Submitted:
19 February 2024
Posted:
20 February 2024
You are already at the latest version
Abstract
Neuronal cells are believed to rely on autophagy to maintain cellular homeostasis. Nevertheless, autophagy may be a therapeutic target for neurodegenerative diseases because of the role it plays in neuronal cell death. Despite Beclin 1 association with autophagy and apoptosis, the molecular mechanisms that underlie Tau-induced vesicular pathology and Tau protein aggregates in humans remain poorly understood. Here, we set out to examine the role of three genes-ATG14L, Beclin 1 and VPS34- in autophagy nucleation failure-associated Tau disorders in fruit fly models. We found that either reduced ATG14L levels improve neuronal outcomes, or transgenic expression of ATG6/Beclin 1 greatly reduced age-dependent neurodegeneration and Tau pathology; whereas, the loss of Beclin 1 exacerbated these effects. Prior to p-Tau accumulation, the neuronal Beclin 1 network is critical for reducing p-Tau accumulation, indicating that the loss of Beclin 1 causes Tau pathology and neurodegeneration. As neurodegeneration progressed, glial cells showed an upregulation of vesicular Beclin 1 immunoreactivities. The reduced VPS34 also resulted in p-Tau accumulation, suggesting that neurons require both Beclin 1 and VPS34 to reduce p-Tau accumulation. Our results provide mechanistic insight into the molecular pathways that cause age-dependent Tau pathology and neurodegeneration due to an excess of autophagy nucleation processes and autophagy activity, and we derive novel inferences on the therapeutic possibilities of rebalanced Beclin 1 network by reducing autophagic specific nucleation component of ATG14L.
Keywords:
Autophagy
; Alzheimer’s disease
; ATG14L
; Beclin 1
; VPS34
; Neurodegenerative disease
; GFP-mCherry-LC3
; Tau
1. Introduction
Tau, as described by Cleveland (1977), is a protein known as microtubule-associated protein tau (MAPT) that regulates the transportation of axonal vesicles and the stability of axonal microtubules [1,2]. In age-dependent neurodegenerative diseases such as Alzheimer’s disease and frontal temporal dementia, progressive accumulation of hyperphosphorylated tau cause neurodegenerative diseases across the late age spectrum referred to as tauopathies [3,4,5,6,7,8,9,10]. Neurons are more vulnerable to disruption of vesicular transport, which is dependent on axonal Tau associated microtubules. Not surprisingly, the widespread autophagy-related vesicle pathology suggests the disruptions of axonal transport in major neurodegenerative diseases. The precise correlation between Tau and the role of autophagy in neurodegenerative diseases remains poorly understood [11,12,13,14,15,16,17].
The molecular mechanism of autophagy has been conserved through evolution and is responsible for recycling damaged cellular components through lysosomal degradation for repair and cellular renovation [18,19,20]. The autophagic process is mediated by double-membrane autophagosomes, which allow for the lysosomal destruction of pathogens (xenophagy), protein aggregates and damaged mitochondria (mitophagy) [21,22,23]. As a result of this recycling into fundamental building blocks, hydrolase enzymes in vacuoles or lysosomes degrade biological components [24]. Selective autophagy is crucial for the survival of post-mitotic neurons that require autophagy to remove protein aggregates and damaged mitochondria [25]. Ohsumi and colleagues had identified that more than 30 ATG genes are required during autophagy processes in yeast. The cup-shaped double membrane phagophore can be formed by the ATG1 (ULK1/2) complex in the absence of nitrogen [22,26]. After initiating nucleation through Beclin 1 complex I of ATG14L-Beclin1-VPS15-VPS34, this double-membrane phagophore elongates and envelopes the enclosed cytoplasmic components, resulting in the formation of an autophagosome [27,28,29,30]. Neuronal cells may endure stress conditions because of autophagy.
One autophagy component that inhibits autophagy and apoptosis is Beclin 1, which is a protein with Bcl-2 homology 3 (BH3) domain [31,32,33,34]. Binding of Beclin 1 to Bcl-2 in the endoplasmic reticulum and mitochondria disrupts the connections among ATG14L, Beclin 1, VPS15, and VPS34 inhibiting autophagy in normal condition [35,36]. During induction, autophagic nucleation process consists of ATG14L-Beclin 1-VPS15-VPS34 complex I at the phagophore is activated by the multiple layer regulation to disrupt Beclin 1 binding with Bcl-2 in response to stress [37,38,39,40,41,42]. In order to activate autophagic progresses, the newly generated nucleation acts as a docking site for PI3P-binding components and facilitates membrane transfer [43]. In order to safeguard neurons, autophagy is enhanced to eliminate damaged mitochondria and protein aggregates [44]. On the other hand, autophagic vesicle pathology and autophagy dysfunction are common in neurodegenerative diseases, leading some to speculate that autophagy failure could exacerbate protein pathology, especially the accumulation of phosphorylated Tau in Alzheimer’s disease [14,45,46]. However, it is incompletely understood the Beclin 1 mechanisms that maintain neuronal survival and regulate their quality control within neurons, which are lost in neurodegenerative diseases that develop with age [47,48,49].
This work aimed to investigate the hypothesis that the upregulated ATG14L-Beclin 1-VPS34 complex I has a negative impact on neurons and contributes to human Tau-induced neurodegeneration by regulating autophagosome formation and autophagic activity. Our investigation revealed that expression of human Tau induced autophagy and leading to a failure in the autophagic processes in Drosophila melanogaster. Investigating the autophagy-specific gene ATG14L using reverse genetics has shown that reduced ATG14L inhibits the development of Tau pathology and neurodegeneration. Conversely, the expression of the Beclin 1 transgene improves neuronal outcomes, instead of reducing Beclin 1 expression. Ultimately, we demonstrated that increased VPS34 is essential for maintaining neuronal quality control by reducing the buildup of p-Tau and altering Tau-induced neurodegeneration. Our findings indicate that neuronal balanced beclin 1 network has the potential to be used as a therapeutic target such as reducing upregulated ATG14L for suppressing neurodegeneration caused by Tau pathology.
2. Materials and Methods
2.1. Husbandry and Genotypes of Flies
The maintenance of fly stocks was authorized by the Bureau of Animal and Plant Health Inspection and Quarantine Council of Agriculture regulation in Taiwan. The fruit flies were housed in a room that was controlled by automation. The room followed a 12-hour cycle of light and darkness. The temperature in the room ranged from 20°C to 25°C, and the humidity was maintained at 70 %. The flies were kept in bottles or vials and cultivated using a cornmeal-yeast-agar medium. The transgenic flies with Tau abnormalities utilized in this investigation were previously generated [50]. The young flies were in the age range of 1-3 days, whereas the old flies were in the age range of 14-21 days. This age difference revealed age-related neurodegeneration, which was determined using a binary expression system called GAL4/UAS system. The fly strains utilized in this investigation are as follows: Canton S; Gmr-Gal4 UAS-Tau; UAS-GFP-mCherry-Atg8; UAS-LacZ; P{TRiP.HMC04086}attP2; M{UAS-Atg6.ORF.3xHA.GW}ZH-86Fb; P{TRiP.HMS00261}attP2.P{TRiP.GL00085}attP2.
2.2. Histochemistry Examination of Transgenic Retinas
Following prior descriptions, the compound eyes of the flies were dissected [51,52]. We fixed the compound eyes in 4% paraformaldehyde (PFA) for 1 hr. The fly compound eyes were washed three times in phosphate-buffered saline containing 0.3% Triton X-100 (PBST); then, they were blocked with 5% goat serum and incubated with primary antibodies together with either rhodamine-conjugated phalloidin (Sigma) or Alexa FluorTM 633 conjugated phalloidin (Thermo Fisher) at 4°C overnight. Next, the retinal tissues were washed three or four times; then, they were stained with immunofluorescent-conjugated secondary antibodies in PBS containing 5% goat serum. Tissues were mounted in VectaShield antifade mounting medium (Vector Laboratories, Burlingame, CA, USA) after three washes with PBST. The mCherry-GFP-Atg8a reporter was used to visualize autophagy in retina. Two anti-human Tau antibodies had been used in this study: one anti-Tau (aa. 243-441; 1:200, Dako) and the other monoclonal antibody AT8 (anti-pSer202, Thr205; Thermo Fisher Scientific). There were two LC3 antibodies tuilized in the study: LC3 and GABARAG. We utilized anti-VPS34 polyclonal antibody and anti-Beclin 1 monoclonal antibody both from invitrogen. The specificity of the antibodies was examined by utilizing transgene expression to knockdown gene expression as a negative control and transgene expression as a positive control.
