2C (amino acids 1–125) interacts with all isoforms of the PP1 catalytic subunit through a PP1-docking motif [69]. One possibility is, therefore, that 1a interferes with this binding thus preventing virus-induced blockade of the NF-κB pathway. On the other hand, binding of the N-terminus of EV71 2C with host protein reticulon 3 (RTN3) was shown to be necessary for the synthesis of viral proteins and replicative double-stranded RNA [70]. The protein site responsible for this interaction was mapped within amino acids 10-27, with isoleucine 25 having the highest importance for binding. This part of 2C, however, is located rather far from the mutated amino acid 109 and 1a binding site. Therefore, alteration of 2C-RTN3 interaction by 1a is of low probability, or indirect. One more possibility is that, since the N-terminal amphipathic helix of 2C binds to cellular lipid droplets to build RO membranes [71], 1a could potentially interfere with this interaction too.
Importantly, data on antioxidant activity indicate that compounds without substituent in the sixth position of the tetrazine ring, as well as compounds with a methoxy group in the aromatic fragment at N1, turned out to be the most effective antioxidants. Interestingly, while possessing the highest antioxidant activity (Figure 2), compounds 1a and 1b also demonstrated the best virus-inhibiting properties toward CVB3 in vitro (Table 1). As shown previously, enteroviruses can use ROS-based signaling and metabolic processes for their efficient propagation within the cell [18,19,20,21]. It cannot be ruled out, therefore, that 1a has a dual mechanism of virus-inhibiting activity, potentially being a multitarget compound. Few compounds, however, have been studied in this regard, and this issue should be addressed in further experiments.
In conclusion, we have identified a novel class of anti-enteroviral compounds, leucoverdazyls, differing in structure and mechanism of action from all previously described viral inhibitors. Further structural and functional studies are necessary to fully understand the mechanism of antiviral activity of leucoverdazyls and the molecular basis of resistance formation.
5. Conclusions
This section is not mandatory but can be added to the manuscript if the discussion is unusually long or complex.
Supplementary Materials
CVB3 specific primers used for cDNA amplification and sequencing are listed in Table S1.
Author Contributions
Conceptualization: VVZ, GNL, TGF, Investigation: ASV, TGF, MSV, VAS, Resources: OIK, NAT, ASG, Visualization: ANG, VVZ, Writing – Original Draft: ASV, TGF, Writing - Review & Editing: VVZ, Supervision: VVZ, GNL, Project administration: ASV, Funding acquisition: TGF, ASV All authors have read and agreed to the published version of the manuscript
Funding
The work was funded within the framework of the state task of the IOS of the Ural Branch of the Russian Academy of Sciences for 2024 and АААА-А21-121030200272-6: Development of novel prophylactic and therapeutic remedies against socially important viral diseases and a grant for young scientists from the St. Petersburg Pasteur Institute.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We express our sincere gratitude to Maria A. Anastasina from Okinawa Institute of Science and Technology, Japan, who helped in molecular modeling. We are also extremely grateful to Edward Ramsay (St. Petersburg Pasteur Institute) for invaluable help in editing and preparation of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Simmonds, P. , Gorbalenya, A.E., Harvala, H., Hovi, T., Knowles, N.J., Lindberg, A.M., Oberste, M.S., Palmenberg, A.C., Reuter, G., Skern, T., Tapparel, C., Wolthers, K.C., Woo, P.C.Y., Zell, R., 2020. Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch Virol. 2020 Mar;165(3):793-797. Erratum in: Arch Virol. Jun;165(6):1515. [CrossRef] [PubMed] [PubMed Central]
- Lugo, D. , Krogstad, P., 2016. Enteroviruses in the early 21st century: new manifestations and challenges. Curr Opin Pediatr. Feb;28(1):107-13. [CrossRef] [PubMed] [PubMed Central]
- Genoni, A. , Canducci, F., Rossi, A., Broccolo, F., Chumakov, K., Bono, G., Salerno-Uriarte, J., Salvatoni, A., Pugliese, A., Toniolo, A., 2017. Revealing enterovirus infection in chronic human disorders: An integrated diagnostic approach. Sci Rep. Jul 10;7(1):5013. [CrossRef] [PubMed] [PubMed Central]
- Nekoua, M.P. , Alidjinou, E.K., Hober, D., 2022. Persistent coxsackievirus B infection and pathogenesis of type 1 diabetes mellitus. Nat Rev Endocrinol 18, 503–516. [CrossRef]
- Chapman, N.M., 2022. Persistent Enterovirus Infection: Little Deletions, Long Infections. Vaccines (Basel). May 12;10(5):770. [CrossRef] [PubMed] [PubMed Central]
- Légeret, C. , Furlano, R., 2021. Oral ulcers in children—A clinical narrative overview. Ital J Pediatr 47, 144. [CrossRef] [PubMed] [PubMed Central]
- Zhang, X. , Zhang, Y., Li, H., Liu, L, 2023. Hand-Foot-and-Mouth Disease-Associated Enterovirus and the Development of Multivalent HFMD Vaccines. Int J Mol Sci 24, 169. [CrossRef] [PubMed] [PubMed Central]
- Frydenberg, A. , Starr, M., 2003. Hand, foot and mouth disease. Aust Fam Physician 32, 594–595. [PubMed]
- Alhazmi, A.; Nekoua, M.P.; Mercier, A.; Vergez, I.; Sane, F.; Alidjinou, E.K.; Hober, D. Combating coxsackievirus B infections. Rev Med Virol e 2406. [CrossRef] [PubMed]
- Bandyopadhyay, A.S. , Garon J., Seib K., Orenstein, W.A., 2015. Polio vaccination: past, present and future. Future Microbiol. 10(5):791-808. [CrossRef] [PubMed]
- Chumakov, K. , Ehrenfeld, E., Agol, V.I, Wimmer, E., 2021. Polio eradication at the crossroads. Lancet Glob Health. Aug;9(8):e1172-e1175. [CrossRef] [PubMed]
- Li, M.L. , Shih, S.R., Tolbert, B.S., Brewer, G., 2021. Enterovirus A71 Vaccines. Vaccines (Basel). Feb 27;9(3):199. [CrossRef] [PubMed] [PubMed Central]
- Baggen, J. , Thibaut, H.J., Strating, J.R.P.M., van Kuppeveld, F.J.M, 2018. The life cycle of non-polio enteroviruses and how to target it. Nat Rev Microbiol. 2018 Jun;16(6):368-381. . Erratum in: Nat Rev Microbiol. May 3;:. [CrossRef] [PubMed]
- Wen, X. , Sun, D., Guo, J., Elgner, F., Wang, M., Hildt, E., Cheng, A., 2019. Multifunctionality of structural proteins in the enterovirus life cycle. Future Microbiol. Sep;14:1147-1157. [CrossRef] [PubMed]
- Bauer, L. , Lyoo, H., van der Schaar, H.M., Strating, J.R., van Kuppeveld F.J., 2017. Direct-acting antivirals and host-targeting strategies to combat enterovirus infections. Curr Opin Virol. Jun;24:1-8. [CrossRef] [PubMed] [PubMed Central]
- Laajala, M. , Reshamwala, D., Marjomäki, V., 2020. Therapeutic targets for enterovirus infections. Expert Opin Ther Targets. Aug;24(8):745-757. [CrossRef] [PubMed]
- Tammaro, C.; Guida, M.; Appetecchia, F.; Biava, M.; Consalvi, S.; Poce, G. , 2023. Direct-Acting Antivirals and Host-Targeting Approaches against Enterovirus B Infections: Recent Advances. Pharmaceuticals, 16, 203. [CrossRef]
- Tao, L. , Lemoff, A., Wang, G., Zarek, C., Lowe, A., Yan, N., Reese, T.A., 2020. Reactive oxygen species oxidize STING and suppress interferon production. Elife. Sep 4;9:e57837. [CrossRef] [PubMed] [PubMed Central]
- Wang, L. , Cao, Z., Wang, Z., Guo, J., Wen, J., 2022. Reactive oxygen species associated immunoregulation post influenza virus infection. Front Immunol. Jul 29;13:927593. [CrossRef] [PubMed] [PubMed Central]
- Cheng, M. L, Weng, S.F., Kuo, C.H., Ho, H.Y., 2014. Enterovirus 71 induces mitochondrial reactive oxygen species generation that is required for efficient replication. PLoS One. Nov 17;9(11):e113234. [CrossRef] [PubMed] [PubMed Central]
- Cheng, M.L. , Wu, C.H., Chien, K.Y., Lai, C.H., Li, G.J., Liu, Y.Y., Lin, G., Ho, H.Y., 2022. Enteroviral 2B Interacts with VDAC3 to Regulate Reactive Oxygen Species Generation That Is Essential to Viral Replication. Viruses. Aug 4;14(8):1717. [CrossRef] [PubMed] [PubMed Central]
- Sander, W.J. , Fourie, C., Sabiu, S., O‘Neill, F.H., Pohl, C.H., O‘Neill, H.G., 2022. Reactive oxygen species as potential antiviral targets. Rev Med Virol.; 32(1):e2240. [CrossRef]
- Filardo, S. , Di Pietro, M., Mastromarino, P., Sessa, R., 2020. Therapeutic potential of resveratrol against emerging respiratory viral infections. Pharmacol Ther. Oct;214:107613. [CrossRef] [PubMed]
- Di Petrillo, A. , Orrù, G., Fais, A., Fantini, M.C., 2022. Quercetin and its derivates as antiviral potentials: A comprehensive review. Phytother Res. Jan;36(1):266-278. [CrossRef] [PubMed] [PubMed Central]
- Wang, Y. , Zhao, S., Chen, Y., Wang, Y., Wang, T., Wo, X., Dong, Y., Zhang, J., Xu, W., Qu, C., Feng, X., Wu, X., Wang, Y., Zhong, Z., Zhao, W., 2019. N-Acetyl cysteine effectively alleviates Coxsackievirus B-Induced myocarditis through suppressing viral replication and inflammatory response. Antiviral Res. 2020 Jul;179:104699. [CrossRef] [PubMed]
- Volobueva, A.S. , Zarubaev, V.V., Fedorchenko, T.G., Lipunova, G.N., Tungusov V.N., Chupakhin,O.N., 2023. Antiviral properties of verdazyls and leucoverdazyls and their activity against group B enteroviruses. Russian Journal of Infection and Immunity, Vol. 13. - N. 1. - P. 107-118. [CrossRef]
- Fedorchenko, T.G. , Lipunova, G.N., Shchepochkin, A.V., Valova, M.S., Tsmokalyuk, A.N., Slepukhin, P.A., Chupakhin, O. N., 2020. Synthesis and Spectral, Electrochemical, and Antioxidant Properties of 2-(5-Aryl-6-R-3-phenyl-5,6-dihydro-4H-1,2,4,5-tetrazin-1-yl)-1,3-benzothiazole. Russian Journal of Organic Chemistry, vol. 56, no. 1, pp. 38–48.].
