Submitted:
26 February 2024
Posted:
27 February 2024
You are already at the latest version
Abstract
Background:
DICER1, cancer predisposition syndrome (CPS), seems to escape timely diagnosis in pediatric patients.
Case report 1:
A 16-year-old female patient was referred to the endocrinology ward due to a large goiter. Her medical history indicated normal sexual maturation, with menarche occurring at 13.5 years. Over the past 2.5 years, she developed pronounced androgenic symptoms, including a deepened male voice, facial, back and neckline acne, hirsutism, and menstrual irregularities leading to secondary amenorrhoea.
A thyroid ultrasound identified a multinodular goiter with cystic-solid lesions containing calcifi-cations. An abdominal ultrasound identified a 5.7x6.9 cm solid mass in the right adnexal region, displacing the uterus to the left. Histopathological examination confirmed a Sertoli-Leydig cell tumor. The patient was subjected to total thyroidectomy. Histopathology revealed benign follicular cell-derived neoplasms. Thyroid follicular nodular disease was diagnosed bilaterally.
DNA analysis using NGS, confirmed by the Sanger method, revealed a pathogenic heterozygotic variant c.2953>T [p.Gln985*] in exon 18 of DICER1 gene.
Case report 2:
A 12-year-old male patient was admitted to the pediatric surgery unit due to a 33 ml goiter. A month prior to admission, the patient discovered a palpable nodule in his neck, accompanied by hoarseness. An ultrasound revealed a multinodular goiter. Molecular analysis revealed a pathogenic heterozygotic variant c.2782C>T [p.Gln928Ter] in exon 17 of DICER1 gene. Subsequently, a total thyroidectomy was performed, and histopathological examination revealed thyroid follicular nodu-lar disease bilaterally.
Conclusion:
Multinodular goiter, although rare in pediatric population, when accompanied by characteristic ultrasound and histopathological features, and by additional features such as androgen excess,
warrants exclusion of DICER1 syndrome.
Keywords:
multinodular goiter
; secondary amenorrhoea
; Sertoli-Leydig cell tumor
; DICER1 syndrome
; euthyroid goiter
1. Introduction
DICER1 Syndrome
DICER1 syndrome is a cancer predisposition syndrome caused by heterozygous germline loss-of-function mutation in the DICER1 gene (localized on 14q32.13)[1]. The enzyme, Dicer, identified by Bernstein et al. in 2001, modifies protein-coding genes by influencing microRNA (guide RNA molecules of about 22 nucleotides in length) biogenesis [2]. DICER1 is essential for embryogenesis and development of many organs and has a distinct role in the oncogenesis of specific tumor types (both benign and malignant) [3]. Germline mutations of DICER1 have been well documented in a wide range of tumors in DICER1 syndrome [4,5,6,7,8,9,10]. The syndrome demonstrates an autosomal dominant inheritance pattern with reduced penetrance, which subsequently decreases the frequency of familial cases [9,10]. The incidence of DICER1 syndrome is unknown. It is believed that pathogenic DICER1 variants might be more prevalent than previously assumed [11]. Kim et al., estimated the prevalence of pathogenic DICER1 variants to be between 1:310 and 1:10,600 in the US population [11]. Stewart et al. reported that the risk of developing a neoplasm is 5.3% before the age of 10 years and of 31.5% before the age of 60 in carriers of pathogenic variant in DICER1 and this incidence is larger than in the general population [9,10].
DICER1 syndrome includes a variety of dysplastic and neoplastic lesions (including the tendency to develop sarcomas) [10]. It is classically associated with pleuropulmonary blastoma (PPB), a rare malignant tumor of the lungs seen primarily in children younger than 6 years of age, as well as multinodular goiter (MNG), cystic nephroma, and ovarian Sertoli-Leydig cell tumors [10].
However, the clinical spectrum is quite broad, age-dependent, and likely still evolving. It involves many sites in the body [1,2,3,4,5,6,7,8,9,10]. We presented age-dependent comorbidities in Figure 1 [1,2,3,4,5,6,7,8,9,10]. The primary central nervous system tumor manifestations of this syndrome include pineoblastoma, pituitary blastoma, embryonal tumor with multilayered rosettes, and DICER1-mutant primary intracranial sarcoma [12]. Head and neck manifestations of this syndrome include nasal chondromesenchymal hamartoma, ciliary body medulloepithelioma, thyroblastomas, multinodular goiter, and thyroid carcinomas [12]. Lesions in the chest include PPB, while in the abdomen they include cystic nephroma, Wilms tumor, and probably also neuroblastoma, as well as lesions in pelvis like ovarian sex cord-stromal tumors and embryonal rhabdomyosarcoma of the ovary, bladder, and cervix [8,13].
Despite a higher risk of malignancy, most patients with the pathogenic germline DICER1 variants live healthy lives, with tumors seen in <20% patients by the 50 years of age [10,14].
This study presents two case reports of adolescent patients from one center in South Poland, carrying pathogenic variants associated with the rare DICER1 syndrome. In this article, we present specific clinical findings, as well as unique ultrasound and histopathological features of the tumors. Our goal is to heighten awareness about this syndrome and underline the importance of point-of-care ultrasonography in the pediatric office, as a tool that can potentially shorten the diagnostic process, especially when a clinician is confronted with a patient presentation pointing clearly to DICER1 syndrome.
2. Methods
The analysis of medical records included the evaluation of thyroid and ovarian function, as well as ultrasound, cytological and histopathological variables in patients with DICER1 syndrome. All the assessments were performed at the Department of Biochemistry at the University Children’s Hospital in Krakow, Poland, and were determined in a single fasting blood sample. TSH, fT3, fT4, LH, FSH, estradiol, testosterone and 17-hydroxyprogesterone (17-OHP) levels were measured using the immunochemistry method with the ADVIA Centaur machine. Thyroperoxidase (TPOAb), thyroglobulin (TgAb), and TSH receptor (TRAb) antibody levels were assessed using the radioimmunoassay method with the Brams machine.
Ultrasonographies (US) of the thyroid gland and pelvis were performed at the University Children’s Hospital by a pediatric endocrinologist and surgeons with experience in pediatric US (DJ, ATN > 20 years, and AKW > 10 years). The thyroid US was performed using a high-resolution Philips iE22 (L11-3 linear transducer) and Samsung HS40 (LA3-16AD transducer). The analysis included ultrasound features of the thyroid gland according to EU-TIRADS PL 2022 classification (Polish update of EU-TIRADS 2017) [15,16].
