Submitted:
25 March 2024
Posted:
26 March 2024
You are already at the latest version
Abstract
Novel therapies for treatment of familial dilated cardiomyopathy (DCM) are lacking. Triggers for the progression of the disorder commonly occur due to specific gene variants that affect the production of sarcomeric/cytoskeletal proteins. Generally these variants cause a decrease in tension by the myofilaments, resulting in signaling abnormalities within the micro-environment which over time result in structural and functional maladaptations leading to heart failure (HF). Current concepts support the hypothesis that the mutant sarcomere proteins induce a causal depression in the tension time integral (TTI) of linear preparations of cardiac muscle. However, molecular mechanisms underlying tension generation particularly in relation to mutant proteins and their impact on sarcomere molecular signaling, are currently the subject of controversy. Thus, there is a need for clarification as to how mutant proteins affect sarcomere molecular signaling in the etiology and progression of DCM. A main topic in this controversy is the control of the number of tension generating myosin heads reacting with the thin filament. One line of investigation proposes that this number is determined by changes in the ratio of myosin heads in a sequestered super-relaxed state (SRX) or in a disordered relaxed state (DRX) poised for force generation upon Ca-activation of the thin filament. Contrasting evidence from nanometer–micrometer-scale x-ray diffraction in intact trabeculae indicates that the SRX/DRX states may have a lesser role. Instead, the proposal is that myosin heads are in a basal OFF state in relaxation then transfer to an ON state through a mechano-sensing mechanism induced during early thin filament activation and increasing thick filament strain. Recent evidence about the modulation of these mechanisms by protein phosphorylation has also introduced a need for reconsidering control of tension. We discuss these mechanisms that lead to different ideas related to how tension is controlled and disturbed by mutant sarcomere proteins linked to DCM including cardiac myosin, myosin-binding protein C, titin filaments, and thin filaments. Resolving the various mechanisms and incorporating them into a unified concept is crucial for gaining a comprehensive understanding of DCM. This deeper understanding is not only important for diagnosis and treatment strategies with small molecules, but also for understanding the reciprocal signaling processes that occur between cardiac myocytes and their micro-environment. By unraveling these complexities, we can pave the way for improved therapeutic interventions for managing DCM.
Keywords:
Sarcomere Disease Genes
; TTI
; Myosin Control Mechanisms
; Sarcomere Activators
; Precision Medicine
1. Introduction
Genetic dilated cardiomyopathy (DCM), which accounts for ~40% of the cases of dilated cardiomyopathy, is a complex multi-factorial cardiac disorder lacking a cure and in need of patient specific therapeutic strategies [1,2,3,4]. Progression of the disorder to heart failure (HF) includes an exacerbating wall thinning, fibrosis, arrythmias, dyspnea, exercise intolerance, and in some cases sudden death. A challenge to treatment strategies is the variability of the disease-causing genes, the variability of penetrance, and the variability of the clinical course. Disease genes include variants expressing proteins involved in sarcomere function, in mechano-transduction, in cell and nuclear membrane function, and mitochondrial metabolic protein energetic processes [1]. Therapies with the objective of maintaining cardiac function in DCM are generally not patient specific and treat symptoms but not the cause. Treatments include use of agents such as angiotensin-neprilysin inhibition, Na-glucose co-transport inhibitors, and invasive approaches such as resynchronization therapies [5,6,7]. With a need for a bridge to cardiac transplant, energy wasting inotropic agents elevating intracellular Ca2+ are also employed with the danger of arrythmias and increased mortality [8].
Our focus here is on the disease genes expressing mutant sarcomere proteins that modify tension generating capability of the sarcomeres triggering disease progression [9]. One rationale for discussion of this form of DCM is that in the context of precision medicine there is a need for deep understanding of the pathological processes, diagnostic approaches, and therapies. A further rationale is that in the case of mutant sarcomere proteins, it appears success is within reach in achieving this understanding and making advances in therapies with small molecules. Success in developing these small molecules provides evidence that this approach is feasible for drug development in the case of omecamtiv mecarbil (OM)and danicamtiv in DCM [10,11,12] and in the clinical use of mavacampten (Camzyos®) and aficamten in patients with obstructive hypertrophic cardiomyopathy (HCM) [13,14]. In contrast, with many DCM disease genes such TTN and LMNA, treatment relies heavily on gene replacement therapy. As detailed in a recent assessment of approaches in gene therapy, the authors concluded that whereas promising techniques are emerging they are in early development and require ethical and safe use [15]. Thus, although they are patient specific, many challenges remain in the use of gene therapy in genetic cardiomyopathies [2,15].
2. Recent Diverse Theories of Control Mechanisms of Tension Development in Cardiac Sarcomeres
An apparently important advance in the understanding of the pathology and treatment strategies in DCM gene variants expressing sarcomere proteins is the identification of a correlation of DCM with a fall in the tension-time integral (TTI) in linear strips of heart muscle [16]. The TTI is the time integral of tension developed in a twitch for example in isolated trabeculae at 30oC and a constant 2.2 µm sarcomere length. The idea that TTI is a critical determinant of adverse signaling in the instigation and the progression of DCM gives rise to the following question: What is the correlation of TTI with muscle function in the beating heart? We think that wall stress is an appropriate variable correlating tension in a linear strip of muscle or cells with the mechanics of the beating heart. As illustrated in the early studies of Sandler and Dodge [17] tension developed by regions of the beating heart is a complex function of chamber pressure and chamber dimensions (size and shape) related to the Law of LaPlace. Early studies by Sandler and Dodge provided a summary of these complexities and a cogent analysis of the determinants of wall stress related explicitly to wall tension [17]. Davis et al. [16] employed computational approaches in the context of various DCM models linked to a variety of gene variants and concluded that the TTI of linear muscle and cardiac myocytes derived from human inducible pluripotent stem cells (hiPSC-CMs) can predict heart thickening in HCM and thinning in DCM. A follow up study by Powers et al. [9] supported this concept in investigations of TTIs in preparations from a mouse model harboring DCM-linked tropomyosin mutant (Tm-D230N). However, clouding the understanding of the effects of TTI in triggering DCM is a lack of a unified theory of tension generation and control in the heart. A related and important issue is that the most common causes of DCM are truncation mutants of titin in which studies with skinned muscle preparations report an increase myofilament response to Ca2+ [18]. Titin truncation mutants have a a complex pathology involving post-translational mechanisms, co-morbidities, and activation of other genes [19] and possible effects of both haploinsufficiency and poison peptides [20]. Recent publications discussed below regarding the generation and control of tension in heart muscle indicate the need to reevaluate recent ideas presented regarding the relation between TTI, heart growth, and adverse remodeling.
Despite the many years of studies investigating mechanisms of tension generation with each the beat of the heart, there is a confluence of new information from integrated molecular/biochemical, cellular, biophysical, and structural studies that demand altered thinking of not only how a DCM point mutation in a sarcomere protein alters the TTI in DCM, but also as to how this mechanical signal is transduced to adverse remodeling leading to symptomatic HF. These more recent studies have influenced concepts of the role of processes in thin, thick, and titin filaments in control tension development with each heartbeat. Results of one line of investigations have altered perceptions of force generation by stressing a novel role for thick filament structural state and signaling than previously appreciated [21,22]. Employing largely biochemical approaches and in vitro methods, these studies with healthy rabbit heart tissue identified and characterized the existence of a population of cross-bridges in an interacting head motif (IHM) promoting a super-relaxed state (SRX) with low ATPase activity and sequestered from participating in force generation [21]. These SRX myosin heads are believed to exist together with a population of myosins in a disordered relaxed state (DRX) available for interaction with thin filament sites upon activation by Ca-binding to cardiac troponin C (cTnC). Variations in the thick filament DRX/SRX populations have been theorized to be responsible for adjustments in inotropic state in physiological conditions such as adrenergic stimulation, and for altered tension development in HF, HCM, and DCM [23,24,25,26,27]. Shifts in SRX/DRX population have also been characterized as being responsible for inotropic effects of myosin activators and inhibitors [23,25,26].
In contrast, earlier and more recent studies employing high resolution nano-meter x-ray analysis have emphasized a transition of myosins from an OFF to an ON state by mechano-sensing with a minimal role for the IHM theory in living skeletal and cardiac muscle preparations [28]. Evidence from the x-ray data supported the conclusion that in relaxation, myosins are in an OFF state with heads in helical order, folded close to the thick filament surface [29,30]. A few myosins were proposed to be scanning for thin filament sites. Mechanical strain on the thick filament induced by the initial binding of force generating cross-bridges with the onset of thin filament activation in the heartbeat was theorized to activate transition of the OFF myosins to a force generating ON state. Additional evidence relating to mechano-sensing of the thick filament has been reported by Caremani et al. [31], who demonstrated that inotropic interventions phosphorylating cardiac troponin I (cTnI) and cardiac myosin binding protein-C (cMyBP-C) had no effect on the state of myosin in relaxed muscle. These findings do not support a role for cMyBP-C phosphorylation previously suggested to modulate and promote the transition from SRX to DRX states [24]. Moreover, Caremani et al. [31] stress that the lack of effect of cMyBP-C phosphorylation on thick filament state in diastole indicates that the regulatory mechanism is down-stream of thin filament activation and fits with a model in which variations in thick filament strain are significant determinants of force generation. Thus, the gain in the interaction energies induced by strain-dependent activation of the thick filament is thought to be a modulated variable, which can be affected by sarcomere length, post-translational modifications (PTM) in thin and thick filaments, as well as shifts in sarcomere protein isoforms associated with altered physiological and pathological remodeling. As discussed below, recent x-ray diffraction investigations testing the effect of OM in porcine trabeculae support the prominent significance of the OFF/ON versus the SRX/DRX state [32]. These studies demonstrated a transition of the resting cross-bridges from OFF to ON states induced by OM, but there was no evidence that this transition involved a modification in SRX/DRX states. Moreover, the authors emphasized that the OFF state and SRX state are not equivalent. These findings indicate the need for more thorough consideration of the mechanisms of activation of thick filament function considering the possibility of different conclusions depending on the methods of approach. Interpretation of the detailed structural information derived from cryo-electron tomography of relaxed native cardiac sarcomeres provides a cogent example of the need for caution in the interpretation of data related to thick filament structure/function relations [33]. Whereas the resolution and imaging are impressive and advance understanding of the three-dimensional structure of myosin, titin, and cMyBP-C, the studies were carried out in isolated myofilaments in the presence of 50 µM mavacampten. The results showed no interaction between titin and cMyBP-C that others have reported [34]. More recently these studies have been repeated in thick filament preparations without mavacamten [35]. These studies without mavacamten identified three population of motors in relaxed thick filaments indicated as equivalent to SRX, DRX, and active myosins [35]. In both these studies discussion of the data included the IHM concept but did not include the ON/OFF concept of thick filament activation derived from x-ray data in preparations with myofilaments functioning in an intact cellular environment. Other examples are recent publicatins on the mechanism of the Anrep effect and the Frank-Starling relation that reviewed the literature concluding that there is a major role of the SRX/DRX transition responding to load with no reference to the ON/OFF mechanisms [36,37].