2.3. Confocal Microscopy Detection of Fluorescence
Imaging was captured using a Zeiss LSM-610 confocal laser-scanning microscope located in the Brain Research Center, as well as a Zeiss LSM-800 confocal laser-scanning microscope manufactured by Carl Zeiss, located in Oberkochen, Germany. The software utilized for this imaging was Zen 2012 SP1. Labeling the F-actin cytoskeleton was done with the rhodamine or Alexa FluorTM 633 fluorescence-conjugated phalloidin (Thermo Fisher Scientific). Donkey anti-rabbit and anti-mouse secondary antibodies, which were fluorescently tagged, were used for fluorescent immunohistochemistry analysis to detect appropriate primary antibody immunoreactivity. The following secondary antibodies were utilized at a dilution of 1:200 dilution: Alexa Fluor 488, AlexFluor 647 (Invitrogen), donkey anti-mouse Cy3-, and anti-rabbit Cy3-, anti-mouse Cy5-conjugated secondary antibodies (Jackson Immuno Research Laboratories).
2.4. Scanning Electron Microscope (SEM) Image Acquisition
We normalized the pixel intensity and recreated the SEM images in greyscale for each fly compound eye examination. Furthermore, blinding analysis validated the representative SEM images, and measurements of changes in eye size were also taken simultaneously in parallel groups [53].
3. Results
3.1. The Impairment of Autophagy Caused by Human Tau in Drosophila
In order to investigate the role of autophagy in neurodegeneration, we utilized a fly model, by which the longest human tau isoform (known as 2N4R with 441 amino acids) transgene is driven by the multiple glass promoter and expressed in the photoreceptor cells and other retinal cells (referred to as the Tau-expressing retina. The compound eye in Drosophila consists of ~780 unit eyes which has a compound eye structure that appears as a hexagonal neurocrystalline observed under a scanning electron microscope [52]. In contrast to normal adult retina, the surface of Tau-expressing eye seemed uneven and absent in most bristles as shown in Figure 1A. Within each unit eye, it contains seven outer sensory neurons. The apical surface of each neuron is structured into thousands of finger-like membrane protrusions that contain axial actin. This specialized region, known as the rhabdomere, exhibited minimal LC3-II immunoreactivity in normal eyes (Figure 1B). However, in the presence of human Tau-induced neurodegeneration, we observed significant autophagic LC3-II puncta associated with the degeneration caused by Tau.
We developed a visualization platform that utilizes GFP-mCherry-LC3 to monitor autophagic activity induced by human Tau. This platform allows for a straightforward visualization of convertible fluorescent transformation from yellow GFP-mCherry-LC3-labeled autophagosomes to red GFP-mCherry-LC3-labeled autolysosomes [54]. In the wild-type retina, neurons and glial cells showed a specific group of autophagosomes marked with yellow GFP-mCherry-LC3 and autolysosomes marked with red GFP-mCherry-LC3. These autolysosomes are responsible for the normal degradation of the inner membrane and its content in the GMR-GAL4/UAS-GFP-mCherry-LC3 retina (Figure 1C-D). In order to study the effect of Tau on autophagy, we employed two transgenic modified fly lines to simultaneously express the UAS-GFP-mCherry-LC3 transgene. Confocal microscopy was used to visualize the fluorescent signals of this reporter. The gl-tau eye produced the elevated amounts of yellow autophagosomes or non-acidic autolysosomes in GMR-Gal4, gl-tau/UAS-GFP-mCherry-LC3 (Figure 1C). In order to examine the malfunction of autophagy that is linked to age-related neurodegeneration, we analyzed changes of autophagy between young and elderly retinas. The expression of Tau resulted in increased autolysosomes, which were characterized by red GFP-mCherry-LC3 in young GMR-GAL4, UAS-Tau/UAS-GFP-mCherry-LC3 retina, compared to normal retina. However, the presence of autophagosomes, marked by yellow red GFP-mCherry-LC3 was rarely observed. The phenotype of the young individuals was transformed into a phenotype, resembling yellow autophagosomes or yellow non-acidic autolysosomes caused by gl-tau, in old GMR-GAL4, UAS-Tau/UAS-GFP-mCherry-LC3 retina (Figure 1D) [7]. The presence of increased autophagy microtubule-associated protein light chain 3 (LC3-II) at a given time can indicate either an increase in autophagy or a malfunction in the process of autophagic flux. Our observations indicate that the neurotoxicity caused by human Tau in Drosophila is linked to a significant impairment in autophagy, as seen by the presence of either red GFP-mCherry-LC3-labeled autolysosomes, or exclusively yellow GFP-mCherry-LC3-labeled autophagosomes and non-acidic autolysosomes.
3.2. The Role of ATG14/Barkor in Regulating Autophagic Activity and Neurodegeneration Triggered by Tau
The specific role of ATG14L, also referred to as Barkor, in human Tau-induced neurodegeneration has not been established, despite its known involvement in autophagy induction and nucleation as part of the ATG14L-Beclin 1-VPS15-VPS34 complex I [55]. To identify whether ATG14L gene implicated in the autophagic failure induced by Tau, we conducted reverse genetic study by reducing the upregulated ATG14L gene expression using Atg14L RNAi transgenic expression in Tau expressing retina. ATG14L plays a crucial role as an autophagy specific component that is necessary component of autophagy nucleation complex. It is responsible for sensing the curvature of the pre-autophagosome membrane, as well as facilitating the tethering and fusion of vesicles. Contrary to our expectations, it indeed improved, rather than worsened, the neurodegeneration caused by Tau. We found that reduction in ATG14L protein involved in autophagy nucleation process, significantly reduced the loss of neurons and decreased the buildup of phosphorylated Tau burdens in neurons in the retina of GMR-GAL4, UAS-Tau/+; UAS-ATG14L-RNAi/+ flies. The rescue effect was confirmed by expression of additional UAS-ATG14L-RNAi transgene in Tau expressing retina (not shown), as well as in another fly model expressing Tau (Figure 2A) from GMR-GAL4, gl-Tau/+; UAS-ATG14L-RNAi/+ retina (Figure 2A). These findings suggest that decreasing the expression of the autophagy-specific protein ATG14L could potentially be used as a therapeutic approach to prevent neurodegeneration caused by Tau.
We subsequently assessed the association between ATG14L expression and autophagy activity. In the Tau-expressing retina, GMR-GAL4, UAS-Tau/+, we observed an increase in autophagy activity, as shown by the strong presence of LC3-II puncta immunoreactivity. This was in contrast to the normal level of LC3-II puncta immunoreactivity observed in the wild-type retina (Figure 2B). In addition, we noticed that the inhibition of upregulated ATG14L led to a decrease in autophagy activity, resulting in a lack of strong LC3-II immunoreactivity induced by human Tau from GMR-GAL4, UAS-Tau/+; UAS-ATG14L-RNAi/+ retina (Figure 2B). This suggests that increased autophagy activity and the presence of autophagic vesicle pathology may be associated with an increased susceptibility of neurons to Tau toxicity. Nevertheless, the beneficial effect found when one copy of the UAS-ATG14L-RNAi transgene was no longer evident when two copies were co-expressed simultaneously. The findings suggest that increased levels of ATG14L are linked to impaired autophagy, while decreasing ATG14L expression can enhance neuronal cell survival by mitigating neurodegeneration caused by human Tau in Drosophila.
