
Submitted:
28 February 2024
Posted:
29 February 2024
You are already at the latest version
Abstract
Zika virus (ZIKV) infection and pathogenesis are linked to the disruption of neurogenesis, congenital Zika syndrome and microcephaly by affecting neural progenitor cells. Nonstructural protein 5 (NS5) is the largest product encoded by ZIKV-RNA and is important for replication and immune evasion. Here, we studied the potential effects of NS5 on microtubules (MTs) and autophagy flux, together with the interplay of NS5 with histone deacetylase 6 (HDAC6). Fluorescence microscopy, biochemical cell-fractionation combined with the use of HDAC6 mutants, chemical inhibitors and RNA interference indicated that NS5 accumulates in nuclear structures and strongly promotes the acetylation of MTs that aberrantly reorganize in nested-like structures. Similarly, NS5 accumulates the p62 protein, an autophagic-flux marker. Therefore, NS5 alters events that are under the control of the autophagic tubulin-deacetylase HDAC6. HDAC6 appears to degrade NS5 by autophagy in a deacetylase- and BUZ domain-dependent manner and to control the cytoplasmic expression of NS5. Moreover, NS5 inhibits RNA-mediated RIG-I interferon (IFN) production, resulting in greater activity when autophagy is inhibited (i.e., effect correlated with NS5 stability). Therefore, it is conceivable that NS5 contributes to cell toxicity and pathogenesis, evading the IFN-immune response by overcoming HDAC6 functions. HDAC6 has emerged as an anti-ZIKV factor by targeting NS5.
Keywords:
ZIKV
; NS5
; aberrant MTs
; MT acetylation
; autophagic p62
; HDAC6
; tubulin deacetylase
; BUZ domain
; NS5 clearance
; inhibition of IFN production
1. Introduction
The Flaviviridae (genus Flavivirus) Zika virus (ZIKV) is a mosquito-borne, positive-sense single-stranded RNA (RNA+) virus [1,2,3,4,5,6,7,8,9,10,11,12]. ZIKV is classified by homology to the Spondweni virus (SPONV) in the Spondweni viral clade or serogroup [2,13,14]. Notably, ZIKV and SPONV viruses were first characterized in Africa in 1947 and 1952 [11,15], respectively. ZIKV was discovered during the search for a potential vector responsible for the cycle of sylvan yellow fever virus (YFV) in Uganda [16] and was found in the serum of a pyrexial rhesus monkey caged in the canopy of the Zika Forest in Uganda [11].
The first confirmed human infections by ZIKV occurred in Nigeria (1954) [17], followed by several cases reported in Uganda (1962-63) [18] and in central Java, Indonesia (1977) [19]. The World Health Organization (WHO) declared Zika an "extraordinary event that needed a coordinated response, constituting a public health emergency of international concern (PHEIC)” [20], due to the description of a large outbreak of rash illness [21,22,23], with short-term and low-grade fever [23], not in all cases, and a cluster of microcephaly in newborns to infected mothers [24,25,26,27,28,29,30,31,32] together with neurological abnormalities and Guillain‒Barré syndrome (GBS) in Brazil [21,33,34,35,36,37,38,39,40].
In fact, ZIKV infection carries the risk of adverse pregnancy outcomes, including increased risk of preterm birth, fetal death and stillbirth, and congenital malformations collectively characterized as congenital Zika syndrome (CZS), including the abovementioned microcephaly, abnormal brain development, limb contractures, eye abnormalities, brain calcifications, and other neurologic manifestations [27,41,42,43,44,45]. ZIKV has been found in the cerebrospinal fluid (CSF) and brain of adults infected by the virus who manifested neurological disorders [34,46,47,48,49]. This flavivirus causes harmful effects in the adult brain, such as GBS [46,48,50,51,52], encephalitis [46,51,52,53], meningoencephalitis [34,54], acute myelitis [46,52,55] and encephalomyelitis [46,49,51,56,57], as well as sensory polyneuropathy [58] and other neurological complications [59,60].
Several ZIKV proteins have been well characterized for their functions in the pathology of the virus, as well as for their biology (i.e., viral infection and transmission) and immune escape [61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87]. Furthermore, microtubules (MTs) play a critical role in ZIKV infection, as in other flavivirus life cycles, and are associated with the modification of the tubulin cytoskeleton and the closely related process of autophagy to the pathogenesis of the virus [88,89,90,91,92,93,94,95]. In this sense, nonstructural protein 5 (NS5) of ZIKV appears to interact with components of the cilium base that are associated with MTs, promoting ciliopathy and premature neurogenesis [96]. However, little is known about the functional involvement of the NS5 protein in altering tubulin cytoskeleton dynamics or sequestosome 1 (SQSTM1)/p62-associated autophagy flux [97,98,99,100,101,102,103], key events for cell permissivity and survival that are regulated by the antiviral factor histone deacetylase 6 (HDAC6) [91,94,98,99,100,101,104,105,106,107,108].
Considering the accepted role of MTs in innate immunity and infection [92,109,110,111,112], their essential roles in intracellular trafficking and cell morphology (reviewed in [113]), the involvement of the ZIKV NS5 protein in virus replication and evasion from the interferon (IFN)-associated immune response, and its association with the centrosome or microtubule (MT) organized center (MTOC) during mitosis and cell division [95,114,115,116], we aimed to study the effect of the ZIKV NS5 protein on MTs to ascertain potential interplay at this level. Our results indicate that the ZIKV NS5 protein mainly accumulates in nuclear structures, as previously reported [64,65,117,118], and promotes the acetylation of MTs that aberrantly reorganize in nested-like structures organized at the cell periphery. Similarly, we observed that the p62 protein, a marker of autophagy flux [97,98,99,100,101,102,103], accumulated in cells overexpressing the ZIKV NS5 protein. Notably, these data indicate that NS5 alters cell events that are under the control of the antiviral tubulin deacetylase and the autophagy-associated enzyme HDAC6 [91,98,99,100,101,104,105,106,107,108]. HDAC6 appears to degrade NS5 by autophagy in a deacetylase- and BUZ (binder of or bound to ubiquitin zinc finger [119,120]) domain-dependent manner and by controlling the cytoplasmic expression of NS5. Similarly, our results indicate that the ZIKV NS5 protein inhibits foreign RNA-mediated retinoic acid-inducible gene I (RIG-I) interferon (IFN) production, resulting in increased activity when autophagy is inhibited.
Taken together, these results suggest that the ZIKV NS5 protein contributes to cell toxicity and pathogenesis by affecting MT dynamics and p62-associated autophagic flux and evades the IFN-immune response by overcoming HDAC6 functions. Therefore, HDAC6 has emerged as an anti-ZIKV factor by targeting the ZIKV NS5 protein.
2. Materials and Methods
2.1. Antibodies and Reagents
Mouse anti-myc (9E10; sc-40), rabbit anti-HDAC6 (H-300; sc-11420), rabbit anti-HA (Y-11; sc-805), mouse anti-p62/SQSTM1 (D3; sc-28359) and mouse anti-Histone 3 (1G1; sc-517576) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-NS5 (GTX133312) was obtained from GeneTex (GeneTex, CA, USA). Mab anti-a-tubulin (T6074), anti-acetylated a-tubulin (T7451), and secondary horseradish peroxidase (HRP)-conjugated Abs specific for any Ab species assays were purchased from Sigma‒Aldrich (Sigma‒Aldrich, St. Louis, MO, USA). Acetyl-a-tubulin (Lys40) (D20G3) XP® rabbit mAb (5335S) was acquired from Cell Signaling Technology (Cell Signaling Technology, Danvers, MA, USA). Alexa Fluor 568-conjugated goat a-mouse IgG1 (g1) (A21124) and Alexa Fluor 488-conjugated goat a-mouse IgG1 (g1) (A21121) were obtained from Life Technologies (Life Technologies Corporation, CA, USA). FuGENE HD Transfection Reagent (E2312) and LipofectamineTM RNAi MAX (13778075) were purchased from Promega (Promega Corporation, WI, USA) and Thermo Fisher (Thermo Fisher Scientific, MA, USA), respectively. Mission® siRNA Universal Negative Control #1 (SIC001), 3-methyladenine (3-MA) (M9281), tubacin (SML0065), 4′,6-Diamidino-2-phenylindole (DAPI), phenylmethylsulfonyl fluoride (PMSF) and poly-D-lysine (P6407) were obtained from Sigma‒Aldrich (Sigma‒Aldrich). A Complete™ protease inhibitor cocktail (11697498001) was obtained from Roche Diagnostics (GmbH, Mannheim, Germany).
2.2. DNA Plasmids and Viral DNA Constructs
The DNA sequence encoding nonstructural protein 5 (NS5) of ZIKV (Zika strain MP1751_East_African (Uganda)) was cloned with the myc or human influenza hemagglutinin (HA) epitope at the C-terminus using the BamHI/XhoI restriction sites and T4 DNA ligation in pcDNATM 3.1(+). HDAC6 constructs were generated by Drs. X.-J. Yang and N. R. Bertos (Molecular Oncology Group, Department of Medicine, McGill University Health Centre, Montreal, QC, Canada) [101,106,121], and the HA epitope was introduced as an N-terminal tag (HA-wt-HDAC6), as previously reported [100,101,106]. When indicated, we used double mutations affecting the HDAC6 catalytic domain (H216A/DD1 mutant and H611A/DD2 mutant (HA-dm-HDAC6)) or deletions in the proautophagic BUZ domain (HA-HDAC6-∆BUZ), which lacks the 1092 residue in the C-terminal region that bears the Cys/His-rich motif [101,106,120,121,122]. These constructs are unable to promote autophagic degradation of some viral proteins, as previously reported [100,101,106]. The wild-type (wt)-HDAC6 encoding the fluorescent protein DsRed was obtained using the AgeI/NotI restriction sites of pDsRed (Clontech, Palo Alto, CA, United States) as previously reported [100]. The pcDNATM 3.1(+) empty vector (Life Technologies) was used as a control for cDNA transfection.
2.3. Cells
HEK-293T cells (cat. number 103, NIH AIDS Research and Reference Reagent Program) and Lucia luciferase reporter HEK-293 cells expressing human RIG-I, HEK-Lucia™ RIG-I, (hkl-hrigi, InvivoGen, CA, USA) were grown at 37°C in a humidified atmosphere with 5% CO2 in DMEM (Lonza, Verviers, Belgium) supplemented with 10% foetal calf serum (FCS) (Lonza), 1% L-glutamine and 1% penicillin–streptomycin (Lonza). The cells were harvested and passaged every 3 days using trypsin-EDTA (L0930-100) (Biowest, FL, USA) or Versene 1x (15040-066; Gibco Chemicals, Thermo Fisher Scientific, MA, USA). HEK-Lucia™ RIG-I cells were cultured to 50-70% confluence in fresh supplemented DMEM 24 h before cell transfection with viral or human DNA constructs and before the induction of interferon (IFN) production. To maintain the stable expression of RIG-I and the luciferase reporter downstream of tandem interferon-stimulated gene 54 (ISG54) promoter elements, 30 mg/mL blasticidin and 100 mg/mL Zeocin™ were added to the HEK-Lucia™ RIG-I reporter cell line. Mycoplasma-free (Mycozap antibiotics, Lonza) or 100 mg/mL Normocin™ was routinely added to each HEK-293T and HEK-Lucia™ RIG-I cell split. Cell viability was quantitatively determined by light microscopic quantitation visualizing trypan blue-stained cells under each experimental condition.
2.4. Messenger RNA Silencing
We used the short interfering RNA (siRNA) oligonucleotides (oligos) specifically generated by Eurogentec (Hampshire, United Kingdom) against the mRNA sequence of HDAC6 to knockdown HDAC6 expression. Transient siRNA transfections were performed using LipofectamineTM RNAi MAX dissolved in Opti-MEM®. A mixture of LipofectamineTM RNAi MAX/oligos (1:1) was gently vortexed, incubated for 5 min at room temperature (RT), and then added to HEK-293T cells cultured in 6-well plates. A total of 25 pmol/well of each commercial scrambled control oligo (Mission® siRNA Universal Negative Control #1) (Sigma‒Aldrich) or HDAC6-specific siRNA oligos spanning nucleotides 193-213, 217-237, and 284-304 were used as previously described [100,101,106]. Twenty-four hours later, the cells were transfected with siRNAs, and the same cells were subjected to a second round of treatment with FuGENE HD Transfection reagent for transient expression of viral NS5 or pcDNATM 3.1(+) empty vector under control conditions and incubated for 24 h to complete a 48 h interval of HDAC6 silencing, when the siRNAs for HDAC6 induced specific interference with protein expression. Cells were then lysed and analysed with specific Abs by Western blotting to validate endogenous HDAC6 silencing, as well as the associated expression of the studied cell and viral proteins, as similarly described [98,99,100,101,106].
2.5. Cell Fractionation
HEK-293T cells (1x106) were harvested 48 h posttransfection by using a cell scraper, washed with PBS and pelleted at 200 × g (1,500 rpm) for 5 min. The cells were resuspended by gentle pipetting in cytosolic extraction buffer (CEB) (10 mM HEPES pH 7.5, 10 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol (DTT), 0.5% Nonidet-40 and 0.5 mM PMSF), usually approximately 80-100 mL (5 times the volume of pellet), and incubated for 20 min on ice (+4°C), pipetting after 10 min to promote the cell lysis process. Centrifugation was performed at 12,000 × g (11,400 rpm) at +4°C for 10 min, and the resulting supernatant containing the cytoplasmic extract was separated into another tube (Eppendorf-like size) and washed three times (centrifugation between washes was performed for 5 min at +4°C at 12,000 × g). The pellet corresponding to the nuclear extract was washed at least twice with CEB to eliminate cytoplasmic debris by centrifugation at 12,000 × g at +4°C for 5 min and resuspended in 50 mL (2 times the volume of pellet, taking into account the nucleus-cytoplasm ratio) of nuclear extraction buffer (NEB) (20 mM HEPES pH 7.5, 400 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF) and incubated on ice for 30 min, vortexing once every 10 min. Finally, the pellet was centrifuged for 15 min at +4°C and 12,000 × g, and the supernatant was collected as the nuclear fraction. For Western blot analysis, a-tubulin was used as the cytoplasmic marker and for control of total protein load, and histone 3 was used as the marker for the nuclear fraction.
2.6. Western Blotting
Protein expression in cell lysates was determined by SDS‒PAGE and Western blotting. HEK-293T or HEK-Lucia™ RIG-I cells were transfected with different cDNA constructs using FuGENE® HD Transfection reagent in a mixture of FuGene/plasmids at a 3:1 ratio, gently vortexed, incubated for 10 min at RT, and then added to the cells in culture. Briefly, 48 h after transfection, the cells were treated with lysis buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate and 1x Complete™ Protease Inhibitor Cocktail) for 30 min and sonicated for 30 s at +4°C. The effects of the different inhibitors were similarly assayed in HEK-293T cells or HEK-Lucia™ RIG-I cells as indicated in each case. To inhibit HDAC6 catalytic activity, cells were treated overnight at 37°C with 1 mM tubacin in DMSO (< 0.1% of the total volume). For the prevention of autophagosome formation and subsequent autophagic degradation, 3-MA (5 mM) in PBS was added for 5 h before cell lysis. Equivalent amounts of protein (30-40 mg), determined using the bicinchoninic acid (BCA) method (Millipore Corporation, Billerica, MA, United States), were resuspended and treated with Laemmli buffer and then separated by 10-12% SDS‒PAGE and electroblotted onto 0.45 mm-vinylidene difluoride (PVDF) membranes (Millipore) using Trans-blot Turbo (Bio-Rad, Hercules, CA, United States). The membranes were blocked with 5% nonfat dry milk in TBST (100 mM Tris, 0.9% NaCl, pH 7.5, 0.1% Tween 200) for 30 min and then incubated with specific Abs. Proteins were detected by luminescence using an enhanced chemiluminescence (ECL) system (Bio-Rad) and analysed using a ChemiDoc MP device and Image LabTM Software, version 5.2 (Bio-Rad).
2.7. Fluorescence Microscopy
HEK-293T cells (1×105 cells) were seeded on poly-D-lysine-coated Ø 18 mm glass coverslips and transfected with NS5-myc or cotransfected with the different pcDNATM 3.1(+) empty vector (control) or wt-HDAC6-DsRed plasmids using FuGENE® HD Transfection Reagent as described above to analyse NS5 expression and distribution. Forty-eight hours posttransfection, the cells were washed three times with PBS, fixed for 15 min in 4% paraformaldehyde (in PBS), permeabilized for 45 min with PBS-T (0.1% Tween 20 in 1× PBS), quenched with 100 mM glycine in 1× PBS for 15 min and treated with blocking solution (5% FCS in PBS-T) for up to 1 h. Mouse a-myc (9E10; sc-40) (1:50) and Alexa Fluor 568 goat a-mouse IgG1 (g1) or Alexa Fluor 488 goat a-mouse IgG1 (g1) (1:500) were subsequently incubated overnight or for 1 h, after which they were diluted in blocking solution. Coverslips were then washed and mounted in Mowiol Antifade (Dako, Glostrup, Denmark), and image acquisition was performed by epifluorescence microscopy (Zeiss 200 M (Zeiss, Jena, Germany)), as previously reported [100,123,124]. The expression and distribution of fluorescent NS5 were analysed via a line scan and quantified using MetaMorph software (Universal Imaging, Downington, PA, United States), as previously described [100,123,124].
