
Submitted:
01 April 2024
Posted:
01 April 2024
You are already at the latest version
Abstract
Lead optimization in drug discovery is a crucial phase where initial hits are refined into compounds with improved pharmacological properties. While traditional methods rely on manual experimentation and modifications, AI-driven techniques have revolutionized this process by leveraging big data and predictive modeling. This review explores how AI-driven approaches accelerate lead optimization, showcasing examples like deep neural networks and reinforcement learning. Integration of multi-omic data and experimental validation further enhances AI-driven strategies. The future lies in refining these AI methods, democratizing tools, and interdisciplinary collaboration to streamline drug discovery and address medical needs efficiently.
Keywords:
Lead optimization
; drug discovery
; artificial intelligence
; machine learning
; deep neural networks
; cheminformatics
; chemical modifications
; drug development
1. Introduction
Recent advancements in AI have significantly accelerated the drug compound identification and testing process, leading to a shorter time to market [1,2]; both highlight the use of AI in drug identification and lead optimization, resulting in a substantial reduction in the clinical trial process from 10-12 years to 3-4 months [3,4], at times, highlighting the predictive capacity of AI in identifying potential drug candidates and optimizing lead compounds, respectively. These advancements lead to the use of AI in high throughput screening and the potential for rapid development of safe and effective therapies [5,6]. A practical example of AI's role in drug discovery is the demonstration of the successful and rapid discovery of drug candidates using a DM-AI drug discovery platform [7].
Lead compounds are initial chemical entities identified during drug discovery that exhibit promising biological activity against a specific target or disease pathway. These compounds are the starting points for further optimization to enhance their potency, selectivity, and pharmacokinetic properties. Lead optimization stands at the critical juncture of drug discovery, where initial hits with promising biological activity are meticulously refined into optimized lead compounds [8,9]. This pivotal phase marks the transition from early-stage research to the development of potential therapeutic agents, requiring a delicate balance between enhancing potency and selectivity while addressing concerns regarding toxicity and pharmacokinetics. At its core, lead optimization encompasses a multifaceted approach, addressing chemical, pharmaceutical, and pharmacological aspects to ensure the development of safe and effective drug candidates. With the escalating demands for novel medicines to address unmet medical needs, the efficiency and effectiveness of lead optimization have become paramount in accelerating the translation of scientific discoveries into clinically viable treatments [10,11,12].
Chemically, optimization involves fine-tuning the molecular structure of lead compounds to enhance their properties, aiming for optimal interaction with the target biomolecule while minimizing off-target effects. In contrast, on the pharmaceutical front, considerations extend to factors such as solubility, stability, and bioavailability, which are crucial for formulation and delivery [9,13,14,15,16]. Furthermore, lead optimization intersects with pharmacological principles, requiring a comprehensive understanding of the target's biological pathways and potential therapeutic implications. While traditional medicinal chemistry methods have historically dominated lead optimization, there's a rising interest in leveraging artificial intelligence (AI) and machine learning (ML) techniques, notably deep neural networks (DNNs), to expedite this process. Such methods harness the vast datasets generated from high-throughput screening, structural biology, and medicinal chemistry to navigate the complex landscape of chemical space and predictively guide molecular modifications with unprecedented accuracy and efficiency. AI-enabled approaches promise to overcome traditional barriers to lead optimization, such as the time-consuming nature of iterative experimentation and the limitations of empirical trial-and-error methods.[13,17,18,19,20,21] These advanced computational techniques have aided in refining compounds by accurately predicting optimal binding poses, estimating binding free energies, and fine-tuning molecular mechanics force fields. This integration of AI not only enhances the efficiency of lead optimization but also reduces the computational burden associated with resource-intensive simulations, ultimately streamlining the journey from lead identification to the development of potent therapeutic candidates [22]. By leveraging advanced algorithms and predictive models, researchers can now rapidly explore and prioritize lead candidates with the highest likelihood of success, reducing the time and resources required for preclinical development. Nonetheless, significant challenges persist, including the requirement for high-quality data, accurate ADMET prediction, and prevention of model overfitting. This review explores the significance of lead optimization across chemical and pharmaceutical dimensions while delving into AI methods' role in advancing this critical stage of drug development [16,23].