Fine needle aspiration biopsy (FNAB) results were classified according to Bethesda criteria [17]. Total thyroidectomy with central lymph node histopathological verification and right ovariectomy were performed in the Departments of Pediatric Surgery and Pathology of the University Children’s Hospital in Krakow. Hematoxylin and eosin (HE) stained tissue slides (deparafinnated, cut to 3.5 µm thickness) were scanned with the NanoZoomer SQ Hamamatsu (x400 magnification). Pictures were taken from the scans, with the scale placed on the left bottom corner of the images.
Molecular analysis was ordered by the Department of Pediatric Genetics on the request of the Department of Pediatric and Adolescent Endocrinology and was performed in the commercial molecular laboratory in Poland (with new generation sequencing, NGS and Sanger methods).
This study was approved by the relevant institutional review board (The Bioethics Committee of the Jagiellonian University opinion number:1072.6120.288.2021). Written informed consent was obtained from all the participants and/or their parents. Written informed consent was obtained from the individuals, and the minors’ legal guardians, for the publication of any potentially identifiable images or data included in this article.
3. Case Report 1
A 16-year-old female patient was referred to the outpatient Pediatric and Adolescent Endocrinology Department for evaluation of her thyroid function due to a large goiter. The patient’s perinatal history was unremarkable (APGAR score of 10 points, birth weight of 3,400g), with normal developmental milestones. Her general health status was good, with no significant diseases reported to date. Lab results showed no abnormalities. However, during conversation with this patient her voice was noticeably deepened, resembling a male voice. Her medical history indicated normal sexual maturation, with menarche occurring at 13.5 years. Over the past 2.5 years, she exhibited pronounced androgenic symptoms of defeminization, including decreased volume of the breasts (evidenced by her need to buy a smaller bra), balding, an enlarged clitoris, progressive deepening of her voice, facial, back and neckline acne, hirsutism score at -18 points according to Ferriman-Gallwey scale (the Polish norm being < 8 points), and menstrual irregularities leading to secondary amenorrhea during the last 12 months. Additionally, she reported consistent genital spotting for the past two months but was not referred to a gynecologist. The patient consulted with an otorhinolaryngologist twice over the past 1.5 years regarding voice changes, and as examination did not reveal any cause, the doctor ordered a testosterone assessment. The hormonal analysis showed elevated testosterone levels on two occasions (6.1 ng/ml and 4.17 ng/ml, with the norm being less than 0.48 ng/ml). But as the latter measurement was lower than the former, the patient did not receive any further referrals. Finally, because of the gradual enlargement of the thyroid, the large goiter started to become visible, and her family doctor referred the patient to a pediatric endocrinologist. This patient did not receive a referral to a gynecologist despite the presence of secondary amenorrhea. The evaluation in the outpatient Pediatric and Adolescent Endocrinology Department revealed that the patient was euthyroid, as both TSH at 1.9 uIU/ml (N 0.3-4.0), and fT4-16.6 pmol/l (N 10-25) were within normal limits. TPOAb, TgAb and TRAbs tests were negative.
Further hormonal testing revealed testosterone at 3.52 ng/ml (N<1), estradiol at 71.3 pg/ml (normal for age), suppressed FSH at <0.3 mIU/ml, and LH at 3.94 mIU/ml (normal for age), 17OHP at 5.04 ng/ml (normal for age).
Point-of care ultrasonography was performed by the consulting endocrinologist. A thyroid ultrasound scan identified a multinodular 68 ml goiter with 15 isoechogenic cystic-solid lesions containing macrocalcifications. Vascularization was not increased in the solid parts of the nodules. An abdominal ultrasound identified a 5.7 x 6.9 cm solid mass in the right adnexal region, displacing the uterus to the left. Magnetic Resonance Imaging findings were consistent with either arrhenoblastoma or dysgerminoma. A right ovariectomy was performed, and histopathology confirmed a moderately differentiated Sertoli-Leydig cell tumor. Ultrasound-histopathological analysis is presented in Figure 2(A-A4).
Hormonal tests performed 11 days after the surgery revealed an increase of estradiol: 104.1 pg/ml, disinhibition of FSH at 4.1 mIU/ml and LH at 8.94 mIU/ml with normalisation of testosterone to 0.30 ng/ml. Menses appeared 34 days after the surgery, with menses being regular at present.
The patient was referred to a speech therapist, whose first attempts to improve the tone of the patient’s voice were not very effective. However, six years after the surgery and consistent voice exercises, the tone of her voice is noticeably higher.
The patient was also subjected to total thyroidectomy for which there were two indications. Firstly, the results of FNAB fulfilled the Bethesda criteria for stage III (risk of malignancy 28%), but above all, the patient complained of dyspnea and dysphagia caused by the large tumor on her neck. Histopathology revealed benign follicular cell-derived neoplasms indicating thyroid follicular nodular disease bilaterally. Ultrasound-histopathological analysis is presented in Figure 3 (A-A3). At present the patient is euthyroid while on levothyroxine.
The presence of multinodular goiter in her family history (mother, grandmother, aunt, and cousin) further indicated the need for genetic evaluation. Molecular analysis using NGS, confirmed with the Sanger method, revealed a pathogenic heterozygotic variant c.2953>T [p.Gln985*] in exon 18 of the DICER1 gene. This variant was also found in the patient’s mother, leading to a DICER1 syndrome diagnosis for both, two and a half year after the onset of the symptoms. Neither ovarian nor any other tumors typical for DICER1 syndrome were found in other members of the family. The patient continues to undergo annual health evaluations as part of medical surveillance. Six years after the surgery she complained of recurrent cysts of the left ovary for which she received oral contraceptives with good outcome.
4. Case Report 2
A 12-year-old male patient was admitted to the outpatient Pediatric and Adolescent Endocrinology Department for diagnostic evaluation of a thyroid lesion. A month prior to admission, the patient discovered a palpable nodule in his neck, accompanied by hoarseness. An ultrasound assessment revealed structural changes throughout the thyroid gland, with multiple isoechogenic cystic-solid lesions in both lobes. His lymph nodes were unremarkable, and levels of thyroid hormones and calcitonin were within normal range. Until then, the patient had been healthy, and there were no reported cases of neoplasms or thyroid issues in the family history. An otorhinolaryngologist’s assessment indicated mild compression of the larynx from the left side, although vocal cord mobility was found to be normal. In the unit, biopsies from four thyroid nodules were taken, which showed atypical cells in all samples, categorized as Bethesda III. In total, ten nodules were identified, and it was decided that the patient would be placed under observation.