3. Cross-Bridge ON/OFF States and Control of Tension/Wall Stress
Implications of the ON/OFF mechanism in various states of cardiac function indicate the need for a reconsideration of previous reviews of how the integrated interactions among sarcomere proteins control gradations in cardiac wall stress/tension and contraction/relaxation dynamics. There is general agreement that control of tension in striated muscle in both relaxation and the activation of tension with sliding of myofilaments require mechanisms involving all three major myofilaments, thin, thick and titin. However, there is not a clear consensus on the fundamental mechanisms. As summarized above, we focus here on compelling evidence provided by the x-ray data highlighting the dominance of the ON/OFF mechanisms in contrast to shifts in the SRX/DRX ratio [31,32]. Integrated interactions among sarcomere proteins controlling function are illustrated in Figure 1 and the reaction flow in Figure 2, based on high resolution structure [38,39] and recent reviews [40,41]. Figure 1 illustrates a snapshot of the sarcomeres of the C-zone where cMyBP-C is localized in an end-diastolic state (relaxing levels of cellular Ca2+) at a basal heart rate with low levels of phosphorylation of key control elements in cTnI, cMyBP-C, and titin. The diagram in Figure 2 is a companion to Figure 1, which depicts these protein-protein interactions as reactions promoting tension and inducing relaxation. In Figure 1, myosin heads are shown uniformly folded close to the thick filament proper. With regulatory sites of cTnC free of bound Ca2+, functional regulatory units localized in the two strands of the thin filament and made up of a 7:1:1 ratio of actins: tropomyosin (Tm) and troponin (Tn) complex establish a thin filament inhibited or so-called B-state with Tm in a configuration sterically blocking the reaction of myosin with actin [42]. We note that the regulatory units in each strand are not in register and not equivalent [39,43]. As shown, the cTn complex extends along the entire functional unit with each end of the complex having functional roles ensuring a thin filament off state. The C-terminal domain of cTnI contains a highly basic inhibitory peptide (Ip) and a switch peptide (SwP) extending from the cTn core domain and interacting with actin/Tm holding Tm in the B-state. Near N-terminal regions of cTnI extending along the cardiac troponin T (cTnT) C-terminal region (I-T arm) position cTnT in a configuration also promoting binding and immobilization of Tm. There are also interactions of regions of cTnT N-terminus with the head to tail overlap of contiguous Tms forming a key element in signaling of the diastolic state and reducing the possibility of cooperative activation along the sarcomere strand [44,45]. In diastole with low levels of intra-cellular Ca2+, there is evidence of modulation of this inhibited state by the interactions of N-terminal domains of cMyBP-C with actin-Tm that move Tm away from the blocking configuration [43,46,47]. This interaction is weakened in high Ca2+ and abolished with phosphorylation of cMyBP-C [48]. It has also been proposed that there are direct interactions between cMyBP-C and cTnI that coordinate effects of phosphorylation of these proteins. [49]. However, it is not clear how these interactions may modulate inhibition. A role for titin must be considered in the establishment of the diastolic state. At end diastole, sarcomere length is relatively long, the stretch of titin elastic domains induces resting tension and diastolic pressure [50]. Interactions of titin with C-terminal domains of cMyBP-C myosin rods may modulate thick filament packing. Specific interactions of 3 C-terminal domains with specific super-repeats on titin establish localization of cMyBPC in its striped configuration in the C-zone of the sarcomere [34]. There are also interactions of cMyBP-C domains with the regulatory light chain (RLC) of myosin in a phosphorylation dependent manner [51]. The stability of the relaxed OFF state of the thick filament may involve these interactions of the RLC, titin and cMyBP-C that modulate head-head interactions as well as tail-tail interactions that deploy myosins in the relaxed state folded close to the thick filament [52].
The transfer from the diastolic to the systolic state involves complex allosteric, steric, and cooperative mechano-sensing mechanisms involving all the major sarcomere proteins in various roles. This ensures a rich array of mechanisms for beat-to-beat tuning of dynamics to HR and modulation of the coupling of cardiac function to the metabolic needs of the cells of the body. Membrane excitation and elevations in intracellular Ca2+ promote allosteric interactions in both thin and thick filaments signaling the potential interactions of cross-bridges with actin triggered by a Ca-dependent release of the steric block imposed by Tm. With Ca2+ localized in its regulatory hydrophobic pocket, cTnC enters a rotation that aids binding of the Sw peptide and translocation of the Ip away from sites of inhibition. Recent cryo-electron microscopy (cryo-EM) evidence also indicates a structural interaction between the cTnC N-terminus and the cTnI-C-terminus [43]. In turn, the mobile cTnI C-terminal domain moves away from a position no longer tethering Tm in a blocking position. The relief of inhibition includes a reconfiguration of the complex that alters interactions of C-terminal regions of cTnT with cTnI, cTnC and Tm that alters interactions of N-terminal regions of cTnT at the overlap of N- and C-terminal Tm regions. These interactions induce a landscape of protein-protein interactions promoting cooperative spread of activation along the strand of sarcomeres. However, despite the significant release from their inhibited state thin filament activation is not complete as full activation requires binding of force-generating cross-bridges [43,53]. A fundamental tenant of the ON/OFF mechanism of the promotion of tension development is that the initial activation of some cross-bridge interactions produce a mechanical strain inducing a transition of other cross-bridges into the force generating cycle. The efficacy of this strain dependent activation is stated to be dependent on the gain of the protein-protein interactions [31]. This “gain” with variations in the energies of interaction among the thick filament components provides a regulatory device critical to control by sarcomere load and length, PTMs, and biochemical environment. Imagining the arrows in Figure 2 to be of variable intensity, the graphic provides a graphic example of the breadth of possible regulatory mechanisms available to maintain a homeostatic range of cardiac function under varying hemodynamic loads. Evidence that the peak tension in a twitch of intact trabeculae involves only ~10% of the available myosin motors in the sarcomere C-zone indicates the extent of the reserve in contractile state [54].
4. Implications of the On/Off Theory in Modulation of Tension and Dynamics by Dominant Myocardial Regulatory Mechanisms
Unraveling the variations in the gain in the detailed mechanisms of the generation and control of tension/wall stress in the heart has wide ranging impact on the understanding of physiological and pathological processes in the heart as well as diagnosis, patient stratification, and application of personalized medicine. Extensive consideration of this impact is beyond the scope of this review/commentary. We choose to focus on the impact of the ON/OFF theory of tension generation in familial DCM with emphasis on modulation of function by length and load on the sarcomeres, and by protein phosphorylation of major sarcomere proteins. Our rationale is also that these mechanisms underpin the search for sarcomere-related therapies in the clinical course of DCM. Figure 1 and Figure 2 illustrate the unfolding of the activation process in the ON/OFF mechanism in the context of a basal physiological environmental and mechanical state with low influence of PTMs. Common sarcomere-related control mechanisms that modulate this basal state include changes in sarcomere length (Frank-Starling mechanism) and neuro-humoral adrenergic stimulation. Thus, it is important to discuss how consideration of signaling in the ON/OFF state transitions alters understanding of these control mechanisms, whether and how they are modified in DCM, and altered by sarcomere activators.
4.1. Regulation by Sarcomere Length and Load
Understanding of mechanisms of modifications in the sarcomere response to Ca2+ as a key element in the Frank-Starling relation is not complete. Yet there is general agreement that the stretch of titin instigates the increase in developed pressure with increased ventricular filling. Possible mechanisms of the effect of titin stretch in diastole include modulation of thin filament and thick filament proteins including cMyBP-C. As indicated in Figure 1 C-zone regions of titin interact with myosin heavy and light chains and cMyBP-C. There is also interaction of the Z-repeats of titin with Z-disk proteins telethonin, alpha-actinin, and actin [19]. Evidence indicates that these interaction sense the compliance of titin and induce signaling resulting in an alteration of the number of force generating cross-bridges. Moreover, DCM associated with truncating mutations in titin have been theorized to be significantly related to altered length-dependent activation (LDA) and modified by titin phosphorylation [18]. Early results of studies of effects of titin strain on LDA concluded an important role for a stretch induced alteration in inter-filament spacing [55]. The idea was that increased compliance of titin blunted an effect to move myosin heads nearer the thin filament. Phosphorylation of cMyBP-C was also proposed to modify LDA by a mechanism shown by low angle x-ray studies of skinned trabeculae to move myosin heads closer to thin filaments [56,57]. However, more recent investigations of intact muscle preparations have not supported this mechanism. From results of the x-ray investigation on the effects of changes in sarcomere length, Caremani et al. [31] and Brunello et al. [54] demonstrated no effects of a sarcomere length change in the diastolic OFF state just before contraction or isoproterenol treatment of myosins in relaxed preparations despite a robust effect of to both interventions on tension development. They note that with preparations treated with isoproterenol, there was an effect on meridional reflections attributed to cMyBP-C and troponin. X-ray analysis reported by Ait-Mou et al. [58] also did not demonstrate changes in inter-filament spacing or a movement of myosin heads toward the thin filament with stretch of electrically stimulated mouse heart muscles. However, other reflections indicated significant structural rearrangement in myosin heads not found by others [31,54]. Other lines of evidence point to a mechanism in which titin strain signals the thin filament complex to promote an increase in tension with an increase in sarcomere length. Evidence for this signaling comes from the data demonstrating that the troponin complex senses length not only by structural rearrangements but also by enhanced cTnC Ca-binding. Employing RBM20 deletion mice that express the N2BA highly compliant titin isoform, Li et al. [59] reported that stretch induced a diminished FRET signal at the cTnI-cTnC interface compared to controls with relatively higher passive tension. The signal was also induced by strong cross-bridge binding. Taken together the data summarized above indicate that a change in sarcomere length signals the thin filament to increase the population of force generating cross-bridges and potentially increase the gain in mechano-sensing affecting thick filament strain. This mechanism supports the ON/OFF theory in that length-dependent-activation occurs down stream of thin filament signaling and depends on the compliance of titin. In support of a role for thin filament and cMyBP-C in signaling in the Frank-Starling relation are data reporting altered LDA independent of a change in titin compliance in sarcomeres controlled by a deletion mutant of cTnT (cTnT-ΔK210), by the mutant cTnT-R171W, and by a mutation in cTnC (cTnC-G159D) [60,61,62]. All these mutations are linked to DCM. However, the cTnT mutations decrease myofilament response to Ca2+, whereas the mutation in cTnC induces an increase in Ca-sensitivity [62].