2.8. RIG-I/ISG54 Activity Assay: RNA-Mediated RIG-I IFN Production
HEK-Lucia™ RIG-I cells were transfected with the NS5 construct or control plasmid as described above for HEK-293T cells. Depending on the experimental conditions, at 48 h posttransfection, some cells were treated with 200 mM 3-MA, an autophagy inhibitor, for 5 h, and PBS (vehicle for 3-MA) was used as a control. HEK-Lucia™ RIG-I cells were then detached using Versene (Gibco), and a fraction of the cells was used for lysis, while the remaining cells were seeded in a 96-well plate at a concentration of 1x105 cells/well in 180 mL of antibiotic-free media (DMEM supplemented with 10% FBS) (six replicates/condition). The RIG-I agonist, 5’ triphosphate hairpin RNA (3p-hpRNA) and RIG-I-like receptor ligands (tlrl-hprna, InvivoGen) were transfected with LyoVec™ (Lyec-2, InvivoGen) following the manufacturer’s instructions, and the cells were incubated overnight to ensure adequate induction. The next day, 20 mL of the supernatant was tested for ISG54-Lucia secretion, whose expression depended on the activation of the IFN-I promoters, using 50 mL of QUANTI-Luc™ Gold (rep-qlcg-2), which contains coelenterazine and allows the Lucia signal to be measured, added by automatic injection into the wells of a 96-well white microplate. Luminescence was measured by obtaining relative light units (RLUs) for each condition at the same time point after injection using a LuminoskanTM Ascent microplate luminometer (Thermo Fisher Scientific, MA, USA).
2.9. Statistical Analysis
Statistical analyses were performed using GraphPad Prism, version 6.0b (GraphPad Software, San Diego, CA, USA). The significance of differences between groups was determined using Student’s t test, as indicated in the figure legends.
3. Results
Considering the above findings, we investigated the effect of the ZIKV NS5 protein on cell MTs, the interplay of ZIKV NS5 with potential associated factors, such as the tubulin deacetylase HDAC6, and specific RNA/RIG-I-triggered IFN production.
3.1. The ZIKV NS5 Protein Mainly Localizes to Nuclear Structures
First, we examined the subcellular localization of the ZIKV NS5 protein in HEK-293T cells, a model for ZIKV and other RNA virus infection and production [98,99,100,101,125,126,127], which were transfected with the NS5-myc construct. At 48 h posttransfection (Figure 1a), ZIKV NS5 protein was mainly localized to the nucleus, as observed by biochemical analysis of cell fractioning (Figure 1b) and by immunofluorescence of transfected cells (Figure 1c).
Therefore, ZIKV NS5 appears to be restricted to nuclear structures, as previously reported [64,65,117,118], according to fluorescence microscopy analysis (Figure 1c). This observation was confirmed by cell-fraction biochemical analysis of cells overexpressing the ZIKV NS5 protein, which is mainly distributed in the nuclear fractions and is weakly detected in the cytoplasmic fractions (Figure 1b). Hence, we observed that ZIKV NS5 localized to discrete punctate nuclear bodies. NS5 appears as regular 20-50 punctate spot-shaped structures that vary in size from one (Figure 1c, type i) to several (Figure 1c, type ii) micrometers in diameter. Notably, mostly irregularly shaped structures that range in length form ring-like structures that we define as “speckles” (Figure 1c, type iii). The diameter observed for the different NS5 structures (i.e., line scan analysis), their frequency per cell (%) and the amount of NS5 detected are shown (Figure 1d, 1e and 1f, respectively). Considerably, this punctate localization observed by immunofluorescence microscopy presented appreciably less intense DAPI staining in the structure core, suggesting the absence of any DNA colocalization, as similarly reported [65]. Although the cells were synchronously transfected, the ZIKV NS5 protein selectively formed diverse structural patterns in different cells, showing its highly dynamic nature and governed by processes that are independent of time, as previously reported in studies carried out with infectious viruses in which diversity was attributed to distinct phases of the viral cycle [64].
Under these experimental conditions, our results indicate that the NS5 protein mainly accumulated in the nucleus and behaved in a very dynamic manner, residing in entities whose number and size varied, suggesting that nuclear accumulation of the ZIKV NS5 protein is an integral part of the viral life cycle.
3.2. The ZIKV NS5 Protein Promotes the Acetylation of Microtubules That Reorganize into a Nested-Like Structure at the Cell Periphery
MTs have been reported to support ZIKV and other flavivirus infections [88,95,128]. In fact, the ZIKV NS5 protein was reported to be localized to the MTOC or centrosome during mitosis and cell division [95,114,115,116]. Hence, we aimed to study the effect of the ZIKV NS5 protein on MTs to ascertain the potential interplay at this level.
Fluorescence microscopy studies of HEK-293T cells overexpressing the NS5-myc construct indicated that the presence of the ZIKV protein promoted the aberrant reorganization of MTs, which form a caged or nested-like structure at the periphery of cells (Figure 2a and 2b). The MTs of this caged structure are highly acetylated at the Lys40 residue of the a-tubulin subunits of MTs (Figure 2a and 2b), a posttranslational modification associated with stable MTs [105,106,129,130,131,132,133]. HDAC6 is an antiviral cytoplasmic enzyme [91,98,100,101,104,105,106] that regulates MT dynamics by deacetylating the Lys40 residue of the a-tubulin subunit of stabilized MTs [107,108,134]. We therefore compared the effect of a specific HDAC6 deacetylase inhibitor, tubacin [107,108,134,135,136], with that of NS5 on the acetylation of MTs (Figure 2b). The results obtained indicate that tubacin-mediated inhibition of the HDAC6 tubulin-deacetylase promotes acetylation of MTs, as similarly observed with the ZIKV NS5 protein. Notably, tubacin does not induce the aberrant reorganization of MTs in nested-like peripheric structures that occurs in cells expressing NS5 (Figure 2b). Biochemical analysis of these NS5-positive cells revealed that NS5 promoted the acetylation of MTs in a dose-dependent manner (Figure 2c, quantified in the associated histograms). These data indicate that NS5 stably and aberrantly reorganizes cellular MTs, provoking their acetylation and overcoming the potential protective effect of the endogenous tubulin deacetylase HDAC6.
It has been reported that HDAC6 exerts its antiviral activity against several key human immunodeficiency virus type 1 (HIV-1) factors (i.e., envelope complex (Env), Vif and Pr55Gag proteins [98,99,100,101,105,106]) through its tubulin-deacetylase activity and the C-terminal BUZ domain (also known as polyubiquitin associated zinc finger (PAZ) [119] or ubiquitin carboxyl-terminal hydrolase-like zinc finger, ubiquitin-specific protease (ZnF-UBP) domain [137]), both of which are necessary for autophagy-mediated viral protein clearance [100,101]. We investigated whether the ZIKV NS5 protein could disturb autophagy, as measured by the stability of the p62 protein, a marker of autophagic p62 flux that interacts and works with HDAC6 in the autophagic clearance of proteins, where p62 also fades [97,98,99,100,101,102,103]. The ZIKV NS5 protein negatively altered autophagic p62 flux since cells expressing NS5 accumulated p62 protein (Figure 2d), indicating that NS5 had a dose-dependent effect on the accumulation of the autophagy marker p62 (Figure 2e).
3.3. HDAC6 Targets the ZIKV NS5 Protein by Autophagy, which Requires Its Deacetylase Activity and BUZ Domain for NS5 Clearance
Considering the above data, the ZIKV NS5 protein appears to alter HDAC6-associated substrates that are key for the antiviral functions of the enzyme, such as MT dynamics (i.e., organization and acetylation) and the stability of the autophagic p62 factor [91,98,99,100,101,105,106].
We next studied the effect of HDAC6 on the ZIKV NS5 protein. Biochemical (Figure 3a) and fluorescence microscopy (Figure 3b) analyses of HEK-293T cells overexpressing both HDAC6 (N-terminal HA-tagged wyld-type (wt)-HDAC6 (HA-wt-HDAC6) [100,101,106] or wt-HDAC6-DsRed [100]) and NS5-myc constructs showed that functional tubulin-deacetylase HDAC6 targets NS5-myc (Figure 3a, quantified in the associated histogram, and 3b). Notably, overexpression of the NS5 construct stabilized the acetylated MTs (Figure 3a, see Ac-tub in lane 2 compared with control lane 1). However, when functional HDAC6 was overexpressed together with NS5, we observed a significant decrease in the level of expression of the ZIKV NS5 protein (Figure 3a and 3b) concomitant with the decrease in the acetylation level of the a-tubulin subunit of MTs (Figure 3a, see NS5 (quantified in associated histograms) and Ac-tub in lane 4 compared with lane 2). Cells overexpressing functional HDAC6 lost the nuclear pattern of ZIKV NS5 protein expression (Figure 3b), and the NS5 protein was not detected in cells overexpressing HDAC6 by fluorescence microscopy.
To determine the degradative pathway used by HDAC6 to target the ZIKV NS5 protein, we inhibited two key functions of HDAC6 involved in its autophagy antiviral activity-deacetylase activity, which resides in two functional deacetylase domains [136,137,138,139,140,141] and the BUZ domain [91,94,100,101,106]. As in the experiment presented in Figure 2b, we first inhibited the deacetylase domains of endogenous HDAC6 by using the specific chemical inhibitor of the enzyme tubacin (Figure 3c). Remarkably, in cells overexpressing NS5-myc, tubacin-mediated inhibition of the endogenous tubulin-deacetylase HDAC6 allows the ZIKV NS5 protein to be expressed not only in the nucleus (as observed for the different nuclear NS5 structures in the control nontreated cells) (Figure 3c) but also widely in the cytoplasm (Figure 3c). Moreover, we analysed the effect exerted on the ZIKV NS5 protein by the well-characterized HDAC6 double deacetylase-dead mutant (dm) (N-terminal HA-tagged (HA-dm-HDAC6)), which is not able to deacetylate HDAC6 substrates and negatively affects the autophagy function of the enzyme [100,101,106]. Thus, biochemical analysis of cells overexpressing both the ZIKV NS5 protein and the dm-HDAC6 mutant revealed that the NS5 protein is more stable and more than 6-fold more highly expressed in these cells (Figure 3d, lane 6 (quantified in the associated histograms)) than in cells expressing only the ZIKV NS5 protein (Figure 3d, lane 2 (quantified in the associated histograms)), suggesting that the catalytic activity of HDAC6 is key for the degradation of the ZIKV NS5 protein. This dm-HDAC6-associated “protective” effect on the NS5 protein could be explained by the competitive action exerted by the overexpressed dm-HDAC6 mutant on the endogenous HDAC6 that avoids HDAC6-mediated NS5 degradation. Therefore, these data confirmed that endogenous HDAC6 restricts ZIKV NS5 protein expression and is preserved from the antiviral enzyme in the nucleus (Figure 1 and 2 and Figure 3b and 3c (control conditions)).
Once again, functional wt-HDAC6 promoted the clearance of the ZIKV NS5 protein (Figure 3d, lane 5 (quantified in the associated histograms)) concomitantly with the deacetylation of MTs (Figure 3d, lane 5). Thus, the ZIKV NS5 protein could not promote the acetylation of MTs in cells overexpressing functional HDAC6 (Figure 3d, lane 5). Taken together, these data indicate that HDAC6 controls the level of expression and the cellular distribution of the ZIKV NS5 protein and requires its deacetylase activity, which is key for the autophagy function of HDAC6 [100,101].
A well-characterized HDAC6 construct lacking the C-terminal BUZ domain (HA-HDAC6-DBUZ), which is involved in autophagy activation and its degradative activity [100,101,106], was not able to promote clearance of the ZIKV NS5 protein (Figure 4 a, comparing lanes 2 and 4 (quantified in the histogram)). This HA-HDAC6-DBUZ construct could deacetylate MTs (Figure 4 a, comparing lanes 3 and 4 with lanes 1 and 2) and promote the stabilization of the autophagy marker p62 (Figure 4 a, comparing lanes 3 and 2 with control lane 1) due to the absence of the BUZ region of the enzyme. Notably, the ZIKV NS5 protein negatively alters p62-autophagy flux, as observed by the accumulation of p62 in cells overexpressing NS5 compared with that in control cells (Figure 4 a, comparing lanes 2 and 1) and in cells coexpressing the HA-HDAC6-DBUZ construct (Figure 4 a, comparing lanes 2 and 4 with control lane 1). These observations indicate that the ZIKV NS5 protein could negatively influence autophagic p62 flux to protect itself from fading and accomplishing its viral functions. In fact, the interplay between autophagy and different ZIKV proteins in the context of virus infection and transmission is a very complex subject of research (reviewed in [142,143]).
We further analysed the effect of the chemical inhibition of the autophagy-associated process exerted by 3-methyladenine (3-MA) on HDAC6-mediated ZIKV NS5 degradation in HEK-293T cells. The 3-MA inhibitor was used as previously reported [101] to inhibit the formation of aggresomes and the associated HDAC6-triggered autophagic degradation of targeted proteins [100,101,144,145,146]. 3-MA inhibits the autophagic sequestration of cytoplasmic proteins, as it was previously reported to block HDAC6-mediated autophagic degradation of the HIV-1 Vif and Pr55Gag proteins [98,99,100,101].
Thus, in 3-MA-treated cells, the blockade of the autophagic process is signaled by the stabilization of the NS5 protein in the presence of overexpressed functional HDAC6 compared with that in the control conditions (PBS vehicle control), where NS5 is faded by the HDAC6 degradative effect (Figure 4c, 3-MA vs PBS NS5 bands in associated Western blot lanes 4, compared with their associated control lane 2; quantified in associated histograms). Furthermore, the ability of HDAC6 to degrade the ZIKV NS5 protein was validated in cells in which endogenous HDAC6 was specifically silenced by short interfering RNA (siRNA). HDAC6 knockdown provoked a significant increase in the expression of the ZIKV NS5 protein compared with that in control, scrambled-treated cells (Figure 4d, HDAC6 silencing and concomitant NS5 stabilization are quantified in the associated histograms).
Therefore, these data suggest that HDAC6 controls the expression of ZIKV NS5 by targeting the viral protein via autophagy, which requires both its deacetylase activity and the BUZ domain of HDAC6.
3.4. Autophagy Controls the Stability of the ZIKV NS5 Protein and Its Regulatory Immune Activity on RNA-Mediated RIG-I IFN Production
It has been reported that NS5 allows ZIKV evasion of IFN-mediated innate immunity [61,62,66,68,71,82,147,148,149,150,151]. The ZIKV NS5 protein targets each level of the IFN activation axis in host cells, particularly impairing genomic RNA sensing at its 5′ untranslated region (UTR) capped by RIG-I (retinoic acid-inducible gene I), a cytosolic pattern recognition receptor (PRR) that mediates the induction of a type-I IFN (IFN1) response [152,153,154]. In fact, NS5 represses RIG-I K63-linked polyubiquitination by means of the NS5-MTase (methyltransferase) domain, but MTase functions independently, thereby preventing RIG-I from activating IRF3 and therefore IFN-β production [63] or by acting as a barrier to IFN activation (reviewed in [82]).
Furthermore, we analysed the effect of the ZIKV NS5 protein on RNA-mediated RIG-I IFN production and its ability to inhibit IFN production in cells in which autophagy-mediated NS5 clearance was abrogated. For this purpose, we could not use either a tubacin inhibitor or HDAC6-overexpressing mutants altered in the autophagic-degratative functions of the antiviral enzyme since RIG-I is a substrate for HDAC6 [155,156,157]. Thus, specific chemical inhibition or mutation of the deacetylase domains of HDAC6, as well as the use of the HDAC6-DBUZ mutant, which is still deacetylase active, could alter the functional status of RIG-I, which directly depends on its acetylation state [155,156,158]. Moreover, as we are characterizing here, the degradative effect exerted by HDAC6 on the NS5 protein through its functions (i.e., deacetylases and BUZ-HDAC6 domains) could directly affect NS5 protein expresion and the associated IFN interference. Hence, we assayed 3-MA in a HEK-based cell model for IFN production (HEK-LuciaTM RIG-I cells; see Materials and methods) to inhibit the formation of aggresomes and the associated HDAC6-triggered autophagic degradation of targeted proteins, as reported [100,101,144,145,146].
We observed that the ZIKV NS5 protein inhibited RNA-mediated RIG-I IFN production in HEK-Lucia™ RIG-I cells (Figure 5a, compared with cells not expressing NS5), a model for measuring IFN production in a 5’ triphosphate hairpin RNA (3p-hpRNA) RIG-I agonist-dependent manner [159,160,161,162]. In the presence of 3-MA, NS5 completely abrogated the IFN response mediated by the RIG-I agonist 3p-hpRNA (Figure 5a). Treatment of cells not expressing NS5 with 3-MA decreased the capacity of the RIG-I agonist 3p-hpRNA to promote IFN production. This effect could be a consequence of the complex interplay between autophagy and RIG-I/IFN responses (reviewed in [163]). Likewise, the IFN response mediated by the RIG-I agonist 3p-hpRNA was inhibited by the overexpression of NS5 in 3-MA-treated cells, correlating this neutralizing effect of the ZIKV NS5 protein on IFN production (Figure 5b) with the stability of the NS5 protein after autophagy inhibition (Figure 5b, compare NS5 bands in Western blot lanes 4 and 3). Similarly, biochemical analysis of NS5-expressing HEK-Lucia™ RIG-I cells revealed the stabilization of MTs, as indicated by the increase in the acetylation of the a-tubulin subunits of MTs (Figure 5b, Ac-tub bands were compared between lanes 3 and 4 and lanes 1 and 2).
Taken together, these data suggest that HDAC6-mediated autophagy targeting the ZIKV NS5 protein regulates NS5 cell distribution and localization and that the associated aberrant effects on the tubulin cytoskeleton control the ability of NS5 to interfere with the IFN response mediated by the RIG-I pathway activated by foreign RNA agonists.