The patient’s perinatal history was unremarkable, with APGAR scores of 9 and 10 and a birth weight of 4,130g. The patient’s psychomotor development was normal. No genetic conditions were reported in the family’s medical history. However, given the clinical findings, a genetic evaluation targeting DICER1 was initiated. Molecular analysis using the NGS method, confirmed with the Sanger method, revealed a pathogenic heterozygotic variant c.2782C>T [p.Gln928Ter] in exon 17 of DICER1 gene. This variant has not been previously described in the Ensembl and ClinVar genetic databases. In silico analysis using MutationTaster2021 indicated the pathogenic nature of this variant, a finding further corroborated by Varsome. This variant was absent in both of the patient’s parents. Subsequently, a total thyroidectomy was performed, and the patient began thyroid hormone replacement therapy with biannual health evaluation. Histopathology revealed benign follicular cell-derived neoplasms indicating thyroid follicular nodular disease bilaterally. Ultrasound-histopathological analysis is presented in Figure 3 (B-B4, C-C3).
5. Discussion
In this work we presented the unique ultrasound and histopathological features of benign thyroid nodules in adolescent patients with DICER1 syndrome. The typical picture of multinodular goiter in DICER1 syndrome presented previously by Niedziela et al. is useful in ultrasonography point-of care in patient evaluation [18]. The lesions we discovered were mostly cystic-solid and isoechogenic and this typical imaging picture initiated reevaluation of other adolescent patients with MNG from our department (unpublished data). We also observed, similar to Niedziela et al., that the benign nodules in patients with DICER1 syndrome, have a unique and distinguishable pattern: multiple lesions (≥3), predominantly mixed solid and cystic nodules, isoechogenic with macrocalcifications, with lack of blood flow or blood flow only in solid lesions and no suspicious cervical lymph nodes [18]. This contrasts with malignant thyroid lesions which are predominantly solitary, solid, and hypoechogenic, frequently with subcapsular localization and irregular margins, taller than wider with microcalcifications and with high intranodular blood flow, with pathological cervical lymph nodes showing up frequently in children [15,16,18,19,20,21]. In this study we presented these differences in Figure 4 and Figure 5 where we reported a typical ultrasound picture of papillary thyroid carcinoma confirmed by histopathology in two adolescent females, contrasting with the ultrasound-histopathological picture of lesions in DICER1 syndrome presented in Figure 3.
Both study patients exhibited multinodular goiter. While MNG often correlates with iodine deficiency, genetic factors might contribute to its occurrence in iodine-rich areas [1]. Euthyroid multinodular goiter before 30 years of age strongly suggests a hereditary cause and as presented in the literature, a pathogenic DICER1 variant is the cause in a majority of cases involving young patients [18]. However, differential diagnosis must also exclude other syndromes, such as intestinal polyposis syndromes, PTEN hamartoma tumor syndrome, Carney complex, Werner’s syndrome, and familial medullary and papillary thyroid carcinomas among others [1,18,22].
Research involving 53 individuals from five families identified pathogenic DICER1 variants in 37, strengthening the DICER1-MNG connection [25]. The coexistence of familial MNG and ovarian sex cord-stromal tumors was found to be independent of PPB [25]. A separate study highlighted a higher MNG prevalence among DICER1 carriers, with 75% of female carriers and one-sixth of male carriers developing benign MNG [26]. The estimated penetrance of MNG in DICER1 carriers is around 10-20% [26,27]. DICER1 carriers also exhibit a 16 to 24-fold increase in thyroid carcinoma risk due to benign thyroid nodules that accumulate supplementary genetic abnormalities over time and eventually develop into malignant growths [26].
While it is not practical to suggest that every MNG patient has the DICER1 tumor predisposition syndrome, in regions with iodized salt in their diet, this association certainly warrants consideration.
By presenting the first Patient we would like to highlight the symptoms of androgen excess in adolescent patients such as: acne, hirsutism, irregular menses, balding, clitoromegaly, decrease of breast tissue, and deepening of the voice as was seen in the presented patient. Differential diagnosis should exclude non-classic congenital adrenal hyperplasia, the most common etiology, and should subsequently evaluate possible adrenal or gonadal sources of androgens.
Ovarian sex-cord stromal cell tumors (SCSTs) include a group of distinct subtypes with variable clinical presentations and prognosis, such as germ cell tumors and Sertoli-Leydig cell tumors (SLCTs) [28,29,30]. The Sertoli-Leydig cell tumors, constitute less than 1% of all primary ovarian tumors [28,29,30]. These tumors can present at any age, from infancy to later adulthood, but around 75% of cases are detected in women under 30 years of age [1]. As reported in the literature, the median diagnosis age of DICER1-related SLCTs was 13 years, considerably earlier than median age of onset at 19 years in sporadic SLCTs [25,31].
The Sertoli-Leydig cell tumors (SLCTs) are currently classified into 3 molecular pathogenic subtypes: DICER1 variant (younger patient age), FOXL2 variant, and DICER1/FOXL2 wildtype variant [32]. Yang et al. presented data showing that all pediatric SLCTs examined in their institution belonged to the DICER1-variant molecular subtype, highlighting that somatic hotspot DICER1 variant detection had high sensitivity (100%) for diagnosis of pediatric SLCTs (age ≤ 16 y) [32].
However, the authors found that some pediatric SLCTs harbor molecular genetic aberrations other than the DICER1 mutation, so further research on their significance is ongoing [32].
Given that SLCTs can be categorized into three groups based on endocrine functions (androgen excess, estrogen excess, and no endocrine function) DICER1 hotspot mutations may be more prevalent in patients with androgenic symptoms [33].
From the point of view of a pediatric endocrinologist it is interesting to understand the mechanism of formation of Sertoli and Leydig cells in the ovary. The molecular mechanism underlying the unusual differentiation of Sertoli and Leydig cells in the SLCTs of the ovary is largely unknown [3]. Research groups have demonstrated that DICER1 hotspot mutation could alter the expression of key genes controlling the fate of ovarian pregranulosa cells by the downregulation of several key ovarian development genes, such as FST and CYP19A1, and the upregulation of two key Sertoli cell differentiation genes, FGF9 and FGFR2, that are known to stimulate Sertoli cell differentiation by sustaining the expression of SOX9 [3,34,35].
As interpreted by Wang et al., the imbalance of gonadal gene expression may therefore shift the differentiation of the primordial gonadal cells towards a pseudotesticular cell differentiation in the ovary [3]. The selective accumulation of hotspot mutations in the DICER1 gene in SLCTs causes defective miRNA biogenesis resulting in altered expression in key gonad development genes, along with potentiation of proliferation, which may ultimately rewire differentiation and drive oncogenesis in ovarian gonads [3].
The presented cases underscore the elusive nature of the DICER1 tumor predisposition syndrome. It can manifest as asymptomatic, as seen in the first patient’s family, or produce symptoms that diminish the patient’s quality of life (large goiter, androgenization, voice change) or even pose a lethal risk. Failing to recognize the syndrome can diminish the likelihood of effective treatment for DICER1 syndrome patients, who face a higher risk of developing neoplasms compared to the general population.