2. Traditional Methods vs AI-driven Lead Optimization:
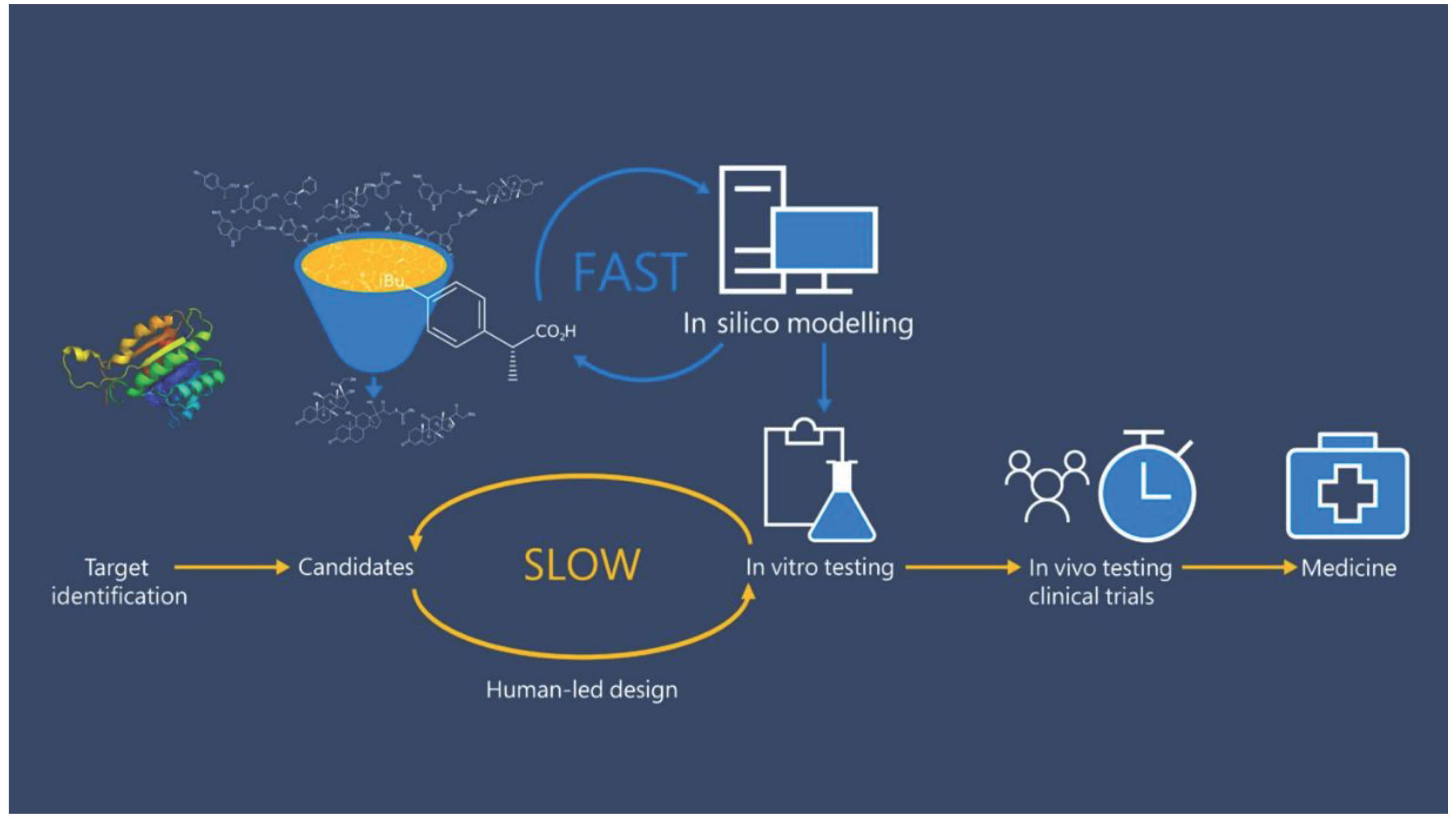
Figure 1 shows a comparative perspective of the AI-driven lead compound discovery and the discovery process pre-AI era.
Traditional lead optimization methods typically rely on empirical approaches, where medicinal chemists iteratively design and synthesize analogs of lead compounds based on intuition, prior knowledge, and experimental data. These methods involved extensive laboratory experimentation, including synthesis, biochemical assays, and pharmacological testing, to evaluate candidate compounds' potency, selectivity, and pharmacokinetic properties. From the discovery of aspirin's analgesic and anti-inflammatory properties over a century ago to the development of statins like Lipitor for managing cholesterol levels, these medications have stood the test of time and remain indispensable in modern healthcare [24,25,26]. While traditional methods have successfully produced many clinically valuable drugs, they are often time-consuming, resource-intensive, and limited by the capacity to explore vast chemical space comprehensively. Researchers have enhanced these drugs' efficacy, safety, and pharmacokinetic profiles through iterative chemical modifications and empirical testing, demonstrating the enduring value of traditional lead optimization approaches in pharmaceutical innovation [27,28,29,30].
