
Submitted:
25 March 2024
Posted:
26 March 2024
You are already at the latest version
Abstract
To maintain an optimal body content of phosphorus throughout postnatal life, variable phosphate absorption from food must be finely matched with urinary excretion. This amazing feat is accomplished through synchronised phosphate transport by myriads of ciliated cells lining the renal proximal tubules. These respond in real time to changes in the phosphate and composition of the renal filtrate, and to hormonal instructions. How they do this has stimulated decades of research. New analytical techniques, coupled with incredible advances in computer technology, have opened new avenues for investigation at a sub-cellular level. There has been a surge of research into different aspects of the process. These have verified long held beliefs and are also dramatically extending our vision of the intense, integrated, intracellular activity which mediates phosphate absorption. Already, some have indicated new approaches for pharmacological intervention to regulate phosphate in common conditions, including chronic renal failure and osteoporosis, as well as rare inherited biochemical disorders. It is a rapidly evolving field. The aim here is to provide an overview of our current knowledge, to show where it is leading, and where there are uncertainties. Hopefully this will raise questions and stimulate new ideas for further research.
Keywords:
RGS14
; FGF23
; PTH/PTHrP receptor
; NHERF1
; NaPi-2a
; kidney stones
; Tumor Induced Osteomalacia
; hyperphosphatemia
; hypophosphatemia
; allosteric
1. Introduction
Phosphate is essential for bone synthesis and turnover and numerous cellular processes including biosynthesis of structural molecules, metabolism, generation of ATP and intracellular signalling. In healthy adults, almost 80% of total body phosphate is in the skeleton, and only around 0.1% in the ECF [1,2,3,4,5]. The body content is maintained by balancing phosphate absorption from food with removal by renal excretion. A balanced diet supplies approximately 1g of phosphorus per day, but more with high meat or dairy consumption. Trans-cellular absorption from the intestine is facilitated by 1,25 [OH]2D, but on a normal diet, most influx is via the paracellular route. As this is not tightly regulated, absorption increases with phosphate consumption [6,7,8]. Fine regulation of body phosphorus is entirely dependent on the kidneys, instructed by circulating hormones (notably PTH, FGF23, dopamine), IGF1, phosphate load, and other agents [8,9,10,11,12,13]. This is a daunting task. The human kidney has around 1.3 x 106 functioning units [nephrons]. Each day 160-170L of plasma ultrafiltrate passes across the glomerulus into the kidney at a rate of approximately 1.5 nL /sec per nephron [14]. The ratio of filtered phosphate to plasma phosphate concentration is between 0.89-0.96 [1]. On a normal diet filtered phosphate is roughly 170-180 mmol/24h. Between 85% -95% is subsequently reabsorbed.
Clinically, the importance of the renal regulation of body phosphate is only too apparent in chronic renal failure and from rare inherited disorders when the process fails [8,9,12,15,16,17,18,19,20]. A frequently encountered problem is reduced phosphate reabsorption in many individuals who form common calcium kidney stones, which is still unexplained [8,21,22]. Treatment is empiric with a risk of causing calcium phosphate precipitation in the kidneys [9]. We need a better understanding of the cellular mechanisms of phosphate transport to improve management. These have been researched intensively for decades. With the very powerful analytical tools now available, the complexity, speed and amazing co-ordination of the processes involved have become increasingly apparent [11,12,23,24]. Already, findings have shown new avenues for pharmacological intervention to regulate phosphate in common conditions, including chronic renal failure and osteoporosis, as well as rare inherited biochemical disorders [12,19,23,25,26,27,28,29,30,31,32,33].
This review focuses on the cellular mechanisms of phosphate reabsorption in proximal renal tubular cells. The aims are to integrate findings from research of the many aspects of the process, to get a cohesive view of our current knowledge and to highlight areas of uncertainty. A brief overview of the anatomy of the proximal tubular lining and its role in phosphate reabsorption is followed by descriptions of the chief executers, the amino acid transporters and NHERF1. Next, their regulation by hormones and dietary phosphate are described, and then possible mechanisms of phosphaturic hormones produced by tumours.
2. The Proximal Renal Tubule
Amazingly, almost all renal phosphate reabsorption occurs in the first segments of the nephrons, the short proximal convoluted tubules (PCTs), which have an average length in humans of only 14 mm [10]. The PCTs have three morphologically distinct segments: S1 (pars convoluta), S2, and S3, which are associated with differences in functional activity. The pars convoluta is lined by columnar cells with long tightly packed microvilli which form a tall brush border, lateral cell processes which interdigitate extensively with adjacent cells, and a prominent endocytic-lysosomal system [35,36]. Each microvillus has a central actin core, scaffolding or PDZ proteins to link membrane proteins to the actin cytoskeleton, and myosin VI [37]. This segment absorbs most of the phosphate and much of the fluid from the glomerular filtrate [38,39], and transports H+ ions into the tubular lumen via the sodium–hydrogen exchanger 3 (NHE3) transporter. In rats, the pH falls along the proximal tubule from 7.25 in the glomerular filtrate to approximately 6.70 [40]. The villi are shorter in S2 and endocytic vacuoles are smaller, and the structure of S3 is simpler [35,36]. Recent investigations have found variation in transcriptomes [41] and metabolic autofluorescence signals along the PCT [42].
3. Executors of Phosphate Reabsorption: Renal Phosphate Transporters and NHERF1 (Na+/H+ Exchange Regulatory Cofactor 1)
Two essential executors of the phosphate absorption process are a responsive transport system to transfer phosphate from the renal tubular lumen into the cells, and an intracellular systems operator, NHERF1, to co-ordinate phosphate uptake and the cellular response.
3.1. The Renal Phosphate Transporters
Phosphate reabsorption is mediated by at least three sodium-linked transporters belonging to solute carrier families SLC34 and SLC20, NaPi-2a (NPT2A, Npt2a, SLC34A1), NaPi-2c (NaPi-2c, SLC34A3), and Pit-2 (Npt3, Ram-1, SLC20A2) sited in the apical brush border membrane (BBM) [12,24,43]. They actively transport phosphate into the cells. The driving force is the inwardly directed electrochemical gradient of Na+ ions established by Na+/K+- ATPase located in the cell basal membranes. There is uncertainty about which proteins transport phosphate out of the cells across the basolateral membrane [4,15,24,44,45,46,47,48]. mRNA abundance of Slc34a1 was about ten times higher than Slc34a3 in mouse kidney [49], and in mouse and rat kidneys was >50-fold higher than slc20a2 mRNA [50]. Immunoreactive NaPi-2a was demonstrated in the BBM and subapical vesicles of the S1 section of rat proximal tubules and decreased gradually along the straight S2 section [43].
3.1.1. NaPi-2a
NaPi-2a accounts for around 70–80% of overall renal phosphate reabsorption [47]. The gene was identified and cloned in 1993 [24,51]. Human NaPi-2a [Uniprot Q06495] has 639 amino acids. It is predicted to contain two sets of transmembrane-spanning domains separated by a large extracellular loop, and cytoplasmic carboxyl and amino terminals. The first intracellular and third extracellular loops may be part of a permeation pore [12,52,53,54,55]. The three carboxyl-terminal residues comprise a PDZ (PSD-95/Discs-large/ZO1)-binding domain which can bind the carrier to proteins containing sequences of 80–90 amino acid residues that typify PDZ domains [44,55,56]. NaPi-2a binds to the PDZ scaffolding protein NHERF-1 (Na+/H+ exchange regulatory cofactor 1), and to other cytosolic scaffolding proteins (Section 3.2). HPO42- is the preferred substrate. Transport is inhibited by PTH, FGF23, and dopamine (Section 4, Section 5 and Section 6).
NaPi-2a proteins probably assemble in the apical membrane as dimers, but the two protomers function independently [12,24]. The rate of phosphate reabsorption is proportional to the number of active transporters on the cell surface and is not dependent on functional changes in the transporter itself [24]. Availability of Na+ and phosphate, pH and membrane voltage influence transport rate. Phosphate affinity is voltage dependent. At neutral pH and with 1mM phosphate, the affinity for phosphate is 50–100 μM and for Na+ around 50mM [44,53,57,58]. pH modulates transport by altering the HPO42- /H2PO 41- ratio, by reducing sodium affinity at one or more binding sites and by directing the orientation of the empty carrier between inward and outward-facing. Reducing extracellular pH over the range 8.0–6.2 reduced Na-dependent phosphate uptake significantly (up to 80%) [44,59]. In the proximal tubule, the pH can fall from approximately 7.4 in the glomerular filtrate to around 6.6, decreasing the ratio of HPO4 2- / H2PO4- from 4.0 to 1.6 [50]. Phosphate is transported with a 3:1 ratio of Na+ to HPO42-and hence each transport cycle carries net positive charge across the membrane [12,24,44,52,53,57,58]. In a proposed model, phosphate transport by NaPi-2a is viewed as a kinetic cyclic process comprising a sequence of transitions between conformationally distinct states of the protein [44,52,60].
NaPi-2a Partitioning Into Lipid Rafts.
Around 80% of apical NaPi-2a is localised in lipid rafts in the plasma membrane which are domains enriched in cholesterol, sphingomyelin and GM1 glycosphingolipids [12,61]. In vitro, an increase in the cholesterol content decreases NaPi-2a activity of the BBM and is associated with a parallel reduction of membrane fluidity [62]. This situation is observed in ageing rats who have impaired renal tubular phosphate transport which is not fully explained by reduced NaPi-2a mRNA and protein abundance [63,64]. Cholesterol-depletion reverses these abnormalities in renal cell cultures and BBM. In K+ deficiency NaPi-2a partitions in rafts enriched with sphingomyelin, glucosylceramide and ganglioside GM3. Reduced fluidity of the BBM and lateral mobility of NaPi-2a are associated with decreased transport activity. The abundance of NaPi-2a in the BBM is increased [65,66] and NaPi-2a forms pentamers rather than dimers [61]. Normally, within minutes after PTH stimulation of the apical PTH receptor, NaPi-2a moves laterally in the BBM before being endocytosed for destruction in lysosomes [67]. Perhaps an increase in membrane lipids hinders turnover of membrane-bound NaPi-2a. An increase in BBM cholesterol may partially account for the observation that tubular phosphate reabsorption was significantly lower in men over 50y attending a renal stone clinic than in younger men [21].
3.1.2. NaPi-2c
NaPi-2c was first cloned from human and rat kidney in 2002 [68] and mouse kidney in 2003 [69]. It is found exclusively in mammals [59]. In mice it is predicted to mediate 15–30% of phosphate reabsorption [3,70,71], but it may have a more important role in humans [47,72,73]. The affinities for Na+ and phosphate are 30-50 mM and 0.07 mM, respectively [68,74]. Transport is electroneutral with 2:1 Na+: HPO4 2- stoichiometry. NaPi-2c is very sensitive to pH but transport is insensitive to membrane voltage [53]. Acidosis reduces mRNA expression but not membrane abundance, and probably has a direct inhibitory effect on phosphate transport [75]. Although NaPi-2c lacks a canonical C-terminal PDZ-binding domain, it binds to the scaffolding proteins NHERF1 and NHERF3 (PDZK1). In contrast to NaPi-2a, NaPi-2c has a much stronger affinity for NHERF3 than for NHERF1. This is thought to contribute to its strong tethering to the apical membranes of the proximal tubular microvilli, where NaPi-2c and NHERF3 are co-expressed [76,77]. NaPi-2c is internalized through a microtubule-dependent pathway which requires myosin VI [76,77]. Unlike NaPi-2a it is not degraded, but accumulates in a subapical compartment, possibly including recycling endosomes, but this requires proof [77].
High and low phosphate intakes, respectively, decrease and increase activity but at a much slower rate than observed for NaPi-2a [47,77,78]. Potassium deficiency decreases mRNA expression and in addition sequesters the transporter in intracellular vesicles, resulting in reduced apical expression and phosphaturia [65]. NaPi-2c transport is inhibited by PTH and FGF23 [47,77,79,80]. The response to PTH is very slow and occurs over several hours in rodents [47,77]. In opossum kidney (OK) cells siRNA inactivation of NaPi-2c significantly suppressed the expression of NaPi-IIa protein and mRNA, suggesting that NaPi-2c is important for the expression of NaPi-2a [81]. Expression of NaPi-2c is increased in NaPi-2a deficient mice [75,80]. Bi-allelic loss-of-function mutations in SLC34A3 cause hypophosphatemic rickets with hypercalciuria (HHRH) [82], a disorder with renal Pi wasting, rickets and kidney stones. An unusual family with apparent HHRH had digenic inheritance of dominant heterozygous mutations in SLC34A1 and SLC34A3. Individuals with both mutations had significantly more severe disease than those with only one of the mutations, suggesting a gene dosage effect [73].
3.1.3. PiT-2
SLC20A1 (PiT-1) and SLC20A2 (PiT-2) are Type 3 sodium-dependent phosphate symporters. Both are expressed in kidneys, but only PiT-2 is localized at the apical membrane of proximal tubular epithelia [50], where it closely overlaps NaPi-2c [44,45,50]. PiT-2 transports two Na+ ions for each phosphate. The affinities for Na+ and phosphate are ~50 mM and 100 µM, respectively [10,74]. Since the preferred phosphate species is monovalent H2PO4 -, transport is electrogenic. The rate of phosphate transport is affected by external pH because this affects the ratio of H2PO4- to HPO4 2-. The apparent affinity for phosphate is lowest in the pH range 6.2–6.8 but fourfold higher at pH 5.0. PiT-2 may contribute only moderately (~5%) to renal transepithelial Pi transport but was upregulated in NaPi-2a- /- mice during metabolic acidosis and may have a compensatory role [75]. High and low phosphate intakes [50], potassium deficiency [65] and PTH [83] regulate mRNA expression and the abundance of PiT-2 in the brush border of renal proximal tubules.
3.2. NHERF1-the Systems Manager
3.2.1. NHERF1
NHERF1 was first identified in 1993 and cloned from rabbit kidneys in 1998 [84,85]. Others found it as an ezrin-binding partner and named it ezrin-binding phosphoprotein of 50 kDa (EBP50) [86]. NHERF1 (SLC9A3 Regulator 1) is from a family of cytoplasmic scaffolding proteins. Their main role is to assemble membrane-associated proteins and signalling molecules, kinases, phosphatases and trafficking proteins transiently in complexes to direct cell signalling or transport activities [11,86]. They are concentrated at the apical surface of polarized epithelial cells. Apical targeting requires microtubule function [86]. Four members of the NHERF family are expressed in the proximal tubules: NHERF1 is predominant and is expressed at high levels, NHERF2 (SLC9A3 Regulator 2) which has very weak expression, and NHERF3 (NaPi-Cap1, PDZK1) and NHERF4 (NaPi-Cap2, PDZK2) whose roles in phosphate reabsorption are of uncertain significance [56,87,88].
NHERF1 is a multifunctional protein which scaffolds membrane-bound proteins to the sub-apical actin cytoskeleton. This stabilises them at the cell surface and promotes their incorporation into intracellular signalling complexes [11,89,90]. Amongst numerous membrane proteins bound by NHERF1 are NaPi-2a and NaPi-2c, CFTR (cystic fibrosis transmembrane conductance regulator), NHE3, PTH1R (the receptor for PTH and PTH-related peptide) and β2-AR (the β2 adrenergic receptor). Cytosolic proteins bound by NHERF1 include RGS14 (regulator of G protein signalling 14), and the phosphokinases PKCα and GRK6A [47,55,56,91,92,93]. It is estimated that 35-50% of apical membrane NaPi-2a is bound to NHERF1 [94]. However, the concentration of NHERF1 greatly exceeds the total amount of membrane-bound NaPi-2a and GRK6A [95]. NHERF1 null mice have hypophosphatemia, increased renal phosphate excretion, and decreased NaPi-2a in apical membranes [94]. Humans with loss of function NHERF1 mutations similarly have hyper phosphaturia and low plasma phosphate [96].
Structure
Human and rabbit NHERF1 have 358 amino acids and share 84% identity over the entire length [85,89]. NHERF1 has five features which are essential for its versatile function (Figure 1).
First, NHERF1 has two PDZ domains, PDZ1 and PDZ2. These are binding sites for target proteins. Second, it has a C-terminal ezrin-binding domain (EBD). Through binding to ezrin, NHERF1 is tethered to the subapical actin cytoskeleton [86]. Binding of NHERF1 to ezrin allosterically increases the affinity of the PDZ domains for ligands [97,98]. Third, it is a phosphoprotein with 31 ser and 9 thr residues, of which 17 are in the linker connecting PDZ2 to the EBD. Phosphorylation/ dephosphorylation of selected residues changes the conformation of NHERF1 and impacts on its interaction with bound proteins [11,89,93,94,99]. Fourth, almost 30% of the protein chain is intrinsically disordered and hence NHERF1 is flexible. Altered phosphorylation leads to conformational changes in NHERF1 which may cause allosteric disturbances in the distant PDZ2 and C-terminal domains [11,89]. Fifth, it has a PDZ-binding motif (FSNL) at its C-terminal. In the absence of a regulatory hormone, the C-terminal folds back to bind loosely with PDZ2, so forming a closed molecular structure and blocking access of binding proteins to PDZ2 and the PDZ2-EBD linker. The C-terminal/PDZ2 link is broken by hormone-induced conformational changes, so releasing inhibition [Graphical Abstract]. Purified NHERF-1 forms a dimer and heterodimers with NHERF2 and PDZK1 which could form an extended NHERF scaffold, but this is still speculative [86].
3.3. Hormonal Regulation of Phosphate Transport by NaPt-2a
Four hormones regulate renal phosphate transport by NaPt-2a. It is decreased by PTH/PTHrP, dopamine, and FGF23 which promote phosphaturia, and is increased by growth hormone, acting via IGF1, which promotes phosphate reabsorption. The three phosphaturic hormones, their receptors, and signalling are described individually. The mechanisms by which they decrease phosphate transport by NaPt-2a are then considered collectively.
4. Regulation of NaPi-2a by Parathyroid Hormone (PTH) and Parathyroid Hormone -Related Protein PTHrP
4.1. PTH
PTH is synthesized in the parathyroid glands as a precursor, pre-pro-parathyroid hormone (pre-pro-PTH). The 25-residue ‘pre’ signal sequence and 6 residue ‘pro’ sequences are cleaved off sequentially, leaving mature PTH (1-84). This is concentrated in secre tory vesicles and granules, and released into the circulation, together with inactive carboxy-terminal fragments cleaved by proteases during storage. PTH (1-84) is cleared rapidly from the circulation and has a plasma half-life of approximately 2 min. Approximately 70% is extensively degraded in the liver. Around 20% of intact PTH (1-84) is filtered by the renal glomerulus. Most is then bound by megalin in the proximal tubular membrane, internalized and degraded [9]. Carboxy-terminal fragments are also cleared by glomerular filtration. Because their half-life is longer, their plasma concentrations are several-fold higher than those of PTH (1-84). Regulation of PTH secretion has been investigated and reviewed extensively [8,9]. In brief, acute (minute to minute) regulation is by the calcium sensing receptor [CaSR] in response to circulating ionised Ca2+ [2,8,100,101,102]. Longer term regulation is by altering transcription of the PTH gene. This is increased by raised plasma phosphate concentrations acting directly [9,102,103], or indirectly by lowering blood calcium and 1,25 (OH)2D levels, or by increasing cell proliferation [102]. PTH transcription and expression are suppressed by 1,25 (OH)2D [102,104] and isolated hypophosphatemia, which also decreases cell proliferation [102]. FGF23 activation of FG receptor 1 with its co-receptor, α-klotho inhibits PTH production [94,100,105,106] and, in contrast to the kidneys, increases CYP27B1 expression in the parathyroid glands [107]. α-Klotho may also have a direct action on the parathyroid glands to stimulate PTH secretion [9,100].
4.2. PTHrP
PTHrP is a secreted neuroendocrine peptide which is widely expressed in normal human tissues, and by some tumours, and is essential for fetal development and bone formation. It acts mainly as an autocrine or paracrine hormone to regulate cell prolif eration and differentiation and epithelial calcium ion transport [108]. Canonical human PTHrP [Uniprot P12272] has 177 amino acids, with a signal pro-peptide (residues 1-24). There are 3 principal secretory forms, PTHrP [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36], PTHrP [38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94], and PTHrP 107-139 (osteostatin), produced by endoproteolytic cleavage, and splice variants [108,109]. Further protease cleavage generates multiple protein fragments, some of which may have endocrine activity when released into the circulation. The amino terminal of human PTHrP shares close homology with hPTH, with 8 of the first 13 residues being identical, and binds the peptide to the PTH receptor [110]. Hence PTHrP can simulate most of the actions of PTH including proximal tubular phosphate transport. The peptide sequence from residue 14 has little similarity to PTH.
4.3. The PTH/PTHrP Receptor (PTH1R)
4.3.1. Structure
PTH1R is a class B1 (secretin) G protein coupled receptor [111,112]. It is expressed at high levels in kidney and in osteo blasts of bone and in a wide variety of tissues where it may be a target for PTHrP rather than for PTH [9,113]. PTH1R receptors are expressed in both apical and basolateral membranes of renal proximal tubules and are proposed to bind filtered and circulating PTH, respectively [94,114,115].
Class B1 GPCRs have a large extracellular domain (ECD), also named the N-terminal domain, which is required for ligand binding [23,111,112,116,117]. This has ~ 120 residues and a conserved fold composed of an N-terminal α-helix and two pairs of antiparallel β-sheets flanked by a long and a short a-helical segment stabilized by three disulfide bridges. N-linked glycosylation sites in the ECD regulate receptor trafficking and ligand binding [117,118,119]. The heptahelical transmembrane domain (TMD) section has ~260 residues and the intracellular C-terminal ~70 residues [23,116,117]. The transmembrane helices are linked by three extracellular loops and three intracellular loops [23,120]. Crystal structures are reported for the N-terminal [111,121,122] but are not achievable for full length PTH1R. Single particle cryo-electron microscopy (cryo-EM) has revolutionized membrane protein structural biology, with resolution below 2 Å achievable for rigid protein sequences. However, structures of intrinsically disordered sequences cannot generally be resolved [117,123]. An engineered peptide (LA-PTH) with a high affinity for PTH1R was used to solve cryo-EM structures of PTH1R in complex with GS [23].
4.3.2. PTH/PTHrP Binding
Most binding and signalling studies have used N-terminal 1-34 PTH and PTHrP fragments and truncated hPTH analogues. The N-terminal portion, especially the most N-terminal residues, is critical for PTH1R signalling. Deletion of PTH Ser1 and Val2 significantly reduces cAMP production. The C-terminal amino acids of the fragments (residues 15-34) bind to the PTH1R ECD and have a critical role in receptor selectivity and affinity [23]. A two-step activation model has been proposed [23,116,117,124]. Regions near the C-termini of the hormone ligands interact with the receptor’s ECD. This directs the amino-terminal portions of the ligands to interact with the receptor transmembrane domain. A change in the conformation of the sixth transmembrane domain (TM6) occurs, resulting in a large outward movement of TM6 associated with a substantial kink in its centre. This exposes an intracellular pocket which enables binding of the Gα subunit via its C-terminal and activation of G-protein signalling [23,117,124]. PTH1R activation is relatively slow. The initial N-terminal binding step has a time constant of ∼140 ms and the following interaction of the ligand with the transmembrane core, has a time constant of ∼1 s [125,126].
4.3.3. Signalling Pathways and Signalling Bias
PTH1R signals primarily via Gs which stimulates adenylyl cyclase activity but can also couple to Gq/11 which activates phospholipase C (PLC), G12/13 which regulates Rho guanine nucleotide exchange factors, and Gi/o, which inhibits adenylyl cyclase activity and interacts with, and signals, via β-arrestins [124,127,128,129]. The Gγ subunit can couple to diverse transducer proteins, such as a wide array of G proteins, kinases, and arrestin proteins [130,131] (Figure 2).
Various N-terminal PTH and PTHrP fragments and truncated hPTH (1–34) analogues selectively stimulate Gs, Gq or G protein-independent signalling pathways. These all have modifications in the N-terminal sequences, which interact directly with the transmembrane domain of the receptor [116,128]. There is evidence that binding of the C-terminal of PTH/PTHrP peptides to the ECD can also bias signalling. The C-terminal tips of the C-PTH-type and C-PTHrP-type 1-34 peptides diverge to opposite sides of the PTH1R ECD [23]. In addition, protease cleavage of the ECD enhanced coupling efficacy of the receptor to the Gs pathway, while reducing Gq-coupling at the same time and resulted in a signalling bias [116]. An important manifestation of bias introduced at the receptor is that PTH can induce prolonged signalling from intracellular sites, while PTHrP signals exclusively from the cell surface [129] (Section 4).
4.3.4. Termination of Signalling
Within seconds after agonist binding, GPCR kinase-2 (GRK2) initiates receptor desensitization by preventing G protein coupling [132], and by phosphorylating serine residues in the receptor C terminal which then then engages β-arrestins 1 and 2. This leads to rotation of the N- and C-terminal domains by 20°, binding of clathrin and the clathrin adaptor AP2 and internalization of the receptor–arrestin complex into endosomes [23,129,133,134,135]. Downstream signalling is terminated, and the G-protein subunits reassociate into the heterotrimeric complex [136]. Within the endosomes the agonist and receptor are dissociated. The receptor is then targeted to lysosomes and degraded, or dephosphorylated and recycled back to the plasma membrane [137,138]. In addition, PTH secretion by the parathyroid glands is suppressed by negative feedback when plasma Ca2+ is restored [137]. PTH activation of PLC signalling is suppressed by receptor phosphorylation and by phosphorylation of PLCβ3 by PKA [139]. Two additional GPCR regulators may be implicated in desensitisation of PTH1R.