- World Health Organization (WHO). 2004. Manual for the virological investigation of polio, 4th ed.; WHO: Geneva, Switzerland, pp. 1-112.
- Nix, W.A.; Oberste, M.S.; Pallansch, M.A. , 2006. Sensitive, Seminested PCR Amplification of VP1 Sequences for Direct Identification of All Enterovirus Serotypes from Original Clinical Specimens. J. Clin. Microbiol., 44, 2698–2704.
- Mosmann, T. , 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 65(1-2), 55-63. [CrossRef]
- Shetnev, A.A. , Volobueva, A.S., Panova, V.A., Zarubaev, V.V., Baykov, S.V., 2022. Design of 4-Substituted Sulfonamidobenzoic Acid De-rivatives Targeting Coxsackievirus B3. Life. 12, 1832. [CrossRef]
- Daelemans, D. , Pauwels, R., De Clercq, E., Pannecouque, C., 2011. A time-of-drug addition approach to target identification of antiviral compounds. Nat Protoc. Jun;6(6):925-33. [CrossRef] [PubMed] [PubMed Central]
- Liu, B. , Li, Z., Xiang, F., Li, F., Zheng, Y., Wang, G., 2014. The whole genome sequence of coxsackievirus B3 MKP strain leading to myocarditis and its molecular phylogenetic analysis. Virol J. Feb 21;11:33. [CrossRef] [PubMed] [PubMed Central]
- Okonechnikov, K. , Golosova, O., Fursov, M., 2012. UGENE team. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. Apr 15;28(8):1166-7. [CrossRef] [PubMed]
- Available online: https://web.expasy.org/translate/ (accessed on 12 February 2022).
- Available online: http://hexserver.loria.fr/(accessed on).
- Schmidtke, M. , Hammerschmidt, E., Schüler, S., Zell, R., Birch-Hirschfeld, E., Makarov, V.A., Riabova, O.B., Wutzler, P., 2005. Susceptibility of coxsackievirus B3 laboratory strains and clinical isolates to the capsid function inhibitor pleconaril: antiviral studies with virus chimeras demonstrate the crucial role of amino acid 1092 in treatment. J Antimicrob Chemother. Oct;56(4):648-56. [CrossRef] [PubMed]
- Fu, X. , Wan, Z., Li, Y. et al. National Epidemiology and Evolutionary History of Four Hand, Foot and Mouth Disease-Related Enteroviruses in China from 2008 to 2016. Virol. Sin. 35, 21–33 (2020). [CrossRef]
- Zhuang ZC, Kou ZQ, Bai YJ, Cong X, Wang LH, Li C, Zhao L, Yu XJ, Wang ZY, Wen HL. Epidemiological Research on Hand, Foot, and Mouth Disease in Mainland China. Viruses. 2015 Dec 7;7(12):6400-11. [CrossRef] [PubMed] [PubMed Central]
- Andreoni AR, Colton AS. Coxsackievirus B5 associated with hand-foot-mouth disease in a healthy adult. JAAD Case Rep. 2017 Mar 27;3(2):165-168. [CrossRef] [PubMed] [PubMed Central]
- Barton DJ, Flanegan JB. Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J Virol. 1997 Nov;71(11):8482-9. [CrossRef] [PubMed] [PubMed Central]
- Salmikangas, S. ., Laiho, J.E., Kalander, K., Laajala, M., Honkimaa, A., Shanina, I., Oikarinen, S., Horwitz, M.S., Hyöty, H., Marjomäki, V., 2020. Detection of Viral-RNA and +RNA strands in Enterovirus-infected cells and tissues. Microorganisms, vol. 8, no. 12: 1928. [CrossRef]
- Li, X. , Wang, M., Cheng, A., Wen, X., Ou, X., Mao, S., Gao, Q., Sun, D., Jia, R., Yang, Q., Wu, Y., Zhu, D., Zhao, X., Chen, S., Liu, M., Zhang, S., Liu, Y., Yu, Y., Zhang, L., Tian, B., Pan, L., Chen, X., 2020. Enterovirus Replication Organelles and Inhibitors of Their Formation. Front Microbiol. Aug 20;11:1817. [CrossRef] [PubMed] [PubMed Central]
- Domanska, A. , Guryanov, S., Butcher, S.J., 2021. A comparative analysis of parechovirus protein structures with other picornaviruses. Open Biol. Jul;11(7):210008. [CrossRef]
- Lee, H.C. , Chathuranga, K., Lee, J.S., 2019. Intracellular sensing of viral genomes and viral evasion. Exp Mol Med. Dec 11;51(12):1-13. [CrossRef]
- Belov, G.A. , 2016. Dynamic lipid landscape of picornavirus replication organelles. Curr Opin Virol. Aug;19:1-6. [CrossRef]
- Wolff, G. , Melia, C.E., Snijder, E.J., Bárcena, M., 2020. Double-Membrane Vesicles as Platforms for Viral Replication. Trends Microbiol. Dec;28(12):1022-1033. [CrossRef]
- Lai, M. , De Carli, A., Filipponi, C., Iacono, E., La Rocca, V., Lottini, G., Piazza, C.R., Quaranta, P., Sidoti, M., Pistello, M., Freer, G., 2022. Lipid balance remodelling by human positive-strand RNA viruses and the contribution of lysosomes. Antiviral Res. Oct;206:105398. [CrossRef]
- Li, X. , Wang, M., Cheng, A., Wen, X., Ou, X., Mao, S., Gao, Q., Sun, D., Jia, R., Yang, Q., Wu, Y., Zhu, D., Zhao, X., Chen, S., Liu, M., Zhang, S., Liu, Y., Yu, Y., Zhang, L., Tian, B., Pan, L., Chen, X., 2020. Enterovirus Replication Organelles and Inhibitors of Their Formation. Front Microbiol. Aug 20;11:1817. [CrossRef]
- Kadare, G. , Haenni, A.L., 1997 Virus-encoded RNA helicases. J. Virol. 71, 2583–2590. [CrossRef]
- Wang, S.H. , Wang, K., Zhao, K., Hua, S.C., Du, J., 2020. The Structure, Function, and Mechanisms of Action of Enterovirus Non-structural Protein 2C. Front Microbiol. Dec 14;11:615965. [CrossRef] [PubMed] [PubMed Central]
- Singleton, M.R. , Dillingham, M.S., Wigley, D.B. 2007. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50. [CrossRef]
- Xia, H. , Wang, P., Wang, G.C., Yang, J., Sun, X., Wu, W., Qiu, Y., Shu, T., Zhao, X., Yin, L., Qin, C.F., Hu, Y., Zhou, X., 2015. Human Enterovirus Nonstructural Protein 2CATPase Functions as Both an RNA Helicase and ATP-Independent RNA Chaperone. PLoS Pathog. Jul 28;11(7):e1005067. [CrossRef]