3.3. The Expression of the Beclin 1 Transgene Protects Neurons Against the Accumulation of p-Tau and Age-Related Neurodegeneration
The intricate regulatory role of ATG14L in autophagy activity indicates that increased levels of ATG14L have a multifaceted and contradictory regulatory function in Tau-induced neurodegeneration. Based on these findings, we proposed the hypothesis that the increase in ATG14L might potentially have a detrimental effect on neurons via decreasing other Beclin 1 complexes and VPS15-VPS34 complexes. This, in turn, could have an effect on both healthy and disease development [56]. The Beclin 1-Bcl-2 Complex exerts a negative regulatory effect on Beclin 1 complex I and autophagy. Consequently, we examined whether the increased activity of autophagy nucleation process of the ATG14-Beclin 1-VPS15-VPS34 activity led to impaired autophagy by diminishing the inhibition of the Beclin 1 complex. If the loss of Beclin 1 network regulation is the cause, we hypothesized that the expression of Beclin 1 transgene may have a beneficial effect on the regulation of autophagy and neuronal survival in a reverse manner [57]. To clarify the function of Beclin 1 and autophagy, we crossed human Tau transgenic flies, GMR-GAL4, UAS-Tau, with UAS-ATG6/VPS30 transgenic flies. The expression of the Beclin 1 transgen from GMR-GAL4, UAS-Tau/+; UAS-Beclin 1/ATG6/VPS30/+ retina, had a suppressive effect on human Tau-induced neurodegeneration, as observed in Figure 3A. The expression of the Beclin 1 transgene indicates that the Beclin 1 network has a positive impact on the regulation of neuronal protein quality control and neuronal viability. Given that human Tau-induced progressive neurodegeneration during aging, we proceeded to investigate the impact of Beclin 1 transgenic expression on Tau-induced neurodegeneration at two additional time points: 10 days and 20 days post-eclosion. The number of visual sensory neuronal degeneration in the old retina was higher compared to the young retina from GMR-GAL4, UAS-Tau/+ retina. Conversely, the introduction of the Beclin 1 transgene inhibited the degeneration of neurons caused by Tau in both young and old retinas. There were seven visual sensory neurons present in the unit eyes of both young and aged retinas from GMR-GAL4, UAS-Tau/+; UAS-Beclin 1/+ retina. This indicates that the Beclin 1 network plays a critical role in preventing age-dependent neurodegeneration produced by Tau (Figure 3A). In order to ascertain how Beclin 1 transgene expression protects neurons from Tau toxicity, we conducted an analysis of the p-Tau immunoreactivity. The expression of Beclin 1 transgene is sufficient to both prevent the buildup of p-Tau in neurons and the progressive neurodegeneration (Figure 3B). This indicates that Beclin 1 plays a vital role in maintaining a low level of p-Tau and promoting neuronal survival.
3.4. The loss of Beclin 1 Promotes Neuronal Accumulation of p-Tau and Exacerbates Age-Related Neurodegeneration
To clarify the mechanism by which Beclin 1 may inhibit Tau-induced neurodegeneration, we proceeded to investigate the consequences of reducing Beclin 1 gene expression and analyzing its effects on the Tau-induced phenotype. Tau-induced neurodegeneration was strongly related with a significant decrease of neuronal Beclin 1. Aligned with the pattern of connection between the absence of Beclin 1 and the development of neurodegeneration caused by Tau, we noted that reducing the expression of Beclin 1 gene increased Tau-induced phenotype. Neurodegeneration was detected in the young retina of GMR-GAL4, UAS-Tau/+; UAS-Beclin 1-RNAi/+ flies, in contrast to the absence of significant neurodegeneration in the control retina of the same age (Figure 4). However, the confirmation of this improved phenotype was not always confirmed by the utilization of all distinct UAS-Beclin 1-RNAi transgenes (not shown). This indicats that the impact of Beclin 1 loss on neurons is influenced by both beneficial and detrimental effects, and is contingent upon the dosage. Given the prominent role of Tau aggregates associated with neurodegeneration, we investigated whether the loss of Beclin 1 was associated with any changes of Tau pathology. To do this, we utilized the UAS-Beclin 1-RNAi transgene. During the initial time period of young (1-7 day), a significant amount to Tau pathology was observed to be insoluble and formed aggregates in neurons and glial cells of the GMR-GAL4, UAS-Tau/+; UAS-Beclin 1-RNAi/+ retina (Figure 4). In contrast, the young control retina without Beclin 1 knockdown did not exhibit any Tau aggregates. This suggests that the loss of Beclin 1 plays a critical role prior to the accumulation of phosphorylated Tau in neurons, leading to result in Tau aggregate associated neurodegeneration.
3.5. Beclin 1 in Neurons is Essential for Neuronal Protection, Whereas Beclin 1 in Degenerative Illness is Associated with Glial Cells
We found that reduced levels of mitochondrial or ER Beclin 1 network in neurons were associated with enhanced activity of autophagy-related processes in fly models that expressed human Tau. Figure 5 shows that glial cells surrounding the unit eyes of the GMR-GAL4, UAS-Tau/+ retina that showed an increase in Beclin 1 positive vesicles as a result of Tau-induced neurodegenerative process. Once again, neuronal loss of Beclin 1 during progressive neurodegeneration was correlated to an increased Beclin 1 immunoreactivity contained in the majority of glial cells. Figure 5 shows that this association was enhanced in the reduced Beclin 1 gene expression in the GMR-GAL4, UAS-Tau/UAS-Beclin 1-RNAi retina, which was accompanied with densely packed glial cell Beclin 1 positive vesicles. This provides more direct genetic evidence that glial cell adaptation during Tau-induced neurodegeneration involves the loss of Beclin 1 in neurons, more sensitive to the loss of Beclin 1, and the subsequent rise of Beclin 1-positive vesicles in glial cells. After the Beclin 1 transgene was expressed, the phenotype of autophagy dysregulation was mitigated. Beclin 1 transgenic expression was associated with increased neuronal Beclin 1 expression, but did not lead to the formation of Beclin 1 positive vesicles in hexagonal glial cells in GMR-GAL4, UAS-Tau/+; UAS-Beclin 1/+ retina (Figure 5). This indicates that neuronal Beclin 1 appears to have a crucial function in preventing the buildup of p-Tau.
3.6. The Association between Buildup of Phosphorylated Tau and Age-Dependent Neurodegeneration is Influenced by the Absence of VPS34
Given that Beclin-dependent autophagic process relies on VPS34 lipid signal for transporting vesicles, we investigated whether VPS34 plays a role in controlling the accumulation of p-Tau following the loss of Beclin 1. In that case, the absence of VPS34 would lead to a comparable phenotype. In order to assess this, we conducted a thorough examination of the impact of reducing the expression of the VPS34 gene. We next analyzed the autophagy activity induced by Tau and the levels of p-Tau which may potentially be altered due to the absence of VPS34. The relationship between p-Tau accumulation and age-dependent neurodegeneration was found to be influenced by the loss of VPS34, as anticipated. Phosphorylated-Tau was shown to accumulate in both neurons and glial cells of young GMR-GAL4, UAS-Tau/+; UAS-VPS34-RNAi/+ retina, but no accumulation of p-Tau was observed in the young control retina (Figure 6). Consistent with this, p-Tau buildup in neurons was observed multiple times utilizing various UAS-VPS34-RNAi transgene (not shown).These data indicate that the decrease in p-Tau level in neurons is dependent on the presence of both Beclin and VPS34. The association of age-dependent p-Tau accumulation and neurodegeneration were found to be increased by the absence of Beclin 1. On the other hand, the loss of VPS34 from GMR-GAL4, UAS-Tau/+; UAS-VPS34-RNAi/+ retina, had a significant impact to modify the relationship between p-Tau buildup and age-dependent neurodegeneration, compared to control retina without VPS34 knockdown (Figure 6). Surprisingly, the connection between the buildup of p-Tau in neurons and neurodegeneration was influenced in distinct ways by the absence of VPS34, resulting in a partial correlation. Remarkably, while p-Tau exhibited significant accumulation in neurons when VPS34 gene expression was reduced, it did not lead to an increase in Tau aggregates. This is in contrast to the distinct phenotype of Tau aggregates in young neurons when Beclin 1 gene expression was reduced (Figure 4 and Figure 6). These findings suggest that the effects of p-Tau accumulation on neurons vary depending on the specific disease conditions, such as the loss of Beclin 1 and the loss of VPS34.
4. Discussion
Our investigations on genetic reversal of human Tau-induced neurodegeneration offer valuable insights into the intricate molecular mechanisms behind the failure of autophagy, which is a crucial factor in the neurodegeneration triggered by Tau in Drosophila. The common characteristic observed in both human AD or FTD patients and animal models is the malfunction of autophagy and the presence of autophagy-related vesicle pathology. The connection between neurodegenerative illnesses and impairment of autophagy, which leads to mitochondrial stress response and protein pathology, has been established indirectly. Specific autophagic abnormalities have been found to be linked to different states of neurodegeneration in the primary neurodegenerative disorders. Our findings suggest that an excessive increase in autophagy, through the upregulation of ATG14L, does not have a beneficial effect on the neuronal protein quality control mechanism, leading to neurodegeneration caused by human Tau accumulation in neurons. This is likely owing to the loss of neuronal Beclin 1 and the disruption of Beclin 1 inhibitory regulation of autophagy and apoptosis. Diminishing the expression of the ATG14L gene in Drosophila resulted in a reduction of neurotoxicity caused by human Tau. By targeting the expression of the Beclin 1 transgen, it is possible to safeguard neurons against the accumulation of p-Tau and decrease the connection between autophagy and death. To summarize, we have identified that targeting the expression of Beclin 1 transgene or reducing the expression of ATG14L gene are two potential therapeutic strategies that might specifically mitigate the disruption of autophagy regulation and neurotoxicity caused by Tau.
