Submitted:
10 February 2024
Posted:
12 February 2024
You are already at the latest version
Abstract
Proteolysis Targeting Chimeras (PROTACs) describe compounds that bind to and induce degradation of a target by simultaneously binding to a ubiquitin ligase. More generally referred to as bifunctional degraders, PROTACs have led the way in the field of targeted protein degradation (TPD) with several compounds currently undergoing clinical testing. Alongside bifunctional degraders, single-moiety compounds, or molecular glue degraders (MGDs), are increasingly being considered as a viable approach for development of therapeutics, driven by advances in rational discovery approaches. This review focusses on drug discovery with respect to bifunctional and molecular glue degraders within the ubiquitin proteasome system, including analysis of mechanistic concepts and discovery approaches, with an overview of current clinical and pre-clinical degrader status in oncology, neurodegenerative and inflammatory disease.
Keywords:
TPD
; PROTAC
; molecular glue degrader
; heterobifunctional degrader
; Ubiquitin
; proteasome
; targeted protein degradation
1.1. General Concepts in TPD Drug Discovery
The concept of pharmacologically modulating turnover of a specific target protein by leveraging the ubiquitin-proteasome system (UPS) was first realized by Ray Deshaies, Craig Crews and others in a landmark study from 2001 [1], where the authors coined the term Proteolysis Targeting Chimeras (PROTACs). PROTACs, or more formally, bifunctional degraders, are composed of two separate moieties, a ubiquitin ligase (also called E3) recruiter, and a target recruiter, with a linker connecting the two. In this way, bifunctional degraders can bind to the ligase and target at the same time, effectively putting the ligase into close proximity of the protein-of-interest (POI), leading to its ubiquitination and proteasomal proteolysis (Figure 1).
Molecular glue degraders (MGDs), in contrast, consist of a single moiety and have a conceptually distinct mode of generating the ternary complex. Whereas bifunctional degraders rely on two separate ligands for ligase and POI recruitment, and can independently and separately bind both, glue degraders must first bind to one of the proteins, which modulates the surface properties of the bound protein to induce binding to the second. Most reported MGDs are ligase ligands, where compound binding induces interaction with a neo-substrate, a protein that otherwise would not interact with or be degraded by the ligase. An MGD can also bind the target protein, with resulting induced binding to a ligase, as exemplified by degraders for Cyclin K [2,3,4,5] and BRD4 [6,7,8]. In addition to their therapeutic potential, degraders represent valuable tools for exploration of fundamental cell biology, where the functional consequences of acute protein ablation can provide novel insights into cell physiology and the intricate regulatory networks that govern cellular functions.
Due to being composed of two ligands and a linker, the molecular weight and physicochemical properties of bifunctional degraders are outside of the rule of five [9], which poses significant challenges in developing these compounds for an oral route of administration. In comparison, molecular glue degraders are generally compact, with a low molecular weight and the potential for favourable drug-like physiochemical properties, simplifying the late-stage optimization process. The challenge with MGDs lies largely in the identification of compounds, where the field is poised to encounter significant challenges for the rational identification of glues between a selected ligase-target pair.
Both MGDs and bifunctional degraders have event-driven pharmacology. In contrast, conventional small molecule inhibitors function by occupying a binding pocket on the target protein, for example an enzyme active site. Degraders circumvent this requirement by acting catalytically, where a single degrader molecule turns over multiple target protein molecules in a sub-stoichiometric mode [10]. This event-driven mechanism of action is dramatically different from occupancy-based inhibitors, where the catalytic property may allow lower or less frequent dosing.
In this context, it is worth noting that degraders may be developed as covalent compounds, and interestingly the first PROTAC was based on the covalent target ligand ovalicin [1]. Since the catalytic advantage is lost for degraders that covalently bind the target, covalent bifunctional degraders have largely been explored for ligands reacting with the ligase [11,12,13,14]. Importantly, covalent E3 recruiters cannot be inhibitory of ligase function since this would invalidate their utility in a TPD setting, and thus avoiding reaction with active site residues in ligases that have enzymatic activity is essential. The concept of target-covalent degraders has been explored for BTK, utilizing both reversible and irreversible covalent ibrutinib derivatives [15,16,17]. Since ibrutinib has high non-covalent binding affinity for BTK, covalent degraders based on this ligand will fundamentally be expected to reduce potency due to loss of catalytic function [16]. Similarly, degraders for KRASG12C have been generated from existing covalent ligands [18,19]. Although these examples are based on ligands that were developed as covalent inhibitors, the concept of covalency on the target side will apply equally to ligands developed specifically as binders for degrader purposes, where a covalent route has the potential advantage of identification of ligands for otherwise un-ligandable targets.
In contrast to inhibitors, the pharmacology of degraders can lead to a disconnect between pharmacokinetic (PK) and pharmacodynamic (PD) properties. This is, in part, related to the re-synthesis time of the target, where slowly re-synthesized proteins may remain absent for a significant time after the degrader has been cleared from circulation. Exemplifying a PK/PD disconnect, degraders for the protein RIPK2 show a prolonged functional response after dosing [20]. This is an advantage shared with covalent inhibitors, however degraders have the additional benefit of catalytic functionality.
A further advantage with degraders is that they are not required to bind to an active site in a target. This opens up the potential for developing therapies where the target active site is either un-ligandable or absent. In these situations, identification of a small molecule binder for the target protein is required, which may still pose a considerable challenge.
The parameters to assess in vitro potency of degraders in drug discovery approaches are principally DC50 and Dmax, which correspond to the compound concentration at half-maximal degradation and the maximum degradation, respectively. More comprehensive compound characterization at the screening stage may also include assessment of rate of degradation [21] and ternary complex formation (vide infra).
1.2. Ubiquitin ligases in TPD drug discovery
Ubiquitin ligases, or E3s, mediate the post-translation modification of substrates with the small protein ubiquitin. Iterative ubiquitination of ubiquitin itself leads to protein poly-ubiquitination, which mediates different downstream functions depending on the ubiquitin chain architecture; largely, lysine 48 poly-ubiquitinated proteins are degraded by the 26S proteasome [22].
Out of the approximately 600 ubiquitin ligases in the human proteome, only a small number have currently been explored in TPD approaches, principally due to the paucity of existing E3 ligands. The most utilized ligases are CRBN and VHL, where bifunctional degraders based on both have entered the clinic. Other E3s that have been demonstrated to function in a degrader approach include MDM2, IAPs, KEAP1, DCAF11, DCAF15, DCAF16, RNF4, RNF114, Ahr, FEM1B, DCAF1 and KLHDC2 [23,24,25,26,27].
An interesting concept is the identification of essential ligases, which may reduce the occurrence of resistance mechanisms due to loss of ligase expression or mutations that reduce ligase activity. The DCAF1 ligase is reported to be essential and has recently been demonstrated to be active in the context of bifunctional degraders [27].
Utilizing ligases that are critical or upregulated in a certain disease condition may allow for effective target degradation primarily in affected tissues, for example in oncology [28]. To enhance tumour specificity, bifunctional degraders have been coupled with antibodies recognising specific tumour types. Antibody–drug conjugates (ADCs) are an interesting approach to reduce side effects when using degraders that recruit ubiquitously expressed ligases [29].
Similarly, taking advantage of differential tissue expression of E3s may allow preferential degradation in the tissue that is relevant to the disease, and an early example of this is the BCL-XL degrader DT2216, which has reduced toxicity due to low VHL expression in platelets [30].