- Chen, P. , Li, Z., Cui, S., 2021. Picornaviral 2C proteins: A unique ATPase family critical in virus replication. Enzymes.;49:235-264. [CrossRef]
- Chen, P. , Wojdyla, J.A., Colasanti, O., Li, Z., Qin, B., Wang, M., Lohmann, V., Cui, S., 2022. Biochemical and structural characterization of hepatitis A virus 2C reveals an unusual ribonuclease activity on single-stranded RNA. Nucleic Acids Res. Sep 9;50(16):9470-9489. [CrossRef]
- Li, Q. , Zheng, Z., Liu, Y., Zhang, Z., Liu, Q., Meng, J., Ke, X., Hu, Q., Wang, H., 2016. 2C Proteins of Enteroviruses Suppress IKKβ Phosphorylation by Recruiting Protein Phosphatase 1. J Virol. Apr 29;90(10):5141-5151. [CrossRef]
- Du, H. , Yin, P., Yang, X., Zhang, L., Jin, Q., Zhu, G., 2015. Enterovirus 71 2c Protein Inhibits NF-KappaB Activation by Binding to RelA(P65). Sci Rep 5:14302. [CrossRef]
- Li, L. , Fan, H., Song, Z., Liu, X., Bai, J., Jiang, P., 2019. Encephalomyocarditis Virus 2C Protein Antagonizes Interferon-Beta Signaling Pathway Through InteractionWithMDA5. Antiviral Res 161:70–84. [CrossRef]
- Li, Z. , Ning, S., Su, X., Liu, X., Wang, H., Liu, Y., Zheng, W., Zheng, B., Yu, X.F., Zhang, W., 2018. Enterovirus 71 antagonizes the inhibition of the host intrinsic antiviral factor A3G. Nucleic Acids Res. Nov 30;46(21):11514-11527. [CrossRef]
- Guan, H. , Tian, J., Qin, B., Wojdyla, J.A., Wang, B., Zhao, Z., Wang, M., Cui, S., 2017. Crystal structure of 2C helicase from enterovirus 71. Sci Adv. Apr 28;3(4):e1602573. [CrossRef] [PubMed] [PubMed Central]
- Guan, H. , Tian, J., Zhang, C., Qin, B., Cui, S., 2018. Crystal structure of a soluble fragment of poliovirus 2CATPase. PLoS Pathog. Sep 19;14(9):e1007304. [CrossRef] [PubMed] [PubMed Central]
- Hurdiss, D.L. , El Kazzi, P., Bauer, L., Papageorgiou, N., Ferron, F.P., Donselaar, T., van Vliet, A.L.W., Shamorkina, T.M., Snijder, J., Canard, B., Decroly, E., Brancale, A., Zeev-Ben-Mordehai, T., Förster, F., van Kuppeveld, F.J.M., Coutard, B., 2022. Fluoxetine targets an allosteric site in the enterovirus 2C AAA+ ATPase and stabilizes a ring-shaped hexameric complex. Sci Adv. Jan 7;8(1):eabj7615. [CrossRef]
- Adams, P. , Kandiah, E., Effantin, G., Steven, A.C., Ehrenfeld, E., 2009. Poliovirus 2C protein forms homo-oligomeric structures required for ATPase activity. J Biol Chem. Aug 14;284(33):22012-22021. [CrossRef] [PubMed] [PubMed Central]
- Sweeney, T.R. , Cisnetto, V., Bose, D., Bailey, M., Wilson, J.R., Zhang, X., Belsham, G.J., Curry, S., 2010. Foot-and-mouth disease virus 2C is a hexameric AAA+ protein with a coordinated ATP hydrolysis mechanism. J Biol Chem. 2010 Aug 6;285(32):24347-59. [CrossRef] [PubMed] [PubMed Central]
- Papageorgiou, N. , Coutard, B., Lantez, V., Gautron, E., Chauvet, O., Baronti, C., Norder, H., de Lamballerie, X., Heresanu, V., Ferté, N., Veesler, S., Gorbalenya, A.E., Canard, B., 2010. The 2C putative helicase of echovirus 30 adopts a hexameric ring-shaped structure. Acta Crystallogr D Biol Crystallogr. 2010 Oct;66(Pt 10):1116-20. [CrossRef] [PubMed]
- Klein, M. , Hadaschik, D., Zimmermann, H., Eggers, H.J., Nelsen-Salz, B., 2000. The picornavirus replication inhibitors HBB and guanidine in the echovirus-9 system: the significance of viral protein 2C. J Gen Virol.Apr;81(Pt 4):895-901. [CrossRef] [PubMed]
- De Palma, A.M. , Heggermont, W., Lanke, K., Coutard, B., Bergmann, M., Monforte, A.M., Canard, B., De Clercq, E., Chimirri, A., Pürstinger, G., Rohayem, J., van Kuppeveld, F., Neyts, J., 2008. The thiazolobenzimidazole TBZE-029 inhibits enterovirus replication by targeting a short region immediately downstream from motif C in the nonstructural protein 2C. J Virol. May;82(10):4720-30. [CrossRef]
- Shimizu, H. , Agoh, M., Agoh, Y., Yoshida, H., Yoshii, K., Yoneyama, T., Hagiwara, A., Miyamura, T., 2000. Mutations in the 2C region of poliovirus responsible for altered sensitivity to benzimidazole derivatives. J Virol. May;74(9):4146-54. [CrossRef]
- Swain, S.K. , Panda, S., Sahu, B.P., Sarangi, R., 2022. Activation of Host Cellular Signaling and Mechanism of Enterovirus 71 Viral Proteins Associated with Hand, Foot and Mouth Disease. Viruses. Oct 4;14(10):2190. [CrossRef]
- Tang, W.F. , Yang, S.Y., Wu, B.W., Jheng, J.R., Chen, Y.L., Shih, C.H., Lin, K.H., Lai, H.C., Tang, P., Horng, J.T., 2007. Reticulon 3 binds the 2C protein of enterovirus 71 and is required for viral replication. J Biol Chem. Feb 23;282(8):5888-98. [CrossRef]
- Laufman, O. , Perrino, J., Andino, R., 2019. Viral Generated Inter-Organelle Contacts Redirect Lipid Flux for Genome Replication. Cell. Jul 11;178(2):275-289.e16. [CrossRef] [PubMed] [PubMed Central]
Figure 1.
Structures of leucoverdazyls tested in the study and reference compounds: pleconaril and guanidine hydrochloride. Pleconaril was kindly provided by dr. V.A. Makarov (Research Center of Biotechnology RAS, 33-1 Leninsky prospect, 119071, Moscow, Russia). Guanidine hydrochloride was bought from Dia-M ltd (Moscow, Russia).
Figure 1.
Structures of leucoverdazyls tested in the study and reference compounds: pleconaril and guanidine hydrochloride. Pleconaril was kindly provided by dr. V.A. Makarov (Research Center of Biotechnology RAS, 33-1 Leninsky prospect, 119071, Moscow, Russia). Guanidine hydrochloride was bought from Dia-M ltd (Moscow, Russia).