RGS14 (Regulator of G Protein Signalling 14)
RGS14 is expressed in the proximal and distal renal tubules. RGS proteins control signalling through heterotrimeric G proteins by accelerating the intrinsic GTPase activity of Gα subunits, typically resulting in inhibition of downstream G protein signalling pathways (Section 8).
Receptor-Activity-Modifying Proteins (RAMPs).
RAMPS are ubiquitously expressed single-pass trans-membrane proteins that interact with class B1 GPCRs, including PTH1R, and modulate their function. There are three RAMPs in mammals [117,124]. PTH1R-and RAMP2 are highly co-expressed in kidneys, and PTH1R expressed in HEK293 cells has clear preference for RAMP2. It is unknown whether, or how, RAMP proteins affect PTH1R function. RAMP2 was observed to shift PTH1R bound to PTH (but not to PTHrP) to a preactivated state. This increased Gs and Gi3 activation kinetics in response to PTH, increased both PTH- and PTHrP-triggered β-arrestin2 recruitment to PTH1R which paralleled the overshoot in Gs activation, and increased PTH-mediated Gi3 signalling sensitivity [124].
4.3.5. Endosomal Signalling
Förster resonance energy transfer (FRET)-based studies of ligand–PTH1R interaction and cAMP production showed that PTHrP1-36/ PTH1R binding was fully reversible. cAMP increased transiently and ceased after ligand washout. However, PTH1-34, remained bound to PTH1R and cAMP generation continued after ligand washout [23]. This protracted cAMP response deviates from the conventional model of GPCR desensitization (Section 4.3.4.). It was found that PTH mediates sustained cAMP signalling from early endosomes following internalization of a PTH1R–Gβγ−β-arrestin complex [140]. PTH1R has two distinct active PTH1R conformations (RG and R0). The RG conformation is G protein dependent and is associated with transient cAMP responses from the plasma membrane. It is stabilized by PTH1-34 and PTHrP1-36 indistinguishably. The R0 state is stabilized preferentially by PTH. It is not altered by G protein coupling and maintains cAMP production after the receptor is internalized [131,132,133,134]. In the endosomes, the PTH/ PTH1R, β-arrestin, Gβγ complex (1) promotes the activation cycle of Gs from endosomes [140], (2) activates extracellular signal–regulated protein kinase 1/2 (ERK1/2) via β-arrestins and thereby decreases cAMP degradation by phosphodiesterase PDE4 [141], and (3) activates adenylate cyclase type 2 via Gβγ [142]. It was shown that formation of the ternary PTH1R–Gβγ−arrestin complex is determined by PTH activation of Gq. Gβγ released upon Gq activation stimulates phosphoinositide 3-kinase β (PI3Kβ) conversion of PI(4,5)P2 to PI(3,4,5)P3. This promotes β-arrestin recruitment to PTH1R and formation of the ternary receptor complex [143]. At the acid pH in the endosomes, PTH dissociates from PTH1R–Gβγ–arrestin. Inactive PTH1R is released, assembled with the retromer complex and trafficked to the Golgi apparatus [141,144], and/or recycled to the plasma membrane. Endosomal cAMP signalling is prolonged by an engineered peptide referred to as LA-PTH, which maintains a higher affinity binding to the PTHR R0 conformation than PTH [23,142,145].
4.3.6. Inherited Defects of PTH/PTHrP Receptor Signalling
4.4. Epac1 (Exchange Protein Directly Activated by cAMP 1)
Two intracellular cAMP receptors, protein kinase A, alias cAMP-dependent protein kinase, and exchange protein directly activated by cAMP (Epac), alias cAMP regulated guanine nucleotide exchange factor (cAMP-GEF), mediate the effects of cAMP generated by adenylyl cyclase in response to GPCR stimulation [146,147,149]. Both have a regulatory domain which binds cAMP with equal affinity [150]. By sensing intracellular concentrations of cAMP, this domain acts as a molecular switch to control diverse cell activities. Dependent on intracellular location and prevailing intracellular conditions, PKA and Epac may act synergistically, as in inactivation of NHE3 [151], or antagonistically. Having two cAMP receptors enables differential regulation of generated cAMP spatially and temporally [146,150]. There are two isoforms of Epac, Epac1 (gene RAPGEF3) and Epac2. EPAC1 is the isoform expressed in the kidneys and is detected immunochemically in most of the nephron. In the renal proximal tubules, protein expression was largely restricted to the brush border [152]. Compared with PKA, the role of Epac1 in regulation of proximal tubular function has been largely neglected.
4.4.1. Structure
Epac1 contains an N-terminal regulatory region and a C-terminal catalytic region (Figure 3). The regulatory region comprises a Dishevelled/Egl-10/pleckstrin (DEP) domain and the cAMP-binding [CBD] domain. The DEP domain is involved in locating Epac1 at the plasma membrane. The catalytic region consists of a RAS exchange motif (REM) domain which stabilizes the active conformation of Epac, a protein interaction motif, the RAS association domain, and a cell division cycle 25 -homology domain (CDC25HD) which promotes the exchange of GDP for GTP on Rap GTPases [146,147,153]. In the absence of cAMP, the CBD associates closely with the catalytic region and prevents activation. cAMP binding increases to the CBD domain causes a conformational change which removes the autoinhibition of the catalytic domain [147,150].
4.4.2. Actions of Epac1 in the Proximal Tubules
Epac1 is one of the guanine-nucleotide exchange factors (GEFs) which activate the small GTPases Rap1, and Rap2 in a protein kinase A (PKA)-independent manner [148,149,154,155]. Small GTPases function as molecular switches which regulate signalling events that control numerous cell processes [155]. To date there are only limited data on the functions of Epac1 in the proximal tubules. Epac signalling was shown to promote expression and trafficking of the Na+-glucose cotransporter type 1 via a mechanism involving caveolin-1 and F-actin in proximal renal tubular cells [151,153]. In the only reported comparison of PKA and Epac on tubular transport, opossum kidney (OK) cells and murine kidney slices were treated with cAMP analogues which selectively activated PKA or Epac. Both PKA and Epac were shown to inhibit NHE3 activity without changing the expression of NHE3 in the BBM. The Epac effect was independent of PKA and, unlike PKA, did not increase NHE3 phosphorylation. However, in contrast to PKA, Epac activation had no effect on NaPi-2a activity and did not induce retrieval of NaPi-2a from the BBM [151]. In an Epac1-deficient mouse model, NHE-3 expression in the proximal tubule decreased by 75% and the mice had a phenotype consistent with NHE3 deficiency. It was suggested that one action of Epac1 might be to stabilize its interaction with the cytoskeleton at the brush border membrane via a mechanism involving NHERF1[156].
From the above findings [151], Epac does not have a role in regulating phosphate transport by NaPi-2a. However, OK cells do not express RGS14, and RGS14 in rodent cells lacks the C-terminal extension expressed in human renal cells which was recently reported to bind to NHERF1. RGS14 has a Ras /Rap-binding domain and is a Ras effector [157] (Section 8). It would be interesting to know whether the same results are found in human proximal tubular cells stimulated by PTH.
5. Dopamine
Dopamine is a tyrosine derivative made in sympathetic system neurons and in non-neuronal tissues including the kidneys. In the kidneys, dopamine inhibits Na+ transport by autocrine/paracrine actions on the sodium-dependent transporters NHE1, NHE3, NaPi-2a, sodium bicarbonate cotransporter 1 (NBCe1), Na+/K+-ATPase and probably the Na+Cl- symporter (NCC) [168,169,170]. The major determinants of the renal tubular synthesis/release of dopamine are probably sodium intake and intracellular sodium [168].
5.1. Sources and Degradation
The concentration of free dopamine in plasma is low (< 1nmol/L) [168], and dopamine entering the kidneys by glomerular filtration makes little contribution to the urinary excretion which is in the micromolar range. Up to ~ 30% of this derives from renal dopaminergic nerves [171,172,173,174,175]. However, most is produced by cells lining the renal tubules by decarboxylation of the dopamine precursor L-dihydroxyphenylalanine (L-DOPA) by aromatic amino acid decarboxylase (AADC) [176,177,178]. This is a circulating intermediate mainly released from sympathetically innervated tissues [179,180]. L-DOPA is transferred from the glomerular filtrate and circulation by SLC7A8 (LAT-2) and SLC7A9/SLC3A1 amino acid transporters in the apical and basolateral membranes [181,182]. The proximal tubules have the highest expression of AADC and contribute most to renal production [168,183]. Dopamine then enters the tubular lumen and activates dopamine 1 receptors (D1R) on the apical membranes in an autocrine/paracrine manner (Figure 4). Paradoxically, in inherited AADC deficiency in which brain dopamine is low and circulating L-DOPA is raised, urinary dopamine excretion is normal or high, indicative of residual renal AADC activity [184].
Dopamine is degraded in renal tissues by deamination via monoamine oxidase (MAO), methylation via catechol- O -methyltransferase (COMT) [185,186] and oxidation by renalase. Renalase is a secreted flavin adenine dinucleotide (FAD)-dependent amine oxidase. It is synthesized in the renal glomeruli and proximal tubules and released into the blood and renal tubular lumen [183,187]. Renalase expression and activity are downregulated by an increase in dietary phosphate and might explain why feeding a diet high in phosphate diet increases renal dopamine excretion [173,188,189,190]. Urinary phosphate excretion is increased in renalase knockout mice [191,192].
5.2. Dopamine Receptors and Signalling.
Dopamine interacts with two families of G protein-coupled membrane receptors, D1 receptors comprising D1R and D5R, and D2 receptors comprising D2R, D3R, and D4R. D1R receptors are highly expressed in the proximal tubules, on both apical and basolateral membranes, and on the renal vasculature, but the subtypes differ [169,193,194]. Dopamine secreted into the tubular lumen acts mainly via D1R in an autocrine/paracrine manner to regulate ion transport in the proximal and distal nephron.
Dopamine binding to the receptor triggers dissociation of the attached trimeric G protein into Gα and Gβγ subunits. Depending on the dopamine receptor subtype, the Gα subunit either activates or inhibits adenylyl cyclase. The βγ subunit recruits G protein-coupled receptor kinases (GRKs) which phosphorylate serine and threonine residues. This promotes binding of arrestins and disrupts the interaction of D1R with G protein subunits [168,195,196,197]. The D1R/β-arrestin complex undergoes endocytosis/internalization via clathrin-coated pits. The dopamine receptors are sorted into recycling endosomes and returned to the cell membrane or transported through the endosome system and degraded in lysosomes. It is likely that D1-like receptors undergo time-dependent desensitization [198]. In humans D1R and D5R receptors stimulate adenylyl cyclase and activate PKA [199,200,201]. D1R, but not D5R, couples to Go [202]. D1R are also linked to Gαq and activation of PKC [203,204,205,206].
6. FGF23 and FGFR1C/KLOTHO Receptor
Unlike the majority of FGFs which bind to cells locally and have confined autocrine/paracrine activities, FGF23 synthesised by bone osteocytes and osteoblasts and by rare mesenchymal tumours enters the circulation and has hormonal actions [207,208]. Low expression has been reported in other tissues but is of unknown significance. FGF23 targets the proximal kidney tubules where it decreases phosphate reabsorption and reduces 1,25 (OH)2D production [209], and the distal tubules where it increases calcium reabsorption from the luminal fluid [8,9,12,16,18,19,210,211]. Individuals with inherited disorders and animal models with increased expression or activity of FGF23 generally have bone abnormalities, hyperphosphaturia, and low plasma concentrations of 1,25 [OH]2D and phosphate, unless renal function is impaired. Individuals with inherited disorders causing deficiencies of FGF23 or of its actions have ecto pic mineralization of soft tissues, including the kidney, impaired renal phosphate excretion, hyperphosphatemia and hypercalcemia due to high levels of 1,25 (OH)2D [8,212,213,214] (Table 2). Targeted ablation of FGF23 in mice results in premature death [213].
6.1. Physiological Factors Increasing/Decreasing FGF23 Production
Circulating FGF23 levels are increased physiologically by dietary phosphorus, raised serum phosphorus, 1,25 [OH]2D, leptin and other factors [100,219,220,221,222,223]. Activation of FGFR receptors in osteoblasts by locally produced growth factors FGF1 and FGF2 increases FGF23 production [224]. This may be a central pathway for regulating FGF23 expression in bone [8,100,224]. The major systemic regulating factor is 1,25 (OH)2D which acts via vitamin receptor (VDR)-dependent and independent signalling pathways [222]. Dietary phosphate has variable and modest effects on FGF23 in humans [3,100,225]. FGF23 was suppressed in NaPi-2a null mice on a low phosphate diet and normalized by a high phosphate intake [226]. PTH increases FGF23 expression and transcription in vitro and in vivo, but also increases furin cleavage and the effects are variable [100,227,228]. FGF23 is increased in patients with primary hyperparathyroidism, or activating mutations of PTH or the PTH receptor, and by continuous PTH infusion, but is decreased by intermittent PTH infusion to promote bone anabolism, and sometimes in hypoparathyroidism. A PTH-FGF23 feedback loop has been proposed in which increased PTH increases FGF23 which then decreases PTH [100].
6.2. Structure
Nascent canonical FGF23 has 251 amino acids. The first 24 residues are a signalling peptide which is removed [210]. A key feature is a proteolytic cleavage site [R176HTR179] which is hydrolysed by a subtilisin-like proprotein convertase, furin, [209,210,229] intracellularly to yield inactive secreted N-terminal and C-terminal fragments with 156 and 71 amino acids respectively [210,229,230,231,232]. Extracellular proteases of the plasminogen activation system and plasmin may cleave FGF23 at this site, and at other sites [210]. The site is protected from cleavage by glycosylation of Thr178 by GALNT3 [N-acetylgalactosaminyl transferase 3], whereas proteolysis is promoted by phosphorylation of Ser 175 by extracellular kinase family member 20C [FAM20] [210,233].
6.3. Renal Receptors and α-klotho
FGF23 interacts with the FGF receptors FGFR1c, 2c, 3c and FGFR4 [79]. In the proximal tubules, FGF23 binds preferentially to FGFR1c at the basolateral membrane. The single-pass transmembrane protein α-klotho is an essential partner for binding and signal transduction [210,234,235]. The receptor is a dimer of two units in each of which the ectodomain of receptor FGFR1c is bound to the ectodomain of the co-receptor α-klotho. This forms a binding pocket for the C-terminal region of c-FGF23 [236]. Heparan sulfate (HS) which has a weak affinity to FGF23 itself, facilitates the formation of a 2:2:2:2 FGF23– FGFR1c–klotho–HS signal transduction unit [236,237]. There is evidence that the circulating free C-terminal fragment of FGF23 produced by furin cleavage competes for binding to the receptor complex and may block the phosphaturic response to intact FGF23 [29,238].
6.3.1. α-Klotho
In normal proximal tubules (mice) α-klotho is expressed at the mRNA and protein level together with FGFR1, 3, and 4 [239]. In addition to full length α-klotho there are two soluble forms: a truncated isoform produced by alternative gene splicing, and the extracellular domain of full length α-klotho released through cleavage by metalloproteinases ADAM 10 and ADAM 17 at the cell surface. Soluble α-klotho is secreted into blood, CSF and urine and functions as an endocrine/paracrine substance. The kidneys are the main source of circulating soluble α-klotho [100,240]. Intact α-klotho is too large for glomerular filtration [100,240,241].
6.4. Signal Transduction and Signalling
FGFs signal through the four FGF tyrosine kinase receptors (FGFR1–4) and thereby activate the RAS-MAPK and the PI3K-AKT pathways [242]. The phosphaturic effects of FGF23 in the kidney tubules are klotho dependent. In the proximal renal tubules, FGF23 reduces the membrane abundance of NaPi-2a and NaPi-2c. The internalization and degradation of these phosphate transporters was shown to depend on the activation of ERK1/2 and serum/glucocorticoid-regulated kinase-1 (SGK1) pathways, [239] resulting in phosphorylation of NHERF1 [243]. FGF23 stimulates both fibroblast growth factor receptor substrate 2 (FRS2) and ERK1/2 in the proximal tubule [239,244] (Figure 5). Targeted ablation of proximal tubule klotho increased serum Pi and reduced urine Pi, suggesting that FGF23 regulates re-absorption of Pi in a klotho-dependent manner in the proximal tubules [245]. In the absence of klotho high concentrations of FGF23 can activate FGFR4. This induced PLC-catalysed production of diacylglycerol and inositol 1,4,5-trisphosphate which increased cytoplasmic calcium levels [246]. Calcium activation of calcineurin dephosphorylates the transcription factor NFAT, which permits its translocation into the nucleus to modulate the expression of specific target genes [247]. The transcription factor EGR1 [early growth response factor 1] is a downstream marker of MAPK signalling [244,248]. In Phex-deficient mice MAPK/ERK1/2 signalling was activated. A selective MEK inhibitor blocked this response [249,250].
7. Mechanisms of Hormonal Regulation of Phosphate Transport by NaPt-2a
From a review of studies up to 2012 to investigate the mechanisms of action of the three phosphaturic hormones on NaPi-2a transport, it was proposed that they use a common route which leads to severance of a link between NaPi-2a and NHERF1 [94]. Binding of NaPi-2a to this scaffolding normally stabilises the transporter in the apical plasma membrane. When the link is broken, detached NaPt-2a is endocytosed and degraded, the number of transporters at the cell surface decreases and phosphate absorption is reduced. The process is initiated by phosphorylation of NHERF1. The kinases in the PTH and dopamine pathway are PKA and PKC, and probably MAPK in the FGF23 pathway.
The evidence for this proposal was based on the culmination of years of research. (1) Phosphate transport correlates with the amount of NaPi-2a expressed on the apical membranes of proximal renal tubules [34,251,252]. (2) Expression is decreased by clathrin-mediated endocytosis of NaPi-2a and targeting to lysosomes for degradation [67,252,253]. Trafficking of the endocytic vesicles to lysosomes is via microtubules and requires myosin VI and a dynamic actin skeleton [254,255]. (3) Surface expression is decreased by PTH, dopamine and FGF2, and phosphate transport is inhibited within 30-45 min [34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257,258,259]. (4) NHERF1 null mice have increased phosphate excretion, low plasma phosphate, and decreased NaPi-2a abundance in the apical membrane [260]. (5) NHERF1 phosphorylation is the leading event in dissociation of NaPi-2a from NHERF1. NaPi-2a binds to the PDZ1 motif in NHERF1. Ser77 is the major phosphate acceptor in the PDZ1 domain for all three hormones. Ser77 phosphorylation decreases NaPi-2a binding to NHERF1 causing dissociation. Thr95 is a secondary acceptor for PTH and dopamine stimulation [261]. However, thr95 phosphorylation blocks the actions of FGF23 on phosphate transport [259]. (6) PTH is expressed both on apical and basolateral membranes of proximal tubular cells. Apically, the PTH receptor binds to NHERF1 PDZ1 and activates PKC [262,263]. However, in the absence of NHERF1 at the basolateral membrane, PTH stimulates cAMP/PKA but still reduces phosphate absorption [264]. In vitro PKC, but not PKA, phosphorylates ser77 directly, suggesting that PKA acts indirectly. (7) At low concentrations PTH and FGF23 were synergistic, with increased activation of PKC and PKA, but not MAPK [259].
Observations from subsequent studies support the proposal. The capacity to probe cell signalling pathways and determine molecular structures and interactions is revealing an amazing dynamic regulatory process. Knowledge of PDZ domains and PDZ-binding partners and their promiscuity has increased [264]. Flexible unstructured protein sequences have gained prominence because of their roles in allosteric regulation of the interaction between domains, and in modifying protein molecular conformation and interaction of amino acid residues [89,265,266].
Using a panel of kinase inhibitors, acute down-regulation of NaPi-2a transport by PTH was shown to be mediated by PKA, PKC, and ERK1/2 (MAPK) down-stream of PKC, whereas SGK1 (serum and glucocorticoid-activated kinase) mediated down-regulation by FGF23. NHERF1 knockdown prevented both PTH and FGF23 actions, indicating that the different PTHR and FGFR1c signalling pathways converge at the level of NHERF1 phosphorylation [267]. The PTH receptor PTH1Rwas shown to have a C-terminal PDZ-binding motif [ETVM] which was predicted to interact with both NHERF1 PDZ1 and 2 domains. Determinants outside the PDZ-ligand pocket enhanced formation of the NHERF1-PTH1R complex [93,268,269,270].
In a study of NHERF1 residues phosphorylated by PTH stimulation, Ser290 in the intrinsically disordered PDZ2-EBD linker displayed a conspicuous transient de-phosphorylation [11]. Ser290 phosphorylation decreased by 90% at 1min after PTH but was fully restored at 5 min. Dephosphorylation was associated with changes in NHERF1 conformation at critical positions for binding NaPi-2a and in the EBD at the C-terminal tail. These were reversed by re-phosphorylation. De-phosphorylation was attributed to a novel NHERF1 binding partner, protein phosphatase 1 (PP1), bound at V257PF259 a putative VxF/W motif. Re-phosphorylation of Ser290 was mediated by GRK6A bound to phosphorylated Ser162 in PDZ2 [11]. Ser162 is known as a PKCα phosphorylation site in human NHERF and is essential for hormone-sensitive phosphate uptake [95]. In the unstimulated state, binding of the NHERF1 C-terminal to PDZ2 would block access of GRK6.
From these findings, it was proposed that activation of PKC by PTH triggers rapid dynamic de-phosphorylation/ re-phosphorylation cycling at Ser290, leading to allosteric conformational changes in NHERF1. These disrupt binding of NaPi-2a to NHERF1 resulting in internalisation and destruction of NaPi-2a and reduced phosphate absorption [11] (graphical abstract). An interesting observation was that after PTH addition at normal or high extracellular phosphate concentrations in the presence of sodium, there was a transient burst of intracellular phosphate for ~10 min followed by a decline to resting levels. The Authors commented that this was consistent with uptake by NaPt-2a but offered no explanation for NaPi-2a activation. Perhaps one possibility to consider is an increase in extracellular pH [44,45] by PTH inhibition of NHE3.
In addition to its C-terminal TRL PDZ-binding motif, NaPt-2a, human NaPt-2a has an internal T494RL motif which has not been fully characterised [93]. Two disease-associated mutations in NHERF1were identified in this motif, R495H [271] and R495C [272]. Affected individuals had increased phosphate and cAMP excretion and low serum phosphate. Three mutations located in PDZ2 and in the PDZ1-PDZ2 linker were identified in seven patients with decreased phosphate reabsorption. In OK cells the PTH-stimulated cAMP response was significantly higher in cells transfected with mutant NHERF1 cDNA than with wild-type cDNA and was associated with a significantly greater decrease of phosphate uptake. There was no difference in intracellular Ca2+ concentration or inositol trisphosphate production [96].
FGF23 induces phosphaturia by decreasing the abundance of NaPi-2a in proximal tubular cell membranes through activation of the ERK1/2-SGK1 signalling pathway [96,239,267] (Section 6.4.). This action requires NHERF1 and synergizes with PTH [239,259]. It seems likely that FGF23 triggers a dynamic de-phosphorylation/ re-phosphorylation episode which causes allosteric conformational changes in NHERF1 and disrupts NaPi-2a/ NHERF1 binding as observed for PTH. However, this may not involve Ser290 which is constitutively phosphorylated by GRK6 [11]. There are closely sited serine residues which are alternative potential substrates for phosphorylation by SGK1. The observation that, in contrast to PTH and dopamine, thr95 phosphorylation inhibits the phosphaturic response to FGF23 may indicate a difference between SGK1 and GRK6 in the conformational changes induced by PTH and FGF23, respectively.
8. Regulator of G protein Signalling 14 (RGS14)
Genome-wide association studies [GWAS] to look for common gene variants associated with calcium kidney stones [273,274,275,276,277] and chronic renal failure [278,279] identified significant associations with single intronic nucleotide polymorphisms (SNPs) in the gene encoding one of the Regulator of G protein Signalling proteins, RGS14. These were associated with circulating plasma phosphate concentrations. Recent studies demonstrating that RGS14 binds to NHERF1 in the human renal proximal tubules and prevents inactivation of NaPi-2a by PTH [157] may help to explain this.
RGS proteins control signalling through heterotrimeric G proteins by accelerating the intrinsic GTPase activity of Gα subunits, typically resulting in an inhibition of downstream G protein signalling pathways. RGS proteins are primarily regulated by mechanisms that control their local concentration at the site of signalling. The expression of RGS proteins is highly dynamic and is regulated by epigenetic, transcriptional and post-translational mechanisms [280]. RGS14 is expressed in the proximal and distal renal tubules. In human proximal tubular cells expressing protein at endogenous levels RGS14 co-localises with NHERF1 [157].