Important aspects to consider when developing a degrader approach is the tissue expression profile and subcellular localization of the E3. For a degrader to be successful, the target and ligase are required to be present in the same cellular compartment, and for clinical application, the ligase must be expressed, and active, in the tissue that is relevant for the indication. VHL and CRBN are widely expressed and have been reported to degrade both nuclear and cytosolic targets [31]. Other ligases may have activity that is restricted to a certain compartment in the cell, such as DCAF16, reported to be limited to the nucleus [32]. Plasma membrane bound ubiquitin ligases, such as RNF43, have also been shown to be possible to hijack for TPD purposes, which opens up an interesting prospect of targeting transmembrane proteins for degradation via the use of degraders that act extracellularly [33,34].
While the vast majority of characterized ubiquitin ligases transfer ubiquitin to lysine residues, E3s that act on Serine and Threonine have been described and could in principle be used in TPD approaches [35], which may open up for degrading proteins recalcitrant to ubiquitylation on lysines.
Key to the use of novel ligases for bifunctional degraders is the identification of E3 ligands with attractive physicochemical properties, such as a low molecular weight and a low number of hydrogen bond donors [36].
1.3. Ternary Complex Assessment for Compound Development
The activity of both bifunctional and molecular glue degraders is reliant on the formation of a productive ternary complex, where the compound is bridging the ubiquitin ligase with the target protein in a spatial orientation that is compatible with transfer of ubiquitin to lysines in the target protein. It is worth noting that formation of a ternary complex does not necessarily translate into ubiquitination and degradation [37,38] as the target protein, while recruited successfully to the ligase, may not be positioned to allow ubiquitin transfer to lysines.
Ternary complex assessment can be performed both in cell-based assays or using purified proteins and can provide both qualitative and quantitative information that can aid compound development. An emerging key parameter of ternary complex formation is cooperativity. Cooperativity, denoted as α, measures how the affinity of the degrader for the ligase is affected by simultaneous binding to the target, or vice versa, and is defined as the binary affinity divided by the ternary affinity [39] (Figure 1). Cooperativity can be positive, non-cooperative, or negative. Positively cooperative degraders bind with higher affinity to the ligase in presence of the target protein and α is > 1. Conversely, if presence of the target protein decreases the binding affinity to the ligase, α is < 1, and the degrader exhibits negative cooperativity. Compounds that are not affected in either direction, with α = 1, are termed non-cooperative. Molecular glues typically have high positive cooperativity with α > 1, while bifunctional compounds range from negatively to positively cooperative.
Positive cooperativity suggests that the ternary complex is stabilized by energetically favourable de novo interactions between the ligase and the target protein [40] but may also derive from more unexpected sources, as recently exemplified by Geiger et al. [41]. Here, the authors describe a potent VHL-based degrader for FKBP51, where the VHL and FKBP51 ligands act in a molecular glue-like manner with their respective non-partner protein. In a similar manner, linkers have been shown to contribute to ternary complex formation [42,43], and optimization of linker-protein interactions can enhance cooperativity, as shown for SMARCA2 degraders [42].
Negative cooperativity may be due to steric clashes that are counterproductive to complex formation, or to other properties of the compounds that shift the equilibrium towards binary complexes. For example, an FKBP12 degrader with negative cooperativity was recently described [41]. This compound (6a2) showed high binary affinity for FKBP12 and VHL respectively, however bound dramatically less well to VHL in the presence of FKBP12. Structural studies revealed that the compound collapses onto itself, forming intramolecular contacts in a horse-shoe shape while bound to FKBP12. In this binary complex, the VHL ligand forms contacts with the FKBP12 protein that likely further stabilize the folded conformation, thus preventing the formation of a ternary complex with VHL.
Importantly, cooperativity has been reported to correlate with increased potency of degradation [39], and a recent study by from Amgen [44] has illustrated that both ternary complex affinity and cooperativity contribute to the potency of bifunctional degraders. This study assessed VHL based degraders for SMARCA2 and BRD4 using surface plasmon resonance, and the results highlight that cooperativity is highly correlated to potency of degradation. In contrast, ternary complex half-life, i.e., the stability of the induced complex between the ligase and target protein, showed target-dependent correlation with potency of degradation. While BRD4 degraders with more stable complexes were more effective degraders, this correlation was weak for SMARCA2 degraders, highlighting the challenges in building predictive models for bifunctional degraders.
Of note, bifunctional degraders that are non-cooperative or have negative cooperativity may still be highly potent [45,46], calling into question the value of cooperativity as a key parameter in early degrader discovery. However, highly cooperative lead compounds may be advantageous starting points, as they likely will be more permissive to structural changes that optimize DMPK and physicochemical properties at the cost of reduced ligand affinities. As such, a compound series with strong cooperativity may make room for the medicinal chemist to balance potency with properties that progress the compounds towards the clinic.
A well-described property of bifunctional degraders is their capacity to form binary complexes with the target protein and the ligase at saturating concentrations, leading to a decrease in activity – this generates the characteristic “hook” appearance in a concentration-response curve (Figure 1). Note that the hook-effect is not observed for MGDs, since these compounds do not bind to the target and ligase individually. Highly cooperative bifunctional degraders have lower propensity to form binary complexes and a reduced tendency for hook effect at high compound concentrations, which is an advantage for clinical development.
2.1. Directly recruiting E3s for TPD
Engaging E3 ligases is a critical step in degraders’ mode of action. Although three groups are now recognized, E3 Ubiquitin ligases were classically divided into two distinct classes based on their conserved hallmark catalytic domain and ubiquitin transfer mechanism: really interesting new gene-type E3s (RINGs) and homologous to the E6AP carboxyl terminus-type E3s (HECTs) [47]
While RING family ligases catalyze a direct ubiquitin transfer from the E2-ubiquitin complex to a substrate protein [48], HECT E3s initially transfer ubiquitin to an internal catalytic cysteine residue before transferring it to a protein [49]. A third group: RING-in-between-RING (RBR) combines features of both RING and HECT families, the N-terminal RING domain first recruits E2-Ub complex, then transfers ubiquitin onto another RING domain, before transfer to the substrate protein [50,51] (Figure 2). RINGs represent the most prevalent class of human E3 ligases, with approximately 600 members, surpassing the numbers of HECTs (28 members) and RBRs (14 members) [52].
To date, only RING ligases have been leveraged in TPD approaches [28]. Members of this family are subclassified into two categories: Cullin–RING ligases (CRLs) and non-CRL ligases. CRLs are modular protein complexes consisting of four variable subunits: a RING box protein (RBX1 or RBX2), a cullin (CUL) protein, an adaptor protein and a substrate-recognizing receptor (Figure 2A). CUL proteins are scaffolds that bind an RBX protein through its C-terminus, while its N-terminus binds to an adaptor protein linked to a substrate receptor [53].
CRLs are classified based on the cullin subunit used. The mammalian family comprises eight members: CUL1, CUL2, CUL3, CUL4A, CUL4B, CUL5, CUL7 and CUL9 [54], each with their cognate substrate adaptor and receptor proteins. For instance, CRL1 is a complex consisting of a variable component F-box protein, as adaptor protein, and three invariable components: the S phase kinase-associated protein 1 (SKP1), CUL1 and RBX1 [55].
CRL2 and CRL5 ligases recruit both elongins B and C as adaptor proteins while variable suppressors of cytokine signaling (SOCS) box proteins are used as substrate receptors [56,57]. CRL2 is important in a TPD context, due to the ligase complex based on the Von Hippel-Lindau (VHL) substrate receptor. VHL and SOCS-box proteins contain a common motif, known as the BC-box, essential for binding to the elongin BC complex [57].
A single bric-a-brac-tramtrack-broad complex (BTB) protein is necessary for linking the CUL3 domain to a substrate protein [58,59].