The molecular mechanism responsible for autophagic failure linked with Tau pathology is not fully understood, despite substantial studies on autophagic failure in various neurodegenerative illnesses [58]. Indeed, the failure of autophagy produced by Tau could result in a complicated and different disease conditions connected to autophagy. This pathology involves various components involved in vesicle trafficking including autophagy components as VPS30/ATG6 and VPS34 [59]. The regulation of autophagy gene expression and protein activities indicates that VPS30/ATG6 and VPS34 are necessary for neurons to decrease the buildup of p-Tau. Nevertheless, the impact of suppressing ATG14 is different from the impact of suppressing VPS30 and VPS34, indicating that the intricate coordination of Beclin 1 network is essential. Alternatively, the decrease in p-Tau levels may be influenced by an additional non-autophagic pathway, such as endosomal pathways. Moreover, there is a distinction between Beclin 1/VPS30 complex I, and complex II or III in terms of their divergence between the autophagic and endocytic trafficking routes. This divergence has been extensively studies and is known to lead to a greater affinity to ATG14L in the autophagic pathway, compared to the endosome UVRAG-containing Beclin 1 complex II pathway. Autophagy, namely in the formation of the
ATG14L-VPS30/Beclin 1-VPS15-VPS34 complex I, may be connected to the endocytic pathway through the sharing of similar molecular machinery involved in vesicular nucleation and lysosome fusion processes [60]. The upregulation of ATG14L-Beclin 1-VPS34 complex I indicates autophagic failure. This suggests that the beneficial effects of reducing ATG14 gene expression may be due to the suppression of excessive failure of autophagic activity and the restoration of Beclin 1 network inhibitory regulation of autophagy and apoptosis [61]. To test this possibility, it will be necessary to identify the exact pathways that are dependent on Beclin 1 or independent of Beclin 1 for VPS34 [62,63]. Currently, we can utilize fly genetics to investigate potential autophagy-related pathways involved in the development of Tau -induced neurodegeneration. Specifically, we can examine the impact of decreased levels of ATG14L, Beclin 1 and VPS34, on the progression of Tau-induced neurodegeneration.
5. Conclusions
Our experimental findings demonstrate that the buildup of p-Tau neurons is associated with a hierarchical system including the regulation of autophagy nucleation components ATG14, Beclin 1, and VPS34. This regulation is driven by expression of human Tau and leads to neurodegeneration. While it may be challenging to point to the exact defect in a particular autophagic mechanism, our findings indicate that the absence of Beclin 1 plays a role in the age-related buildup of p-Tau in neurons and upregulated autophagy activity which leads to subsequent neurodegeneration. However, our data indicate that autophagic failure occurs due to a process in which neurons increase autophagy, or vice versa, leading to autophagic failure, the formation of Tau aggregates, and neurodegeneration. This model elucidates the dispensability of inhibitory regulation of autophagy in degenerative neurons. It unveils novel molecular pathways behind Tau aggregation and neurodegeneration, which may have significant implications for the development of autophagy-targeted treatment strategy.
Author Contributions
CL.C. and HY.C. carried the reverse genetic studies and confocal imaging of retina; YH.T., and YR.L. acquired confocal imaging data and SEM data; H.C. and TK.S. analyzed data; HY.C. and TK.S. supervise the studies. All authors contributed to the manuscript in writing.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Raw data are available on request in this study from tauthors.
Acknowledgments
We express our gratitude to the members of the Chang laboratory and our colleagues for their invaluable discussions. We thank the support from the Imaging Core of the Brain Research Center. This work received partial funding from National Science and Technology Council of Taiwan. Yi-Han Tu received financial support from a NTHU scholarship specifically for young scientists.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes: Chia-Lung Chuang’s present address is St. Jude Children’s Research Hospital. Department of Developmental Neurobiology, 262 Danny Thomas Place, DNB, D2038C, MS324, Memphis, TN 38105, USA
Abbreviations
| MAPT | microtubule-associated protein tau |
| LC3 | microtubule-associated protein 1 light chain 3 |
| VPS15 | Serine/threonine-protein kinase VPS15 |
| VPS34 | vacuolar protein sorting 34 |
| Barkor | Beclin-1 associated autophagy-related key regulator |
| AD | Alzheimer’s disease |
| FTD | frontotemporal dementia |
References
- Cleveland, D.W., S. Y. Hwo, and M.W. Kirschner, Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol, 1977, 116, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Kadavath, H. , et al., Folding of the Tau Protein on Microtubules. Angew Chem Int Ed Engl, 2015, 54, 10347–10351. [Google Scholar] [CrossRef] [PubMed]
- Braak, H. , et al., Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol, 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M., R. A. Crowther, and M.G. Spillantini, Tau mutations cause frontotemporal dementias. Neuron, 1998, 21, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M. Goedert, and J.Q. Trojanowski, Neurodegenerative tauopathies. Annu Rev Neurosci, 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K. , et al., Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res, 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.R. , et al., Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron, 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Mazanetz, M.P. and P.M. Fischer, Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov, 2007, 6, 464–479. [Google Scholar] [CrossRef]
- Chang, H.Y., T. K. Sang, and A.S. Chiang, Untangling the Tauopathy for Alzheimer’s disease and parkinsonism. J Biomed Sci, 2018, 25, 54. [Google Scholar] [CrossRef]
- Chi, H. Y. Chang, and T.K. Sang, Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int J Mol Sci, 2018, 19. [Google Scholar] [CrossRef]
- Blennow, K. , et al., Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol, 1995, 26, 231–245. [Google Scholar] [CrossRef]
- Buerger, K. , et al., CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain, 2006, 129 Pt 11, 3035–3041. [Google Scholar] [CrossRef]
- Whiteman, I.T. , et al., Rapid changes in phospho-MAP/tau epitopes during neuronal stress: cofilin-actin rods primarily recruit microtubule binding domain epitopes. PLoS One, 2011, 6, e20878. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. and D.S. Yang, Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol Dis, 2011, 43, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M., S. W. Ryter, and B. Levine, Autophagy in human health and disease. N Engl J Med, 2013, 368, 1845–1846. [Google Scholar] [CrossRef]
- Nixon, R.A. , The role of autophagy in neurodegenerative disease. Nat Med, 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Aman, Y. , et al., Autophagy in healthy aging and disease. Nat Aging, 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Mizushima, N. and M. Komatsu, Autophagy: renovation of cells and tissues. Cell, 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Parzych, K.R. and D.J. Klionsky, An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal, 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Glick, D., S. Barth, and K.F. Macleod, Autophagy: cellular and molecular mechanisms. J Pathol, 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Yang, Z. and D.J. Klionsky, An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol, 2009, 335, 1–32. [Google Scholar] [PubMed]
- Ohsumi, Y. , Historical landmarks of autophagy research. Cell Res, 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L. , et al., Molecular definitions of autophagy and related processes. EMBO J, 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. , Autophagy revisited: a conversation with Christian de Duve. Autophagy, 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M. , et al., Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron, 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K. , et al., Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells, 2007, 12, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. , Autophagy: process and function. Genes Dev, 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Itakura, E. and N. Mizushima, Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy, 2010, 6, 764–776. [Google Scholar] [CrossRef]