8.1. Structure
In common with other RGS proteins, RGS14 has an RGS domain in the N-terminal which binds to Gαi G-protein subunits and blocks G protein signalling. In addition, RGS14 has two flexible tandem Ras/Rap-binding domains, RBD1 and RBD2 (Figure 6). Initial in vitro studies identified Rap1 and Rap2 and activated Rap2 as binding partners for the RBD domain, suggesting that RGS14 might be an effector for activated Rap proteins. However further investigation demonstrated that RGS14 bound selectively to activated H-Ras and not to Rap isoforms [281]. The RGS14/H-Ras association could assemble a multiprotein complex with components of the ERK MAPK pathway [281]. In the RGS14 C-terminal, a G-protein regulator (GPR, alias GoLoco) motif binds to inactive Gαi1/3 to anchor RGS14 at the cell membrane where it has GDI (GDP- dissociation inhibition) activity at the Gαi subunits. This prevents exchange of GDP for GTP and activation of G protein [282,283]. The GPR motif also blocks the association of Gα and Gβγ, potentially leading to prolonged Gβγ signalling [284]. Notably, unlike most other genotyped animals including rodents, RGS14 of humans and primates has a 21- residue extension to the C-terminal which terminates in a PDZ-binding motif (DSAL). This binds to the PDZ2 motif of NHERF1. RGS14 is the only RGS protein that has a canonical PDZ-recognition motif [157].
8.2. RGS14 Inhibition of PTH Regulation of NaPi-2a
In human primary kidney cells RGS14 knock-down unmasked PTH-sensitive Pi transport. However, RGS14 did not affect PTHR-Gs coupling or cAMP production, which indicated an action at a post-receptor site. RGS14 expression in human renal proximal tubule epithelial cells blocked the effects of PTH and FGF23 and stabilized the NaPi-2a–NHERF1 complex [157]. It was proposed that binding of the C-terminal PDZ ligand of RGS14 to PDZ2 of NHERF1 confers a tonic inhibition of the PTH and FGF23 actions. Against this is the dynamic expression of RGS14, and previous observations in unstimulated hippocampal neurons that RGS14 is most abundant in the cytosol but is recruited to the plasma membrane following stimulation [285]. An alternative scenario might be that cAMP generated by PTH/PTH1R signalling activates Epac and thereby activates Rap1 and/or Rap2 (Section 4.4.). Activated Rap might then bind with RGS14 and promote migration of the RGS14 C-terminal to NHERF1 and binding to PDZ2. This would then inhibit the actions of PTH and FGF23 as proposed above. Could Epac/RGS14 interaction be an additional mechanism for regulating phosphate absorption?
8.3. RGS14 Polymorphisms in GWAS Studies
Three of the intronic SNPs identified in the GWAS studies rs4074995 [278,279], rs56235845 [274] and rs11746443 [275,277] are in the RBD domain of RGS14 (Figure 7). The fourth, rs12654812 [273,276], is in the RGS domain. Because the next down-stream gene from RGS14 is SLC34A which encodes NaPi-2a, it has been thought previously that the RGS14 SNPs were picking up a mutation which decreases NaPi-2a production or transport activity. The alternative is that they are ‘sensing’ a dysfunctional mutation in RGS14. This is an attractive proposition. A mutated RGS14 that cannot effectively block the inhibitory action of PTH on NaPi-2a could produce biochemical disturbances of hyperparathyroidism without increased serum PTH levels. A high proportion of idiopathic calcium stone formers have an increased fractional excretion of phosphate [8,18,21]. In 6% of men attending a stone clinic this was associated with a low plasma phosphate concentration. PTH was normal [21]. The fact that RGS14 is under epigenetic control makes it an attractive candidate for a common disorder in which environmental and dietary factors play a large part [8,286].
9. Growth Hormone and Insulin-Like Growth Factor-1 (IGF-1)
Growth hormone [GH] stimulates phosphate absorption by the proximal renal tubules and reduces phosphate excretion. Plasma phosphate concentrations are increased during growth. They are raised in infants and children compared with adults [287], and in acromegaly [9]. GH treatment of children with GH deficiency increases plasma phosphate and renal tubular reabsorption of phosphate [288,289]. GH acts on target tissues mainly via stimulation of the insulin-like growth factor-1 (IGF-1), a 7.6 kDa protein synthesised predominantly in liver. In the circulation more than 99% of IGF-1 is bound to IGF-binding proteins (IGFBPs), mostly to IGFBP-3. Some IGF-1 is synthesised in the kidneys, and some derives from the circulation [290].
GH stimulation of phosphate reabsorption in the proximal tubules is mediated by up-regulation of NaPi-2a by IGF-1 bound to high affinity IGFR-1 receptors on the apical and basolateral membranes [291,292,293,294]. Expression and stability of the transporter in the cell membrane were increased, but not NaPi-2a synthesis [293]. IGF-I stimulation of OK cells resulted in a dose-dependent increase in tyrosine phosphorylation of the p95 kDa P-subunit of the IGF-I receptor β-subunit. Tyrosine phosphorylation recruits
SH2 domain-containing adapter proteins which activate signalling via the PI3-pathways and the MAPK cascade. However signalling to these pathways did not appear to be sufficient to induce Pi transport stimulation [294].
10. Dietary Phosphate
Dietary phosphate intake varies widely. With high intakes, intestinal absorption is mainly by the paracellular route and is poorly regulated [7,295]. Normally functioning kidneys respond to oscillations in the filtered phosphate load by adjusting the amount reabsorbed.
10.1. Acute Changes in Dietary Phosphate
10.1.1. Low Phosphate Intake:
Adaptation to a low phosphate diet involves increased insertion of NaPi-2a into the brush border membrane and requires NHERF1. This is followed by increased apical expression of NaPi-2c, and eventually of PIT2 [50,68,296,297,298]. Whereas phosphate depletion increased apical expression of NaPi-2a in rats and wild type (wt) and vitamin D receptor null mice [2,58,65,70], this response was absent in Nherf1-deficient mice [299,300]. In rats fed a 0.1% Pi diet the abundance of NaPi-2a, NaPi-2c, and Pit-2 in kidney was 100% higher than in rats on a 0.6% Pi diet. Pit-1 was not modified. The increase was pH dependent. In BBM vesicles, adaptation to the 0.1% Pi diet was accompanied by a 65% increase in the V (max) of Pi transport at pH 7.5, compared to the 0.6% Pi diet. At pH 6.0 the increase was only 11%. Metabolic acidosis increased the expression of NaPi-2c and Pit-2 in animals adapted to the low Pi diet. NaPi-2a RNA was estimated to contribute contributed 95% to the total mRNA of the Pi transporters, and that NaPi-2a accounted for 97% of Pi transport at pH 7.5 and 60% of Pi transport at pH 6.0, with little contribution from PiT-2 [301].
10.1.2. High Phosphate Intake:
In rats and mice apical expression of NaPi-2a is rapidly downregulated by a high dietary phosphate intake [3,65,296,302], followed by decreases in NaPi-2Ic and PIT2 [50]. This occurs independently of PTH and FGF23 [303]. Acute variations in dietary Pi levels do not alter RNA levels of the transporters [12]. Of several hormones analysed in rats, only PTH seems to be necessary for the early adaptation of renal phosphate transport to high dietary Pi [304]. In healthy humans challenged by acute intragastric or intravenous Pi loading, the increases in phosphaturia were similar and occurred at a similar time post-load. This was preceded by increases in plasma phosphate and PTH. Plasma FGF23 level increased after the onset of phosphaturia [305].
10.2. Chronic Changes in Dietary Phosphate
Persistent changes in serum Pi concentration resulting from chronic alterations in dietary phosphorus intake primarily modulate the gene expression of NaPi-2a in the proximal tubule [306]. This may be mediated in part by changes in plasma concentrations of hormones including PTH, FGF23, and 1,25(OH)2 D. In mice, a phosphate-response element has been identified in the NaPi-2a gene which binds the mouse transcription factor muE3 (TFE3). Renal TFE3 expression was upregulated in mice fed a low phosphorus diet [307], suggesting that this activates NaPi-2a transcription on a low intake. Physiologically, adaptive hormonal regulation of the abundance of NaPi-2a on the apical membrane proximal tubule may be more important than transcriptional regulation of NaPi-2a by TFE3 [308,309,310]. The activator protein 1 (AP1), nuclear factor erythroid 2-related factor 2 (Nrf2), and early growth response 1 (EGR1) transcription factors are upregulated in mammalian cell lines in response to increased extracellular phosphate concentration [311,312,313,314]. Increased extracellular Pi, activates the Raf/MEK/ERK pathway and leads to translocation of these transcription factors into the nucleus where they regulate the expression of phosphate-responsive genes.
10.3. High Dietary Phosphate and Renal Dopamine Excretion
Feeding a high phosphate diet to rats and mice [189,190] increased the renal excretion of dopamine. In mice this was attributable to increased dopamine synthesis through upregulation of AADC, and to significant decreases in renalase and other enzymes which degrade dopamine [94,190,191] (Section 5.1). PKA and PKC were activated, perhaps indicative of dopamine signalling via D1-like receptors. Carbidopa administration to inhibit dopamine synthesis decreased the phosphaturic response to a high-phosphate diet [190]. Collectively, the animal studies indicate that an increase in endogenous dopamine has an important role in acute adaptation to a high dietary phosphate intake [94]. A study of 884 humans similarly found that high dietary phosphate was associated with higher urine dopamine. However, unlike animal models, fractional excretion of phosphate was reduced. This questions the significance of urine dopamine in the acute adaptive response to dietary phosphate in humans [315].
11. Tumour-Induced Osteomalacia (TIO)
Tumour-induced osteomalacia (TIO) is a rare paraneoplastic syndrome characterised by renal phosphate wasting and hypophosphatemic osteomalacia. The causative tumours are usually mesenchymal, commonly hemangiopericytomas and giant cell tumours, often benign and frequently difficult to localise [27,316,317,318]. TIO results from over-production of phosphaturic factors. FGF23 is most often the causative agent, but other proteins may be responsible, notably MEPE (matrix extracellular phosphoglycoprotein) and, infrequently, secreted frizzled-related protein 4 (SFRP4) and the growth factor FGF7 [319,320,321,322,323]. In 50 well-characterized phosphaturic mesenchymal tumours excessive FGF23 production was attributable to a fibronectin (FN1)–FGFR1 fusion gene in 21 (42%) tumours and in 3 (6%) to a FN1-FGF1 fusion gene [322,324]. Unlike native FGFR1, the chimeric receptors seem to function independently of klotho co-receptor. In one patient with TIO raised FGF23 production was attributed to a novel NIPBL-BEND2 fusion gene (fused Nipped-B gene and a gene for a protein with two BEN domains). The NIPBL fusion gene promoted cell proliferation possibly via the MYC pathway [325]. Only half of the above 51 tumours had one of these fusion-genes and other causes remain to be identified.
11.1. MEPE
MEPE was first cloned and characterised in three tumours causing TIA [326]. Expression of MEPE was 104 to 105 times higher in TIA associated tumours than in control tumours [320]. MEPE is a secreted matrix extracellular phosphoglycoprotein belonging to a family of small integrin- binding ligand proteins, N-linked glycoproteins (SIBLING proteins) [327,328,329]. The predominant isoform expressed in humans has 525 amino acids. It is a flexible N-glycosylated protein with two defined functional domains (Figure 8).
The Ac-100 domain includes an integrin-binding RGD motif and a glycosaminoglycans (SGDG)-binding motif. The ASARM domain is the short carboxy-terminal peptide (21 residues). It is acidic because of its five aspartate and two glutamate residues. It contains 8 serine residues, of which five are phosphorylation sites for casein kinase (FAM20C). The phosphorylated ASARM peptide is released from the parent MEPE molecule by extracellular cathepsins at closely located cleavage sites and possibly by PHEX in bone [330].
MEPE is expressed in bone osteoblasts and osteocytes and in the brush border membranes of proximal renal tubules [331,332,333]. In bone MEPE interacts with membrane-bound PHEX (Phosphate Regulating Endopeptidase X-Linked) and DMP1 (Dentin Matrix Acidic Phosphoprotein) to control FGF23 transcription [329]. In the early stages of osteogenesis, MEPE stimulates bone formation. This requires the AC-100 motif which probably activates integrin receptors and thereby triggers downstream signalling [327,328]. In the later stages of osteogenesis, cleaved phosphorylated ASARM peptide binds to hydroxyapatite released from osteocytes and inhibits mineralisation [329,333]. Both MEPE and the cleaved ASARM peptide are released from bone into the circulation in normal individuals. Table 3 summarises studies to investigate the renal actions of MEPE on phosphate excretion.
Collectively, the findings from the infusion, perfusion and cell studies show that MEPE increases the fractional excretion of phosphate FEPO4 by decreasing the rate of phosphate transport (V max) by Npt-2a but not the affinity for phosphate. This was explained by decreased expression of Npt-2a protein in the brush border membrane (BBM). Plasma 1,25 (OH)2D was only measured in one of these studies and was decreased, comparable with FGF23. Over-expression of mepe in transgenic mice resulted in extensive renovascular changes which affected sodium balance, with activation of the renin / aldosterone system. The compensatory increase in NaPi-2a expression increased phosphate absorption and plasma phosphate was raised, apparently over-riding MEPE activity [333].
Like MEPE, infusion of the ASARM peptide increased FEPO4 and decreased serum phosphate. Plasma 1,25 (OH)2D increased. In transgenic mice with over-expressed ASARM peptide [337], changes in the biochemistry reflected a large increase in FGF23. This, coupled with an increase in PTH, makes it impossible to gauge the contribution of ASARM to the hyperphosphaturia.
11.1.1. Mechanism Causing Hyper-Phosphaturia
How does MEPE cause hyper-phosphaturia? From the observed decrease in expression in the BBM, this may be via NHERF1, as for PTH and FGF23 (Sections 7. and 8.2.). So far, no MEPE receptor has been identified in the kidneys. Circulating MEPE could associate with proximal tubular cells through attachment of the AC-100 domain to integrin(s) and carbohydrates in the basolateral membranes of the proximal tubules. This would then activate signalling via the integrin C-terminal and perhaps could lead to ser290 phosphorylation in NHERF1 by SGRK. This merits investigation. Integrins αvß3 and αvß5 are expressed in renal tubules and possible binding partners. The second partner might be a heparan-sulfate containing mucoprotein. A peptide sequence in the syndecan-1 core interacts with αvß3 and αvß5 and modulates cell adhesion [340]. An additional/alternative mechanism is direct inactivation of apical NaPi-2a by the ASARM peptide followed by internalisation, as has been reported for the reactive compound phosphonoformic acid [341]. The peptide could be released by proteolytic cleavage of MEPE in the bone or circulation, or even from MEPE synthesised in the renal tubules. It is unknown whether the highly charged ASARM peptide is filtered at the glomerulus.
11.2. sFRP4 Secreted Frizzled-Related Protein 4 (sFRP4)
sFRP4 was cloned from tumours associated with TI0 [323]. In rats, sFRP4 infusion increased urine phosphate and FEPO4 and reduced serum phosphate by PTH-independent mechanisms. Renal NaPi-2a mRNA levels were unchanged, renal beta-catenin concentration was reduced, and phosphorylated beta-catenin concentration increased. sFRP4 inhibited sodium-dependent transport of cultured OK cells [342]. However, in a study of hemangiopericytomas and various control cell lines, FGF7 and sFRP-4 were widely expressed in all studied cell lines and tissues and were not tumour-specific [343]. sFrp4 modulates extracellular signalling by soluble secreted Wingless integrated (Wnt) glycoproteins. Wnts bind to Frizzled (FZ) class F GPCRs that mediate Wnt signalling which has wide-ranging functions [344,345,346,347,348,349]. After binding to the receptor, the signal is transduced to the cytoplasmic adapter phosphoprotein dishevelled (Dsh/Dvl) which transiently recruits signalling complexes. Dsh directs the Wnt signal into at least three major cascades, canonical, and non-canonical planar cell polarity (PCP) and calcium pathways [344,345,348,350,351,352]. Canonical signalling is mediated by β-catenin and activates T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors to regulate the expression of target genes. The non-canonical pathways act independently of β-catenin. The Wnt/Ca2+ pathway activates phospholipase C, increases intracellular Ca2+ which in turn activates CamKII (Ca2+/calmodulin-dependent protein kinase II) and PKC and the nuclear transcription factors NFkB, CREB and NFAT [345,350,353].
sFRP4 has a cysteine-rich domain in the N-terminus which is 30–40% identical to the Wnt ligand-binding domain of the Frizzled receptors [347,354,355]. It binds secreted Wnts and act as a Wnt decoy receptor. This reduces signalling by both canonical and noncanonical Wnt signalling pathways [323,342,346,348,351,352]. In the rat studies, sFRP4 suppressed canonical wnt signalling in vivo [342] and by inference TCF/LEF transcription. There is no clear direct connection between this and the decreased NaPi-2a observed, although secondary disturbances in cell signalling might have been responsible. There was no increase in PTH or cAMP, to incriminate PKA activation. An alternative is that PKC was activated by non-canonical Wnt/Ca2+ signalling. This could then phosphorylate NHERF1 and inactivate NaPi-2a as described for PTH and dopamine [Section 7]. However sFRP4 would be predicted to decrease Wnt/Ca2+ signalling. It may be that sFRP4 has a weak affinity for non-canonical Wnts and does not displace them from their receptors and interrupt their signalling.
11.3. Fibroblast Growth Factor 7 (FGF7)
FGF7 (keratinocyte growth factor KGF) was identified in tumours associated with TIO [356], and FGF7 and FGF23 were increased in blood draining the tumour site of a patient with TIO [315]. FGF7 inhibited phosphate uptake by renal tubular epithelial cells in vitro [356], and increased phosphate excretion in rats [357]. However, FGF7 was expressed in a range of cell lines not associated with TIO [343]. In a cross-sectional study of patients with chronic renal failure there was no significant correlation between serum immunoreactive iFGF7 and phosphate, iFGF23, PTH or 1,25(OH)2D [358]. FGF7 expression was upregulated in cysts from kidneys of patients with autosomal dominant polycystic kidney disease. In vitro FGF7 stimulated the proliferation of cyst-lining epithelial cell by regulating the expression of cyclin D1 and P21 genes [359]. In summary, there is experimental evidence that FGF7 causes hyper-phosphaturia, but its association with TIO tumours may be as a mediator of tumour growth rather than as a phosphotonin.
FGF7 is a paracrine-acting FGF which activates the “b” isoforms of FGFR1, FGFR1b and FGFR2b and contributes to organ development [360]. The FGF receptors (FGFRs) are single-pass transmembrane tyrosine kinase receptors with an intracellular tyrosine kinase domain. Heparan sulphate glycosaminoglycans (HSGAG) expressed on the cell surface are essential co-receptors. They promote the formation of a symmetric 2:2 FGF: FGFR dimer on the cell surface and activation of the receptors [360]. The mechanism by which FGF7 causes hyper-phosphaturia has not been investigated. It might be by activating GRSK, phosphorylation of NHERF1 Ser 290 and inactivation of NaPi-2a as shown for FGF23 (Section 6.3, 6.4), with heparan sulfate substituting for klotho as co-receptor.
12. Clinical Applications of the Expanding Knowledge of Phosphate Transport
Already, knowledge of the cellular mechanisms has indicated potentially useful ways to treat conditions with disordered phosphate metabolism. Treatment with Burosomab, an FGF23 antibody is now an approved therapy for X-linked hypophosphatemic rickets and inoperable TIO, to tackle persistent hypophosphatemia due to a primary increase in FGF23 production [32]. In cystic fibrosis due to F508del CFTR, the CFTR corrector VX-809 increases the binding affinity between NHERF1 PDZ1 and the mutant protein and its membrane stability [361,362]. Perhaps small molecules with a similar action may have a place in controlling hypophosphatemia. In chronic renal failure (CRF), very high levels of FGF23 are secondary to phosphate retention and probably contribute to increased cardiovascular morbidity and mortality [31]. Here various interventions are being explored to reduce plasma phosphate in advanced CRF. Tenapanor, an agent which inhibits NHE3 and reduces paracellular phosphate absorption from the intestine [7] now has FDA approval for use in patients with end-stage renal failure receiving dialysis. The calcimimetic cinacalcet which binds to the CaSR and decreases PTH secretion reduces phosphate concentrations indirectly. Another approach under investigation is to increase FGF23 clearance by inhibiting 0-glycosylation by GALNT3, which normally protects FGF23 from proteolysis. Use of FGF23 C-terminal fragments to block interaction of FGF23 with FGFR1c-klotho and of other agents to inhibit FGFR signalling are also being considered [32]. The demonstration that PTH signalling continues after PTH/PTH1R internalisation [23] is driving research to develop a PTHrP derivative with extended activity which will enhance the benefits of intermittent treatment with abaloparatide, a human parathyroid hormone-related peptide analogue, on bone mineral density in osteoporosis [9,23,112]. An important, but less obvious, benefit of the molecular findings is that they will guide selection of genes and pathways for interrogation in the genomic data generated by on-going GWAS studies.
13. Summary and Conclusions
Years of research have established the physiology of phosphate regulation. The introduction and availability of a bewildering array of new technologies has enabled detailed exploration of the cellular mechanisms that mediate this at a molecular level. The findings are revealing an amazing, intricately regulated, system within individual cells which co-ordinates with neighbouring cellular activity. Some findings which particularly merit highlighting are: (1) the incredible versality of NHERF1 (see below), (2) the essential functions of inherently disordered protein sequences in regulating interaction of distant PDZ domains with their PDZ-binding partners, (3) the increasing evidence of the promiscuity of the PDZ/PDZ-binding partnerships, (4) the prolonged activity of PTH after endocytic uptake of the liganded PTH1R receptor, (5) the discovery that the primate isoform of RGS14 binds to NHERF1 and inhibits phosphate transport.
How does NHERF1 coordinate the simultaneous activities of a multiplicity of membrane-bound proteins? One NHERF1 PDZ domain can only bind one protein at a time, and even a raft of NHERF1 proteins could only accommodate a handful of proteins. Factors which may contribute to the explanation are first that there appears to be a large surfeit of NHERF1 relative to the amounts of competing proteins competing for binding, hence many PDZ domains may be unoccupied. Second, interactions with NHERF1 are transitory and very brief. Third, selection of a binding ligand for the PDZ domains depends on interaction of amino acid side chains in and around the NHERF1 PDZ domains with those of residues upstream of the ligand C-terminal [93]. Allosteric conformational induced by phosphorylation/ dephosphorylation of NHERF1 [11] will change the orientation of side chains of amino acids in and around the PDZ domains. This will change the selection of protein ligand bound at the PDZ domain.
Inevitably as our vision of the cellular processes involved in phosphate absorption expands, so does the list of questions/issues to resolve. Amongst them are:
- (1)
- Whether interaction of full length PTH the PTH1R receptor and G-signalling are the same as for PTH1-34 fragment.
- (2)
- Clearer definition of the signalling pathway via cAMP and PKA to NHERF1. It appears speculative at present.
- (3)
- Clearer definition of the signalling pathway of IGFR which increases renal phosphate absorption.
- (4)
- The form of PTH which normally activates proximal tubule apical PTH1R. Activation by filtered intact PTH or N-terminal PTH fragments seems an imprecise regulatory mechanism for such a finely controlled reabsorption system. How far is locally produced PTHrP involved?
- (5)
- The function of PTHrP in the proximal tubules post-natally.
- (6)
- How PTH signalling at the apical and basolateral membranes in the proximal tubule are co-ordinated
- (7)
- The location of NHERF1 at the BBM? Logically it should be in the (long) cilia close to apically-sited NaPi-2a, but findings conflict.
- (8)
- The roles of RAMPS in PTH/PTH1R signalling.
- (9)
- The function of the internal PDZ-binding motif of NaPi-2a.
- (10)
- Whether RGS14 has roles in regulating phosphate transport through inactivation of PTH/PTH1R signalling, and/or in humans through blocking NaPi-2a inactivation by PTH.
- (11)
- Whether Epac is stimulated in parallel with PKA by PTH/PTH1R. If it is, do PKA and Epac operate an activation/inhibitory partnership to regulate PTH activity?
- (12)
- The dysfunctional protein activity which is being highlighted in GWAS studies of calcium stone formers.
- (13)
- Whether MEPE has a physiological role in the kidneys.
- (14)
- The on-going research into the molecular mechanisms of renal phosphate absorption is revealing an amazingly intricate system which has evolved over millions of years. The emerging findings are providing greater insight into failures of the process, and indicating new avenues for therapeutic intervention.
Author Contributions
VW is the sole author. VW conceived the project, undertook a literature search, reviewed the publications, wrote and corrected the manuscript, and devised and drew the figures. VW personally approved the review. VW is personally accountable for the content of the review.
Funding
This review received no external funding. Processing and publication charges to be paid by VW from personal income with a small contribution from MDPI Reviewer’s vouchers (400-Swiss francs).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable. All the data used was available on the Internet.
Acknowledgments
The author would like to thank Professor Sarah Ennis for guidance with genomic issues, Professor Peter Rowe for sharing thoughts about MEPE and Dr Paul Cook for clinical discussions.
Conflicts of interest
The author declares no conflict of interest.
References
- Knochel JP. Hypophosphatemia and phosphorus deficiency. In The Kidney, 4TH ed.; Brenner BM, Rector FC, Jr., Eds.; W.B. Saunders Company, Harcourt Brace Jovanovich, Inc.: West Philadelphia, United States of America, 1991; Volume 1 Chapter 21, pp. 888-915.