CRL4A and CRL4B are highly conversed (82% identity) [60], and both recruit DNA damage-binding protein 1 (DDB1) as adaptor and DDB1- and CUL4-associated factor (DCAF) proteins, such as CRBN, as substrate receptors [61,62].
Atypical CRL7 recruits a single F-box protein: F-box and WD40 domain 8 (FBXW8) as substrate receptor and SKP1 as adaptor [63]. The substrate adaptors for another atypical cullin family member, CUL9, also described as p53-associated parkin-like cytoplasmic protein (PARC), are not known [63].
CRLs are highly dynamically regulated, activated or decommissioned depending on substrate availability. Both processes depend on the small ubiquitin-like modifier NEDD8, nearly 60% identical to ubiquitin but with distinct targets and functions. Covalent modification of a C-terminal lysine in cullins with NEDD8 induces a structural change stabilizing an open active conformation facilitating the access of the substrates for ubiquitination. Conversely, deneddylation mediated by the constitutive photomorphogenesis 9 (COP9) signalosome (CSN) enables the exchange of substrate receptors from CRLs via the scaffold protein CAND1 [64].
While CRL are multi-subunit E3s, non-CRL ligases are single-subunit entities. They may have a U-box domain instead of a RING domain, which shares a similar structure but lacks the characteristic coordination of zinc ions. Both domains serve the same purpose in facilitating the transfer of ubiquitin from the Ub-E2 conjugated to the substrate protein. Non-CRL ligases can exist in different forms—either as monomeric, homodimeric, or heterodimeric, with the latter being specific to RING-containing non-CRLs only [65].
The first E3 ligands were designed by mimicking specific peptide motifs in E3 substrates, referred to as degrons [23]. Some degrons are linear sequence motifs, whereas structural degrons depend on the spatial conformation of the folded protein, or on a conformational change generated by a posttranslational modification [66]. For example, Protac-1 developed by Crews and Deshaies in 2001 recruits the F-box protein β-TRCP associated with the CUL1-RBX1-SKP1 E3 complex through an IκBα phosphopeptide [1].
Similarly, three years later, the substrate receptor VHL of the CUL2-RBX1-ElonginB-ElonginC complex, known as CRL2VHL, was successfully recruited using the seven amino acid ALAPYIP sequence derived from the HIF-1α substrate [67]. However, these early peptide PROTACs had moderate binding affinity and low cell permeability, resulting in a weak activity. Based on the interaction of HIF-1α with VHL, high affinity synthetic VHL ligands, such as VH032 and later VH298, were developed via considerable efforts of the Ciulli group, paving the way for successful CUL2VHL-based PROTACs [66,68,69]. It is worth noting that while these ligands have high affinity for VHL, their physicochemical properties make them challenging for use in the context of generating orally bioavailable bifunctional degraders.
Hiroshi Handa and collaborators discovered that thalidomide and its analogues, referred to as immunomodulatory imide drugs (IMiDs), more recently known as Cereblon E3 ligase modulators (CELMoDs), bind to CRBN, the substrate receptor of CUL4–RBX1–DDB1–CRBN complex identified as CRL4CRBN [70].
A highly conserved hydrophobic pocket on CRBN allows binding of thalidomide and its derivatives, forming a new molecular surface capable of novel protein–protein interactions (PPIs) and thus recruitment of neo-substrates for degradation [71]. Initial characterisation of thalidomide, lenalidomide and pomalidomide showed degradation of the transcription factors IKZF1 and IKZF3 [72,73]. Further work by Elbert et al. showed that lenalidomide also led to the degradation of casein kinase 1 alpha 1 (CK1α), but that thalidomide had no effect on CK1α levels [74], demonstrating that changes around the IMiD core leads to different neo-substrate target selectivity.
Molecular glues can also trigger degradation by promoting protein polymerization; BI-3802, targeting the oncogenic transcription factor B cell lymphoma 6 (BCL6), illustrates this mechanism [75]. Binding of BI-3802 to the BTB domain of BCL6 triggers its homodimerization and subsequent higher-order assembly into filaments. BI-3802-induced polymerization enhances the interaction between BCL6 and SIAH1, an E3 ligase that recognizes a VxP motif distal to the drug-binding site [76], leading to accelerated ubiquitination and proteasomal degradation of BCL6. In comparison, previously discovered CRBN-based heterobifunctional BCL6 degraders did not achieve complete BCL6 degradation and failed to induce a significant phenotypic response in diffuse large B-cell lymphoma (DLBCL) [77].
By linking a POI warhead to IMiD/CELMoD compounds, molecular glues can be converted into PROTACs [78]. In the past few years, a wave of CRBN-based PROTACs has emerged [79]. It is noteworthy that bifunctional degraders based on IMiDs may retain their MGD-dependent ability to trigger the degradation of Ikaros (IKZF1), Helios (IKZF2), Aiolos (IKZF3) or other targets [80]. This property has been exploited by Nurix Therapeutics with their combined BTK and IKZF degrader NX-2127, currently in phase 1 clinical trials.
2.2. Recruiting Alternative UPS components for TPD
Most of the approaches used for degraders so far have focused on the recruitment of E3 ligases, particularly on the substrate receptors of CRLs, such as VHL, CRBN or DCAF15. As mentioned above, substrate receptors are interchangeable subunits which are docked to the scaffold cullin through an adaptor protein. Hijacking and reprogramming CRL activity is an effective strategy used by several viruses and there is evidence of direct association of different viral proteins with the adaptor protein DDB1, which induces the degradation of host proteins such as the STAT1 transcription factors. DDB1 contains three β-propeller domains, the BPB domain docks the N-terminal of the cullin scaffold, whereas domains BPA and BPC binds DCAFs proteins. Some virus proteins have evolved to functionally mimic DCAFs and bind to the adaptor protein DDB1, thus, recruiting E3 ligase activity to direct the degradation of factors relevant to the immune response and escape its effects, as occurs with hepatitis B virus X protein [81] among others.
Learning from these viral molecular strategies, it has been proposed that molecular glues could mimic viral hijacking of CRL components. This concept has been demonstrated for the E3 component DDB1, where molecular glue degraders rely on the activity of the CRL4B ligase, but is mechanistically independent of substrate receptors, with MGDs acting directly via the DDB1 adaptor to induce POI ubiquitination [2,4] (Figure 2). Recently, covalent ligands for DDB1 and another CRL substrate adaptor, SKP1, have been identified and successfully used to generate bifunctional degraders, further establishing that induced degradation is possible via a cullin adaptor protein [82,83]. As mentioned above, cullin-RING E3 adaptor proteins are associated with various substrate receptors, and consequently, these proteins are essential for cell viability, as their removal or dysfunction would lead to impaired functions across a broad range of E3 ligases. As such, exploiting CRL substrate adaptors to build degraders may lessen the probability of resistance mutations that render degraders inactive.
Although ligase recruitment is currently the most well-explored modality to induce target degradation, other components of the ubiquitin proteasome system (UPS) can also be harnessed. Given that E3 ligases depend on the action of E2 conjugating enzymes, some research groups have sought small-molecule binders for E2s. Very recently, by library screening and chemoproteomic approaches, King and collaborators discovered a covalent molecular glue degrader, EN450, that impairs leukaemia cell viability in a NEDDylation and proteasome-dependent way. EN450 was found to target an allosteric cysteine on the E2 UBE2D and induced the proximity of the E2 with the transcription factor NFKB1 to induce its degradation in leukaemia cells [84]. Shortly after, with this approach, the same group demonstrated that core proteins within UPS machinery, such as E2s, can be exploited for PROTAC applications [85] (Figure 2).