4. Discussion
The ZIKV NS5 protein is required for virus genome replication, and one of the ZIKV proteins involved in immune evasion of the IFN-mediated innate response [61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82]. In this work, we aimed to study potential cell structures and functions that could be affected by the ZIKV NS5 protein and considered potential mechanisms for cell toxicity and pathogenesis associated with ZIKV infection. Our results obtained in HEK-293T cells, a model for ZIKV and other RNA virus infection and production [98,99,100,101,125,126,127], suggested that the ZIKV NS5 protein could aberrantly reorganize the tubulin cytoskeleton by promoting nested-like structures of acetylated MTs organized at the periphery of the cell. Although the caged reorganization of MTs has been reported in ZIKV-infected cells [88], which surround the viral replication factory close to the endoplasmic reticulum (ER), we believe that this is the first time that this caged effect on MTs has been reported to be associated with the ZIKV NS5 protein. These kinds of nested structures have also been described for Bunyamwera virus (BUNV) but for virus-triggered aberrant actin cytoskeleton reorganization [164]. Notably, BUNV belongs to another family of emergent RNA enveloped viruses that can cause severe episodes of encephalitis and haemorrhagic fevers in humans [165,166,167,168,169].
The ZIKV NS5 protein appears to be located mainly in nuclear structures, as previously reported [64,65,117,118], according to fluorescence microscopy analysis. This observation was confirmed by biochemical analysis of cell fractions from cells overexpressing the ZIKV NS5 protein, which was mainly distributed in the nuclear fraction and was only slightly detected in the cytoplasmic fraction. Some studies have associated the ZIKV NS5 protein with the MTOC or centrosome during mitosis and cell division [95,114,115,116]. It is conceivable that the cytoplasmic pool of the ZIKV NS5 protein could alter MT dynamics and stability. However, we cannot rule out that the ZIK NS5 protein must drive MT reorganization from the nucleus, as reported for nuclear elements that can establish transient or stable connections with MTs (reviewed in [170]), where aberrant crosstalk between MTs and the nucleus may contribute to a plethora of human developmental defects and other diseases [171,172,173,174,175]. Likewise, abnormal tubulin cytoskeleton organization and functions could be responsible for a plethora of named primary tubulinopathies and neurodegenerative diseases [176]. As described in the introductory section of this work, a hallmark of ZIKV infection is CZS (i.e., including microcephaly) together with abnormal brain development and other neurologic manifestations [27,41,42,43,44,45] and disorders [34,46,47,48,49], such as GBS [46,48,50,51,52], encephalitis [46,51,52,53], meningoencephalitis [34,54], acute myelitis [46,52,55] and encephalomyelitis [46,49,51,56,57], as well as sensory polyneuropathy [58] and other neurological complications [59,60]. Similarly, it has been recently reported that the ZIKV NS5 protein interacts with components of the cilium base, promoting ciliopathy and premature neurogenesis [96]. Therefore, the NS5-induced nested-like rearrangement of MTs at the periphery of the cell could be considered a cell anomaly that, in part, could be responsible for cell toxicity, abnormal tissue development and/or function and ZIKV infection pathology.
Although it is a unique flavivirus that impacts cellular architecture, it is also exceptional in that it controls and restricts its NS5 protein in nuclear discrete bodies, as we described here and previously reported [64,65,117,118]. Consequently, this peculiar way of organizing could mechanistically explain how the virus supports infection [88]. In fact, ZIKV seems to require MTs and the MTOC for viroplasm organization and virus production [95]. Remarkably, the authors observed that viroplasm formation and ZIKV production are not negatively affected when infected cells have no centrosomes or an impaired MTOC, suggesting that viroplasm acts as a potential site for MT organization and efficient virus production [95]. It is well established that MTs are commonly hijacked by viruses to traffic to sites of replication after entry and to promote egress from infected cells (reviewed in [92,111]). Therefore, the NS5 protein could assure efficient viral production and egress by organizing nested-like MT platforms located at the periphery of the cells, as we observed.
In this regard, one of the main cellular functions that regulates viral infection and is closely related to the organization of MTs is the autophagy process [89,90,91,92,93,94]. Although the complexity of the interplay between autophagy and different ZIKV proteins in the context of virus infection and transmission has been reported [142,143,177,178], little is known about the ZIKV NS5 protein. Our observations indicate that the ZIKV NS5 protein negatively alters p62-autophagy flux in a dose-dependent manner, as indicated by the accumulation of p62 in cells overexpressing NS5. Therefore, the ZIKV NS5 protein affects cell structures and events that are key for cell permissivity to viral infection and survival, such as tubulin cytoskeleton dynamics or p62-associated autophagy flux [97,98,99,100,101,102,103], which are functions regulated by the antiviral factor HDAC6 [91,94,98,99,100,101,104,105,106,107,108], presumably to protect itself from fading and accomplishing its viral functions. Remarkably, p62 and HDAC6 are associated with aggresomes and the autophagy-derived pathway [97,102,122,179,180], which has been reported to act against ZIKV infection [181]. In this study, the authors compete for HDAC6-mediated aggresome formation by using small synthetic “designed ankyrin repeat proteins” (DARPins) that impair the interaction between HDAC6 and ubiquitin. This DARPins-associated action blocked infection by ZIKV and the influenza A virus (IAV) at the uncoating step of the viral cycle [181].
In this context, our results indicate that HDAC6 targets the NS5 protein via autophagy in a deacetylase- and BUZ domain-dependent manner and controls the cytoplasmic expression of the ZIKV NS5 protein. Similarly, by using HDAC6 mutants lacking key functions responsible for the autophagy activity of antiviral enzymes, such as deacetylases and BUZ domains, the results confirmed that the ZIKV NS5 protein is under the degradative control of HDAC6. Functional HDAC6 targets NS5 by autophagy, as the degradative effect of HDAC6 on NS5 is blocked by the 3-MA aggresome/autophagy inhibitor or by the overexpression of the deacetylase dead-mutant (dm-HDAC6) or DBUZ mutant (HDAC-DBUZ) of HDAC6. siRNA-mediated knockdown of endogenous HDAC6 indicated that the endogenous enzyme targeted the ZIKV NS5 protein. Remarkably, fluorescence microscopy revealed that the ZIKV NS5 protein is present both in the nucleus and in the cytoplasm of cells when the deacetylase activity of HDAC6 is specifically inhibited by tubacin. Of note, tubacin-mediated inhibition of HDAC6 deacetylase activity induces MT acetylation but does not cause the caged reorganization of MTs observed by the ZIKV NS5 protein. Therefore, it is conceivable that the effect of NS5 on aberrant MT rearrangement does not involve an inhibitory action of the viral protein on the tubulin deacetylase activity of endogenous HDAC6. Taken together, these data suggest that functional HDAC6 negatively regulates the protein expression of NS5 in the cytoplasm, which is predominantly detected in the nucleus, and prevents the aberrant acetylation of MTs and p62 accumulation observed when the ZIKV NS5 protein is overexpressed. Thus, it is plausible that, as occurs with HIV, IAV and other viruses [91,92,94,98,99,100,101,104,105,106,182,183], HDAC6 could also be involved in the control of ZIKV NS5-driven viral RNA replication and virus production at the late stages of the viral cycle and that the ZIKV NS5 protein targets the p62/HDAC6 aggresome pathway to ensure viral persistence.
Moreover, our results indicate that the ZIKV NS5 protein inhibits RNA-mediated RIG-I IFN production in HEK-Lucia RIG-I-adapted cells and that the inhibitory activity of the viral protein increases when cell autophagy is inhibited by the chemical inhibitor 3-MA. 3-MA inhibits the formation of aggresomes and the associated HDAC6-triggered autophagic degradation of targeted proteins [100,101,144,145,146]. In fact, 3-MA impairs autophagic sequestration and HDAC6-mediated degradation of the HIV-1 Vif and Pr55Gag proteins [98,99,100,101]. Thus, the results obtained with 3-MA for NS5 inhibition of RIG-I-mediated IFN production, triggered by the RIG-I agonist 3p-hpRNA (5’ triphosphate hairpin RNAs), are in accordance with the increase observed in the level of ZIKV NS5 protein expression when endogenous HDAC6-associated autophagy on NS5 is blocked by 3-MA.
Notably, HDAC6 mediates the deacetylation of the C-terminal region of RIG-I, thereby promoting the viral RNA-sensing activity of RIG-I and the associated IFN response [155]. Therefore, similar to the acetylation of MTs (a substrate of HDAC6) promoted by NS5, it is conceivable that the ZIKV NS5 protein may act on the RIG-I substrate of HDAC6, affecting its deacetylation state and activity to inhibit IFN production. The ZIKV NS5 protein targets each level of the IFN activation axis in host cells, particularly impairing genomic RNA sensing at its 5’ untranslated region (UTR) capped by RIG-I by repressing RIG-I polyubiquitination by means of the NS5-MTase (methyltransferase) domain but independent of MTase function, thereby preventing RIG-I from activating IFN regulatory factor 3 (IRF3) and therefore IFN-β production [63] or by acting as a barrier to IFN activation. Hence, NS5 abrogates IRF3 and nuclear factor kappa B (NF-κB) signalling [71], as it is able to interact with IRF3, preventing its activation [184], or bind to inhibitor of kappa-B kinase epsilon (IKKε), affecting IKKε protein levels and phosphorylation, which ultimately results in IRF3 inactivation [151]. Furthermore, the NS5 protein antagonizes IFN production by impairing the activation of TANK-binding kinase 1 (TBK1) and, therefore, IRF3, a TBK1 substrate for phosphorylation [61]. In addition, NS5 competitively binds to the ubiquitin-like domain of TBK1, affecting its interaction with tumor necrosis factor (TNF)-associated factor 6 (TRAF6). This complex is required for TBK1-mediated IRF3 phosphorylation and activation, thus interfering with type I and III IFN transcription [61]. Additionally, in IFN-induced human cells, ZIKV NS5 expression results in proteasomal degradation of the IFN-regulated transcriptional activator STAT2 (signal transducer and activator of transcription 2) in humans and affects STAT1 phosphorylation levels, suppressing INF-mediated Janus kinase (JAK)/STAT signal transduction [62,149], which shows that ZIKV-induced disease takes advantage of NS5-promoted IFN deficiency. Moreover, although the role that NS5 plays in the nucleus remains enigmatic, recent studies have suggested that the subcellular localization of NS5 is important for its function in innate immune suppression, which provides new insight into ZIKV pathogenesis [65,118,185]. Thus, the inhibition of the RIG-I/IFN response by ZIKV NS5 is thought to be key to ensuring the viral cycle and the infection of cells found in immunoprivileged sites, such as the brain or placenta [71,186], thereby allowing ZIKV evasion of IFN-mediated innate immunity. Therefore, the RIG-I/HDAC6 interplay could be key for the control of the IFN-mediated immune response against ZIKV by acting on NS5.
Taken together, these results suggest that the ZIKV NS5 protein contributes to cell toxicity and pathogenesis by affecting MT dynamics and p62-associated autophagic flux and evades the IFN-immune response by overcoming HDAC6 functions. Moreover, through autophagy targeting of the ZIKV NS5 protein, HDAC6 has emerged as a key anti-ZIKV factor that can control NS5-mediated functions in cells, such as aberrant alteration of the tubulin cytoskeleton and inhibition of autophagic p62 flux; thus, HDAC6 is a potential protective factor against cell toxicity and associated ZIKV infection pathogenesis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figures S1A and S1B: Western blots associated with Figures 1a and 1b; Figures S2C, S2D and S2E: Western blots associated with Figures 2c, 2d and 2e; Figures S3A and S3D: Western blots associated with Figures 3a and 3d; Figures S4A, S4C and S4D: Western blots associated with Figures 4a, 4c and 4d; Figure S5B: Western blot associated with Figure 5b.
Author Contributions
J-EH wrote the paper and performed the research and supervised the experimental design; S-PY, I-LG, and J-GL performed the research and supervised the experimental design; R-TG performed the statistical analysis and the research. A-VF wrote the paper, conceived, and designed the research and laboratory experiments, and supervised the experimental design, analysis and interpretation of the data. All the authors have read and approved the final manuscript.
Funding
This research was funded by PID2021-123031OB-I00 (MCIN (“Ministerio de Ciencia e Innovación”)/AEI (“Agencia Española de Investigación”)/10.13039/501100011033/FEDER, UE) (Spain), UNLL10-3E-783 (the European Regional Development Fund (ERDF) and “Fundación CajaCanarias”) and “SEGAI-ULL”. RC-R was funded by ProID2020010093 (“Agencia Canaria de Investigación, Innovación y Sociedad de la Información” and European Social Fund”). SP-Y was funded by “Contrato Predoctoral Ministerio-ULL Formación de Doctores” (2019 Program) (“Ministerio de Ciencia, Innovación y Universidades”, Spain). JG-L was funded by the “Juan de la Cierva de Incorporación” Spanish Program IJC2019-038902-I (“Ayudas Juan de la Cierva de incorporación; AEI/MCIN”). JE-H was funded by the Cabildo of Tenerife “Agustin de Betancourt” 2017 Program (ref. 2021/0000866) and “Plan de incentivación de la actividad investigadora de la Universidad de La Laguna 2023, Proyectos dirigidos por noveles investigadores; ref. 2022/20149”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data availability statement
All presented data are available upon request.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Marchette, N.J.; Garcia, R.; Rudnick, A. Isolation of Zika virus from Aedes aegypti mosquitoes in Malaysia. The American journal of tropical medicine and hygiene 1969, 18, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Kuno, G.; Chang, G.J. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Archives of virology 2007, 152, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.V.; Tesh, R.B.; Vasilakis, N. The emergence of arthropod-borne viral diseases: A global prospective on dengue, chikungunya and zika fevers. Acta tropica 2017, 166, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Diallo, D.; Sall, A.A.; Diagne, C.T.; Faye, O.; Faye, O.; Ba, Y.; Hanley, K.A.; Buenemann, M.; Weaver, S.C.; Diallo, M. Zika virus emergence in mosquitoes in southeastern Senegal, 2011. PloS one 2014, 9, e109442. [Google Scholar] [CrossRef]
- Faye, O.; Faye, O.; Diallo, D.; Diallo, M.; Weidmann, M.; Sall, A.A. Quantitative real-time PCR detection of Zika virus and evaluation with field-caught mosquitoes. Virology journal 2013, 10, 311. [Google Scholar] [CrossRef] [PubMed]
- Weinbren, M.P.; Williams, M.C. Zika virus: further isolations in the Zika area, and some studies on the strains isolated. Transactions of the Royal Society of Tropical Medicine and Hygiene 1958, 52, 263–268. [Google Scholar] [CrossRef]
- Haddow, A.J.; Williams, M.C.; Woodall, J.P.; Simpson, D.I.; Goma, L.K. TWELVE ISOLATIONS OF ZIKA VIRUS FROM AEDES (STEGOMYIA) AFRICANUS (THEOBALD) TAKEN IN AND ABOVE A UGANDA FOREST. Bulletin of the World Health Organization 1964, 31, 57–69. [Google Scholar]
- Berthet, N.; Nakouné, E.; Kamgang, B.; Selekon, B.; Descorps-Declère, S.; Gessain, A.; Manuguerra, J.C.; Kazanji, M. Molecular characterization of three Zika flaviviruses obtained from sylvatic mosquitoes in the Central African Republic. Vector borne and zoonotic diseases (Larchmont, N.Y.) 2014, 14, 862–865. [Google Scholar] [CrossRef]
- McCrae, A.W.; Kirya, B.G. Yellow fever and Zika virus epizootics and enzootics in Uganda. Transactions of the Royal Society of Tropical Medicine and Hygiene 1982, 76, 552–562. [Google Scholar] [CrossRef]
- Grard, G.; Caron, M.; Mombo, I.M.; Nkoghe, D.; Mboui Ondo, S.; Jiolle, D.; Fontenille, D.; Paupy, C.; Leroy, E.M. Zika virus in Gabon (Central Africa)--2007: a new threat from Aedes albopictus? PLoS neglected tropical diseases 2014, 8, e2681. [Google Scholar] [CrossRef]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Transactions of the Royal Society of Tropical Medicine and Hygiene 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Enfissi, A.; Codrington, J.; Roosblad, J.; Kazanji, M.; Rousset, D. Zika virus genome from the Americas. Lancet 2016, 387, 227–228. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerging infectious diseases 2008, 14, 1232–1239. [Google Scholar] [CrossRef]
- Casals, J. Viruses: the versatile parasites; the arthropod-borne group of animal viruses. Transactions of the New York Academy of Sciences 1957, 19, 219–235. [Google Scholar] [CrossRef]
- Haddow, A.D.; Woodall, J.P. Distinguishing between Zika and Spondweni viruses. Bulletin of the World Health Organization 2016, 94, 711–711a. [Google Scholar] [CrossRef]
- Brès, P. [Recent data from serological surveys on the prevalence of arbovirus infections in Africa, with special reference to yellow fever]. Bulletin of the World Health Organization 1970, 43, 223–267. [Google Scholar] [PubMed]
- Macnamara, F.N. Zika virus: a report on three cases of human infection during an epidemic of jaundice in Nigeria. Transactions of the Royal Society of Tropical Medicine and Hygiene 1954, 48, 139–145. [Google Scholar] [CrossRef]
- Wikan, N.; Smith, D.R. Zika virus: history of a newly emerging arbovirus. Lancet Infect Dis 2016, 16, e119–e126. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.G.; Ksiazek, T.G.; Suhandiman; Triwibowo. Zika virus, a cause of fever in Central Java, Indonesia. Transactions of the Royal Society of Tropical Medicine and Hygiene 1981, 75, 389–393. [CrossRef]
- virus., W.I.E.C.o.Z. WHO statement on the first meeting of the International Health Regulations (2005) (IHR 2005) Emergency Committee on Zika virus and observed increase in neurological disorders and neonatal malformations. https://www.who.int/news/item/01-02-2016-who-statement-on-the-first-meeting-of-the-international-health-regulations-(2005)-(ihr-2005)-emergency-committee-on-zika-virus-and-observed-increase-in-neurological-disorders-and-neonatal-malformations. 2016.