- Fukumoto S. Phosphate metabolism and vitamin D. Bonekey Rep. 2014 Feb 5;3:497. [CrossRef]
- Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab. 2006 Aug;91(8):3144-9. [CrossRef]
- Berndt T, Kumar R. Phosphatonins and the regulation of phosphate homeostasis. Annu Rev Physiol. 2007;69:341-59. [CrossRef]
- Gaasbeek A, Meinders AE. Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005 Oct;118(10):1094- 101. [CrossRef]
- Marks J, Debnam ES, Unwin RJ. Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Renal Physiol. 2010 Aug;299(2):F285-96. [CrossRef]
- King AJ, Siegel M, He Y, Nie B, Wang J, Koo-McCoy S, Minassian NA, Jafri Q, Pan D, Kohler J, et al. Inhibition of sodium/hydrogen exchanger 3 in the gastrointestinal tract by tenapanor reduces paracellular phosphate permeability. Sci Transl Med. 2018 Aug 29;10(456):eaam6474. [CrossRef]
- Walker V. Phosphaturia in kidney stone formers: Still an enigma. Adv Clin Chem. 2019;90:133-96. [CrossRef]
- Bringhurst FR, Demay MB, Kronenberg HM. Hormones and Disorders of Mineral Metabolism. In Williams Textbook of Endocrinology, 14th ed.; Melmed S, Koenig R, Rosen CJ, Auchus RJ, Goldfine AB., Eds.; Elsevier: Amsterdam, Netherlands, 2019; Chapter 29, pp. 1196-1255.
- Curthoys NP, Moe OW. Proximal tubule function and response to acidosis. Clin J Am Soc Nephrol. 2014 Sep 5;9(9):1627-38. [CrossRef]
- Zhang Q, Xiao K, Paredes JM, Mamonova T, Sneddon WB, Liu H, Wang D, Li S, McGarvey JC, Uehling D, et al. Parathyroid hormone initiates dynamic NHERF1 phosphorylation cycling and conformational changes that regulate NPT2A-dependent phosphate transport. J Biol Chem. 2019 Mar 22;294(12):4546-71. [CrossRef]
- Levi M, Gratton E, Forster IC, Hernando N, Wagner CA, Biber J, Sorribas V, Murer H. Mechanisms of phosphate transport. Nat Rev Nephrol. 2019 Aug;15(8):482-500. [CrossRef]
- Wagner CA. The basics of phosphate metabolism -Nephrol Dial Transplant. 2023 Sep 2: gfad188. doi: 0.1093/ndt/gfad188.
- Silverman M, Turner RJ (1979). The Renal Proximal Tubule. In: Manson, L.A. (eds) Biomembranes. Biomembranes, vol 10. Springer, Boston, MA. [CrossRef]
- Wagner CA, Rubio-Aliaga I, Biber J, Hernando N. Genetic diseases of renal phosphate handling. Nephrol Dial Transplant. 2014 Sep;29 Suppl 4:iv45-54. [CrossRef]
- Wagner CA, Rubio-Aliaga I, Hernando N. Renal phosphate handling and inherited disorders of phosphate reabsorption: an update. Pediatr Nephrol. 2019 Apr;34(4):549-59. [CrossRef]
- Alizadeh Naderi AS, Reilly RF. Hereditary disorders of renal phosphate wasting. Nat Rev Nephrol. 2010 Nov;6(11):657-65. [CrossRef]
- Prié D, Friedlander G. Genetic disorders of renal phosphate transport. N Engl J Med. 2010 Jun 24;362(25):2399-409. [CrossRef]
- Gohil A, Imel EA. FGF23 and Associated Disorders of Phosphate Wasting. Pediatr Endocrinol Rev. 2019 Sep;17(1):17-34. [CrossRef]
- Sayer JA. Progress in Understanding the Genetics of Calcium-Containing Nephrolithiasis. J Am Soc Nephrol. 2017 Mar;28(3):748-59. [CrossRef]
- Walker V, Stansbridge EM, Griffin DG. Demography and biochemistry of 2800 patients from a renal stones clinic. Ann Clin Biochem. 2013 Mar;50(Pt 2):127-39. [CrossRef]
- Prié D, Ravery V, Boccon-Gibod L, Friedlander G. Frequency of renal phosphate leak among patients with calcium nephrolithiasis. Kidney Int. 2001 Jul;60(1):272-6. [CrossRef]
- Vilardaga JP, Clark LJ, White AD, Sutkeviciute I, Lee JY, Bahar I. Molecular Mechanisms of PTH/PTHrP Class B GPCR Signaling and Pharmacological Implications. Endocr Rev. 2023 May 8;44(3):474-91. [CrossRef]
- Murer H, Biber J, Forster IC, Werner A. Phosphate transport: from microperfusion to molecular cloning. Pflugers Arch. 2019 Jan;471(1):1-6. [CrossRef]
- Wagner CA. Coming out of the PiTs-novel strategies for controlling intestinal phosphate absorption in patients with CKD . Kidney Int 2020; 98 :273–5. [CrossRef]
- Wagner CA. Pharmacology of mammalian Na + -dependent transporters of inorganic phosphate. Handb Exp Pharmacol 2023;. [CrossRef]
- Montanari A, Pirini MG, Lotrecchiano L, Di Prinzio L, Zavatta G. Phosphaturic mesenchymal tumors with or without Phosphate Metabolism Derangements. Curr Oncol. 2023 Aug 8;30(8):7478-88. [CrossRef]
- Rowe PS, McCarthy EM, Yu AL, Stubbs JR. Correction of vascular calcification and hyperphosphatemia in CKD Rats treated with ASARM peptide. Kidney360. 2022 Aug 30;3(10):1683-98. [CrossRef]
- Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, et al. Isolated C- terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A. 2010 Jan 5;107(1):407-12. [CrossRef]
- Doshi SM, Wish JB. Past, present, and future of phosphate management. Kidney Int Rep 2022; 7: 688–98. [CrossRef]
- Verbueken D, Moe OW. Strategies to lower fibroblast growth factor 23 bioactivity. Nephrol Dial Transplant. 2022 Sep 22;37(10):1800-07. [CrossRef]
- Marques JVO, Moreira CA, Borba VZC. New treatments for rare bone diseases: hypophosphatemic rickets/osteomalacia. Arch Endocrinol Metab. 2022 Nov 11;66(5):658-65. [CrossRef]
- Zhang MY, Ranch D, Pereira RC, Armbrecht HJ, Portale AA, Perwad F. Chronic inhibition of ERK1/2 signaling improves disordered bone and mineral metabolism in hypophosphatemic (Hyp) mice. Endocrinology. 2012 Apr;153(4):1806-16. [CrossRef]
- Biber J, Hernando N, Forster I, Murer H. Regulation of phosphate transport in proximal tubules. Pflugers Arch. 2009 May;458(1):39-52. [CrossRef]
- Tisher CC, Madsen KM. Anatomy of the kidney. In The Kidney, 4TH ed.; Brenner BM, Rector FC, Jr., Eds.; W.B. Saunders Company, Harcourt Brace Jovanovich, Inc.: West Philadelphia, United States of America, 1991; Volume 1 Chapter 1, pp. 3-75 (proximal tubule pp. 22-35).
- McDonough AA. Motoring down the microvilli. Focus on "PTH-induced internalization of apical membrane NaPi2a: role of actin and myosin VI". Am J Physiol Cell Physiol. 2009 Dec;297(6):C1331-2. [CrossRef]
- Blaine J, Okamura K, Giral H, Breusegem S, Caldas Y, Millard A, Barry N, Levi M. PTH-induced internalization of apical membrane NaPi2a: role of actin and myosin VI. Am J Physiol Cell Physiol. 2009 Dec;297(6):C1339-46. [CrossRef]
- Schuh CD, Polesel M, Platonova E, Haenni D, Gassama A, Tokonami N, Ghazi S, Bugarski M, Devuyst O, Ziegler U, et al. Combined Structural and Functional Imaging of the Kidney Reveals Major Axial Differences in Proximal Tubule Endocytosis. J Am Soc Nephrol. 2018 Nov;29(11):2696-2712. [CrossRef]
- Maddox DA, Gennari FJ. The early proximal tubule: a high-capacity delivery-responsive reabsorptive site. Am J Physiol. 1987 Apr;252(4 Pt 2):F573-84. [CrossRef]
- DuBose TD Jr, Pucacco LR, Lucci MS, Carter NW. Micropuncture determination of pH, PCO2, and total CO2 concentration in accessible structures of the rat renal cortex. J Clin Invest. 1979 Aug;64(2):476-82. [CrossRef]
- Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol. 2015 Nov;26(11):2669-77. [CrossRef]
- Hato T, Winfree S, Day R, Sandoval RM, Molitoris BA, Yoder MC, Wiggins RC, Zheng Y, Dunn KW, Dagher PC. Two-Photon Intravital Fluorescence Lifetime Imaging of the Kidney Reveals Cell-Type Specific Metabolic Signatures. J Am Soc Nephrol. 2017 Aug;28(8):2420-30. [CrossRef]
- Custer M, Lötscher M, Biber J, Murer H, Kaissling B. Expression of Na-P(i) cotransport in rat kidney: localization by RT-PCR and immunohistochemistry. Am J Physiol. 1994 May;266(5 Pt 2):F767-74. [CrossRef]
- Virkki LV, Biber J, Murer H, Forster IC. Phosphate transporters: a tale of two solute carrier families. Am J Physiol Renal Physiol. 2007 Sep;293(3):F643-54. [CrossRef]
- Forster IC, Hernando N, Biber J, Murer H. Phosphate transport kinetics and structure-function relationships of SLC34 and SLC20 proteins. Curr Top Membr. 2012;70:313-56. [CrossRef]
- Biber J, Hernando N, Forster I. Phosphate transporters and their function. Annu Rev Physiol. 2013;75:535-50. [CrossRef]
- Lederer E. Renal phosphate transporters. Curr Opin Nephrol Hypertens. 2014 Sep;23(5):502-6. [CrossRef]
- Carpenter TO. Primary Disorders of Phosphate Metabolism. [Updated 2022 Jun 8]. In: Feingold KR, Anawalt B, Blackman MR, et al., eds. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279172/.
- Moser SO, Haykir B, Küng CJ, Bettoni C, Hernando N, Wagner CA. Expression of phosphate and calcium transporters and their regulators in parotid glands of mice. Pflugers Arch. 2023 Feb;475(2):203-16. [CrossRef]
- Villa-Bellosta R, Ravera S, Sorribas V, Stange G, Levi M, Murer H, Biber J, Forster IC. The Na+-Pi cotransporter PiT-2 (SLC20A2) is expressed in the apical membrane of rat renal proximal tubules and regulated by dietary Pi. Am J Physiol Renal Physiol. 2009 Apr;296(4):F691-9. [CrossRef]
- Magagnin S, Werner A, Markovich D, Sorribas V, Stange G, Biber J, Murer H. Expression cloning of human and rat renal cortex Na/Pi cotransport. Proc Natl Acad Sci U S A. 1993 Jul 1;90(13):5979-83. [CrossRef]
- Forster IC. The molecular mechanism of SLC34 proteins: insights from two decades of transport assays and structure-function studies. Pflugers Arch. 2019 Jan;471(1):15-42. [CrossRef]
- Fenollar-Ferrer C, Patti M, Knöpfel T, Werner A, Forster IC, Forrest LR. Structural fold and binding sites of the human Na⁺- phosphate cotransporter NaPi-II. Biophys J. 2014 Mar 18;106(6):1268-79. [CrossRef]
- de La Horra C, Hernando N, Forster I, Biber J, Murer H. Amino acids involved in sodium interaction of murine type II Na(+)-P(i) cotransporters expressed in Xenopus oocytes. J Physiol. 2001 Mar 1;531(Pt 2):383-91. [CrossRef]
- Murer H. Functional domains in the renal type IIa Na/P(i)-cotransporter. Kidney Int. 2002 Aug;62(2):375-82. [CrossRef]
- Shenolikar S, Voltz JW, Cunningham R, Weinman EJ. Regulation of ion transport by the NHERF family of PDZ proteins. Physiology (Bethesda). 2004 Dec;19:362-9. [CrossRef]
- Forster I, Hernando N, Biber J, Murer H. The voltage dependence of a cloned mammalian renal type II Na+/Pi cotransporter (NaPi-2). J Gen Physiol. 1998 Jul;112(1):1-18. [CrossRef]
- Busch A, Waldegger S, Herzer T, Biber J, Markovich D, Hayes G, Murer H, Lang F. Electrophysiological analysis of Na+/Pi cotransport mediated by a transporter cloned from rat kidney and expressed in Xenopus oocytes. Proc Natl Acad Sci U S A. 1994 Aug 16;91(17):8205-8. [CrossRef]
- Werner A, Patti M, Hany S Zinad HS, Fearn A, Laude A, Forster I. Molecular determinants of transport function in zebrafish Slc34a Na-phosphate transporters. Am J Physiol Regul Integr Comp Physiol. 2016 Dec;311(6):R1213-22. [CrossRef]
- Patti M, Fenollar-Ferrer C, Werner A, Forrest LR, Forster IC. Cation interactions and membrane potential induce conformational changes in NaPi-IIb. Biophys J. 2016 Sep 6;111(5):973-88. [CrossRef]
- Inoue M, Digman MA, Cheng M, Breusegem SY, Halaihel N, Sorribas V, Mantulin WW, Gratton E, Barry NP, Levi M. Partitioning of NaPi cotransporter in cholesterol-, sphingomyelin-, and glycosphingolipid-enriched membrane domains modulates NaPi protein diffusion, clustering, and activity. J Biol Chem. 2004 Nov 19;279(47):49160-71. [CrossRef]
- Levi M, Baird BM, Wilson PV. Cholesterol modulates rat renal brush border membrane phosphate transport. J Clin Invest. 1990 Jan;85(1):231-7. [CrossRef]
- Alcalde AI, Sarasa M, Raldúa D, Aramayona J, Morales R, Biber J, Murer H, Levi M, Sorribas V. Role of thyroid hormone in regulation of renal phosphate transport in young and aged rats. Endocrinology. 1999 Apr;140(4):1544-51. [CrossRef]
- Sorribas V, Lötscher M, Loffing J, Biber J, Kaissling B, Murer H, Levi M. Cellular mechanisms of the age-related decrease in renal phosphate reabsorption. Kidney Int. 1996 Sep;50(3):855-63. [CrossRef]
- Breusegem SY, Takahashi H, Giral-Arnal H, Wang X, Jiang T, Verlander JW, Wilson P, Miyazaki-Anzai S, Sutherland E, Caldas Y, eta al. Differential regulation of the renal sodium-phosphate cotransporters NaPi-IIa, NaPi-IIc, and PiT-2 in dietary potassium deficiency. Am J Physiol Renal Physiol. 2009 Aug;297(2):F350-61. [CrossRef]
- Zajicek HK, Wang H, Puttaparthi K, Halaihel N, Markovich D, Shayman J, Béliveau R, Wilson P, Rogers T, Levi M. Glycosphingolipids modulate renal phosphate transport in potassium deficiency. Kidney Int. 2001 Aug;60(2):694-704. [CrossRef]
- Weinman EJ, Steplock D, Shenolikar S, Blanpied TA. Dynamics of PTH-induced disassembly of Npt2a/NHERF-1 complexes in living OK cells. Am J Physiol Renal Physiol. 2011 Jan;300(1):F231-5. [CrossRef]
- Segawa H, Kaneko I, Takahashi A, Kuwahata M, Ito M, Ohkido I, Tatsumi S, Miyamoto K (2002) Growth-related renal type II Na/Pi cotransporter. J Biol Chem 277:19665–72 . [CrossRef]
- Ohkido I, Segawa, Hiroko & Yanagida, R & Nakamura, M & Miyamoto, K. (2003). Cloning, gene structure and dietary regulation of the type-IIc Na/Pi cotransporter in the mouse kidney. Pflügers Archiv: European journal of physiology. 446. 106-15. 10.1007/s00424-003-1010-6.
- Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U S A. 1998 Apr 28;95(9):5372-7. [CrossRef]
- Segawa H, Onitsuka A, Furutani J, Kaneko I, Aranami F, Matsumoto N, Tomoe Y, Kuwahata M, Ito M, Matsumoto M, Li M, Amizuka N, Miyamoto K. Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am J Physiol Renal Physiol. 2009 Sep;297(3):F671-8. [CrossRef]
- Jaureguiberry G, Carpenter TO, Forman S, Jüppner H, Bergwitz C. A novel missense mutation in SLC34A3 that causes hereditary hypophosphatemic rickets with hypercalciuria in humans identifies threonine 137 as an important determinant of sodium-phosphate cotransport in NaPi-IIc. Am J Physiol Renal Physiol. 2008 Aug;295(2):F371-9. doi: 0.1152/ajprenal.00090.2008.
- Gordon RJ, Li D, Doyle D, Zaritsky J, Levine MA. Digenic heterozygous mutations in SLC34A3 and SLC34A1 cause dominant hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab. 2020 Jul 1;105(7):2392–400. [CrossRef]
- Forster IC, Hernando N, Biber J, Murer H Phosphate transporters of the SLC20 and SLC34 families. Molecular Aspects of Medicine 2013; 34(2-3): 386-95. [CrossRef]
- Nowik M, Picard N, Stange G, Capuano P, Tenenhouse HS, Biber J, Murer H, Wagner CA. Renal phosphaturia during metabolic acidosis revisited: molecular mechanisms for decreased renal phosphate reabsorption Pflugers Arch. 2008 Nov;457(2):539-49. [CrossRef]
- Giral H, Lanzano L, Caldas Y, Blaine J, Verlander JW, Lei T, Gratton E, Levi M. Role of PDZ domain containing 1 (PDZK1) in apical membrane expression of renal Na-coupled phosphate (Na/Pi) transporters. J Biol Chem 2011; 286: 15032–42. [CrossRef]
- Segawa H, Kaneko I, Shiozaki Y, Ito M, Tatsumi S, Miyamoto K-i. Molecular control of growth-related sodium-phosphate co- transporter (SLC34A3). Current Molecular Biology Reports 2019; 5: 26-33. 10.1007/s40610-019-0112-7.
- Segawa H, Yamanaka S, Ito M, Kuwahata M, Shono M, Yamamoto T, Miyamoto K. Internalization of renal type IIc Na-Pi cotransporter in response to a high-phosphate diet. Am J Physiol Renal Physiol. 2005 Mar;288(3):F587-96. [CrossRef]
- Hori M, Shimizu Y, Fukumoto S. Minireview: fibroblast growth factor 23 in phosphate homeostasis and bone metabolism. Endocrinology. 2011 Jan;152(1):4-10. [CrossRef]
- Tomoe Y, Segawa H, Shiozawa K, Kaneko I, Tominaga R, Hanabusa E, Aranami F, Furutani J, Kuwahara S, Tatsumi S, et al. Phosphaturic action of fibroblast growth factor 23 in Npt2 null mice. Am J Physiol Renal Physiol. 2010 Jun;298(6):F1341-50. [CrossRef]
- Fujii T, Shiozaki Y, Segawa H, Nishiguchi S, Hanazaki A, Noguchi M, Kirino R, Sasaki S, Tanifuji K, Koike M,et al. Analysis of opossum kidney NaPi-IIc sodium-dependent phosphate transporter to understand Pi handling in human kidney. Clin Exp Nephrol. 2019 Mar;23(3):313-24. [CrossRef]
- Bergwitz C, Miyamoto KI. Hereditary hypophosphatemic rickets with hypercalciuria: pathophysiology, clinical presentation, diagnosis and therapy. Pflugers Arch. 2019 Jan;471(1):149-63. [CrossRef]
- Picard N, Capuano P, Stange G, Mihailova M, Kaissling B, Murer H, Biber J, Wagner CA. Acute parathyroid hormone differentially regulates renal brush border membrane phosphate cotransporters. Pflugers Arch. 2010 Aug;460(3):677-87. [CrossRef]
- Weinman EJ, Steplock D, Shenolikar S. CAMP-mediated inhibition of the renal brush border membrane Na+-H+ exchanger requires a dissociable phosphoprotein cofactor. J Clin Invest. 1993 Oct;92(4):1781-6. [CrossRef]
- Weinman EJ, Shenolikar S. The Na-H exchanger regulatory factor. Exp Nephrol 1997 5(6):449-52. PMID: 9438172.
- Reczek D, Berryman M, Bretscher A. Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J Cell Biol. 1997 Oct 6;139(1):169-79. [CrossRef]
- Ardura JA, Friedman PA. Regulation of G protein-coupled receptor function by Na+/H+ exchange regulatory factors. Pharmacol Rev. 2011 Dec;63(4):882-900. [CrossRef]
- Hernando N, Gisler SM, Pribanic S, Déliot N, Capuano P, Wagner CA, Moe OW, Biber J, Murer H. NaPi-IIa and interacting partners. J Physiol. 2005 Aug 15;567(Pt 1):21-6. [CrossRef]
- Bhattacharya S, Stanley CB, Heller WT, Friedman PA, Bu Z. Dynamic structure of the full-length scaffolding protein NHERF1 influences signaling complex assembly. J Biol Chem. 2019 Jul 19;294(29):11297-310. [CrossRef]
- Hernando N, Wagner CA, Gisler SM, Biber J, Murer H. Hernando N, Wagner CA, Gisler SM, Biber J, Murer H. PDZ proteins and proximal ion transport. Curr Opin Nephrol Hypertens. 2004 Sep;13(5):569-74. [CrossRef]
- He J, Bellini M, Inuzuka H, Xu J, Xiong Y, Yang X, Castleberry AM, Hall RA. Proteomic analysis of beta1-adrenergic receptor interactions with PDZ scaffold proteins. J Biol Chem. 2006 Feb 3;281(5):2820-7. [CrossRef]
- Hall RA, Premont RT, Chow CW, Blitzer JT, Pitcher JA, Claing A, Stoffel RH, Barak LS, Shenolikar S, Weinman EJ, et al. The beta2-adrenergic receptor interacts with the Na+/H+-exchanger regulatory factor to control Na+/H+ exchange. Nature. 1998 Apr 9;392(6676):626-30. [CrossRef]
- Mamonova T, Friedman PA. Noncanonical Sequences Involving NHERF1 Interaction with NPT2A Govern Hormone-Regulated Phosphate Transport: Binding Outside the Box. Int J Mol Sci. 2021 Jan 22;22(3):1087. [CrossRef]
- Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol. 2012 Aug 1;303(3):F321-7. [CrossRef]
- Vistrup-Parry M, Sneddon WB, Bach S, Strømgaard K, Friedman PA, Mamonova T. Multisite NHERF1 phosphorylation controls GRK6A regulation of hormone-sensitive phosphate transport. J Biol Chem. 2021 Jan-Jun;296:100473. [CrossRef]
- Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, Silve C, Planelles G, Urena-Torres P, Grandchamp B, et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med. 2008 Sep 11;359(11):1128-35. [CrossRef]
- Li J, Callaway DJ, Bu Z. Ezrin induces long-range interdomain allostery in the scaffolding protein NHERF1. J Mol Biol. 2009 Sep 11;392(1):166-80. [CrossRef]
- Farago B, Li J, Cornilescu G, Callaway DJ, Bu Z. Activation of nanoscale allosteric protein domain motion revealed by neutron spin echo spectroscopy. Biophys J. 2010 Nov 17;99(10):3473-82. [CrossRef]
- Weinman EJ, Steplock D, Zhang Y, Biswas R, Bloch RJ, Shenolikar S. Cooperativity between the phosphorylation of Thr95 and Ser77 of NHERF-1 in the hormonal regulation of renal phosphate transport. J Biol Chem. 2010 Aug 13;285(33):25134-8. [CrossRef]
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012 Jan;92(1):131-55. [CrossRef]
- Kumar R, Thompson JR. The regulation of parathyroid hormone secretion and synthesis. J Am Soc Nephrol. 2011 Feb;22(2):216-24. [CrossRef]
- Naveh-Many T, A Sela-Brown, J Silver, Protein-RNA interactions in the regulation of PTH gene expression by calcium and phosphate. Nephrol. Dial. Transplant. 1999; 14 : 811–13. [CrossRef]
- Almaden Y, Canalejo A, Hernandez A, Ballesteros E, Garcia-Navarro S, Torres A, Rodriguez M. Direct effect of phosphorus on PTH secretion from whole rat parathyroid glands in vitro. J Bone Miner Res. 1996 Jul;11(7):970-6. [CrossRef]
- Taketani Y, Segawa H, Chikamori M, Morita K, Tanaka K, Kido S, Yamamoto H, Iemori Y, Tatsumi S, Tsugawa N, et al. Regulation of type II renal Na+-dependent inorganic phosphate transporters by 1,25-dihydroxyvitamin D3. Identification of a vitamin D-responsive element in the human NAPi-3 gene. J Biol Chem. 1998 Jun 5;273(23):14575-81. [CrossRef]
- Silver J, Naveh-Many T. FGF23 and the parathyroid. Adv Exp Med Biol. 2012;728:92-9. [CrossRef]
- Naveh-Many T, Silver J. The Pas de Trois of Vitamin D, FGF23, and PTH. J Am Soc Nephrol. 2017 Feb;28(2):393-5. [CrossRef]
- Chanakul A, Zhang MY, Louw A, Armbrecht HJ, Miller WL, Portale AA, Perwad F. FGF-23 regulates CYP27B1 transcription in the kidney and in extra-renal tissues. PLoS One. 2013 Sep 3;8(9):e72816. [CrossRef]
- Maeda S, Sutliff RL, Qian J, Lorenz JN, Wang J, Tang H, Nakayama T, Weber C, Witte D, Strauch AR, et al. Targeted overexpression of parathyroid hormone-related protein (PTHrP) to vascular smooth muscle in transgenic mice lowers blood pressure and alters vascular contractility. Endocrinology. 1999 Apr;140(4):1815-25. [CrossRef]
- Strewler GJ. The physiology of parathyroid hormone-related protein. N Engl J Med. 2000 Jan 20;342(3):177-85. [CrossRef]
- Pioszak AA, Parker NR, Gardella TJ, Xu HE. Structural basis for parathyroid hormone-related protein binding to the.
