Submitted:
24 March 2024
Posted:
26 March 2024
Read the latest preprint version here
Abstract
In Virology, the term reverse genetics refers to a set of methodologies in which changes are introduced into the viral genome and their effects on the generation of infectious viral progeny and their phenotypic features are assessed. Reverse genetics emerged thanks to advances in recombinant DNA technology which made the isolation, cloning, and modification of genes through mutagenesis possible. Most virus reverse genetics studies depend on our capacity to rescue an infectious wild-type virus progeny from cell cultures transfected with an “infectious clone”. This infectious clone generally consists of a circular DNA plasmid containing a functional copy of the full-length viral genome, under the control of an appropriate RNA polymerase promoter. For most DNA viruses, reverse genetics systems are very straightforward, since DNA virus genomes are relatively easy to handle and modify and are also (with few notable exceptions) infectious per se. This is not true for RNA viruses, whose genomes need to be reverse transcribed into cDNA before any modification -such as restriction enzyme digestion, gene knock-out or knock-in, site directed mutagenesis, ligation, etc.- can be done. Establishing reverse genetics systems for members of the Caliciviridae has proven exceptionally challenging, due to the low number of members of this family that propagate in cell cultures. Despite the successful rescue of Feline calicivirus (FCV) from cDNA containing plasmid more than 20 years ago, reverse genetics methods are not routine procedures than can be easily extrapolated to other caliciviruses. Reports of calicivirus reverse genetics systems have been few and far between, in this review, we discuss the main pitfalls, failures, and delays behind several successful calicivirus infectious clones.
Keywords:
calicivirus
; reverse genetics
; infectious clone
; RNA virus
1. Introduction
Unlike classical genetics, in which efforts are made to discern an unknown genotype from the observation of the phenotype, reverse genetics relies on the direct alteration of the genotype and the study of the effects of such alterations on the phenotype. In the molecular virology field, the term reverse genetics refers to a set of methodologies in which changes are introduced into the viral genome and their effects on the generation of infectious viral progeny and their phenotypic features are assessed in terms of viability, gene essentiality, virulence, attenuation, tropism, host range, resistance to antivirals, etc.
Reverse genetics emerged thanks to recombinant DNA technology which made possible the isolation, cloning of genes, and their modification through mutagenesis. Most viral reverse genetics studies depend on the capability of rescuing an infectious wild-type viral progeny from cell cultures transfected with an “infectious clone”. An infectious clone generally consists of a circular DNA plasmid containing a functional copy of the full-length viral genome, under the control of an appropriate RNA polymerase promoter. For most DNA viruses, reverse genetics systems are straightforward, since DNA virus genomes are relatively easy to handle and modify, and are also (with few notable exceptions) infectious per se. This is not true for RNA viruses, whose genomes always need to be reverse transcribed into cDNA before any modification -such as restriction enzyme digestion, gene knock-out or knock-in, site-directed mutagenesis, ligation, etc.- can be done, as this molecular toolbox is not fully available for RNA alteration [1].
Even though fully functional infectious clones from ribosome-ready, positive-sense RNA viruses are theoretically simpler to obtain compared to those for negative-sense or double-stranded RNA genomes, reverse genetics systems for the Caliciviridae are exceptionally challenging. In part this may be due to the small number of members of this family that propagate in cell cultures. Despite success in the rescue of Feline calicivirus (FCV) from cDNA clones [2] more than 20 years ago, reverse genetics methods are not routine procedures than can be quickly extrapolated to other caliciviruses. Obtaining the RNA transcript from a cDNA clone of the viral genome is essential, but it does not ensure its biological activity, which is why reports of calicivirus reverse genetics system have been few and far between [1,3,4].
The methodology used for infectious clone rescue seeks to reproduce the effects of viral infection, by transfecting permissive cells either with a cDNA vector (generally a plasmid) containing the viral genome under control of appropriate promoter, or with synthetic RNA produced through in vitro transcription of the former. Regardless of the strategy followed, obtaining an infectious clone provides a powerful tool for the use of reverse genetics techniques. Thus allowing the manipulation of the viral genome and the study of the effects of certain gene changes (point mutations, deletions, insertions, inversions or translocations) on the biology of viruses, their replication cycle, the role of viral proteins in pathogenicity or in the interactions between viruses and the immune response components [3,5,6].
Additionally, reverse genetics opens new avenues for vaccine development based on the possibility of specific directed attenuation of viruses and the use of recombinant replicons or defective viruses as vectors for the expression of proteins with potential biotechnological application. Replicons are RNAs derived from viral genomes that retain the ability to replicate autonomously in the cytoplasm. Usually, replicons harbor partial or complete deletion of the genes encoding the structural proteins to prevent the formation of infectious particles. Such deletions also allow the replicon system to accept the insertion of foreign genes of interest without exceeding its coding capacity that ultimately could compromise RNA replication. Sometimes, the supplementation of virion structural proteins in trans allows packaging of the replicon within viral particles. These de novo produced viral particles are defective in their ability to produce progeny, since their genomes lack the sequence for structural proteins, but can be engineered to express foreign genes of interest for a single round of infection. Except for retroviruses, replicons derived from positive-sense RNA viruses do not integrate exogenous genetic information into the host cell genome. Several systems have been described for heterologous gene expression based on infectious cDNA clones of picornavirus [7], flavivirus [8], alphavirus [9] or coronavirus [6,10,11].
2. The Caliciviridae: Genome Organization, Gene Expression and Replication Strategies
The family Caliciviridae comprises viruses that infect vertebrates, such as birds, reptiles and mammals, including humans [reviewed in [12]]. In recent years, the number of genera making up the family increased from 5 to 11, including a total of 13 recognized species [4,13], which are shown in Figure 1.
Calicivirus have a linear, single-stranded, positive-sense RNA genome of 7.3 to 8.5 kb in length. The genome is flanked by well-conserved untranslated regions (UTR) at both the 5′ and 3′ termini [14]. The 5′-UTR (ranging from 4 to 19 nucleotides) always starts with the 5′-pGpU-3′ sequence [15], and the most 5′-G is covalently linked to a nucleotidylated tyrosine residue of a small basic virus-encoded polypeptide, namely “viral protein genome-linked” (VPg) [16]. A polycistronic coding region is followed by a downstream 3′-UTR spanning 46 to 108 nucleotides and a variable-length poly-A tail [15].
A subgenomic RNA (sgRNA), which is 3′ co-terminal with the genomic RNA (gRNA) is produced during replication and packaged into progeny virions along with the gRNA. The sgRNA has a similar organization in all genera, being equivalent to the last third of its corresponding genome and is also VPg-linked at the 5′ terminus [17].
The genome organization has established itself as a distinctive feature of each genus within the Caliciviridae (Figure 2). Two clearly different models exist based on the number of open reading frames (ORFs) present within the coding region of the genome. Some calicivirus genomes are made up of two main ORFs (genera Bavo-, Lago-, Mino-, Naco-, Nebo-, Salo-, Valo- and Sapovirus) (Figure 2A), while other comprise three (genera Reco-, Vesi- and Norovirus) (Figure 2B). However, an additional nested ORF has been found within the major capsid protein (VP1) coding sequence in Sapovirus and Norovirus genomes (represented in Figure 2 with dashed lines). These cryptic ORFs have been designated as ORF3 and ORF4, respectively [13], and their putative functions will be briefly mentioned elsewhere in this review.
For all Caliciviridae members, ORF1 encodes a large precursor polyprotein, which is co- and post-translationally processed into precursors and mature polypeptides. The extent of proteolytic processing and therefore the mature products display remarkable differences between calicivirus genera [18,19]. During translation of viral RNA, the 5’most nonstructural gene products (NS1–NS7) appear first followed by the major structural protein (VP1) and the minor structural protein (VP2) [3].
In the “two-ORFs” model of genome organization, VP1 is the most C-terminal portion of the ORF1-encoded polyprotein. Therefore, theoretically VP1 could be released from the polyprotein upon NS6-mediated proteolytic cleavage following translation [20]. However, VP1 is mainly produced from the subgenomic RNA during late stages of infection, when higher amounts of the capsid protein are required for packaging [21]. For the viruses with organization following the “three-ORFs” model, the ORF1-encoded polyprotein ends with NS7 and VP1 is only produced during sgRNA translation. In both models, the ORF closest to the 3′-end is the smallest and contains information for the synthesis of the minor capsid protein VP2. This ORF generally overlaps to some extent with the previous ORF [22,23].
The roles of all non-structural proteins have not yet been fully elucidated, however, it is clear that NS3 possesses NTPase/helicase activity; NS5 (also referred to as VPg) is a paradigm-breaking polypeptide, since it acts as a protein primer for RNA synthesis initiation [25]. VPg also serves for the recruitment of translation initiation factors (such as eIF4A, eIF4E, eIF4G) onto ribosomes during viral RNA translation [26,27,28,29], a function which when absent is reportedly recoverable by simply adding a regular eukaryotic 7-methylguanosine cap structure to the 5′ end of viral RNA [2,30]. NS6 is a protease very similar to the picornavirus 3C cysteine protease, which is why the former is referred to as 3C-like protease in the literature. Calicivirus 3C-like cysteine proteases (NS6s) are considered members of a family of chymotrypsin-like serin proteases that contain a cysteine instead of serine as the nucleophile in the active site [18,20]. NS7 is an RNA-dependent RNA polymerase (RdRp), also known as viral replicase (reviewed in [12]). NS1/2 and NS4 have not been fully characterized but it has been hypothesized that, as they contain membrane-spanning hydrophobic domains, they might be involved in the rearrangement of cell organelle membranes during the assembly of membrane-associated virus replication factories [31]. It has also been suggested that they act as virulence factors (e.g., viroporins and antiviral immune response suppressors) thus playing roles in viral pathogenicity and epidemiological fitness (for a comprehensive review of calicivirus non-structural proteins and their analogs in picornaviruses, see the article by Smertina et al. [32]).
Although the taxonomy of caliciviruses is mainly based on sequence comparisons, there are some genome peculiarities within some of the genera regarding gene expression and processing of precursor proteins. For example, the ORF2 of all members of the Vesivirus genus produces a precursor VP1 protein that is further cleaved by the viral protease, removing a small N-terminal peptide and yielding the mature major capsid protein [18,33]. The small peptide released upon VP1 processing was named ‘leader of the capsid protein’ (LC) and has some ability to promote viral replication [34]. In FCV, the LC is considered essential for infection in vitro, and for the production of the characteristic virus-induced cytopathic effects [35].
Additional genus-specific extraordinary features come from the members of the Vesivirus, whose proteolytic and RNA polymerase enzymatic activities are exerted by a unique bi-functional polypeptide (referred to as NS6-7Pro-Pol), as no further proteolytic cleavage has been reported in their NS6 and NS7 junctions. Differences also exist in the processing of the NS1/2 junctions, with the Naco-, Reco- and Valovirus genera members showing no processing; Vesi-, Lago-, Nebo- and Sapovirus’ NS1/2 being processed by the viral NS6; while Norovirus members’ NS1/2 junctions are processed by the caspase-3 cellular protease (reviewed in [32]).
As previously stated, an additional ORF has been identified in sapovirus (ORF3) and murine norovirus (ORF4), which appears nested within the sequence coding for VP1. While the function of sapovirus ORF3 remain elusive [36], MNV’s ORF4 encodes a protein named as virulence factor 1 (VF1), with a potential role in regulating the innate immune response and apoptosis during the infection [19,37].
3. Calicivirus Replication Cycle
Following adsorption and entry into a susceptible and permissive cell, the viral genome is released in the cytoplasm and the translation of the ORF1 starts [3]. The VPg protein is crucial in this step, as it functions as a proteinaceous cap substitute for translation factors and ribosome positioning, in contrast to the eukaryotic mRNAs that require a 5′-cap structure for eIF4F cap-binding complex recruiting and protein synthesis initiation. The first product synthesized is the large polyprotein encoded by ORF1, which includes NS1-5, the cis-acting cysteine protease NS6 for its own processing and the viral replicase NS7.
Following translation, the genomic RNA (starting from its 3′-end) serves as a template for NS7-driven de novo RNA synthesis, yielding a transient double-strand RNA (dsRNA) intermediate [3,38]. The newly synthesized negative-sense RNA strand then serves as a template for the VPg-primed synthesis of multiple copies of both genome-length and subgenomic RNA that, in turn, are used as the substrate for several rounds of translation, allowing the accumulation of all viral proteins, especially the structural proteins VP1 and VP2 [3,17]. The VP2-encoding ORF is the second ORF within the sgRNA, just downstream the VP1-encoding ORF. Because the eukaryotic translation machinery only reads and translates the first ORF, most eukaryotic mRNAs are monocistronic, the translation of VP2 in the context of bicistronic caliciviral sgRNA is challenging. The mechanism underlying VP2 synthesis has not been fully elucidated in all caliciviruses, but several studies suggest a termination/reinitiation mechanism, where the ribosome backtracks from the first ORF’s stop codon to the next ORF’s initiating AUG. Apparently, this phenomenon strictly depends on the presence of the above-mentioned terminal codon and a specific signal sequence called termination upstream ribosome binding site (TURBS), which encompasses approximately the 80 nucleotides upstream the stop codon [22,23,39,40].
When critical levels of structural proteins and genomic RNA are reached, capsid assembly, genome packaging and release of progeny virions take place, ultimately leading to cell lysis. As for most viruses with isometric (e.g., icosahedral) symmetry, capsid self-assembly is thermodynamically favored and spontaneously occurs under specific conditions [41]. A role for VP2 in this process has been reported [42]. It is noteworthy that the VPg-linked sgRNA is packaged together with the VPg-linked genomic RNA inside progeny virions, although the biological significance of this phenomenon remains unknown [17]. A schematic representation of the replication cycle of a hypothetical calicivirus can be seen in Figure 3. Further details of the replication cycle of a model calicivirus are comprehensively reviewed in [3,43].