- Miranda-Filho Dde, B.; Martelli, C.M.; Ximenes, R.A.; Araújo, T.V.; Rocha, M.A.; Ramos, R.C.; Dhalia, R.; França, R.F.; Marques Júnior, E.T.; Rodrigues, L.C. Initial Description of the Presumed Congenital Zika Syndrome. American journal of public health 2016, 106, 598–600. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Jamieson, D.J.; Powers, A.M.; Honein, M.A. Zika Virus. N Engl J Med 2016, 374, 1552–1563. [Google Scholar] [CrossRef]
- Brasil, P.; Pereira, J.P., Jr.; Moreira, M.E.; Ribeiro Nogueira, R.M.; Damasceno, L.; Wakimoto, M.; Rabello, R.S.; Valderramos, S.G.; Halai, U.A.; Salles, T.S.; et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N Engl J Med 2016, 375, 2321–2334. [Google Scholar] [CrossRef]
- Kleber de Oliveira, W.; Cortez-Escalante, J.; De Oliveira, W.T.; do Carmo, G.M.; Henriques, C.M.; Coelho, G.E.; Araújo de França, G.V. Increase in Reported Prevalence of Microcephaly in Infants Born to Women Living in Areas with Confirmed Zika Virus Transmission During the First Trimester of Pregnancy - Brazil, 2015. MMWR. Morbidity and mortality weekly report 2016, 65, 242–247. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Jamieson, D.J.; Honein, M.A.; Petersen, L.R. Zika Virus and Birth Defects--Reviewing the Evidence for Causality. N Engl J Med 2016, 374, 1981–1987. [Google Scholar] [CrossRef]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jääskeläinen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N Engl J Med 2016, 374, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Martines, R.B.; Bhatnagar, J.; Keating, M.K.; Silva-Flannery, L.; Muehlenbachs, A.; Gary, J.; Goldsmith, C.; Hale, G.; Ritter, J.; Rollin, D.; et al. Notes from the Field: Evidence of Zika Virus Infection in Brain and Placental Tissues from Two Congenitally Infected Newborns and Two Fetal Losses--Brazil, 2015. MMWR. Morbidity and mortality weekly report 2016, 65, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljšak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodušek, V.; et al. Zika Virus Associated with Microcephaly. N Engl J Med 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Schuler-Faccini, L.; Ribeiro, E.M.; Feitosa, I.M.; Horovitz, D.D.; Cavalcanti, D.P.; Pessoa, A.; Doriqui, M.J.; Neri, J.I.; Neto, J.M.; Wanderley, H.Y.; et al. Possible Association Between Zika Virus Infection and Microcephaly - Brazil, 2015. MMWR. Morbidity and mortality weekly report 2016, 65, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Soares de Araújo, J.S.; Regis, C.T.; Gomes, R.G.; Tavares, T.R.; Rocha Dos Santos, C.; Assunção, P.M.; Nóbrega, R.V.; Pinto, D.F.; Bezerra, B.V.; Mattos, S.D. Microcephaly in north‒east Brazil: a retrospective study on neonates born between 2012 and 2015. Bulletin of the World Health Organization 2016, 94, 835–840. [Google Scholar] [CrossRef]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.O.; Sampaio, S.A.; de Filippis, I.; Fabri, A.; Araujo, E.S.M.; de Sequeira, P.C.; de Mendonça, M.C.L.; de Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Dyer, O. Zika virus spreads across Americas as concerns mount over birth defects. BMJ (Clinical research ed.) 2015, 351, h6983. [Google Scholar] [CrossRef]
- WHO, O.g.e.r. WHO Situation report. Zika virus microcephaly Guillain‒Barré syndrome. http://reliefweb.int/report/world/zika-virus-microcephaly-and-guillain-barr-syndrome-situation-report-10-march-2017 2017, March 10, 2017. Data as of 9 March 2017.
- Carteaux, G.; Maquart, M.; Bedet, A.; Contou, D.; Brugières, P.; Fourati, S.; Cleret de Langavant, L.; de Broucker, T.; Brun-Buisson, C.; Leparc-Goffart, I.; et al. Zika Virus Associated with Meningoencephalitis. N Engl J Med 2016, 374, 1595–1596. [Google Scholar] [CrossRef]
- Barbi, L.; Coelho, A.V.C.; Alencar, L.C.A.; Crovella, S. Prevalence of Guillain‒Barré syndrome among Zika virus infected cases: a systematic review and meta-analysis. The Brazilian journal of infectious diseases: an official publication of the Brazilian Society of Infectious Diseases 2018, 22, 137–141. [Google Scholar] [CrossRef]
- Costello, A.; Dua, T.; Duran, P.; Gülmezoglu, M.; Oladapo, O.T.; Perea, W.; Pires, J.; Ramon-Pardo, P.; Rollins, N.; Saxena, S. Defining the syndrome associated with congenital Zika virus infection. Bulletin of the World Health Organization 2016, 94, 406–406a. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, T.; Rodriguez, A.; Almiron, M.; Sanhueza, A.; Ramon, P.; de Oliveira, W.K.; Coelho, G.E.; Badaró, R.; Cortez, J.; Ospina, M.; et al. Zika Virus and the Guillain‒Barré Syndrome - Case Series from Seven Countries. N Engl J Med 2016, 375, 1598–1601. [Google Scholar] [CrossRef] [PubMed]
- Hoen, B.; Schaub, B.; Funk, A.L.; Ardillon, V.; Boullard, M.; Cabié, A.; Callier, C.; Carles, G.; Cassadou, S.; Césaire, R.; et al. Pregnancy Outcomes after ZIKV Infection in French Territories in the Americas. N Engl J Med 2018, 378, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Control., E.C.f.D.P.a. Zika virus disease epidemic: potential association with microcephaly and Guillain‒Barré syndrome (first update). European Centre for Disease Prevention and Control, Stockholm, Sweden. 2016, http://ecdc.europa.eu/en/publications/Publications/rapid-risk-assessment-zika-virus-first-update-jan-2016.pdf.
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Dudley, D.M.; Van Rompay, K.K.; Coffey, L.L.; Ardeshir, A.; Keesler, R.I.; Bliss-Moreau, E.; Grigsby, P.L.; Steinbach, R.J.; Hirsch, A.J.; MacAllister, R.P.; et al. Miscarriage and stillbirth following maternal Zika virus infection in nonhuman primates. Nat Med 2018, 24, 1104–1107. [Google Scholar] [CrossRef]
- van der Eijk, A.A.; van Genderen, P.J.; Verdijk, R.M.; Reusken, C.B.; Mögling, R.; van Kampen, J.J.; Widagdo, W.; Aron, G.I.; GeurtsvanKessel, C.H.; Pas, S.D.; et al. Miscarriage Associated with Zika Virus Infection. N Engl J Med 2016, 375, 1002–1004. [Google Scholar] [CrossRef]
- Sarno, M.; Sacramento, G.A.; Khouri, R.; do Rosário, M.S.; Costa, F.; Archanjo, G.; Santos, L.A.; Nery, N., Jr.; Vasilakis, N.; Ko, A.I.; et al. Zika Virus Infection and Stillbirths: A Case of Hydrops Fetalis, Hydranencephaly and Fetal Demise. PLoS neglected tropical diseases 2016, 10, e0004517. [Google Scholar] [CrossRef]
- Schaub, B.; Monthieux, A.; Najihoullah, F.; Harte, C.; Césaire, R.; Jolivet, E.; Voluménie, J.L. Late miscarriage: another Zika concern? European journal of obstetrics, gynecology, and reproductive biology 2016, 207, 240–241. [Google Scholar] [CrossRef]
- Leisher, S.H.; Balalian, A.A.; Reinebrant, H.; Shiau, S.; Flenady, V.; Kuhn, L.; Morse, S.S. Systematic review: fetal death reporting and risk in Zika-affected pregnancies. Tropical medicine & international health: TM & IH 2020. [Google Scholar] [CrossRef]
- da Silva, I.R.F.; Frontera, J.A.; Bispo de Filippis, A.M.; Nascimento, O. Neurologic Complications Associated With the Zika Virus in Brazilian Adults. JAMA neurology 2017, 74, 1190–1198. [Google Scholar] [CrossRef]
- Rozé, B.; Najioullah, F.; Signate, A.; Apetse, K.; Brouste, Y.; Gourgoudou, S.; Fagour, L.; Abel, S.; Hochedez, P.; Cesaire, R.; et al. Zika virus detection in cerebrospinal fluid from two patients with encephalopathy, Martinique, February 2016. Euro Surveill 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.S.; Araujo, M.T.; Martins Filho, A.J.; Oliveira, C.S.; Nunes, B.T.; Cruz, A.C.; Nascimento, A.G.; Medeiros, R.C.; Caldas, C.A.; Araujo, F.C.; et al. Zika virus epidemic in Brazil. I. Fatal disease in adults: Clinical and laboratorial aspects. Journal of clinical virology: the official publication of the Pan American Society for Clinical Virology 2016, 85, 56–64. [Google Scholar] [CrossRef]
- Alves-Leon, S.V.; Lima, M.D.R.; Nunes, P.C.G.; Chimelli, L.M.C.; Rabelo, K.; Nogueira, R.M.R.; de Bruycker-Nogueira, F.; de Azeredo, E.L.; Bahia, P.R.; Rueda Lopes, F.C.; et al. Zika virus found in brain tissue of a multiple sclerosis patient undergoing an acute disseminated encephalomyelitis-like episode. Multiple sclerosis (Houndmills, Basingstoke, England) 2019, 25, 427–430. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jiménez-Arango, J.A.; Zea-Vera, A.F.; González-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuñiga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain‒Barré Syndrome Associated with Zika Virus Infection in Colombia. N Engl J Med 2016, 375, 1513–1523. [Google Scholar] [CrossRef]
- Brito Ferreira, M.L.; Antunes de Brito, C.A.; Moreira Á, J.P.; de Morais Machado, M.; Henriques-Souza, A.; Cordeiro, M.T.; de Azevedo Marques, E.T.; Pena, L.J. Guillain‒Barré Syndrome, Acute Disseminated Encephalomyelitis and Encephalitis Associated with Zika Virus Infection in Brazil: Detection of Viral RNA and Isolation of Virus during Late Infection. The American journal of tropical medicine and hygiene 2017, 97, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, L.S.; Barreras, P.; Pardo, C.A. Zika Virus-Associated Neurological Disease in the Adult: Guillain‒Barré Syndrome, Encephalitis, and Myelitis. Seminars in reproductive medicine 2016, 34, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Soares, C.N.; Brasil, P.; Carrera, R.M.; Sequeira, P.; de Filippis, A.B.; Borges, V.A.; Theophilo, F.; Ellul, M.A.; Solomon, T. Fatal encephalitis associated with Zika virus infection in an adult. Journal of clinical virology: the official publication of the Pan American Society for Clinical Virology 2016, 83, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Schwartzmann, P.V.; Ramalho, L.N.; Neder, L.; Vilar, F.C.; Ayub-Ferreira, S.M.; Romeiro, M.F.; Takayanagui, O.M.; Dos Santos, A.C.; Schmidt, A.; Figueiredo, L.T.; et al. Zika Virus Meningoencephalitis in an Immunocompromised Patient. Mayo Clinic proceedings 2017, 92, 460–466. [Google Scholar] [CrossRef]
- Mécharles, S.; Herrmann, C.; Poullain, P.; Tran, T.H.; Deschamps, N.; Mathon, G.; Landais, A.; Breurec, S.; Lannuzel, A. Acute myelitis due to Zika virus infection. Lancet 2016, 387, 1481. [Google Scholar] [CrossRef]
- Niemeyer, B.; Niemeyer, R.; Borges, R.; Marchiori, E. Acute Disseminated Encephalomyelitis Following Zika Virus Infection. European neurology 2017, 77, 45–46. [Google Scholar] [CrossRef] [PubMed]
- Galliez, R.M.; Spitz, M.; Rafful, P.P.; Cagy, M.; Escosteguy, C.; Germano, C.S.; Sasse, E.; Gonçalves, A.L.; Silveira, P.P.; Pezzuto, P.; et al. Zika Virus Causing Encephalomyelitis Associated With Immunoactivation. Open forum infectious diseases 2016, 3, ofw203. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.T.; England, J.D.; Lorenzana, I.; Medina-Montoya, M.; Alvarado, D.; De Bastos, M.; Fontiveros, S.; Sierra, M.; Contreras, F. Zika virus associated with sensory polyneuropathy. Journal of the neurological sciences 2016, 369, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Nicastri, E.; Castilletti, C.; Balestra, P.; Galgani, S.; Ippolito, G. Zika Virus Infection in the Central Nervous System and Female Genital Tract. Emerging infectious diseases 2016, 22, 2228–2230. [Google Scholar] [CrossRef] [PubMed]
- Bido-Medina, R.; Wirsich, J.; Rodríguez, M.; Oviedo, J.; Miches, I.; Bido, P.; Tusen, L.; Stoeter, P.; Sadaghiani, S. Impact of Zika Virus on adult human brain structure and functional organization. Annals of clinical and translational neurology 2018, 5, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yang, S.; He, J.; Guest, J.D.; Ma, Z.; Yang, L.; Pierce, B.G.; Tang, Q.; Zhang, Y.J. Zika virus NS5 protein antagonizes type I interferon production by blocking TBK1 activation. Virology 2019, 527, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Hertzog, J.; Dias Junior, A.G.; Rigby, R.E.; Donald, C.L.; Mayer, A.; Sezgin, E.; Song, C.; Jin, B.; Hublitz, P.; Eggeling, C.; et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signalling. European journal of immunology 2018, 48, 1120–1136. [Google Scholar] [CrossRef]
- Li, A.; Wang, W.; Wang, Y.; Chen, K.; Xiao, F.; Hu, D.; Hui, L.; Liu, W.; Feng, Y.; Li, G.; et al. NS5 Conservative Site Is Required for Zika Virus to Restrict the RIG-I Signalling. Frontiers in immunology 2020, 11, 51. [Google Scholar] [CrossRef]
- Tan, M.J.A.; Chan, K.W.K.; Ng, I.H.W.; Kong, S.Y.Z.; Gwee, C.P.; Watanabe, S.; Vasudevan, S.G. The Potential Role of the ZIKV NS5 Nuclear Spherical-Shell Structures in Cell Type-Specific Host Immune Modulation during ZIKV Infection. Cells 2019, 8. [Google Scholar] [CrossRef]
- Ng, I.H.W.; Chan, K.W.; Tan, M.J.A.; Gwee, C.P.; Smith, K.M.; Jeffress, S.J.; Saw, W.G.; Swarbrick, C.M.D.; Watanabe, S.; Jans, D.A.; et al. Zika Virus NS5 Forms Supramolecular Nuclear Bodies That Sequester Importin-α and Modulate the Host Immune and Pro-Inflammatory Response in Neuronal Cells. ACS infectious diseases 2019, 5, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Elshahawi, H.; Syed Hassan, S.; Balasubramaniam, V. Importance of Zika Virus NS5 Protein for Viral Replication. Pathogens (Basel, Switzerland) 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ketkar, H.; Geng, T.; Lo, E.; Wang, L.; Xi, J.; Sun, Q.; Zhu, Z.; Cui, Y.; Yang, L.; et al. Zika Virus Nonstructural Protein 4A Blocks the RLR-MAVS Signalling. Frontiers in microbiology 2018, 9, 1350. [Google Scholar] [CrossRef]
- Best, S.M. The Many Faces of the Flavivirus NS5 Protein in Antagonism of Type I Interferon Signalling. Journal of virology 2017, 91. [Google Scholar] [CrossRef]
- Hu, Y.; Dong, X.; He, Z.; Wu, Y.; Zhang, S.; Lin, J.; Yang, Y.; Chen, J.; An, S.; Yin, Y.; et al. Zika virus antagonizes interferon response in patients and disrupts RIG-I-MAVS interaction through its CARD-TM domains. Cell & bioscience 2019, 9, 46. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell discovery 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signalling. EMBO reports 2016, 17, 1766–1775. [Google Scholar] [CrossRef]