Throughout the development of PROTACs and molecular glues, it has been observed that a limitation is that non-native E3 substrates must be efficiently ubiquitinated by the selected ligase. The efficiency of degradation of the POI is determined by the ubiquitination, and this in turn depends on the existence and availability of a ubiquitin acceptor site on the POI. To overcome these constraints, an alternative strategy would be targeting proteins directly to the 26S proteasome, and thus in principle skipping the requirement for ubiquitination. Bashore et al. explored this strategy, sampling a wide range of chemical space by using mRNA display technology. They found a macrocyclic ligand against the 26S subunit PSMD2. By synthesizing heterobifunctional molecules that bind the proteasome subunit on one side and the protein BRD4 on the other, they were able to successfully degrade BRD4 proteins; they referred to these compounds as chemical inducers of degradation (CIDEs; Figure 2). To confirm that this strategy avoids CUL3 ligase (CRL3) mediation through the KLHL15 adaptor protein, the authors generated KLHL15-knockout clones using CRISPR-Cas9 protocols. Their results showed that degradation occurs independent of this E3 ligase through direct recruitment to the proteasome [86]. This direct-to-proteasome approach has been further explored by the Ciulli group and others, with induced degradation achieved by leveraging ligands for proteasome associated components such as UCHL5, RPN11, USP14 and RPN13 [87,88,89,90], as well as modified proteasome inhibitors [91], and it will be interesting to see how this strategy compares to ligase recruitment in terms of target scope and efficacy.
3.1. TPD as a therapeutic strategy
A fundamental question in a TPD drug discovery campaign is whether the target and indication are suitable for a degrader approach. Arguably, many therapeutically relevant targets may be effectively addressed by adopting a traditional occupancy-based strategy, in particular for enzymes, with a likely faster and more straightforward development route. When considering a degrader approach, it is also worth noting that targets that are rapidly resynthesized may yield less therapeutic benefit, compared to slowly resynthesized proteins. However, there are circumstances where a degrader strategy is advantageous, and these are discussed below.
3.1.1. Selectivity
Selectivity of inhibitors against related proteins is sometimes required to limit toxicity, but not possible to achieve due to high active-site or ligand binding pocket homology. Interestingly, degraders offer two potential routes around selectivity challenges. The first relies on identification of a novel ligand that binds the desired target in a domain outside of the conserved areas, and thus will only degrade the intended target. This requires significant time and financial investment, and the success will rely on the nature of the ligand and its suitability for use in bifunctional compounds. Similarly, a molecular glue degrader approach may be investigated limiting the glue to a non-conserved domain, however MGD identification is currently challenging.
The second route takes advantage of the observation that bifunctional degraders based on existing non-selective target ligands can exhibit selective degrader activity. For example, the non-selective ATP-competitive fibroblast growth factor receptor (FGFR) inhibitor BGJ398 was utilized to generate a selective FGFR1/2 degrader, DGY-09-192, sparing FGFR3/4 [12], and selective degraders for STAT3 and STAT5, respectively, have been generated from relatively non-selective ligands [43,92]. Selectivity in this context likely stems from structural differences that dictate how a target interacts with or can be accessed by the ubiquitin ligase, such that ternary complex formation is either unproductive or does not take place efficiently for a subset of proteins within a group ligandable by the degrader. From this argument follows that different ubiquitin ligases may produce different selectivity profiles, as indeed shown for CRBN and VHL in large-scale proteomics studies using promiscuous kinase ligands [37]. Another example of ligase-driven selectivity is the use of the RNF114 ligase with the non-selective tyrosine kinase inhibitor dasatanib as target recruiter. Although linker chemotype and exit vector geometry will likely impact selectivity, data indicate that VHL and CRBN favour c-ABL degradation, whereas RNF114 preferentially drives degradation of BCR-ABL [93,94]. The expansion of ligases validated for TPD approaches will undoubtedly provide further opportunities to achieve ligase-driven selectivity based on use of non-selective target ligands.
3.1.2. Degraders for Non-Enzymatic Functions
Degraders may provide a way to develop effective drugs for proteins driving biology through scaffolding functions or protein-protein interactions (PPIs), instead of enzymatic activity. TPD is seen as a promising approach for challenging targets like transcription factors, scaffolding proteins, and receptors. However, despite this potential, addressing traditionally undruggable targets with degraders presents significant challenges, with a key consideration being whether identifying a novel ligand for the target is feasible.
Many clinically relevant enzymes have been attempted to target using inhibitors, and existing ligands can be leveraged to build degraders that address both enzymatic and non-enzymatic functions. Examples here include the IRAK4 and BTK kinases, where both enzymatic and scaffolding functions mediate signalling [95,96,97]. Whereas inhibitors can only address the active-site functionality of these targets, degraders have been demonstrated to be more effective due to removing all functionalities of the targeted protein [98,99].
Inhibiting PPIs is generally considered difficult, where interacting proteins often share a relatively large and flat binding interface with little opportunity for ligandable pockets [100]. While the concept of targeting PPIs with degraders has been demonstrated utilizing existing PPI inhibitors for BCL family proteins [30,101], identification of ligands that bind outside of PPI interfaces opens up for addressing a broader variety of PPIs via degraders, and it will be interesting to see if the maturing degrader field will exploit this possibility.
A highly attractive property of degraders is that they may offer a route for removal of aggregated proteins, a hallmark of many neurodegenerative disorders (discussed in detail below). In this scenario, a degrader strategy can offer clear advantages, however identification of aggregate specific ligands and access of the degrader to the CNS pose significant challenges. In particular, bifunctional degraders are difficult to optimize for blood brain barrier (BBB) penetration, and as such MGDs are an interesting option for degraders that act within the CNS.
3.1.3. Resistance mutations
Resistance mutations in cancer treatments pose a substantial clinical challenge, where cells undergo genetic alterations that confer resistance to initially successful therapeutic interventions, such as upregulation of expression levels or point mutations. In cases where resistance mutations occur in the binding site of small-molecule inhibitors, degraders may offer an advantage. Since degraders do not rely on binding to the active site of the target protein, they can potentially overcome mutations that interfere with traditional inhibitor binding. Moreover, due to their event-based pharmacology and potential for cooperative ternary complexes, degraders may be more forgiving to affinity-reducing mutations in the ligand binding site of the target [102].
An example where degraders have been shown to have activity against resistance associated mutants is BCR-ABL1, present in approximately 95% of chronic myelogenous leukaemia (CML) cases. The introduction of imatinib, an ATP-competitive tyrosine kinase inhibitor, marked a significant milestone in treatment of CML [103], which was followed by a number of second-generation inhibitors, including dasatanib [104]. Unfortunately, resistance to both imatinib and second-generation compounds inevitably arise upon treatment. The concept of a degrader approach for BCR-ABL1 has been demonstrated utilizing both ATP-competitive inhibitors and compounds binding allosterically, where bifunctional degraders have activity against treatment resistant BCR-ABL1 mutants [105,106,107]. In particular, the dasatanib based degrader SIAIS056 was shown to efficiently degrade BCR-ABL1 mutants associated with dasatanib resistance [106]. Likely this is due to the event driven pharmacology of the degrader, where resistance mutations may decrease ligand affinity [108], and consequently reduce efficacy of an occupancy-based therapy, but still allow a significant level of induced degradation. It remains to be seen whether specific BCR-ABL1 degraders will be of value for clinical development to supplement the inhibitor pipeline.