4. In Vitro Study of Caliciviruses
4.1. The challenges of Reverse Genetics
A permissive and productive cell culture system is a major step towards the functional analysis of viral proteins and opens up the possibility of efficiently recovering (rescuing) viruses, a pivotal prerequisite for reverse genetics studies [3]. A few caliciviruses are cultivable in vitro. Some notable examples include murine norovirus, which replicates well in murine macrophages, RAW264, BV-2 and WEHI cells, etc. [45,46]; feline calicivirus, in Crandell-Rees feline kidney (CRFK) cells [47]; and recovirus A in monkey kidney (LLC-MK2) cell line [48]. However, for many members of the family, a robust and reproducible cell-culture system has not yet been reported, and this has hampered the study of the calicivirus replication cycle and has delayed the development of reverse genetics systems [3]. This is especially true for human noroviruses, whose rescue from BJAB cells –a B lymphocyte-derived cell line– has been described, though the success of such virion rescue is rather limited, due to the poor virus recovery yield [49].
Calicivirus genomes are infectious per se and are immediately translated by the eukaryotic ribosomes, following entry into the host cell. This means that, if introduced into permissive cells, full-length viral RNA should ultimately lead to productive infection and the concomitant generation of infectious virions (virus rescue or recovery). Although the term virus rescue has systematically been employed as a synonym of reverse genetics, the latter more properly refers to the attempts of recovering virions from mutated or genetically altered viral genomes, provided that the rescue of wild-type virions from unaltered genomes has previously been achieved in a reproducible manner, so that it could be set up as a parallel control assay in every reverse genetics experiment.
As previously mentioned, the advent of recombinant DNA technology provided a vast toolbox for DNA alteration (such as hundreds of different restriction endonucleases, DNA modifying enzymes, ligases, etc.). However, because RNA molecule modification is not as straightforward, for RNA viruses a complete genome-length cDNA clone, obtained through reverse transcription (RT), must be assembled into an appropriate expression vector before any genetic alteration (reverse genetics study) can be done. While advances in RT technology mean that the synthesis of a full-length cDNA is relatively simple, the choice of a final sequence after cloning may not be so: due to the lack of proofreading activity of RdRps, most RNA viruses’ stocks are reportedly a mix of slightly different (polymorphic) genomes, often referred to as virus quasispecies. The first challenge encountered during the setup of an RNA virus reverse genetics system is the fact that a single cDNA clone only represents a unique genome sequence within the RNA quasispecies. Because the cloning procedure does not assess sequence quality in terms of virus fitness, this selected sequence may harbor lethal point mutations that render it replication-incompetent [50,51]. Such defective RNA molecules would most probably be naturally removed from the pool in subsequent rounds of replication but, they may end up being the chosen cDNA picked from bacterial colonies during cloning [1].
Depending on the viral genome size, the construction of a genome-length cDNA vector supporting the synthesis of infectious transcripts can be long and tedious [6,52,53], but once a reproducible workflow is established from genotype (viral genome) to the phenotype (rescued virions), it represents a major leap for virus research. An established and reproducible infectious clone provides a powerful tool for reverse genetics experiments, in which researchers directly manipulate the viral genome (for example introducing point mutations, deletions, insertions, inversions or translocations) and further assess the effects of such manipulations on the phenotype in terms of fitness, replication competence (virus yield, attenuation), tropism, host range, virulence, pathogenicity, immunogenicity, etc. Also, reverse genetics finds applications in the study of viral proteins functions and vaccine development [5].
For many viral genomes, another relevant challenge is the intrinsic instability of large full-length constructs and their toxicity for bacteria, which make the preparative purification of genome-length cDNA-containing plasmids, a very tricky task. Uncontrolled sequence rearrangements and mutations that render the derived RNA transcripts non-functional, have been systematically reported [54,55,56]. Cloning the genomic cDNA of interest into an expression vector, trying a different bacterial strain, lowering the culture temperature (for example, using 30-32°C instead of 37°C), or even increasing the bacterial culture volume to compensate for the reduced plasmid yield during maxi-preps, could eventually help [30].
Figure 4 summarizes the two main strategies for the recovery of infectious virus from a full genome-encoding cDNA. The permissive cell culture can be transfected with either synthetic genome-emulating, potentially infectious RNA obtained through in vitro transcription (strategy A); or with the genome-length cDNA, properly cloned into an expression vector under the control of an appropriate RNA polymerase promoter (strategy B). The simultaneous occurrence of the genome-length cDNA-expressing vector and the relevant RNA polymerase will drive transcription. Regardless of the strategy used, the presence of infectious genome-length RNA will ultimately lead to productive infection: viral protein synthesis, viral genome replication, sgRNA synthesis, virus assembly, maturation, and virion progeny release (virus rescue).
When cloning a viral genome in the form of cDNA into an expression vector or plasmid, several prokaryotic or eukaryotic RNA polymerase promoters can be used to drive transcription. SP6, T3 and T7 bacteriophages RNA polymerase promoters are the most popular prokaryotic promoters and are usually used when in vitro transcription and further RNA transfection is the goal (Figure 4, strategy A), although eukaryotic promoter-based RNA synthesis systems are also available (e.g., CMV promoter-driven in vitro transcription kit).
The synthesis of cDNA is performed by RT using viral genomic RNA as a template, and an oligonucleotide primer annealing within the 3’-end of the viral genome (usually oligo-dT, since calicivirus RNAs are 3′-polyadenylated). Secondary structures are a distinctive feature of ssRNA. When present in viral genomic RNA, highly stable and strong variants of such structures may pose a major obstacle for RT fidelity, sometimes leading to sequence gaps due to reverse transcriptase sequence jumps while copying, or other types of mutations that ultimately compromise the recovery of viable viruses [56]. After ribonuclease H-mediated removal of viral RNA, single-stranded first DNA is amplified by PCR using a second primer. In general, the larger the genome size, the lower the probability of obtaining an error-free, genome-length cDNA copy in a row. Therefore, sometimes this goal is achieved by assembling several smaller cDNA pieces into the whole genomic cDNA, by means of ligation following small PCRs and enzymatic cut within unique endonucleases restriction sites [30,56] or through Gibson assembly [57].
Overall, for a cDNA-derived RNA transcript to mimic as closely as possible the viral genome and trigger a productive infection, extreme care should be taken during the cDNA construct design, regarding the selection of the expression vector, the cloning strategy, the choice of promoter, delivery method, etc. Not only do the coding regions of the genomic sequence need to be accurate, but also both the 5′ and 3′ ends, due to their crucial roles in translation and replication processes, respectively. It is generally accepted that the presence of non-viral nucleotides upstream the 5′-end of RNA transcripts (potentially coming from the vector multiple cloning site or promoter) drastically reduces (or completely abolish) infectivity, which jeopardizes virus recovery.
Regarding this approach, it is worth recalling that in vitro synthesized transcripts need to be “translation-ready”, a quality naturally conferred to calicivirus RNAs by the multifaceted VPg protein. However, the in vitro covalent linkage of synthetic transcripts to VPg is technically challenging [28] therefore, the generation of a capped 5′-end (as a VPg substitute) is required for translation initiation of synthetic RNAs. A synthetic cap structure has been used (m7G [5′]ppp[5′]G) at the 5′-end of in vitro transcribed RNA allowing the recovery of infectious viruses [2], albeit sometimes with low efficiency [58]. The 5′-cap structure can be either co-transcriptionally incorporated into nascent RNA, or post-transcriptionally added to RNA. Alternatively, the introduction of an internal ribosome entry site (IRES) within the 5′ UTR of RNA transcripts bypasses the requirement for a covalently linked VPg or a 5′-cap structure. In vitro translation of synthetic RNA can be attempted as a complementary assay to evaluate the translation-worthiness of transcripts [59].
For the transfection of vectors containing transcription-ready cDNA (Figure 4, strategy B), eukaryotic promoters such as the CMV, SV40 or the EF-1α promoters are often used. The synthesis of genome-emulating RNA transcripts takes places in the nucleus and is catalyzed by the eukaryotic cell RNA polymerase II. If a eukaryotic promoter cannot be used or a prokaryotic promoter is preferred, the transcription of viral genome-like cDNA can alternatively be controlled by a prokaryotic promoter such as that of T7 RNA polymerase, provided that this phage’s transcriptase, which is naturally absent in the eukaryotic cell, is provided in trans. The supplementation of eukaryotic cells with bacteriophage RNA polymerase in trans to drive transcription of calicivirus cDNA can be achieved by infecting the cells with a “helper virus”, which is usually a recombinant poxvirus expressing T7 RNA polymerase, such as vaccinia virus-T7 (rVV-T7), Ankara-modified vaccinia-T7 (rMVA-T7) or fowlpoxvirus-T7 (rFPV-T7). The infection with helper virus is generally performed prior to cDNA transfection.
Some advantages of the use of helper poxviruses in calicivirus reverse genetics include the high levels of expression of the heterologous RNA polymerase which in turns guarantees a high transcription rate for calicivirus cDNA, and the fact that poxviruses encode their own RNA capping enzymatic complex to make caliciviral transcripts translation-ready. In addition, the complete poxvirus replication cycle occurs in the cytoplasm, thus avoiding potential deleterious effects due to interaction of calicivirus RNA transcripts with the nucleus. The major disadvantages associated with helper viruses’ usage during calicivirus reverse genetics consist of their toxicity for the host cells, the difficulties to distinguish between the helper virus-associated cytopathic effects (CPEs) and those CPEs potentially attributable to calicivirus rescue; as well as the need for specific methods for helper virus removal in case of successful calicivirus recovery. In this regard, fowlpoxvirus-based helper viruses are preferred over vaccinia ones because of its abortive replication cycle in mammalian cells, which prevent an FPV progeny being formed (reviewed in [60]).
The effectiveness of a specific helper virus in the recovery of a particular calicivirus is not guaranteed. Instead, the degree of success achieved has varied from one virus system to another and is rather the result of several trial-and-error experiments. For example, the rescue of porcine enteric calicivirus (PEC) from a cDNA clone systematically failed when using rMVA-T7 as a helper virus because of the strong CPE recorded in the host cell line [61], while rMVA-T7 was apparently useful to achieve some degree of human norovirus replication in 293T [62]. rMVA-T7 did not allow murine norovirus recovery upon transfection of RAW264.7 cells, however the virus rescue was successful when the helper virus rFPV-T7 was used instead [58]. Finally, the viral genome-encoding cDNA vector can also be transfected into a recombinant cell line expressing the T7 phage RNA polymerase, bypassing the need for a helper virus.
4.2. The Hallmarks of a Promising Virus-Expressing cDNA
Figure 5 is a schematic diagram showing the design of a functional calicivirus T7 promoter-based infectious clone, emphasizing aspects of the regulatory elements surrounding the viral genomic sequence. As previously mentioned, the T7 phage RNA polymerase promoter is by far the most commonly used system, as it tolerates well a minor 2-nt truncation of its 3′-end, which is required to avoid adding these two non-viral nucleotides to the transcript 5′-end, without a significant yield decrease. By doing so, one the first virus-genome nucleotide can be engineered to coincide with the transcription start site. Similarly, there is evidence that a single point mutation in the 3’-UTR can thwart virus rescue in a reverse genetics system. Chaudhry et al. found that a single nucleotide change at the last residue of murine norovirus-1 (MNV-1) 3’-UTR was sufficient for its inactivation [58].
Figure 5.
Schematic representation of a calicivirus, genome-length cDNA-expressing vector, showing the main genetic regulatory elements that should be included in an ideal infectious clone to generate functional “genome-emulating” RNA transcripts of a defined length with a precise 5′ terminus and a sufficiently long, polyadenylated free 3′ end. See further details in the text (Adapted from [30]).
Figure 5.
Schematic representation of a calicivirus, genome-length cDNA-expressing vector, showing the main genetic regulatory elements that should be included in an ideal infectious clone to generate functional “genome-emulating” RNA transcripts of a defined length with a precise 5′ terminus and a sufficiently long, polyadenylated free 3′ end. See further details in the text (Adapted from [30]).