In contrast, AI-driven approaches in lead optimization leverage computational techniques and ML algorithms to augment and accelerate the drug discovery process. These methods harness the power of big data and predictive modeling to analyze large datasets of chemical structures, biological activities, and experimental results. By learning patterns and relationships from these data, AI algorithms can predictively guide lead optimization efforts, prioritize promising candidates, and even propose novel chemical scaffolds with enhanced properties [31].
One significant advantage of AI-driven approaches is their ability to explore chemical space more comprehensively and efficiently than traditional methods. Through virtual screening, molecular docking, and quantitative structure-activity relationship (QSAR) modeling, AI algorithms can rapidly evaluate millions of compounds, thereby accelerating the identification of lead candidates with desirable properties. Additionally, AI helps optimize lead compounds by predicting their binding affinity, ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties, and relevant pharmacological parameters, reducing the need for extensive experimental validation. Moreover, AI-driven approaches offer the potential for automation and scalability, allowing researchers to expedite lead optimization campaigns and explore larger chemical libraries with minimal manual intervention. By integrating AI into the drug discovery workflow, organizations streamline decision-making, reduce costs, and increase the likelihood of identifying successful drug candidates[32,33,34,35,36].
In recent research endeavors, the BIOVIA Generative Therapeutics Design (GTD) application has emerged as a powerful tool for addressing lead finding and optimization challenges. This platform harnesses 3D structural models of ligand-protein interactions, incorporating pharmacophoric representations of desired features. For instance, in a study involving the discovery of SYK inhibitors entospletinib and lanraplenib, GTD effectively tackled common issues in lead optimization. By retrospectively re-identifying drug candidate molecules based on data from intermediate project stages and applying chemical space constraints, researchers demonstrated GTD's capability to refine lead compounds. Moreover, the GTD platform was configured to generate molecules incorporating features from multiple unrelated molecule series, showcasing its application of AI/ML to drug discovery and lead optimization [37,38,39].
Similarly, the Query-based Molecule Optimization (QMO) framework has been proposed as a versatile solution for improving the properties of input molecules based on efficient queries guided by molecular property predictions and evaluation metrics. Outperforming existing methods, QMO excelled in optimizing small organic molecules for drug-likeness and solubility under similarity constraints. Its effectiveness was further demonstrated in tasks such as optimizing potential SARS-CoV-2 main protease inhibitors and enhancing antimicrobial peptides, showcasing its potential in addressing real-world discovery problems and facilitating material optimization with design constraints[40].
Furthermore, the integration of AlphaFold2 and Chemistry42 has proven instrumental in discovering Cyclin-Dependent Kinase 20 (CDK20) inhibitors. Leveraging AlphaFold2's protein structure prediction and Chemistry42's generative chemistry capabilities, researchers navigated challenges posed by the absence of structural information for the protein and reported tool compounds. Through the generative pipeline, numerous molecules were produced, with hit optimization resulting in the discovery of primary hits with improved potency. This synergy between AI methods in target identification, protein folding, and generative chemistry showcases a promising approach to lead optimization [18,19,41,42,43].
Lastly, ZairaChem has emerged as an AI- and ML-based tool for modeling quantitative structure-activity/property relationships (QSAR/QSPR), streamlining data-driven drug discovery processes. Deployed at the H3D Centre for Malaria and Tuberculosis Drug Discovery, ZairaChem has facilitated the development of a virtual screening cascade for critical decision-making assays. This tool represents a significant advancement in enabling streamlined drug discovery processes, informing compound progression, and facilitating lead identification in resource-limited settings. Through real-world prospective studies, ZairaChem has demonstrated its efficacy in identifying lead-like compounds, showcasing its potential to revolutionize drug discovery efforts [44].
3. Key Future Optimization Strategies:
Key strategies are pivotal for maximizing the effectiveness and efficiency of lead optimization endeavors. Data integration and harmonization are foundational, ensuring that diverse datasets from experimental assays, chemical databases, and literature are combined cohesively. Feature engineering and representation learning play critical roles in capturing compound properties and biological activities, with techniques like graph-based representations and molecular embeddings extracting meaningful features automatically. Model selection is paramount, with various architectures like Deep neural networks (DNN) and ensemble methods offering unique advantages. Active learning and experimental design strategies iteratively select informative compounds for testing, accelerating the exploration of chemical space. Interpretability and explainability are vital for understanding model predictions and fostering collaborative methods. Transfer learning and domain adaptation techniques enhance model generalization, while integration with experimental validation ensures real-world applicability. In addition to the foundational strategies mentioned, several advanced techniques optimize lead optimization using AI. One such strategy uses Bayesian optimization, which efficiently selects compounds based on predictive models' uncertainty estimates for experimental testing. Bayesian optimization iteratively explores the chemical space by balancing the exploitation of known regions with the exploration of uncertain areas, thereby maximizing the discovery of promising lead candidates [45,46,47].