- Riedl, W.; Acharya, D.; Lee, J.H.; Liu, G.; Serman, T.; Chiang, C.; Chan, Y.K.; Diamond, M.S.; Gack, M.U. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell host & microbe 2019, 26, 493–503e496. [Google Scholar] [CrossRef]
- Serman, T.M.; Gack, M.U. Evasion of Innate and Intrinsic Antiviral Pathways by the Zika Virus. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Cervantes-Salazar, M.; Gutiérrez-Escolano, A.L.; Reyes-Ruiz, J.M.; del Angel, R.M. The Nonstructural Proteins 3 and 5 from Flavivirus Modulate Nuclear-Cytoplasmic Transport and Innate Immune Response Targeting Nuclear Proteins. bioRxiv 2018, 375899. [Google Scholar] [CrossRef]
- Ding, Q.; Gaska, J.M.; Douam, F.; Wei, L.; Kim, D.; Balev, M.; Heller, B.; Ploss, A. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proceedings of the National Academy of Sciences of the United States of America 2018, 115, E6310–e6318. [Google Scholar] [CrossRef]
- Ngueyen, T.T.N.; Kim, S.J.; Lee, J.Y.; Myoung, J. Zika Virus Proteins NS2A and NS4A Are Major Antagonists that Reduce IFN-β Promoter Activity Induced by the MDA5/RIG-I Signalling Pathway. Journal of microbiology and biotechnology 2019, 29, 1665–1674. [Google Scholar] [CrossRef]
- Sager, G.; Gabaglio, S.; Sztul, E.; Belov, G.A. Role of Host Cell Secretory Machinery in Zika Virus Life Cycle. Viruses 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.P.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. The EMBO journal 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Strange, D.P.; Jiyarom, B.; Pourhabibi Zarandi, N.; Xie, X.; Baker, C.; Sadri-Ardekani, H.; Shi, P.Y.; Verma, S. Axl Promotes Zika Virus Entry and Modulates the Antiviral State of Human Sertoli Cells. mBio 2019, 10. [Google Scholar] [CrossRef]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell reports 2017, 18, 324–333. [Google Scholar] [CrossRef]
- Sirohi, D.; Kuhn, R.J. Zika Virus Structure, Maturation, and Receptors. The Journal of infectious diseases 2017, 216, S935–s944. [Google Scholar] [CrossRef]
- Estevez-Herrera, J.; Perez-Yanes, S.; Cabrera-Rodriguez, R.; Marquez-Arce, D.; Trujillo-Gonzalez, R.; Machado, J.D.; Madrid, R.; Valenzuela-Fernandez, A. Zika Virus Pathogenesis: A Battle for Immune Evasion. Vaccines 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, R.; Kaur, G.; Seth, P. Molecular mechanisms of zika virus pathogenesis: An update. Indian J Med Res 2021, 154, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Ojha, C.R.; Rodriguez, M.; Lapierre, J.; Muthu Karuppan, M.K.; Branscome, H.; Kashanchi, F.; El-Hage, N. Complementary Mechanisms Potentially Involved in the Pathology of Zika Virus. Frontiers in immunology 2018, 9, 2340. [Google Scholar] [CrossRef]
- Gorshkov, K.; Shiryaev, S.A.; Fertel, S.; Lin, Y.W.; Huang, C.T.; Pinto, A.; Farhy, C.; Strongin, A.Y.; Zheng, W.; Terskikh, A.V. Zika Virus: Origins, Pathological Action, and Treatment Strategies. Frontiers in microbiology 2018, 9, 3252. [Google Scholar] [CrossRef]
- Christian, K.M.; Song, H.; Ming, G.L. Pathophysiology and Mechanisms of Zika Virus Infection in the Nervous System. Annu Rev Neurosci 2019, 42, 249–269. [Google Scholar] [CrossRef]
- Fujimura, K.; Guise, A.J.; Nakayama, T.; Schlaffner, C.N.; Meziani, A.; Kumar, M.; Cheng, L.; Vaughan, D.J.; Kodani, A.; Van Haren, S.; et al. Integrative systems biology characterizes immune-mediated neurodevelopmental changes in murine Zika virus microcephaly. iScience 2023, 26, 106909. [Google Scholar] [CrossRef]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell reports 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Mackeh, R.; Perdiz, D.; Lorin, S.; Codogno, P.; Pous, C. Autophagy and microtubules - new story, old players. J Cell Sci 2013, 126, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Gonzalez, J.; Garcia-Exposito, L.; Puigdomenech, I.; de Armas-Rillo, L.; Machado, J.D.; Blanco, J.; Valenzuela-Fernandez, A. Viral infection: Moving through complex and dynamic cell-membrane structures. Commun Integr Biol 2011, 4, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Rodriguez, R.; Perez-Yanes, S.; Estevez-Herrera, J.; Marquez-Arce, D.; Cabrera, C.; Espert, L.; Blanco, J.; Valenzuela-Fernandez, A. The Interplay of HIV and Autophagy in Early Infection. Frontiers in microbiology 2021, 12, 661446. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Rodriguez, R.; Perez-Yanes, S.; Lorenzo-Sanchez, I.; Trujillo-Gonzalez, R.; Estevez-Herrera, J.; Garcia-Luis, J.; Valenzuela-Fernandez, A. HIV Infection: Shaping the Complex, Dynamic, and Interconnected Network of the Cytoskeleton. International journal of molecular sciences 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- de Armas-Rillo, L.; Valera, M.S.; Marrero-Hernandez, S.; Valenzuela-Fernandez, A. Membrane dynamics associated with viral infection. Reviews in medical virology 2016, 26, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernandez, A.; Cabrero, J.R.; Serrador, J.M.; Sanchez-Madrid, F. HDAC6: a key regulator of cytoskeleton, cell migration and cell‒cell interactions. Trends Cell Biol 2008, 18, 291–297. [Google Scholar] [CrossRef]
- Buchwalter, R.A.; Ogden, S.C.; York, S.B.; Sun, L.; Zheng, C.; Hammack, C.; Cheng, Y.; Chen, J.V.; Cone, A.S.; Meckes, D.G., Jr.; et al. Coordination of Zika Virus Infection and Viroplasm Organization by Microtubules and Microtubule-Organizing Centers. Cells 2021, 10. [Google Scholar] [CrossRef]
- Saade, M.; Ferrero, D.S.; Blanco-Ameijeiras, J.; Gonzalez-Gobartt, E.; Flores-Mendez, M.; Ruiz-Arroyo, V.M.; Martinez-Saez, E.; Ramon, Y.C.S.; Akizu, N.; Verdaguer, N.; et al. Multimerization of Zika Virus-NS5 Causes Ciliopathy and Forces Premature Neurogenesis. Cell stem cell 2020, 27, 920–936 e928. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark T Fau - Johansen, T.; Johansen, T. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. 2006.
- Cabrera-Rodriguez, R.; Perez-Yanes, S.; Lorenzo-Sanchez, I.; Estevez-Herrera, J.; Garcia-Luis, J.; Trujillo-Gonzalez, R.; Valenzuela-Fernandez, A. TDP-43 Controls HIV-1 Viral Production and Virus Infectiveness. International journal of molecular sciences 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Rodriguez, R.; Perez-Yanes, S.; Montelongo, R.; Lorenzo-Salazar, J.M.; Estevez-Herrera, J.; Garcia-Luis, J.; Inigo-Campos, A.; Rubio-Rodriguez, L.A.; Munoz-Barrera, A.; Trujillo-Gonzalez, R.; et al. Transactive Response DNA-Binding Protein (TARDBP/TDP-43) Regulates Cell Permissivity to HIV-1 Infection by Acting on HDAC6. International journal of molecular sciences 2022, 23. [Google Scholar] [CrossRef]
- Marrero-Hernandez, S.; Marquez-Arce, D.; Cabrera-Rodriguez, R.; Estevez-Herrera, J.; Perez-Yanes, S.; Barroso-Gonzalez, J.; Madrid, R.; Machado, J.D.; Blanco, J.; Valenzuela-Fernandez, A. HIV-1 Nef Targets HDAC6 to Assure Viral Production and Virus Infection. Frontiers in microbiology 2019, 10, 2437. [Google Scholar] [CrossRef] [PubMed]
- Valera, M.S.; de Armas-Rillo, L.; Barroso-Gonzalez, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernandez, S.; Borel, S.; Garcia-Exposito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, H.; Wei, C.; Xiang, Y.; Liang, X.; Phang, C.W.; Jiao, R. HDAC6 regulates lipid droplet turnover in response to nutrient deprivation via p62-mediated selective autophagy. J Genet Genomics 2019, 46, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Rodriguez, R.; Hebmann, V.; Marfil, S.; Pernas, M.; Marrero-Hernandez, S.; Cabrera, C.; Urrea, V.; Casado, C.; Olivares, I.; Marquez-Arce, D.; et al. HIV-1 envelope glycoproteins isolated from Viremic Non-Progressor individuals are fully functional and cytopathic. Sci Rep 2019, 9, 5544. [Google Scholar] [CrossRef]
- Casado, C.; Marrero-Hernandez, S.; Marquez-Arce, D.; Pernas, M.; Marfil, S.; Borras-Granana, F.; Olivares, I.; Cabrera-Rodriguez, R.; Valera, M.S.; de Armas-Rillo, L.; et al. Viral Characteristics Associated with the Clinical Nonprogressor Phenotype Are Inherited by Viruses from a Cluster of HIV-1 Elite Controllers. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernandez, A.; Alvarez, S.; Gordon-Alonso, M.; Barrero, M.; Ursa, A.; Cabrero, J.R.; Fernandez, G.; Naranjo-Suarez, S.; Yanez-Mo, M.; Serrador, J.M.; et al. Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol Biol Cell 2005, 16, 5445–5454. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. The EMBO journal 2002, 21, 6820–6831. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Peng, D.; Cao, H.; Yang, X.; Li, S.; Qiu, H.J.; Li, L.F. The Host Cytoskeleton Functions as a Pleiotropic Scaffold: Orchestrating Regulation of the Viral Life Cycle and Mediating Host Antiviral Innate Immune Responses. Viruses 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.H.; Walsh, D. Microtubule Regulation and Function during Virus Infection. Journal of virology 2017, 91. [Google Scholar] [CrossRef]
- Seo, D.; Gammon, D.B. Manipulation of Host Microtubule Networks by Viral Microtubule-Associated Proteins. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Oliva, M.A.; Tosat-Bitrian, C.; Barrado-Gil, L.; Bonato, F.; Galindo, I.; Garaigorta, U.; Alvarez-Bernad, B.; Paris-Ogayar, R.; Lucena-Agell, D.; Gimenez-Abian, J.F.; et al. Effect of Clinically Used Microtubule Targeting Drugs on Viral Infection and Transport Function. International journal of molecular sciences 2022, 23. [Google Scholar] [CrossRef]
- Akhmanova, A.; Kapitein, L.C. Mechanisms of microtubule organization in differentiated animal cells. Nat Rev Mol Cell Biol 2022, 23, 541–558. [Google Scholar] [CrossRef]
- Kesari, A.S.; Heintz, V.J.; Poudyal, S.; Miller, A.S.; Kuhn, R.J.; LaCount, D.J. Zika virus NS5 localizes at centrosomes during cell division. Virology 2020, 541, 52–62. [Google Scholar] [CrossRef]
- Kodani, A.; Knopp, K.A.; Di Lullo, E.; Retallack, H.; Kriegstein, A.R.; DeRisi, J.L.; Reiter, J.F. Zika virus alters centrosome organization to suppress the innate immune response. EMBO reports 2022, 23, e52211. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell reports 2016, 16, 2576–2592. [Google Scholar] [CrossRef]
- Ji, W.; Luo, G. Zika virus NS5 nuclear accumulation is protective of protein degradation and is required for viral RNA replication. Virology 2020, 541, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Tao, M.; Han, W.; Fan, Z.; Imran, M.; Cao, S.; Ye, J. Nuclear localization of Zika virus NS5 contributes to suppression of type I interferon production and response. The Journal of general virology 2019. [CrossRef] [PubMed]
- Hook, S.S.; Orian, A.; Cowley, S.M.; Eisenman, R.N. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proceedings of the National Academy of Sciences of the United States of America 2002, 99, 13425–13430. [Google Scholar] [CrossRef]
- Pai, M.T.; Tzeng, S.R.; Kovacs, J.J.; Keaton, M.A.; Li, S.S.; Yao, T.P.; Zhou, P. Solution structure of the Ubp-M BUZ domain, a highly specific protein module that recognizes the C-terminal tail of free ubiquitin. J Mol Biol 2007, 370, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Bertos, N.R.; Gilquin, B.; Chan, G.K.; Yen, T.J.; Khochbin, S.; Yang, X.J. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J Biol Chem 2004, 279, 48246–48254. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Exposito, L.; Barroso-Gonzalez, J.; Puigdomenech, I.; Machado, J.D.; Blanco, J.; Valenzuela-Fernandez, A. HIV-1 requires Arf6-mediated membrane dynamics to efficiently enter and infect T lymphocytes. Mol Biol Cell 2011, 22, 1148–1166. [Google Scholar] [CrossRef]
- Barroso-Gonzalez, J.; Machado, J.D.; Garcia-Exposito, L.; Valenzuela-Fernandez, A. Moesin regulates the trafficking of nascent clathrin-coated vesicles. J Biol Chem 2009, 284, 2419–2434. [Google Scholar] [CrossRef]
- Kim, Y.C.; Dumoux, M.; Owens, R.J.; Reyes-Sandoval, A. Optimization of Small-Scale Production of Zika Virus Envelope Glycoprotein by Transient Expression in HEK293 Cells for ELISA. Methods Mol Biol 2020, 2142, 103–112. [Google Scholar] [CrossRef]
- Horibata, S.; Teramoto, T.; Vijayarangan, N.; Kuhn, S.; Padmanabhan, R.; Vasudevan, S.; Gottesman, M.; Padmanabhan, R. Host gene expression modulated by Zika virus infection of human-293 cells. Virology 2021, 552, 32–42. [Google Scholar] [CrossRef]
- Kim, Y.C.; Garcia-Larragoiti, N.; Lopez-Camacho, C.; Viveros-Sandoval, M.E.; Reyes-Sandoval, A. Production and Purification of Zika Virus NS1 Glycoprotein in HEK293 Cells. Methods Mol Biol 2020, 2142, 93–102. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, W.; Li, J.; Wu, W.; Jiu, Y. The Role of Host Cytoskeleton in Flavivirus Infection. Virol Sin 2019, 34, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Howes, S.C.; Alushin Gm Fau - Shida, T.; Shida T Fau - Nachury, M.V.; Nachury Mv Fau - Nogales, E.; Nogales, E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure.
- Bulinski, J.C.; Richards, J.E.; Piperno, G. Posttranslational modifications of alpha tubulin: detyrosination and acetylation differentiate populations of interphase microtubules in cultured cells. J Cell Biol 1988, 106, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Piperno, G.; LeDizet, M.; Chang, X.J. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol 1987, 104, 289–302. [Google Scholar] [CrossRef]
- LeDizet, M.; Piperno, G. Cytoplasmic microtubules containing acetylated alpha-tubulin in Chlamydomonas reinhardtii: spatial arrangement and properties. J Cell Biol 1986, 103, 13–22. [Google Scholar] [CrossRef] [PubMed]
- De Brabander Mj Fau - Van de Veire, R.M.; Van de Veire Rm Fau - Aerts, F.E.; Aerts Fe Fau - Borgers, M.; Borgers M Fau - Janssen, P.A.; Janssen, P.A. The effects of methyl (5-(2-thienylcarbonyl)-1H-benzimidazol-2-yl) carbamate, (R 17934; NSC 238159), a new synthetic antitumoral drug interfering with microtubules, on mammalian cells cultured in vitro.