In a clinical setting, a recent study has focused on BTK in the treatment of B cell cancers like chronic lymphocytic leukemia (CLL). This report characterizes two types of drug resistance mutations in BTK: kinase proficient and kinase impaired. Kinase-impaired mutants, exemplified by L528W, retain downstream BCR signaling despite reduced BTK kinase activity. NX-2127 from Nurix Therapeutics (Table 1) is a potent BTK degrader that is currently in phase 1 clinical trials, where it has demonstrated over 80% BTK degradation and positive clinical responses in 79% of evaluated CLL patients, irrespective of mutant BTK genotypes, showing promise for overcoming BTK resistance mutations [97].
A further clinical example of where degraders have been used to mitigate resistance mutations is the androgen receptor (AR), an important target in prostate cancer (PC). The AR protein is a transcription factor that promotes cell-proliferation when activated by binding to the androgen hormones. In the initial phases of the disease, PC often relies on androgens, and surgical/chemical castration or AR antagonists are employed to deprive the tumour of AR activity [109]. Unfortunately, various mechanisms lead to resistance to androgen-based therapy, including AR overexpression or mutations that convert antagonist activity into an agonist activity, as reported for the clinical AR antagonist enzalutamide [110]. Given the nature of resistance mechanisms for the AR, adopting a degrader approach can provide new routes for treatment options, and pre-clinical examples include the VHL-based AR degrader ARCC-4, utilizing enzalutamide as an AR recruiter. This degrader was demonstrated to effectively degrade the AR F876L antagonist-agonist conversion mutant [111]. Further pre-clinical examples are ARD-61 [112] and MTX-23 [113], where the latter was developed utilizing a ligand for the AR DNA binding domain (DBD), exemplifying the use of ligands that bind outside of the androgen binding pocket to facilitate degradation. ARV-110 (Bavdegalutamide), developed by Arvinas, was the first AR degrader to be evaluated in patients, and currently there are four additional bifunctional AR degraders in clinical trials (ARV-766, CC-94676, AC176 and HP518; Table 1).
4.1. Discovery Approaches for Bifunctional Degraders
A key requirement for development of a bifunctional degrader is to have access to a ligand for the POI (assuming use of one of the known ligase ligands). Therefore, some of the most important current efforts for development of heterobifunctional degraders include the search for new high-specificity POI binders (Figure 3).
A second area of great importance for PROTAC development is the search for new molecules recruiting distinct ubiquitin ligases, which is the preoccupation of many public and private laboratories. There are several reasons justifying the need for expansion of the E3 binders’ toolbox. One of the most important aspects is the rapid emergence of resistance, particularly in cancer. If mutations occur in ligases such as CRBN or VHL, or if there is downregulation of the ubiquitin conjugation machinery, current PROTACs may become non-functional [114,115,116].
Moreover, it is particularly important to have the choice of using ligases that might give better results in terms of proteolytic activity or if combinatorial treatments are suited. Also, it is important not to hijack all the activity of a ligase, which may block crucial endogenous functions, resulting in unwanted toxicity. These instances underscore the necessity of identifying and developing new binders for alternative ubiquitin ligases.
4.1.1. Aspects to Consider for POI Ligand Development
As mentioned above, the most common strategy used to block the function of a protein has been the development of small chemical inhibitors to target its active site. This effort resulted in generation of hundreds of molecules specifically recognising a target, but in many cases failing to inhibit functional activity.
With a PROTAC strategy, these molecules can be successfully re-employed to build bifunctional degraders. However, when targeting proteins previously considered “undruggable”, new small molecules still need to be specifically designed for degrader development.
Usually, traditional small-molecule research focuses on developing high-affinity inhibitors targeting an enzyme through its active site to neutralise its effect by an occupancy-driven mechanism. Since heterobifunctional degraders act in an event-driven mechanism where they only need to interact transiently with their target [117], compounds with moderate POI affinity (≥1–500 nM) may be sufficient. Alongside this, ternary complex cooperativity (vide supra), can allow for relatively weak binary ligand affinity.
Recently, a framework has been created to assess whether human proteins are “PROTACtable”. This system considers the sub-cellular location of the protein, its ubiquitination site(s), its half-life, and the availability of one or more small-molecule ligands. The workflow is integrated into the Open Targets Platform (https://platform.opentargets.org/).
Despite “PROTACtable” proteins not requiring an enzyme active site, a small-molecule ligand-binding remains necessary. In the case of scaffolding proteins, the selection of a binding site is particularly challenging since the POI might be only partially exposed within a given complex. Unfortunately, structural information on scaffolding proteins or multimeric complexes is still limited for use as a systematic approach. This is mainly due to the complex architecture of scaffolding proteins, making them difficult to produce individually or to integrate into a functional multimeric complex.
Many currently available degraders are based on existing small molecule inhibitors, and it is worth noting that inefficient inhibitors can be successfully repurposed to build efficient heterobifunctional degraders, exemplified by ligands developed for TRIM24 [118]. Building upon these early tool compounds, the validation of novel targets using degraders is becoming an increasingly popular strategy in drug discovery.
For targets where there are no existing inhibitors, it is necessary to identify a ligand for the POI. As the only requirement for compounds is binding to the protein in question, screening technologies based on DNA encoded libraries (DEL) or Affinity selection-mass spectrometry (AS-MS) are highly suited for identification of novel ligands for challenging targets, for either a POI or an E3.
A DEL is a collection of diverse chemical compounds, each uniquely tagged with DNA sequences, which enables high-throughput screening of vast compound libraries to identify compounds that bind the POI. From a TPD perspective, an advantage with a DEL approach is that the DNA attachment point to the library compound may also serve as a linker exit vector. AS-MS integrates compound binding with mass spectrometric analysis for compound isolation and identification, enabling rapid screening of large compounds collections to identify protein binders [119].
An example of a DEL approach for TPD is the identification of a novel binder for the oestrogen receptor α, which was incorporated into a bifunctional degrader [120]. A DEL approach was also utilized to identify a binder of the GID4 ubiquitin ligase [121], which interestingly was also addressed by AS-MS to identify a novel ligase binder [122], exemplifying the use of two different high-throughput technologies to identify structurally different protein binders.
4.1.2. Aspects to Consider for E3 Ligand Development
The search for new E3 ligases to be used in TPD strategies should consider the ubiquitous (or specific) presence of a ligase in different tissues and cell lines, which may offer benefits in terms of limiting off-target toxicities [30]. As new E3 ligase binders emerge, it will remain important to characterise their scope and applicability, to ensure that the degradation efficiency is at least equal to or better than that observed with VHL or CRBN [19]. Another factor to consider is the subcellular localisation of an E3 ligase, since the effect of the degrader could be reduced if its access is limited, for instance, to the nuclear compartment [32].
In addition, several ligases exist in an inactive state or are auto-inhibited in the absence of an activating post-translational modification or binding partner [117]. Using an auto-inhibited ligase may pose additional challenges when considering them for PROTAC development. Naturally, it is also critical that the ligase binder does not inhibit the actual ligase function itself!
As mentioned above, some ligases are only expressed in specific cell types or are over-expressed in certain pathologies, and this can potentially be leveraged to develop more specific therapies. If the tumour enrichment of an E3 ligase aligns with the dependence of the tumour on the expression of that ligase, this approach could be suitable. In this regard, certain E3 ligases and other UPS components, such as the WD repeat-containing protein 82 (WDR82), have been identified as promising targets essential for various cancer cell types [114]. This dependence may reduce the chances of developing resistance, as observed with some CRBN and VHL-based degraders [114,115,116].