6. Conclusion
Multinodular goiter, although rare in the pediatric population, when accompanied by characteristic ultrasound and histopathological features, and by additional features such as androgenization, warrants exclusion of DICER1 syndrome. DICER1 syndrome is widely known to endocrinologists and oncologists but as the spectrum of this syndrome is broad there is a need to present it to all specialists. Doctors from all specialties should be familiar with androgen excess symptoms in adolescents and refer patients for further hormonal and imaging evaluations.
Funding
This research received no external funding.
Informed Consent Statement
Informed and signed written consent was obtained from all individual participants and their parents included in the study.
Conflicts of interest
The authors declare no conflicts of interest.
References
- González IA, Stewart DR, Schultz KAP, Field AP, Hill DA, Dehner LP. DICER1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma. Mod Pathol 2022; 35(1):4–22. [CrossRef]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001; 409(6818):363-6. [CrossRef] [PubMed]
- Wang Y, Chen J, Yang W, et al. The oncogenic roles of DICER1 RNase IIIb domain mutations in ovarian Sertoli-Leydig cell tumors. Neoplasia 2015;17:650–660. [CrossRef]
- Slade I, Bacchelli C, Davies H, et al. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet 2011; 48: 273–278. [CrossRef]
- Schultze-Florey RE, Graf N, Vorwerk P, Koscielniak E, Schneider DT, Kratz CP. DICER1 syndrome: a new cancer syndrome. Klin Padiatr 2013; 225(3):177-8. Epub 2013 Apr 26. [CrossRef] [PubMed]
- de Kock L, Sabbaghian N, Druker H, et al. Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol 2014; 128: 583–595. [CrossRef]
- de Kock L, Sabbaghian N, Plourde F, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol 2014;128(1):111-22. PMID: 24839956; PMCID: PMC4129448. [CrossRef]
- Robertson JC, Jorcyk CL, Oxford JT. DICER1 Syndrome: DICER1 Mutations in Rare Cancers. Cancers (Basel) 2018;10(5):143. PMID: 29762508; PMCID: PMC5977116. [CrossRef]
- Stewart DR, Best AF,Williams GM, et al. Neoplasm Risk Among Individuals With a Pathogenic Germline Variant in DICER1. J Clin Oncol 2019; 37(8):668–76. [CrossRef]
- Caroleo AM, De Ioris MA, Boccuto L, et al. DICER1 Syndrome and Cancer Predisposition: From a Rare Pediatric Tumor to Lifetime Risk. Front Oncol 2021;10:614541. PMID: 33552988; PMCID: PMC7859642. [CrossRef]
- Kim J, Field A, Schultz KAP, Hill DA, Stewart DR. The prevalence of DICER1 pathogenic variation in population databases. Int J Cancer 2017;141(10):2030-2036. Epub 2017 Aug 21. PMID: 28748527; PMCID: PMC5749397. [CrossRef]
- Agarwal A, Bathla G, Soni N, et al. Newly Recognized Genetic Tumor Syndromes of the CNS in the 5th WHO Classification: Imaging Overview with Genetic Updates. AJNR Am J Neuroradiol 2024; 45(2):128-138. [CrossRef] [PubMed]
- Saskin A, de Kock L, Sabbaghian N, et al. A case of neuroblastoma in DICER1 syndrome: Chance finding or noncanonical causation? Pediatr Blood Cancer 2018;65(1). Epub 2017 Aug 2. [CrossRef] [PubMed]
- Schultz KA, Williams GM, Kamihara J, et al. DICER1 and associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res 2018; 24:2251–61. pmid:29343557. [CrossRef]
- Jarząb B, Dedecjus M, Lewiński A, et al. Diagnosis and treatment of thyroid cancer in adult patients - recommendations of polish scientific societies and the national oncological strategy. 2022 update. Endokrynol Pol 2022; 73(2):173–300. [CrossRef]
- Russ G, Bonnema SJ, Erdogan MF, Durante C, Ngu R, Leenhardt L. European Thyroid association guidelines for ultrasound malignancy risk stratification of thyroid nodules in adults: The EU-TIRADS. Eur Thyroid J 2017; 6(5):225–37. [CrossRef]
- Cibas ES, Ali SZ. The Bethesda system for reporting thyroid cytopathology. Thyroid 2009 19:1159–65. [CrossRef]
- Niedziela M, Muchantef K, Foulkes WD. Ultrasound features of multinodular goiter in DICER1 syndrome. Sci Rep 2022;12(1):15888. PMID: 36151231; PMCID: PMC9508228. [CrossRef]
- Niedziela M, Handkiewicz-Junak D, Małecka-Tendera E, et al. Diagnostics and treatment of differentiated thyroid carcinoma in children - guidelines of polish national societies. Endokrynol Pol 2016; 67(6):628–42. [CrossRef]
- Francis GL, Waguespack SG, Bauer AJ, et al. American Thyroid Association Guidelines Task Force. Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2015; 25(7):716-59. PMID: 25900731; PMCID: PMC4854274. [CrossRef]
- Januś D, Wójcik M, Taczanowska-Niemczuk A, et al. Ultrasound, laboratory and histopathological insights in diagnosing papillary thyroid carcinoma in a pediatric population: a single centre follow-up study between 2000-2022. Front Endocrinol (Lausanne) 2023;14:1170971. PMID: 37274328; PMCID: PMC10233204. [CrossRef]
- Niedziela, M. Thyroid nodules. Best Pract Res Clin Endocrinol Metab 2014;28(2):245-77. Epub 2013 Aug 30. [CrossRef] [PubMed]
- Bignell GR, Canzian F, Shayeghi M, et al. Familial nontoxic multinodular thyroid goiter locus maps to chromosome 14q but does not account for familial nonmedullary thyroid cancer. Am J Hum Genet 1997;61(5):1123-30. PMID: 9345104; PMCID: PMC1716029. [CrossRef]
- Capon F, Tacconelli A, Giardina E, et al. Mapping a dominant form of multinodular goiter to chromosome Xp22. Am J Hum Genet 2000;67(4):1004-7. Epub 2000 Sep 11. PMID: 10986044; PMCID: PMC1287870. [CrossRef]
- Frio TR, Bahubeshi A, Kanellopoulou C, et al. DICER1 Mutations in Familial Multinodular Goiter With and Without Ovarian Sertoli-Leydig Cell Tumors. JAMA 2011;305(1):68–77. [CrossRef]
- Khan NE, Bauer AJ, Schultz KAP, et al. Quantification of thyroid cancer and multinodular goiter risk in the DICER1 syndrome: A family-based cohort study. J Clin Endocrinol Metab 2017;102(5):1614–22. [CrossRef]
- de Kock L, Sabbaghian N, Soglio DB, et al. Exploring the association Between DICER1 mutations and differentiated thyroid carcinoma. J Clin Endocrinol Metab 2014;99(6):E1072-7. Epub 2014 Mar 11. [CrossRef] [PubMed]
- Shah SP, Kobel M, Senz J, et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med 2009; 360: 2719–2729. [CrossRef]
- Kommoss S, Anglesio MS, Mackenzie R, et al. FOXL2 molecular testing in ovarian neoplasms:diagnostic approach and procedural guidelines. Mod Pathol 2013;26: 860–867. [CrossRef]
- Heravi-Moussavi A, Anglesio MS, Cheng SW, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 2012; 366:234–242. [CrossRef]
- Young RH, Scully RE. Ovarian Sertoli-Leydig cell tumors. Am J Surg Pathol 1985; 9(8):543-5693911780. [CrossRef]
- Yang B, Chour W, Salazar CG, et al. Pediatric Sertoli-Leydig Cell Tumors of the Ovary: An Integrated Study of Clinicopathological Features, Pan-cancer-Targeted Next-generation Sequencing and Chromosomal Microarray Analysis From a Single Institution. Am J Surg Pathol 2024;48(2):194-203. [CrossRef]
- Gui T, Cao D, Shen K, et al. A clinicopathological analysis of 40 cases of ovarian Sertoli-Leydig cell tumors. Gynecol Oncol 2012;127: 384–389. [CrossRef]
- Ross AJ, Capel B. Signaling at the crossroads of gonad development.Trends Endocrinol Metab 2005; 16: 19–25. [CrossRef]
- Eggers S, Sinclair A. Mammalian sex determination-insights from humans and mice. Chromosome Res 2012; 20: 215–238. [CrossRef]
Figure 1.