4.2. Regulation by Sarcomere Protein Phosphorylation
Among the many sites and types of PTMs in sarcomere proteins, phosphorylations of cTnI and cMyBP-C at their unique N-terminal domains have taken on prominent roles in tuning tension and dynamics to common physiological demands on the heart. Both mechanisms have been well-studied and discussed for some time but have been challenged by recent data [31,49]. In the case of cMyBP-C, we mentioned above that a predicted shift in SRX/DRX ratio with phosphorylation was not detected in the intact electrically stimulated trabeculae studied by nano-meter x-ray diffraction [31]. Previous studies concluded that in relaxed sarcomeres following adrenergic stimulation cMyBP-C moves from it binding to the thin filament interacts with the S-2 region of myosin engaging in a process shifting SRX to DRX myosins [63]. In the ON/OFF mechanism there is a shift in myosin heads with adrenergic stimulation, but not in relaxation. Thus, thin filament activation with an increase in thick filament strain precedes a downstream shift in cMyBP-C binding to myosin. Although Caremani et al, [31] noted no modifications of the OFF state with adrenergic stimulation, there were altered reflections of both cMyBP-C and cTnI in these experiments.
Apart from these findings in the x-ray analysis, the detailed mechanisms, and effects of cTnI phosphorylation at its N-terminus Ser residues have also been challenged in the literature [49]. For nearly 50 years, although there have been reports of differences in the altered molecular signaling with cTnI phosphorylation at S23, S24, there has been general agreement that the phosphorylation is a negative regulator of myofilament response to Ca2+ and an important element in the lusitropic response to β-adrenergic stimulation [64,65]. Results of studies in the natural cellular environment in preparations with and without expression of phospholamban demonstrated that β-adrenergic stimulation enhanced relaxation in mechanisms associated with desensitization myofilament response to Ca2+ [66,67]. Current theories are driven by the dominant idea that cTnI phosphorylation induces lusitropy in cardiac dynamics important in tuning cardiac dynamics to HR and offsetting effects of HCM and DCM [65,68,69]. An example of proposed molecular mechanism was reported by Pavadai et al. [70] in a simulation of protein-protein interactions employing molecular dynamics and molecular docking. They concluded that at sub-maximum myocyte Ca2+ concentrations unphosphorylated cTnI is highly mobile. With pseudo-phosphorylation at Ser 23 and Ser 24, they predict the unique N-terminal domain interacts, with both the N-lobe of cTnC interfering with Sw peptide binding and with Tm resulting in promotion of the off state of the thin filament. In stark contrast to these predictions, studies by Sevrieva et al. [49] employing isolated myofibrils and skinned trabeculae preparations with authentically phosphorylated cTnI and cMyBP-C reported that cTnI phosphorylation alone increased Ca-sensitivity of trabeculae force generation at short and long sarcomere lengths. When both cTnI and cMyBP-C were phosphorylated, compared to controls there was an increase in force only at long sarcomere lengths. Moreover, other in vitro studies reported a direct phosphorylation dependent interaction between the N-terminal domains of cMyBP-C and cTnI. In their paper, Sevrieva et al. [49] indicate that pseudo-phosphorylation does not faithfully reproduce the effect of authentic phosphorylation without a direct test of this assertion in cTnI. In addition, there was no mention of the criticism of skinned fiber preparations by Caremani et al. [31] in relation to mechanisms of effects cMyBP-C phosphorylation. These criticisms are relevant to a series of publications emphasizing the importance uncoupling of protein phosphorylation and sarcomere mechanics as modifier of DCM progression [69]. There are several issues regarding the conclusion reported by Sevrieva et al. [49], which indicate a need for a reevaluation of their conclusions. A major issue is the lack of evidence for their findings in the integrated in situ regulation of the heartbeat. In Figure S2 the authors reported a relatively low efficiency of exchange (35-50%) of bis-phosphorylated cTnI, and the native cTnI appears to be highly phosphorylated (Figure 1A in the paper). This suggests that they might be assessing the effect of only a small additional increase in cTnI phosphorylation. Additionally, the isolated trabeculae did not demonstrate RLC phosphorylation (Figure S6 in their manuscript), which is not explained. This lack of RLC phosphorylation may impact the results and conclusions from the fibers exchanged with bis-phosphorylated TnI. Earlier work reported by Sevrieva et al. [71] concluded a role of MLCK in complex mechanisms affecting the relative effects of cTnI and RLC phosphorylation. Moreover, fully dephosphorylated myofilament preparations were treated with PKA but there was no thorough analysis of sites of phosphorylation that are apparent in high molecular regions of the gels. Finally, some degradation of cTnI was noted in Figure 2C of their manuscript and this may be a cause for concern.
5. Research Challenges in the Quest to Develop Sarcomere Targeted Drugs Treating DCM
Landim-Viera and Knollman [72] made the following statement in a recent editorial: “The search for small molecules that increase contractile function by directly targeting the muscle of the failing heart can be considered the holy grail in drug development.” Research challenges in the search for agents effective in DCM therapy include the lack of a unified theory of the control of tension development, evaluation of the role of TTI as a predictor of DCM, and differing views of effects of myofilament protein phosphorylation. Challenges also include some data that mechanistic conclusions from studies with detergent extracted fibers and fiber bundles may not obtain in the intact myocardium. There are criticisms of commonly employed approaches including pseudo-phosphorylation and substitution of Ala residues for Ser/Thr phosphorylation [49]. We discuss these research challenges related to understanding of disease mechanisms and the quest for development of sarcomere-targeted treatments in DCM. We have limited the discussion to agents targeting thin filaments and thick filaments including cMyBP-C.
5.1. Targeting Thin Filaments in DCM Therapy
Although many recent papers continue to refer to “modulation of sarcomere contractility” with small molecules acting at sarcomere proteins as a new or recent therapeutic opportunity [73,74,75,76], research in this area began more than 40 years ago. Solaro and Ruegg introduced a research direction in development of agents with the idea that small molecules can enter the intracellular space and bind directly to myofilament regulatory proteins inducing an increase in sarcomere response to Ca2+ [77]. Support of this hypothesis came with experiments reported by Solaro et al. reported enantiomeric separation of the effects of the thiadiazinone (+/-) EMD57033 with only the (+) enantiomer active in increasing myocyte contractility, myofilament response to Ca2+, cTnC Ca-binding, and thin filament sliding in the motility assay [78]. These data provided proof of principle for targeting sarcomere proteins in HF [78]. Solution nuclear magnetic resonance (NMR) studies of stereospecific interactions correlated the enantiomeric separation by demonstrating functionally significant key interactions of only the EMD (+) chiral group deep in a hydrophobic of cTnC [79]. These agents were originally termed “Ca-sensitizers” and more recently as “sarcomere activators.” [80,81]. The proposed advantages of this approach included an increase in tension with little or no change in the Ca-transient. This action was described as an energy sparing effect with an increase in tension with no change in oxygen consumption related to the reduction in the load on the sarcotubular Ca-pump. Moreover, unlike conventional inotropes that increase intracellular Ca2+, the sensitizers were expected to be non-arrhythmogenic. However, this idea has been challenged in studies reporting elevated myofilament Ca-sensitivity may indeed be arrhythmogenic [82],. Examples of agents targeted to cTnC that progressed successfully through clinical trials are levosimendan (Simdax) developed by Orion Pharma/Abbott Labs [83,84] and pimobendan (Acardi; Vetmedin) developed by Boehringer Ingelheim/Nippon [85]. However, these Ca-sensitizers screened for cTnC binding have off target effects in their pharmacological profile, especially inhibition of phospho- diesterase III (PDE III) [85,86,87,88]. This pleiotropic property led to designation of pimobendan and levosimendan as inodilators [89,90]. However, with PDE III inhibition there are elevations in HR, which has the concern of being arrhythmogenic, especially in compromised cardiac function [8]. In view of this concern, continued efforts to develop sarcomere activators ruled out agents with PDE III inhibition. Reports in the literature have generated mixed results on the efficacy of pimobendan and levosimendan in DCM. The inodilator pimobendan was reported to be of benefit in the cTnT-ΔK210 DCM mouse model despite the lack of evidence that there was an increase in myofilament Ca-response [91,92]. In a recent study employing this model [91], there was a prevention of cardiac remodeling in compensated HF with a significant increase in life span regardless of the stage of treatment. However, at end-stage there was an increase in sudden death. In contrast, pimobendan (marketed as Vetmedin®) is a common treatment meeting with success in dogs with DCM of various etiologies [93]. Whatever the case, with the lack of a consensus on the effects of cAMP dependent phosphorylation of cMyBP-C and cTnI the risk/benefit of the inodilators is not clear.
5.2. Targeting the Troponin Complex Avoiding PDE III Inhibition
The quest to find a troponin activator without the negative effects of PDEIII inhibition continues with early successes in moving forward in development of small molecules presumably acting at the cTnI-cTnC interface. Improvement of DCM phenotype in mice expressing variants of cTnC and cTnI that increase myofilament response to Ca2+ provided proof of principle that this approach may be successful. We have employed genetic modification of thin filament regulation to inform potential approaches to DCM therapy in which there is an increase in myofilament Ca-response by expressing slow skeletal TnI (ssTnI) to alter the cTnC-cTnI interaction [94]. Compared to cTnI-myofilaments, myofilaments controlled by ssTnI demonstrate an increased response to Ca2+ [65,95]. Double TG mice expressing DCM-linked Tm-E54K and ssTnI had significantly improved response to Ca2+ and improvements in the phenotype of the DCM model. Our studies modifying cardiac troponin isoform expression are related to screening for Tn activators employing a TnI-TnC chimera thus probing the modulation of this interaction [96,97]. In a similar approach, Powers et al. [9] tested whether expression of a modified cTnC (cTnC-L48Q), which enhances myofilament Ca-response and increases tension, can improve maladaptive effects of expression of a DCM linked mutant Tm-D230N in a mouse model. Compared to controls, double transgenic mice expressing both the mutant Tm and cTnC-L48Q showed significantly elevated TTI and improvements in the maladaptive effects of DCM.