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. The EMBO journal 2003, 22, 1168–1179. [Google Scholar] [CrossRef]
- Cabrero, J.R.; Serrador, J.M.; Barreiro, O.; Mittelbrunn, M.; Naranjo-Suarez, S.; Martin-Cofreces, N.; Vicente-Manzanares, M.; Mazitschek, R.; Bradner, J.E.; Avila, J.; et al. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Mol Biol Cell 2006, 17, 3435–3445. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proceedings of the National Academy of Sciences of the United States of America 2003, 100, 4389–4394. [Google Scholar] [CrossRef]
- Seigneurin-Berny, D.; Verdel, A.; Curtet, S.; Lemercier, C.; Garin, J.; Rousseaux, S.; Khochbin, S. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signalling pathways. Mol Cell Biol 2001, 21, 8035–8044. [Google Scholar] [CrossRef]
- Verdel, A.; Curtet, S.; Brocard, M.P.; Rousseaux, S.; Lemercier, C.; Yoshida, M.; Khochbin, S. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr Biol 2000, 10, 747–749. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proceedings of the National Academy of Sciences of the United States of America 1999, 96, 4868–4873. [Google Scholar] [CrossRef]
- Jose, B.; Okamura, S.; Kato, T.; Nishino, N.; Sumida, Y.; Yoshida, M. Toward an HDAC6 inhibitor: synthesis and conformational analysis of cyclic hexapeptide hydroxamic acid designed from alpha-tubulin sequence. Bioorg Med Chem 2004, 12, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gilquin, B.; Khochbin, S.; Matthias, P. Two catalytic domains are required for protein deacetylation. J Biol Chem 2006, 281, 2401–2404. [Google Scholar] [CrossRef] [PubMed]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus research 2018, 254, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Gratton, R.; Agrelli, A.; Tricarico, P.M.; Brandao, L.; Crovella, S. Autophagy in Zika Virus Infection: A Possible Therapeutic Target to Counteract Viral Replication. International journal of molecular sciences 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proceedings of the National Academy of Sciences of the United States of America 1982, 79, 1889–1892. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chan, J.F.; Tee, K.M.; Choi, G.K.; Lau, S.K.; Woo, P.C.; Tse, H.; Yuen, K.Y. Comparative genomic analysis of preepidemic and epidemic Zika virus strains for virological factors potentially associated with the rapidly expanding epidemic. Emerging microbes & infections 2016, 5, e22. [Google Scholar] [CrossRef]
- Wang, B.; Thurmond, S.; Hai, R.; Song, J. Structure and function of Zika virus NS5 protein: perspectives for drug design. Cellular and molecular life sciences: CMLS 2018, 75, 1723–1736. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signalling. Cell host & microbe 2016, 19, 882–890. [Google Scholar] [CrossRef]
- Dar, H.A.; Zaheer, T.; Paracha, R.Z.; Ali, A. Structural analysis and insight into Zika virus NS5 mediated interferon inhibition. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases 2017, 51, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, R.; Melén, K.; Westenius, V.; Jiang, M.; Österlund, P.; Khan, H.; Vapalahti, O.; Julkunen, I.; Kakkola, L. Zika Virus Non-Structural Protein NS5 Inhibits the RIG-I Pathway and Interferon Lambda 1 Promoter Activation by Targeting IKK Epsilon. Viruses 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Tschopp, J.; Karin, M. Intracellular pattern recognition receptors in the host response. Nature 2006, 442, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. Journal of virology 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Lee, H.C.; Kim, J.H.; Park, S.Y.; Kim, T.H.; Lee, W.K.; Jang, D.J.; Yoon, J.E.; Choi, Y.I.; Kim, S.; et al. HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. The EMBO journal 2016, 35, 429–442. [Google Scholar] [CrossRef]
- Liu, H.M.; Jiang, F.; Loo, Y.M.; Hsu, S.; Hsiang, T.Y.; Marcotrigiano, J.; Gale, M., Jr. Regulation of Retinoic Acid Inducible Gene-I (RIG-I) Activation by the Histone Deacetylase 6. EBioMedicine 2016, 9, 195–206. [Google Scholar] [CrossRef]
- Li, Z.; Xiao, W.; Yang, Z.; Guo, J.; Zhou, J.; Xiao, S.; Fang, P.; Fang, L. Cleavage of HDAC6 to dampen its antiviral activity by nsp5 is a common strategy of swine enteric coronaviruses. Journal of virology 2024, 98, e0181423. [Google Scholar] [CrossRef] [PubMed]
- Visan, I. Deacetylation for viral sensing. Nature immunology 2016, 17, 229–229. [Google Scholar] [CrossRef]
- Chang, A.Y.; Zhou, Y.J.; Iyengar, S.; Pobiarzyn, P.W.; Tishchenko, P.; Shah, K.M.; Wheeler, H.; Wang, Y.M.; Loria, P.M.; Loganzo, F.; et al. Modulation of SF3B1 in the pre-mRNA spliceosome induces a RIG-I-dependent type I IFN response. J Biol Chem 2021, 297, 101277. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Tan, C.; Yang, X.; Wang, J.; Li, Q.; Xu, L.; Wang, Z.; Mao, J.; Wang, J.; Cheng, K.; et al. Transcriptome Analysis of Retinoic Acid-Inducible Gene I Overexpression Reveals the Potential Genes for Autophagy-Related Negative Regulation. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Gao, M.; Gao, X.; Zhu, B.; Huang, J.; Tu, X.; Kim, W.; Zhao, F.; Zhou, Q.; Zhu, S.; et al. Reciprocal regulation of RIG-I and XRCC4 connects DNA repair with RIG-I immune signalling. Nat Commun 2021, 12, 2187. [Google Scholar] [CrossRef]
- Magdalena, W.; Jacek, S.; Ivan, T.; Zara, N.; Agnieszka, B.; Nila Roy, C.; Tola, T.; Ceren, K.; Elżbieta, N.; Christos, S.; et al. RNA 5ʹ terminal nucleotide determines the strength of the RIG-I/IFN signalling pathway. bioRxiv 2023, 2023.2012.2022.573000. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, M.L.; Zhao, J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. LID - 10.3390/v11020132 [doi] LID - 132. 2019.
- Fontana, J.; Lopez-Montero, N.; Elliott, R.M.; Fernandez, J.J.; Risco, C. The unique architecture of Bunyamwera virus factories around the Golgi complex. Cellular microbiology 2008, 10, 2012–2028. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M. Emerging viruses: the Bunyaviridae. Mol Med 1997, 3, 572–577. [Google Scholar] [CrossRef]
- Dutuze, M.F.; Nzayirambaho, M.; Mores, C.N.; Christofferson, R.C. A Review of Bunyamwera, Batai, and Ngari Viruses: Understudied Orthobunyaviruses With Potential One Health Implications. Front Vet Sci 2018, 5, 69. [Google Scholar] [CrossRef]
- Noronha, L.E.; Wilson, W.C. Comparison of two zoonotic viruses from the order Bunyavirales. Curr Opin Virol 2017, 27, 36–41. [Google Scholar] [CrossRef]
- Bridgen, A.; Weber, F.; Fazakerley, J.K.; Elliott, R.M. Bunyamwera bunyavirus nonstructural protein NSs is a nonessential gene product that contributes to viral pathogenesis. Proceedings of the National Academy of Sciences of the United States of America 2001, 98, 664–669. [Google Scholar] [CrossRef]
- Weber, F.; Bridgen, A.; Fazakerley, J.K.; Streitenfeld, H.; Kessler, N.; Randall, R.E.; Elliott, R.M. Bunyamwera bunyavirus nonstructural protein NSs counteracts the induction of alpha/beta interferon. Journal of virology 2002, 76, 7949–7955. [Google Scholar] [CrossRef]
- Shokrollahi, M.; Mekhail, K. Interphase microtubules in nuclear organization and genome maintenance. Trends Cell Biol 2021, 31, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Mejat, A.; Misteli, T. LINC complexes in health and disease. Nucleus 2010, 1, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.F. LINC complex proteins in development and disease. Curr Top Dev Biol 2014, 109, 287–321. [Google Scholar] [CrossRef] [PubMed]
- Stroud, M.J.; Banerjee, I.; Veevers, J.; Chen, J. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ Res 2014, 114, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.L.; Burke, B. LINC complexes and nuclear positioning. Seminars in cell & developmental biology 2018, 82, 67–76. [Google Scholar] [CrossRef]
- Janin, A.; Bauer, D.; Ratti, F.; Millat, G.; Mejat, A. Nuclear envelopathies: a complex LINC between nuclear envelope and pathology. Orphanet J Rare Dis 2017, 12, 147. [Google Scholar] [CrossRef]
- Cyske, Z.; Gaffke, L.; Pierzynowska, K.; Wegrzyn, G. Tubulin Cytoskeleton in Neurodegenerative Diseases-not Only Primary Tubulinopathies. Cell Mol Neurobiol 2023, 43, 1867–1884. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, G.; Jabeen, S.; Landazuri Vinueza, J.; Ghosh Roy, S.; Lockshin, R.A.; Zakeri, Z. Zika virus triggers autophagy to exploit host lipid metabolism and drive viral replication. Cell Commun Signal 2023, 21, 114. [Google Scholar] [CrossRef]
- Bindu; Pandey, H.S.; Seth, P. Interplay Between Zika Virus-Induced Autophagy and Neural Stem Cell Fate Determination. Mol Neurobiol 2023. [CrossRef]
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PloS one 2013, 8, e76016. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. The EMBO journal 2010, 29, 969–980. [Google Scholar] [CrossRef]
- Wang, L.; Moreira, E.A.; Kempf, G.; Miyake, Y.; Oliveira Esteves, B.I.; Fahmi, A.; Schaefer, J.V.; Dreier, B.; Yamauchi, Y.; Alves, M.P.; et al. Disrupting the HDAC6-ubiquitin interaction impairs infection by influenza and Zika virus and cellular stress pathways.
- Qu, M.; Zhang, H.; Cheng, P.; Wubshet, A.K.; Yin, X.; Wang, X.; Sun, Y. Histone deacetylase 6’s function in viral infection, innate immunity, and disease: latest advances.
- Husain, M.; Cheung, C.Y. Histone deacetylase 6 inhibits influenza A virus release by downregulating the trafficking of viral components to the plasma membrane via its substrate, acetylated microtubules. Journal of virology 2014, 88, 11229–11239. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat Commun 2018, 9, 414. [Google Scholar] [CrossRef]
- Conde, J.N.; Schutt, W.R.; Mladinich, M.; Sohn, S.Y.; Hearing, P.; Mackow, E.R. NS5 Sumoylation Directs Nuclear Responses That Permit Zika Virus To Persistently Infect Human Brain Microvascular Endothelial Cells. Journal of virology 2020, 94. [Google Scholar] [CrossRef]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T., Jr.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell host & microbe 2016, 19, 705–712. [Google Scholar] [CrossRef]
Figure 1.
Characterization of the expression pattern of the ZIKV NS5 protein. (a) Quantitative Western blot analysis of the C-terminal myc-tagged NS5 (NS5-myc) viral protein construct (1.5 µg cDNA) and total α-tubulin protein in a viral packaging HEK-293T cell model compared to those in control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA). α-Tubulin was used as the control for total protein loading. A representative of two experiments is shown (see associated data in Figure S1A). (b) Left, Quantitative Western blot analysis of the cell fraction of HEK-293T cells overexpressing NS5-myc (1.5 µg cDNA) and its distribution. Histone 3 was used as the protein control for the nuclear fraction, whereas α-tubulin was used for the cytoplasmic fraction and total protein load. A representative of two experiments is shown (see associated data in Figure S1B). Right, histograms quantifying the distribution of nuclear and cytoplasmic NS5-myc in lysates from (b) experiments, normalized to total α-tubulin. NS5 is mostly found in the nucleus. The data are presented as the means ± S.E.M.s of two independent experiments. (c) Immunofluorescence microscopy images of HEK-293T cells overexpressing NS5-myc (1.5 µg cDNA) and fixed 48 h posttransfection. The subcellular localization of NS5 was determined using a specific α-myc mAb (green). DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 100x objective and analysed with the MetaMorph program. NS5 is found in the nucleus and forms 3 types of structures: uniform punctate shapes ranging from one (type i) to several (type ii) micrometers in diameter and spheroidal and ring-shaped shapes with longer sizes (type iii). Arrowheads denote the lack of DNA and NS5 colocalization. (d) Line scanning of the different structures containing the NS5 viral protein (α-myc mAb (green)) (type i-black, type ii-green, and type iii-pink lines). (e) Quantification of the frequency of the presence of different NS5 structures (type i-black, type ii-green, and type iii-pink dots) and statistical analysis. (f) Average fluorescence intensity plot of NS5 spots in different structures (type i, black bar; type ii, green bar; and type iii, pink bar). In this figure, when indicated, the p values are ***p ≤ 0.001, **p ≤ 0.01 and *p ≤ 0.05; ns indicates not statistically significant (p > 0.05). The p value is the comparison of the means between the two groups using the parametric Student’s t test.
Figure 1.
Characterization of the expression pattern of the ZIKV NS5 protein. (a) Quantitative Western blot analysis of the C-terminal myc-tagged NS5 (NS5-myc) viral protein construct (1.5 µg cDNA) and total α-tubulin protein in a viral packaging HEK-293T cell model compared to those in control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA). α-Tubulin was used as the control for total protein loading. A representative of two experiments is shown (see associated data in Figure S1A). (b) Left, Quantitative Western blot analysis of the cell fraction of HEK-293T cells overexpressing NS5-myc (1.5 µg cDNA) and its distribution. Histone 3 was used as the protein control for the nuclear fraction, whereas α-tubulin was used for the cytoplasmic fraction and total protein load. A representative of two experiments is shown (see associated data in Figure S1B). Right, histograms quantifying the distribution of nuclear and cytoplasmic NS5-myc in lysates from (b) experiments, normalized to total α-tubulin. NS5 is mostly found in the nucleus. The data are presented as the means ± S.E.M.s of two independent experiments. (c) Immunofluorescence microscopy images of HEK-293T cells overexpressing NS5-myc (1.5 µg cDNA) and fixed 48 h posttransfection. The subcellular localization of NS5 was determined using a specific α-myc mAb (green). DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 100x objective and analysed with the MetaMorph program. NS5 is found in the nucleus and forms 3 types of structures: uniform punctate shapes ranging from one (type i) to several (type ii) micrometers in diameter and spheroidal and ring-shaped shapes with longer sizes (type iii). Arrowheads denote the lack of DNA and NS5 colocalization. (d) Line scanning of the different structures containing the NS5 viral protein (α-myc mAb (green)) (type i-black, type ii-green, and type iii-pink lines). (e) Quantification of the frequency of the presence of different NS5 structures (type i-black, type ii-green, and type iii-pink dots) and statistical analysis. (f) Average fluorescence intensity plot of NS5 spots in different structures (type i, black bar; type ii, green bar; and type iii, pink bar). In this figure, when indicated, the p values are ***p ≤ 0.001, **p ≤ 0.01 and *p ≤ 0.05; ns indicates not statistically significant (p > 0.05). The p value is the comparison of the means between the two groups using the parametric Student’s t test.
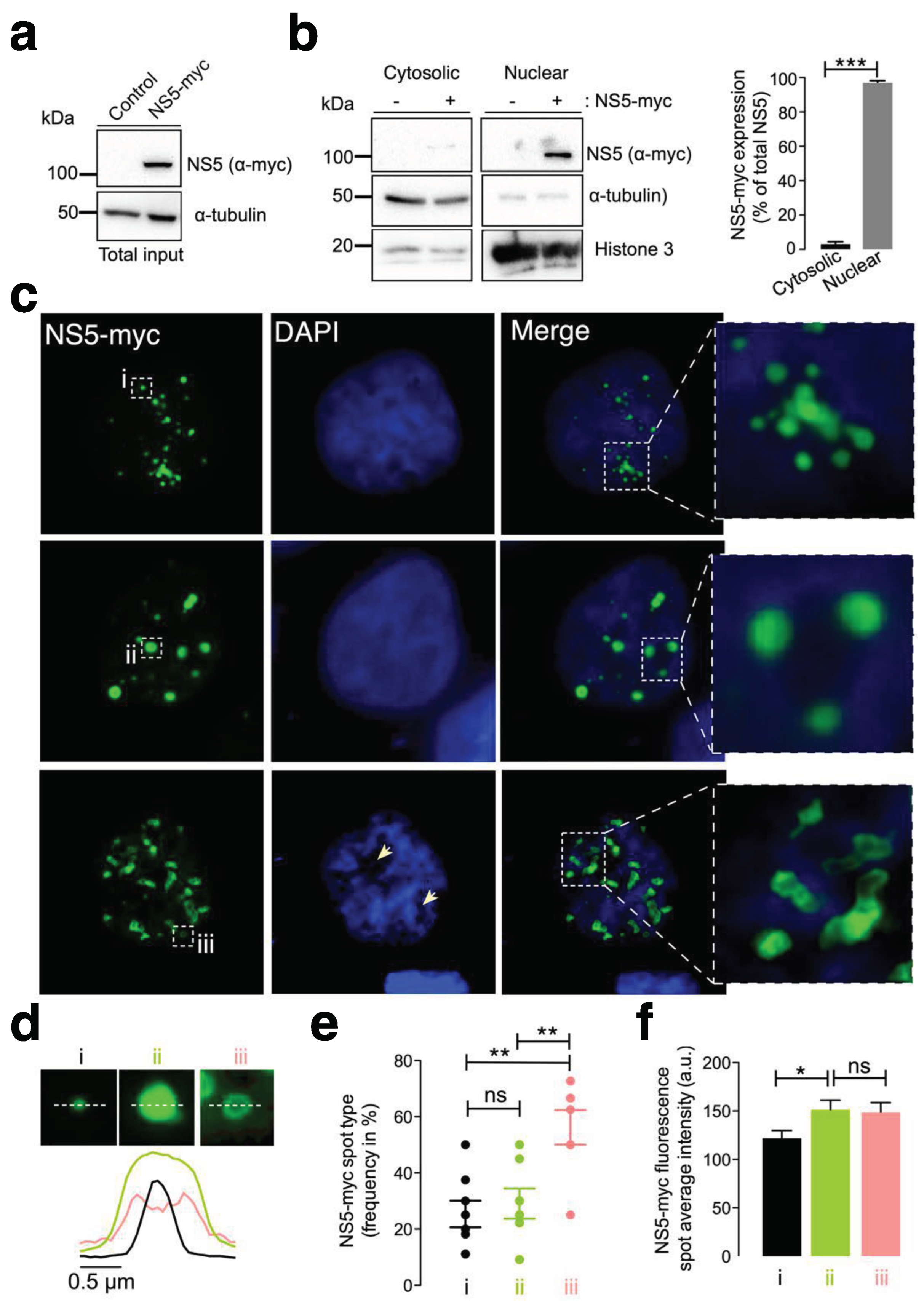
Figure 2.
Characterization of the effect of the ZIKV NS5 protein on microtubule stabilization and reorganization and on the accumulation of the autophagy marker p62. (a) Immunofluorescence microscopy analysis of the effect of NS5 on microtubules (MTs) in HEK-293T cells overexpressing NS5-myc (1.5 µg cDNA) compared to control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA). In this representative experiment, cells were processed for immunostaining with antibodies against the acetylated Lys40 residue in a-tubulin (green) and/or NS5 (using an anti-myc mAb) (red). DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 40x objective and analysed with the MetaMorph program. (b) Immunofluorescence microscopy images showing the exacerbated nested-like structure of acetylated MTs promoted in cells overexpressing NS5 (1.5 µg cDNA). As a control for the acetylation of MTs, cells were treated with tubacin (1 mM), a specific inhibitor of the deacetylase activity of HDAC6. This image also serves as a control for the NS5-mediated acetylation of MTs images presented in panel (a). In these representative experiments, cells were processed for immunostaining with antibodies against the acetylated Lys40 residue in a-tubulin (green) and/or NS5 (using an anti-myc mAb) (red). DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 100x objective and analysed with the MetaMorph program. (c) Quantitative Western blot analysis of the dose-response effect of NS5 overexpression on MT stabilization, as monitored by the acetylation of MTs at Lys40 α-tubulin residues. a-Tubulin was used as the loading control protein. A representative experiment of 6 independent experiments is shown (see associated data in Figure S2C). Histograms quantifying the amount of acetylated α-tubulin induced by NS5 in stabilized MTs from six replicates of the experiment shown in panel (c). The data were normalized to the total α-tubulin protein. (d) Western blot analysis of the effect of the NS5 viral protein (1.5 µg cDNA) on the accumulation of the autophagic protein p62 in HEK-293T cells. NS5, p62 and total a-tubulin are shown. Histograms quantifying the amount of p62 stabilized by NS5 from the experiments shown in panel (d). The data were normalized to the total α-tubulin protein. The data are presented as the means ±S.E. of 3 independent experiments (see associated data in Figure S2D). (e) Quantitative Western blot analysis of the dose‒response effect of NS5 overexpression on the accumulation of the autophagic protein p62 in HEK-293T cells overexpressing NS5. NS5, p62 and total a-tubulin are shown. The data are presented as the means ±S.E. of 4 independent experiments (see associated data in Figure S2E). In panels (c), (d) and (e), when indicated, the p values are **p ≤ 0.01 and *p ≤ 0.1; ns indicates not statistically significant (p > 0.05). The p value is the comparison of the means between the two groups using the parametric Student’s t test.