- Rubinsztein, D.C., T. Shpilka, and Z. Elazar, Mechanisms of autophagosome biogenesis. Curr Biol, 2012, 22, R29–R34. [Google Scholar] [CrossRef]
- Suzuki, K. , et al., The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J, 2001, 20, 5971–5981. [Google Scholar] [CrossRef]
- Marquez, R.T. and L. Xu, Bcl-2:Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res, 2012, 2, 214–221. [Google Scholar]
- Kang, R. , et al., The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ, 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F. , et al., Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature, 2018, 558, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, I.A. , et al., Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene, 2009, 28, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S. , et al., Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell, 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P., J. B. Parys, and G. Bultynck, Regulation of the autophagic bcl-2/beclin 1 interaction. Cells, 2012, 1, 284–312. [Google Scholar] [CrossRef] [PubMed]
- Diao, J. , et al., ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature, 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Obara, K. and Y. Ohsumi, Atg14: a key player in orchestrating autophagy. Int J Cell Biol 2011, 2011, 713435. [Google Scholar] [CrossRef] [PubMed]
- Fogel, A.I. , et al., Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol Cell Biol, 2013, 33, 3675–3688. [Google Scholar] [CrossRef]
- Hurley, J.H. and L.N. Young, Mechanisms of Autophagy Initiation. Annu Rev Biochem 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Funderburk, S.F., Q. J. Wang, and Z. Yue, The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol, 2010, 20, 355–362. [Google Scholar] [CrossRef]
- Wang, C.W. and D.J. Klionsky, The molecular mechanism of autophagy. Mol Med, 2003, 9, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C., P. Codogno, and E. Morel, Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J, 2017, 284, 1267–1278. [Google Scholar] [CrossRef]
- Stavoe, A.K.H. and E.L.F. Holzbaur, Autophagy in Neurons. Annu Rev Cell Dev Biol 2019, 35, 447–500. [Google Scholar] [CrossRef]
- Wong, E. and A.M. Cuervo, Autophagy gone awry in neurodegenerative diseases. Nat Neurosci, 2010, 13, 805–811. [Google Scholar] [CrossRef]
- Nixon, R.A. , et al., Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol, 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Salminen, A. , et al., Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog Neurobiol 2013, 106-107, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F. , et al., The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest, 2008, 118, 2190–2199. [Google Scholar] [PubMed]
- Jaeger, P.A. and T. Wyss-Coray, Beclin 1 complex in autophagy and Alzheimer disease. Arch Neurol, 2010, 67, 1181–1184. [Google Scholar] [CrossRef]
- Wu, T.H. , et al., Loss of vesicular dopamine release precedes tauopathy in degenerative dopaminergic neurons in a Drosophila model expressing human tau. Acta Neuropathol, 2013, 125, 711–725. [Google Scholar] [CrossRef]
- Chang, H.Y. and D.F. Ready, Rescue of photoreceptor degeneration in rhodopsin-null Drosophila mutants by activated Rac1. Science, 2000, 290, 1978–1980. [Google Scholar] [CrossRef]
- Sang, T.K. and D.F. Ready, Eyes closed, a Drosophila p47 homolog, is essential for photoreceptor morphogenesis. Development, 2002, 129, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.L. , et al., Genetic dissection reveals that Akt is the critical kinase downstream of LRRK2 to phosphorylate and inhibit FOXO1, and promotes neuron survival. Hum Mol Genet, 2014, 23, 5649–5658. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M. and A. Thorburn, Sorting cells for basal and induced autophagic flux by quantitative ratiometric flow cytometry. Autophagy, 2014, 10, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q. , et al., Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A, 2008, 105, 19211–19216. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S. and B. Levine, The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene 2008, 27 Suppl 1, S137–S148. [Google Scholar] [CrossRef] [PubMed]
- McKnight, N.C. , et al., Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet, 2014, 10, e1004626. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M. Di Domenico, and D.A. Butterfield, mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis 2015, 84, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A. and S. Gandy, Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to pathogenesis. Neuron, 2006, 52, 15–31. [Google Scholar] [CrossRef]
- Zhao, Y.G., P. Codogno, and H. Zhang, Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol, 2021, 22, 733–750. [Google Scholar] [CrossRef]
- Xiong, X. , et al., The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem, 2012, 287, 39107–39114. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y., S. Sinha, and B. Levine, Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy, 2008, 4, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K. , et al., Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol, 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The control of impaired autophagy and the development of neurodegeneration caused by human Tau in Drosophila. (A) Scanning electron microscope images of Drosophila compound eyes are shown. The left SEM image was of wild-type Canton S (left); whereas the right SEM image was a transgenic eye that carries a human Tau transgene driven by an eye-specific promoter, w; Gmr-Gal4 UAS-Tau/+. Anterior-posterior axis: left to right. (B) Confocal images of ommatidia (horizontal section) double-stained using antibodies targeting phosphorylated Tau and LC3, along with phalloidin labelling F-actin. The F-actin was stained with rhodamine-conjugated phalloidin, appearing as magenta. The anti-phosphorylated Tau (AT8) was labeled with Alexa Fluor 488-conjugated anti-mouse IgG, appearing as green. The sample was stained with a red fluorescent dye, specifically anti-LC3 labeled with Cy3-conjugated anti-rabbit IgG. Genotypes identical to A. (C) The confocal images show a GFP-mCherry-Atg8a reporter in the control retina of w; Gmr-Gal4/ UAS-GFP-mCherry-Atg8 (left), and the Tau-induced phenotype of GFP-mCherry-Atg8a in the retina of w; Gmr-Gal4, gl-Tau/ UAS-GFP-mCherry-Atg8 (right). The blue color represents the presence of anti-phospho-tau, and Cy5-conjugated anti-mouse IgG. (D) The confocal images show a GFP-mCherry-Atg8a reporter in the retina of young control, w; Gmr-Gal4/ UAS-GFP-mCherry-Atg8 (left), as well as in the retina with Tau-induced phenotype at a young age (middle) and at an elderly age (right) from w; Gmr-Gal4, UAS-Tau/ UAS-GFP-mCherry-Atg8 flies. Magenta: F-actin stained with rhodamine-conjugated phalloidin. A: scale bar, 100 µm; B-D: scale bars, 10 µm.
Figure 1.
The control of impaired autophagy and the development of neurodegeneration caused by human Tau in Drosophila. (A) Scanning electron microscope images of Drosophila compound eyes are shown. The left SEM image was of wild-type Canton S (left); whereas the right SEM image was a transgenic eye that carries a human Tau transgene driven by an eye-specific promoter, w; Gmr-Gal4 UAS-Tau/+. Anterior-posterior axis: left to right. (B) Confocal images of ommatidia (horizontal section) double-stained using antibodies targeting phosphorylated Tau and LC3, along with phalloidin labelling F-actin. The F-actin was stained with rhodamine-conjugated phalloidin, appearing as magenta. The anti-phosphorylated Tau (AT8) was labeled with Alexa Fluor 488-conjugated anti-mouse IgG, appearing as green. The sample was stained with a red fluorescent dye, specifically anti-LC3 labeled with Cy3-conjugated anti-rabbit IgG. Genotypes identical to A. (C) The confocal images show a GFP-mCherry-Atg8a reporter in the control retina of w; Gmr-Gal4/ UAS-GFP-mCherry-Atg8 (left), and the Tau-induced phenotype of GFP-mCherry-Atg8a in the retina of w; Gmr-Gal4, gl-Tau/ UAS-GFP-mCherry-Atg8 (right). The blue color represents the presence of anti-phospho-tau, and Cy5-conjugated anti-mouse IgG. (D) The confocal images show a GFP-mCherry-Atg8a reporter in the retina of young control, w; Gmr-Gal4/ UAS-GFP-mCherry-Atg8 (left), as well as in the retina with Tau-induced phenotype at a young age (middle) and at an elderly age (right) from w; Gmr-Gal4, UAS-Tau/ UAS-GFP-mCherry-Atg8 flies. Magenta: F-actin stained with rhodamine-conjugated phalloidin. A: scale bar, 100 µm; B-D: scale bars, 10 µm.