Moreover, reinforcement learning algorithms can guide compound design by learning from past actions and rewards, enabling AI systems to generate compounds with desired properties through trial and error iteratively. Adopting cloud-based AI platforms and high-performance computing infrastructures enables scalable and parallelized lead optimization workflows, facilitating rapid model training, validation, and deployment. Furthermore, implementing robust model validation and uncertainty quantification techniques, such as bootstrap resampling and Monte Carlo dropout, enhances the reliability and robustness of AI predictions, ensuring their applicability in real-world drug discovery scenarios [48,49,50,51]. Researchers can leverage AI to expedite lead optimization by employing these optimization strategies, facilitating the discovery of novel therapeutics with enhanced efficacy and safety profiles.
In modern drug discovery, in silico-driven optimization of compound properties related to pharmacokinetics, pharmacodynamics, and safety is paramount. DNN frameworks have also emerged as predictive models, with various model architectures differing in their applicability and performance in lead optimization projects. One study compares three established DNN-based methods - multilayer perceptron (MLP), graph convolutional network (GCN), and Mol2Vec - for predicting key ADME property trends and biological activity. From a statistical perspective, MLP and GCN outperformed Mol2Vec, with GCN-based predictions demonstrating superior stability over time. Additionally, DNNs prove valuable in guiding local structure-activity relationships (SAR), illustrating their significance in pharmaceutical research projects [27,52,53,54,55,56].
Furthermore, new methods for automated compound design against profiles of multiple properties are of great value in lead optimization. A fragment-based reinforcement learning approach, which utilizes bidirectional long short-term memory (LSTM) networks, has been developed to generate novel molecules with optimal properties. This AI method learns to generate compounds with desired properties by improving upon initial lead molecules through fragment replacement. As demonstrated in a case study, this approach yields chemically valid molecules satisfying targeted objectives, showcasing its potential in lead optimization efforts as well [57]. Moreover, multi-parameter optimization (MPO) poses a significant challenge in lead optimization. Recent advancements in deep learning generative models offer promise for addressing MPO in drug discovery projects. In one study, this AI method is applied to accelerate the obtention of lead compounds, meeting multiple biological activity objectives simultaneously. Using a ligand-based de novo design technology with deep learning generative models, QSAR models were built for 11 objectives, generating virtual compounds predicted as active on all objectives. Synthesized and tested, these AI-designed compounds significantly outperformed initial molecules, demonstrating the efficacy of AI algorithms in optimizing compounds for multiple parameters relevant to lead optimization [58,59,60].
In the realm of lead optimization, these examples showcase the diverse applications of AI-driven approaches in guiding compound design, predicting compound properties, and accelerating the discovery of lead candidates with enhanced efficacy and safety profiles. From leveraging DNNs for predicting key ADME property trends to utilizing reinforcement learning and DL generative models for automated compound design and multi-parameter optimization, AI holds immense potential in revolutionizing the drug discovery process and facilitating the development of novel therapeutics.
4. Conclusions
In conclusion, lead optimization is a critical phase in drug discovery, where initial hits are refined into optimized lead compounds with enhanced potency, selectivity, and pharmacokinetic properties. Traditional methods have historically relied on empirical approaches involving iterative experimentation and manual chemical modifications guided by intuition and prior knowledge. While effective, these methods are often time-consuming and resource-intensive, presenting limitations in exploring vast chemical space comprehensively. However, the advent of AI, ML, and DL techniques has revolutionized lead optimization by leveraging big data and predictive modeling to expedite the identification and refinement of lead candidates. Through advanced algorithms and predictive models, AI facilitates comprehensive exploration of chemical space, guiding molecular modifications with unprecedented accuracy and efficiency. Examples such as the BIOVIA GTD application, QMO framework, and Chemistry42 integration illustrate the diverse applications and transformative potential of AI-driven approaches in lead optimization. By integrating AI into drug discovery workflows, researchers can streamline decision-making, reduce costs, and increase the likelihood of identifying successful drug candidates, ultimately accelerating the translation of scientific discoveries into clinically viable treatments. As the demands for novel medicines continue to escalate, the efficiency and effectiveness of AI-driven lead optimization have become paramount in addressing unmet medical needs and advancing pharmaceutical innovation.