- parathyroid hormone receptor and design of conformation-selective peptides. J Biol Chem. 2009 Oct 9;284(41):28382-91. [CrossRef]
- Ehrenmann J, Schöppe J, Klenk C, Plückthun A. New views into class B GPCRs from the crystal structure of PTH1R. FEBS J. 2019 Dec;286(24):4852-60. [CrossRef]
- Zhai X, Mao C, Shen Q, Zang S, Shen DD, Zhang H, Chen Z, Wang G, Zhang C, Zhang Y,et al. Molecular insights into the distinct signaling duration for the peptide-induced PTH1R activation. Nat Commun. 2022 Oct 21;13(1):6276. [CrossRef]
- Alexander RT, Dimke H. Effects of parathyroid hormone on renal tubular calcium and phosphate handling. Acta Physiol (Oxf). 2023 May;238(1):e13959. doi: 10.1111/apha.13959.Traebert M, Völkl H, Biber J, Murer H, Kaissling B. Luminal and contraluminal action of 1-34 and 3-34 PTH peptides on renal type IIa Na-P(i) cotransporter. Am J Physiol Renal Physiol. 2000 May;278(5):F792-8. doi: 10.1152/ajprenal.2000.278.5.F792.
- Weinman EJ, Lederer ED. PTH-mediated inhibition of the renal transport of phosphate. Exp Cell Res. 2012 May 15;318(9):1027-32. [CrossRef]
- Klenk C, Hommers L, Lohse MJ. Proteolytic cleavage of the extracellular domain affects signaling of parathyroid hormone 1 receptor. Front Endocrinol (Lausanne). 2022 Feb 22;13:839351. [CrossRef]
- Cary BP, Zhang X, Cao J, Johnson RM, Piper SJ, Gerrard EJ, Wootten D, Sexton PM. New Insights into the Structure and Function of Class B1 GPCRs. Endocr Rev. 2023 May 8;44(3):492-517. [CrossRef]
- Lee SM, Jeong Y, Simms J, Warner ML, Poyner DR, Chung KY, Pioszak AA. Calcitonin Receptor N-Glycosylation Enhances Peptide Hormone Affinity by Controlling Receptor Dynamics. J Mol Biol. 2020 Mar 27;432(7):1996-2014. [CrossRef]
- Bisello A, Greenberg Z, Behar V, Rosenblatt M, Suva LJ, Chorev M. Role of glycosylation in expression and function of the human parathyroid hormone/parathyroid hormone-related protein receptor. Biochemistry. 1996 Dec 10;35(49):15890-5. [CrossRef]
- Harmar AJ. Family-B G-protein-coupled receptors. Genome Biol. 2001;2(12):REVIEWS3013. [CrossRef]
- Gardella TJ, Luck MD, Fan MH, Lee C. Transmembrane residues of the parathyroid hormone (PTH)/PTH-related peptide receptor that specifically affect binding and signaling by agonist ligands. J Biol Chem. 1996;271(22):12820–25. [CrossRef]
- Sheikh SP, Vilardarga JP, Baranski TJ, Lichtarge O, Iiri T, Meng EC, Nissenson RA, Bourne HR. Similar structures and shared switch mechanisms of the beta2-adrenoceptor and the parathyroid hormone receptor. Zn(II) bridges between helices III and VI block activation. J Biol Chem. 1999 Jun 11;274(24):17033-41. [CrossRef]
- Fernandez-Leiro R, Scheres SH. Unravelling biological macromolecules with cryo-electron microscopy. Nature. 2016 Sep 15;537(7620):339-46. [CrossRef]
- Nemec K, Schihada H, Kleinau G, Zabel U, Grushevskyi EO, Scheerer P, Lohse MJ, Maiellaro I. Functional modulation of PTH1R activation and signaling by RAMP2. Proc Natl Acad Sci U S A. 2022 Aug 9;119(32):e2122037119. [CrossRef]
- Vilardaga JP, Bünemann M, Krasel C, Castro M, Lohse MJ. Measurement of the millisecond activation switch of G protein- coupled receptors in living cells. Nat Biotechnol. 2003 Jul;21(7):807-12. [CrossRef]
- Castro M, Nikolaev VO, Palm D, Lohse MJ, Vilardaga JP. Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc Natl Acad Sci U S A. 2005 Nov 1;102(44):16084-9. [CrossRef]
- Syme CA, Friedman PA, Bisello A. Parathyroid hormone receptor trafficking contributes to the activation of extracellular signal-regulated kinases but is not required for regulation of cAMP signaling. J Biol Chem. 2005 Mar 25;280(12):11281-8. [CrossRef]
- Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, et al. Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006 Apr 21;281(16):10856-64. [CrossRef]
- Vilardaga JP, Romero G, Feinstein TN, Wehbi VL. Kinetics and dynamics in the G protein-coupled receptor signaling cascade. Methods Enzymol. 2013; 522, 337–63. [CrossRef]
- Wootten D, Miller LJ, Koole C, Christopoulos A, Sexton PM. Allostery and Biased Agonism at Class B G Protein-Coupled Receptors. Chem Rev. 2017 Jan 11;117(1):111-38. [CrossRef]
- Yuan L, Barbash S, Kongsamut S, Eishingdrelo A, Sakmar TP, Eishingdrelo H. 14-3-3 signal adaptor and scaffold proteins mediate GPCR trafficking. Sci Rep. 2019 Aug 1;9(1):11156. [CrossRef]
- Dicker F, Quitterer U, Winstel R, Honold K, Lohse MJ. Phosphorylation-independent inhibition of parathyroid hormone receptor signaling by G protein-coupled receptor kinases. Proc Natl Acad Sci U S A. 1999 May 11;96(10):5476-81. [CrossRef]
- Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, Gardella TJ, Vilardaga JP. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009 Oct;5(10):734-42. [CrossRef]
- Maeda A, Okazaki M, Baron DM, Dean T, Khatri A, Mahon M, Segawa H, Abou-Samra AB, Jüppner H, Bloch KD, et al. Critical role of parathyroid hormone (PTH) receptor-1 phosphorylation in regulating acute responses to PTH. Proc Natl Acad Sci U S A. 2013 Apr 9;110(15):5864-9. [CrossRef]
- Cheloha RW, Gellman SH, Vilardaga JP, Gardella TJ. PTH receptor-1 signalling-mechanistic insights and therapeutic prospects. Nat Rev Endocrinol. 2015 Dec;11(12):712-24. [CrossRef]
- Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov. 2004 Sep;3(9):801-8. [CrossRef]
- Lee M, Partridge NC. Parathyroid hormone signaling in bone and kidney. Curr Opin Nephrol Hypertens. 2009 Jul;18(4):298- 302. [CrossRef]
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006 Sep;5(9):730-9. [CrossRef]
- Tawfeek HA, Abou-Samra AB. Negative regulation of parathyroid hormone (PTH)-activated phospholipase C by PTH/PTH- related peptide receptor phosphorylation and protein kinase A. Endocrinology. 2008 Aug;149(8):4016-23. [CrossRef]
- Wehbi VL, Stevenson HP, Feinstein TN, Calero G, Romero G, Vilardaga JP. Noncanonical GPCR signaling arising from a PTH receptor-arrestin-Gβγ complex. Proc Natl Acad Sci U S A. 2013 Jan 22;110(4):1530-5. [CrossRef]
- Feinstein TN, Wehbi VL, Ardura JA, Wheeler DS, Ferrandon S, Gardella TJ, Vilardaga JP. Retromer terminates the generation of cAMP by internalized PTH receptors. Nat Chem Biol. 2011 May;7(5):278-84. [CrossRef]
- Jean-Alphonse FG, Wehbi VL, Chen J, Noda M, Taboas JM, Xiao K, Vilardaga JP. β2-adrenergic receptor control of endosomal PTH receptor signaling via Gβγ. Nat Chem Biol. 2017 Mar;13(3):259-61. [CrossRef]
- White AD, Jean-Alphonse FG, Fang F, Peña KA, Liu S, König GM, Inoue A, Aslanoglou D, Gellman SH, Kostenis E, Xiao K, Vilardaga JP. Gq/11-dependent regulation of endosomal cAMP generation by parathyroid hormone class B GPCR. Proc Natl Acad Sci U S A. 2020 Mar 31;117(13):7455-60. [CrossRef]
- Gidon A, Al-Bataineh MM, Jean-Alphonse FG, Stevenson HP, Watanabe T, Louet C, Khatri A, Calero G, Pastor-Soler NM, Gardella TJ, et al. Endosomal GPCR signaling turned off by negative feedback actions of PKA and v-ATPase. Nat Chem Biol. 2014 Sep;10(9):707-9. [CrossRef]
- Noda H, Okazaki M, Joyashiki E, Tamura T, Kawabe Y, Khatri A, Jueppner H, Potts JT Jr, Gardella TJ, Shimizu M. Optimization of PTH/PTHrP Hybrid Peptides to Derive a Long-Acting PTH Analog (LA-PTH). JBMR Plus. 2020 May 30;4(7):e10367. [CrossRef]
- Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai). 2008 Jul;40(7):651-62. [CrossRef]
- Bouvet M, Blondeau J-P, Lezoualc’h F. The Epac1 protein: pharmacological modulators, cardiac signalosome and pathophysiology. Cells 2019, 8: 1543. [CrossRef]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide- exchange factor directly activated by cyclic AMP. Nature. 1998 Dec 3;396(6710):474-7. [CrossRef]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998 Dec 18;282(5397):2275-9. [CrossRef]
- Tomilin VN, Pochynyuk O. A peek into Epac physiology in the kidney. Am J Physiol Renal Physiol 2019 327; F1094-97. [CrossRef]
- Honegger KJ, Capuano P, Winter C, Bacic D, Stange G, Wagner CA, Biber J, Murer H, Hernando N. Regulation of sodium- proton exchanger isoform 3 (NHE3) by PKA and exchange protein directly activated by cAMP (EPAC). Proc Natl Acad Sci U S A. 2006 Jan 17;103(3):803-8. [CrossRef]
- Li Y, Konings IB, Zhao J, Price LS, de Heer E, Deen PM. Renal expression of exchange protein directly activated by cAMP (Epac) 1 and 2. Am J Physiol Renal Physiol 295: F525–33, 2008. [CrossRef]
- Lee K. Epac: new emerging cAMP-binding protein. BMB Rep. 2021 Mar;54(3):149-56. [CrossRef]
- de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL. Mechanism of regulation of the Epac family of cAMP- dependent RapGEFs. J Biol Chem. 2000;275:20829–36. [CrossRef]
- Frische EW, Zwartkruis FJ. Rap1, a mercenary among the Ras-like GTPases. Dev Biol. 2010; 340: 1–9. [CrossRef]
- Cherezova A, Tomilin V, Buncha V, Zaika O, Ortiz PA, Mei F, Cheng X, Mamenko M, Pochynyuk O. Urinary concentrating defect in mice lacking Epac1 or Epac2. FASEB J 33: 2156–70, 2019. [CrossRef]
- Friedman PA, Sneddon WB, Mamonova T, Montanez-Miranda C, Ramineni S, Harbin NH, Squires KE, Gefter JV, Magyar CE, Emlet DR, et al. RGS14 regulates PTH- and FGF23-sensitive NPT2A-mediated renal phosphate uptake via binding to the NHERF1 scaffolding protein. J Biol Chem. 2022 May;298(5):101836. [CrossRef]
- Turan S, Bastepe M. The GNAS complex locus and human diseases associated with loss-of-function mutations or epimutations within this imprinted gene. Horm Res Paediatr. 2013;80(4):229-41. [CrossRef]
- Lee JH, Davaatseren M, Lee S. Rare PTH Gene Mutations Causing Parathyroid Disorders: A Review. Endocrinol Metab (Seoul). 2020 Mar;35(1):64-70. [CrossRef]
- Lemos MC, Thakker RV. GNAS mutations in Pseudohypoparathyroidism type 1a and related disorders. Hum Mutat. 2015 Jan;36(1):11-9. [CrossRef]
- Yu S, Yu D, Lee E, Eckhaus M, Lee R, Corria Z, Accili D, Westphal H, Weinstein LS. Variable and tissue-specific hormone resistance in heterotrimeric Gs protein alpha-subunit (Gsalpha) knockout mice is due to tissue-specific imprinting of the gsalpha gene. Proc Natl Acad Sci U S A. 1998 Jul 21;95(15):8715-20. [CrossRef]
- Bastepe M, Raas-Rothschild A, Silver J, Weissman I, Wientroub S, Jüppner H, Gillis D. A form of Jansen’s metaphyseal chondrodysplasia with limited metabolic and skeletal abnormalities is caused by a novel activating parathyroid hormone (PTH)/PTH-related peptide receptor mutation. J Clin Endocrinol Metab. 2004 Jul;89(7):3595-600. [CrossRef]
- Schipani E, Kruse K, Jüppner H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995 Apr 7;268(5207):98-100. [CrossRef]
- Savoldi G, Izzi C, Signorelli M, Bondioni MP, Romani C, Lanzi G, Moratto D, Verdoni L, Pinotti M, Prefumo F, et al. Prenatal presentation and postnatal evolution of a patient with Jansen metaphyseal dysplasia with a novel missense mutation in PTH1R. Am J Med Genet A. 2013 Oct;161A(10):2614-9. [CrossRef]
- Parkinson DB, Thakker RV. A donor splice site mutation in the parathyroid hormone gene is associated with autosomal recessive hypoparathyroidism. Nat Genet. 1992 May;1(2):149-52. [CrossRef]
- Chase LR, Melson GL, Aurbach GD. Pseudohypoparathyroidism: defective excretion of 3’,5’-AMP in response to parathyroid hormone. J Clin Invest. 1969 Oct;48(10):1832-44. [CrossRef]
- Albright F, Burnett CH, Smith PH, Parson W. Pseudohypoparathyroidism – An example of ‘Seabright-Bantam syndrome’ Endocrinology. 1942; 30: 922–32.
- Ines Armando, Van Anthony M. Villar, and Pedro A. Jose. Dopamine and Renal Function and Blood Pressure Regulation Comp Physiol. 2011 July; 1(3): 1075–117. [CrossRef]
- Jose PA, Soares-da-Silva P, Eisner GM, Felder RA. Dopamine and G protein-coupled receptor kinase 4 in the kidney: role in blood pressure regulation. Biochim Biophys Acta. 2010 Dec;1802(12):1259-67. [CrossRef]
- Harris RC, Zhang MZ. Dopamine, the kidney, and hypertension. Curr Hypertens Rep. 2012 Apr;14(2):138-43. [CrossRef]
- Adam WR, Adams BA. Production and excretion of dopamine by the isolated perfused rat kidney. Ren Physiol. 1985;8(3):150-8. [CrossRef]
- Baines AD. Effects of salt intake and renal denervation on catecholamine catabolism and excretion. Kidney Int. 1982 Feb;21(2):316-22. [CrossRef]
- Berndt TJ, Khraibi AA, Thothathri V, Dousa TP, Tyce GM, Knox FG. Effect of increased dietary phosphate intake on dopamine excretion in the presence and absence of the renal nerves. Miner Electrolyte Metab. 1994; 20(3): 158-62.
- Stephenson RK, Sole MJ, Baines AD. Neural and extraneural catecholamine production by rat kidneys. Am J Physiol 1982; 242: F261–6. [CrossRef]
- Wang ZQ, Siragy HM, Felder RA, Carey RM. Intrarenal dopamine production and distribution in the rat. Physiological control of sodium excretion. Hypertension 1997 ;29(1 Pt 2):228-34. [CrossRef]
- Suzuki H, Nakane H, Kawamura M, Yoshizawa M, Takeshita E, Saruta T. Excretion and metabolism of dopa and dopamine by isolated perfused rat kidney. Am J Physiol. 1984; 247(3 Pt 1): E285-90. [CrossRef]
- Wolfovitz E, Grossman E, Folio CJ, Keiser HR, Kopin IJ, Goldstein DS. Derivation of urinary dopamine from plasma dihydroxyphenylalanine in humans. Clin Sci (Lond) 1993; 84(5): 549-57. [CrossRef]
- Zimlichman R, Levinson PD, Kelly G, Stull R, Keiser HR, Goldstein DS. Derivation of urinary dopamine from plasma dopa. Clin Sci (Lond). 1988 Nov;75(5):515-20. [CrossRef]
- Eldrup E, Hetland ML, Christensen NJ. Increase in plasma 3,4-dihydroxyphenylalanine (DOPA) appearance rate after inhibition of DOPA decarboxylase in humans. Eur J Clin Invest. 1994 Mar;24(3):205-11. [CrossRef]
- Grossman E, Hoffman A, Armando I, Abassi Z, Kopin IJ, Goldstein DS. Sympathoadrenal contribution to plasma dopa (3,4- dihydroxyphenylalanine) in rats. Clin Sci (Lond) 1992; 83: 65–74. [CrossRef]
- Soares-Da-Silva P, Serrão MP, Vieira-Coelho MA. Apical and basolateral uptake and intracellular fate of dopamine precursor L-dopa in LLC-PK1 cells. Am J Physiol 1998; 274(2): F243-51. [CrossRef]
- Gomes P, Soares-da-Silva P. Na+-independent transporters, LAT-2 and b0,+, exchange L-DOPA with neutral and basic amino acids in two clonal renal cell lines. J Membr Biol 2002; 186(2): 63-80. [CrossRef]
- de Toledo FG, Beers KW, Berndt TJ, Thompson MA, Tyce GM, Knox FG, Dousa TP. Opposite paracrine effects of 5-HT and dopamine on Na(+)-Pi cotransport in opossum kidney cells. Kidney Int. 1997 Jul;52(1):152-6. [CrossRef]
- Wassenberg T, Monnens LA, Geurtz BP, Wevers RA, Verbeek MM, Willemsen MA. The paradox of hyperdopaminuria in aromatic L-amino Acid deficiency explained. JIMD Rep. 2012;4:39-45. [CrossRef]
- Fernandes MH, Pestana M, Soares-da-Silva P. Deamination of newly-formed dopamine in rat renal tissues. Br J Pharmacol. 1991 Mar;102(3):778-82. [CrossRef]
- Guimarães JT, Soares-da-Silva P. The activity of MAO A and B in rat renal cells and tubules. Life Sci. 1998;62(8):727-37. [CrossRef]
- Xu J, Li G, Wang P, Velazquez H, Yao X, Li Y, Wu Y, Peixoto A, Crowley S, Desir GV. Renalase is a novel, soluble monoamine oxidase that regulates cardiac function and blood pressure. J Clin Invest. 2005 May;115(5):1275-80. [CrossRef]
- Isaac J, Berndt TJ, Chinnow SL, Tyce GM, Dousa TP, Knox FG. Dopamine enhances the phosphaturic response to parathyroid hormone in phosphate-deprived rats. J Am Soc Nephrol. 1992 Mar;2(9):1423-9. [CrossRef]
- Berndt TJ, MacDonald A, Walikonis R, Chinnow S, Dousa TP, Tyce GM, Knox FG. Excretion of catecholamines and metabolites in response to increased dietary phosphate intake. J Lab Clin Med. 1993 Jul;122(1):80-4.
- Weinman EJ, Biswas R, Steplock D, Wang P, Lau YS, Desir GV, Shenolikar S. Increased renal dopamine and acute renal adaptation to a high-phosphate diet. Am J Physiol Renal Physiol. 2011 May;300(5):F1123-9. [CrossRef]
- Sizova D, Velazquez H, Sampaio-Maia B, Quelhas-Santos J, Pestana M, Desir GV. Renalase regulates renal dopamine and phosphate metabolism. Am J Physiol Renal Physiol. 2013 Sep 15;305(6):F839-44. [CrossRef]
- Quelhas-Santos J, Serrão MP, Soares-Silva I, Fernandes-Cerqueira C, Simões-Silva L, Pinho MJ, Remião F, Sampaio-Maia B, Desir GV, Pestana M. Renalase regulates peripheral and central dopaminergic activities. Am J Physiol Renal Physiol. 2015 Jan 15;308(2):F84-91. [CrossRef]
- O’Connell DP, Botkin SJ, Ramos SI, Sibley DR, Ariano MA, Felder RA, Carey RM. Localization of dopamine D1A receptor protein in rat kidneys. Am J Physiol. 1995 Jun;268(6 Pt 2):F1185-97. [CrossRef]
- Felder CC, McKelvey AM, Gitler MS, Eisner GM, Jose PA. Dopamine receptor subtypes in renal brush border and basolateral membranes. Kidney Int. 1989 Aug;36(2):183-93. [CrossRef]
- Jackson A, Iwasiow RM, Chaar ZY, Nantel MF, Tiberi M. Homologous regulation of the heptahelical D1A receptor responsiveness: specific cytoplasmic tail regions mediate dopamine-induced phosphorylation, desensitization and endocytosis. J Neurochem. 2002 Aug;82(3):683-97. [CrossRef]
- Kim OJ, Gardner BR, Williams DB, Marinec PS, Cabrera DM, Peters JD, Mak CC, Kim KM, Sibley DR. The role of phosphorylation in D1 dopamine receptor desensitization: Evidence for a novel mechanism of arrestin association. J Biol Chem 279: 7999–8010, 2004. 10.1074/jbc.M308281200.
- Tsao P, Cao T, von Zastrow M. Role of endocytosis in mediating downregulation of G-protein-coupled receptors. Trends Pharmacol Sci. 2001 Feb;22(2):91-6. [CrossRef]
- Weinman EJ, Biswas R, Steplock D, Douglass TS, Cunningham R, Shenolikar S. Sodium-hydrogen exchanger regulatory factor 1 (NHERF-1)transduces signals that mediate dopamine inhibition of sodium-phosphate co-transport in mouse kidney. J Biol Chem2010; 285: 13454–60. [CrossRef]
- Jose PA, Eisner GM, Felder RA. Renal dopamine and sodium homeostasis. Curr Hypertens Rep. 2000 Apr;2(2):174-83. [CrossRef]
- Holmes A, Lachowicz JE, Sibley DR. Phenotypic analysis of dopamine receptor knockout mice; recent insights into the functional specificity of dopamine receptor subtypes. Neuropharmacology. 2004 Dec;47(8):1117-34. [CrossRef]
- Shultz PJ, Sedor JR, Abboud HE. Dopaminergic stimulation of cAMP accumulation in cultured rat mesangial cells. Am J Physiol. 1987 Aug;253(2 Pt 2):H358-64. [CrossRef]
- Kimura K, White BH, Sidhu A. Coupling of human D-1 dopamine receptors to different guanine nucleotide binding proteins. Evidence that D-1 dopamine receptors can couple to both Gs and G(o). J Biol Chem. 1995 Jun 16;270(24):14672-8. [CrossRef]
- Felder CC, Jose PA, Axelrod J. The dopamine-1 agonist, SKF 82526, stimulates phospholipase-C activity independent of adenylate cyclase. J Pharmacol Exp Ther. 1989 Jan;248(1):171-5.
- Jin LQ, Wang HY, Friedman E. Stimulated D(1) dopamine receptors couple to multiple Galpha proteins in different brain regions. J Neurochem. 2001 Sep;78(5):981-90. [CrossRef]
- Vyas SJ, Eichberg J, Lokhandwala MF. Characterization of receptors involved in dopamine-induced activation of phospholipase-C in rat renal cortex. J Pharmacol Exp Ther. 1992 Jan;260(1):134-9.
- Armando I, Villar VA, Jose PA. Dopamine and renal function and blood pressure regulation. Compr Physiol. 2011 Jul;1(3):1075-117. [CrossRef]
- Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001 May 22;98(11):6500-5. [CrossRef]
- White KE, Larsson TE, Econs MJ. The roles of specific genes implicated as circulating factors involved in normal and disordered phosphate homeostasis: frizzled related protein-4, matrix extracellular phosphoglycoprotein, and fibroblast growth factor 23. Endocr Rev. 2006 May;27(3):221-41. [CrossRef]
- Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002 Aug;143(8):3179-82. [CrossRef]
- Agoro R, Ni P, Noonan ML, White KE. Osteocytic FGF23 and Its Kidney Function. Front Endocrinol (Lausanne). 2020 Aug 28;11:592. [CrossRef]
- Thomas SM, Li Q, Faul C. Fibroblast growth factor 23, klotho and heparin. Curr Opin Nephrol Hypertens. 2023 Jul 1;32(4):313-23. [CrossRef]
- Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004 Mar;19(3):429-35. [CrossRef]
- Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004 Feb;113(4):561-8. [CrossRef]
- Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Jüppner H, Lanske B. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004 Nov;23(7):421-32. [CrossRef]
- Holbrook L, Brady R. McCune-Albright Syndrome. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan- [Updated 2023 Jul 10].Available from: https://www.ncbi.nlm.nih.gov/books/NBK537092/.