Promising troponin activating compounds have been developed through high throughput assay employing ATPase activity of cardiac myofibrils by investigators at Cytokinetics [98]. These investigators have resported detailed findings in the development of CK-963 and discuss in their recent paper next steps in clinical trial with follow up compound [98] Studies reported by He et al. [75] provide an example of advances in development of small molecules acting as a thin filament activators without PDE III inhibition. They reported that activation of troponin with a small molecule called TA1 increased myofilament Ca-sensitivity, increased tension in intact myocardial preparations, and elevated developed pressure with a reduction in HR and left ventricular end-diastolic pressure. In contrast with the inotrope dobutamine that increased cAMP and was energy wasting, TA1 had no effects on phosphocreatine/ATP ratio, or ΔG~ATP. Another study developed a high throughput assay probing effects of chemical libraries on the cTnI-cTnC interaction with fluorescent probes [99]. Results identified NS5806 as a compound that forms a complex with the N-lobe of cTnC promoting Sw peptide binding without affect Ca-binding. It remains unclear whether these effects of NS5806 occur in situ. Another compound identified in NMR investigations as a troponin activator is RPI-194 [100]. Although studies with RPI-194 provide clues to approaches, its effects are not restricted to cardiac myofilaments.
5.3. Myosin Heavy Chain as a Drug Target
More recently biotech and pharma have made significant progress in shifting emphasis to targeting myosin heavy chain with small molecules as a therapy in HCM, acquired HF, and DCM [73,81,101] Investigators as Cytokinetics, Inc led the way the way with development of OM, an agent activating sarcomere tension by complex mechanisms discussed below [81]. Although trials did not achieve end points for transition to the clinic, the evidence that OM may be an effective therapy in acquired HF has stimulated more research. This further effort resulted in the development of danicamtiv (MYK-491 ) an agent in current clinical trials proposed to have a similar profile as OM, but different pharmacokinetics. [72,73]. Danicamtiv has been proposed to recruit myosin motors from the OFF state and increase myofilament Ca-sensitivity, but this needs to be reevaluated with techniques such as high-resolution x-ray diffraction in intact muscle. Moreover, studies need to be carried out in a variety of DCM mouse models in addition to the one employed in the study expressing a cTnC-I61Q variant with reduced Ca-binding, but which is not linked to DCM gene variants. It is of interest that in the clinical trial effectiveness of treatment with MYK-491 includes patients with DCM gene variants expressing variants of myosin heavy chain as well as titin*.
There are some issues and considerations about the potential use of myosin activators in DCM. An important question is whether emerging myosin activators such as OM and danicamtiv are effective in genetic DCM progression. Agents targeted to activate myosin cross-bridges have gone through clinical trials showing some beneficial effects in human HF, but the population of patients in the cohorts with genetic DCM is unclear [26]. An issue that needs to be considered is whether there are long term effects reducing the progress of the clinical course. Although more evidence is required, there are data indicating that long term effects of OM administration may have palliative effects [102]. Seven days following a single infusion of OM in healthy adult rats, El Oumeiri et al. [102] compared cardiac function and gene expression in the hearts with controls. Hearts of OM injected rats had prolonged ejection time and shortened pre-ejection period, with no changes in ejection fraction, cardiac output, heart rate, and stroke volume. The data indicated a possible beneficial long-term effect via anti-apoptotic and anti-oxidative stress signaling. However, there was an increase in fatty acid oxidation with potential for increased oxygen consumption, and an increase in expression of angiotensin II receptors-1 (AT-1) and AT-2 with a slight change in the ratio of the two receptors. There were no significant changes in proteins regulating Ca movements, except for an increased expression of left ventricular calcium/calmodulin-dependent protein kinase II delta, a regulator of expression of a subunit of voltage dependent Ca channels (Cacna1c),. In view of a lack of effect of OM on intracellular Ca-transients, the functional significance of this change is not clear. Moreover, tests of effects of OM in living tissue slices from human HF patients showed that although there was an increase in contractility and duration of contraction time, relaxation was impaired [103]. The relaxation prolongation worsened with increased beating rates and was suggested to possibly promote arrhythmias. However, in vitro studies reported a low probability of OM being pro-arrhythmic [104]. There is also a consideration emphasized by Tang et al. [11] in relation to mutations in myosin linked to DCM. They reported that OM had a reduced effect to stimulate cross-bridge function in preparations containing β-myosin heavy chain with a DCM-linked mutation, F764L. These investigators concluded that mutation-specific modifications in sarcomere activation by OM need to be considered.
There are other views of the precise mechanism of action of OM in promoting tension development that are relevant to its therapeutic application in altering effects of disease-causing sarcomere genes. Potential effects on thin filament function indicate that myosin activators may be effective in DCM with diminished tension not associated with shifts in the myosin SRX/DRX populations. We reported in vitro studies that demonstrated that OM increased the Ca-sensitivity of Tm-E54K myofilaments [105]. Although we and others had previously reported an effect of OM to increase sarcomere Ca-sensitivity [106,107], and modify cooperative activation, this effect is not generally discussed in considering the mechanism of action. Kieu et al. [108] reported that at physiological temperatures there is a prominent effect of OM in increasing skinned fiber sub-maximal tensions in the force-Ca relation with no effect on maximum tension. Kieu et al. [108] attributed this effect to a mechanism in which the prolonged duty cycle associated with OM enhances cooperative thin filament activation, which is maximized at physiological temperatures disallowing an effect at saturating Ca2+ concentrations. We have also tested the effects of OM on cardiac myocytes harboring a DCM-linked mutation human inducible pluripotent stem cells (hiPSC-CMs) generated from patients expressing cTnT-R173W [109]. We found that OM improved function and promoted F-actin assembly and content in the hiPSC-CMs. OM also reversed a depression in cross-bridge entry into the force generating state in preparations reconstituted with either wild-type cTnT or recombinant cTnT-R173W. Whether myosin activators are therapeutic in DCM remains an open question. However, studies of Tm variants (Tm-T237S) by Barrick et al. [110] linked to DCM reported a decrease in the Ca-sensitivity of the ATPase activity in reconstituted preparations compared to controls. Treatment of these preparations with OM increased Ca-sensitivity indicating that OM may be a useful agent in DCM. Moreover, Lehman et al. [111] have suggested the potential efficacy of danicamtiv and OM in DCM based on their effects in HF studies. It will be of interest to determine early and long-term effects in DCM progression of administration of these more recent sarcomere activators.
A question relating to the therapeutic efficacy of OM and other myosin activators is whether their use not only increases tension in DCM but also reduces fibrosis. [4,112]. This is an important issue as fibrosis has been known for some time to be a predictor of mortality and hospitalization in DCM [1,113,114]. In view of lack of evidence that myosin activators reverse fibrosis in systolic HF, it has been argued that a fibroblast specific therapeutic approach is necessary [4]. This make sense as fibroblast proliferation occurs with an avoidance of cell death Even so the per cent of patients with mid-wall fibrosis, the most common manifestation, is less than half the population. The argument has been made that inhibition in fibrosis by expression of p38 in fibroblasts indicates that there is feedback by which loss of tension at the sarcomere level is reduced in a DCM model [4]. This is not surprising as it is expected that mechano signaling occurs not only within cells but from cell to cell. The proposal that a fibroblast specific therapy should be an adjunct therapy in DCM requires patient specific therapy with supporting information whether fibrosis is present or not. An editorial providing a balanced view of the role of fibrosis in genetic cardiomyopathies has been provided by Frangogiannis [115]. * https://www.mayo.edu/research/clinical-trials/cls-20519537
5.4. Cardiac Myosin Binding Protein C as a Drug Target
In view of the versatility of cMyBP-C actions in controlling cardiac contraction and dynamics, it is not surprising that efforts are underway to modulate its function with small molecules. Initial studies defining high throughput approaches with use fluorescence lifetime measurements of the actin-cMyBP-C interaction have been reported [76]. Early evidence provides an approach with identification of lead compounds with specificity for cardiac N-terminal C0-C2 regions of cMyBP-C with or without phosphorylation. The effect to modify in vitro function required high concentrations of the agents indicating that much work remains.
6. Future Directions and Concluding Remarks
Diverse concepts of control of sarcomere tension discussed here indicate a need for re-examination of the causes and progression of DCM linked to sarcomere mutations as well as therapeutic approaches. It is apparent that this re-examination requires emphasis on studies integrating the myocyte protein activities and interactions in living muscle preparations. Caution has been indicated about the use of pseudo-phosphorylation and removal and replacement of Ser/Thr residues in assessing mechanism. The use of pig hearts that is increasing in the literature should also be emphasized. Lessons from the use of high-resolution x-ray diffraction techniques demand that hypotheses based only on biochemical, biochemical, and structural studies with myofilaments and myofilament proteins isolated from the cellular environment need to be tested in functioning myocardium ideally in a basal and stimulated state with and without regulation by DCM mutant proteins and with and without treatment with candidate therapies. Advances in transcriptomic and proteomic approaches provide a snapshot of a particular state but information on stages of the progression DCM with and without therapeutic interventions to test for pleiotropic effects would be ideal. Although not described here owing to limitations there is a need for consideration of reciprocal interactions of the myocytes with the mosaic of changes within micro-environment [116,117]. Fibrosis, for example, has been well documented confirming non-invasive measurements and validating a link with DCM severity. [114]. Clinical determinations of within the micro-environment are close to reality with emerging high-resolution time resolved approaches including four- dimensional flow cardiac magnetic resonance imaging (CMR), diffusion tensor imaging (DTI), and hybrid positron-emission tomography-MRI [118]. Moreover, clinical workflows for precision medicine in genetic cardiomyopathy remain a work in progress with some papers outlining the needs in the field in the general population [119] and in ethnic racial groups [120].