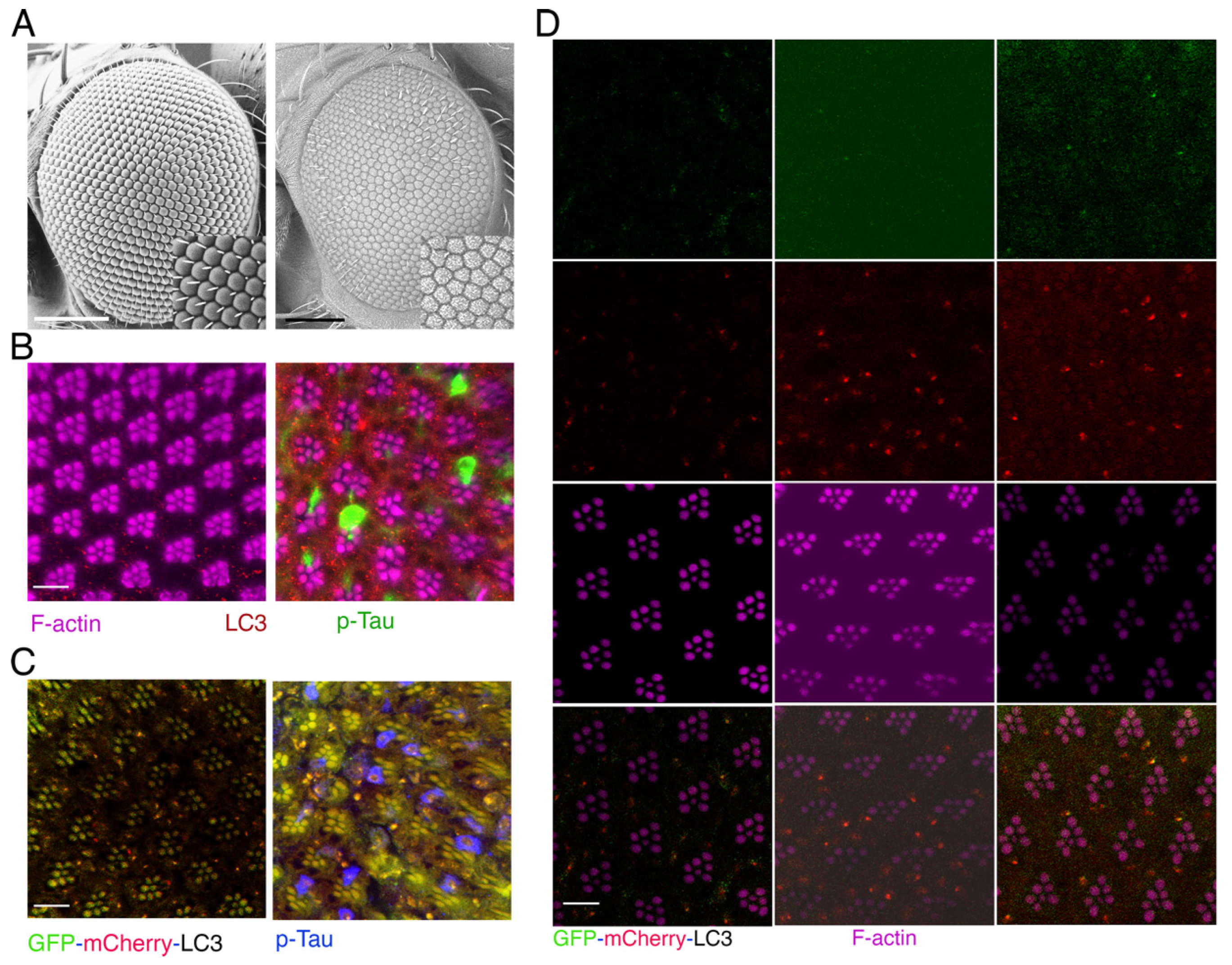
Figure 2.
The neurodegenerative process caused by Tau is improved when the expression of ATG14 gene is decreased. (A) Scanning electron microscopes were used to examine the Drosophila compound eyes with the following genotypes: w; Gmr-Gal4, gl-Tau/+ (first), w; Gmr-Gal4, gl-Tau/+; UAS-ATG14-RANi/+ (second), w; Gmr-Gal4, UAS-Tau/+; UAS-LacZ/+ (third), w; Gmr-Gal4, UAS-Tau/+; UAS-ATG14-RNAi/+ (fourth). (B) Confocal images of ommatidia (horizontal section) double-stained with antibodies targeting phosphorylated Tau and LC3, along with phalloidin labelling F-actin. Magenta: F-actin stained rhodamine-conjugated phalloidin; Green: anti-phosphorylated Tau antibody (AT8) labeled with Alexa Fluor 488-conjugated anti-mouse IgG; Red: anti-LC3 antibody labeled with Cy3-conjugated anti-rabbit IgG. The genotypes are as follows: The upper panels show the wild-type Canton S strain. The middle panels show the w; Gmr-Gal4, UAS-Tau/+; UAS-LacZ/+ strain. The bottom panels show the w; Gmr-Gal4, UAS-Tau/+; UAS-ATG14-RNAi/+ strain. A: the scale bars, 100 µm; B: the scale bars, 10 µm.
Figure 2.
The neurodegenerative process caused by Tau is improved when the expression of ATG14 gene is decreased. (A) Scanning electron microscopes were used to examine the Drosophila compound eyes with the following genotypes: w; Gmr-Gal4, gl-Tau/+ (first), w; Gmr-Gal4, gl-Tau/+; UAS-ATG14-RANi/+ (second), w; Gmr-Gal4, UAS-Tau/+; UAS-LacZ/+ (third), w; Gmr-Gal4, UAS-Tau/+; UAS-ATG14-RNAi/+ (fourth). (B) Confocal images of ommatidia (horizontal section) double-stained with antibodies targeting phosphorylated Tau and LC3, along with phalloidin labelling F-actin. Magenta: F-actin stained rhodamine-conjugated phalloidin; Green: anti-phosphorylated Tau antibody (AT8) labeled with Alexa Fluor 488-conjugated anti-mouse IgG; Red: anti-LC3 antibody labeled with Cy3-conjugated anti-rabbit IgG. The genotypes are as follows: The upper panels show the wild-type Canton S strain. The middle panels show the w; Gmr-Gal4, UAS-Tau/+; UAS-LacZ/+ strain. The bottom panels show the w; Gmr-Gal4, UAS-Tau/+; UAS-ATG14-RNAi/+ strain. A: the scale bars, 100 µm; B: the scale bars, 10 µm.
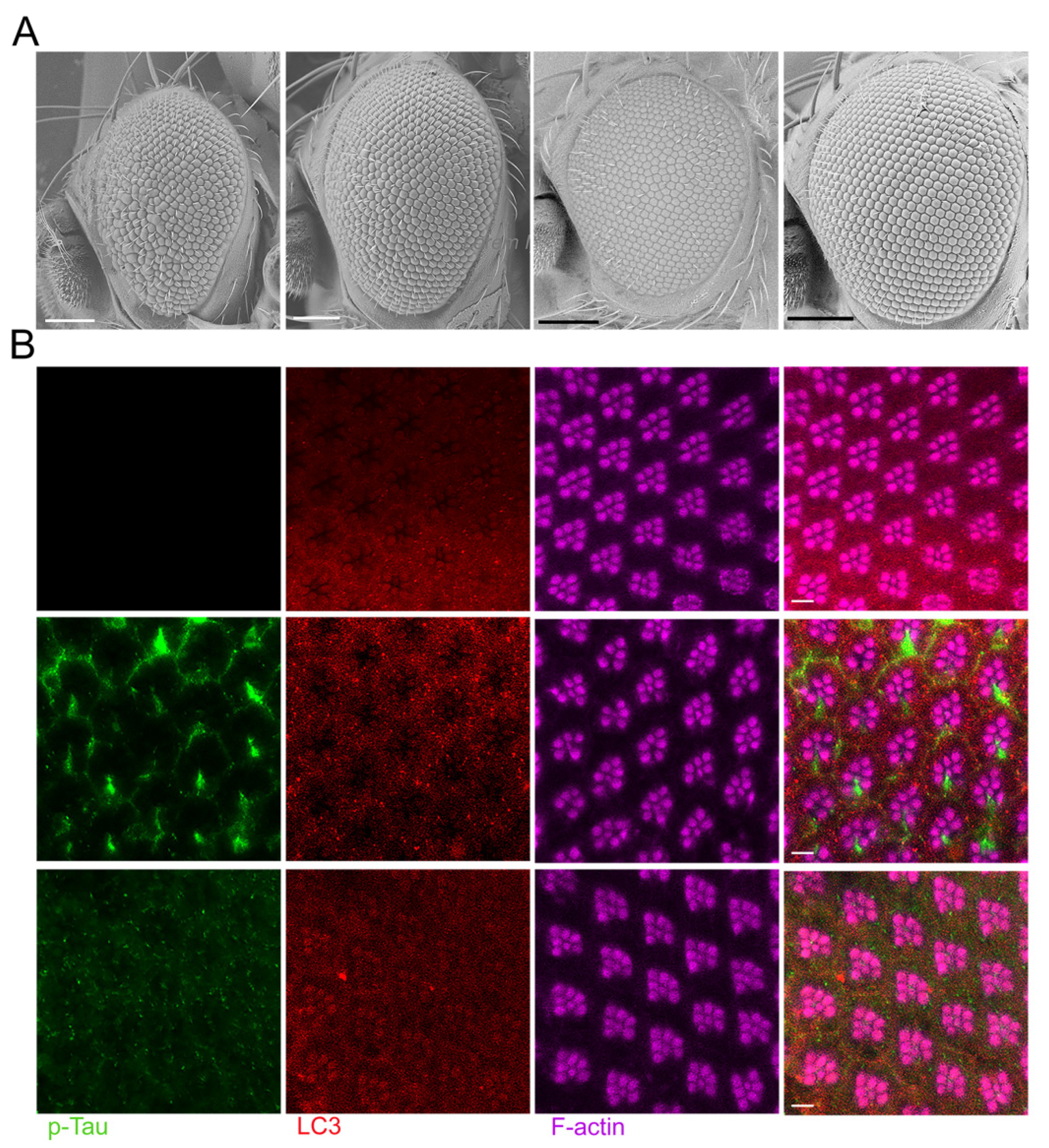
Figure 3.