The future direction of lead optimization lies in further integration and advancement of AI-driven approaches with traditional methodologies. Continued refinement of AI algorithms, including deep learning models and reinforcement learning techniques, will enhance their predictive capabilities and enable more accurate and efficient compound design. Moreover, developing hybrid AI models combining data-driven approaches with expert medicinal chemistry and pharmacology knowledge will further optimize lead optimization strategies. Additionally, there is a growing emphasis on integrating multi-omic data into AI-driven lead optimization workflows. By incorporating comprehensive biological datasets, researchers can gain deeper insights into disease mechanisms and drug-target interactions, leading to the identification of more effective lead compounds.
Furthermore, democratizing AI tools and platforms will play a crucial role in expanding access to advanced computational techniques for lead optimization. Efforts to develop user-friendly interfaces and cloud-based solutions will empower researchers across academia and industry to leverage AI in their drug discovery efforts, driving innovation and accelerating the development of new therapeutics. In terms of experimental validation, there will be a continued focus on integrating AI predictions with high-throughput screening assays and in vitro testing protocols. Researchers can more robustly and efficiently validate AI-driven lead optimization strategies by combining computational predictions with experimental data, increasing confidence in the identified lead candidates.
Overall, the future of lead optimization will be characterized by a synergistic relationship between AI-driven approaches and traditional methodologies fuelled by technological advances, data availability, and interdisciplinary collaboration. By harnessing the full potential of AI, researchers can expedite the discovery and development of safe and effective drugs to address the ever-evolving healthcare challenges of the future.
Author Contributions
ZM conceptualized and wrote; SKN conceptualized, formalized, and edited the paper.
Funding
No funding was taken.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Biswas, R.; Basu, A.; Nandy, A.; Deb, A.; Haque, K.M.G.; Chanda, D. Drug Discovery and Drug Identification using AI. 2020 Indo – Taiwan 2nd International Conference on Computing, Analytics and Networks (Indo-Taiwan ICAN) 2020, 49-51.
- McNair, D. Artificial Intelligence and Machine Learning for Lead-to-Candidate Decision-Making and Beyond. Annual review of pharmacology and toxicology 2022. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.C.; Pal, R.; Chaudhary, M.J.; Nath, R. Artificial intelligence revolutionizing drug development: Exploring opportunities and challenges. Drug Development Research 2023, 84, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Unogwu, O.J.; Ike, M.; Joktan, O.O. Employing Artificial Intelligence Methods in Drug Development: A New Era in Medicine. Mesopotamian Journal of Artificial Intelligence in Healthcare 2023. [Google Scholar] [CrossRef] [PubMed]
- Schwardt, O.; Kolb, H.C.; Ernst, B. Drug discovery today. Current topics in medicinal chemistry 2003, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zieliński, A. AI and the future of pharmaceutical research. arXiv 2021. arXiv, 2107; arXiv:2107.03896.
- Xie, W.; Cheng, X.; Ding, Z.; Deng, R.; Gu, D. Abstract 278: Artificial intelligence accelerate drug discovery. Cancer Chemistry 2021. [Google Scholar] [CrossRef]
- Kenakin, T. Predicting therapeutic value in the lead optimization phase of drug discovery. Nat Rev Drug Discov 2003, 2, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, L.; Muller, C.; Roche, P.; Morelli, X. Chemistry-driven Hit-to-lead Optimization Guided by Structure-based Approaches. Mol Inform 2018, 37, e1800059. [Google Scholar] [CrossRef]
- Broccatelli, F.; C, E.C.A.H.; Wright, M. Strategies to optimize drug half-life in lead candidate identification. Expert Opin Drug Discov 2019, 14, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Braggio, S.; Montanari, D.; Rossi, T.; Ratti, E. Drug efficiency: a new concept to guide lead optimization programs towards the selection of better clinical candidates. Expert Opin Drug Discov 2010, 5, 609–618. [Google Scholar] [CrossRef]
- Das, B.; Baidya, A.T.K.; Mathew, A.T.; Yadav, A.K.; Kumar, R. Structural modification aimed for improving solubility of lead compounds in early phase drug discovery. Bioorg Med Chem 2022, 56, 116614. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Ahamad, S.; Gupta, D.; Mathur, P. Lead optimization, pharmacophore development and scaffold design of protein kinase CK2 inhibitors as potential COVID-19 therapeutics. J Biomol Struct Dyn 2023, 41, 1811–1827. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Khandavilli, S.; Ashokraj, Y.; Jain, A.; Dhanikula, A.; Sood, A.; Thomas, N.S.; Pillai, O.; Sharma, P.; Gandhi, R.; et al. Biopharmaceutic classification system: a scientific framework for pharmacokinetic optimization in drug research. Curr Drug Metab 2004, 5, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Pedreira, J.G.B.; Franco, L.S.; Barreiro, E.J. Chemical Intuition in Drug Design and Discovery. Curr Top Med Chem 2019, 19, 1679–1693. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kogej, T.; Engkvist, O. Cheminformatics in Drug Discovery, an Industrial Perspective. Mol Inform 2018, 37, e1800041. [Google Scholar] [CrossRef] [PubMed]
- AxDrug. AI & Computational Chemistry powered Drug Discovery Platform as a Service - Immunocure CRO. Availabe online: (accessed on 30-01-2024).