In the same vein as for POI ligands, existing ligands for E3s can sometimes be repurposed for use in bifunctional degraders. In the quest to recruit other ubiquitin ligases, the RING ligase mouse double minute 2 homologue (MDM2) was investigated in a TPD approach. Building on the small-molecule PPI inhibitor nutlin-3, which disrupts the interaction of MDM2 with p53, a bifunctional degrader of the androgen receptor was successfully generated [123].
To profile novel (or established) ligases in TPD context, approaches include HaloPROTACs and TAG platforms (dTAG and aTAG) [124,125]. These strategies require engineering of a tagged POI, where the tag provides a handle that can be bound by the degrader, allowing evaluation of a ligase across multiple targets via the tag [126].
While the lack of binders for both POI and E3 is not a standalone limitation, it does signify the need for additional efforts before initiating the development of a bifunctional degrader. Various approaches, with library screening and structure-based methods being the most prevalent, are employed to identify new binders.
Phenotypic screening is a powerful approach to discover small molecules targeting proteins involved in the regulation of cell physiology and pathology. Several chemical libraries integrating simple or high-complexity molecules have been used by private and public research laboratories [127]. Once integrated into PROTACs, the functionality of the identified molecules as degraders can be screened phenotypically. The chemical properties necessary for drug discovery and the selection of de novo substrates can be defined in the context of phenotypic alterations in cells. Interestingly, this approach can also be used to explore the temporal relationships associated with disease development and response/resistance to treatments.
Rational design of heterobifunctional degraders based on crystal structures and computational chemistry has been used to optimise molecules that will efficiently bind to the POI or E3 ligase. This has also provided information on the formation of the ternary complex, which further allows degrader optimization. Several examples illustrate the success of this approach and have been recently reviewed [127,128]. When crystal structure data is available, this technique can be relatively fast. However, in the absence of such knowledge, two possibilities can be considered: generating the knowledge or using artificial intelligence (AI)-driven tertiary structure prediction models such as DeepMind [129] and RoseTTAFold [130]. These and other approaches become popular if free access exists, like Alphafold from DeepMind, which generates high-quality predicted models of the proteome (https://alphafold.ebi.ac.uk/).
One of the first successful computational workflows for PROTACs development was reported in 2018 [46]. In this work, authors evaluate the linear linkers using a steric scoring scheme. A more recent, practical, in silico tool for PROTAC development considered the contribution of the three components in the ternary complex [131,132]. Since then, several groups have reported tools, some of which are freely available, including the Rosetta-based protocols (https://prosettac.weizmann.ac.il/) [133,134,135]. While some of these methods may be of utility, more work is needed to understand the mechanisms that allow efficient design of heterobifunctional degraders. Artificial intelligence will certainly contribute to better model predictions in the future.
4.1.3. Linker Optimization
The overall degradation efficiency of a heterobifunctional degrader does not simply rely on the affinities of the chemical molecules binding the E3 and the POI. The way in which these two molecules are connected by a linker, allowing the formation of a functional ternary complex (TC) able to induce ubiquitination and degradation of the POI, is as crucial [136]. The length and composition of the linker are critical in the generation of an efficient and specific bifunctional degrader. These design considerations contribute to the efficient formation of a TC [137,138], highlighting their importance for degrader optimization [39,40]. Evidence also clearly indicates the importance of the specific attachment point, or exit vector, to the molecules binding the POI and E3s, which must be optimized for each degrader. Unfortunately, this is not generally predictable due to the structural complexity and dynamics of the TC.
Despite the importance of the linker, well-established strategies for design, resulting in high efficiency, are scarce. Often, stepwise optimization through synthetic alteration of the linker is followed by a degradation activity test, utilising short and structurally simple alkyl or PEG chains as starting points. Most linkers have consisted of combinations of a few chemical motifs [136,139], the most common being PEG and alkyl chains of varying lengths, which appear in approximately 55% and 30% of linkers, respectively. Around 65% of heterobifunctional degraders contain both an alkyl and PEG segment. Only 15% use glycol units, incorporating additional methylene moieties to access different chain lengths. Other less-used motifs include alkynes (7%), triazoles (6%), and saturated heterocycles such as piperazine and piperidine (4% each). Several strategies have been used to improve chemical functionalities of PROTACs, such as photo-switches, conformational locks, and covalent binding. Recent reviews have summarised some aspects of linker chemistry and design strategy [136,140].
To accelerate the screening of bifunctional degraders containing different linkers, direct-to-biology (D2B) approaches, where compounds are assessed for activity in the absence of a purification step, are becoming increasingly popular. This approach allows miniaturization and more rapid cycles of compound testing; however, care must be taken to assess non-specific impact of crude reagent mixtures on cell-viability. Using a D2B approach, SAR around linkers, POI ligands, exit vectors and ligase recruiters can be rapidly explored to identify lead compounds for further refinement, and this strategy has been employed by Janssen, GSK and AstraZeneca in recent reports [141,142,143].
Critically, linker optimisation can make dramatic changes to the overall pharmacokinetic properties of the degrader. Whilst optimisation of shape, rigidity, and length impact upon the ability to form a productive ternary complex and, therefore, the degree of degradation observed, these changes can also significantly alter metabolic stability, solubility and permeability. Factors such as aromaticity, 3-D shape and polarity deliver subtle changes to the shape and nature of the linker itself but can deliver significantly different behaviour in in vivo assays. In addition, the nature of the chemistry used to attach the linker to the POI and E3 ligase binders can also be further optimised, often transitioning from the ubiquitous amide bond (which often limits cellular permeability) to a less problematic linking functionality.
4.2. Discovery Approaches for Molecular Glue Degraders
Molecular glue degrader discovery is challenging. Unlike bifunctional degraders, where rational design based on ligase and POI ligands is relatively straightforward, molecular glues modulate protein surfaces to induce interactions in ways that are difficult to predict.
For this reason, molecular glue discovery has historically been serendipitous, with mechanisms of action identified later. Here, we focus on the discovery of molecular glues which drive novel interactions between a target protein and components of the ubiquitin-proteasome pathways and therefore induce degradation of these target proteins.
4.2.1. Serendipity
The first known molecular glue degrader, thalidomide, was marketed from the mid 1950′s to treat insomnia and morning sickness. At the time its mechanism of action was unknown and sadly led to the birth of many children with limb defects due to its unidentified teratogenic effects. In 1965 thalidomide and its derivatives, lenalidomide and pomalidomide, were reinvestigated with renewed interest after it was discovered they had immunomodulatory, anti-inflammatory, and anti-tumorigenic properties. However, it was not until 2014 that these immunomodulatory imide drugs (IMiDs) were finally discovered to bind to CRBN, a substrate receptor of the CUL4-RBX1-DDB1-CRBN ubiquitin ligase complex [70,144].
A second compound with a mechanism of action analogous to the IMiDs, Indisulam, was originally discovered in the 1990′s by screening a library of sulfonamides for cancer cell growth inhibition. Indisulam was shown to cause G1/S cell cycle arrest by flow cytometry and to have in vivo efficacy in human tumour xenograft models [145]. The exact mechanism of action was not elucidated until more than 15 years later, when it was demonstrated that the anti-proliferative effect was due to an induced ternary complex between the mRNA splicing factor RNA binding motif protein 39 (RBM39) and a member of the CUL4 ubiquitin ligase complex called DDB1 and CUL4 associated factor 15 (DCAF15). Formation of this ternary complex leads to the polyubiquitination and degradation of RBM39 [146]. Structural studies showed that indisulam binds DCAF15 and creates a novel surface which enhances RBM39 binding and induces novel protein-protein interactions [147].