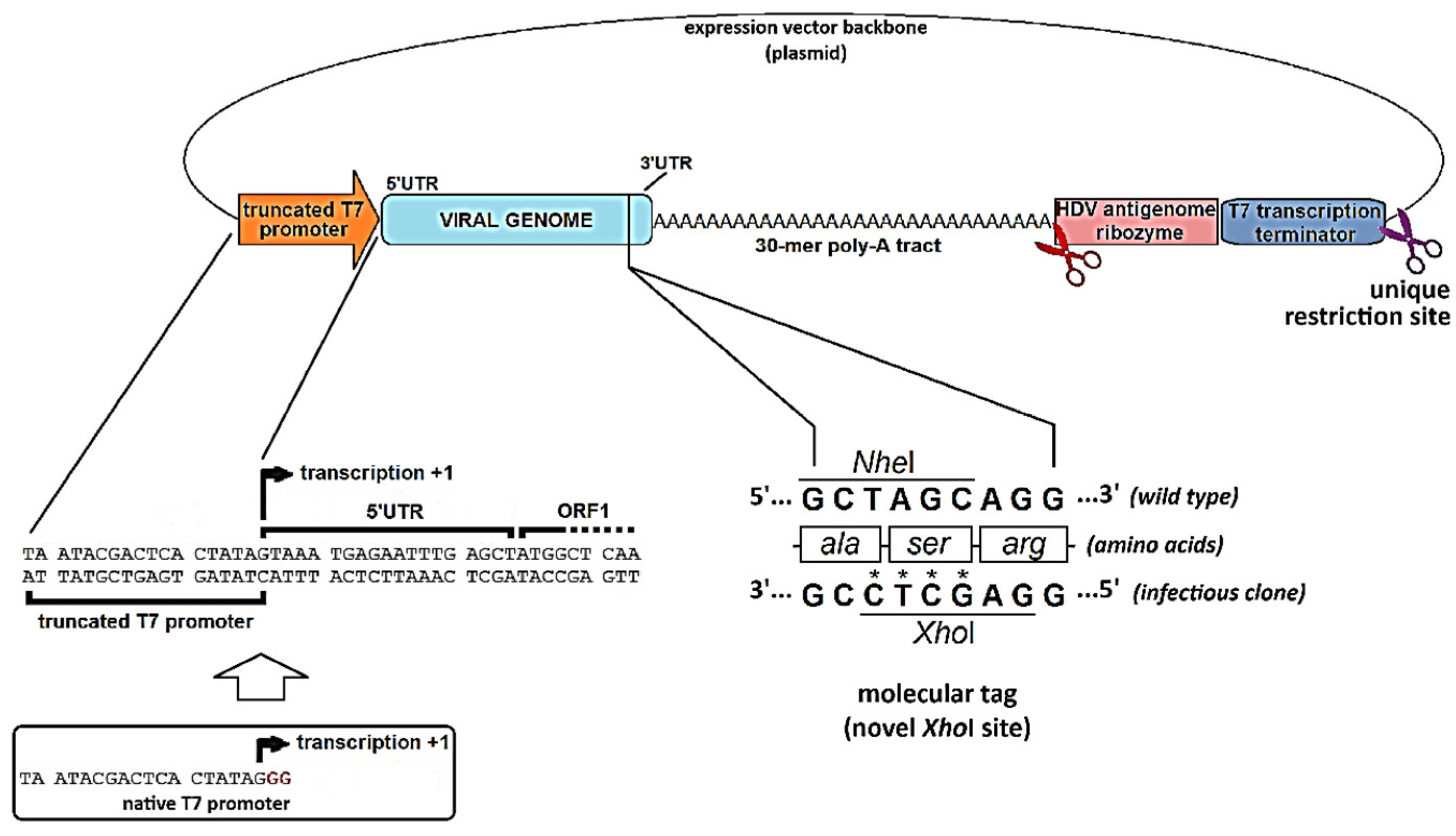
It is highly desirable for a viral genome-emulating RNA to possess a long poly-A tail downstream the 3′-UTR, as described for naturally occurring calicivirus genomes. The poly-A tail is an important determinant of RNA stability: it positively contributes to the RNA half-life and prevents 3′-exoribonucleases reaching the coding region before replication is completed, thus supporting virus recovery. Interestingly, the poly-A tail has also been found to be essential for positive-sense RNA viruses’ translation initiation, as the genome circularizes in a non-covalent fashion through protein-protein interactions that take place between poly-A interacting proteins and the translation initiation factors bound to the 5′-UTR (reviewed in [15]). Many successful reverse genetics systems included in their design a poly-A tail at least 30 nt-long [19,30,61].
The strategy followed to generate and deliver RNA transcripts, it must ensure these transcripts are of a defined length. For instance, if the cDNA vector is to be used in in vitro transcription using T7 phage RNA polymerase, a T7 terminator sequence at the end of the construct can be used to stop transcription in the right place. If a termination sequence is not available for the RNA polymerase used, then a unique restriction site can be placed following the sequence to be transcribed, in order to produce a linear cDNA of defined length, as the template for transcription. The RNA polymerase will fall off the linearized template, terminating the polymerization, a technique often referred to as run-off. Some successful infectious clone designs combine both strategies: a cDNA expressing vector is first linearized with the aid of a unique restriction enzyme, but the RNA polymerase is not expected to reach that region because transcription is supposed to stop at the upstream terminator (Figure 5) [30]. However, if in vitro transcription and further RNA delivery is not an option, and cDNA vector will be transfected instead, it is usually delivered in its circular form, thus transcription termination mostly relies on terminator sequences. An accurate polyadenylated free 3′-end can also be generated by including an autocatalytic ribozyme sequence immediately downstream the poly-A tract (such as the hepatitis delta virus ribozyme, HDV). This sequence will fold and adopt a specific secondary structure capable of cis-acting autocatalytic cleavage, producing a break in the phosphodiester bond right upstream its most 5′ nucleotide, which ultimately separates it from the preceding poly-A tail (Figure 5) [30].
Finally, the introduction of a molecular tag within the viral genome sequence is strongly recommended as it allows the cDNA-derived recovered virus to be unambiguously distinguished from a casuistic contamination of the reverse genetics experiment with wild-type virus. This molecular tag should consist of an innocuous and easy-to-track short sequence alteration, such as the introduction or removal of a notable restriction site that can be recognized in cDNA copies of the rescued viral sequence. Figure 4 (bottom, right) shows an ingenuous molecular tag within a calicivirus reverse genetics system that yields a virus progeny harboring a novel XhoI restriction site (which is absent in wild-type virus) and lacking an NheI site (which is present in wild-type). This tag consisted of four single nucleotide changes within VP2-encoding region (ORF3), elegantly engineered in a way that the amino acid sequence of VP2 remained unchanged [30].
4.3. When Things Go Wrong: Interrogating the Viral Genome for the Occurrence of Replication-Critical Events
Despite the success in the rescue of many viruses from cDNA clones, reverse genetics methods are not routine procedures for all positive-polarity RNA viruses. The synthetic RNA must be recognized by the cellular machinery to produce viral proteins and subsequently interact with them appropriately to complete the viral replication cycle. There is also a need for the existence of permissive cell lines that should desirably be easily transfectable as well. The guidelines compiled in this section, based on the close examination of calicivirus replication cycle, intends to bring a closer look to the events where virus rescue is more likely to fail, and provide a practical approach for their troubleshooting.
For any reverse genetics system to succeed in generating infectious viral particles, every single event of the viral replication cycle should be timely and efficiently achieved. In this regard, the assessment of proper performance of viral cDNA constructs can be conducted through tailored assays designed to directly or indirectly monitor the occurrence of such individual events. Table 1 summarizes the critical steps in calicivirus replication that should be controlled during the replication cycle to rescue infectious virions using different reverse genetics strategies, and under the control of various promoters.
After transfection with synthetic transcripts or cDNA-expressing vectors (Figure 4), the initial analytical approach entailed the microscopic examination of transfected cell monolayers (namely “passage 0”) to identify any cytopathic effects (CPE) that could be attributed to calicivirus’ replication, as a result of successful virus recovery. In general, it must be considered that most of the transfection reagents available may cause cytotoxic effect to some extent, and that such effect could sometimes be morphologically indistinguishable from virus-induced CPE observed in positive controls. Reportedly, the use of helper viruses such as rFPV-T7 or rMVA-T7 to deliver RNA polymerase in trans, may further increase the chance of observing misleading CPE, easily confounded with inexistent calicivirus recovery [30,58,59,63,64].
The difficulty in establishing the occurrence of viral rescue at passage 0 based solely on optical microscopy highlights the need for evaluating the infectivity of the supernatants from these cells (passage 0 supernatant), by inoculating them into new monolayers of susceptible and permissive cells. These inoculated cell cultures (passage 1) neither receive the toxic effect of the transfection reagent, which is diluted more than 10-fold in fresh culture medium, nor are they infected with the helper virus, as supernatants from passage 0 can be filtered before their subsequent inoculation into permissive cells. These helper poxviruses usually are larger than 0.2 μm in diameter [65], therefore being mostly retained in the filters. Furthermore, in the case of FPV-T7, the only viral particles that could be present in passage 0 supernatants are those from the initial inoculum because this avian virus displays an abortive replication in mammalian cells and does not produce viral progeny [66] which also contributes to the fact that passage 1 cultures are not usually infected.
If no signs of CPE can be seen in cell cultures at passage 1, the reasons could be related to specific reverse genetics features, the type of transfected nucleic acid, the cell line serving as the setting for passage 0, the promoters governing the transcription of the expression vectors, or subcellular localization of RNA transcripts. In other words, when genome-length constructs fail to generate an infectious calicivirus progeny, efforts should be targeted at pinpointing the molecular factors responsible for their inability for virus recovery.
After cell transfections, the presence of intact full-length RNA in the cytosol is critical for subsequent events in the cycle to occur. A preliminary approach for troubleshooting will be an initial detection and analysis of the integrity of such RNAs, through RT-PCR and 5′-RACE assays, respectively. Even if RNA quantities in the cytosol were low, these assays are extremely sensitive and will most likely detect any RNA in the order of picograms. Low concentration of internalized synthetic RNA could be due to massive RNA degradation during the transfection process (action of contaminating exogenous RNases or endogenous RNases activated as a cellular defense mechanism against the introduction of foreign RNA, especially if the capping procedure has not been efficient). 5′RACE (rapid amplification of cDNA 5′-ends) is a valuable technique for investigating the precise sequence of RNA most 5′ end. The procedure involves utilizing PCR to amplify regions between the known segments of the sequence and non-specific tags attached to the ends of the cDNA [67]
RNAs that mimic the viral genome produced in the cytosol or introduced into it, either from the outside or from the cell nucleus, should be capable of translation, resulting in the viral polyprotein that contains the non-structural polypeptides. The detection of some of these polypeptides by Western blot analysis allows the demonstration, on one hand, that genomic RNAs present in the cytosol are functional for translation, and on the other hand, that the region with protease activity is also operative and could process the non-structural viral polyprotein. However, Western blot is limited not only by the availability of specific antibodies but also by the viral protein yield obtained during RNA translation. Thus, if such yield is not sufficient to be detected through Western blot, the use of a more sensitive detection technique should be applied, e.g., radioactive labeling followed by autoradiography.
If the replicative cycle proceeds normally, translation of ORF 1 should be followed by the synthesis of a negative-sense intermediate RNA (negative strand) catalyzed by the viral RdRp. The presence of these negative strands can be assayed by Northern blot. From an internal promoter on this negative strand, the viral RdRp synthesizes the sgRNA that gives rise to the capsid proteins (VP1 and VP2). Additionally, from the 3’-end of the negative strand, the same enzyme produces multiple copies of the viral genome that will be packaged into newly produced viral capsids, forming the progeny.
The functionality of viral RdRp could indirectly be studied by means of the 5BR assay, which was initially developed for HCV [68] and subsequently adapted for calicivirus [69]. This assay utilizes components of one of the best-known signaling pathways involved in the innate antiviral response to RNA viral infections: the IFN-β synthesis pathway, activated by RIG-I [69]. During viral RNA replication, RdRps transiently generate dsRNA intermediates which are susceptible of being captured by the helicase domain of RIG-I C-terminal region. Through the caspase activation and recruitment domain (CARD) in its N-terminal region, the activated RIG-I protein can interact with another CARD domain present in MAVS, a mitochondrial transmembrane protein. This interaction, in turn, activates a series of kinases (TBK1, IKKε) that phosphorylate a cluster of serines in the C-terminal region of the transcription factor IRF3, promoting its dimerization and translocation to the nucleus. IRF3, together with NF-κB and ATF-2-c-Jun, forms a multiprotein complex or enhanceosome on the IFN-β promoter enhancer, triggering its transcription [70].
The 5BR assay artificially replicates this pathway using an expression vector based on the IFN-β promoter, in which the coding sequence for this cytokine has been replaced by the sequence of a luciferase. This vector is co-transfected simultaneously with a vector expressing the RIG-I protein, and the vector expressing the polymerase whose functionality is to be evaluated. The recorded luciferase activity is directly proportional to the amount of dsRNA and, therefore, to the RdRp activity.
5. Chronology of the Calicivirus Reverse Genetics
From the very first attempts of establishing a calicivirus reverse genetics system, the viral genome-length cDNA has been cloned into plasmid vectors in the context of several genetic regulatory elements, aiming at producing high quality, genome-length, and potentially infectious RNA transcripts. These elements were briefly introduced in Section 4.2, and include the choice of a RNA polymerase promoter, a poly-A tract flanking the genome by its 3′ side, a transcription termination signal for the polymerase, whenever available, or a unique restriction site for endonuclease cut to make fixed-length transcripts via run-off. Additional elements for RNA 3′-end processing may optionally be added, which help to produce a consistent pool of RNA molecules with an accurate 3′-end, for example the autocatalytic ribozyme.