The expression of ATG6/Beclin 1 in Drosophila helps to safeguard neurons against age-related neurodegeneration linked with p-Tau. (A) At three distinct ages, confocal images of ommatidia (horizontal section) showing phalloidin-tagged-F-actin. Retinas of flies that were 1, 10, and 20 days old were studied. Retinas from flies with the following genotypes were displayed in the figure: upper panels: w; Gmr-Gal4, UAS-Tau/+; UAS-LacZ/+, and lower panels: w; Gmr-Gal4, UAS-Tau/+; UAS-ATG6/+. (B) Confocal images of ommatidia (horizontal section) labeled with an antibody specific to phosphorylated Tau along with phalloidin labeled F-actin. Magenta: rhodamine-conjugated phalloidin stained F-actin; Green: anti-phosphorylated Tau (AT8) that is associated with Alexa Fluor 488-conjugated anti-mouse IgG. Top and bottom panels show genotypes that are the same as in A for. A: the scale bars, 5 µm; B: the scale bars, 10 µm.
Figure 3.
The expression of ATG6/Beclin 1 in Drosophila helps to safeguard neurons against age-related neurodegeneration linked with p-Tau. (A) At three distinct ages, confocal images of ommatidia (horizontal section) showing phalloidin-tagged-F-actin. Retinas of flies that were 1, 10, and 20 days old were studied. Retinas from flies with the following genotypes were displayed in the figure: upper panels: w; Gmr-Gal4, UAS-Tau/+; UAS-LacZ/+, and lower panels: w; Gmr-Gal4, UAS-Tau/+; UAS-ATG6/+. (B) Confocal images of ommatidia (horizontal section) labeled with an antibody specific to phosphorylated Tau along with phalloidin labeled F-actin. Magenta: rhodamine-conjugated phalloidin stained F-actin; Green: anti-phosphorylated Tau (AT8) that is associated with Alexa Fluor 488-conjugated anti-mouse IgG. Top and bottom panels show genotypes that are the same as in A for. A: the scale bars, 5 µm; B: the scale bars, 10 µm.
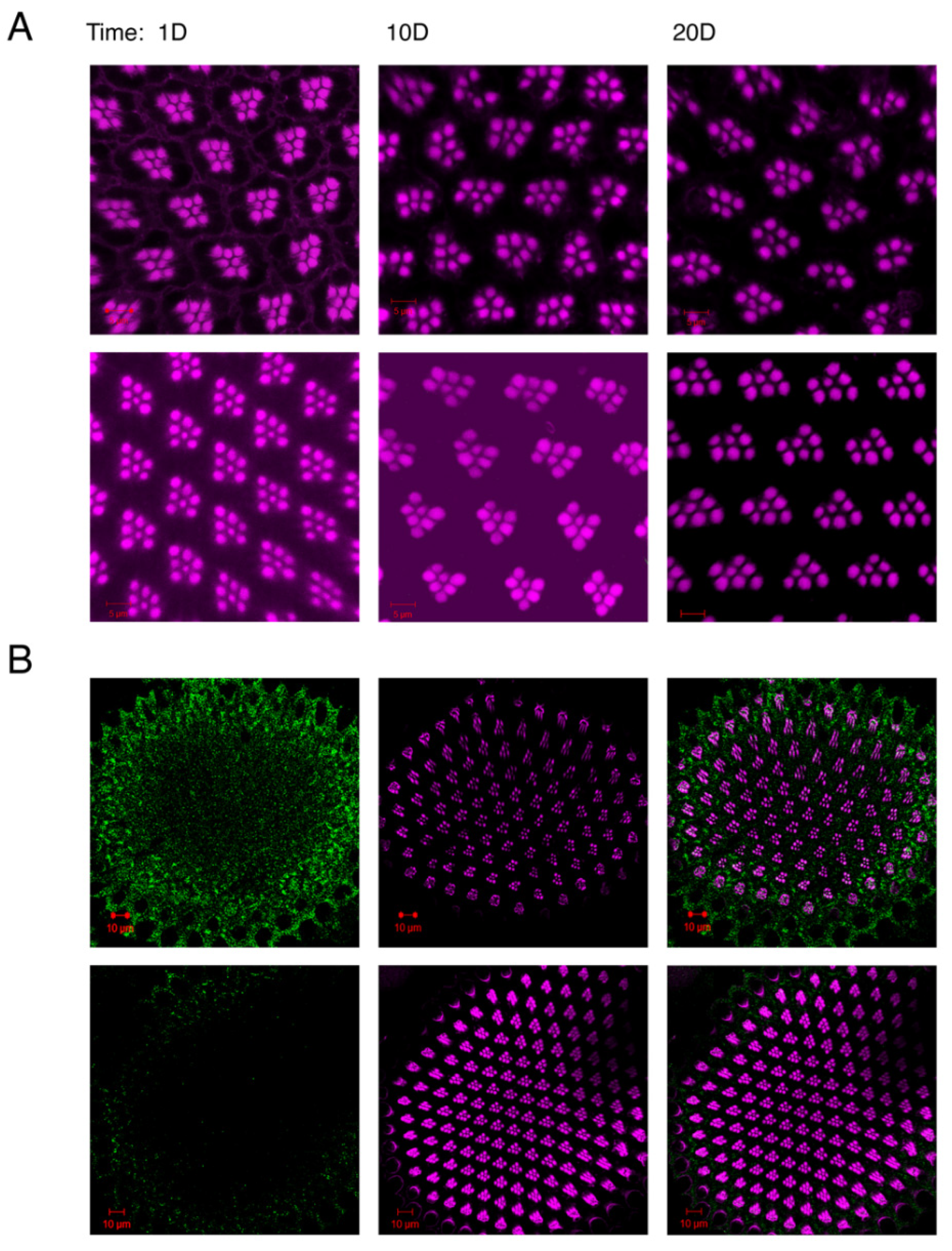
Figure 4.
Loss of ATG6/Beclin 1 in Drosophila enhances both phosphorylated p-Tau in neurons and p-Tau associated neurodegeneration. The confocal images show the retina of w; Gmr-Gal4, UAS-Tau/+; UAS-LacZ/+, and a retina of w; Gmr-Gal4, UAS-Tau/UAS-ATG6-RNAi. The images were processed in a horizontal section and were stained with an antibody against phosphorylated Tau and phalloidin labeled F-actin. Magenta: rhodamine-conjugated phalloidin stained F-actin; Green: anti-phosphorylated Tau (AT8) with Alexa Fluor 488-conjugated anti-mouse IgG. The scale bars, 5 µm.
Figure 4.
Loss of ATG6/Beclin 1 in Drosophila enhances both phosphorylated p-Tau in neurons and p-Tau associated neurodegeneration. The confocal images show the retina of w; Gmr-Gal4, UAS-Tau/+; UAS-LacZ/+, and a retina of w; Gmr-Gal4, UAS-Tau/UAS-ATG6-RNAi. The images were processed in a horizontal section and were stained with an antibody against phosphorylated Tau and phalloidin labeled F-actin. Magenta: rhodamine-conjugated phalloidin stained F-actin; Green: anti-phosphorylated Tau (AT8) with Alexa Fluor 488-conjugated anti-mouse IgG. The scale bars, 5 µm.