As molecular glue degrader discovery has historically been serendipitous, it is reasonable to assume there may be small molecules in development or the clinic for which the molecular glue mechanism has not yet been identified. Ebert et al. used database mining to look for a correlation between the cytotoxicity of 4518 clinal or preclinical small molecules and E3 ligase expression in hundreds of human cancer cell lines [2]. These studies identified a correlation between CR8 (CDK inhibitor) and DDB1 (CUL4 substrate adaptor protein) expression. An X-ray crystal structure revealed that CR8 bound to CDK12 had a solvent exposed pyridyl moiety which induced complex formation of CDK12 with Cyclin K and DDB1(PDB: 6TD3). This ternary complex formation leads to ubiquitination of cyclin K and downstream degradation by the proteasome [2].
4.2.2. Cellular Screening Approaches
Using cell-based assays to screen for compounds with MGD properties has the potential advantage that the complete cellular machinery is present, allowing ubiquitination and subsequent degradation of a target protein. In contrast, the challenge when utilizing cells for compound screening is the extensive hit deconvolution required for validation of MGD mechanism.
To help focus this approach, BMS/Cellgene screened a library of CRBN interacting compounds, with the advantage that half the prospective interaction was already defined and proteomics could be used to identify the target protein. This study was directed towards identification of a molecular glue for the treatment of acute myeloid leukaemia (AML), and screening for compounds with antiproliferative potency was performed using ten AML cell lines. This led to the development of CC-90009, a selective GTPS1 degrader, the first CRBN based molecular glue degrader to enter clinical trials since CRBN was identified to be the primary target of thalidomide [144]. At the time of writing, CC-90009 is in phase II clinical trials for the treatment of AML (Table 2).
Further expansion of MGDs beyond CRBN requires methods with broader scope. Most of the cullin RING ubiquitin ligases require conjugation to NEDD8 for activity, and therefore differential screening in cells with normal and deficient neddylation can identify compounds that require uninterrupted neddylation, inferring that activity is driven by a cullin RING ubiquitin ligase.
Mayor-Ruiz et al. used this approach to screen 2000 cytostatic or cytotoxic small molecules, with identification of differentially active compounds. The results were followed up by analysis in CRISPR knockout cell lines of all known Cullin RING ligases and quantitative expression proteomics of treated cells to identify the relevant ligase and target protein respectively. This strategy identified three compounds which induced degradation of Cyclin K by the CRL4 ligase complex [4]. Analysis showed that although these compounds are structurally different from CR8 (identified by data mining described above), they act by a similar mechanism of binding CDK12 and recruiting Cyclin K and DDB1 [148].
To limit a MGD to a desired ligase, morphological screening approaches using cell lines with different ligase expression levels can be used. Cell painting assays can be used to assess hundreds of parameters including cell viability, morphology and the detection of multiple organelles or cellular components [149], and Ng et al. used this approach in isogenic cell lines expressing different levels of CRBN. A screen of 132 CRBN binders (assessed by a CRBN fluorescence polarization competition assay first) identified FL2-14 as a GSPT2 molecular glue degrader [150].
4.2.3. Biophysical Screening Approaches
As a complementary approach to cell-based systems, purified proteins can be used in biophysical assays to measure the induction of a ternary complex between a POI and ligase. This has the advantage of fully controlling the selection of the E3 and the target. Biophysical assays give a direct read out of ternary complex formation and can therefore help build SAR and drive development of compounds with improved molecular glue properties. However, without the cellular context, biophysical assays cannot give information on target protein degradation and any downstream effects. Of importance for biophysical screening approaches is identifying a target and ligase pairing which is suitable for development, with consideration needed for the target and E3 cellular locations and tissue and disease specific expression profiles.
Although uncommon, a known weak compound-induced interaction provides an advantageous start point. Pomalidomide induces an interaction between CRBN and the transcription factor IKZF1 which leads to the degradation of the latter. Novartis further identified a weak interaction between Pomalidomide, CRBN and IKZF2 but this interaction did not lead to IKZF2 degradation. This suggests there is a recruitment threshold which needs to be met before molecular glue induced interactions are strong enough to lead to target degradation. The Novartis discovery campaign was driven initially by interaction assays between CRBN-IKZF2 and CRBN-IKZF1, allowing optimisation for selectivity before the interaction threshold for target degradation had been met [151]. Another recent study from Novartis has highlighted the use of biophysical approaches to identify the first VHL based molecular glue degrader, where the glue induces binding and degradation of cysteine dioxygenase 1 (CDO1) [152].
An alternative approach is to take advantage of a known physiological E3 and POI pair by focussing on a mutated POI where the ability of native ligase to bind and ubiquitinate has been lost or impaired. In this scenario, a molecular glue could be utilised to re-establish the interaction between the POI and ligase to restore the normal degradation pathway. β-catenin is an effector protein in the Wnt signalling pathway which normally interacts with the β -TrCP ubiquitin ligase, leading to β-catenin degradation. Mutant β-catenin, found in some colorectal cancers, has an impaired ability to bind β-TrCP, leading to enhanced oncogenic transcription. Nurix Therapeutics developed an FP competition assay using β-TrCP with BODIPY-TMR labelled β-catenin phosphodegron peptides. Discovery was focused on the Serine 37 mutant as this is a hotspot for β-catenin mutations. Using an HTS approach, 350,000 compounds were screened through the FP assay to identify enhancers of the β-TrCP and β-catenin peptide interaction. Promising compounds were validated by orthogonal TR-FRET and surface plasmon resonance (SPR) assays [153]. Using native binding E3-POI partners provides the advantage that there is already an interaction surface between the two proteins, with lysine residues available for ubiquitination. It also avoids any issues that may arise from hijacking unrelated E3s to POIs, such as incompatible expression profiles or subcellular localizations.
Establishing a rational and unbiased screening approach that is broadly applicable for identification of novel molecular glues across different protein pairs is extremely challenging. Significant efforts have focussed around redirecting CRBN towards new targets either by expanding around the IMiDs or looking for new chemical matter. Whilst efforts to expand the IMiD chemical repertoire have been successful, with multiple compounds heading towards the clinic, expanding to look at more E3 ligases may yet open up a range of novel target proteins. Phenotypic screening approaches have identified novel glues, but methods require thorough follow up for target deconvolution and hit validation. Exploiting known weak interactions or, in the case of mutant proteins, using its native E3 ligase may provide advantageous starting points due to an already known protein-protein interfaces and lysine residues available for ubiquitination by the E3 ligase. Molecular glues, although challenging to identify, provide an opportunity hijack the protein degradation pathways using compounds with more favourable physicochemical properties than bifunctional molecules.
4.3. Target Validation Via Degraders
In addition to direct clinical application, chemical degraders represent a valuable strategy for characterizing the role and importance of a protein in cellular physiology and disease development. Many candidate targets have been selected based on a vital role in the processes they impact, such as oncogenesis, neurodegenerative disorders or inflammation. Validating new targets in a more efficient way represents a crucial step to justify investing efforts in drug development. While transient (siRNA) or permanent (knock-out) genetic approaches can be utilized for target validation experiments, these approaches can have drawbacks. Generally, genetic methods, particularly knock-out systems, may lead to compensatory mechanisms, such as upregulation of redundant pathways, that may reduce the phenotypic effect related to the absence of the target protein. In contrast, degraders act rapidly, similar to how drugs act on patients, allowing the assessment of how acute depletion of the target affects the relevant readout. There is also a benefit of degrader use to establish pharmacology in situations where catalytic site inhibition does not match the result of siRNA knock-down.