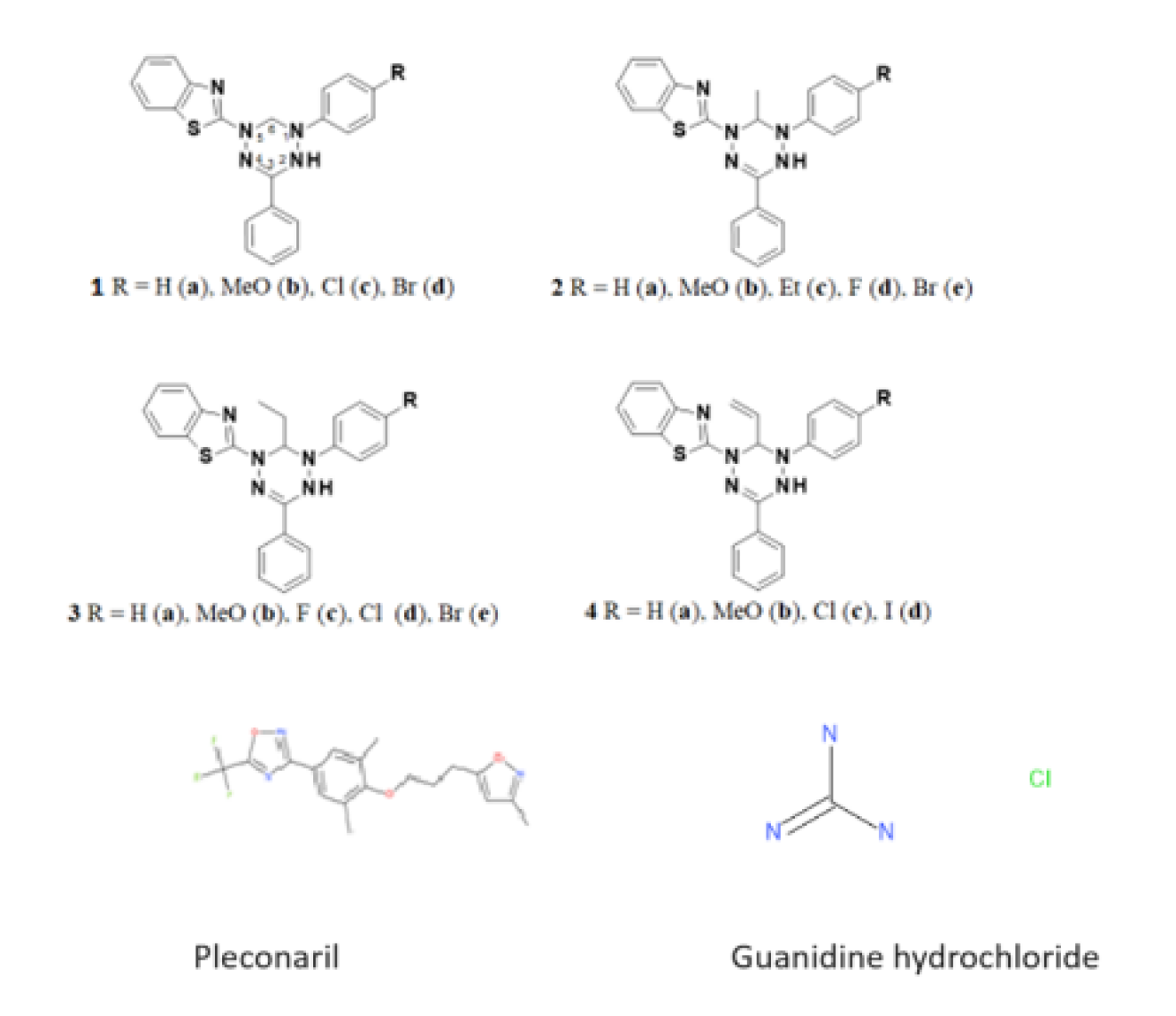
Figure 2.
Antioxidant activity of selected dihydrotetrazines. Presented are IC50 values (µM) for antioxidant activity of the tested compounds by DPPH assay. Vitamin C was used as a reference.
Figure 2.
Antioxidant activity of selected dihydrotetrazines. Presented are IC50 values (µM) for antioxidant activity of the tested compounds by DPPH assay. Vitamin C was used as a reference.
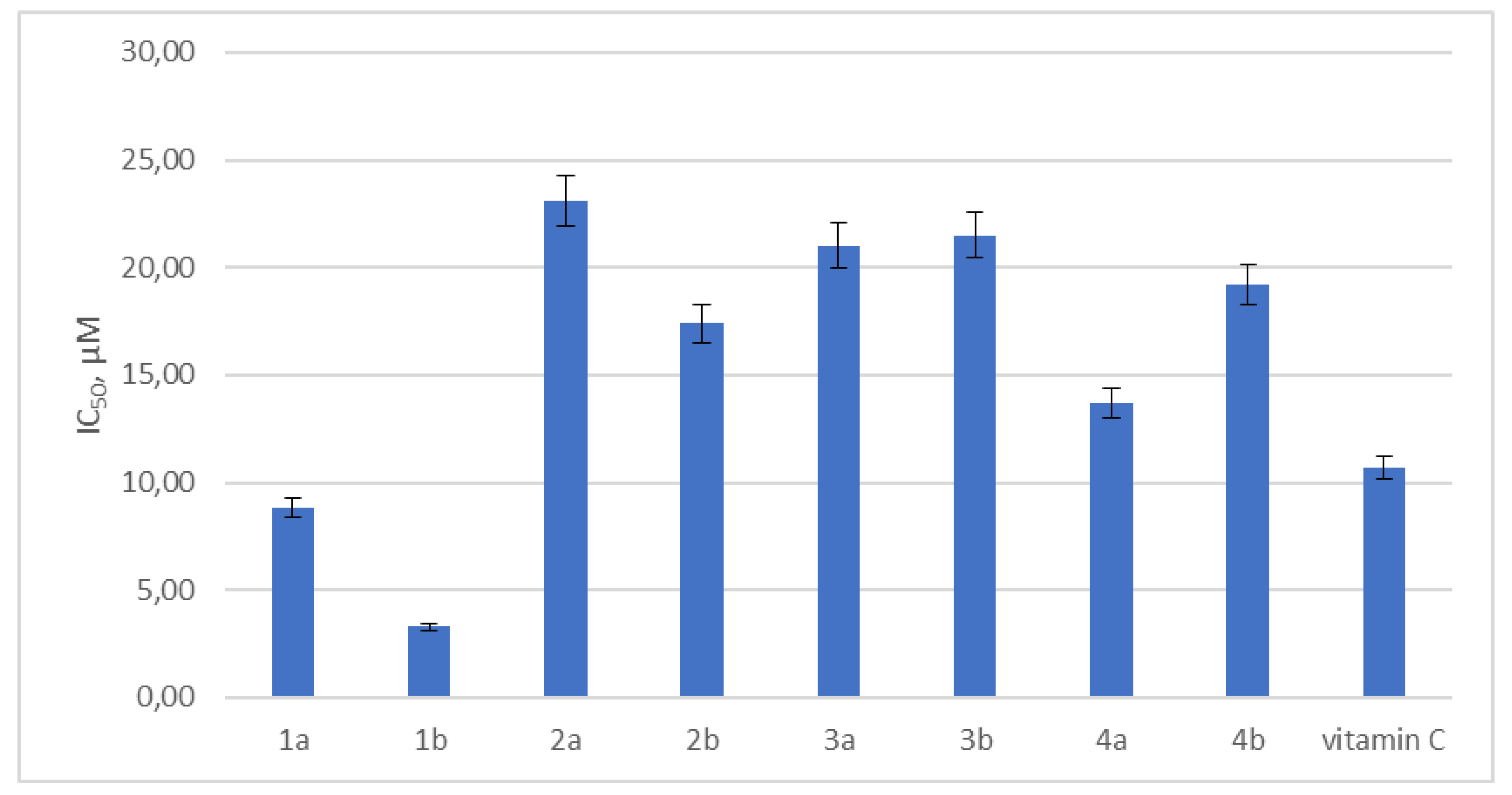
Figure 3.
Thermostabilizing properties of 1a in comparison to those of pleconaril. Values are the mean ± SD of three independent experiments. The legend shows the concentration of each compound tested. The asterisk indicates the significance of the difference in viral titer for pleconaril at 51°C and 55.2°C relative to the virus control, p < 0.05 by Mann–Whitney U-test.
Figure 3.
Thermostabilizing properties of 1a in comparison to those of pleconaril. Values are the mean ± SD of three independent experiments. The legend shows the concentration of each compound tested. The asterisk indicates the significance of the difference in viral titer for pleconaril at 51°C and 55.2°C relative to the virus control, p < 0.05 by Mann–Whitney U-test.