Author Contributions
Conceptualization, R.J.S, B.M.W.; Writing – Original Draft Preparation, R.J.S.; Writing – Review & Editing, R.J.S, B.M.W, P.H.G.; Funding Acquisition, R.J.S, B.M.W, P.H.G.. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are supported by National Institutes of Health, NHLBI, RO1HL 158634 (BMW, PHG, and RJS).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McNally EM, Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res. 2017;121(7):731-48.
- Fatkin D, Huttner IG, Kovacic JC, Seidman JG, Seidman CE. Precision Medicine in the Management of Dilated Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;74(23):2921-38.
- Koslow M, Mondaca-Ruff D, Xu X. Transcriptome studies of inherited dilated cardiomyopathies. Mammalian Genome. 2023;34(2):312-22. [CrossRef]
- Bretherton RC, Reichardt IM, Zabrecky KA, Goldstein AJ, Bailey LRJ, Bugg D, et al. Correcting dilated cardiomyopathy with fibroblast-targeted p38 deficiency. bioRxiv. 2023.
- Merlo M, Cannatá A, Vitagliano A, Zambon E, Lardieri G, Sinagra G. Clinical management of dilated cardiomyopathy: current knowledge and future perspectives. Expert Rev Cardiovasc Ther. 2016;14(2):137-40. [CrossRef]
- von Lewinski D, Gasser R, Rainer PP, Huber MS, Wilhelm B, Roessl U, et al. Functional effects of glucose transporters in human ventricular myocardium. Eur J Heart Fail. 2010;12(2):106-13. [CrossRef]
- Chandra Mohan N, Foley P, Chandrasekaran B. A case report of successful physiological pacing in a patient with lamin A/C cardiomyopathy. Eur Heart J Case Rep. 2022;6(8):ytac324. [CrossRef]
- Fairuz S, Ang CW, Mraiche F, Goh JK. Current Targets and Future Directions of Positive Inotropes for Heart Failure. Curr Med Chem. 2023. [CrossRef]
- Powers JD, Kooiker KB, Mason AB, Teitgen AE, Flint GV, Tardiff JC, et al. Modulating the tension-time integral of the cardiac twitch prevents dilated cardiomyopathy in murine hearts. JCI Insight. 2020;5(20). [CrossRef]
- Teerlink JR, Diaz R, Felker GM, McMurray JJV, Metra M, Solomon SD, et al. Cardiac Myosin Activation with Omecamtiv Mecarbil in Systolic Heart Failure. N Engl J Med. 2021;384(2):105-16.
- Tang W, Unrath WC, Desetty R, Yengo CM. Dilated cardiomyopathy mutation in the converter domain of human cardiac myosin alters motor activity and response to omecamtiv mecarbil. J Biol Chem. 2019;294(46):17314-25. [CrossRef]
- Kooiker KB, Mohran S, Turner KL, Ma W, Flint G, Qi L, et al. Danicamtiv increases myosin recruitment and alters the chemomechanical cross bridge cycle in cardiac muscle. bioRxiv. 2023.
- Nag S, Gollapudi SK, Del Rio CL, Spudich JA, McDowell R. Mavacamten, a precision medicine for hypertrophic cardiomyopathy: From a motor protein to patients. Sci Adv. 2023;9(30):eabo7622. [CrossRef]
- Coats CJ, Maron MS, Abraham TP, Olivotto I, Lee MMY, Arad M, et al. Exercise Capacity in Patients With Obstructive Hypertrophic Cardiomyopathy: SEQUOIA-HCM Baseline Characteristics and Study Design. JACC Heart Fail. 2023.
- Paratz ED, Mundisugih J, Rowe SJ, Kizana E, Semsarian C. Gene therapy in cardiology: is a cure for hypertrophic cardiomyopathy on the horizon? Can J Cardiol. 2023.
- Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, et al. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell. 2016;165(5):1147-59. [CrossRef]
- Sandler H, Dodge HT. LEFT VENTRICULAR TENSION AND STRESS IN MAN. Circ Res. 1963;13:91-104. [CrossRef]
- Vikhorev PG, Vikhoreva NN, Yeung W, Li A, Lal S, Dos Remedios CG, et al. Titin-truncating mutations associated with dilated cardiomyopathy alter length-dependent activation and its modulation via phosphorylation. Cardiovasc Res. 2022;118(1):241-53. [CrossRef]
- LeWinter MM, Granzier HL. Titin is a major human disease gene. Circulation. 2013;127(8):938-44.
- Solaro RJ. Advances in understanding the state of titin truncation variants in dilated cardiomyopathy. Pflugers Arch. 2022;474(3):265-6. [CrossRef]
- Hooijman P, Stewart MA, Cooke R. A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys J. 2011;100(8):1969-76.
- Nag S, Trivedi DV. To lie or not to lie: Super-relaxing with myosins. eLife. 2021;10. [CrossRef]
- Anderson RL, Trivedi DV, Sarkar SS, Henze M, Ma W, Gong H, et al. Deciphering the super relaxed state of human β-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc Natl Acad Sci U S A. 2018;115(35):E8143-e52. [CrossRef]
- McNamara JW, Singh RR, Sadayappan S. Cardiac myosin binding protein-C phosphorylation regulates the super-relaxed state of myosin. Proc Natl Acad Sci U S A. 2019;116(24):11731-6. [CrossRef]
- Schmid M, Toepfer CN. Cardiac myosin super relaxation (SRX): a perspective on fundamental biology, human disease and therapeutics. Biol Open. 2021;10(2). [CrossRef]
- Day SM, Tardiff JC, Ostap EM. Myosin modulators: emerging approaches for the treatment of cardiomyopathies and heart failure. J Clin Invest. 2022;132(5). [CrossRef]
- Kawana M, Spudich JA, Ruppel KM. Hypertrophic cardiomyopathy: Mutations to mechanisms to therapies. Front Physiol. 2022;13:975076. [CrossRef]
- Linari M, Brunello E, Reconditi M, Fusi L, Caremani M, Narayanan T, et al. Force generation by skeletal muscle is controlled by mechanosensing in myosin filaments. Nature. 2015;528(7581):276-9. [CrossRef]
- Reconditi M, Linari M, Lucii L, Stewart A, Sun YB, Narayanan T, et al. Structure-function relation of the myosin motor in striated muscle. Ann N Y Acad Sci. 2005;1047:232-47. [CrossRef]
- Park-Holohan SJ, Brunello E, Kampourakis T, Rees M, Irving M, Fusi L. Stress-dependent activation of myosin in the heart requires thin filament activation and thick filament mechanosensing. Proc Natl Acad Sci U S A. 2021;118(16). [CrossRef]
- Caremani M, Pinzauti F, Powers JD, Governali S, Narayanan T, Stienen GJM, et al. Inotropic interventions do not change the resting state of myosin motors during cardiac diastole. J Gen Physiol. 2019;151(1):53-65. [CrossRef]
- Ma W, Jani VP, Song T, Gao C, Gong H, Sadayappan S, et al. The structural OFF and ON states of myosin can be decoupled from the biochemical super-relaxed and disordered-relaxed states. bioRxiv. 2023. [CrossRef]
- Tamborrini D, Wang Z, Wagner T, Tacke S, Stabrin M, Grange M, et al. Structure of the native myosin filament in the relaxed cardiac sarcomere. Nature. 2023. [CrossRef]
- Tonino P, Kiss B, Gohlke J, Smith JE, 3rd, Granzier H. Fine mapping titin's C-zone: Matching cardiac myosin-binding protein C stripes with titin's super-repeats. J Mol Cell Cardiol. 2019;133:47-56.
- Chen L, Liu J, Rastegarpouyani H, Janssen PML, Pinto JR, Taylor KA. Structure of mavacamten-free human cardiac thick filaments within the sarcomere by cryoelectron tomography. Proc Natl Acad Sci U S A. 2024;121(9):e2311883121. [CrossRef]
- Sequeira V, Maack C, Reil GH, Reil JC. Exploring the Connection Between Relaxed Myosin States and the Anrep Effect. Circ Res. 2024;134(1):117-34. [CrossRef]
- dos Remedios CG, Law KYC, McNamara JW, Kraft T, Peckham M, van der Velden J, et al., editors. The Molecular Basis of the Frank-Starling Law of the Heart: A Possible Role for PIEZO1?2024; Cham: Springer International Publishing.
- Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424(6944):35-41. [CrossRef]
- Yamada Y, Namba K, Fujii T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat Commun. 2020;11(1):153. [CrossRef]
- Brunello E, Fusi L. Regulating Striated Muscle Contraction: Through Thick and Thin. Annu Rev Physiol. 2023. [CrossRef]
- Solís C, Solaro RJ. Novel insights into sarcomere regulatory systems control of cardiac thin filament activation. J Gen Physiol. 2021;153(7). [CrossRef]
- Rynkiewicz MJ, Pavadai E, Lehman W. Modeling Human Cardiac Thin Filament Structures. Front Physiol. 2022;13:932333. [CrossRef]
- Risi CM, Belknap B, Atherton J, Leite Coscarella I, White HD, Bryant Chase P, et al. Troponin structural dynamics in the native cardiac thin filament revealed by cryo electron microscopy. Journal of Molecular Biology. 2024:168498. [CrossRef]
- Risi C, Schäfer LU, Belknap B, Pepper I, White HD, Schröder GF, et al. High-Resolution Cryo-EM Structure of the Cardiac Actomyosin Complex. Structure. 2021;29(1):50-60.e4. [CrossRef]
- Tobacman LS. Troponin Revealed: Uncovering the Structure of the Thin Filament On-Off Switch in Striated Muscle. Biophys J. 2021;120(1):1-9.
- Mun JY, Previs MJ, Yu HY, Gulick J, Tobacman LS, Beck Previs S, et al. Myosin-binding protein C displaces tropomyosin to activate cardiac thin filaments and governs their speed by an independent mechanism. Proc Natl Acad Sci U S A. 2014;111(6):2170-5. [CrossRef]
- Risi C, Belknap B, Forgacs-Lonart E, Harris SP, Schröder GF, White HD, et al. N-Terminal Domains of Cardiac Myosin Binding Protein C Cooperatively Activate the Thin Filament. Structure. 2018;26(12):1604-11.e4. [CrossRef]
- Ponnam S, Sevrieva I, Sun YB, Irving M, Kampourakis T. Site-specific phosphorylation of myosin binding protein-C coordinates thin and thick filament activation in cardiac muscle. Proc Natl Acad Sci U S A. 2019;116(31):15485-94. [CrossRef]
- Sevrieva IR, Ponnam S, Yan Z, Irving M, Kampourakis T, Sun YB. Phosphorylation-dependent interactions of myosin-binding protein C and troponin coordinate the myofilament response to protein kinase A. J Biol Chem. 2023;299(1):102767. [CrossRef]
- Loescher CM, Breitkreuz M, Li Y, Nickel A, Unger A, Dietl A, et al. Regulation of titin-based cardiac stiffness by unfolded domain oxidation (UnDOx). Proc Natl Acad Sci U S A. 2020;117(39):24545-56. [CrossRef]
- Ratti J, Rostkova E, Gautel M, Pfuhl M. Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J Biol Chem. 2011;286(14):12650-8.