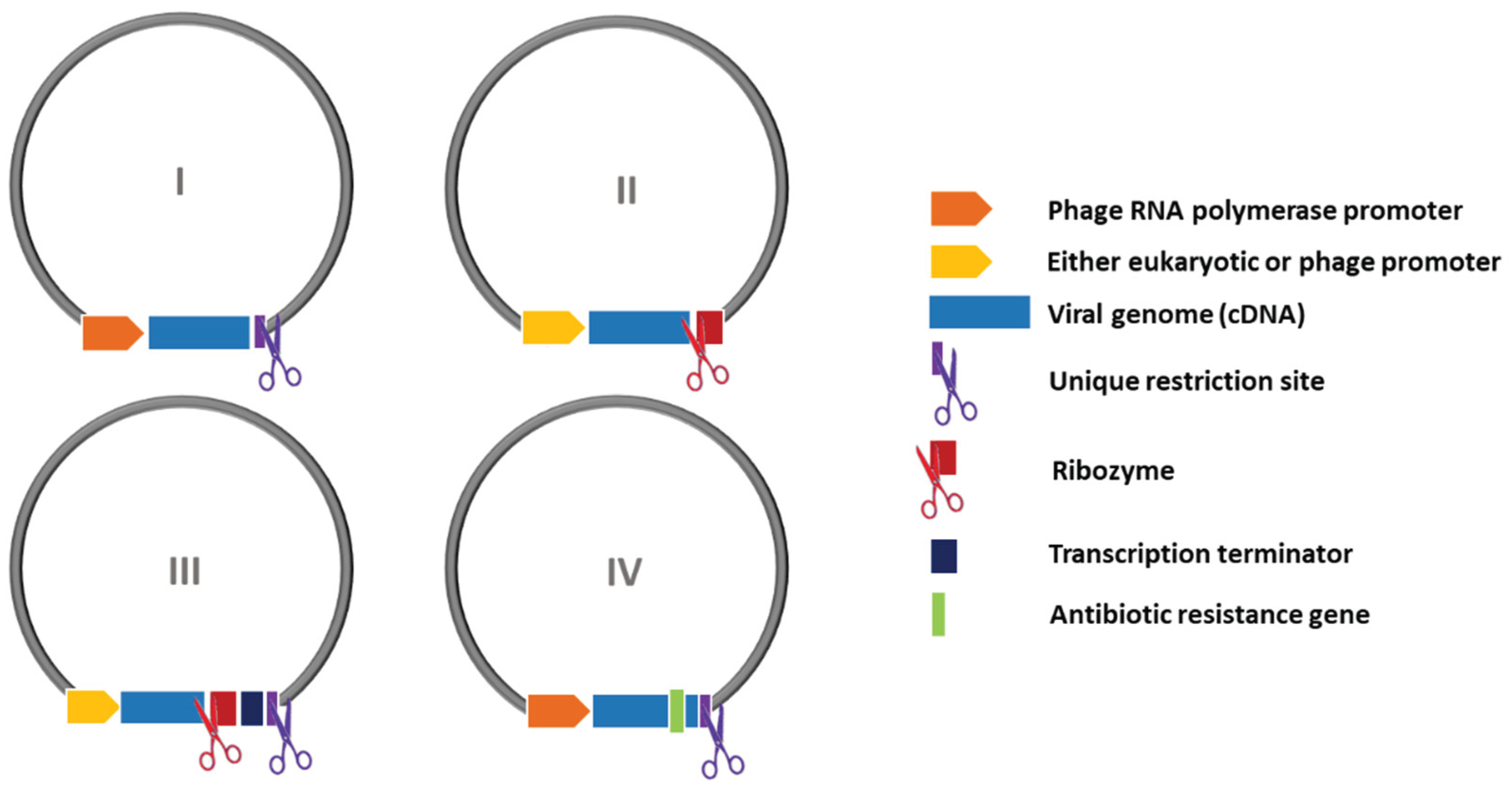
All these regulatory sequences have gradually been incorporated to improve the expression context of viral cDNA, and the overall quality of RNA transcripts. Currently, most reliable reverse genetics systems for caliciviruses combine several of such improvements, arranged according to one out of four possible combinations (I-IV), that are schematically summarized in Figure 6. In addition, a brief review of chronology of the different calicivirus reverse genetics are gathered in Table 2.
Figure 5.
Schematic representation of four distinct arrangements (designs I-IV) of regulatory elements surrounding viral cDNA within expression vectors, observed among the reported calicivirus reverse genetics systems. Designs are numbered in increasing order of complexity. See further details in the text.
Figure 5.
Schematic representation of four distinct arrangements (designs I-IV) of regulatory elements surrounding viral cDNA within expression vectors, observed among the reported calicivirus reverse genetics systems. Designs are numbered in increasing order of complexity. See further details in the text.
The first infectious clone of a calicivirus was established for a cultivable strain (Urbana) of Feline calicivirus (FCV) [2]. The construction of a cDNA clone was achieved from a library, by sequentially assembling three fragments into one genome-length copy. The design of the complete vector included the 5′ region of the viral genome juxtaposed to the promoter sequence of T7 bacteriophage RNA polymerase so that the transcription start point of the T7 RNA polymerase matched with the first nucleotide of the FCV genome. In the 3′ region, the sequence recognized by the restriction enzyme NotI was placed following the poly-A tail of the FCV cDNA sequence. This construction provides linear templates for in vitro synthesis of genome-length transcripts, but it involved the addition of two nucleotides of non-viral origin downstream the poly-A tail. Transfection of these synthetic transcripts in CRFK cells resulted in an identical infectious process to that caused by RNA purified from virions. To confirm that the rescued virus came from the vector expressing the genome of cloned FCV, site-directed mutagenesis of the cDNA clone was performed to replace a StuI site with a HindIII restriction site, as a readily detectable molecular tag.
The synthetic RNA derived from this modified vector was also infectious and produced recombinant FCV virions whose packaged genomic RNA contained the introduced mutation. The authors stressed the need for a cap analog added to the in vitro transcription since transcripts without a cap were not infectious. The FCV infectious clone was then used to investigate the proteolytic processing of capsid protein precursor [18] and the polyprotein encoded in ORF-1, by introducing point mutations [71], as well as for determining the VPg residue responsible for binding to viral RNA [64]. These reverse genetics techniques have also been used to generate chimeric viruses in order to study antigenic variation in FCV [72], and to provide valuable information about virus replication mechanisms [34,35,73,74,75].
Following a similar approach, infectious clones were described for another FCV, vaccine strain 2024 [76], for Porcine enteric calicivirus (PEC) [61], and for Tulane virus (TV) [77]. In all cases, the expression vector contained the corresponding sequence of cDNA from the viral genome flanked by the T7 RNA polymerase promoter and by a unique restriction site to obtain linear templates for in vitro transcription. The incorporation of 5′-cap was essential for the functionality of synthetic RNA derived from these infectious clones, though their infectivity were about 100 to 1000-fold lower than those of RNA purified from virions [76]. For FCV and PEC, an alternative approach also allowed the rescue of infectious viruses: a recombinant vaccinia virus expressing the T7 phage RNA polymerase (rMVA-T7) was used to provide the polymerase in trans. The ulterior transfection of a plasmid vector expressing the viral cDNA, generated genome-length RNA transcripts in the cytoplasm. This strategy is thought to provide a higher number of RNA copies available to initiate the viral infection [63,72,76].
This approach has also been used to study the replication and packaging of the Human norovirus (formerly Norwalk virus, NV). The full-length viral cDNA from two different human norovirus isolates were independently produced using an identical approach, producing genome-length transcripts lacking nucleotides of non-viral origin [62,78]. Both cDNA clones were flanked by the T7 promoter (in their 5′-ends), and the sequence of hepatitis delta virus ribozyme (HDV) followed by the T7 terminator signal at their 3′-ends. As mentioned elsewhere, the HDV ribozyme possesses auto-catalytic endoribonuclease activity, cleaving the RNA at the phosphodiester bond preceding its own sequence [58,62,78]. So, when placed downstream the poly-A tract, no additional nucleotides appear in the transcripts, once the ribozyme has self-cleaved and released. Although human noroviruses appear reluctant to completing their infectious cycle in vitro, the expression of these clones in cells infected with the vaccinia helper virus showed limited evidence of replication, such as the production of the non-structural proteins and the subgenomic RNA. The rescue of viral particles was only possible when the system was supplemented with a plasmid expressing a cDNA copy of the viral subgenomic RNA. In any case, such particles were not able to reproduce infection in new cell cultures, probably due to the lack of functional receptors (non-susceptible cells) [79]. The HDV ribozyme has proven effective in rescuing other caliciviruses from cDNA, such as Murine norovirus-1 (MNV-1) [80] and FCV strain F4 [81], and in rescuing members of other viral families with different genome types, like Influenzavirus B (Orthomyxoviridae) [82] and rotaviruses (Reoviridae) [83].
Intracellular expression of a cDNA clone was also used to obtain an innovative reverse genetics system for MNV-1 [80]. The strategy chosen in this case is complex and involved the use of two baculoviruses. The first one contains the cDNA of the MNV-1 genome inserted between an inducible promoter for RNA polymerase II and the HDV ribozyme sequence. The second baculovirus expresses a transactivator of the inducible promoter, which allows transcription of the MNV-1 genome [80]. Thus, RNA transcription occurs in the nucleus and is subsequently processed and exported to the cytoplasm, where translation occurs for the synthesis of viral proteins and subsequent viral genomic replication. Although the exact position in which the polymerase begins transcription is not known, the virus recovered from the inducible baculovirus system showed the correct sequence at its 5′-end. This is important for the authenticity of the rescued virus since the manipulations carried out by reverse genetics should not include uncontrolled changes in the 5′-end, which could cause unknown effects. In this sense, strategies using promoters for eukaryotic polymerases can be less efficient when compared with systems using a bacteriophage promoter, which allows complete control over the first nucleotide in the RNA transcript.
Of the in vivo cDNA expression systems, the most common are those using Vaccinia virus helper MVA/T7 for two main reasons: 1) the T7 promoter ensures a controlled transcription initiation; and 2) the cytoplasmic localization of the RNA, produced by the MVA/T7 RNA polymerase, prevents possible modifications arising from exposure to the splicing and/or the nuclear-cytoplasmic transport machinery. As aforementioned, the rescue of FCV, PEC and NV-1 from their respective cDNA clones with the help of MVA-T7 virus has been possible [61,63,78]. However, studies by Chaudhry et al. [58] showed that MVA/T7 virus has negative effects on the replication of MNV-1. Instead, they found that recombinant fowlpox rFPV-T7, which also expresses the T7 RNA polymerase, did not hamper replication of RNA purified from MNV-1 virions. Moreover, they employed this rFPV-T7 as a helper virus to rescue MNV-1 using a vector where the MNV-1 cDNA clone is inserted between the T7 promoter and the HDV ribozyme sequence. These authors noted the importance of the sequence of RNA ends derived from a cDNA clone, since mutation of a single nucleotide preceding the poly-A tail has crucial effects on the functionality of the transcripts [58]. This infectious clone enabled a reverse genetics system useful for the study of the influence of viral RNA secondary structures in replication of MNV [84,85] and to investigate factors that determine their virulence [86,87].
Initial studies with an MNV infectious clone indicated that, unlike the RNA purified from virions, transfection of synthetic transcripts (with or without a cap) did not result in the rescue of viral particles [58]. Nevertheless, in following studies the optimization of a reverse genetics system based on RNA transfection was described [19]. The system consists of post-transcriptionally capping of in vitro-synthesized transcripts with a recombinant guanylyl transferase from Vaccinia virus, which ensures efficiency close to 100% in the addition of a cap structure to the 5′-end of RNAs, much higher than the traditional procedure. Transfection of this RNA produced an infective process in cell culture, increasing the recovery of viral progeny in the order of 10 to 100 times in comparison to intracellular transcriptional systems aided by a helper virus [50] and a baculovirus system [59], respectively. This reverse genetics system using optimized synthetic transcripts has allowed studies on the functional domains of the various MNV genomic regions [88] and on other aspects of norovirus biology [38,89]. The greater persistence of the virus in infected animals has been associated with secondary structures affecting the entire genome. The modification of these structures by reverse genetics has altered the persistence of the virus without modifying the kinetics of viral replication [87].
Some of these calicivirus infectious clones have served as a basis for replicons that have allowed the expression of exogenous genes in eukaryotic cells. The GFP gene was inserted in the VP1 coding region of FCV RNA without affecting the ability of this rescued virus to replicate and the rescue of the replicon was possible by providing the capsid protein in trans. The resulting viral particles are capable of infecting a cell line susceptible to FCV, starting a new cycle of replication and expressing the fluorescent protein [76]. Later on, FCV LC region was found to tolerate foreign insertions such as AcGFP, DsRedFP [74], or mCherry [90] without hampering viral viability. In human norovirus, a GFP reporter construct containing the GFP gene in ORF1 produced complete virions that contain VPg-linked RNA, establishing a complete reverse genetics system expressed from cDNA with EF-1α mammalian promoter and without the need of a helper virus [91].
Several replicons based on Tulane virus (TV) have been described, disclosing viral regions encoding structural proteins that are dispensable for RNA replication. A chimeric replicon, in which the TV VP1 gene has been replaced by the equivalent sequence of the VP1 gene in NV, has also been obtained. This replicon causes cytopathic effects in transfected cells, but despite expressing the capsid protein, NV exogenous capsid is unable to produce infectious TV virions [77]. Not all regions of the calicivirus genome are likely to incorporate exogenous sequences. The insertion of the GFP gene at the start of ORF1 of TV completely abolished the infectivity of the transcribed RNA, which is not capable of expressing the fluorescent protein either [77]. This is why, in recent years, studies have been conducted to identify regions that tolerate insertions [88] and allow greater effectiveness in obtaining labeled replicon reporter genes [74,90]. The case of Norwalk virus replicons require special mention since a cell line stably expressing the replicon has been established [92]. For this, a fragment of the coding region of the VP1 gene was replaced by the gene for neomycin resistance, providing the mechanism for selecting cells that have incorporated the replicon. This system has further allowed studies on the replication of this non-cultivable virus [93,94] and the evaluation of specific inhibitors [95,96].
During the COVID-19 pandemic, unveiling SARS-CoV-2 behavior became crucial. Thus, a versatile reverse genetics platform for this virus was developed based on circular polymerase extension reaction (CPER) methodology. By generating overlapping cDNA fragments from viral RNA and assembling them along with a linker fragment containing a CMV promoter, a circular full-length viral cDNA is formed in a single reaction. Upon transfection of this circular cDNA into mammalian cells, infectious SARS-CoV-2 virus could be recovered. This system has been extended to Casuarina virus, Ross River virus, and Human and Murine noroviruses [11].
5.1. The Rabbit Vesivirus Reverse Genetics Journey
Rabbit vesivirus (RaV) was first isolated from domestic rabbits’ feces in Portland (Oregon, USA) and further identified and characterized in our laboratory [31,97]. Since then, the development of a reverse genetics system for RaV has been a primary goal in our laboratory for many years. Multiple obstacles had to be overcome in the pursuit of consistent infectious virus recovery. Most of the available reverse genetics’ strategies have been attempted, including the transfection of permissive cells with either synthetic genome-length RNA transcripts obtained in vitro or genome-encoding cDNA vectors ready for in vivo transcription, thereby minimizing RNA manipulation, potential exposure to contaminating RNases, and undesired degradation.