- Leet AI, Collins MT. Current approach to fibrous dysplasia of bone and McCune-Albright syndrome. J Child Orthop. 2007 Mar;1(1):3-17. [CrossRef]
- Kinoshita Y, Hori M, Taguchi M, Fukumoto S. Functional analysis of mutant FAM20C in Raine syndrome with FGF23-related hypophosphatemia. Bone. 2014 Oct;67:145-51. [CrossRef]
- Palma-Lara I, Pérez-Ramírez M, García Alonso-Themann P, Espinosa-García AM, Godinez-Aguilar R, Bonilla-Delgado J, López- Ornelas A, Victoria-Acosta G, Olguín-García MG, Moreno J, et al. FAM20C Overview: classic and novel targets, pathogenic variants and Raine Syndrome phenotypes. Int J Mol Sci. 2021 Jul 27;22(15):8039. [CrossRef]
- Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, Ogata E, Segawa H, Miyamoto K, Fukushima N. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005 Jan 28;280(4):2543-9. [CrossRef]
- Yu X, Sabbagh Y, Davis SI, Demay MB, White KE. Genetic dissection of phosphate- and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone. 2005 Jun;36(6):971-7. [CrossRef]
- Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005 Dec;146(12):5358-64. [CrossRef]
- Courbebaisse M, Lanske B. Biology of Fibroblast Growth Factor 23: From Physiology to Pathology. Cold Spring Harb Perspect Med. 2018 May 1;8(5):a031260. [CrossRef]
- Tsuji K, Maeda T, Kawane T, Matsunuma A, Horiuchi N. Leptin stimulates fibroblast growth factor 23 expression in bone and suppresses renal 1alpha,25-dihydroxyvitamin D3 synthesis in leptin-deficient mice. J Bone Miner Res. 2010 Aug;25(8):1711-23. [CrossRef]
- Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol. 2007 Jun;18(6):1637-47. [CrossRef]
- Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005 Mar;90(3):1519-24. [CrossRef]
- Tenenhouse HS, Gauthier C, Chau H, St-Arnaud R. 1alpha-Hydroxylase gene ablation and Pi supplementation inhibit renal calcification in mice homozygous for the disrupted Npt2a gene. Am J Physiol Renal Physiol. 2004 Apr;286(4):F675-81. [CrossRef]
- Knab VM, Corbin B, Andrukhova O, Hum JM, Ni P, Rabadi S, Maeda A, White KE, Erben RG, Jüppner H, et al. Acute parathyroid hormone injection increases C-terminal but not intact fibroblast growth factor 23 levels. Endocrinology. 2017 May 1;158(5):1130-39. [CrossRef]
- Meir T, Durlacher K, Pan Z, Amir G, Richards WG, Silver J, Naveh-Many T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014 Dec;86(6):1106-15. [CrossRef]
- White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001 Dec;60(6):2079-86. [CrossRef]
- Ho BB, Bergwitz C. FGF23 signalling and physiology. J Mol Endocrinol. 2021 Feb;66(2):R23-R32. [CrossRef]
- Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, Koller A, Nizet V, White KE, Dixon JE. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A. 2014 Apr 15;111(15):5520-5. [CrossRef]
- Edmonston D, Wolf M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat Rev Nephrol. 2020 Jan;16(1):7-19. [CrossRef]
- Kato K, Jeanneau C, Tarp MA, Benet-Pagès A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem. 2006 Jul 7;281(27):18370-7. [CrossRef]
- Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010 Sep;24(9):3438-50. [CrossRef]
- Gattineni J, Alphonse P, Zhang Q, Mathews N, Bates CM, Baum M. Regulation of renal phosphate transport by FGF23 is mediated by FGFR1 and FGFR4. Am J Physiol Renal Physiol. 2014 Feb 1;306(3):F351-8. [CrossRef]
- Chen G, Liu Y, Goetz R, Fu L, Jayaraman S, Hu MC, Moe OW, Liang G, Li X, Mohammadi M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature. 2018 Jan 25;553(7689):461-6. [CrossRef]
- Xu D, Esko JD. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem. 2014;83:129-57. [CrossRef]
- Agrawal A, Ni P, Agoro R, White KE, DiMarchi RD. Identification of a second Klotho interaction site in the C terminus of FGF23. Cell Rep. 2021 Jan 26;34(4):108665. [CrossRef]
- Andrukhova O, Zeitz U, Goetz R, Mohammadi M, Lanske B, Erben RG. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone. 2012 Sep;51(3):621-8. [CrossRef]
- Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, Ravikumar P, Gillings N, Bian A, Sidhu SS, et al. Renal production, uptake, and handling of circulating αKlotho. J Am Soc Nephrol. 2016 Jan;27(1):79-90. [CrossRef]
- Han X, Yang J, Li L, Huang J, King G, Quarles LD. Conditional Deletion of Fgfr1 in the Proximal and distal tubule identifies distinct roles in phosphate and calcium transport. PLoS One. 2016 Feb 3;11(2):e0147845. [CrossRef]
- Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015 May-Jun;4(3):215- 66. [CrossRef]
- Déliot N, Hernando N, Horst-Liu Z, Gisler SM, Capuano P, Wagner CA, Bacic D, O’Brien S, Biber J, Murer H. Parathyroid hormone treatment induces dissociation of type IIa Na+-P(i) cotransporter-Na+/H+ exchanger regulatory factor-1 complexes. Am J Physiol Cell Physiol. 2005 Jul;289(1):C159-67. [CrossRef]
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006 Dec 7;444(7120):770-4. [CrossRef]
- Ide N, Olauson H, Sato T, Densmore MJ, Wang H, Hanai JI, Larsson TE, Lanske B. In vivo evidence for a limited role of proximal tubular Klotho in renal phosphate handling. Kidney Int. 2016 Aug;90(2):348-62. [CrossRef]
- Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, Li J, Shehadeh LA, Hare JM, David V, et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015 Dec 1;22(6):1020-32. [CrossRef]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002 Apr;109 Suppl:S67-79. [CrossRef]
- Farrow EG, Davis SI, Summers LJ, White KE. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol. 2009 May;20(5):955-60. [CrossRef]
- Zhang MY, Ranch D, Pereira RC, Armbrecht HJ, Portale AA, Perwad F. Chronic inhibition of ERK1/2 signaling improves disordered bone and mineral metabolism in hypophosphatemic (Hyp) mice. Endocrinology. 2012 Apr;153(4):1806-16. [CrossRef]
- Ranch D, Zhang MY, Portale AA, Perwad F. Fibroblast growth factor 23 regulates renal 1,25-dihydroxyvitamin D and phosphate metabolism via the MAP kinase signaling pathway in Hyp mice. J Bone Miner Res. 2011 Aug;26(8):1883-90. [CrossRef]
- Murer H, Lötscher M, Kaissling B, Levi M, Kempson SA, Biber J. Renal brush border membrane Na/Pi-cotransport: molecular aspects in PTH-dependent and dietary regulation. Kidney Int. 1996 Jun;49(6):1769-73. [CrossRef]
- Murer H, Hernando N, Forster I, Biber J. Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev. 2000 Oct;80(4):1373-409. [CrossRef]
- Keusch I, Traebert M, Lötscher M, Kaissling B, Murer H, Biber J. Parathyroid hormone and dietary phosphate provoke a lysosomal routing of the proximal tubular Na/Pi-cotransporter type II. Kidney Int. 1998 Oct;54(4):1224-1232. [CrossRef]
- Lötscher M, Scarpetta Y, Levi M, Halaihel N, Wang H, Zajicek HK, Biber J, Murer H, Kaissling B. Rapid downregulation of rat renal Na/P(i) cotransporter in response to parathyroid hormone involves microtubule rearrangement. J Clin Invest. 1999 Aug;104(4):483-94. [CrossRef]
- Tumbarello DA, Kendrick-Jones J, Buss F. Myosin VI and its cargo adaptors - linking endocytosis and autophagy. J Cell Sci. 2013 Jun 15;126(Pt 12):2561-70. [CrossRef]
- Cunningham R, E X, Steplock D, Shenolikar S, Weinman EJ. Defective PTH regulation of sodium-dependent phosphate transport in NHERF-1-/- renal proximal tubule cells and wild-type cells adapted to low-phosphate media. Am J Physiol Renal Physiol. 2005 Oct;289(4):F933-8. [CrossRef]
- Cunningham R, Biswas R, Brazie M, Steplock D, Shenolikar S, Weinman EJ. Signaling pathways utilized by PTH and dopamine to inhibit phosphate transport in mouse renal proximal tubule cells. Am J Physiol Renal Physiol. 2009 Feb;296(2):F355-61. [CrossRef]
- Lederer ED, Sohi SS, McLeish KR. Dopamine regulates phosphate uptake by opossum kidney cells through multiple counter- regulatory receptors. J Am Soc Nephrol. 1998 Jun;9(6):975-85. [CrossRef]
- Weinman EJ, Steplock D, Shenolikar S, Biswas R. Fibroblast growth factor-23-mediated inhibition of renal phosphate transport in mice requires sodium-hydrogen exchanger regulatory factor-1 (NHERF-1) and synergizes with parathyroid hormone. J Biol Chem. 2011 Oct 28;286(43):37216-21. [CrossRef]
- Shenolikar S, Voltz JW, Minkoff CM, Wade JB, Weinman EJ. Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc Natl Acad Sci U S A. 2002 Aug 20;99(17):11470-5. [CrossRef]
- Voltz JW, Brush M, Sikes S, Steplock D, Weinman EJ, Shenolikar S. Phosphorylation of PDZ1 domain attenuates NHERF-1 binding to cellular targets. J Biol Chem. 2007 Nov 16;282(46):33879-87. [CrossRef]
- Mahon MJ, Donowitz M, Yun CC, Segre GV. Na(+)/H(+ ) exchanger regulatory factor 2 directs parathyroid hormone 1 receptor signalling. Nature. 2002 Jun 20;417(6891):858-61. [CrossRef]
- Wheeler D, Garrido JL, Bisello A, Kim YK, Friedman PA, Romero G. Regulation of parathyroid hormone type 1 receptor dynamics, traffic, and signaling by the Na+/H+ exchanger regulatory factor-1 in rat osteosarcoma ROS 17/2.8 cells. Mol Endocrinol. 2008 May;22(5):1163-70. [CrossRef]
- Lee HJ, Zheng JJ. PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun Signal. 2010 May 28;8:8. [CrossRef]
- Dicks M, Kock G, Kohl B, Zhong X, Pütz S, Heumann R, Erdmann KS, Stoll R. The binding affinity of PTPN13’s tandem PDZ2/3 domain is allosterically modulated. BMC Mol Cell Biol. 2019 Jul 8;20(1):23. [CrossRef]
- Walker V, Vuister GW. Biochemistry and pathophysiology of the Transient Potential Receptor Vanilloid 6 (TRPV6) calcium channel. Adv Clin Chem. 2023;113:43-100. [CrossRef]
- Sneddon WB, Ruiz GW, Gallo LI, Xiao K, Zhang Q, Rbaibi Y, Weisz OA, Apodaca GL, Friedman PA. Convergent Signaling Pathways Regulate Parathyroid Hormone and Fibroblast Growth Factor-23 Action on NPT2A-mediated Phosphate Transport. J Biol Chem. 2016 Sep 2;291(36):18632-42. [CrossRef]
- Mahon MJ, Segre GV. Stimulation by parathyroid hormone of a NHERF-1-assembled complex consisting of the parathyroid hormone I receptor, phospholipase Cbeta, and actin increases intracellular calcium in opossum kidney cells. J Biol Chem. 2004 May 28;279(22):23550-8. [CrossRef]
- Mamonova T, Zhang Q, Chandra M, Collins BM, Sarfo E, Bu Z, Xiao K, Bisello A, Friedman PA. Origins of PDZ Binding Specificity. A Computational and Experimental Study Using NHERF1 and the Parathyroid Hormone Receptor. Biochemistry. 2017 May 23;56(20):2584-93. [CrossRef]
- Mamonova T, Kurnikova M, Friedman PA. Structural basis for NHERF1 PDZ domain binding. Biochemistry. 2012 Apr 10;51(14):3110-20. [CrossRef]
- Rajagopal A, Braslavsky D, Lu JT, Kleppe S, Clément F, Cassinelli H, Liu DS, Liern JM, Vallejo G, Bergadá I, Gibbs RA, Campeau PM, Lee BH. Exome sequencing identifies a novel homozygous mutation in the phosphate transporter SLC34A1 in hypophosphatemia and nephrocalcinosis. J Clin Endocrinol Metab. 2014 Nov;99(11):E2451-6. [CrossRef]
- Kang SJ, Lee R, Kim HS. Infantile hypercalcemia with novel compound heterozygous mutation in SLC34A1 encoding renal sodium-phosphate cotransporter 2a: a case report. Ann Pediatr Endocrinol Metab. 2019 Mar;24(1):64-67. [CrossRef]
- Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, Haraldsdottir E, Eyjolfsson GI, Sigurdardottir O, Olafsson I, Masson G, Holm H, Gudbjartsson DF, Thorsteinsdottir U, Indridason OS, Palsson R, Stefansson K. Common and rare variants associated with kidney stones and biochemical traits. Nat Commun. 2015 Aug 14;6:7975. [CrossRef]
- Howles SA, Wiberg A, Goldsworthy M, Bayliss AL, Gluck AK, Ng M, Grout E, Tanikawa C, Kamatani Y, Terao C, Takahashi A, Kubo M, Matsuda K, Thakker RV, Turney BW, Furniss D. Genetic variants of calcium and vitamin D metabolism in kidney stone disease. Nat Commun. 2019 Nov 15;10(1):5175. [CrossRef]
- Urabe Y, Tanikawa C, Takahashi A, Okada Y, Morizono T, Tsunoda T, Kamatani N, Kohri K, Chayama K, Kubo M, Nakamura Y, Matsuda K. A genome-wide association study of nephrolithiasis in the Japanese population identifies novel susceptible Loci at 5q35.3, 7p14.3, and 13q14.1. PLoS Genet. 2012;8(3):e1002541. [CrossRef]
- Chen WC, Chou WH, Chu HW, Huang CC, Liu X, Chang WP, Chou YH, Chang WC. The rs1256328 (ALPL) and rs12654812 (RGS14) Polymorphisms are Associated with Susceptibility to Calcium Nephrolithiasis in a Taiwanese population. Sci Rep. 2019 Nov 21;9(1):17296. [CrossRef]
- Tanikawa C, Kamatani Y, Terao C, Usami M, Takahashi A, Momozawa Y, Suzuki K, Ogishima S, Shimizu A, Satoh M, Matsuo K, Mikami H, Naito M, Wakai K, Yamaji T, Sawada N, Iwasaki M, Tsugane S, Kohri K, Yu ASL, Yasui T, Murakami Y, Kubo M, Matsuda K. Novel Risk Loci Identified in a Genome-Wide Association Study of Urolithiasis in a Japanese Population. J Am Soc Nephrol. 2019 May;30(5):855-64. [CrossRef]
- Kestenbaum B, Glazer NL, Köttgen A, Felix JF, Hwang SJ, Liu Y, Lohman K, Kritchevsky SB, Hausman DB, Petersen AK, et al. Common genetic variants associate with serum phosphorus concentration. J Am Soc Nephrol. 2010 Jul;21(7):1223-32. [CrossRef]
- Laster ML, Rowan B, Chen HC, Schwantes-An TH, Sheng X, Friedman PA, Ikizler TA, Sinshiemer JS, Ix JH, Susztak K, et al. Genetic Variants Associated With Mineral Metabolism Traits in Chronic Kidney Disease. J Clin Endocrinol Metab. 2022 Aug 18;107(9):e3866-e3876. [CrossRef]
- Alqinyah M, Hooks SB. Regulating the regulators: Epigenetic, transcriptional, and post-translational regulation of RGS proteins. Cell Signal. 2018 Jan;42:77-87. [CrossRef]
- Willard FS, Willard MD, Kimple AJ, Soundararajan M, Oestreich EA, Li X, Sowa NA, Kimple RJ, Doyle DA, Der CJ, Zylka MJ, Snider WD, Siderovski DP. Regulator of G-protein signaling 14 (RGS14) is a selective H-Ras effector. PLoS One. 2009;4(3):e4884. [CrossRef]
- Kimple RJ, De Vries L, Tronchère H, Behe CI, Morris RA, Gist Farquhar M, Siderovski DP. RGS12 and RGS14 GoLoco motifs are G alpha(i) interaction sites with guanine nucleotide dissociation inhibitor Activity. J Biol Chem. 2001 Aug 3;276(31):29275-81. [CrossRef]
- Siderovski DP, Harden TK. The RGS Protein Superfamily. In Handbook of Cell Signaling, Bradshaw R.A., Dennis E.A., Eds.;Academic Press (Elsevier Inc): Amsterdam, Netherlands, 2003, Volume 2, pp. 631-8 . [CrossRef]
- Sjögren B. The evolution of regulators of G protein signalling proteins as drug targets - 20 years in the making: IUPHAR Review 21. Br J Pharmacol. 2017 Mar;174(6):427-37. [CrossRef]
- Brown NE, Goswami D, Branch MR, Ramineni S, Ortlund EA, Griffin PR, Hepler JR. Integration of G protein α (Gα) signaling by the regulator of G protein signaling 14 (RGS14). J Biol Chem. 2015 Apr 3;290(14):9037-49. [CrossRef]
- Worcester EM, Coe FL. Clinical practice. Calcium kidney stones. N Engl J Med. 2010 Sep 2;363(10):954-63. [CrossRef]
- Adeli K, Higgins V, Nieuwesteeg M, Raizman JE, Chen Y, Wong SL, Blais D. Biochemical marker reference values across pediatric, adult, and geriatric ages: establishment of robust pediatric and adult reference intervals on the basis of the Canadian Health Measures Survey. Clin Chem. 2015 Aug;61(8):1049-62. [CrossRef]
- Saggese G, Baroncelli GI, Bertelloni S, Cinquanta L, Di Nero G. Effects of long-term treatment with growth hormone on bone and mineral metabolism in children with growth hormone deficiency. J Pediatr. 1993 Jan;122(1):37-45. [CrossRef]
- Boot AM, Engels MA, Boerma GJ, Krenning EP, De Muinck Keizer-Schrama SM. Changes in bone mineral density, body composition, and lipid metabolism during growth hormone (GH) treatment in children with GH deficiency. J Clin Endocrinol Metab. 1997 Aug;82(8):2423-8. [CrossRef]
- Haffner D, Grund A, Leifheit-Nestler M. Renal effects of growth hormone in health and in kidney disease. Pediatr Nephrol. 2021 Aug;36(8):2511-30. [CrossRef]
- Quigley R, Baum M. Effects of growth hormone and insulin-like growth factor I on rabbit proximal convoluted tubule transport. J Clin Invest. 1991 Aug;88(2):368-74. [CrossRef]
- Hirschberg R, Ding H, Wanner C. Hirschberg R, Ding H, Wanner C. Effects of insulin-like growth factor I on phosphate transport in cultured proximal tubule cells. J Lab Clin Med. 1995 Nov;126(5):428-34.
- Jehle AW, Forgo J, Biber J, Lederer E, Krapf R, Murer H. IGF-I and vanadate stimulate Na/Pi-cotransport in OK cells by increasing type II Na/Pi-cotransporter protein stability. Pflugers Arch. 1998 Dec;437(1):149-54. [CrossRef]
- Palmer G, Bonjour JP, Caverzasio J. Stimulation of inorganic phosphate transport by insulin-like growth factor I and vanadate in opossum kidney cells is mediated by distinct protein tyrosine phosphorylation processes. Endocrinology. 1996 Nov;137(11):4699-705. [CrossRef]
- Marks J, Debnam ES, Unwin RJ. The role of the gastrointestinal tract in phosphate homeostasis in health and chronic kidney disease. Curr Opin Nephrol Hypertens. 2013 Jul;22(4):481-7. [CrossRef]
- Capuano P, Bacic D, Roos M, Gisler SM, Stange G, Biber J, Kaissling B, Weinman EJ, Shenolikar S, Wagner CA, Murer H. Defective coupling of apical PTH receptors to phospholipase C prevents internalization of the Na+-phosphate cotransporter NaPi- IIa in Nherf1-deficient mice. Am J Physiol Cell Physiol. 2007 Feb;292(2):C927-34. [CrossRef]
- Murer H, Forster I, Biber J. The sodium phosphate cotransporter family SLC34. Pflugers Arch. 2004 Feb;447(5):763-7. [CrossRef]
- Levi M, Lötscher M, Sorribas V, Custer M, Arar M, Kaissling B, Murer H, Biber J. Cellular mechanisms of acute and chronic adaptation of rat renal P(i) transporter to alterations in dietary P(i). Am J Physiol. 1994 Nov;267(5 Pt 2):F900-8. [CrossRef]
- Weinman EJ, Boddeti A, Cunningham R, Akom M, Wang F, Wang Y, Liu J, Steplock D, Shenolikar S, Wade JB. NHERF-1 is required for renal adaptation to a low-phosphate diet. Am J Physiol Renal Physiol. 2003 Dec;285(6):F1225-32. [CrossRef]
- Cunningham R, Steplock D, Wang F, Huang H, E X, Shenolikar S, Weinman EJ. Defective parathyroid hormone regulation of NHE3 activity and phosphate adaptation in cultured NHERF-1-/- renal proximal tubule cells. J Biol Chem. 2004 Sep 3;279(36):37815-21. [CrossRef]
- Villa-Bellosta R, Sorribas V. Compensatory regulation of the sodium/phosphate cotransporters NaPi-IIc (SCL34A3) and Pit-2 (SLC20A2) during Pi deprivation and acidosis. Pflugers Arch. 2010 Feb;459(3):499-508. [CrossRef]
- Capuano P, Bacic D, Stange G, Hernando N, Kaissling B, Pal R, Kocher O, Biber J, Wagner CA, Murer H. Expression and regulation of the renal Na/phosphate cotransporter NaPi-IIa in a mouse model deficient for the PDZ protein PDZK1. Pflugers Arch. 2005 Jan;449(4):392-402. [CrossRef]
- Takahashi F, Morita K, Katai K, Segawa H, Fujioka A, Kouda T, Tatsumi S, Nii T, Taketani Y, Haga H, Hisano S, Fukui Y, Miyamoto KI, Takeda E. Effects of dietary Pi on the renal Na+-dependent Pi transporter NaPi-2 in thyroparathyroidectomized rats. Biochem J. 1998 Jul 1;333 ( Pt 1)(Pt 1):175-81. [CrossRef]
- Thomas L, Bettoni C, Knöpfel T, Hernando N, Biber J, Wagner CA. Acute Adaption to Oral or Intravenous Phosphate Requires Parathyroid Hormone. J Am Soc Nephrol. 2017 Mar;28(3):903-14. [CrossRef]
- Scanni R, vonRotz M, Jehle S, Hulter HN, Krapf R. The human response to acute enteral and parenteral phosphate loads. J Am Soc Nephrol. 2014 Dec;25(12):2730-9. [CrossRef]
- Kritmetapak K, Kumar R. Phosphate as a Signaling Molecule. Calcif Tissue Int. 2021 Jan;108(1):16-31. [CrossRef]
- Kido S, Miyamoto K, Mizobuchi H, Taketani Y, Ohkido I, Ogawa N, Kaneko Y, Harashima S, Takeda E. Identification of regulatory sequences and binding proteins in the type II sodium/phosphate cotransporter NPT2 gene responsive to dietary phosphate. J Biol Chem. 1999 Oct 1;274(40):28256-63. [CrossRef]
- Beck L, Tenenhouse HS, Meyer RA, Meyer MH, Biber J, Murer H. Renal expression of Na+-phosphate cotransporter mRNA and protein: effect of the Gy mutation and low phosphate diet. Pflugers Arch. 1996 Apr;431(6):936-41. [CrossRef]
- Hoag HM, Martel J, Gauthier C, Tenenhouse HS. Effects of Npt2 gene ablation and low-phosphate diet on renal Na(+)/phosphate cotransport and cotransporter gene expression. J Clin Invest. 1999 Sep;104(6):679-86. [CrossRef]
- Kilav R, Silver J, Biber J, Murer H, Naveh-Many T. Coordinate regulation of rat renal parathyroid hormone receptor mRNA and Na-Pi cotransporter mRNA and protein. Am J Physiol. 1995 Jun;268(6 Pt 2):F1017-22. [CrossRef]
- Conrads KA, Yi M, Simpson KA, Lucas DA, Camalier CE, Yu LR, Veenstra TD, Stephens RM, Conrads TP, Beck GR Jr. A combined proteome and microarray investigation of inorganic phosphate-induced pre-osteoblast cells. Mol Cell Proteomics. 2005 Sep;4(9):1284-96. [CrossRef]
- Julien M, Magne D, Masson M, Rolli-Derkinderen M, Chassande O, Cario-Toumaniantz C, Cherel Y, Weiss P, Guicheux J. Phosphate stimulates matrix Gla protein expression in chondrocytes through the extracellular signal regulated kinase signaling pathway. Endocrinology. 2007 Feb;148(2):530-7. [CrossRef]
- Nishino J, Yamazaki M, Kawai M, Tachikawa K, Yamamoto K, Miyagawa K, Kogo M, Ozono K, Michigami T. Extracellular Phosphate Induces the Expression of Dentin Matrix Protein 1 Through the FGF Receptor in Osteoblasts. J Cell Biochem. 2017 May;118(5):1151-63. [CrossRef]
- Camalier CE, Yi M, Yu LR, Hood BL, Conrads KA, Lee YJ, Lin Y, Garneys LM, Bouloux GF, Young MR, Veenstra TD, Stephens RM, Colburn NH, Conrads TP, Beck GR Jr. An integrated understanding of the physiological response to elevated extracellular phosphate. J Cell Physiol. 2013 Jul;228(7):1536-50. [CrossRef]
- Bansal N, Hsu CY, Whooley M, Berg AH, Ix JH. Relationship of urine dopamine with phosphorus homeostasis in humans: the heart and soul study. Am J Nephrol. 2012;35(6):483-90. [CrossRef]
- Boland JM, Tebben PJ, Folpe AL. Phosphaturic mesenchymal tumors: what an endocrinologist should know. J Endocrinol Invest. 2018 Oct;41(10):1173-84. [CrossRef]
- Minisola S, Fukumoto S, Xia W, Corsi A, Colangelo L, Scillitani A, Pepe J, Cipriani C, Thakker RV. Tumor-induced Osteomalacia: A Comprehensive Review. Endocr Rev. 2023 Mar 4;44(2):323-53. [CrossRef]
- Folpe AL. Phosphaturic mesenchymal tumors: A review and update. Semin Diagn Pathol. 2019 Jul;36(4):260-8. [CrossRef]
- Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, Oudet CL. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics. 2000 Jul 1;67(1):54-68. [CrossRef]
- Imanishi Y, Hashimoto J, Ando W, Kobayashi K, Ueda T, Nagata Y, Miyauchi A, Koyano HM, Kaji H, Saito T, Oba K, Komatsu Y, Morioka T, Mori K, Miki T, Inaba M. Matrix extracellular phosphoglycoprotein is expressed in causative tumors of oncogenic osteomalacia. J Bone Miner Metab. 2012 Jan;30(1):93-9. [CrossRef]
- Imel EA, Peacock M, Pitukcheewanont P, Heller HJ, Ward LM, Shulman D, Kassem M, Rackoff P, Zimering M, Dalkin A, Drobny E, Colussi G, Shaker JL, Hoogendoorn EH, Hui SL, Econs MJ. Sensitivity of fibroblast growth factor 23 measurements in tumor-induced osteomalacia. J Clin Endocrinol Metab. 2006;91:2055–61. [CrossRef]
- Lee JC, Jeng YM, Su SY, Wu CT, Tsai KS, Lee CH, et al.. Identification of a novel FN1-FGFR1 genetic fusion as a frequent event in phosphaturic mesenchymal tumour. J Pathol (2015) 235:539–45. [CrossRef]
- Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, Finnegan R,et al. Secreted frizzled-related protein 4 is a potent tumor- derived phosphaturic agent. J Clin Invest. 2003 Sep;112(5):785-94. [CrossRef]
- Lee JC, Su SY, Changou CA, Yang RS, Tsai KS, Collins MT, et al.. Characterization of FN1-FGFR1 and novel FN1-FGF1 fusion genes in a large series of phosphaturic mesenchymal tumors. Mod Pathol (2016) 29:1335–46. [CrossRef]
- Sakai T, Okuno Y, Murakami N, Shimoyama Y, Imagama S, Nishida Y. Case report: Novel NIPBL-BEND2 fusion gene identified in osteoblastoma-like phosphaturic mesenchymal tumor of the fibula. Front Oncol. 2023 Jan 5;12:956472. [CrossRef]
- Rowe PS, Kumagai Y, Gutierrez G et al. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 2004; 34: 303-19.