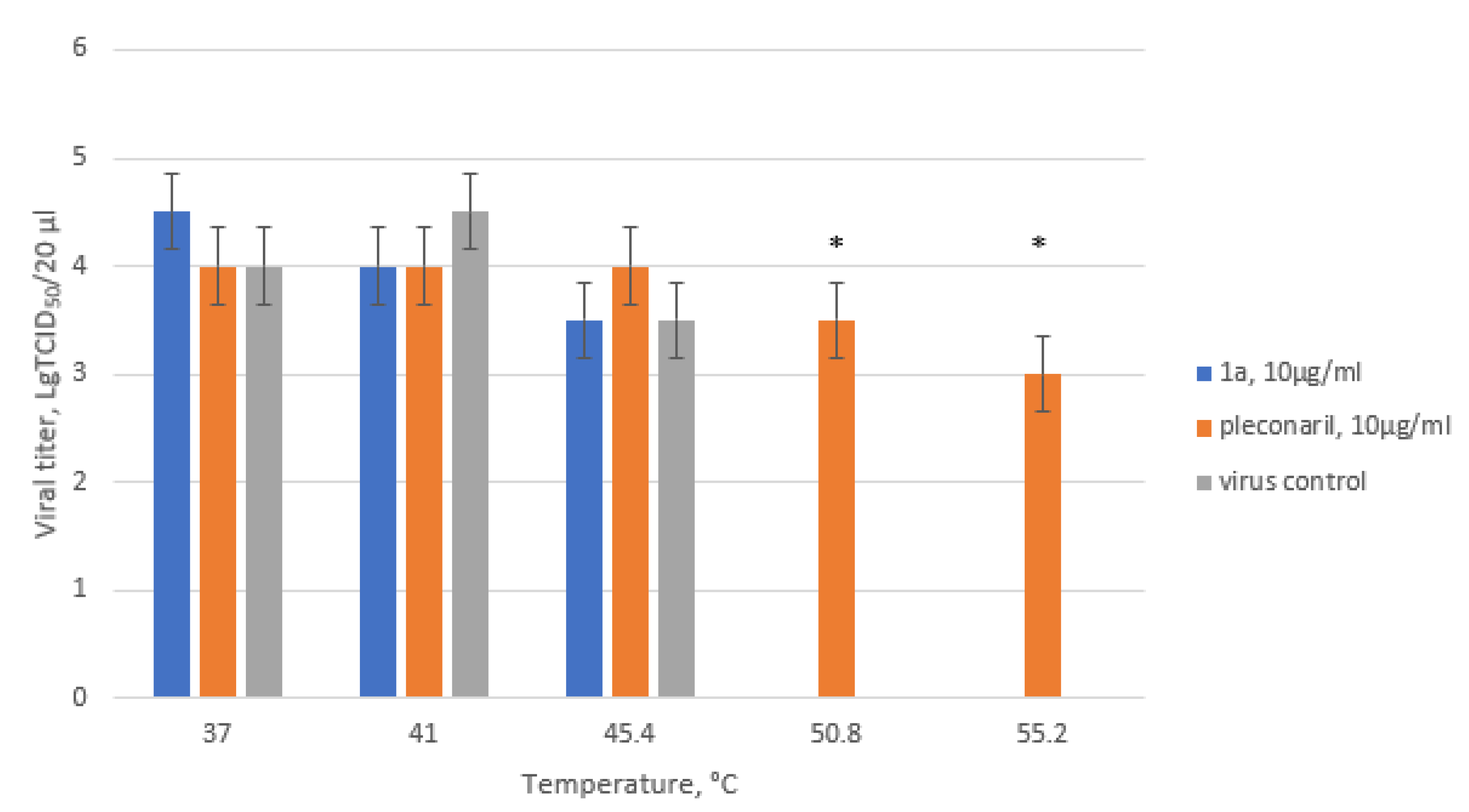
Figure 4.
Results of time-of-addition assay for 1a. The activity of compound 1a against the Coxsackie B4 virus (Powers strain) depending on the time of addition to a permissive cell line upon CVB4 infection. Vero cells were infected with CVB4 (–1 h), and 1a (5 ug/ml) was added at the indicated time points (in hours) either before the virus (–2 h), concomitantly with the virus (–1 h), or after (0, 2, 4, 6 h) infection, where 0 corresponds to the moment of completed virus absorption on the cell surface. The infectious activity of the viral progeny was evaluated by end-point titration in the Vero cells in lg TCID50/0.2 ml. Pleconaril (10 ug/ml) was used as a reference compound. Values are presented as the mean ± SD of three independent experiments. Asterisk indicates significance of difference in viral titer for 1a and pleconaril relative to the virus control, p < 0.05 by Mann–Whitney U-test.
Figure 4.
Results of time-of-addition assay for 1a. The activity of compound 1a against the Coxsackie B4 virus (Powers strain) depending on the time of addition to a permissive cell line upon CVB4 infection. Vero cells were infected with CVB4 (–1 h), and 1a (5 ug/ml) was added at the indicated time points (in hours) either before the virus (–2 h), concomitantly with the virus (–1 h), or after (0, 2, 4, 6 h) infection, where 0 corresponds to the moment of completed virus absorption on the cell surface. The infectious activity of the viral progeny was evaluated by end-point titration in the Vero cells in lg TCID50/0.2 ml. Pleconaril (10 ug/ml) was used as a reference compound. Values are presented as the mean ± SD of three independent experiments. Asterisk indicates significance of difference in viral titer for 1a and pleconaril relative to the virus control, p < 0.05 by Mann–Whitney U-test.