Viral progeny from RaV was successfully rescued once from a plasmid containing the RaV genome under the T7 phage RNA polymerase promoter, and a XhoI restriction site (not found in the wild-type virus) as a molecular tag. This cDNA vector was named pTA23/Xh and, together with the superinfecting helper virus rFPV-T7 allowed the recovery of a progeny of RaV confirmed to harbor the XhoI molecular tag (unpublished data). Regrettably, despite this early successful RaV rescue, subsequent attempts failed to yield additional infectious viral particles from the same construct. Ensuing RaV genome-expressing vectors were derived from the genomic RNA extracted from the XhoI-tagged virus, rescued on that occasion (e.g., pT7-RaV and pT7-RaV/Xh).
Multiple RaV genome-encoding cDNA vectors were designed that gradually incorporated improvements aimed at producing viral genomic transcripts with an authentic 5’-end and a defined 3’-end, flanked only by the poly-A tail which is naturally present in the Caliciviridae. These vectors carried different RNA polymerase promoters (SP6, T7, CMV) and, in cases where the sequence of such promoters spanned beyond the +1 transcription site, truncated versions of the promoters were produced by removing the nucleotides following the +1 site, thus preventing the introduction of non-viral nucleotides at the 5’ end of the RNA. A ribozyme was placed at the 3′-end for the generation of authentic 3’ ends. Despite these improvements, no CPE was observed in passage 1 cultures in the very first attempts, suggesting the lack of a successful virus rescue. Since VP1 protein could not be detected in cell monolayers from passage 0 (transfected cells), it was assumed that the absence of capsid assembly was the cause behind the failed rescues. Several auxiliary plasmids constitutively expressing each of the mature peptides derived from the ORF1-encoded wild-type RaV polyprotein, as well as wild-type VP1 and VP2, were separately or combinedly co-transfected with the full-length genome-expressing cDNA vector to address whether these gene products could somehow resume virus recovery, when provided in trans. No virus recovery was achieved during these experiments, suggesting the occurrence of additional defects in the RaV infectious clone that render it rather non-infectious.
Following a meticulous analysis of sequence alignments including all RaV constructs assayed in our laboratory, we found a single nucleotide change consistently present among constructs that systematically failed to generate infectious RaV. This nucleotide change consisted of an A-to-G transition within the 3′-UTR at position 8288, i.e., exactly eight nucleotides upstream the first A of the poly-A tail. While all RaV infectious cDNA clones tested in our laboratory (including pTA23/Xh, pT7-RaV and pT7-RaV/Xh) contain a G nucleotide, the original wild-type RaV contains an A at that position. More interestingly, the virus rescued once from the pTA23/Xh construct also contains an A, suggesting the occurrence of a unique reversion event that allowed such virus recovery just one time. When the correct nucleotide (A) was placed at position 8288 of the genome within the constructs pT7-RaV and pT7-RaV/Xh, those cDNA clones became infectious and the recovery of RaV from such clones became consistently reproducible [30]. In addition to this correction, we also doubled the length of poly-A tail of RaV infectious clones, which ultimately increased rescued virus yields probably due to increased virus RNA stability and half-time. This achievement was accomplished by transfecting permissive cells with plasmids that encodes the full genome-length cDNA, driven by the T7 phage RNA polymerase. This enzyme was provided in trans by infecting the cells with a helper recombinant poxvirus rFPV-T7, prior to transfection. Likewise, infectious RaV was also successfully recovered when the transcription step was conducted in vitro followed by synthetic genome-length RNA transfection, provided that a 5′-cap structure was added to the 5′-end of the synthetic genome-length RNAs, either co- or post-transcriptionally [30].
Long Story Short: Concluding Remarks
The recovery of infectious virions from genomic cDNA is not a routine procedure in the laboratory. Unfortunately, there are not universal protocols or fixed rules applicable to all caliciviruses. The striking diversity of reverse genetics strategies attempted by researchers, the intensive use of genetic engineering techniques to modify promoters, achieve precise transcription termination, or promote proper transcript processing, are just a glimpse on the complexity of this methodology. All the techniques summarized in this review, based on the replication cycle of caliciviruses, have contributed to address which replication events proceed normally and which ones do not, bringing us closer to identifying the putative reasons behind the failure of viral rescue, and to establishing guidelines for future experiments.
Reverse genetics systems play a pivotal role in various aspects of virus-related research endeavors. For highly contagious viruses, biosafe replicons derived from viral reverse genetics systems offer a secure and convenient research platform in standard laboratories, alleviating biosafety concerns. While many significant questions remain unanswered, the increasing availability of diverse reverse genetics systems and potential animal models has significantly bolstered our capacity to address these questions. This fact has led to novel insights into the biological mechanisms of these pathogens, marking a substantial step forward in our understanding of caliciviruses.
Many insights in this review stem from our laboratory practice in constructing full-length viral cDNA clones using various strategies. While some perspectives may be subjective, this straightforward depiction aims to assist researchers unfamiliar with the calicivirus field in selecting the most suitable reverse genetics system for their studies.
Author Contributions
All authors have contributed substantially to the work: Conceptualization, ALA and JMMA; Literature search, ALA, AA, FAAS, AGM, IN, KPD and JMMA; Data analisis, AGM, AA, FAAS, IN, KPD; Original draft writing, AGM and JMMA; Further writing, review and editing, ALA, AA, KPD, FP and JMMA; Figure preparation, ALA, AA and FAAS; Funding acquisition, ALA, KPD, FP and JMMA (co-principal investigators). All authors have read and agreed to the published version of the manuscript.
Funding
Part of the research reviewed in this article and its APC were funded by Principado de Asturias (grant number AYUD/2021/51288, principal investigator ALA); Ministerio de Ciencia e Innovación (grant number MCINN-23-CPP-008433, principal investigator FP) and Agencia Estatal de Investigación (grant number PID2020-120349RB-I00, principal investigators KPD and JMMA).
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- J.C. Boyer, and A.L. Haenni, Infectious transcripts and cDNA clones of RNA viruses. Virology 198 (1994) 415-426. [CrossRef]
- S. Sosnovtsev, and K.Y. Green, RNA transcripts derived from a cloned full-length copy of the feline calicivirus genome do not require VpG for infectivity. Virology 210 (1995) 383-390. [CrossRef]
- Goodfellow, and S. Taube, Chapter 3.2 - Calicivirus Replication and Reverse Genetics. in: L. Svensson, U. Desselberger, H.B. Greenberg, and M.K. Estes, (Eds.), Viral Gastroenteritis, Academic Press, Boston, 2016, pp. 355-378.
- U. Desselberger, Caliciviridae Other Than Noroviruses. Viruses 11 (2019).
- H. Ebihara, A. Groseth, G. Neumann, Y. Kawaoka, and H. Feldmann, The role of reverse genetics systems in studying viral hemorrhagic fevers. Thromb Haemost 94 (2005) 240-53. [CrossRef]
- H.L. Cai, and Y.W. Huang, Reverse genetics systems for SARS-CoV-2: Development and applications. Virol Sin 38 (2023) 837-850.
- R. Andino, D. Silvera, S.D. Suggett, P.L. Achacoso, C.J. Miller, D. Baltimore, and M.B. Feinberg, Engineering poliovirus as a vaccine vector for the expression of diverse antigens. Science 265 (1994) 1448-51. [CrossRef]
- A.A. Khromykh, and E.G. Westaway, Subgenomic replicons of the flavivirus Kunjin: construction and applications. Journal of virology 71 (1997) 1497-1505. [CrossRef]
- P. Liljeström, Alphavirus expression systems. Current Opinion in Biotechnology 5 (1994) 495-500.
- L. Enjuanes, I. Sola, F. Almazan, A. Izeta, J.M. Gonzalez, and S. Alonso, Coronavirus Derived Expression Systems. in: E. Lavi, S.R. Weiss, and S.T. Hingley, (Eds.), The Nidoviruses: Coronaviruses and Arteriviruses, Springer US, Boston, MA, 2001, pp. 309-321.
- A.A. Amarilla, J.D.J. Sng, R. Parry, J.M. Deerain, J.R. Potter, Y.X. Setoh, D.J. Rawle, T.T. Le, N. Modhiran, X. Wang, N.Y.G. Peng, F.J. Torres, A. Pyke, J.J. Harrison, M.E. Freney, B. Liang, C.L.D. McMillan, S.T.M. Cheung, D. Guevara, J.M. Hardy, M. Bettington, D.A. Muller, F. Coulibaly, F. Moore, R.A. Hall, P.R. Young, J.M. Mackenzie, J. Hobson-Peters, A. Suhrbier, D. Watterson, and A.A. Khromykh, A versatile reverse genetics platform for SARS-CoV-2 and other positive-strand RNA viruses. Nat Commun 12 (2021) 3431.
- K.Y. Green, Chapter 20. Caliciviridae: The Noroviruses. in: D.M. Knipe, Howley, P.M., (Ed.), Fields Virology, Philadelphia, PA, 2013, pp. 582-608.
- J. Vinjé, M.K. Estes, P. Esteves, K.Y. Green, K. Katayama, N.J. Knowles, Y. L’Homme, V. Martella, H. Vennema, P.A. White, and I.R. Consortium, ICTV Virus Taxonomy Profile: Caliciviridae. Journal of General Virology 100 (2019) 1469-1470.
- I.N. Clarke, and P.R. Lambden, Organization and expression of calicivirus genes. J.Infect.Dis. 181 (Suppl 2) (2000) S309-S316. [CrossRef]
- B. Alhatlani, S. Vashist, and I. Goodfellow, Functions of the 5’ and 3’ ends of calicivirus genomes. Virus Res. (2015).
- T.P. Herbert, I. Brierley, and T.D.K. Brown, Identification of a protein linked to the genomic and subgenomic mRNAs of feline calicivirus and its role in translation. Journal of General Virology 78 (1997) 1033-1040. [CrossRef]
- G. Meyers, C. Wirblich, and H.J. Thiel, Genomic and subgenomic RNAs of rabbit hemorrhagic disease virus are both protein-linked and packaged into particles. Virology 184 (1991) 677-686. [CrossRef]
- S.V. Sosnovtsev, S.A. Sosnovtseva, and K.Y. Green, Cleavage of the feline calicivirus capsid precursor is mediated by a virus-encoded proteinase. J.Virol. 72 (1998) 3051-3059. [CrossRef]
- M.A. Yunus, L.M. Chung, Y. Chaudhry, D. Bailey, and I. Goodfellow, Development of an optimized RNA-based murine norovirus reverse genetics system. J.Virol.Methods 169 (2010) 112-118. [CrossRef]
- B. Boniotti, C. Wirblich, M. Sibilia, G. Meyers, H.J. Thiel, and C. Rossi, Identification and characterization of a 3C-like protease from rabbit hemorrhagic disease virus, a calicivirus. Journal of Virology 68 (1994) 6487-6495. [CrossRef]
- F. Parra, J.A. Boga, M.S. Marin, and R. Casais, The amino terminal sequence of VP60 from rabbit hemorrhagic disease virus supports its putative subgenomic origin. Virus Research 27 (1993) 219-228. [CrossRef]
- G. Meyers, Translation of the minor capsid protein of a calicivirus is initiated by a novel termination-dependent reinitiation mechanism. J.Biol.Chem. 278 (2003) 34051-34060. [CrossRef]
- T.A. Poyry, A. Kaminski, E.J. Connell, C.S. Fraser, and R.J. Jackson, The mechanism of an exceptional case of reinitiation after translation of a long ORF reveals why such events do not generally occur in mammalian mRNA translation. Genes Dev. 21 (2007) 3149-3162. [CrossRef]
- I.N. Clarke, M.K. Estes, K.Y. Green, G.S. Hansman, N.J. Knowles, M.K. Koopmans, D.O. Matson, G. Meyers, J.D. Neill, A. Radford, A.W. Smith, M.J. Studdert, H.J. Thiel, and J. Vinje, Family Caliciviridae. in: A.M.Q. King, M.J. Adams, E.B. Carstens, and E.J. Lefkowitz, (Eds.), Virus Taxonomy. Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses, Elsevier, San Diego, 2011, pp. 977-986.