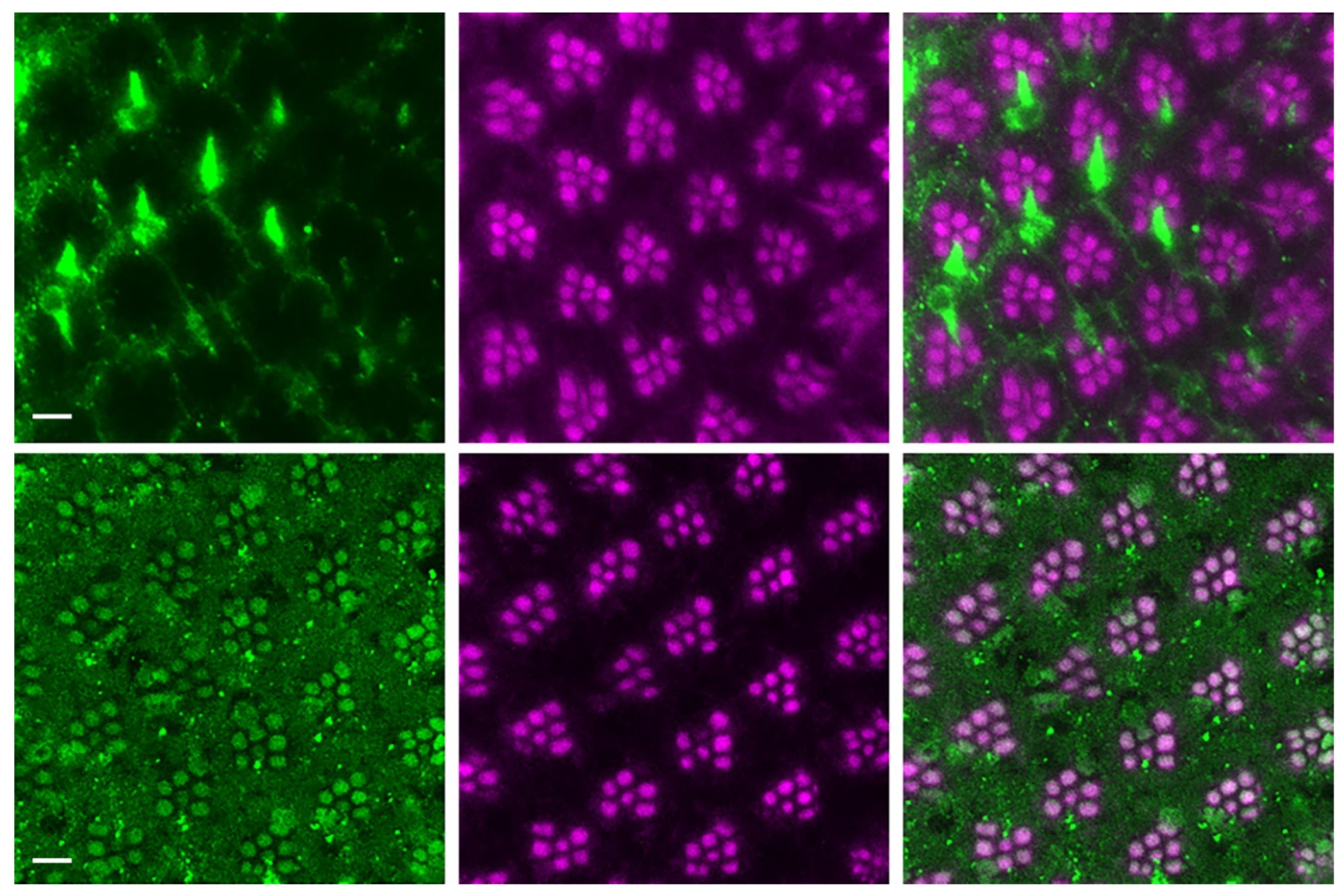
Figure 5.
Knocking down the ATG6 gene expression in Drosophila enhances the loss of neuronal ATG6 and elevation of glial cell ATG6 induced by human Tau; whereas, ATG6 transgenic expression restores the neuronalATG6 network and prevents Tau-induced neurodegeneration. Horizontal slice of confocal images of ommatidia stained with Beclin 1 antibody and phalloidin labeled F-actin for w; Gmr-Gal4, UAS-Tau/+; UAS-ATG6/+ (left), middle section of w; Gmr-Gal4, UAS-Tau/+ (middle), and right section of w; Gmr-Gal4, UAS-Tau/UAS-ATG6-RNAi retina (right). Green: anti-Beclin 1 antibody with Alexa Fluor 488-conjugated anti-mouse IgG; Magenta: rhodamine-conjugated phalloidin stained F-actin. The scale bars, 5 µm.
Figure 5.
Knocking down the ATG6 gene expression in Drosophila enhances the loss of neuronal ATG6 and elevation of glial cell ATG6 induced by human Tau; whereas, ATG6 transgenic expression restores the neuronalATG6 network and prevents Tau-induced neurodegeneration. Horizontal slice of confocal images of ommatidia stained with Beclin 1 antibody and phalloidin labeled F-actin for w; Gmr-Gal4, UAS-Tau/+; UAS-ATG6/+ (left), middle section of w; Gmr-Gal4, UAS-Tau/+ (middle), and right section of w; Gmr-Gal4, UAS-Tau/UAS-ATG6-RNAi retina (right). Green: anti-Beclin 1 antibody with Alexa Fluor 488-conjugated anti-mouse IgG; Magenta: rhodamine-conjugated phalloidin stained F-actin. The scale bars, 5 µm.
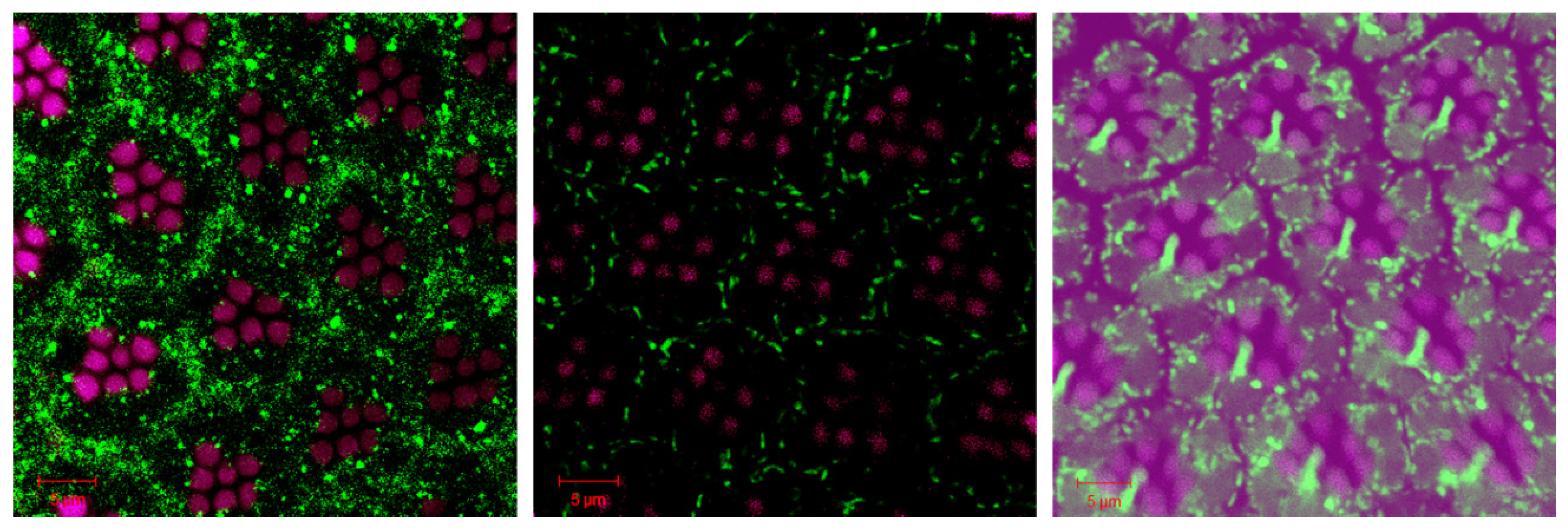
Figure 6.
Loss of VPS34 differently regulates neuronal phosphorylated Tau accumulation and p-Tau associated neurodegeneration. The confocal images show the retina of a retina of w; Gmr-Gal4, UAS-Tau/+; UAS-LacZ/+ (upper panels) and a retina of w; Gmr-Gal4, UAS-Tau/+;UAS-VPS34-RNAi/+ (lower panels) stained with an antibody against phosphorylated Tau and phalloidin labeled F-actin. Magenta: rhodamine-conjugated phalloidin stained F-actin; Green: anti-phosphorylated Tau (AT8) with Alexa Fluor 488-conjugated anti-mouse IgG. The scale bars, 5 µm.
Figure 6.
Loss of VPS34 differently regulates neuronal phosphorylated Tau accumulation and p-Tau associated neurodegeneration. The confocal images show the retina of a retina of w; Gmr-Gal4, UAS-Tau/+; UAS-LacZ/+ (upper panels) and a retina of w; Gmr-Gal4, UAS-Tau/+;UAS-VPS34-RNAi/+ (lower panels) stained with an antibody against phosphorylated Tau and phalloidin labeled F-actin. Magenta: rhodamine-conjugated phalloidin stained F-actin; Green: anti-phosphorylated Tau (AT8) with Alexa Fluor 488-conjugated anti-mouse IgG. The scale bars, 5 µm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