Age-dependent clinical manifestation of DICER1 syndrome. The colors represent the age of onset for various conditions as detailed in the accompanying legend. Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Figure 1.
Age-dependent clinical manifestation of DICER1 syndrome. The colors represent the age of onset for various conditions as detailed in the accompanying legend. Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
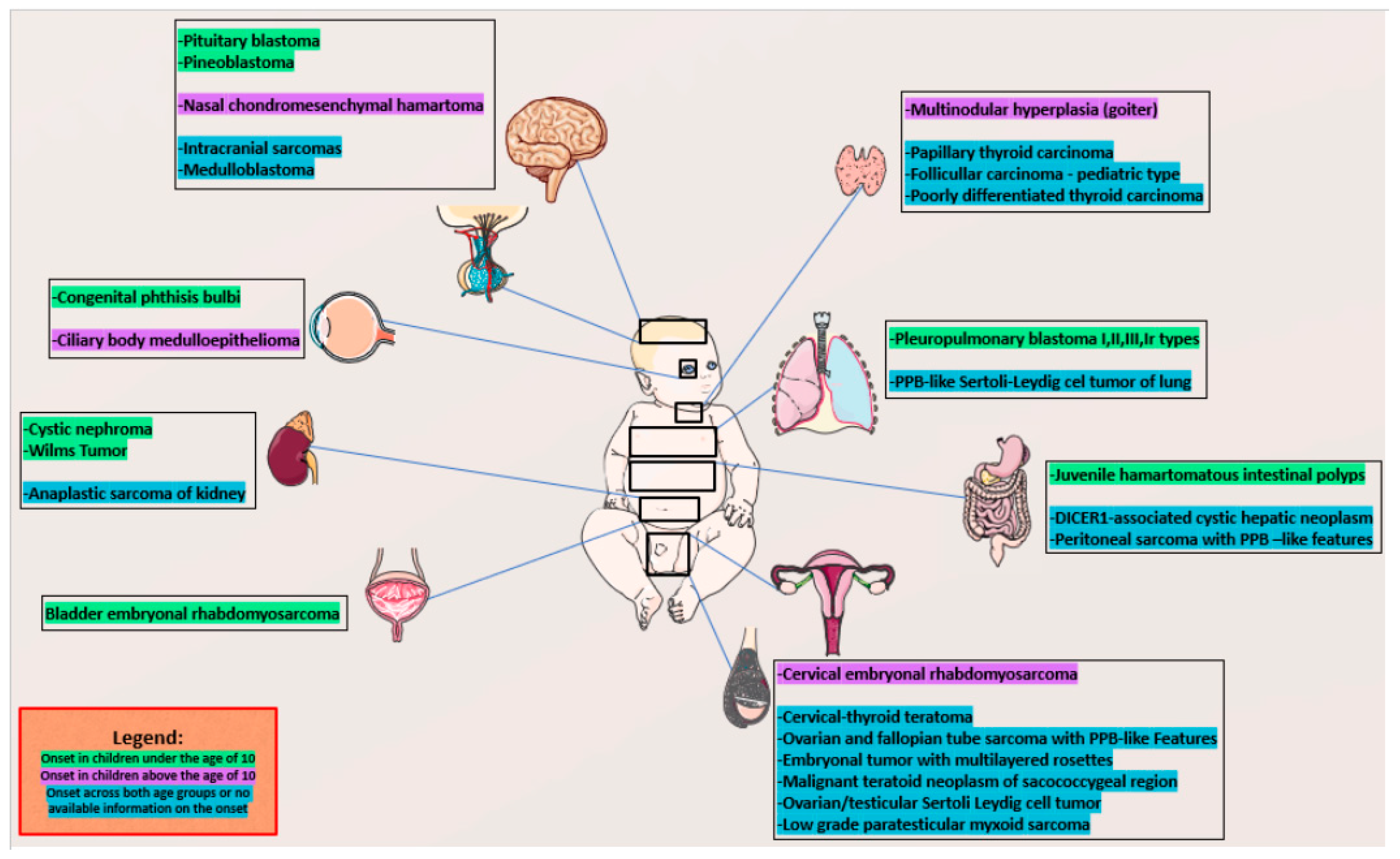
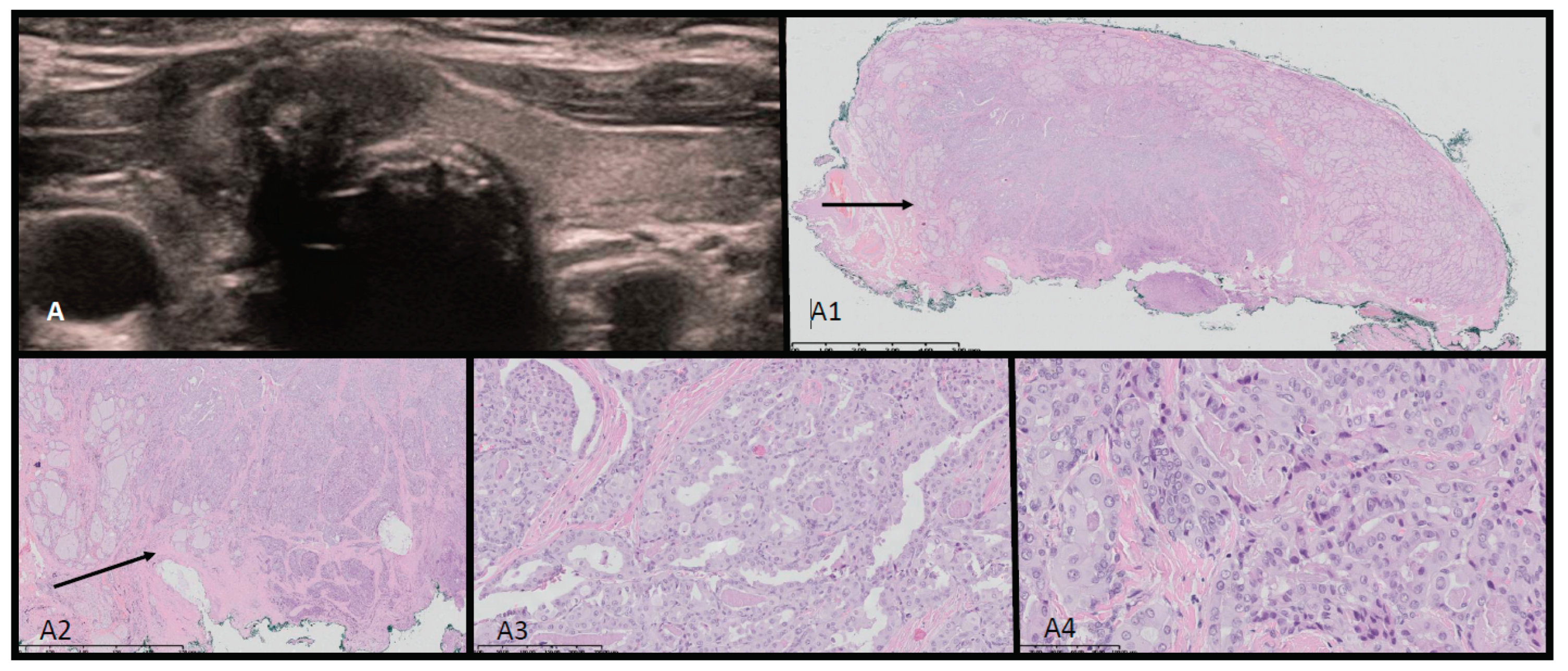
Figure 2.
Ultrasound-histopathological assessment of SLCT moderately differentiated in Patient 1. A- 5.7x6.9 cm solid mass in the right adnexal region seen on US scan. A1-A4: HE histopathological assessment. The SLCT is composed of cords, solid and microcystic areas of small Sertoli cells [red arrows], as well as scattered and clustered Leydig cells with abundant eosinophilic bright-pink cytoplasm and round nucleus [yellow arrows]. Fibrous stroma [black arrows] with lymphocytic infiltration is visible. The low magnification (A1) shows hypo- and hypercellular nodules predominantly built by Sertoli cells presenting different architecture. The tumor was confirmed with immunohistochemistry [CK AE1/AE3 positive in Sertoli cells, vimentin [+], and calretinin, CD99, WT1 – positive in the part of cells, and also EMA and CD34 – negative [data not shown]. The proliferation index of Ki67 is approx. 