- Machin, J.M. Martin Alonso, and F. Parra, Identification of the amino acid residue involved in rabbit hemorrhagic disease virus VPg uridylylation. J.Biol.Chem. 276 (2001) 27787-27792. [CrossRef]
- Goodfellow, Y. Chaudhry, I. Gioldasi, A. Gerondopoulos, A. Natoni, L. Labrie, J.F. Laliberte, and L. Roberts, Calicivirus translation initiation requires an interaction between VPg and eIF 4 E. EMBO Rep. 6 (2005) 968-972. [CrossRef]
- Y. Chaudhry, A. Nayak, M.E. Bordeleau, J. Tanaka, J. Pelletier, G.J. Belsham, L.O. Roberts, and I.G. Goodfellow, Caliciviruses differ in their functional requirements for eIF4F components. J.Biol.Chem. 281 (2006) 25315-25325. [CrossRef]
- Goodfellow, The genome-linked protein VPg of vertebrate viruses - a multifaceted protein. Curr Opin Virol 1 (2011) 355-62. [CrossRef]
- K.F. Daughenbaugh, C.E. Wobus, and M.E. Hardy, VPg of murine norovirus binds translation initiation factors in infected cells. Virol.J. 3 (2006) 33. [CrossRef]
- Á.L. Álvarez, A. García-Manso, K.P. Dalton, J.M. Martín-Alonso, I. Nicieza, A. Podadera, M. Acosta-Zaldívar, D. De Llano, and F. Parra, Reverse Genetics System for Rabbit vesivirus. Frontiers in Microbiology 11 (2020). [CrossRef]
- R. Casais, L.G. Molleda, A. Machin, B.G. del, A.G. Manso, K.P. Dalton, A. Coto, J.M. Alonso, M. Prieto, and F. Parra, Structural and functional analysis of virus factories purified from Rabbit vesivirus-infected Vero cells. Virus Res. 137 (2008) 112-121. [CrossRef]
- E. Smertina, R.N. Hall, N. Urakova, T. Strive, and M. Frese, Calicivirus Non-structural Proteins: Potential Functions in Replication and Host Cell Manipulation. Front Microbiol 12 (2021) 712710. [CrossRef]
- Y. Matsuura, Y. Tohya, M. Onuma, F. Roerink, M. Mochizuki, and T. Sugimura, Expression and processing of the canine calicivirus capsid precursor. Microbiology 81 (2000) 195-199. [CrossRef]
- K.O. Chang, D.W. George, J.B. Patton, K.Y. Green, and S.V. Sosnovtsev, Leader of the capsid protein in feline calicivirus promotes replication of Norwalk virus in cell culture. J.Virol. 82 (2008) 9306-9317. [CrossRef]
- E.J. Abente, S.V. Sosnovtsev, C. Sandoval-Jaime, G.I. Parra, K. Bok, and K.Y. Green, The feline calicivirus leader of the capsid protein is associated with cytopathic effect. J.Virol. 87 (2013) 3003-3017. [CrossRef]
- G.S. Hansman, T. Oka, K. Katayama, and N. Takeda, Human sapoviruses: genetic diversity, recombination, and classification. Rev.Med Virol. 17 (2007) 133-141. [CrossRef]
- N. McFadden, D. Bailey, G. Carrara, A. Benson, Y. Chaudhry, A. Shortland, J. Heeney, F. Yarovinsky, P. Simmonds, A. Macdonald, and I. Goodfellow, Norovirus Regulation of the Innate Immune Response and Apoptosis Occurs via the Product of the Alternative Open Reading Frame 4. PLoS Pathogens 7 (2011) e1002413. [CrossRef]
- C.V. Subba-Reddy, M.A. Yunus, I.G. Goodfellow, and C.C. Kao, Norovirus RNA synthesis is modulated by an interaction between the viral RNA-dependent RNA polymerase and the major capsid protein, VP1. Journal of Virology 86 (2012) 10138-10149. [CrossRef]
- C.J. McCormick, O. Salim, P.R. Lambden, and I.N. Clarke, Translation termination reinitiation between open reading frame 1 (ORF1) and ORF2 enables capsid expression in a bovine norovirus without the need for production of viral subgenomic RNA. J.Virol. 82 (2008) 8917-8921. [CrossRef]
- R. Wennesz, C. Luttermann, F. Kreher, and G. Meyers, Structure–function relationship in the ‘termination upstream ribosomal binding site’ of the calicivirus rabbit hemorrhagic disease virus. Nucleic Acids Research 47 (2019) 1920-1934.
- K. Geissler, K. Schneider, A. Fleuchaus, C.R. Parrish, G. Sutter, and U. Truyen, Feline calicivirus capsid protein expression and capsid assembly in cultured feline cells. J.Virol. 73 (1999) 834-838. [CrossRef]
- B. Di Martino, and F. Marsilio, Feline calicivirus VP2 is involved in the self-assembly of the capsid protein into virus-like particles. Res.Vet.Sci. 89 (2010) 279-281. [CrossRef]
- L.G. Thorne, and I.G. Goodfellow, Norovirus gene expression and replication. The Journal of general virology 95 Pt 2 (2014) 278-91.
- S.J. Flint, L.W. Enquist, R.M. Krug, V.R. Racaniello, and A.M. Skalka, Picornaviruses, Principles of Virology. Molecular Biology, Pathogenesis and Control, ASM, Washington, 2000, pp. 752.
- C.E. Wobus, S.M. Karst, L.B. Thackray, K.-O. Chang, S.V. Sosnovtsev, G. Belliot, A. Krug, J.M. Mackenzie, K.Y. Green, and H.W.I.V. Virgin, Replication of Norovirus in Cell Culture Reveals a Tropism for Dendritic Cells and Macrophages. PLOS Biology 2 (2004) e432. [CrossRef]
- C. Cox, S. Cao, and Y. Lu, Enhanced detection and study of murine norovirus-1 using a more efficient microglial cell line. Virol J 6 (2009) 196. [CrossRef]
- L.C. Kreutz, B.S. Seal, and W.L. Mengeling, Early interaction of feline calicivirus with cells in culture. Arch Virol 136 (1994) 19-34. [CrossRef]
- T. Farkas, K. Sestak, C. Wei, and X. Jiang, Characterization of a rhesus monkey calicivirus representing a new genus of Caliciviridae. J.Virol. 82 (2008) 5408-5416. [CrossRef]
- M.K. Jones, K.R. Grau, V. Costantini, A.O. Kolawole, M. de Graaf, P. Freiden, C.L. Graves, M. Koopmans, S.M. Wallet, S.A. Tibbetts, S. Schultz-Cherry, C.E. Wobus, J. Vinjé, and S.M. Karst, Human norovirus culture in B cells. Nat Protoc 10 (2015) 1939-47. [CrossRef]
- L.C. van Dinten, J.A. den Boon, A.L. Wassenaar, W.J. Spaan, and E.J. Snijder, An infectious arterivirus cDNA clone: identification of a replicase point mutation that abolishes discontinuous mRNA transcription. Proc Natl Acad Sci U S A 94 (1997) 991-6. [CrossRef]
- G. van Marle, L.C. van Dinten, W.J. Spaan, W. Luytjes, and E.J. Snijder, Characterization of an equine arteritis virus replicase mutant defective in subgenomic mRNA synthesis. J Virol 73 (1999) 5274-81. [CrossRef]
- F. Almazan, I. Sola, S. Zuniga, S. Marquez-Jurado, L. Morales, M. Becares, and L. Enjuanes, Coronavirus reverse genetic systems: infectious clones and replicons. Virus Res 189 (2014) 262-70. [CrossRef]
- R. Casais, V. Thiel, S.G. Siddell, D. Cavanagh, and P. Britton, Reverse genetics system for the avian coronavirus infectious bronchitis virus. J Virol 75 (2001) 12359-69. [CrossRef]
- C.J. Lai, B.T. Zhao, H. Hori, and M. Bray, Infectious RNA transcribed from stably cloned full-length cDNA of dengue type 4 virus. Proc.Natl.Acad.Sci.U.S.A 88 (1991) 5139-5143. [CrossRef]
- N. Ruggli, J.D. Tratschin, C. Mittelholzer, and M.A. Hofmann, Nucleotide sequence of classical swine fever virus strain Alfort/187 and transcription of infectious RNA from stably cloned full-length cDNA. J.Virol. 70 (1996) 3478-3487. [CrossRef]
- M. Yanagi, R.H. Purcell, S.U. Emerson, and J. Bukh, Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc.Natl.Acad.Sci.U.S.A 94 (1997) 8738-8743. [CrossRef]
- D.G. Gibson, L. Young, R.-Y. Chuang, J.C. Venter, C.A. Hutchison, and H.O. Smith, Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods 6 (2009) 343-345.
- Y. Chaudhry, M.A. Skinner, and I.G. Goodfellow, Recovery of genetically defined murine norovirus in tissue culture by using a fowlpox virus expressing T7 RNA polymerase. J.Gen.Virol. 88 (2007) 2091-2100. [CrossRef]
- C. Sandoval-Jaime, K.Y. Green, and S.V. Sosnovtsev, Recovery of murine norovirus and feline calicivirus from plasmids encoding EMCV IRES in stable cell lines expressing T7 polymerase. J.Virol.Methods 217 (2015) 1-7. [CrossRef]
- Bridgen, and R.M. Elliot, Chapter 9. Reverse genetics of RNA viruses. in: A.J. Cann, (Ed.), RNA viruses: a practical approach, Oxford University Press, New York, 2000, pp. 201-227.
- K.O. Chang, S.V. Sosnovtsev, G. Belliot, Q. Wang, L.J. Saif, and K.Y. Green, Reverse genetics system for porcine enteric calicivirus, a prototype sapovirus in the Caliciviridae. J.Virol. 79 (2005) 1409-1416. [CrossRef]
- K. Katayama, G.S. Hansman, T. Oka, S. Ogawa, and N. Takeda, Investigation of norovirus replication in a human cell line. Arch.Virol. 151 (2006) 1291-1308. [CrossRef]
- S.V. Sosnovtsev, S.A. Sosnovtseva, and K.Y. Green, Recovery of feline calicivirus from plasmid DNA containing a full-length copy of the genome. in: D. Chasey, R.M. Gaskell, and I.N. Clarke, (Eds.), The first international symposium on calicivirus, European Society for Veterinary Virology and Central Veterinary Laboratory, Reading, United Kingdom, 1996, pp. 125-130.
- T. Mitra, S.V. Sosnovtsev, and K.Y. Green, Mutagenesis of tyrosine 24 in the VPg protein is lethal for feline calicivirus. J.Virol. 78 (2004) 4931-4935. [CrossRef]
- C. Morgan, S.A. Ellison, H.M. Rose, and D.H. Moore, Structure and development of viruses observed in the electron microscope: II. vaccinia and fowl pox viruses. The Journal of Experimental Medicine 100 (1954) 301-310.
- P. Somogyi, J. Frazier, and M.A. Skinner, Fowlpox virus host range restriction: gene expression, DNA replication, and morphogenesis in nonpermissive mammalian cells. Virology 197 (1993) 439-444. [CrossRef]
- E. Scotto-Lavino, G. Du, and M.A. Frohman, 5’ end cDNA amplification using classic RACE. Nat Protoc 1 (2006) 2555-62.
- C.T. Ranjith-Kumar, Y. Wen, N. Baxter, K. Bhardwaj, and C.C. Kao, A cell-based assay for RNA synthesis by the HCV polymerase reveals new insights on mechanism of polymerase inhibitors and modulation by NS5A. PLoS.One. 6 (2011) e22575. [CrossRef]
- C.V. Subba-Reddy, I. Goodfellow, and C.C. Kao, VPg-primed RNA synthesis of norovirus RNA-dependent RNA polymerases by using a novel cell-based assay. J.Virol. 85 (2011) 13027-13037. [CrossRef]
- R.B. Seth, L. Sun, and Z.J. Chen, Antiviral innate immunity pathways. Cell Res 16 (2006) 141-147. [CrossRef]
- S.V. Sosnovtsev, M. Garfield, and K.Y. Green, Processing map and essential cleavage sites of the nonstructural polyprotein encoded by ORF1 of the feline calicivirus genome. J.Virol. 76 (2002) 7060-7072. [CrossRef]
- J.D. Neill, S.V. Sosnovtsev, and K.Y. Green, Recovery and altered neutralization specificities of chimeric viruses containing capsid protein domain exchanges from antigenically distinct strains of feline calicivirus. J.Virol. 74 (2000) 1079-1084. [CrossRef]
- S.V. Sosnovtsev, G. Belliot, K.O. Chang, O. Onwudiwe, and K.Y. Green, Feline calicivirus VP2 is essential for the production of infectious virions. J.Virol. 79 (2005) 4012-4024. [CrossRef]
- E.J. Abente, S.V. Sosnovtsev, K. Bok, and K.Y. Green, Visualization of feline calicivirus replication in real-time with recombinant viruses engineered to express fluorescent reporter proteins. Virology 400 (2010) 18-31. [CrossRef]
- Karakasiliotis, S. Vashist, D. Bailey, E.J. Abente, K.Y. Green, L.O. Roberts, S.V. Sosnovtsev, and I.G. Goodfellow, Polypyrimidine tract binding protein functions as a negative regulator of feline calicivirus translation. PLoS.One. 5 (2010) e9562. [CrossRef]
- J.R.O. Thumfart, and G. Meyers, Feline Calicivirus: Recovery of Wild-Type and Recombinant Viruses after Transfection of cRNA or cDNA Constructs. Journal of Virology 76 (2002) 6398-6407. [CrossRef]
- C. Wei, T. Farkas, K. Sestak, and X. Jiang, Recovery of infectious virus by transfection of in vitro-generated RNA from tulane calicivirus cDNA. J.Virol. 82 (2008) 11429-11436. [CrossRef]
- M. Asanaka, R.L. Atmar, V. Ruvolo, S.E. Crawford, F.H. Neill, and M.K. Estes, Replication and packaging of Norwalk virus RNA in cultured mammalian cells. Proc.Natl.Acad.Sci.U.S.A 102 (2005) 10327-10332. [CrossRef]
- S. Guix, M. Asanaka, K. Katayama, S.E. Crawford, F.H. Neill, R.L. Atmar, and M.K. Estes, Norwalk Virus RNA Is Infectious in Mammalian Cells. Journal of Virology 81 (2007) 12238-12248. [CrossRef]
- V.K. Ward, C.J. McCormick, I.N. Clarke, O. Salim, C.E. Wobus, L.B. Thackray, H.W. Virgin, and P.R. Lambden, Recovery of infectious murine norovirus using pol II-driven expression of full-length cDNA. Proc.Natl.Acad.Sci.U.S.A 104 (2007) 11050-11055. [CrossRef]
- T. Oka, H. Takagi, and Y. Tohya, Development of a novel single step reverse genetics system for feline calicivirus. J.Virol.Methods 207 (2014) 178-181. [CrossRef]
- D. Jackson, A. Cadman, T. Zurcher, and W.S. Barclay, A reverse genetics approach for recovery of recombinant influenza B viruses entirely from cDNA. J.Virol. 76 (2002) 11744-11747. [CrossRef]
- S. Komoto, J. Sasaki, and K. Taniguchi, Reverse genetics system for introduction of site-specific mutations into the double-stranded RNA genome of infectious rotavirus. Proc.Natl.Acad.Sci.U.S.A 103 (2006) 4646-4651. [CrossRef]
- P. Simmonds, I. Karakasiliotis, D. Bailey, Y. Chaudhry, D.J. Evans, and I.G. Goodfellow, Bioinformatic and functional analysis of RNA secondary structure elements among different genera of human and animal caliciviruses. Nucleic Acids Res. 36 (2008) 2530-2546. [CrossRef]
- D. Bailey, I. Karakasiliotis, S. Vashist, L.M. Chung, J. Rees, N. McFadden, A. Benson, F. Yarovinsky, P. Simmonds, and I. Goodfellow, Functional analysis of RNA structures present at the 3’ extremity of the murine norovirus genome: the variable polypyrimidine tract plays a role in viral virulence. J.Virol. 84 (2010) 2859-2870. [CrossRef]
- D. Bailey, L.B. Thackray, and I.G. Goodfellow, A single amino acid substitution in the murine norovirus capsid protein is sufficient for attenuation in vivo. J.Virol. 82 (2008) 7725-7728. [CrossRef]
- N. McFadden, A. Arias, I. Dry, D. Bailey, J. Witteveldt, D.J. Evans, I. Goodfellow, and P. Simmonds, Influence of genome-scale RNA structure disruption on the replication of murine norovirus—similar replication kinetics in cell culture but attenuation of viral fitness in vivo. Nucleic Acids Research 41 (2013) 6316-6331. [CrossRef]
- L. Thorne, D. Bailey, and I. Goodfellow, High-resolution functional profiling of the norovirus genome. J.Virol. 86 (2012) 11441-11456. [CrossRef]
- E. Lopez-Manriquez, S. Vashist, L. Urena, I. Goodfellow, P. Chavez, J.E. Mora-Heredia, C. Cancio-Lonches, E. Garrido, and A.L. Gutierrez-Escolano, Norovirus Genome Circularization and Efficient Replication Are Facilitated by Binding of PCBP2 and hnRNP A1. Journal of Virology 87 (2013) 11371-11387. [CrossRef]
- J. Cheng, A. Tang, J. Chen, D. Zhang, C. Meng, C. Li, H. Wei, and G. Liu, A cDNA-based reverse genetics system for feline calicivirus identifies the 3’ untranslated region as an essential element for viral replication. Arch Virol 168 (2023) 33.