Figure 5.
Ultrastructure of Vero cells infected by CVB3 revealed by transmissive electron microscopy. A – intact cell. No vacuoles or replication organelles are visible within cytoplasm. B – CVB3-infected cell. Numerous vacuoles representing virus-specific replication organelles are indicated by arrowheads. C – CVB3-infected cell in the presence of 100 μM compound 1a. No morphological signs of viral replication can be seen.
Figure 5.
Ultrastructure of Vero cells infected by CVB3 revealed by transmissive electron microscopy. A – intact cell. No vacuoles or replication organelles are visible within cytoplasm. B – CVB3-infected cell. Numerous vacuoles representing virus-specific replication organelles are indicated by arrowheads. C – CVB3-infected cell in the presence of 100 μM compound 1a. No morphological signs of viral replication can be seen.
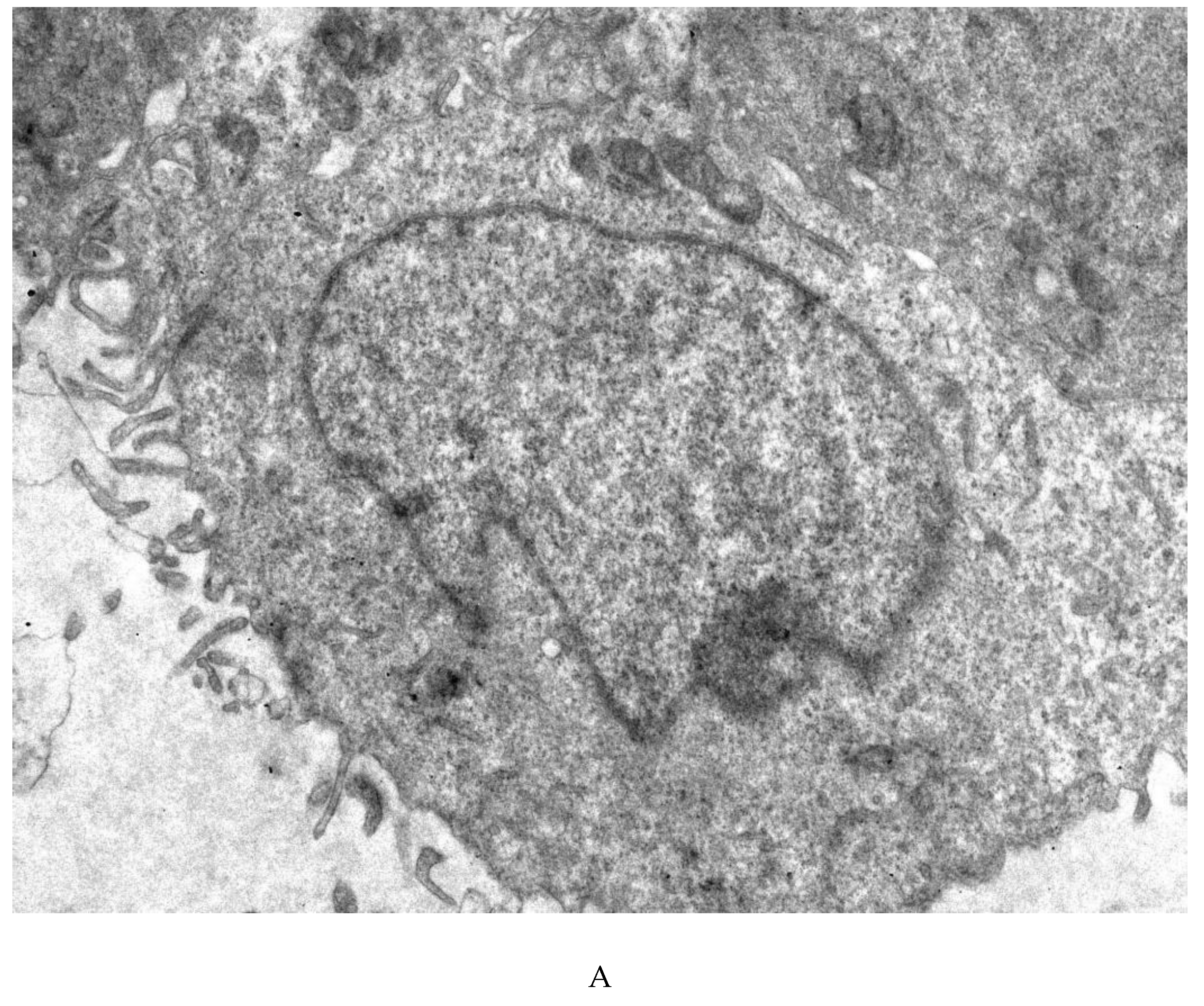
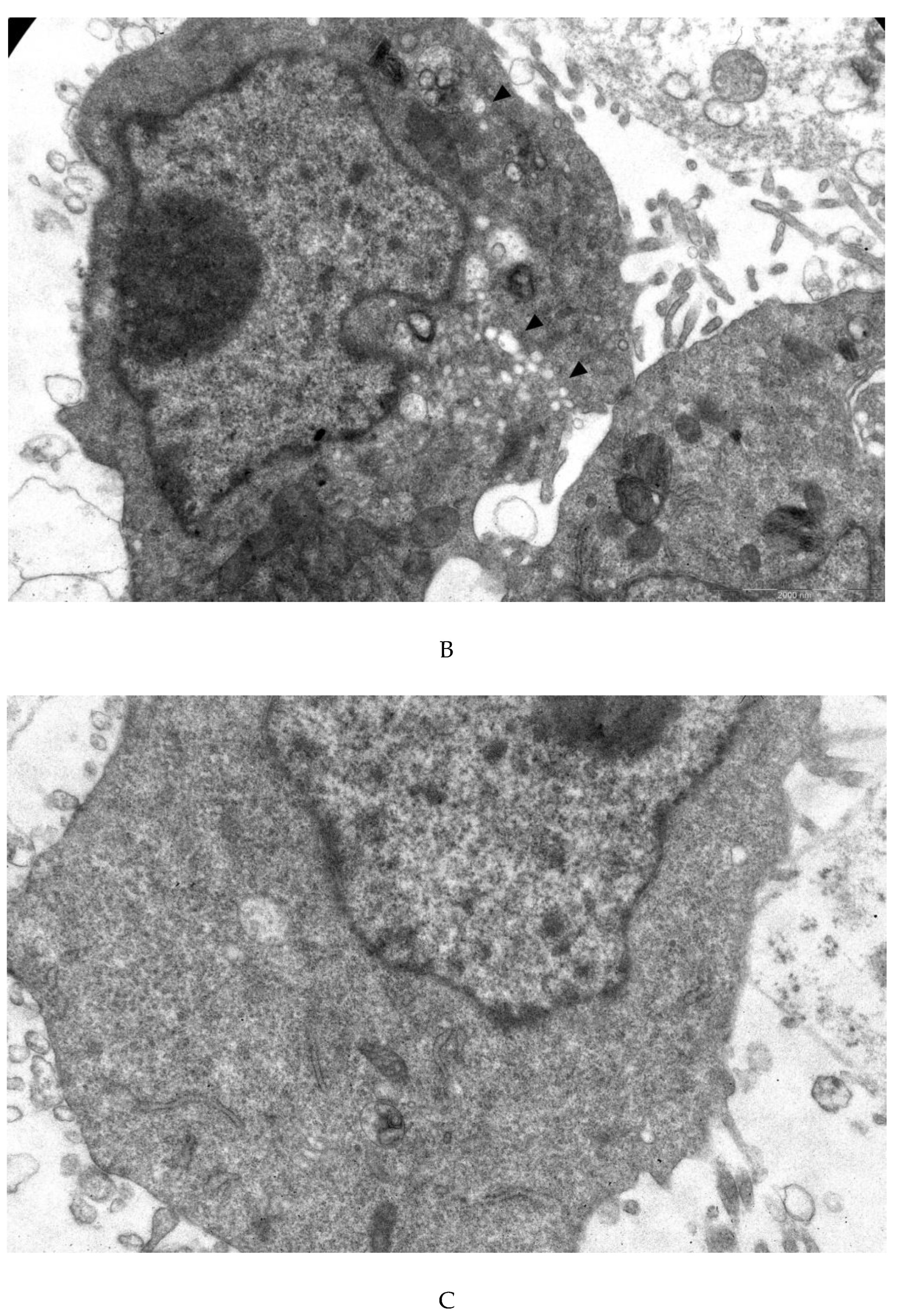
Figure 6.
Comparison of IC50 values for the CVB3 R and CVB3 WT strains. Presented are the results of viral yield reduction assay for two CVB3 strains: wild-type and resistant virus propagated in the presence of 1a. 4PL were fitted using GraphPad Prism 6. Viral titer is represented in % relative to virus control.
Figure 6.
Comparison of IC50 values for the CVB3 R and CVB3 WT strains. Presented are the results of viral yield reduction assay for two CVB3 strains: wild-type and resistant virus propagated in the presence of 1a. 4PL were fitted using GraphPad Prism 6. Viral titer is represented in % relative to virus control.
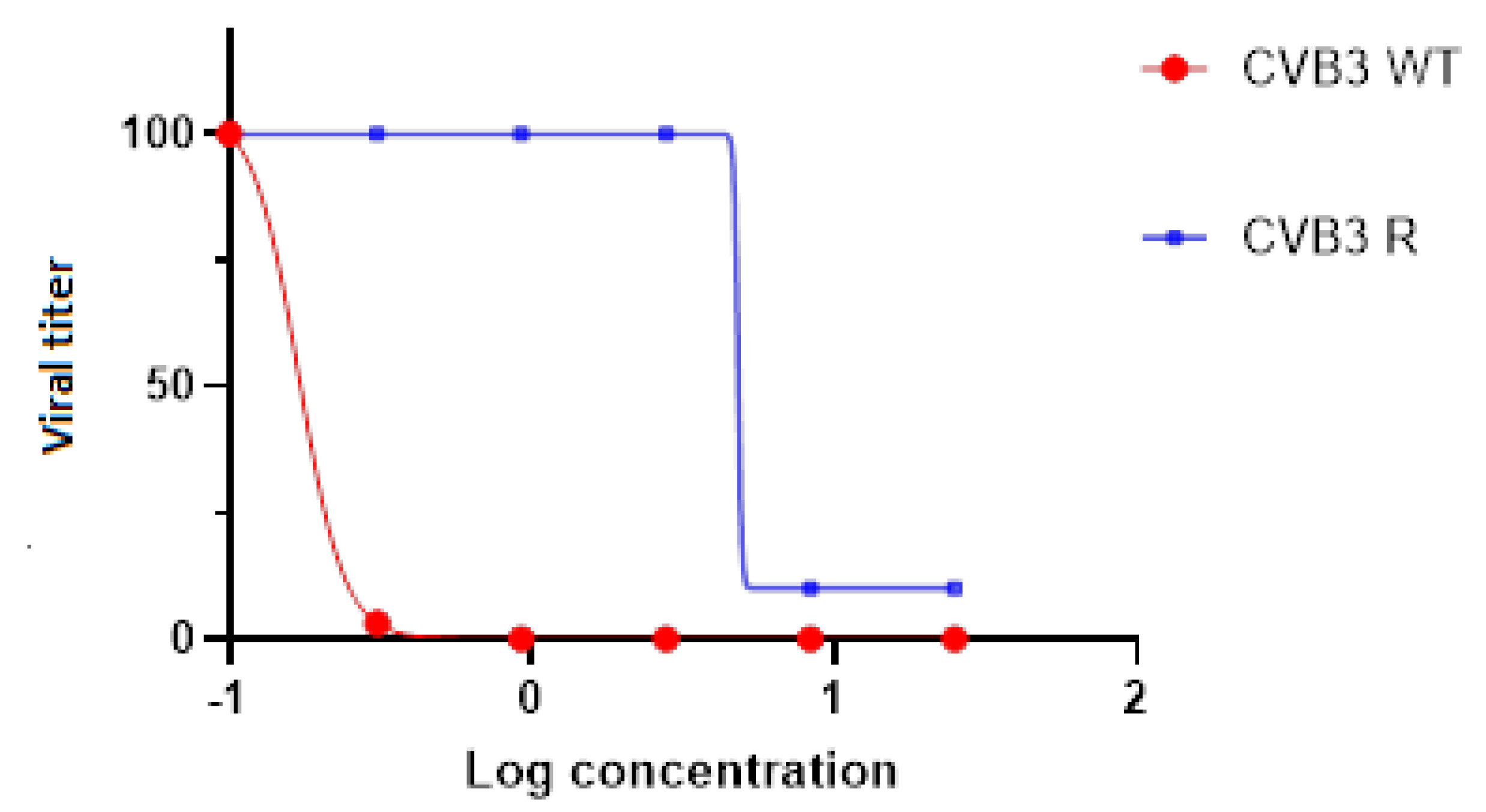
Figure 7.