- Jung HS, Komatsu S, Ikebe M, Craig R. Head-head and head-tail interaction: a general mechanism for switching off myosin II activity in cells. Mol Biol Cell. 2008;19(8):3234-42. [CrossRef]
- Sevrieva I, Knowles AC, Kampourakis T, Sun YB. Regulatory domain of troponin moves dynamically during activation of cardiac muscle. J Mol Cell Cardiol. 2014;75:181-7. [CrossRef]
- Brunello E, Fusi L, Ghisleni A, Park-Holohan SJ, Ovejero JG, Narayanan T, et al. Myosin filament-based regulation of the dynamics of contraction in heart muscle. Proc Natl Acad Sci U S A. 2020;117(14):8177-86. [CrossRef]
- Fukuda N, Wu Y, Farman G, Irving TC, Granzier H. Titin-based modulation of active tension and interfilament lattice spacing in skinned rat cardiac muscle. Pflugers Arch. 2005;449(5):449-57. [CrossRef]
- Colson BA, Bekyarova T, Locher MR, Fitzsimons DP, Irving TC, Moss RL. Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ Res. 2008;103(3):244-51.
- Mamidi R, Gresham KS, Verma S, Stelzer JE. Cardiac Myosin Binding Protein-C Phosphorylation Modulates Myofilament Length-Dependent Activation. Front Physiol. 2016;7:38. [CrossRef]
- Ait-Mou Y, Hsu K, Farman GP, Kumar M, Greaser ML, Irving TC, et al. Titin strain contributes to the Frank-Starling law of the heart by structural rearrangements of both thin- and thick-filament proteins. Proc Natl Acad Sci U S A. 2016;113(8):2306-11. [CrossRef]
- Li KL, Methawasin M, Tanner BCW, Granzier HL, Solaro RJ, Dong WJ. Sarcomere length-dependent effects on Ca(2+)-troponin regulation in myocardium expressing compliant titin. J Gen Physiol. 2019;151(1):30-41. [CrossRef]
- Groen M, López-Dávila AJ, Zittrich S, Pfitzer G, Stehle R. Hypertrophic and Dilated Cardiomyopathy-Associated Troponin T Mutations R130C and ΔK210 Oppositely Affect Length-Dependent Calcium Sensitivity of Force Generation. Front Physiol. 2020;11:516. [CrossRef]
- Reda SM, Chandra M. Dilated cardiomyopathy mutation (R174W) in troponin T attenuates the length-mediated increase in cross-bridge recruitment and myofilament Ca(2+) sensitivity. Am J Physiol Heart Circ Physiol. 2019;317(3):H648-h57. [CrossRef]
- Biesiadecki BJ, Kobayashi T, Walker JS, Solaro RJ, de Tombe PP. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ Res. 2007;100(10):1486-93.
- Colson BA, Patel JR, Chen PP, Bekyarova T, Abdalla MI, Tong CW, et al. Myosin binding protein-C phosphorylation is the principal mediator of protein kinase A effects on thick filament structure in myocardium. J Mol Cell Cardiol. 2012;53(5):609-16.
- Solaro RJ, Moir AJ, Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262(5569):615-7. [CrossRef]
- Solaro RJ, Henze M, Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res. 2013;112(2):355-66. [CrossRef]
- Wolska BM, Stojanovic MO, Luo W, Kranias EG, Solaro RJ. Effect of ablation of phospholamban on dynamics of cardiac myocyte contraction and intracellular Ca2+. Am J Physiol. 1996;271(1 Pt 1):C391-7. [CrossRef]
- Kranias EG, Garvey JL, Srivastava RD, Solaro RJ. Phosphorylation and functional modifications of sarcoplasmic reticulum and myofibrils in isolated rabbit hearts stimulated with isoprenaline. Biochem J. 1985;226(1):113-21. [CrossRef]
- Alves ML, Dias FAL, Gaffin RD, Simon JN, Montminy EM, Biesiadecki BJ, et al. Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins. Circ Cardiovasc Genet. 2014;7(2):132-43. [CrossRef]
- Messer AE, Marston SB. Investigating the role of uncoupling of troponin I phosphorylation from changes in myofibrillar Ca(2+)-sensitivity in the pathogenesis of cardiomyopathy. Front Physiol. 2014;5:315. [CrossRef]
- Pavadai E, Rynkiewicz MJ, Yang Z, Gould IR, Marston SB, Lehman W. Modulation of cardiac thin filament structure by phosphorylated troponin-I analyzed by protein-protein docking and molecular dynamics simulation. Archives of biochemistry and biophysics. 2022;725:109282. [CrossRef]
- Sevrieva IR, Brandmeier B, Ponnam S, Gautel M, Irving M, Campbell KS, et al. Cardiac myosin regulatory light chain kinase modulates cardiac contractility by phosphorylating both myosin regulatory light chain and troponin I. J Biol Chem. 2020;295(14):4398-410. [CrossRef]
- Landim-Vieira M, Knollmann BC. Danicamtiv Recruits Myosin Motors to Aid the Failing Heart. Circ Res. 2023;133(5):444-6. [CrossRef]
- Kooiker KB, Mohran S, Turner KL, Ma W, Martinson A, Flint G, et al. Danicamtiv Increases Myosin Recruitment and Alters Cross-Bridge Cycling in Cardiac Muscle. Circ Res. 2023;133(5):430-43. [CrossRef]
- Hitsumoto T, Tsukamoto O, Matsuoka K, Li J, Liu L, Kuramoto Y, et al. Restoration of Cardiac Myosin Light Chain Kinase Ameliorates Systolic Dysfunction by Reducing Superrelaxed Myosin. Circulation. 2023;147(25):1902-18. [CrossRef]
- He H, Baka T, Balschi J, Motani AS, Nguyen KK, Liu Q, et al. Novel Small-Molecule Troponin Activator Increases Cardiac Contractile Function Without Negative Impact on Energetics. Circ Heart Fail. 2022;15(3):e009195. [CrossRef]
- Bunch TA, Guhathakurta P, Thompson AR, Lepak VC, Carter AL, Thomas JJ, et al. Drug discovery for heart failure targeting myosin-binding protein C. bioRxiv. 2023. [CrossRef]
- Solaro RJ, Rüegg JC. Stimulation of Ca++ binding and ATPase activity of dog cardiac myofibrils by AR-L 115BS, a novel cardiotonic agent. Circ Res. 1982;51(3):290-4. [CrossRef]
- Solaro RJ, Gambassi G, Warshaw DM, Keller MR, Spurgeon HA, Beier N, et al. Stereoselective actions of thiadiazinones on canine cardiac myocytes and myofilaments. Circ Res. 1993;73(6):981-90. [CrossRef]
- Wang X, Li MX, Spyracopoulos L, Beier N, Chandra M, Solaro RJ, et al. Structure of the C-domain of human cardiac troponin C in complex with the Ca2+ sensitizing drug EMD 57033. J Biol Chem. 2001;276(27):25456-66. [CrossRef]
- Kass DA, Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006;113(2):305-15. [CrossRef]
- Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331(6023):1439-43. [CrossRef]
- Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, et al. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118(12):3893-903.
- Sorsa T, Pollesello P, Solaro RJ. The contractile apparatus as a target for drugs against heart failure: interaction of levosimendan, a calcium sensitiser, with cardiac troponin c. Mol Cell Biochem. 2004;266(1-2):87-107. [CrossRef]
- Haikala H, Kaivola J, Nissinen E, Wall P, Levijoki J, Lindén IB. Cardiac troponin C as a target protein for a novel calcium sensitizing drug, levosimendan. J Mol Cell Cardiol. 1995;27(9):1859-66.
- Lee JA, Allen DG. Calcium sensitisers. Bmj. 1990;300(6724):551-2.
- Pollesello P, Papp Z, Papp JG. Calcium sensitizers: What have we learned over the last 25 years? Int J Cardiol. 2016;203:543-8.
- Brunkhorst D, v der Leyen H, Meyer W, Nigbur R, Schmidt-Schumacher C, Scholz H. Relation of positive inotropic and chronotropic effects of pimobendan, UD-CG 212 Cl, milrinone and other phosphodiesterase inhibitors to phosphodiesterase III inhibition in guinea-pig heart. Naunyn Schmiedebergs Arch Pharmacol. 1989;339(5):575-83. [CrossRef]
- Edes I, Kiss E, Kitada Y, Powers FM, Papp JG, Kranias EG, et al. Effects of Levosimendan, a cardiotonic agent targeted to troponin C, on cardiac function and on phosphorylation and Ca2+ sensitivity of cardiac myofibrils and sarcoplasmic reticulum in guinea pig heart. Circ Res. 1995;77(1):107-13.
- Cleland JG, Nikitin N, McGowan J. Levosimendan: first in a new class of inodilator for acute and chronic severe heart failure. Expert Rev Cardiovasc Ther. 2004;2(1):9-19. [CrossRef]
- Remme WJ. Inodilator therapy for heart failure. Early, late, or not at all? Circulation. 1993;87(5 Suppl):Iv97-107.