40% and the mitotic figure count 11/ 10 HPF [high power field]. The tumor was totally resected with minimal margin of 2.7 mm. In the periphery the ovarian cortex with primary and vesicular follicles can be seen [green arrows].
Figure 2.
Ultrasound-histopathological assessment of SLCT moderately differentiated in Patient 1. A- 5.7x6.9 cm solid mass in the right adnexal region seen on US scan. A1-A4: HE histopathological assessment. The SLCT is composed of cords, solid and microcystic areas of small Sertoli cells [red arrows], as well as scattered and clustered Leydig cells with abundant eosinophilic bright-pink cytoplasm and round nucleus [yellow arrows]. Fibrous stroma [black arrows] with lymphocytic infiltration is visible. The low magnification (A1) shows hypo- and hypercellular nodules predominantly built by Sertoli cells presenting different architecture. The tumor was confirmed with immunohistochemistry [CK AE1/AE3 positive in Sertoli cells, vimentin [+], and calretinin, CD99, WT1 – positive in the part of cells, and also EMA and CD34 – negative [data not shown]. The proliferation index of Ki67 is approx. 40% and the mitotic figure count 11/ 10 HPF [high power field]. The tumor was totally resected with minimal margin of 2.7 mm. In the periphery the ovarian cortex with primary and vesicular follicles can be seen [green arrows].