Figure 2.
Characterization of the effect of the ZIKV NS5 protein on microtubule stabilization and reorganization and on the accumulation of the autophagy marker p62. (a) Immunofluorescence microscopy analysis of the effect of NS5 on microtubules (MTs) in HEK-293T cells overexpressing NS5-myc (1.5 µg cDNA) compared to control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA). In this representative experiment, cells were processed for immunostaining with antibodies against the acetylated Lys40 residue in a-tubulin (green) and/or NS5 (using an anti-myc mAb) (red). DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 40x objective and analysed with the MetaMorph program. (b) Immunofluorescence microscopy images showing the exacerbated nested-like structure of acetylated MTs promoted in cells overexpressing NS5 (1.5 µg cDNA). As a control for the acetylation of MTs, cells were treated with tubacin (1 mM), a specific inhibitor of the deacetylase activity of HDAC6. This image also serves as a control for the NS5-mediated acetylation of MTs images presented in panel (a). In these representative experiments, cells were processed for immunostaining with antibodies against the acetylated Lys40 residue in a-tubulin (green) and/or NS5 (using an anti-myc mAb) (red). DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 100x objective and analysed with the MetaMorph program. (c) Quantitative Western blot analysis of the dose-response effect of NS5 overexpression on MT stabilization, as monitored by the acetylation of MTs at Lys40 α-tubulin residues. a-Tubulin was used as the loading control protein. A representative experiment of 6 independent experiments is shown (see associated data in Figure S2C). Histograms quantifying the amount of acetylated α-tubulin induced by NS5 in stabilized MTs from six replicates of the experiment shown in panel (c). The data were normalized to the total α-tubulin protein. (d) Western blot analysis of the effect of the NS5 viral protein (1.5 µg cDNA) on the accumulation of the autophagic protein p62 in HEK-293T cells. NS5, p62 and total a-tubulin are shown. Histograms quantifying the amount of p62 stabilized by NS5 from the experiments shown in panel (d). The data were normalized to the total α-tubulin protein. The data are presented as the means ±S.E. of 3 independent experiments (see associated data in Figure S2D). (e) Quantitative Western blot analysis of the dose‒response effect of NS5 overexpression on the accumulation of the autophagic protein p62 in HEK-293T cells overexpressing NS5. NS5, p62 and total a-tubulin are shown. The data are presented as the means ±S.E. of 4 independent experiments (see associated data in Figure S2E). In panels (c), (d) and (e), when indicated, the p values are **p ≤ 0.01 and *p ≤ 0.1; ns indicates not statistically significant (p > 0.05). The p value is the comparison of the means between the two groups using the parametric Student’s t test.
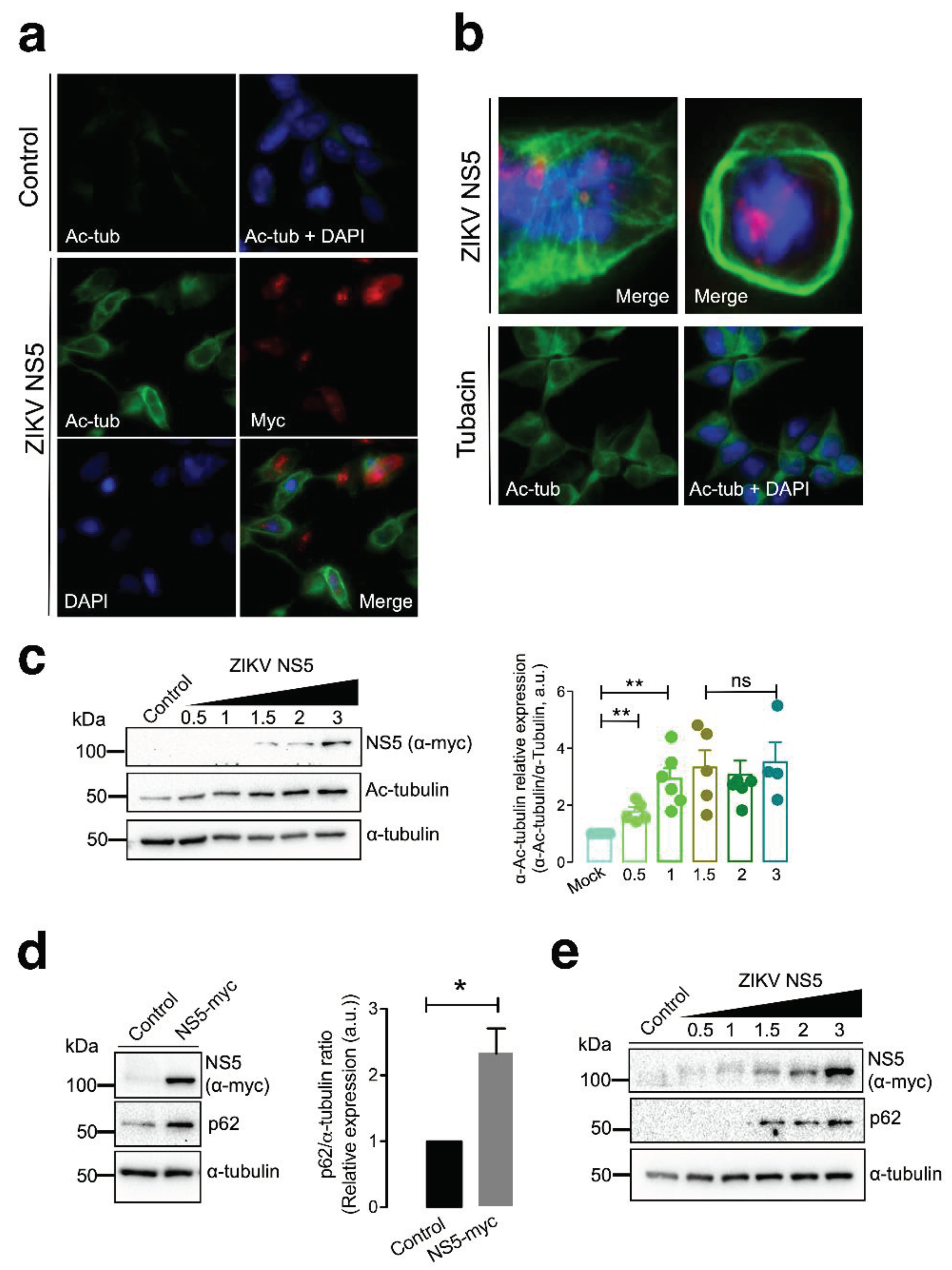
Figure 3.
HDAC6 targets the ZIKV NS5 protein in a deacetylase-dependent manner, regulating the level of expression of NS5 in the cytoplasm. (a) Quantitative Western blot analysis of HEK-293T cells overexpressing NS5-myc (lane 2), N-terminal HA-tagged wt-HDAC6 (HA-wt-HDAC6) (lane 3) or both constructs (lane 4) (1.5 µg cDNA) and comparison to control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA) (lane 1). The negative effects of HA-wt-HDAC6 on NS5-myc expression (lane 4), the stabilization of acetylated MTs by NS5 (lane 2) and the deacetylase activity of HDAC6 on acetylated MTs (lanes 3 and 4) are shown. The α-tubulin protein was used as the loading control protein. A representative experiment of 4 is shown (see associated data in Figure S3A). Histograms quantifying the amount of NS5-myc expressed in the absence (corresponding to lane 2 in the left panel) or presence of overexpressed HDAC6 (corresponding to lane 4 in the left panel) normalized to total α-tubulin. The data are presented as the means ±S.E.M. of 4 independent experiments. (b) Immunofluorescence images showing NS5 labelling 48 h posttransfection in cells cotransfected with HDAC6-DsRed (red), fixed and labelled with an α-myc antibody for NS5 detection (green). NS5 was detected only in cells not overexpressing wt-HDAC6-DsRed (white asterisks). The white dotted lanes indicate cell perimeters of three cells (1-3) that express wt-HDAC6-DsRed in the field. DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 100x objective and analysed with the MetaMorph program. (c) Immunofluorescence microscopy analysis of NS5 cell distribution in cells treated or not treated (control) with tubacin (1 μM), a deacetylase inhibitor of HDAC6, in cells overexpressing NS5-myc (1.5 µg cDNA). The cells were processed for immunostaining with an anti-myc mAb (green). DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 100x objective and analysed with the MetaMorph program. (d) Quantitative Western blot analysis of the effect of functional (HA-wt-HDAC6) or deacetylase inactive (HA-dm-HDAC6) HDAC6 on the stability of the ZIKV NS5 protein (NS5-myc) in HEK-293T cells overexpressing the different constructs (1.5 µg cDNA per construct) and comparison with cells overexpressing either only NS5 or only HA-wt-HDAC6 or HA-dm-HDAC6. The levels of acetylated a-tubulin and total a-tubulin under the different experimental conditions are shown. A representative experiment of 2 is shown (see associated data in Figure S3D). Histograms quantifying the amount of NS5 detected in cells from the experiment (d). The data are presented as the means ±S.E. of 2 independent experiments. For the quantitative analysis of the Western blots, when indicated, the p values are ***p ≤ 0.001 and **p ≤ 0.01. The p value is the comparison of the means between the two groups using the parametric Student’s t test.
Figure 3.
HDAC6 targets the ZIKV NS5 protein in a deacetylase-dependent manner, regulating the level of expression of NS5 in the cytoplasm. (a) Quantitative Western blot analysis of HEK-293T cells overexpressing NS5-myc (lane 2), N-terminal HA-tagged wt-HDAC6 (HA-wt-HDAC6) (lane 3) or both constructs (lane 4) (1.5 µg cDNA) and comparison to control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA) (lane 1). The negative effects of HA-wt-HDAC6 on NS5-myc expression (lane 4), the stabilization of acetylated MTs by NS5 (lane 2) and the deacetylase activity of HDAC6 on acetylated MTs (lanes 3 and 4) are shown. The α-tubulin protein was used as the loading control protein. A representative experiment of 4 is shown (see associated data in Figure S3A). Histograms quantifying the amount of NS5-myc expressed in the absence (corresponding to lane 2 in the left panel) or presence of overexpressed HDAC6 (corresponding to lane 4 in the left panel) normalized to total α-tubulin. The data are presented as the means ±S.E.M. of 4 independent experiments. (b) Immunofluorescence images showing NS5 labelling 48 h posttransfection in cells cotransfected with HDAC6-DsRed (red), fixed and labelled with an α-myc antibody for NS5 detection (green). NS5 was detected only in cells not overexpressing wt-HDAC6-DsRed (white asterisks). The white dotted lanes indicate cell perimeters of three cells (1-3) that express wt-HDAC6-DsRed in the field. DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 100x objective and analysed with the MetaMorph program. (c) Immunofluorescence microscopy analysis of NS5 cell distribution in cells treated or not treated (control) with tubacin (1 μM), a deacetylase inhibitor of HDAC6, in cells overexpressing NS5-myc (1.5 µg cDNA). The cells were processed for immunostaining with an anti-myc mAb (green). DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 100x objective and analysed with the MetaMorph program. (d) Quantitative Western blot analysis of the effect of functional (HA-wt-HDAC6) or deacetylase inactive (HA-dm-HDAC6) HDAC6 on the stability of the ZIKV NS5 protein (NS5-myc) in HEK-293T cells overexpressing the different constructs (1.5 µg cDNA per construct) and comparison with cells overexpressing either only NS5 or only HA-wt-HDAC6 or HA-dm-HDAC6. The levels of acetylated a-tubulin and total a-tubulin under the different experimental conditions are shown. A representative experiment of 2 is shown (see associated data in Figure S3D). Histograms quantifying the amount of NS5 detected in cells from the experiment (d). The data are presented as the means ±S.E. of 2 independent experiments. For the quantitative analysis of the Western blots, when indicated, the p values are ***p ≤ 0.001 and **p ≤ 0.01. The p value is the comparison of the means between the two groups using the parametric Student’s t test.
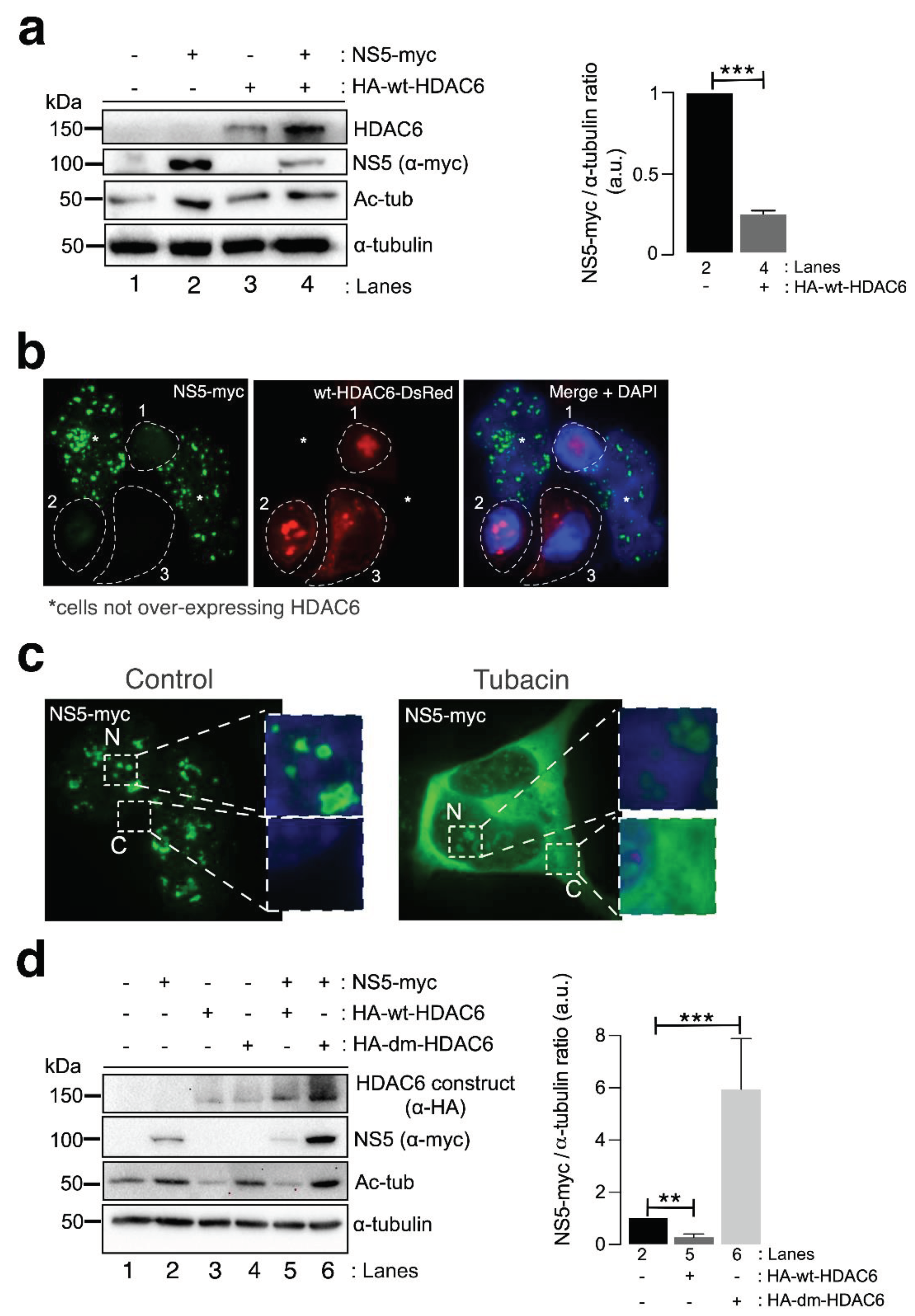
Figure 4.