- Nonaka M, Morimoto S, Murayama T, Kurebayashi N, Li L, Wang YY, et al. Stage-dependent benefits and risks of pimobendan in mice with genetic dilated cardiomyopathy and progressive heart failure. British journal of pharmacology. 2015;172(9):2369-82. [CrossRef]
- Solaro RJ. Translational medicine with a capital T, troponin T, that is. Circ Res. 2007;101(2):114-5. [CrossRef]
- Boyle KL, Leech E. A review of the pharmacology and clinical uses of pimobendan. J Vet Emerg Crit Care (San Antonio). 2012;22(4):398-408. [CrossRef]
- Alves ML, Warren CM, Simon JN, Gaffin RD, Montminy EM, Wieczorek DF, et al. Early sensitization of myofilaments to Ca2+ prevents genetically linked dilated cardiomyopathy in mice. Cardiovasc Res. 2017;113(8):915-25. [CrossRef]
- Arteaga GM, Palmiter KA, Leiden JM, Solaro RJ. Attenuation of length dependence of calcium activation in myofilaments of transgenic mouse hearts expressing slow skeletal troponin I. J Physiol. 2000;3(Pt 3):541-9. [CrossRef]
- Li MX, Mercier P, Hartman JJ, Sykes BD. Structural Basis of Tirasemtiv Activation of Fast Skeletal Muscle. J Med Chem. 2021;64(6):3026-34. [CrossRef]
- Cai F, Kampourakis T, Parijat P, Cockburn KT, Sykes BD. Conversion of a Cardiac Muscle Modulator from an Inhibitor to an Activator. ACS Med Chem Lett. 2023;14(4):530-3. [CrossRef]
- Collibee SE, Romero A, Muci AR, Hwee DT, Chuang C, Hartman JJ, et al. Cardiac Troponin Activator CK-963 Increases Cardiac Contractility in Rats. Journal of Medicinal Chemistry. 2024. [CrossRef]
- Parijat P, Ponnam S, Attili S, Campbell KS, El-Mezgueldi M, Pfuhl M, et al. Discovery of novel cardiac troponin activators using fluorescence polarization-based high throughput screening assays. Sci Rep. 2023;13(1):5216. [CrossRef]
- Mahmud Z, Tikunova S, Belevych N, Wagg CS, Zhabyeyev P, Liu PB, et al. Small Molecule RPI-194 Stabilizes Activated Troponin to Increase the Calcium Sensitivity of Striated Muscle Contraction. Front Physiol. 2022;13:892979. [CrossRef]
- Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351(6273):617-21. [CrossRef]
- El Oumeiri B, Dewachter L, Van de Borne P, Hubesch G, Melot C, Jespers P, et al. Altered Left Ventricular Rat Gene Expression Induced by the Myosin Activator Omecamtiv Mecarbil. Genes (Basel). 2023;14(1). [CrossRef]
- Amesz JH, Langmuur SJJ, Bierhuizen MFA, de Groot NMS, Manintveld OC, Taverne YJHJ. Omecamtiv mecarbil in precision-cut living heart failure slices: A story of a double-edged sword. Journal of Molecular and Cellular Cardiology Plus. 2023:100040. [CrossRef]
- Qu Y, Gao B, Arimura Z, Fang M, Vargas HM. Comprehensive in vitro pro-arrhythmic assays demonstrate that omecamtiv mecarbil has low pro-arrhythmic risk. Clin Transl Sci. 2021;14(4):1600-10. [CrossRef]
- Utter MS, Ryba DM, Li BH, Wolska BM, Solaro RJ. Omecamtiv Mecarbil, a Cardiac Myosin Activator, Increases Ca2+ Sensitivity in Myofilaments With a Dilated Cardiomyopathy Mutant Tropomyosin E54K. J Cardiovasc Pharmacol. 2015;66(4):347-53. [CrossRef]
- Nagy L, Kovács Á, Bódi B, Pásztor ET, Fülöp G, Tóth A, et al. The novel cardiac myosin activator omecamtiv mecarbil increases the calcium sensitivity of force production in isolated cardiomyocytes and skeletal muscle fibres of the rat. Br J Pharmacol. 2015;172(18):4506-18. [CrossRef]
- Halas M, Langa P, Warren CM, Goldspink PH, Wolska BM, Solaro RJ. Effects of Sarcomere Activators and Inhibitors Targeting Myosin Cross-Bridges on Ca(2+)-Activation of Mature and Immature Mouse Cardiac Myofilaments. Mol Pharmacol. 2022;101(5):286-99. [CrossRef]
- Kieu TT, Awinda PO, Tanner BCW. Omecamtiv Mecarbil Slows Myosin Kinetics in Skinned Rat Myocardium at Physiological Temperature. Biophys J. 2019;116(11):2149-60. [CrossRef]
- Broughton KM, Li J, Sarmah E, Warren CM, Lin YH, Henze MP, et al. A myosin activator improves actin assembly and sarcomere function of human-induced pluripotent stem cell-derived cardiomyocytes with a troponin T point mutation. Am J Physiol Heart Circ Physiol. 2016;311(1):H107-17. [CrossRef]
- Barrick SK, Garg A, Greenberg L, Zhang S, Lin CY, Stitziel NO, et al. Functional assays reveal the pathogenic mechanism of a de novo tropomyosin variant identified in patient with dilated cardiomyopathy. J Mol Cell Cardiol. 2023;176:58-67. [CrossRef]
- Lehman SJ, Crocini C, Leinwand LA. Targeting the sarcomere in inherited cardiomyopathies. Nat Rev Cardiol. 2022;19(6):353-63. [CrossRef]
- Lakdawala NK, Dellefave L, Redwood CS, Sparks E, Cirino AL, Depalma S, et al. Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J Am Coll Cardiol. 2010;55(4):320-9. [CrossRef]
- Assomull RG, Prasad SK, Lyne J, Smith G, Burman ED, Khan M, et al. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J Am Coll Cardiol. 2006;48(10):1977-85. [CrossRef]
- Eijgenraam TR, Silljé HHW, de Boer RA. Current understanding of fibrosis in genetic cardiomyopathies. Trends in cardiovascular medicine. 2020;30(6):353-61. [CrossRef]
- Frangogiannis NG. Editorial commentary: Myocardial fibrosis in genetic cardiomyopathies: A cause of dysfunction, or simply an epiphenomenon? Trends in cardiovascular medicine. 2020;30(6):362-3. [CrossRef]
- Solís C, Warren CM, Dittloff K, DiNello E, Solaro RJ, Russell B. Cardiomyocyte external mechanical unloading activates modifications of α-actinin differently from sarcomere-originated unloading. The FEBS journal. 2023;290(22):5322-39. [CrossRef]
- Langa P, Wolska BM, Solaro RJ. The Hippo Signaling Pathway as a Drug Target in Familial Dilated Cardiomyopathy. International Journal of Drug Discovery and Pharmacology. 2022;1(1):4. [CrossRef]
- Chowdhary A, Garg P, Das A, Nazir MS, Plein S. Cardiovascular magnetic resonance imaging: emerging techniques and applications. Heart. 2021;107(9):697-704. [CrossRef]
- Nomura S, Ono M. Precision and genomic medicine for dilated and hypertrophic cardiomyopathy. Frontiers in cardiovascular medicine. 2023;10:1137498. [CrossRef]
- Jordan E, Kinnamon DD, Haas GJ, Hofmeyer M, Kransdorf E, Ewald GA, et al. Genetic Architecture of Dilated Cardiomyopathy in Individuals of African and European Ancestry. Jama. 2023;330(5):432-41. [CrossRef]
Figure 1.
Near-Neighbor C-zone Cardiac Regulatory Units in OFF and ON States. The left regulatory unit is in an end-diastolic state (relaxing levels of cellular Ca2+) with low levels of phosphorylation of key control elements in cTnI, cMyBP-C, and titin. See the text and the legend and diagram in Figure 2 for further discussion. Relaxed myosin heads are folded close to the thick filament in an OFF state with force-generating reactions with actin impeded by Tm. This Tm steric block is by the cTn complex extending along the functional unit with multiple inhibitory interactions involving N- and C-terminal regions of cTnI and cTnT interacting with cTnC, actin and Tm. Head to tail overlap of contiguous Tms interact with regions of cTnT N-terminus reducing the possibility of cooperative activation along the sarcomere strand. The low Ca2+ blocked state of the thin filament is tempered by interactions of N-terminal domains of cMyBP-C with actin-Tm that partially reduce the steric block. This interaction is reduced with elevated Ca2+ and abolished with phosphorylation of cMyBP-C. Not shown is the possibility for direct interactions of cMyBP-C with cTnI, proposed to affect coordinated effects of phosphorylation of these proteins. The stretch of titin elastic domains induces resting tension and diastolic pressure; titin C-zone interactions with cMyBP-C and myosin potentially modulate the OFF state. There are also interactions of cMyBP-C domains with the regulatory light chain (RLC) of myosin in a phosphorylation dependent manner. Complex allosteric, steric, and cooperative mechano-sensing mechanisms involving all the major sarcomere proteins activate sarcomere force and shortening. Acting allosterically, elevations in Ca-binding to cTnC trigger modification of protein-protein interactions releasing nearly complete the Tm steric block and inducing the reaction of force generating cross-bridges with the thin filament. According to the ON/OFF theory of tension control, these initial cross-bridge reactions induce a mechanical strain on the thick filament promoting more cross-bridges to engage and develop tension and further activate the thin filament. Activation of a near-neighbor functional unit occurs by cooperative spread along the strands of sarcomeres aided by effects cross-bridge dependent thin filament activation mechanisms and by Tm-Tm interactions between contiguous sarcomeres. The reactions responsible for the level of the relaxed state and the intensity of active state are variables controlled by the gain in mechano-sensing by load, length and by neuro-humoral induced post-translational mechanisms. These homoeostatic mechanisms are exquisitely sensitive to mutations triggering DCM.
Figure 1.
Near-Neighbor C-zone Cardiac Regulatory Units in OFF and ON States. The left regulatory unit is in an end-diastolic state (relaxing levels of cellular Ca2+) with low levels of phosphorylation of key control elements in cTnI, cMyBP-C, and titin. See the text and the legend and diagram in Figure 2 for further discussion. Relaxed myosin heads are folded close to the thick filament in an OFF state with force-generating reactions with actin impeded by Tm. This Tm steric block is by the cTn complex extending along the functional unit with multiple inhibitory interactions involving N- and C-terminal regions of cTnI and cTnT interacting with cTnC, actin and Tm. Head to tail overlap of contiguous Tms interact with regions of cTnT N-terminus reducing the possibility of cooperative activation along the sarcomere strand. The low Ca2+ blocked state of the thin filament is tempered by interactions of N-terminal domains of cMyBP-C with actin-Tm that partially reduce the steric block. This interaction is reduced with elevated Ca2+ and abolished with phosphorylation of cMyBP-C. Not shown is the possibility for direct interactions of cMyBP-C with cTnI, proposed to affect coordinated effects of phosphorylation of these proteins. The stretch of titin elastic domains induces resting tension and diastolic pressure; titin C-zone interactions with cMyBP-C and myosin potentially modulate the OFF state. There are also interactions of cMyBP-C domains with the regulatory light chain (RLC) of myosin in a phosphorylation dependent manner. Complex allosteric, steric, and cooperative mechano-sensing mechanisms involving all the major sarcomere proteins activate sarcomere force and shortening. Acting allosterically, elevations in Ca-binding to cTnC trigger modification of protein-protein interactions releasing nearly complete the Tm steric block and inducing the reaction of force generating cross-bridges with the thin filament. According to the ON/OFF theory of tension control, these initial cross-bridge reactions induce a mechanical strain on the thick filament promoting more cross-bridges to engage and develop tension and further activate the thin filament. Activation of a near-neighbor functional unit occurs by cooperative spread along the strands of sarcomeres aided by effects cross-bridge dependent thin filament activation mechanisms and by Tm-Tm interactions between contiguous sarcomeres. The reactions responsible for the level of the relaxed state and the intensity of active state are variables controlled by the gain in mechano-sensing by load, length and by neuro-humoral induced post-translational mechanisms. These homoeostatic mechanisms are exquisitely sensitive to mutations triggering DCM.