- Vora, L.K.; Gholap, A.D.; Jetha, K.; Thakur, R.R.S.; Solanki, H.K.; Chavda, V.P. Artificial Intelligence in Pharmaceutical Technology and Drug Delivery Design. Pharmaceutics 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Deng, Z.; Xu, X.; Feng, Y.; Junliang, S. Application of Artificial Intelligence in Drug-Drug Interactions Prediction: A Review. J Chem Inf Model 2023. [CrossRef]
- Li, G.; Lin, P.; Wang, K.; Gu, C.C.; Kusari, S. Artificial intelligence-guided discovery of anticancer lead compounds from plants and associated microorganisms. Trends Cancer 2022, 8, 65–80. [Google Scholar] [CrossRef] [PubMed]
- McNair, D. Artificial Intelligence and Machine Learning for Lead-to-Candidate Decision-Making and Beyond. Annu Rev Pharmacol Toxicol 2023, 63, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Moshawih, S.; Goh, H.P.; Kifli, N.; Idris, A.C.; Yassin, H.; Kotra, V.; Goh, K.W.; Liew, K.B.; Ming, L.C. Synergy between machine learning and natural products cheminformatics: Application to the lead discovery of anthraquinone derivatives. Chem Biol Drug Des 2022, 100, 185–217. [Google Scholar] [CrossRef] [PubMed]
- Saifi, I.; Bhat, B.A.; Hamdani, S.S.; Bhat, U.Y.; Lobato-Tapia, C.A.; Mir, M.A.; Dar, T.U.H.; Ganie, S.A. Artificial intelligence and cheminformatics tools: a contribution to the drug development and chemical science. J Biomol Struct Dyn 2023. [CrossRef]
- Liu, T.; Zuo, R.; Wang, J.; Huangtao, Z.; Wang, B.; Sun, L.; Wang, S.; Li, B.; Zhu, Z.; Pan, Y. Cardiovascular disease preventive effects of aspirin combined with different statins in the United States general population. Sci Rep 2023, 13, 4585. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, Y.; Niu, Z.; Zong, Y.; Wang, M.; Yao, L.; Lu, Z.; Liao, Q.; Zhao, Y. Atorvastatin (Lipitor) attenuates the effects of aspirin on pancreatic cancerogenesis and the chemotherapeutic efficacy of gemcitabine on pancreatic cancer by promoting M2 polarized tumor associated macrophages. J Exp Clin Cancer Res 2016, 35, 33. [Google Scholar] [CrossRef] [PubMed]
- Mahtta, D.; Ramsey, D.J.; Al Rifai, M.; Nasir, K.; Samad, Z.; Aguilar, D.; Jneid, H.; Ballantyne, C.M.; Petersen, L.A.; Virani, S.S. Evaluation of Aspirin and Statin Therapy Use and Adherence in Patients With Premature Atherosclerotic Cardiovascular Disease. JAMA Netw Open 2020, 3, e2011051. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.M.; Jang, Y.; Lee, S.S.; Jin, M.S.; Jun, C.D.; Kim, M.; Kim, Y.C. Discovery of antiviral SARS-CoV-2 main protease inhibitors by structure-guided hit-to-lead optimization of carmofur. Eur J Med Chem 2023, 260, 115720. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Mekonnen, D.; Mohammed, A.; Shi, R.; Jin, T. Potency, Safety, and Pharmacokinetic Profiles of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front Pharmacol 2020, 11, 630500. [Google Scholar] [CrossRef] [PubMed]
- Najmi, A.; Javed, S.A.; Al Bratty, M.; Alhazmi, H.A. Modern Approaches in the Discovery and Development of Plant-Based Natural Products and Their Analogues as Potential Therapeutic Agents. Molecules 2022, 27. [Google Scholar] [CrossRef] [PubMed]
- Karuppasamy, M.K.M.a.M. Fundamental considerations in drug design. Computer Aided Drug Design (CADD): From Ligand-Based Methods to Structure-Based Approaches, 17–55 2022. [CrossRef]
- de Souza Neto, L.R.; Moreira-Filho, J.T.; Neves, B.J.; Maidana, R.; Guimaraes, A.C.R.; Furnham, N.; Andrade, C.H.; Silva, F.P., Jr. In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front Chem 2020, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Grebner, C.; Matter, H.; Hessler, G. Artificial Intelligence in Compound Design. Methods Mol Biol 2022, 2390, 349–382. [Google Scholar] [CrossRef]
- Langevin, M.; Minoux, H.; Levesque, M.; Bianciotto, M. Scaffold-Constrained Molecular Generation. J Chem Inf Model 2020, 60, 5637–5646. [Google Scholar] [CrossRef]