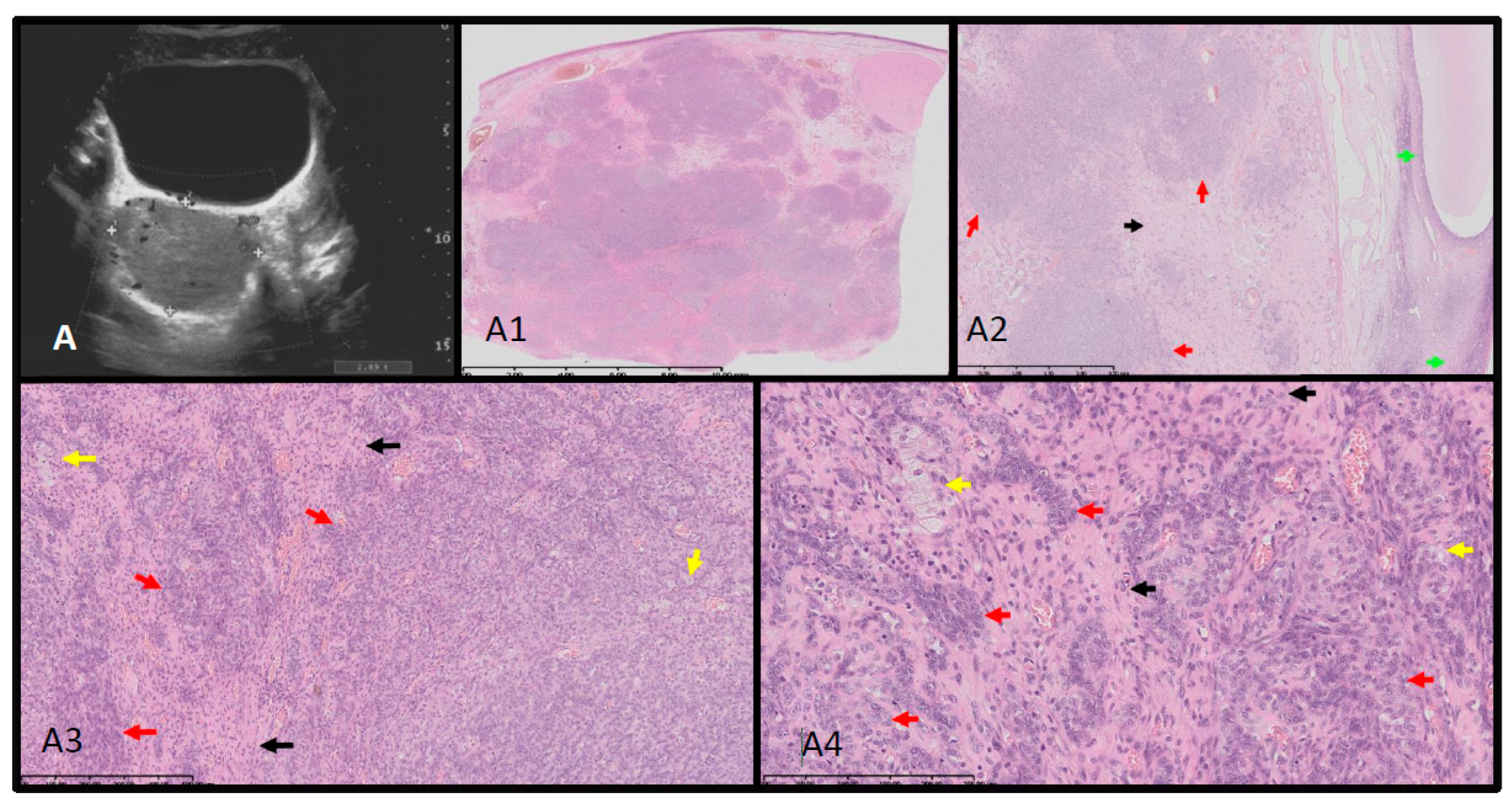
Figure 3.
Ultrasound (US)-histopathological (HP) assessment of multinodular goiter in Patients 1 (A-A3) and 2 (B-B4, C-C3). US shows multinodular goiter composed of isoechogenic solid-cystic nodules. In HP the whole thyroid is built up by many hypocellular nodules filled by pink colloid. The hyperplastic nodules present a small-, medium- and large-vesicular structure and focally papillary arrangement. Some of the nodules show areas of non-specific granulation tissue, fibrosis, single calcifications, and a mixed-cellular inflammatory infiltrate with foamy macrophages containing hemosiderin. The remaining thyroid parenchyma is slightly congested.
Figure 3.
Ultrasound (US)-histopathological (HP) assessment of multinodular goiter in Patients 1 (A-A3) and 2 (B-B4, C-C3). US shows multinodular goiter composed of isoechogenic solid-cystic nodules. In HP the whole thyroid is built up by many hypocellular nodules filled by pink colloid. The hyperplastic nodules present a small-, medium- and large-vesicular structure and focally papillary arrangement. Some of the nodules show areas of non-specific granulation tissue, fibrosis, single calcifications, and a mixed-cellular inflammatory infiltrate with foamy macrophages containing hemosiderin. The remaining thyroid parenchyma is slightly congested.
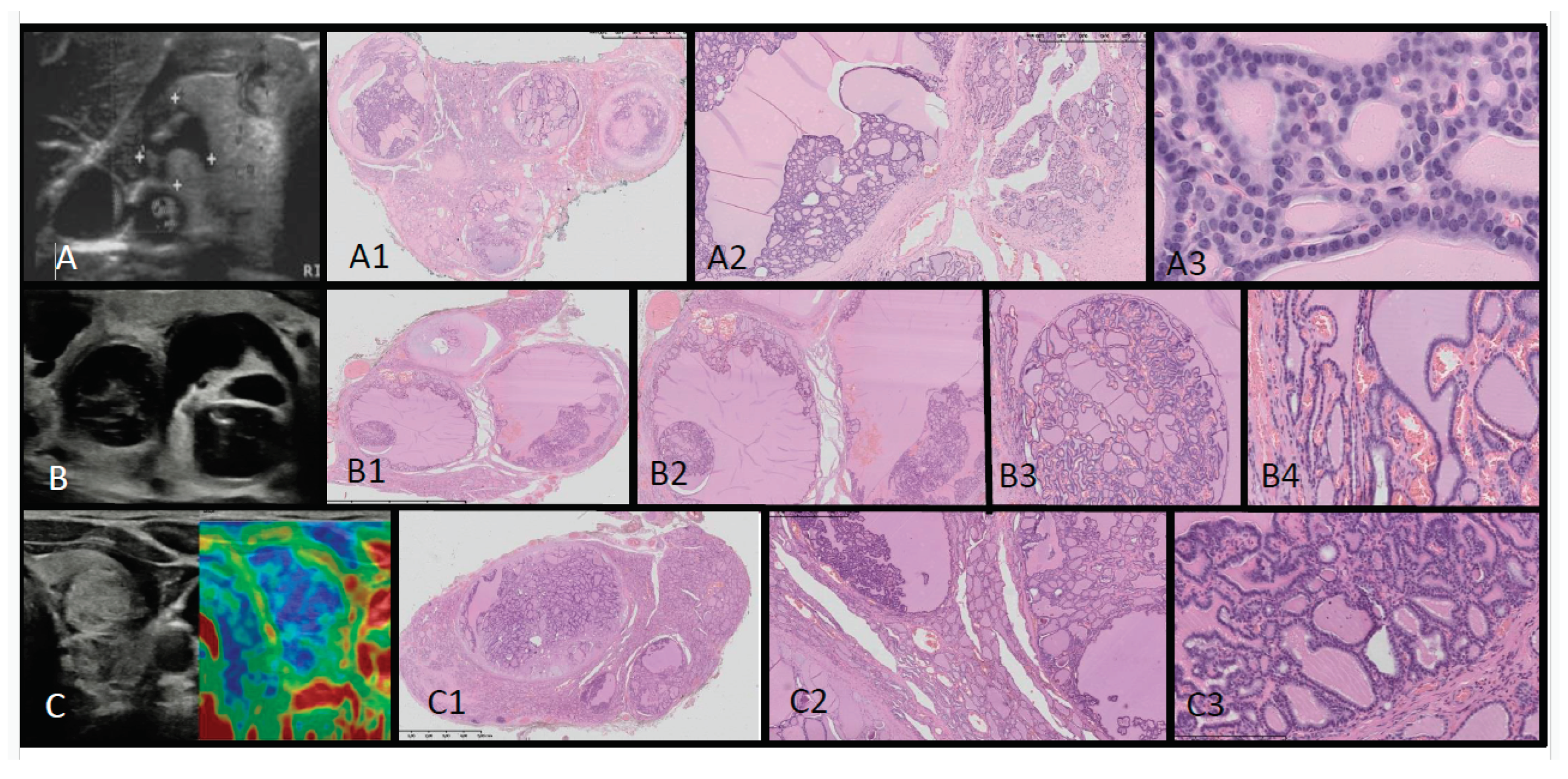
Figure 4.