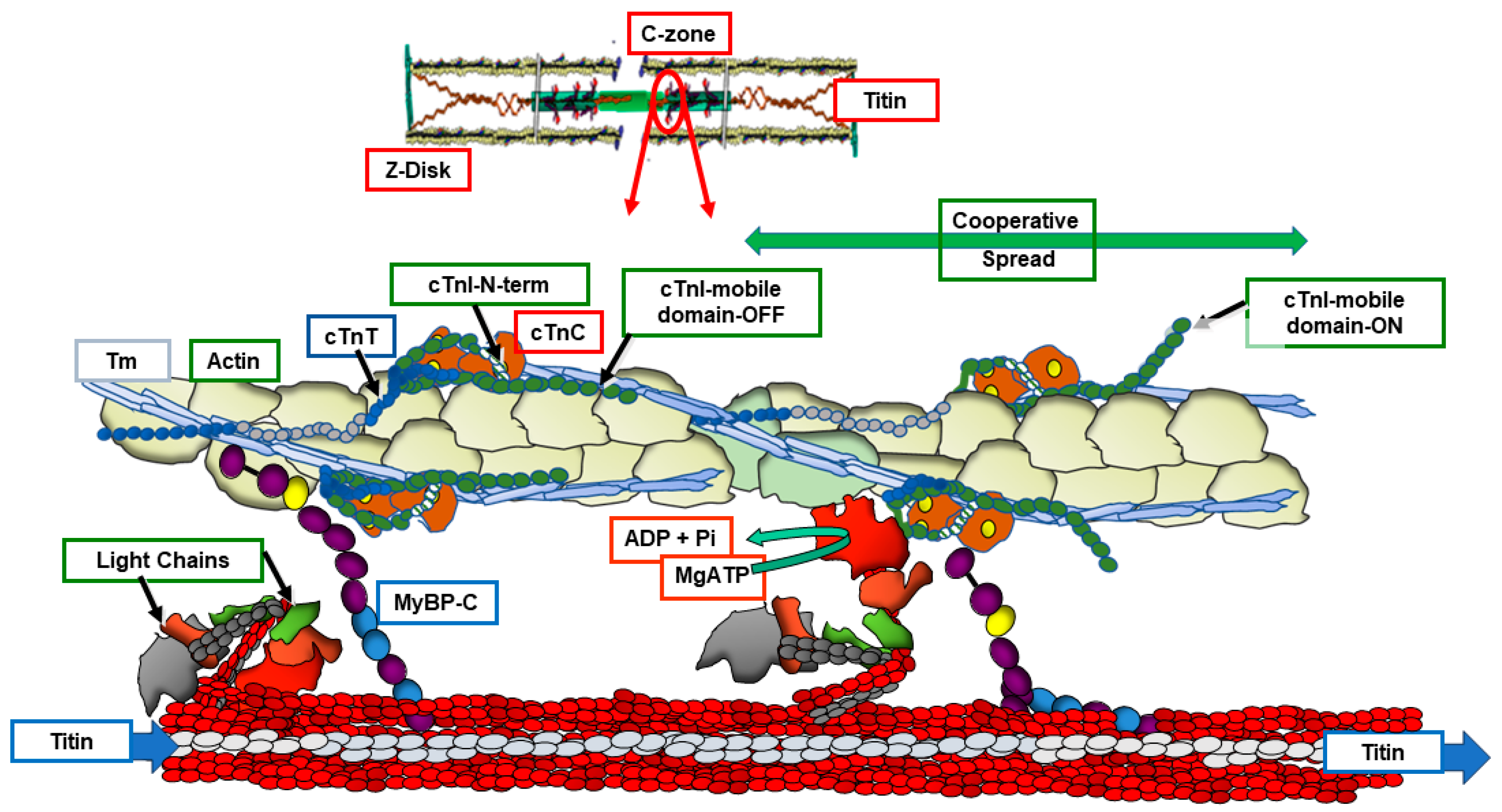
Figure 2.
Sarcomere Protein-Protein Intereactions in the Transition Between Diastole and Systole. The forward (green arrows) and reverse (red arrows) flow of reactions according to the ON/OFF theory of tension generation is initially triggered by Ca-binding to cTnC with generation of tension by few cross-bridges promoting mechano-transduction. The increased strain in the thick filament leads to more force generating cross-bridges and establishing tension during the heartbeat. Modulation of this process is pictured to occur down stream of these interactions that alter the gain in reactions. This variation in gain occurs with alterations in protein-protein interactions in the biochemical environment,, changes in sarcomere length, signaling via post-translational mechanisms, and expression of natural and DCM-linked isoforms. Small molecules that activate tension are also thought to modify this gain. See text and Figure 1 for further discussion and information.
Figure 2.
Sarcomere Protein-Protein Intereactions in the Transition Between Diastole and Systole. The forward (green arrows) and reverse (red arrows) flow of reactions according to the ON/OFF theory of tension generation is initially triggered by Ca-binding to cTnC with generation of tension by few cross-bridges promoting mechano-transduction. The increased strain in the thick filament leads to more force generating cross-bridges and establishing tension during the heartbeat. Modulation of this process is pictured to occur down stream of these interactions that alter the gain in reactions. This variation in gain occurs with alterations in protein-protein interactions in the biochemical environment,, changes in sarcomere length, signaling via post-translational mechanisms, and expression of natural and DCM-linked isoforms. Small molecules that activate tension are also thought to modify this gain. See text and Figure 1 for further discussion and information.
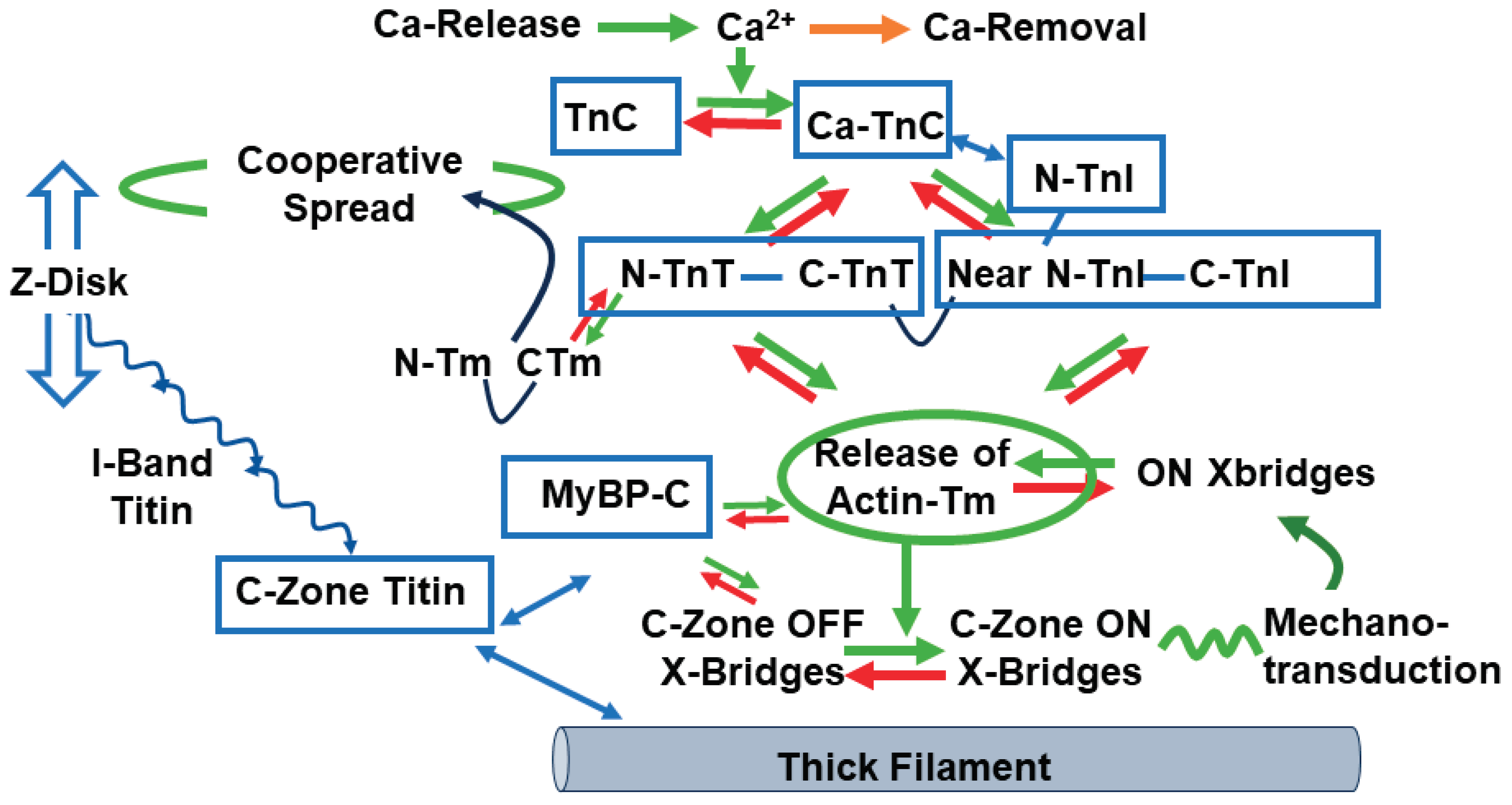
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