- Cavasotto, C.N. Binding Free Energy Calculation Using Quantum Mechanics Aimed for Drug Lead Optimization. Methods Mol Biol 2020, 2114, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Q.; Chen, H.Y.; Dai, W.J.; Lv, Q.J.; Chen, C.Y. Artificial Intelligence Approach to Find Lead Compounds for Treating Tumors. J Phys Chem Lett 2019, 10, 4382–4400. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.P.; Kumar, N. Artificial Intelligence for Autonomous Molecular Design: A Perspective. Molecules 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Loos, N.H.C.; Sparidans, R.W.; Heydari, P.; Bui, V.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. The ABCB1 and ABCG2 efflux transporters limit brain disposition of the SYK inhibitors entospletinib and lanraplenib. Toxicol Appl Pharmacol 2024, 485, 116911. [Google Scholar] [CrossRef] [PubMed]
- Blomgren, P.; Chandrasekhar, J.; Di Paolo, J.A.; Fung, W.; Geng, G.; Ip, C.; Jones, R.; Kropf, J.E.; Lansdon, E.B.; Lee, S.; et al. Discovery of Lanraplenib (GS-9876): A Once-Daily Spleen Tyrosine Kinase Inhibitor for Autoimmune Diseases. ACS Med Chem Lett 2020, 11, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Bleicher, L.S., Van Daelen, T., Honeycutt, J. D., Hassan, M., Chandrasekhar, J., Shirley, W., Tsui, V., & Schmitz, U. . Enhanced utility of AI/ML methods during lead optimization by inclusion of 3D ligand information. Frontiers in Drug Discovery, 2, 1074797. [CrossRef]
- Hoffman, S.C. , Chenthamarakshan, V., Wadhawan, K. et al. Optimizing molecules using efficient queries from property evaluations. Nat Mach Intell 4, 21–31. [CrossRef]
- Zhu, W.; Liu, X.; Li, Q.; Gao, F.; Liu, T.; Chen, X.; Zhang, M.; Aliper, A.; Ren, F.; Ding, X.; et al. Discovery of novel and selective SIK2 inhibitors by the application of AlphaFold structures and generative models. Bioorg Med Chem 2023, 91, 117414. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, Y.A.; Polykovskiy, D.; Bezrukov, D.; Zagribelnyy, B.; Aladinskiy, V.; Kamya, P.; Aliper, A.; Ren, F.; Zhavoronkov, A. Chemistry42: An AI-Driven Platform for Molecular Design and Optimization. J Chem Inf Model 2023, 63, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Ding, X.; Zheng, M.; Korzinkin, M.; Cai, X.; Zhu, W.; Mantsyzov, A.; Aliper, A.; Aladinskiy, V.; Cao, Z.; et al. AlphaFold accelerates artificial intelligence powered drug discovery: efficient discovery of a novel CDK20 small molecule inhibitor. Chem Sci 2023, 14, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Turon, G.; Hlozek, J.; Woodland, J.G.; Kumar, A.; Chibale, K.; Duran-Frigola, M. First fully-automated AI/ML virtual screening cascade implemented at a drug discovery centre in Africa. Nat Commun 2023, 14, 5736. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, H.; Dingfelder, F.; Condado Morales, I.; Patel, B.; Heding, K.E.; Bjelke, J.R.; Egebjerg, T.; Butte, A.; Sokolov, M.; Lorenzen, N.; et al. Design of Biopharmaceutical Formulations Accelerated by Machine Learning. Mol Pharm 2021, 18, 3843–3853. [Google Scholar] [CrossRef] [PubMed]
- Mousaei, M.; Kudaibergenova, M.; MacKerell, A.D., Jr.; Noskov, S. Assessing hERG1 Blockade from Bayesian Machine-Learning-Optimized Site Identification by Ligand Competitive Saturation Simulations. J Chem Inf Model 2020, 60, 6489–6501. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.H.; Long-Boyle, J.; Keizer, R.J. Maximum a posteriori Bayesian methods out-perform non-compartmental analysis for busulfan precision dosing. J Pharmacokinet Pharmacodyn 2024. [CrossRef]
- Lee, J.W.; Maria-Solano, M.A.; Vu, T.N.L.; Yoon, S.; Choi, S. Big data and artificial intelligence (AI) methodologies for computer-aided drug design (CADD). Biochem Soc Trans 2022, 50, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Mishra, A.P.; Panda, B.; Rodriguez, D.C.S.; Gaurav, I.; Majhi, B. Application of Artificial Intelligence in Pharmaceutical and Biomedical Studies. Curr Pharm Des 2020, 26, 3569–3578. [Google Scholar] [CrossRef] [PubMed]