HDAC6 promotes the autophagic clearance of the ZIKV NS5 protein. (a) Quantitative Western blot analysis of HEK-293T cells overexpressing NS5-myc (lane 2) or the N-terminal HA-tagged HDAC6 mutant lacking the C-terminal BUZ domain (HA-HDAC6-DBUZ) (lane 3) or both constructs (lane 4) (1.5 µg cDNA) and comparison to control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA) (lane 1). The absence of a degradative effect of HA-HDAC6-DBUZ on NS5-myc expression (lane 4) and the deacetylation of MTs by HA-HDAC6-DBUZ, deacetylase active (lanes 3 and 4) are shown. NS5 could not stabilize acetylated MTs under this experimental condition (lane 4). The α-tubulin protein was used as the loading control protein. A representative experiment of 3 is shown (see associated data in Figure S4A). Histograms quantifying the amount of NS5-myc expressed in the absence (corresponding to lane 2 in the left panel) or presence of overexpressed HA-HDAC6-DBUZ (corresponding to lane 4 in the left panel), normalized to total α-tubulin. The data are presented as the means ±S.E.M. of 3 independent experiments. (b) Immunofluorescence microscopy analysis of NS5 cell distribution in cells treated or not treated (control) with 3-MA (5 mM), an inhibitor of HDAC6-mediated aggresome formation and further autophagy clearance of targeted proteins, in cells overexpressing NS5-myc (1.5 µg cDNA). The cells were processed for immunostaining with an anti-myc mAb (green). DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 100x objective and analysed with the MetaMorph program. (c) Quantitative Western blot analysis of HEK-293T cells overexpressing NS5-myc (lanes 2), the functional HA-HDAC6 construct (lanes 3) or both constructs (lanes 4) (1.5 µg cDNA) and compared to control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA) (lanes 1) under control conditions (PBS-treated cells, vehicle for 3-MA) or cells treated with 3-MA (5 mM). The absence of a degradative effect exerted by HA-HDAC6 on NS5 in the presence of 3-MA (lane 4 in 3-MA-treated cells compared to lane 4 in PBS-treated cells) and the deacetylation of MTs by HA-HDAC6 are shown. The α-tubulin protein was used as the loading control protein. A representative experiment of 2 is shown (see associated data in Figure S4C). Histograms quantifying the amount of NS5-myc expressed in the absence (lanes 2) or presence of overexpressed HA-HDAC6 (lanes 4), normalized to total α-tubulin, in both PBS- and 3-MA-treated cells are shown. The data are presented as the means ±S.E. of 2 independent experiments. (d) Quantitative Western blot analysis of the effect of siRNA-mediated knockdown of endogenous HDAC6 (siRNA-HDAC6, 25 pmol/well) on the level of NS5 protein expression in HEK-293T cells overexpressing NS5-myc and compared to that in control and scrambled (1 µM)-treated cells. A representative experiment of 4 is shown (see associated data in Figure S4D). Histograms quantifying the amount of endogenous HDAC6 and NS5 detected in cells treated with siRNA-HDAC6 or scrambled oligos, normalized to total α-tubulin. The data are presented as the means ±S.E. of 4 independent experiments. For quantitative analysis of Western blots, when indicated, the p value is *p ≤ 0.1; ns indicates not statistically significant (p > 0.05). The p value is the comparison of the means between the two groups using the parametric Student’s t test.
Figure 4.
HDAC6 promotes the autophagic clearance of the ZIKV NS5 protein. (a) Quantitative Western blot analysis of HEK-293T cells overexpressing NS5-myc (lane 2) or the N-terminal HA-tagged HDAC6 mutant lacking the C-terminal BUZ domain (HA-HDAC6-DBUZ) (lane 3) or both constructs (lane 4) (1.5 µg cDNA) and comparison to control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA) (lane 1). The absence of a degradative effect of HA-HDAC6-DBUZ on NS5-myc expression (lane 4) and the deacetylation of MTs by HA-HDAC6-DBUZ, deacetylase active (lanes 3 and 4) are shown. NS5 could not stabilize acetylated MTs under this experimental condition (lane 4). The α-tubulin protein was used as the loading control protein. A representative experiment of 3 is shown (see associated data in Figure S4A). Histograms quantifying the amount of NS5-myc expressed in the absence (corresponding to lane 2 in the left panel) or presence of overexpressed HA-HDAC6-DBUZ (corresponding to lane 4 in the left panel), normalized to total α-tubulin. The data are presented as the means ±S.E.M. of 3 independent experiments. (b) Immunofluorescence microscopy analysis of NS5 cell distribution in cells treated or not treated (control) with 3-MA (5 mM), an inhibitor of HDAC6-mediated aggresome formation and further autophagy clearance of targeted proteins, in cells overexpressing NS5-myc (1.5 µg cDNA). The cells were processed for immunostaining with an anti-myc mAb (green). DAPI is a blue, fluorescent probe for DNA staining. Images were captured using a 100x objective and analysed with the MetaMorph program. (c) Quantitative Western blot analysis of HEK-293T cells overexpressing NS5-myc (lanes 2), the functional HA-HDAC6 construct (lanes 3) or both constructs (lanes 4) (1.5 µg cDNA) and compared to control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA) (lanes 1) under control conditions (PBS-treated cells, vehicle for 3-MA) or cells treated with 3-MA (5 mM). The absence of a degradative effect exerted by HA-HDAC6 on NS5 in the presence of 3-MA (lane 4 in 3-MA-treated cells compared to lane 4 in PBS-treated cells) and the deacetylation of MTs by HA-HDAC6 are shown. The α-tubulin protein was used as the loading control protein. A representative experiment of 2 is shown (see associated data in Figure S4C). Histograms quantifying the amount of NS5-myc expressed in the absence (lanes 2) or presence of overexpressed HA-HDAC6 (lanes 4), normalized to total α-tubulin, in both PBS- and 3-MA-treated cells are shown. The data are presented as the means ±S.E. of 2 independent experiments. (d) Quantitative Western blot analysis of the effect of siRNA-mediated knockdown of endogenous HDAC6 (siRNA-HDAC6, 25 pmol/well) on the level of NS5 protein expression in HEK-293T cells overexpressing NS5-myc and compared to that in control and scrambled (1 µM)-treated cells. A representative experiment of 4 is shown (see associated data in Figure S4D). Histograms quantifying the amount of endogenous HDAC6 and NS5 detected in cells treated with siRNA-HDAC6 or scrambled oligos, normalized to total α-tubulin. The data are presented as the means ±S.E. of 4 independent experiments. For quantitative analysis of Western blots, when indicated, the p value is *p ≤ 0.1; ns indicates not statistically significant (p > 0.05). The p value is the comparison of the means between the two groups using the parametric Student’s t test.
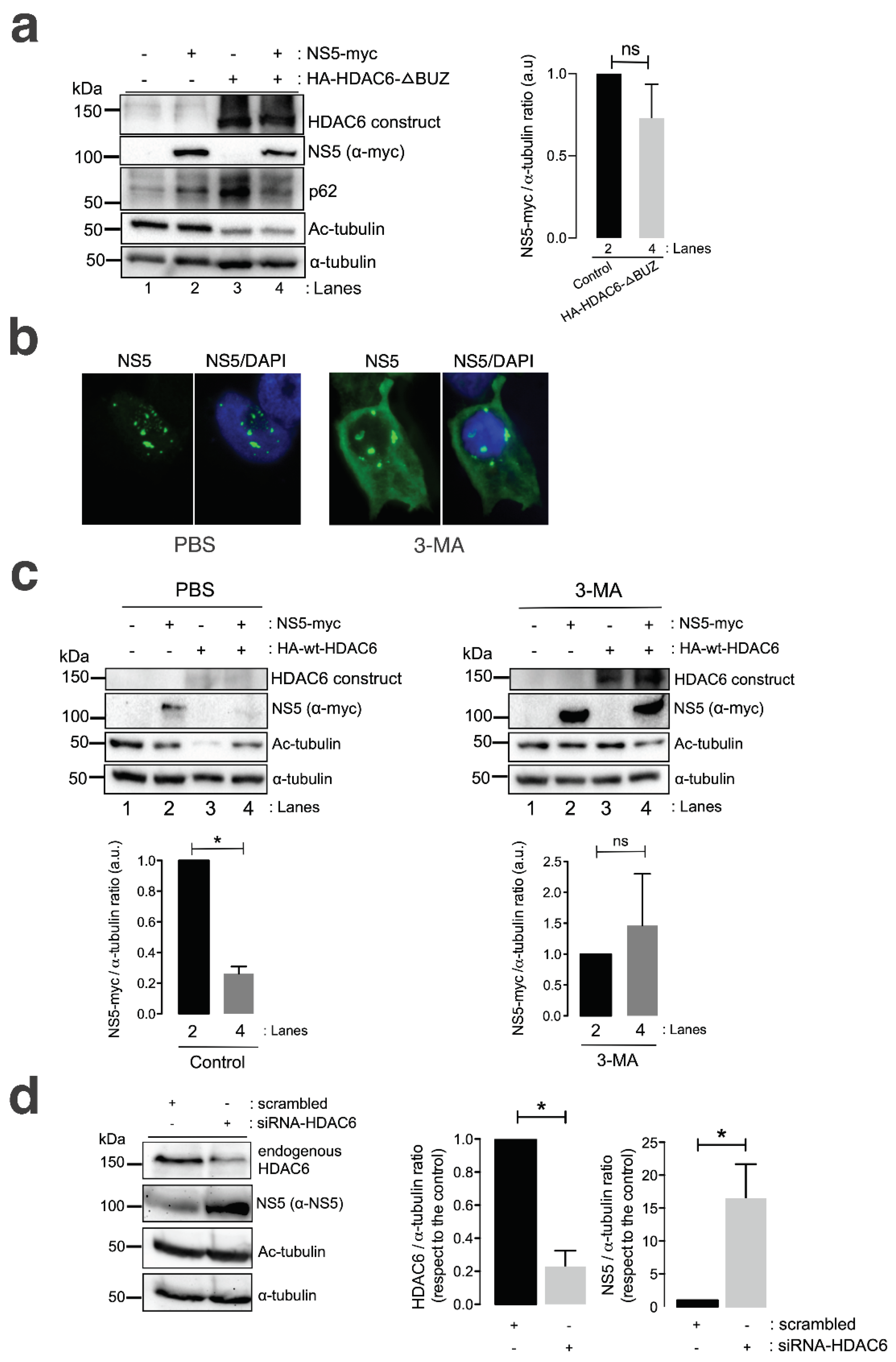
Figure 5.
The ZIKV NS5 protein inhibits RNA-triggered RIG-I-mediated IFN production, and this effect is enhanced by autophagy inhibition. (a) Quantitative assay of the inhibitory effect of NS5 on RNA-triggered RIG-I-mediated IFN production in HEK-LuciaTM RIG-I cells expressing NS5 (1.5 µg cDNA) and treated with 3p-hpRNA (100 nM), an RNA-specific RIG-I agonist, for 24 h before the IFN-I response was quantified by QUANTI-Luc. The data obtained from control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA) and treated with 3p-hpRNA (100 nM) are shown. The effect of 3-MA (5 mM) on the NS5-mediated inhibition of 3p-hpRNA-triggered RIG-I-mediated IFN production is shown. The data are presented as the means ±S.E.M. of 2 independent experiments (n=6). (b) Quantitative Western blot analysis of the effect of 3-MA treatment on HEK-Lucia cells overexpressing NS5-myc (1.5 µg cDNA). This biochemical analysis corresponds to the cells shown in panel (a) for RNA-mediated RIG-I IFN production. Lanes 1 and 2 represent control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA) and treated with PBS (vehicle for 3-MA) or 3-MA (5 mM), respectively. Lanes 3 and 4 represent cells overexpressing NS5 (NS5-HA plasmid, 1.5 µg cDNA) and treated with PBS or 3-MA (5 mM), respectively. The stabilization effect of 3-MA on NS5 (lane 4 in 3-MA-treated cells compared to lane 3 in PBS-treated cells) and the NS5-mediated acetylation of MTs are shown. The α-tubulin protein was used as the loading control protein. A representative experiment of 2 is shown (see associated data in Figure S5B). Histograms quantifying the amount of NS5-HA expressed, normalized to total α-tubulin, in both control (PBS)- and 3-MA-treated cells from the top Western blot experiments. The data are presented as the means ±S.E.M. of 2 independent experiments. In (a), the p values are ***p ≤ 0.001 and *p ≤ 0.1, and in (b), the p value is 0.15. The p value is the comparison of the means between the two groups using the parametric Student’s t test.
Figure 5.
The ZIKV NS5 protein inhibits RNA-triggered RIG-I-mediated IFN production, and this effect is enhanced by autophagy inhibition. (a) Quantitative assay of the inhibitory effect of NS5 on RNA-triggered RIG-I-mediated IFN production in HEK-LuciaTM RIG-I cells expressing NS5 (1.5 µg cDNA) and treated with 3p-hpRNA (100 nM), an RNA-specific RIG-I agonist, for 24 h before the IFN-I response was quantified by QUANTI-Luc. The data obtained from control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA) and treated with 3p-hpRNA (100 nM) are shown. The effect of 3-MA (5 mM) on the NS5-mediated inhibition of 3p-hpRNA-triggered RIG-I-mediated IFN production is shown. The data are presented as the means ±S.E.M. of 2 independent experiments (n=6). (b) Quantitative Western blot analysis of the effect of 3-MA treatment on HEK-Lucia cells overexpressing NS5-myc (1.5 µg cDNA). This biochemical analysis corresponds to the cells shown in panel (a) for RNA-mediated RIG-I IFN production. Lanes 1 and 2 represent control cells transfected with the pcDNATM 3.1(+) plasmid (1.5 µg cDNA) and treated with PBS (vehicle for 3-MA) or 3-MA (5 mM), respectively. Lanes 3 and 4 represent cells overexpressing NS5 (NS5-HA plasmid, 1.5 µg cDNA) and treated with PBS or 3-MA (5 mM), respectively. The stabilization effect of 3-MA on NS5 (lane 4 in 3-MA-treated cells compared to lane 3 in PBS-treated cells) and the NS5-mediated acetylation of MTs are shown. The α-tubulin protein was used as the loading control protein. A representative experiment of 2 is shown (see associated data in Figure S5B). Histograms quantifying the amount of NS5-HA expressed, normalized to total α-tubulin, in both control (PBS)- and 3-MA-treated cells from the top Western blot experiments. The data are presented as the means ±S.E.M. of 2 independent experiments. In (a), the p values are ***p ≤ 0.001 and *p ≤ 0.1, and in (b), the p value is 0.15. The p value is the comparison of the means between the two groups using the parametric Student’s t test.
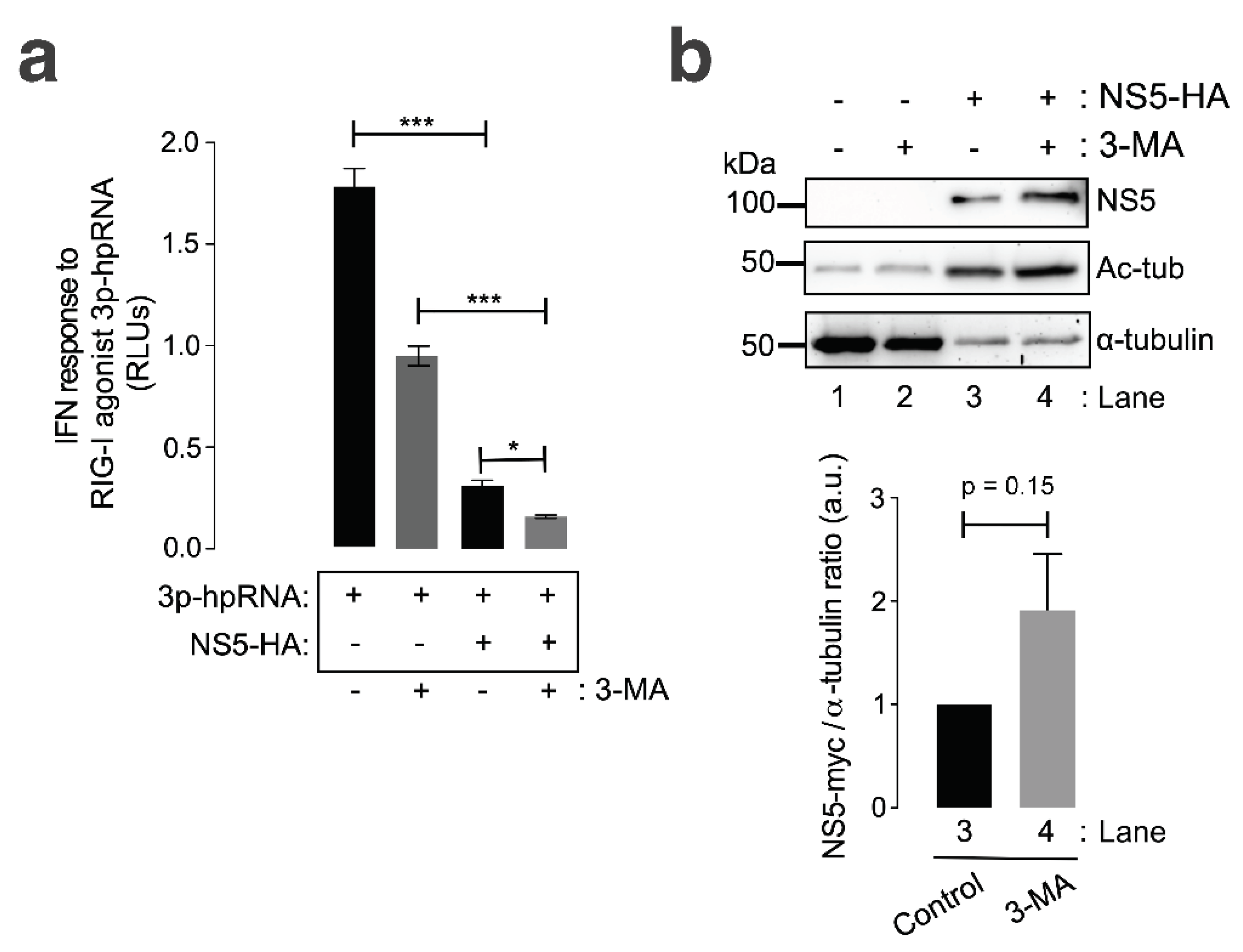
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