- K. Katayama, K. Murakami, T.M. Sharp, S. Guix, T. Oka, R. Takai-Todaka, A. Nakanishi, S.E. Crawford, R.L. Atmar, and M.K. Estes, Plasmid-based human norovirus reverse genetics system produces reporter-tagged progeny virus containing infectious genomic RNA. Proc.Natl.Acad.Sci.U.S.A 111 (2014) E4043-E4052. [CrossRef]
- K.O. Chang, S.V. Sosnovtsev, G. Belliot, A.D. King, and K.Y. Green, Stable expression of a Norwalk virus RNA replicon in a human hepatoma cell line. Virology 353 (2006) 463-73. [CrossRef]
- K.-O. Chang, and D.W. George, Interferons and Ribavirin Effectively Inhibit Norwalk Virus Replication in Replicon-Bearing Cells. Journal of Virology 81 (2007) 12111-12118. [CrossRef]
- K.-O. Chang, Role of Cholesterol Pathways in Norovirus Replication. Journal of Virology 83 (2009) 8587-8595. [CrossRef]
- K. Bok, V.G. Prikhodko, K.Y. Green, and S.V. Sosnovtsev, Apoptosis in murine norovirus-infected RAW264.7 cells is associated with downregulation of survivin. J.Virol. 83 (2009) 3647-3656. [CrossRef]
- K.C. Tiew, G. He, S. Aravapalli, S.R. Mandadapu, M.R. Gunnam, K.R. Alliston, G.H. Lushington, Y. Kim, K.O. Chang, and W.C. Groutas, Design, synthesis, and evaluation of inhibitors of Norwalk virus 3C protease. Bioorg Med Chem Lett 21 (2011) 5315-9. [CrossRef]
- J.M. Martín-Alonso, D.E. Skilling, L. González-Molleda, G. del Barrio, A. Machín, N.K. Keefer, D.O. Matson, P.L. Iversen, A.W. Smith, and F. Parra, Isolation and characterization of a new Vesivirus from rabbits. Virology 337 (2005) 373-383. [CrossRef]
- Arias, L. Urena, L. Thorne, M.A. Yunus, and I. Goodfellow, Reverse genetics mediated recovery of infectious murine norovirus. J.Vis.Exp. (2012).
Figure 1.
Phylogenetic tree showing the 11 established calicivirus genera: Lagovirus, Recovirus, Valovirus, Norovirus, Minovirus, Salovirus, Sapovirus, Babovirus, Nacovirus, Vesivirus and Nebovirus, in accordance with the 2019 classification of the International Committee on Taxonomy of Viruses (ICTV). These genera group 13 recognized species in total. The tree was adapted from [13], and is based on the amino acid sequences of the major capsid protein (VP1). This figure does not make a distinction among genogroups and genotypes included in some of the genera (such as Norovirus). For details, visit the ICTV’s Caliciviridae website (https://ictv.global/report/chapter/caliciviridae/caliciviridae). The silhouettes refer to the hosts of the isolates included in the phylogenetic tree but is not intended to represent all potential hosts.
Figure 1.
Phylogenetic tree showing the 11 established calicivirus genera: Lagovirus, Recovirus, Valovirus, Norovirus, Minovirus, Salovirus, Sapovirus, Babovirus, Nacovirus, Vesivirus and Nebovirus, in accordance with the 2019 classification of the International Committee on Taxonomy of Viruses (ICTV). These genera group 13 recognized species in total. The tree was adapted from [13], and is based on the amino acid sequences of the major capsid protein (VP1). This figure does not make a distinction among genogroups and genotypes included in some of the genera (such as Norovirus). For details, visit the ICTV’s Caliciviridae website (https://ictv.global/report/chapter/caliciviridae/caliciviridae). The silhouettes refer to the hosts of the isolates included in the phylogenetic tree but is not intended to represent all potential hosts.

Figure 2.
Two general models for calicivirus genome organization: the 2-ORFs model (A) and the 3-ORFs model (B). Open reading frames (ORFs) are indicated, as well as the location of regions coding for known enzymatic activities and viral functions. Nterm: N-terminus; NTPase: nucleotidyl-triphosphatase; VPg: viral protein genome-linked; Pro: protease; RdRp: RNA-dependent RNA polymerase; VP1: viral protein 1 (major capsid protein); VP2: viral protein 2 (minor capsid protein). Adapted from [13,24]. See further details in the text.
Figure 2.
Two general models for calicivirus genome organization: the 2-ORFs model (A) and the 3-ORFs model (B). Open reading frames (ORFs) are indicated, as well as the location of regions coding for known enzymatic activities and viral functions. Nterm: N-terminus; NTPase: nucleotidyl-triphosphatase; VPg: viral protein genome-linked; Pro: protease; RdRp: RNA-dependent RNA polymerase; VP1: viral protein 1 (major capsid protein); VP2: viral protein 2 (minor capsid protein). Adapted from [13,24]. See further details in the text.
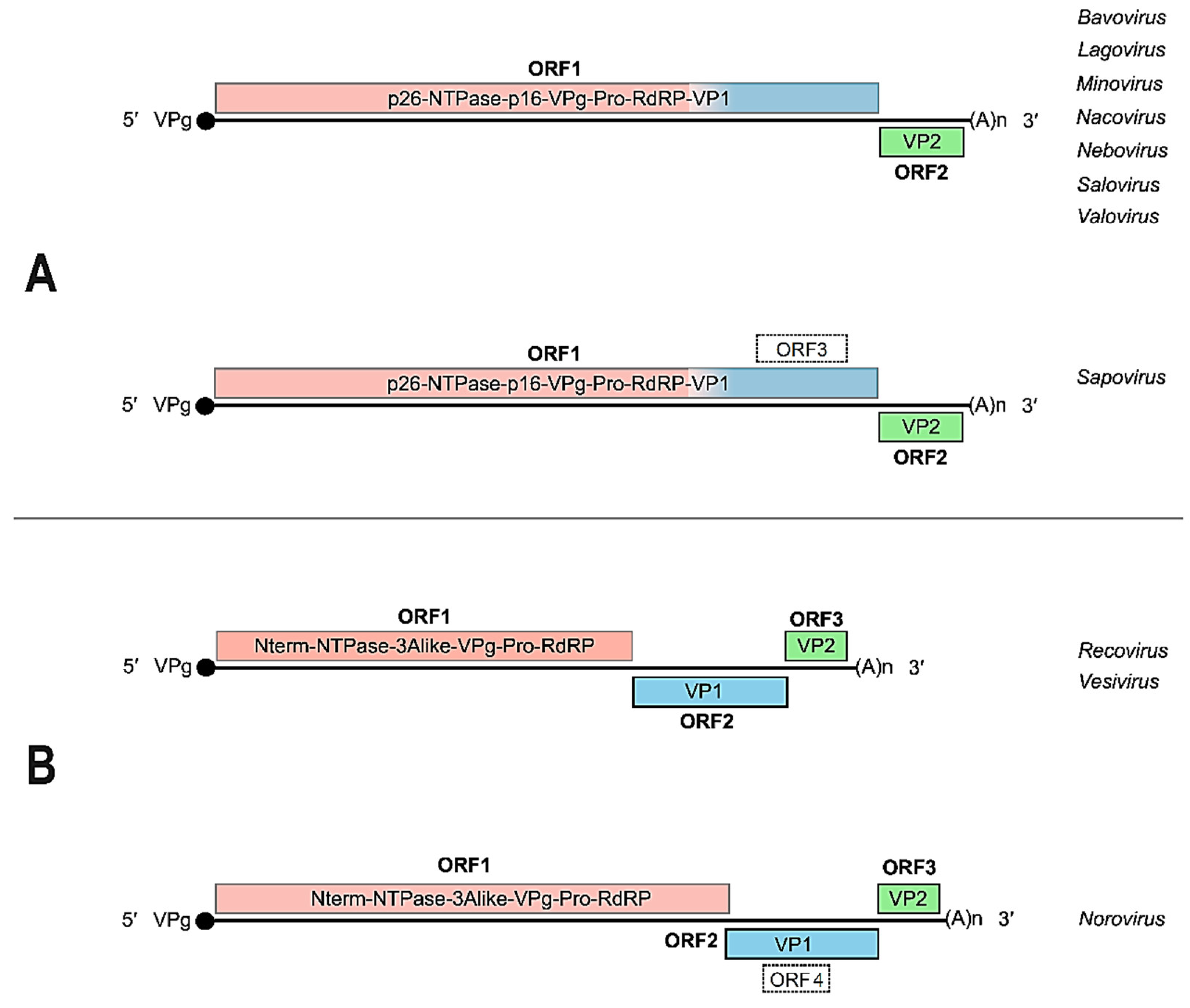
Figure 3.
Schematic representation of the replication cycle of a hypothetical Caliciviridae member. Calicivirus major replication steps include virus attachment or adsorption (1), endocytosis-mediated internalization or entry (2), translocation and uncoating of viral gRNA into the cytoplasm (3), gRNA translation leading to ORF-1-encoded polyprotein synthesis (4) and further autoproteolytic processing that yields the non-structural mature polypeptides (5), synthesis of the genome length antisense RNA intermediate (6) that serves as the template for both: the sgRNA synthesis (7) that is subsequently translated into structural protein (VP1 and VP2) (8), and for the generation of multiple copies of the gRNA (9). The newly synthesized viral components (e.g., capsid proteins gRNA and sgRNA) are put together into the progeny viral particles (10), which are ultimately released from the infected cell (11) during late events, concomitantly associated to cell lysis and death. Inspired by and adapted from [44]. See further details in the text.
Figure 3.
Schematic representation of the replication cycle of a hypothetical Caliciviridae member. Calicivirus major replication steps include virus attachment or adsorption (1), endocytosis-mediated internalization or entry (2), translocation and uncoating of viral gRNA into the cytoplasm (3), gRNA translation leading to ORF-1-encoded polyprotein synthesis (4) and further autoproteolytic processing that yields the non-structural mature polypeptides (5), synthesis of the genome length antisense RNA intermediate (6) that serves as the template for both: the sgRNA synthesis (7) that is subsequently translated into structural protein (VP1 and VP2) (8), and for the generation of multiple copies of the gRNA (9). The newly synthesized viral components (e.g., capsid proteins gRNA and sgRNA) are put together into the progeny viral particles (10), which are ultimately released from the infected cell (11) during late events, concomitantly associated to cell lysis and death. Inspired by and adapted from [44]. See further details in the text.