- Schrauwen I, Valgaeren H, Tomas-Roca L, Sommen M, Altunoglu U, Wesdorp M, Beyens M, Fransen E, Nasir A, Vandeweyer G, et al. Variants affecting diverse domains of MEPE are associated with two distinct bone disorders, a craniofacial bone defect and otosclerosis. Genet Med. 2019 May;21(5):1199-208. [CrossRef]
- Sprowson AP, McCaskie AW, Birch MA. ASARM-truncated MEPE and AC-100 enhance osteogenesis by promoting osteoprogenitor adhesion. J Orthop Res. 2008 Sep;26(9):1256-62. [CrossRef]
- Rowe PS. The wrickkened pathways of FGF23, MEPE and PHEX. Crit Rev Oral Biol Med. 2004 Sep 1;15(5):264-81. [CrossRef]
- Rowe PS. Regulation of bone-renal mineral and energy metabolism: the PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit Rev Eukaryot Gene Expr. 2012;22(1):61-86. [CrossRef]
- Ogbureke KU, Fisher LW. Renal expression of SIBLING proteins and their partner matrix metalloproteinases (MMPs). Kidney Int. 2005 Jul;68(1):155-66. [CrossRef]
- Ogbureke KU, Koli K, Saxena G. Matrix Metalloproteinase 20 Co-expression With Dentin Sialophosphoprotein in Human and Monkey Kidneys. J Histochem Cytochem. 2016 Oct;64(10):623-36. [CrossRef]
- David V, Martin A, Hedge AM, Rowe PS. Matrix extracellular phosphoglycoprotein (MEPE) is a new bone renal hormone and vascularization modulator. Endocrinology. 2009 Sep;150(9):4012-23. [CrossRef]
- Dobbie H, Unwin RJ, Faria NJ, Shirley DG. Matrix extracellular phosphoglycoprotein causes phosphaturia in rats by inhibiting tubular phosphate reabsorption. Nephrol Dial Transplant. 2008 Feb;23(2):730-3. [CrossRef]
- Shirley DG, Faria NJ, Unwin RJ, Dobbie H. Direct micropuncture evidence that matrix extracellular phosphoglycoprotein inhibits proximal tubular phosphate reabsorption. Nephrol Dial Transplant. 2010 Oct;25(10):3191-5. [CrossRef]
- Gowen LC, Petersen DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT, Simmons HA, Crawford DT, Chidsey-Frink KL, Ke HZ, et al. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem. 2003 Jan 17;278(3):1998-2007. [CrossRef]
- Zelenchuk LV, Hedge AM, Rowe PS. Age dependent regulation of bone-mass and renal function by the MEPE ASARM-motif. Bone. 2015 Oct;79:131-42. [CrossRef]
- Marks J, Churchill LJ, Debnam ES, Unwin RJ. Matrix extracellular phosphoglycoprotein inhibits phosphate transport. J Am Soc Nephrol. 2008 Dec;19(12):2313-20. [CrossRef]
- David V, Martin A, Hedge AM, Drezner MK, Rowe PS. ASARM peptides: PHEX-dependent and -independent regulation of serum phosphate. Am J Physiol Renal Physiol. 2011 Mar;300(3):F783-91. [CrossRef]
- Beauvais DM, Ell BJ, McWhorter AR, Rapraeger AC. Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J Exp Med. 2009 Mar 16;206(3):691-705. [CrossRef]
- Tenenhouse HS, Lee J, Harvey N, Potier M, Jette M, Beliveau R. Normal molecular size of the Na(+)-phosphate cotransporter and normal Na(+)-dependent binding of phosphonoformic acid in renal brush border membranes of X-linked Hyp mice. Biochem Biophys Res Commun. 1990 Aug 16;170(3):1288-93. [CrossRef]
- Berndt TJ, Bielesz B, Craig TA, Tebben PJ, Bacic D, Wagner CA, O’Brien S, Schiavi S, Biber J, Murer H, et al. Secreted frizzled- related protein-4 reduces sodium-phosphate co-transporter abundance and activity in proximal tubule cells. Pflugers Arch. 2006 Jan;451(4):579-87. [CrossRef]
- Habra MA, Jimenez C, Huang SC, Cote GJ, Murphy WA Jr, Gagel RF, Hoff AO. Expression analysis of fibroblast growth factor- 23, matrix extracellular phosphoglycoprotein, secreted frizzled-related protein-4, and fibroblast growth factor-7: identification of fibroblast growth factor-23 and matrix extracellular phosphoglycoprotein as major factors involved in tumor-induced osteomalacia. Endocr Pract. 2008 Dec;14(9):1108-14. [CrossRef]
- Zhang Z, Lin X, Wei L, Wu Y, Xu L, Wu L, Wei X, Zhao S, Zhu X, Xu F. A framework for frizzled-G protein coupling and implications to the PCP signaling pathways. Cell Discov. 2024 Jan 5;10(1):3. [CrossRef]
- Azbazdar Y, Karabicici M, Erdal E, Ozhan G. Regulation of Wnt Signaling pathways at the plasma membrane and their misregulation in cancer. Front Cell Dev Biol. 2021 Jan 21;9:631623. [CrossRef]
- Pawar NM, Rao P. Secreted frizzled related protein 4 (sFRP4) update: A brief review. Cell Signal. 2018 May;45:63-70. [CrossRef]
- Cruciat CM, Niehrs C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol.2013;5:a015081. 10.1101/cshperspect.a015081.
- Wawrzak D, Métioui M, Willems E, Hendrickx M, de Genst E, Leyns L. Wnt3a binds to several sFRPs in the nanomolar range. Biochem Biophys Res Commun. 2007 Jun 15;357(4):1119-23. [CrossRef]
- Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014 Oct 3;346(6205):1248012. [CrossRef]
- De A. Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Sin (Shanghai). 2011 Oct;43(10):745-56. [CrossRef]
- Kiper POS, Saito H, Gori F, Unger S, Hesse E, Yamana K, Kiviranta R, Solban N, Liu J, Brommage R, et al. Cortical-Bone Fragility--Insights from sFRP4 Deficiency in Pyle’s Disease. N Engl J Med. 2016 Jun 30;374(26):2553-62. [CrossRef]
- Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003 Jul 1;116(Pt 13):2627-34. [CrossRef]
- Katoh M, Katoh M. Molecular genetics and targeted therapy of WNT-related human diseases (Review). Int J Mol Med. 2017 Sep;40(3):587-606. [CrossRef]
- Finch PW, He X, Kelley MJ, Uren A, Schaudies RP, Popescu NC, Rudikoff S, Aaronson SA, Varmus HE, Rubin JS. Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc Natl Acad Sci U S A. 1997 Jun 24;94(13):6770-5. [CrossRef]
- Rehn M, Pihlajaniemi T, Hofmann K, Bucher P. The frizzled motif: in how many different protein families does it occur? Trends Biochem Sci. 1998 Nov;23(11):415-7. [CrossRef]
- Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. Fibroblast growth factor 7: an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Metab. 2005 Feb;90(2):1012-20. [CrossRef]
- Whyte MP, Zhang F, Wenkert D, Mumm S, Berndt TJ, Kumar R. Hyperphosphatemia with low FGF7 and normal FGF23 and sFRP4 levels in the circulation characterizes pediatric hypophosphatasia. Bone. 2020 May;134:115300. [CrossRef]
- Kritmetapak K, Kumar R. Phosphatonins: From discovery to therapeutics. Endocr Pract. 2023 Jan;29(1):69-79. [CrossRef]
- Mei C, Mao Z, Shen X, Wang W, Dai B, Tang B, Wu Y, Cao Y, Zhang S, Zhao H, et al. Role of keratinocyte growth factor in the pathogenesis of autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2005 Nov;20(11):2368-75. [CrossRef]
- Zinkle A, Mohammadi M. Structural Biology of the FGF7 Subfamily. Front Genet. 2019 Feb 12;10:102. [CrossRef]
- Arora K, Moon C, Zhang W, Yarlagadda S, Penmatsa H, Ren A, Sinha C, Naren AP. Stabilizing rescued surface-localized ΔF508 CFTR by potentiation of its interaction with Na+/H + exchanger regulatory factor 1. Biochemistry. 2014 Jul 1;53(25):4169-79. [CrossRef]
- Loureiro CA, Matos AM, Dias-Alves Â, Pereira JF, Uliyakina I, Barros P, Amaral MD, Matos P. A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Sci Signal. 2015 May 19;8(377):ra48. [CrossRef]
Figure 1.
Schematic of NHERF1 based on findings of Zhang Q et al. J Biol Chem. 2019 Mar 22;294(12):4546-71 [11], and Bhattacharya S et al. J Biol Chem. 2019 Jul 19;294(29):11297-310 [89]. E43: essential residue for NaPi-2a binding; S77, T95, S162, S290: key phosphorylation sites for PTH inactivation of Na-Pi-2a; PP1: protein phosphatase 1; GYGF: core PDZ binding motif.
Figure 1.
Schematic of NHERF1 based on findings of Zhang Q et al. J Biol Chem. 2019 Mar 22;294(12):4546-71 [11], and Bhattacharya S et al. J Biol Chem. 2019 Jul 19;294(29):11297-310 [89]. E43: essential residue for NaPi-2a binding; S77, T95, S162, S290: key phosphorylation sites for PTH inactivation of Na-Pi-2a; PP1: protein phosphatase 1; GYGF: core PDZ binding motif.
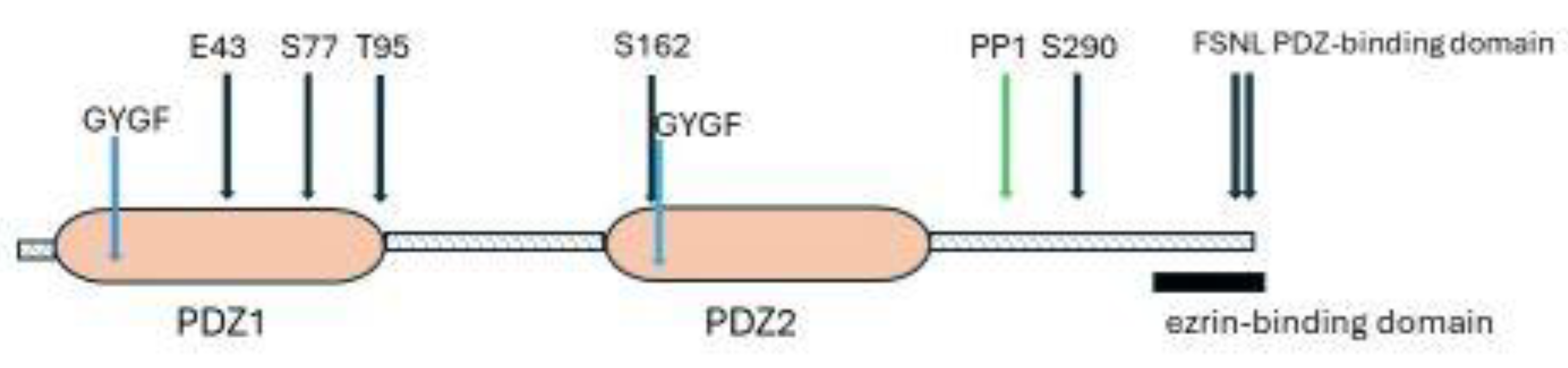
Figure 2.
PTH activation of the PTH1R receptor. 2A: the sequence of receptor activation. 2B: G-protein signaling by PTH/PTH1R. GRK2 G Protein-Coupled Receptor Kinase 2, IP3 inositol trisphosphate, DAG diacylglycerol, CAMKII Ca2+/calmodulin-dependent protein kinase II, epac exchange protein directly activated by c-AMP.
Figure 2.
PTH activation of the PTH1R receptor. 2A: the sequence of receptor activation. 2B: G-protein signaling by PTH/PTH1R. GRK2 G Protein-Coupled Receptor Kinase 2, IP3 inositol trisphosphate, DAG diacylglycerol, CAMKII Ca2+/calmodulin-dependent protein kinase II, epac exchange protein directly activated by c-AMP.

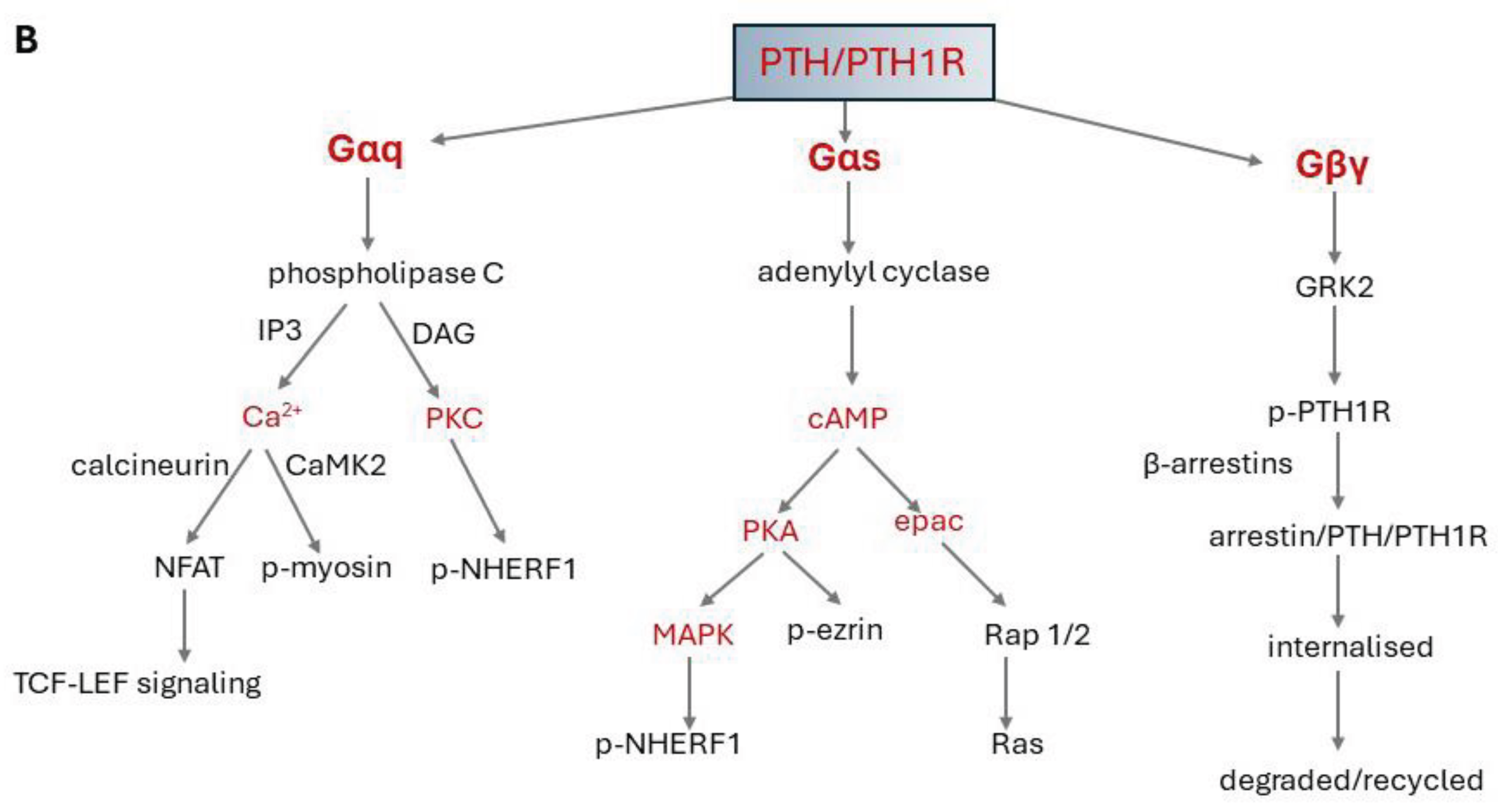
Figure 3.
Schematic of Epac1 (exchange protein directly activated by c-AMP 1). DEP Dishevelled/Egl-10/pleckstrin domain, CBD cAMP-binding domain, REM RAS exchange motif domain, RA RAS association domain, CDC25HD cell division cycle 25 -homology domain.
Figure 3.
Schematic of Epac1 (exchange protein directly activated by c-AMP 1). DEP Dishevelled/Egl-10/pleckstrin domain, CBD cAMP-binding domain, REM RAS exchange motif domain, RA RAS association domain, CDC25HD cell division cycle 25 -homology domain.
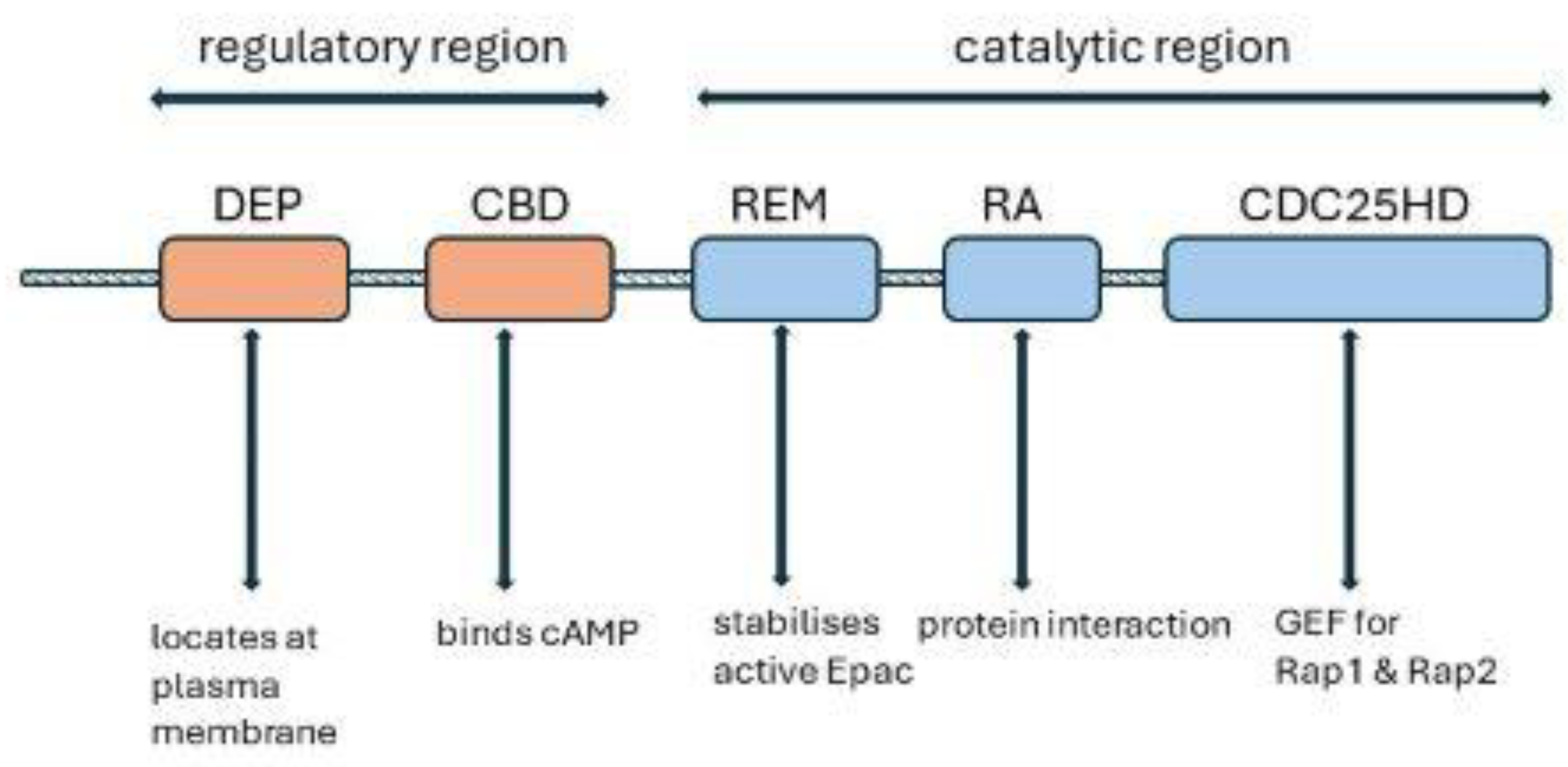
Figure 4.
Dopamine production and metabolism in the proximal renal tubule. L-DOPA 3,4-dihydroxy-L-phenylalanine, DOPAC 3,4-Dihydroxyphenylacetic acid, HVA Homovanillic acid, D1R dopamine 1 receptor, SLC7A8 and SLC7A9/SLC3A1 amino acid transporters, AADC aromatic L-amino acid decarboxylase, MAO monoamine oxidase, COMT catechol-O-methyltransferase.
Figure 4.
Dopamine production and metabolism in the proximal renal tubule. L-DOPA 3,4-dihydroxy-L-phenylalanine, DOPAC 3,4-Dihydroxyphenylacetic acid, HVA Homovanillic acid, D1R dopamine 1 receptor, SLC7A8 and SLC7A9/SLC3A1 amino acid transporters, AADC aromatic L-amino acid decarboxylase, MAO monoamine oxidase, COMT catechol-O-methyltransferase.