Ultrasound (US)-histopathological (HP) assessment of papillary thyroid carcinoma in 18-year old female Patient. US (A) shows hypoechogenic nodule composed of multiple small nodules. In HP (A1-A4), lobulated tumor [black arrow] with fibrotic layers inside invades capsule. The outer part of the whole lesion is of papillary-follicular structure and is surrounded by a dark-blue lymphocytic inflammation [red arrow]. The lesion is composed of polymorphic cells with nuclei of “glassy” clearing and with grooves, invading the capsule (A4).
Figure 4.
Ultrasound (US)-histopathological (HP) assessment of papillary thyroid carcinoma in 18-year old female Patient. US (A) shows hypoechogenic nodule composed of multiple small nodules. In HP (A1-A4), lobulated tumor [black arrow] with fibrotic layers inside invades capsule. The outer part of the whole lesion is of papillary-follicular structure and is surrounded by a dark-blue lymphocytic inflammation [red arrow]. The lesion is composed of polymorphic cells with nuclei of “glassy” clearing and with grooves, invading the capsule (A4).
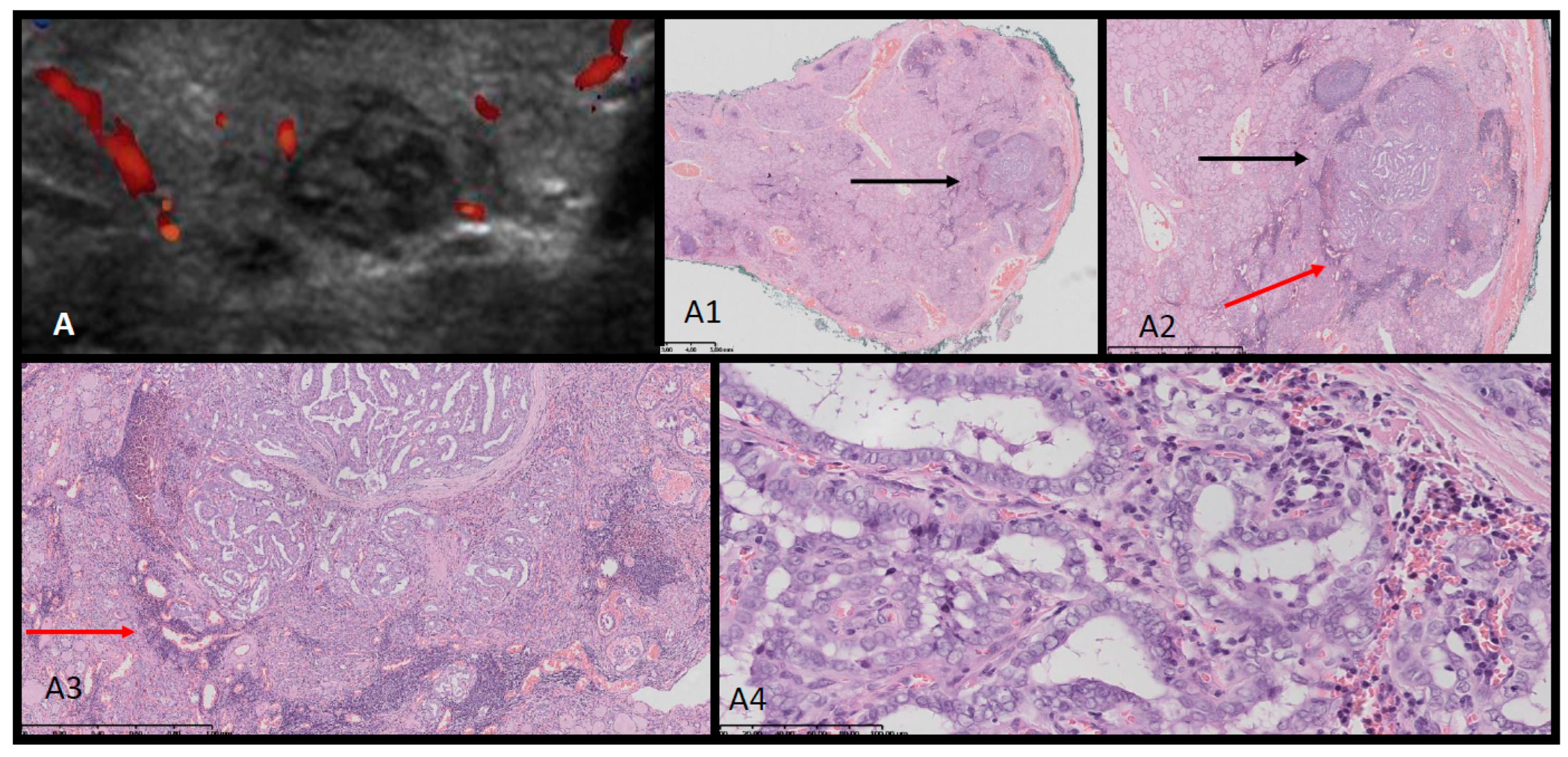
Figure 5.
Ultrasound (US)-histopathological (HP) assessment of papillary thyroid carcinoma in 17-year old female Patient. US (A) shows irregularly contoured, hypoechogenic nodule, invading the capsule. In HE (A1-A4), the lesion is infiltrating the thyroid parenchyma (A1-A2) and is composed of polymorphic cells with nuclei of “glassy” clearing and with grooves (A4).
Figure 5.
Ultrasound (US)-histopathological (HP) assessment of papillary thyroid carcinoma in 17-year old female Patient. US (A) shows irregularly contoured, hypoechogenic nodule, invading the capsule. In HE (A1-A4), the lesion is infiltrating the thyroid parenchyma (A1-A2) and is composed of polymorphic cells with nuclei of “glassy” clearing and with grooves (A4).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