Propagation kinetics of CVB3 WT and CVB3 R strains in Vero cells with and without 1a. Viral progeny titer is plotted versus incubation time. Multistep growth curves are presented. Asterisk indicates significance of difference in virus titer, p<0.05 by Mann–Whitney U-test.
Figure 7.
Propagation kinetics of CVB3 WT and CVB3 R strains in Vero cells with and without 1a. Viral progeny titer is plotted versus incubation time. Multistep growth curves are presented. Asterisk indicates significance of difference in virus titer, p<0.05 by Mann–Whitney U-test.
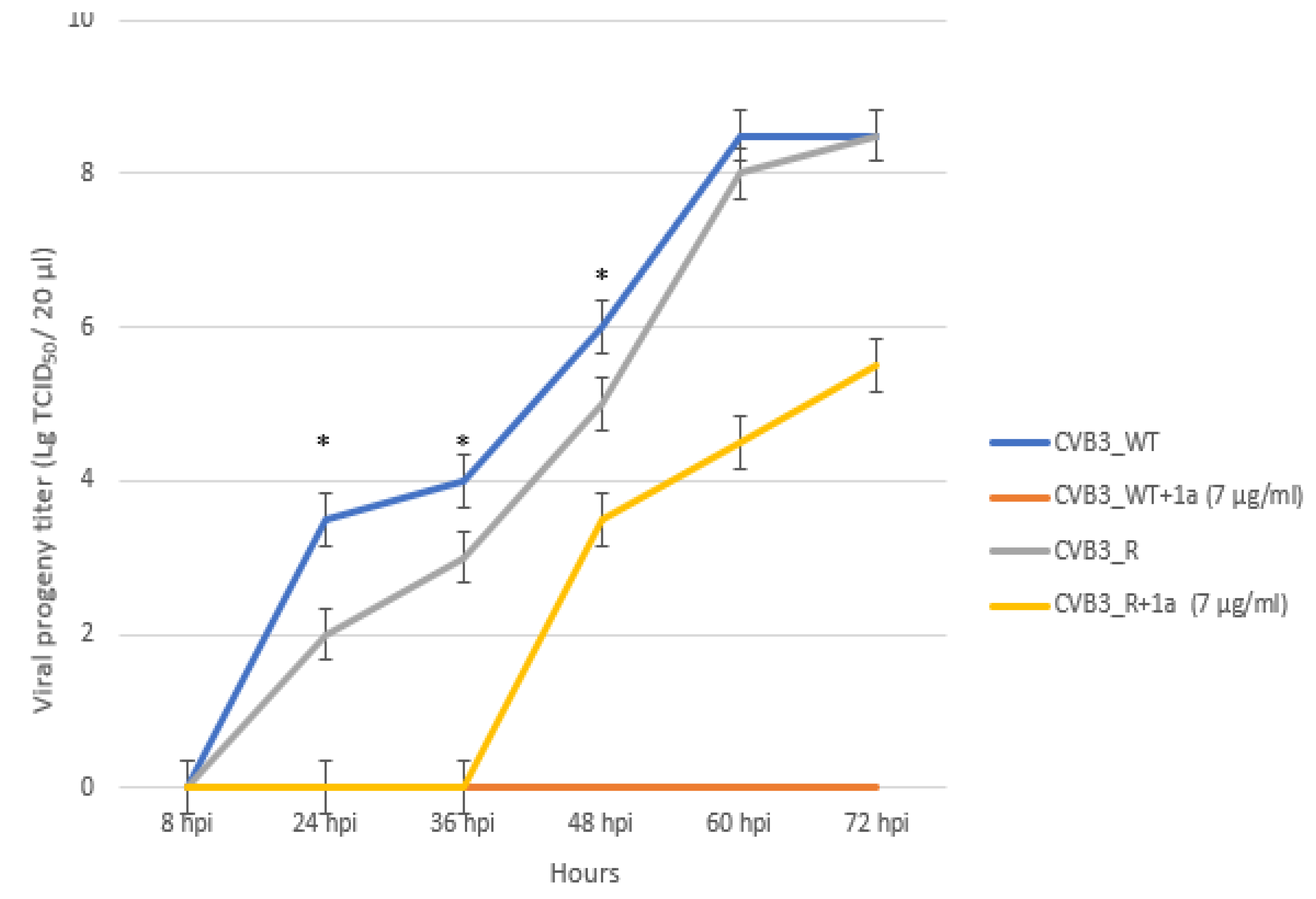
Figure 8.
Position of S109I mutation in 2C protein. The amino acid position is depicted in red. The C- and N- termini of the protein are marked with C and N, respectively.
Figure 8.
Position of S109I mutation in 2C protein. The amino acid position is depicted in red. The C- and N- termini of the protein are marked with C and N, respectively.
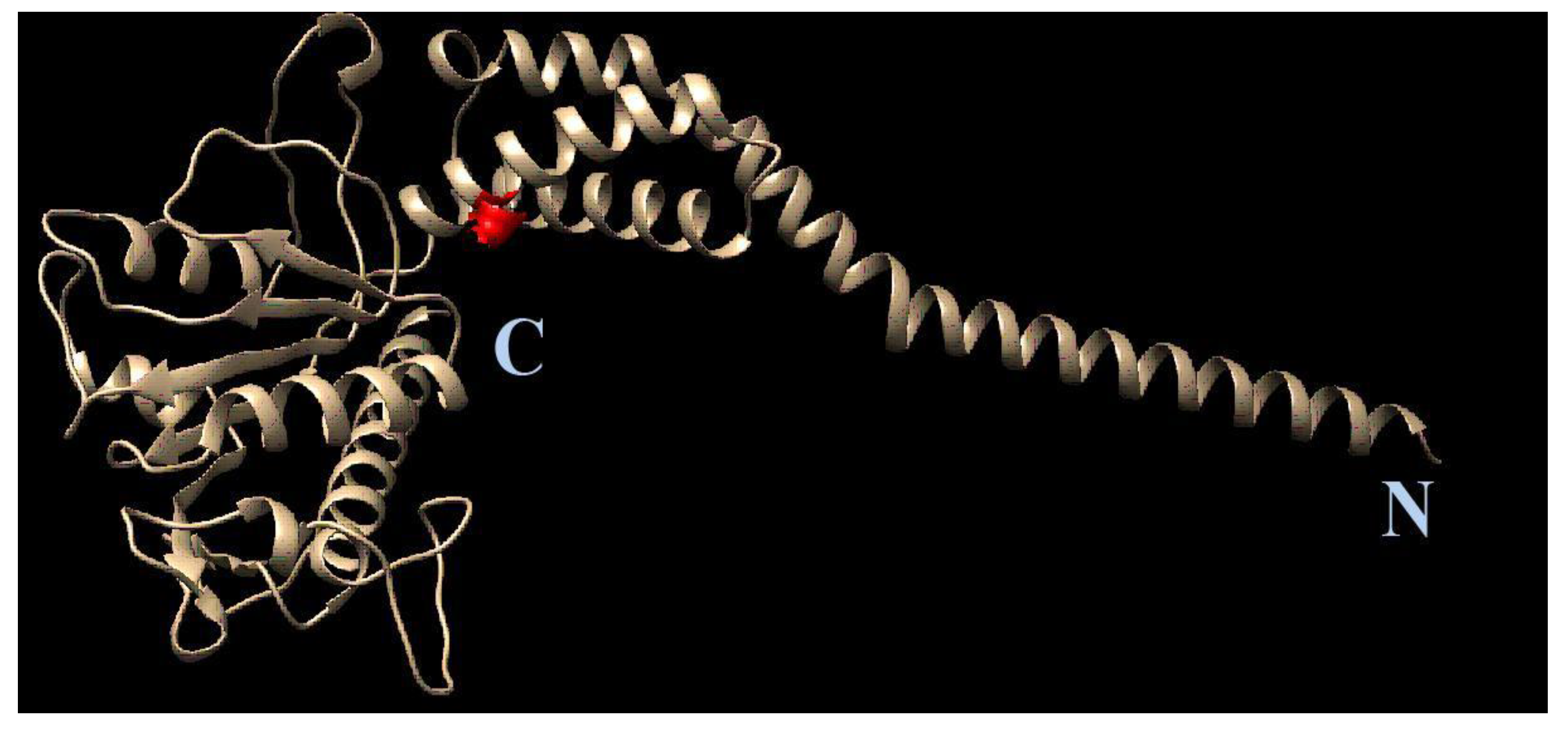
Figure 9.
Colocalization of the 1a binding site and the S109I amino acid substitution in 2C protein of 1a-resistant Coxsackievirus B3. The substitution is indicated by arrow.