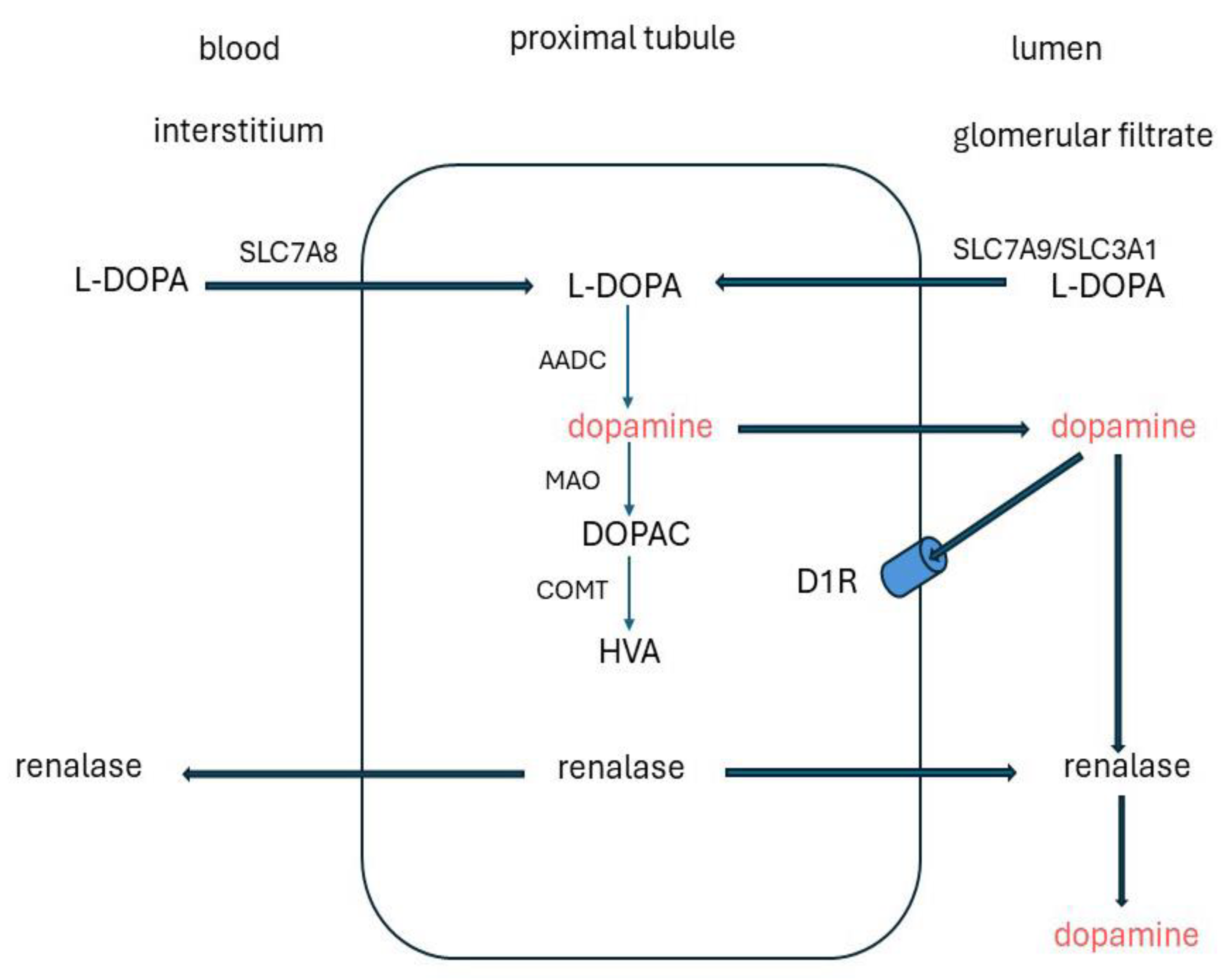
Figure 5.
FGF23/FGFR1c-αklotho signalling pathway which mediates phosphorylation of NHERF1. FGF23 decreases phosphate absorption through activation of the ERK1/2-SGK1 signaling pathway and phosphorylation of NHERF1 [239,244,267]. As for PTH, phosphorylation of Ser 290 is proposed as a central event [11,93]. FRS2 Fibroblast growth factor (FGF) receptor substrate 2, GRB2 Growth factor receptor-bound protein 2, SOS1 Son of sevenless homolog 1 (SOS Ras/Rac Guanine Nucleotide Exchange Factor 1) SGK1 Serum/Glucocorticoid-Regulated Kinase 1.
Figure 5.
FGF23/FGFR1c-αklotho signalling pathway which mediates phosphorylation of NHERF1. FGF23 decreases phosphate absorption through activation of the ERK1/2-SGK1 signaling pathway and phosphorylation of NHERF1 [239,244,267]. As for PTH, phosphorylation of Ser 290 is proposed as a central event [11,93]. FRS2 Fibroblast growth factor (FGF) receptor substrate 2, GRB2 Growth factor receptor-bound protein 2, SOS1 Son of sevenless homolog 1 (SOS Ras/Rac Guanine Nucleotide Exchange Factor 1) SGK1 Serum/Glucocorticoid-Regulated Kinase 1.
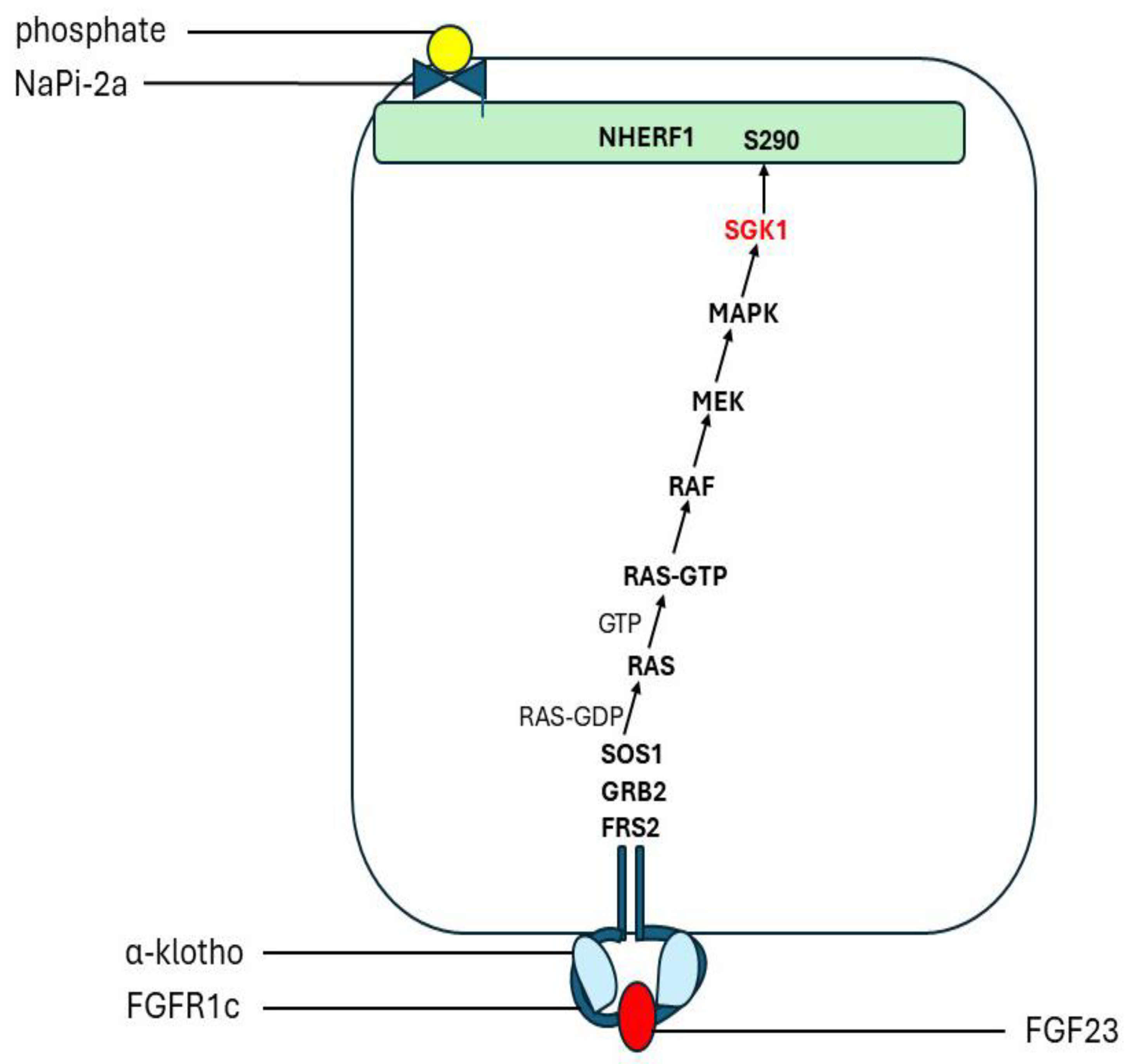
Figure 6.
Schematic of human Regulator of G protein signaling 14 [RGS14]. RGS Regulator of G protein signaling motif, RBD1/RBD2 Ras/Rap-binding domains 1 and 2, GPR G-protein regulator (alias GoLoco) motif, CBD DSAL C-terminal PDZ-binding motif. The C-terminal RGS14 extension found in humans and primates is indicated.
Figure 6.
Schematic of human Regulator of G protein signaling 14 [RGS14]. RGS Regulator of G protein signaling motif, RBD1/RBD2 Ras/Rap-binding domains 1 and 2, GPR G-protein regulator (alias GoLoco) motif, CBD DSAL C-terminal PDZ-binding motif. The C-terminal RGS14 extension found in humans and primates is indicated.
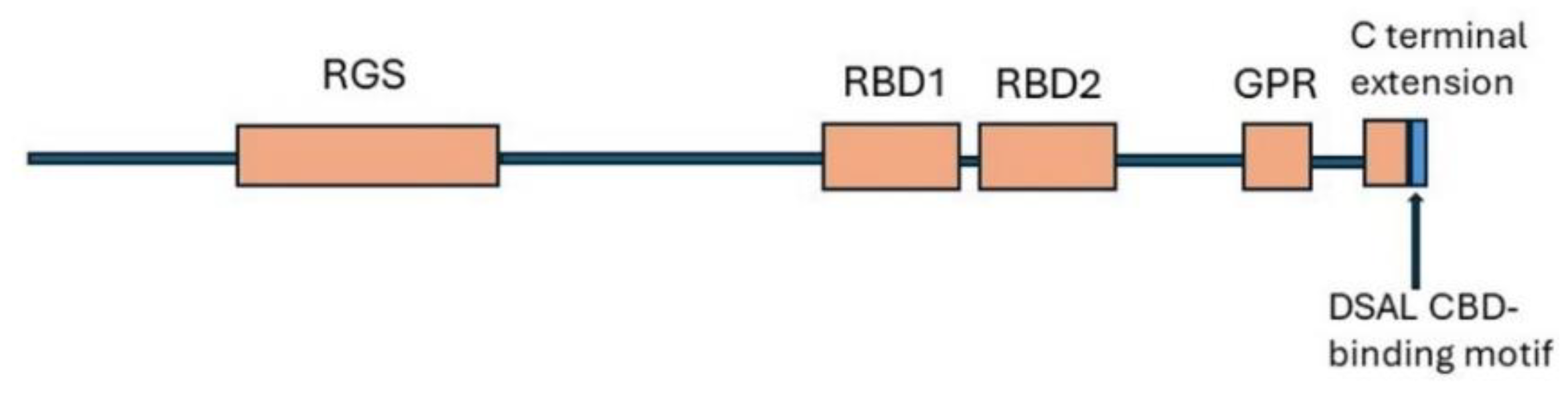
Figure 7.
Single nucleotide polymorphism (SNPs) in the RAP 1 and 2 binding domain of RGS14 identified in genome-wide association studies for kidney stones or plasma phosphate. Studies: Kestenbaum et al. 2010 [278], Laster et al. 2022 [279], Howles et al. 2019 [274], Urabe et al. [2012], Tanikawa et al. [2019]. A fourth SNP was identified up-stream, in the RGS domain of GS14 [273,276].
Figure 7.
Single nucleotide polymorphism (SNPs) in the RAP 1 and 2 binding domain of RGS14 identified in genome-wide association studies for kidney stones or plasma phosphate. Studies: Kestenbaum et al. 2010 [278], Laster et al. 2022 [279], Howles et al. 2019 [274], Urabe et al. [2012], Tanikawa et al. [2019]. A fourth SNP was identified up-stream, in the RGS domain of GS14 [273,276].
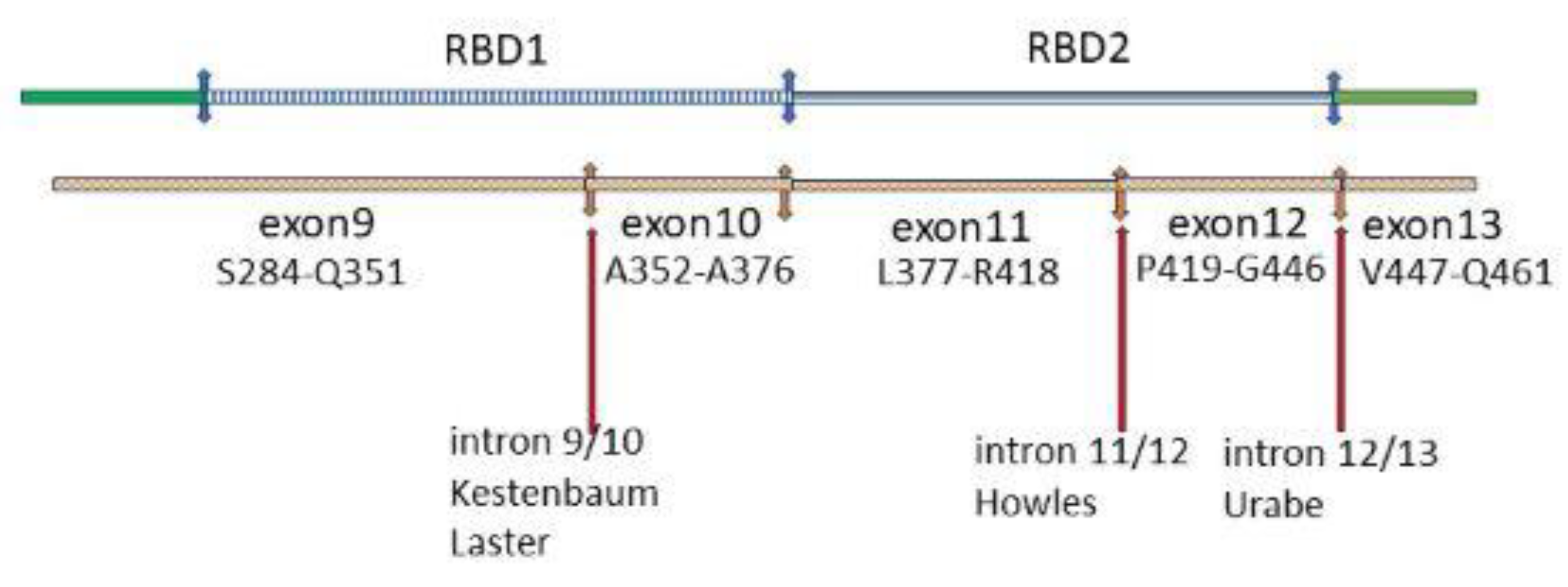
Figure 8.
Structure of MEPE (matrix extracellular phosphoglycoprotein), based on Rowe PSN. Crit Rev Eukaryot Gene Expr 2012; 22: 61-86 [330]. 8A: Schematic of intact MEPE showing locations of the AC-100 domain, ASARM motif and N-glycosylation site. 8B: Amino acid sequence of the AC-100domain. RGD integrin binding motif, SGDG glycosaminoglycans-binding motif. 8C: Proteolytic cleavage sites (arrows). NN N-glycosylation sites, S (red) serine residues, five are casein kinase (FAM2C) phosphorylation sites.
Figure 8.
Structure of MEPE (matrix extracellular phosphoglycoprotein), based on Rowe PSN. Crit Rev Eukaryot Gene Expr 2012; 22: 61-86 [330]. 8A: Schematic of intact MEPE showing locations of the AC-100 domain, ASARM motif and N-glycosylation site. 8B: Amino acid sequence of the AC-100domain. RGD integrin binding motif, SGDG glycosaminoglycans-binding motif. 8C: Proteolytic cleavage sites (arrows). NN N-glycosylation sites, S (red) serine residues, five are casein kinase (FAM2C) phosphorylation sites.
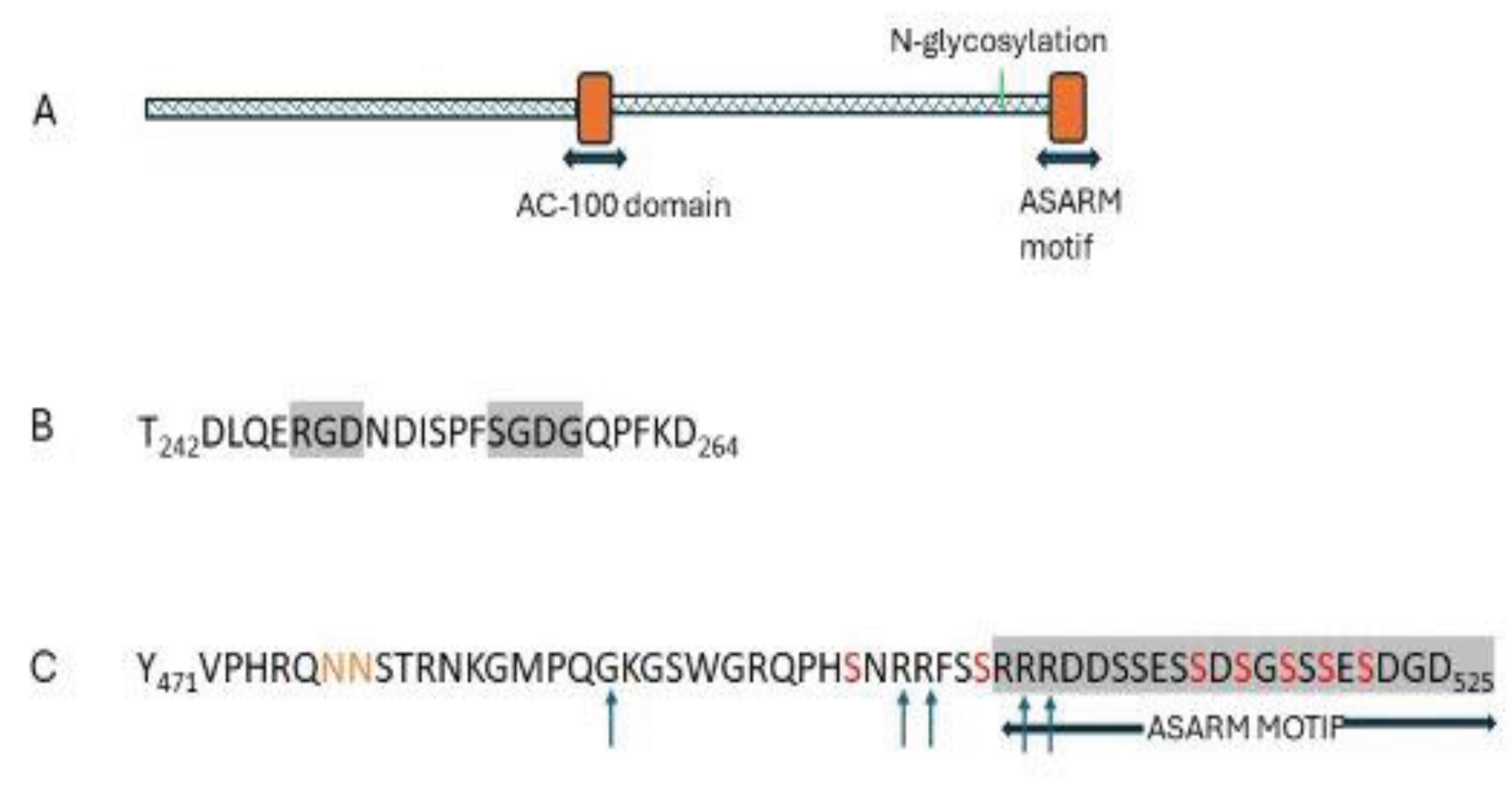

Table 1.
Inherited disorders of PTH/PTHR1 signalling.
| Disorder | Gene | Protein | Plasma Ca2+ |
Pi |
PTH |
FEPO4 | Urine cAMP post PTH | Bone deformity |
| Jansen’s Metaphyseal chondrodysplasia [9,162,163,164] |
PTH1R | PTH/ PTHrP receptor | ↑ | ↓ | low/ ND | ↑ | - | Yes |
| Isolated PTH deficiency [159,165] |
PTH | PTH | ↓ | ↑ | low/ ND | ↓ | - | No |
| ꝉ Pseudohypo- parathyroidism 1A [9,158,160,166,167] |
GNAS from mother | Gsα | ↓ | ↑ | ↑ | ↓ | ↓ | Yes |
| ꝉ Pseudo-pseudo hypoparathyroidism [9,158,160,166,167] |
GNAS from father | Gsα | norm | norm | norm | norm | norm | Yes |
ꝉ Gsα is an imprinted gene. In most tissues, including bone, there is biallelic expression. In the renal cortex, Gsα expression from the paternal allele is silenced. A Gsα mutation inherited from mother causes disturbances in bone and the renal cortex. The same mutation inherited from father has damaging effects on bone, but without a renal disturbance. There are also pseudohypoparathyroidism subclasses 1B and 1C with decreased renal PTH responses due to imprinting abnormalities [Reviewed [9,158,160]]. FEPO4 Fractional excretion of phosphate, norm-normal concentrations, ND not detectable.
Table 2.
Genetic disorders associated with increased circulating FGF23 ꝉ.
| Disorder | Gene defect | Mechanism |
|---|---|---|
| Increased FGF23 synthesis | ||
| Familial X-linked hypophosphatemic rickets (XLH) OMIM 193100 | PHEX: phosphate regulating endopeptidase homolog X-linked | PHEX deficiency |
| Autosomal recessive hypophosphatemia OMIM 241520 |
DMP1 dentin matrix protein 1 | DMP1 deficiency |
| Tumor-induced osteomalacia (TIO) | FN1-FGFR1 fusion gene. | Increased synthesis in tumours |
| Linear nevus sebaceous syndrome OMIM 163200 |
HRAS, KRAS, and NRAS | Increased synthesis in bone |
| McCune-Albright syndrome (fibrous dysplasia) | Activating mutations of GNAS; protein product Gsα | Increased FGF23 synthesis in bone [215,216] |
| Osteoglophonic dysplasia | FGFR1c- activating mutation | Increased FGF23 synthesis-? in bone [19] |
| Non-lethal Raine syndrome | FAM20C: family with sequence similarity 20, member C (Golgi Casein Kinase) | Increased FGF23 synthesis-mechanism unclear [217,218] |
| Osteoglophonic dysplasia | FGFR1c- activating mutation | Increased FGF23 synthesis-? in bone [19] |
| Decreased FGF23 degradation | ||
| Autosomal dominant hypophosphatemic rickets (ADR) OMIM 193100 | N-acetylgalactosaminyl-transferase 3 (GALNT3) | Decreased FGF23 cleavage |
Table 3.
Studies to investigate MEPE and ASARM function in kidneys.
| Study | Procedure | Relevant findings |
|---|---|---|
| MEPE infusion | ||
| Rowe et al 2004 [326] | Wt mice; boluses of human MEPE for 31h | ↓serum phosphate,↓1,25(OH)2 D ↑FEPO4 |
| Dobbie et al 2008 [334] | Wt rats; saline or MEPE infused at three doses for 2h GFR measured with inulin. |
Dose-dependent ↑ FEPO4 |
| Shirley et al 2010 [335] | Wt rats; 2h infusion of vehicle or MEPE; fluid from proximal renal tubules | Significant ↑FEPO4 |
| Animal Models | ||
| Gowen et al 2002 [336] | Mouse mepe KO at 4m and 12m | Homoz -/-, Heteroz -/+: bone mass significant ↑ serum phosphate as wt |
| Zelenchuk et al 2015 [337] | Group 1: Mouse mepe KO up to 22m | at 22m ↑serum phosphate; ↑FEPO4 Kidney mRNA v. wt: ↑NaPi-2a;↑NaPi-2c ↑Bone mass |
| David et al 2009 [333] ꝉ | Transgenic mouse with over-expressed mepe up to 19 weeks normal phosphate & vit D intakes | MEPE protein expressed in bone and kidneys ↑ number of renal blood vessels;↓ diameter ↑serum phosphate; ↓ FEPO4; ↑renin/aldosterone Kidney mRNA ↑ NaPi-2a;↓NaPi-2c ↑VEGFR ↑Bone mass; Bone mineral↓; osteoid ↑ |
| Cell studies | ||
| Renal | ||
| Rowe et al 2004 [326] | 33PO4 uptake: human proximal tubule cells & renal cell line treated with MEPE for 3-4h | significant ↓33PO4 uptake; ↓Vmax Km no change |
| Marks et al 2008 [338] | Rats infused with MEPE for 3h. analyses of BBM vesicles | NaPi-IIa: significant↓- dose-dependent NaPi-IIc: trend for an ↑ |
| ASARM infusion | ||
| David et al 2011 [339] | Wt mice aged 8 to 12w continuous infusion of ASARM or vehicle; normal phosphate diet | Serum:2-fold ↑ASARM & 1,25(OH)2vit D; ↑FGF23 ↓phosphate; PTH no change. ↑FEPO4 |
| Animal Model | ||
| Zelenchuk et al 2015 [337] ꝉꝉ | Group 2: Transgenic Mouse mepe KO with over-expressed ASARM up to 22m | ↓serum phosphate; ↓1,25(OH)2 vit D; ↑PTH; ↑FGF23; ↑FEPO4; Kidney mRNA ↑Npt2a;↑Npt2c;↓bone mineral |
ꝉ Marked renal vasculature changes affecting Na+ balance. ꝉꝉ Significantly reduced body weight (approx. 80%). Raised FGF23 would contribute to increased phosphate excretion and low 1,25(OH)2D. FEPO4 Fractional excretion of phosphate; BBM brush border membrane VEGFR vascular endothelial growth factor receptor.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