- Ryeznik, Y.; Sverdlov, O.; Svensson, E.M.; Montepiedra, G.; Hooker, A.C.; Wong, W.K. Pharmacometrics meets statistics-A synergy for modern drug development. CPT Pharmacometrics Syst Pharmacol 2021, 10, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.M.; MaWhinney, S.; Carlson, N.E.; Kreidler, S. A Bayesian natural cubic B-spline varying coefficient method for non-ignorable dropout. BMC Med Res Methodol 2020, 20, 250. [Google Scholar] [CrossRef] [PubMed]
- Dr. Christoph Grebner, D.H.M., Dr. Daniel Kofink, Dr. Jan Wenzel, Dr. Friedemann Schmidt, Dr. Gerhard Hessler. Application of Deep Neural Network Models in Drug Discovery Programs. Wiley 2021, ChemMedChem, Volume 16, Issue 24. [CrossRef]
- Jaeger, S.; Fulle, S.; Turk, S. Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition. J Chem Inf Model 2018, 58, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhou, D.; Gao, L.; Zha, Y. Prediction of drug response in multilayer networks based on fusion of multiomics data. Methods 2021, 192, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Amendola, G.; Ettari, R.; Previti, S.; Di Chio, C.; Messere, A.; Di Maro, S.; Hammerschmidt, S.J.; Zimmer, C.; Zimmermann, R.A.; Schirmeister, T.; et al. Lead Discovery of SARS-CoV-2 Main Protease Inhibitors through Covalent Docking-Based Virtual Screening. J Chem Inf Model 2021, 61, 2062–2073. [Google Scholar] [CrossRef] [PubMed]
- Gusev, F.; Gutkin, E.; Kurnikova, M.G.; Isayev, O. Active Learning Guided Drug Design Lead Optimization Based on Relative Binding Free Energy Modeling. J Chem Inf Model 2023, 63, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Niclas Ståhl, G.F. , Alexander Karlsson, Gunnar Mathiason, and Jonas Boström. Deep Reinforcement Learning for Multiparameter Optimization in de novo Drug Design. American Chemical Society Publications 2019, 7, 3166–3176. [Google Scholar] [CrossRef]
- Quentin Perron, O.M., Hamza Tajmouati, Adam Skiredj, Anne Rojas, Arnaud Gohier, Pierre Ducrot, Marie-Pierre Bourguignon, Patricia Sansilvestri-Morel, Nicolas Do Huu, Françoise Gellibert, Yann Gaston-Mathé. Deep generative models for ligand-based de novo design applied to multi-parametric optimization. Wiley 2022. [CrossRef]
- Zhong, F.; Xing, J.; Li, X.; Liu, X.; Fu, Z.; Xiong, Z.; Lu, D.; Wu, X.; Zhao, J.; Tan, X.; et al. Artificial intelligence in drug design. Sci China Life Sci 2018, 61, 1191–1204. [Google Scholar] [CrossRef]
- Nayarisseri, A.; Khandelwal, R.; Tanwar, P.; Madhavi, M.; Sharma, D.; Thakur, G.; Speck-Planche, A.; Singh, S.K. Artificial Intelligence, Big Data and Machine Learning Approaches in Precision Medicine & Drug Discovery. Curr Drug Targets 2021, 22, 631–655. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Classic human-led drug design (bottom) is an iterative process of proposing new compounds and testing them in vitro. As this process requires synthesis in the lab, it is very costly and time-consuming. By using computational modeling (top), molecule design can be rapidly performed in silico, with only the most promising molecules promoted to be made in the lab and then eventually tested in vivo. AI-driven drug discovery process, from target identification to selecting drug candidates [https://www.microsoft.com/en-us/research/blog/moler-creating-a-path-to-more-efficient-drug-design/].
Figure 1.
Classic human-led drug design (bottom) is an iterative process of proposing new compounds and testing them in vitro. As this process requires synthesis in the lab, it is very costly and time-consuming. By using computational modeling (top), molecule design can be rapidly performed in silico, with only the most promising molecules promoted to be made in the lab and then eventually tested in vivo. AI-driven drug discovery process, from target identification to selecting drug candidates [https://www.microsoft.com/en-us/research/blog/moler-creating-a-path-to-more-efficient-drug-design/].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