Figure 9.
Colocalization of the 1a binding site and the S109I amino acid substitution in 2C protein of 1a-resistant Coxsackievirus B3. The substitution is indicated by arrow.
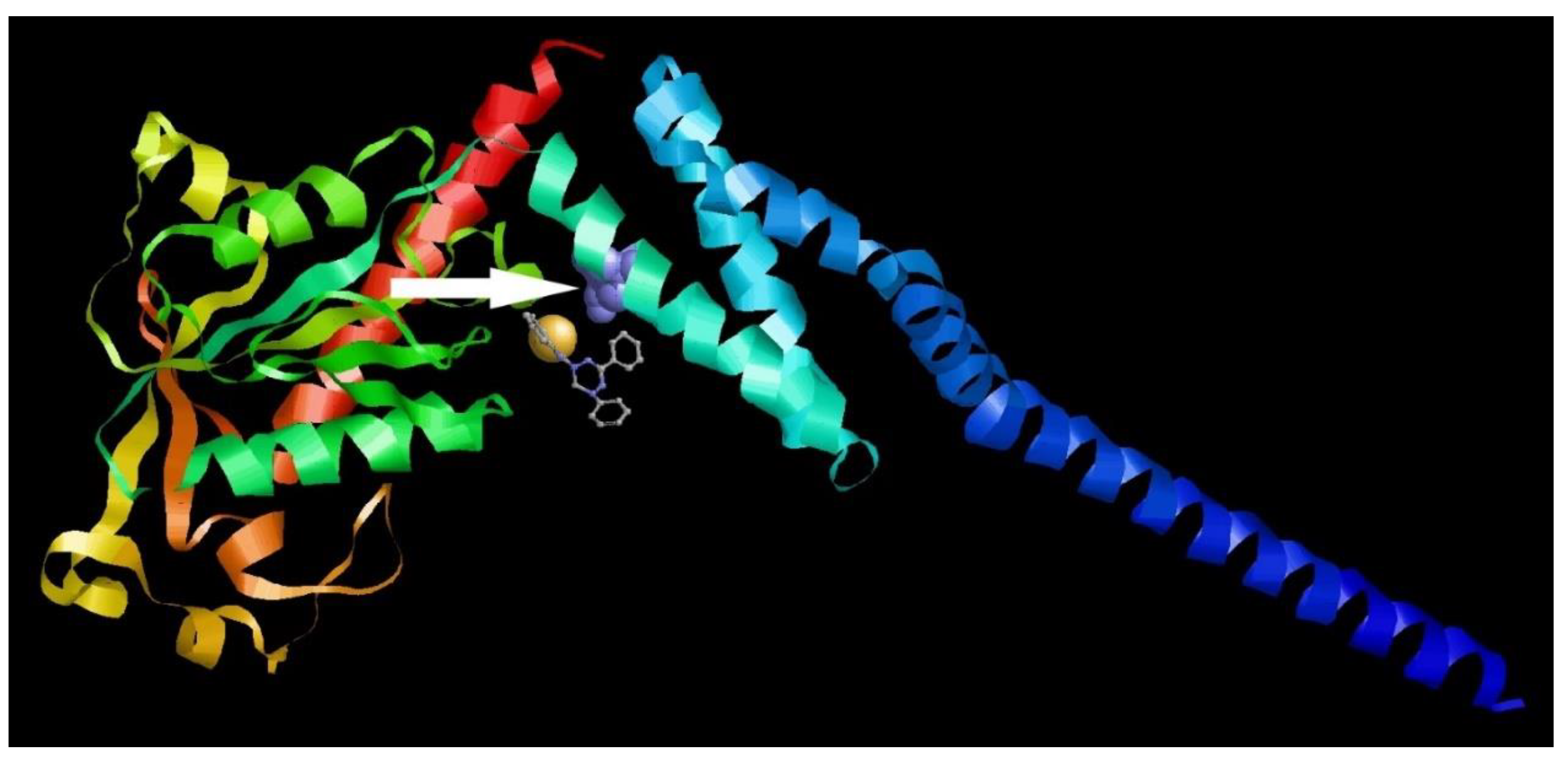

Table 1.
Cytotoxic and virus-inhibiting properties of leucoverdazyls against Coxsackie B3 virus in vitro.
Table 1.
Cytotoxic and virus-inhibiting properties of leucoverdazyls against Coxsackie B3 virus in vitro.
| Compound number | CC50, µM a | IC50 µM b | SI |
|---|---|---|---|
| 1а | 619.9±52.7 | 1.8±0.3 | >230 |
| 1b | >1347 | 5.4±1.1 | >250 |
| 1c | 320.9±28.2 | 7.4±2.2 | 43 |
| 1d | 886.4±90.4 | 6.2±1.9 | 142 |
| 2a | >324.7 | 6.49±0.8 | >50 |
| 2b | 2048.2±198.5 | 24.1±3.2 | 85 |
| 2c | >302.7 | >121.1 | >2 |
| 2d | 49.6±3.7 | >49.6 | <1 |
| 2e | 69.1±5.9 | >69.1 | <1 |
| 2f | >301.2 | 12.1±1.9 | >25 |
| 3a | >313.3 | >125.3 | >2 |
| 3b | 2331.1±25.3 | 60.1±7.2 | >38 |
| 3c | 359.7±41.7 | 71.9±6.4 | 5 |
| 3d | 2309.5±19.7 | >231.0 | >10 |
| 3e | 945.5±89.6 | >209.6 | >4 |
| 4a | >314.8 | >125.9 | >2 |
| 4b | >292.7 | >117.1 | >2 |
| 4c | >290.7 | 27.9±3.3 | 10 |
| 4d | 1529.6±17.2 | 51.6±6.4 | 29 |
a CC50 is the cytotoxic concentration, the concentration resulting in the death of 50% of the cells (CC50 were evaluated after 24 h of incubation with compound only); b IC50 is the 50% virus-inhibiting concentration, the concentration leading to 50% inhibition of virus replication; c SI is the selectivity index, the ratio of CC50/IC50. The data presented are the mean of three independent experiments. The values for CC50 and IC50 are presented as the mean ± error of the experiment.
Table 2.
Activity of 1a against a panel of group A, B, C enteroviruses.
| Virus | CC50, µM a | IC50 µM b | SI |
|---|---|---|---|
| Compound 1a | |||
| CVA16 | 673.3±60.1 | 0.8±0.2 | 841 |
| CVB5 | 619.9±52.7 | 1.5±0.3 | 413 |
| ECHO30 | 673.3±60.1 | 1.6±0.4 | 420 |
| CVA24 | 673.3±60.1 | 0.9±0.3 | 747 |
| CVB4 (strain Powers) | 619.9±52.7 | 1.7 ±0.5 | 364 |
| Guanidine hydrochloride | |||
| CVA16 | >10471.2 | 336.8±28.3 | >31 |
| CVB5 | >5235.6 | 473.7±33.4 | >10 |
| ECHO30 | >10471.2 | 125.6±18.2 | >83 |
| CVA24 | >10471.2 | 314.2±20.1 | >33 |
| CVB4 (strain Powers) | >5235.6 | 495.7±26.5 | >10 |
a CC50 is the cytotoxic concentration, the concentration resulting in the death of 50% of the cells (CC50 were evaluated after 24 h of incubation with compound only); b IC50 is the 50% virus-inhibiting concentration, the concentration leading to 50% inhibition of virus replication; c SI is the selectivity index, the ratio of CC50/IC50. The data presented are the mean of three independent experiments. The values for CC50 and IC50 are presented as the mean ± error of the experiment.
Table 3.
Activity spectra of 1a against RNA and DNA viruses of various structure.
| Virus | CC50, µM a | IC50 µM b | SI |
|---|---|---|---|
| Influenza A/Puerto Rico/8/34 | 850.2±78.3 | 38.7±2.9 | 22 |
| Influenza B/Florida/04/0 6 | 850.2±78.1 | 40.5±5.1 | 21 |
| HSV1 | 450.7±51.2 | 46.3±4.4 | 10 |
| Ad5 | 500.6±45.7 | 43.1±3.8 | 12 |
| SARS-CoV-2 | 656.3±70.4 | 4.5±1.2 | 146 |
a CC50 is the cytotoxic concentration, the concentration resulting in the death of 50% of the cells (CC50 were evaluated after 48 h or 24 h of incubation with compound only depending on the life cycle length of the particular virus tested); b IC50 is the 50% virus-inhibiting concentration, the concentration leading to 50% inhibition of virus replication; c SI is the selectivity index, the ratio of CC50/IC50. The data presented are the mean of three independent experiments. The values for CC50 and IC50 are presented as the mean ± error of the experiment.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