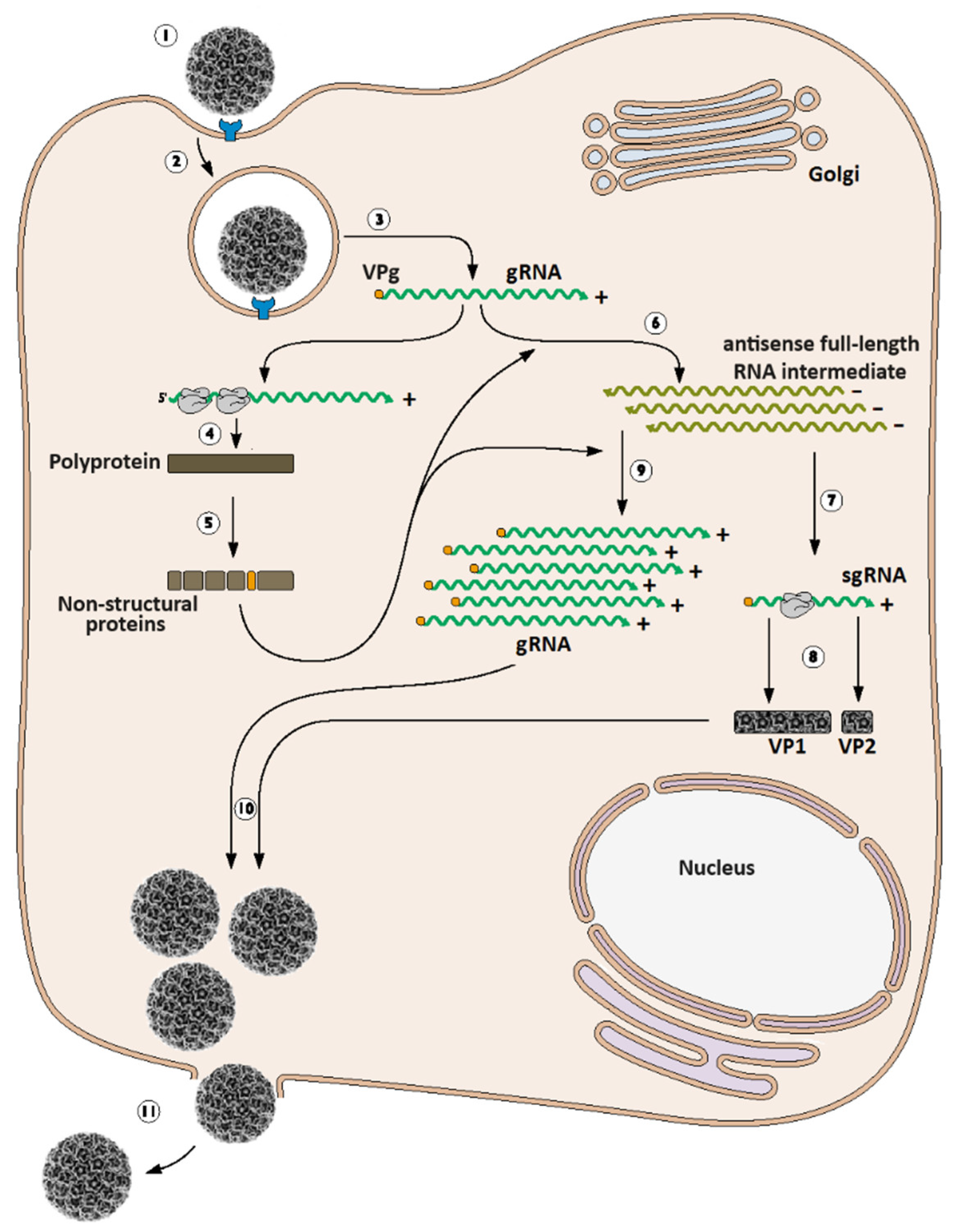
Figure 4.
Flowchart depicting the main strategies for the establishment of a calicivirus reverse genetics system. Strategy A) Transfection with either co- or post-transcriptionally capped in vitro synthetized RNA. Depending on the selected promoter in the cDNA, genome-length RNA transcripts can be produced with any commercially available in vitro transcription system (such as those based on T7 or SP6 bacteriophages RNA polymerases). Strategy B) transfection with cDNA-expressing vector, provided that the appropriate RNA polymerase for cDNA transcription will simultaneously be expressed (or supplemented in trans) within the transfected cells. In vitro translation of synthetic RNA can be attempted as a complementary assay to check translation-worthiness of transcripts. Attempts to revert systematic virus recovery failure can be made by co-transfecting the defective infectious clones with plasmids expressing each of the viral mature peptides. See further details in the text.
Figure 4.
Flowchart depicting the main strategies for the establishment of a calicivirus reverse genetics system. Strategy A) Transfection with either co- or post-transcriptionally capped in vitro synthetized RNA. Depending on the selected promoter in the cDNA, genome-length RNA transcripts can be produced with any commercially available in vitro transcription system (such as those based on T7 or SP6 bacteriophages RNA polymerases). Strategy B) transfection with cDNA-expressing vector, provided that the appropriate RNA polymerase for cDNA transcription will simultaneously be expressed (or supplemented in trans) within the transfected cells. In vitro translation of synthetic RNA can be attempted as a complementary assay to check translation-worthiness of transcripts. Attempts to revert systematic virus recovery failure can be made by co-transfecting the defective infectious clones with plasmids expressing each of the viral mature peptides. See further details in the text.
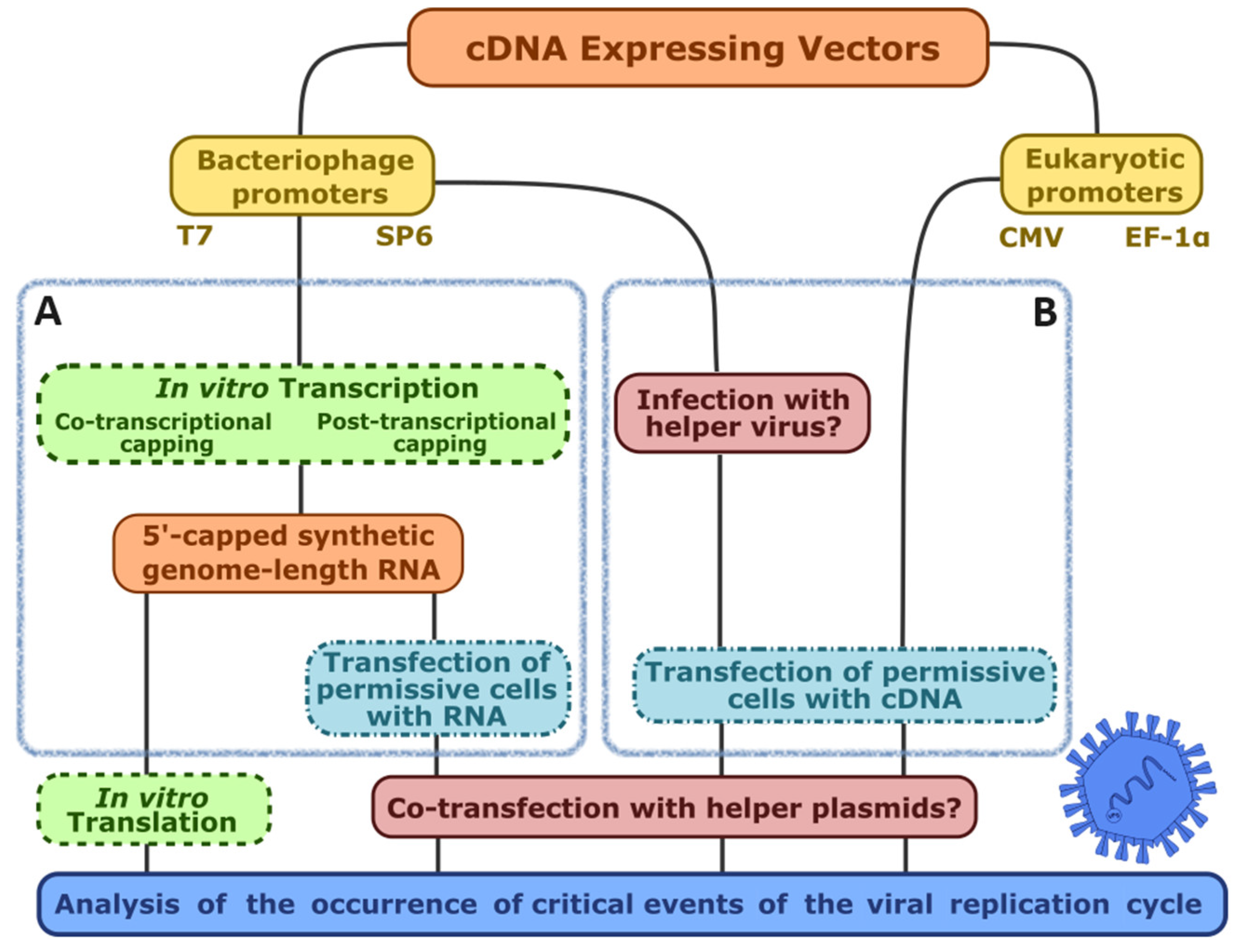

Table 1.
Analyzing the occurrence of critical events in the viral replication cycle.
| Promoter | Bacteriophage (T3, T7, SP6, etc.) | Eukaryotic (CMV, EF-1α, SV40, etc.) |
||
|---|---|---|---|---|
| Reverse genetics strategy | Transfection of in vitro-transcribed RNA | cDNA transfection of helper virus-infected cells | cDNA transfection of phage RNA pol-expressing cell line (no helper virus) | RNA pol II-driven nuclear transcription of cDNA |
| Is there virus-induced CPE in transfected cell monolayers (passage 0)? |
|
|||
| Does the supernatant from transfected cells (passage 0) contain infectious virions? |
|
|||
| Is the genome-emulating RNA transcript present in the cytosol? |
|
|||
| Is there any non-viral sequence added to the 5′-end? |
|
|||
| Is ORF 1 being expressed? |
|
|||
| Is the viral protease functional? |
|
|||
| Is the viral RdRp functional? |
|
|||
| Is the negative strand being synthesized? |
|
|||
| Are VP1 or VP2 being synthesized? |
|
|||
Table 2.
Calicivirus reverse genetics systems. The calicivirus infectious clones are listed chronologically and the fundamentals of each reverse genetics strategy followed, are briefly explained. The third column shows the general pattern observed for each infectious clone’s construction, according to the generalization depicted in Figure 5. Legend: IVT, in vitro transcription; rMVA-T7, Ankara-modified recombinant vaccinia virus expressing T7 RNA polymerase; rVV-T7, recombinant vaccinia virus expressing T7 RNA polymerase; rFPV-T7, recombinant fowlpox virus expressing T7 RNA polymerase; minCMV, minimal cytomegalovirus promoter. See the text for further details.
Table 2.
Calicivirus reverse genetics systems. The calicivirus infectious clones are listed chronologically and the fundamentals of each reverse genetics strategy followed, are briefly explained. The third column shows the general pattern observed for each infectious clone’s construction, according to the generalization depicted in Figure 5. Legend: IVT, in vitro transcription; rMVA-T7, Ankara-modified recombinant vaccinia virus expressing T7 RNA polymerase; rVV-T7, recombinant vaccinia virus expressing T7 RNA polymerase; rFPV-T7, recombinant fowlpox virus expressing T7 RNA polymerase; minCMV, minimal cytomegalovirus promoter. See the text for further details.
| Virus name | Virus recovery strategy and infectious clone features | Design | Year of publication [Reference] |
|---|---|---|---|
| Feline calicivirus | T7 RNA polymerase-driven IVT with co-transcriptional capping, followed by RNA transfection of CRFK cells | I | 1995 {Sosnovtsev, 1995 #1355} |
| Feline calicivirus | T7 RNA polymerase-driven cDNA expression; Poly-(A)32. Two delivery methods:
|
I | 2002 {Thumfart, 2002 #2200} |
| Porcine enteric calicivirus | T7 RNA polymerase-driven cDNA expression. Poly-(A)35. Two delivery methods:
|
I | 2005 {Chang, 2005 #1226} |
| Human norovirus | T7 RNA polymerase promoter: transfection of rMVA-T7-infected 293T cells; Poly-(A)26 | III | 2005 {Asanaka, 2005 #1467} |
| Human norovirus | T7 RNA polymerase promoter: transfection of rVV-T7-infected 293T cells; Poly-(A)30 | III | 2006 {Katayama, 2006 #1468} |
| Human norovirus | No virus rescue: neomycin-resistance gene replacing part of ORF2. Transfection of BHK21 and Huh7 cells with IVT-generated RNA led to the establishment of a VP1-defective replicon that persisted beyond cell passages. Apparently, the replicon further extracted from cells had covalently acquired the 5′-linked VPg. G418 was used for colony selection | IV | 2006 {Chang, 2006 #2207} |
| Murine norovirus-1 | Pol-II-driven: viral cDNA controlled by minCMV promoter; Poly-(A)31; two delivery methods:
|
II | 2007 {Ward, 2007 #1472} |
| Murine norovirus-1 | T7 RNA polymerase-driven cDNA expression. Poly-(A)26. Two helper viruses tested for providing T7 pol:
|
II | 2007 {Chaudhry, 2007 #1117} |
| Tulane virus | T7 RNA polymerase-driven IVT with co-transcriptional capping, followed by RNA transfection of LLC-MK2 cells; poly-(A)17 | I | 2008 {Wei, 2008 #1187} |
| Murine norovirus-1 |
|
II | 2010, 2012 {Yunus, 2010 #1111;Arias, 2012 #2170} |
| Human norovirus | Pol-II-driven cDNA expression: EF-1α promoter. cDNA plasmid was transfected into COS7 cells in the absence of helper virus. Poly-(A)26 | II | 2014 {Katayama, 2014 #1469} |
| Feline calicivirus | Pol-II-driven cDNA expression: EF-1α promoter. cDNA plasmid was transfected into CRFK cells in the absence of helper virus; Poly-(A)30 | II | 2014 {Oka, 2014 #1137} |
| Rabbit vesivirus | T7 RNA polymerase-driven cDNA expression. Poly-(A)30. Two delivery methods:
|
III | 2020 {Álvarez, 2020 #2183} |
| Human norovirus Murine norovirus |
Full-length cDNA with a linker fragment containing CMV promoter synthesized by circular polymerase extension reaction (CPER); transfected in NIH3T3 cells. Poly-(A)30. | II | 2021 {Amarilla, 2021 #2395} |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