4.4. Assays for Validation of Degraders
After developing an initial degrader compound, it is crucial to evaluate parameters for future improvement of critical aspects to obtain an optimal therapeutic molecule. These parameters include binary target engagement, ternary complex formation, efficient polyubiquitination of the target protein, specific proteolysis, pharmacological effects, solubility, stability, and cell permeability.
Various assays can be employed to evaluate physicochemical, pharmacologic, and biologic properties of degraders. This evaluation opens avenues for future improvements of prototype molecules. Techniques like fluorescent polarization [154,155], time-resolved fluorescence resonance energy transfer [46], AlphaLISA [156], surface plasmon resonance (SPR) [39], and calorimetry [42] can be used to assess target engagement and ternary complex formation. Bioluminescence methods like NanoLuc, NanoTag, or NanoBRET are successful for evaluating cellular permeability [157,158].
4.4.1–. Confirmation of Mechanism of Action
Functional assays should determine if POI proteolysis is linked to known cellular mechanisms driven by the ubiquitin-proteasome system (UPS) or autophagy-lysosome system (ALS). A pharmacological approach, such as inhibiting the proteasome (e.g., with bortezomib) or autophagy (e.g., with bafilomycin A), is recommended. Moreover, inhibition of the ubiquitin-activating enzyme using MLN7243 [159] can be utilized to determine ubiquitin-dependence. The NEDD8 activating enzyme inhibitor MLN4924 [160] is also useful to validate dependence on a CRL, if appropriate. Bifunctional degraders can also be assessed for mechanism by competition with isolated ligase or target ligands, or by use of non-binding compound analogues for the E3.
In addition to western blot analysis, POI degradation can be assessed using homogenous time-resolved fluorescence (HTRF), fluorescent reporters such as green fluorescent protein (GFP) or mCherry in imaging or flow cytometry [161,162,163]. However, when using fusion proteins, care should be taken to validate degradation on the endogenous level, as the tag may be contributing to degradation. Expression levels are also highly important when assaying degraders, and simple overexpression systems may not be suitable [158]; if such systems are utilized, again, validation against the endogenous target is important.
Verification of POI ubiquitylation can be done by overexposing western blots of cell extracts treated with proteasome inhibitor. Due to the rapid deconjugation, POI ubiquitylation is more easily observed using ubiquitin traps (also known as Tandem Ubiquitin Binding Entities) to capture ubiquitylated proteins for detection by western blot or protein arrays [164,165]. For evaluating POI ubiquitylation, in vitro assays using recombinant enzymes (E1, E2, E3) in the presence of ATP can be conducted instead [2]. In vitro ubiquitination assays may be challenging to set up, but once functional, these can be adapted to medium-through put techniques.
In addition to confirming that a novel degrader is on-mechanism, it is informative to assess general selectivity. Mass spectrometry can globally evaluate the effect on the proteome of the specific degradation of a POI, ensuring that consequences are restricted to the proteolysis of the target and its known regulated functions [38,166]. Figure 3 summarizes concepts in degrader discovery and validation.
5.1. Therapeutic Areas and Clinical Application
Despite significant anticipated challenges, including metabolic stability, dosing and routes of administration, several degrader projects have already entered clinical evaluation. In line with pre-clinical expectations, oral bioavailability of these large molecules can be challenging to attain. Measured oral bioavailabilities in animal studies, particularly for earlier state derivatives, are generally low, (often in the 3-30% range) [167]. Despite this, the majority of clinically investigated agents have been found suitable for oral dosing. Indeed, for more optimised candidate-stage molecules, bioavailability in mice or rat in vivo PK studies can reach 50-90%. At the current time, little information exists in the public domain to quantify how these bioavailabilities in lower species translate into humans, as the corresponding matched oral/iv dosing studies are rarely undertaken in patients. Understanding and resolving these unknown factors is likely to become important as the field matures. From a patient perspective, low oral bioavailablilty demands a high pill burden, in order to drive sufficient absorption and free drug exposure in the target tissue to deliver therapeutic benefit. Moreover, the potentially large unabsorbed fraction, which may be excreted largely unchanged, has a significant negative impact on the quantities of API required for manufacture and, therefore, on cost of goods. Particularly in cases where degraders are competing with small molecule inhibitors of the same target or pathway, improvements here may be critical to satisfy cost/benefit analyses prior to approval.
5.1.1. Heterobifunctional Degraders in Oncology
As of the time of writing, there are 20 clinical assets in the oncology heterobifunctional space (Table 1). Of these, 70% are dosed orally, and 30% intravenously. One compound (ARV-471) is now in advanced Phase III clinical trials which are not expected to conclude until 2028.
For the majority of clinical degraders, the proteins of interest have already been investigated with significant numbers of small molecule agents. Examples here include the oestrogen receptors (ER) in breast cancer (BC), targeted with selective oestrogen receptor degrader small molecules (SERDs), the androgen receptor (AR) in metastatic castrate-resistant prostate cancer (mCRPC), targeted with selective androgen receptor degrader small molecules (SARDs), and kinases such as mtBRAF and BTK, targeted with both covalent and non-covalent inhibitors. As these trials progress, it will be interesting to see where similarities and differences occur in terms of patient responses and outcomes.
To date, the limited emerging clinical data from those agents progressing beyond Phase I remains relatively modest in terms of response rates. In the case of ARV-471, Phase II data suggested an overall survival benefit of around 3-4 months, and median protein degradation of around 70% [168]. In the AR setting, in Phase I studies, ARV-110 showed >50% decrease in Prostate Specific Antigen (PSA) levels in around 16% of the patient population and 2/7 partial responses in the evaluable patient cohort [169]. Of note, in patients with known resistance mutations, such as T878X and H875Y, these responses were somewhat higher, suggesting a role for degraders in settings where tumour heterogeneity or tumour evolution has led to therapeutic resistance with small molecule inhibitors.
Anecdotal reports have suggested that the modest responses in the clinic may be due to a variety of factors, including:
- The aforementioned low bioavailability and, therefore, potentially sub-therapeutic exposure at target
- Emergent resistance due to loss of function, or decreased expression of the cognate E3 ligase
- Elevated rates of protein re-expression in response to treatment, counteracting active degradation by the heterobifunctional agent.
It is important to state that these agents are only the first few to report early clinical data, and as the field matures, and collective wisdom and expertise widens, it is likely that our understanding of the degree of bioavailability, the importance of protein re-expression rates and appropriate clinical settings will help to drive improvements in efficacy and, ultimately, patient benefit. So far, many of the targeted proteins seem to have a rapid resynthesis rate measured in a few hours (around 3 hours and 4 hours for the AR [170], and ER [171], respectively). Anecdotally, this rapid resynthesis seems to overcome the rate at which the heterobifunctional degraders can eradicate the POI from the cell. In light of this, targeting proteins with a slower rate of compensatory re-expression may become a preferred application for heterobifunctional degraders. Alongside rapid re-expression of the target protein, clinical resistance has also been observed to arise from alterations of the E3 ligase system, or up-regulated expression of multidrug resistance efflux pumps [172].
Beyond slowly re-expressed proteins, heterobifunctional degraders may offer other opportunities where more traditional modalities have struggled to gain traction. Of specific note in the oncology space, the emerging degraders of the BRM protein offer a compelling instance where a degrader delivers benefit over a small molecule inhibitor. Based on the concept of collateral vulnerability [173], both experimental [174], and bioinformatic [175], studies demonstrated that certain lung tumors undergo a collateral loss of the SMARCA4 gene, encoding for the BRG1 protein, an essential part of the SWI/SNF chromatin remodelling complex. This deletion is not a loss of function mutation which drives tumor growth, but a coincidental loss as part of a wider allelic deletion. However, this leaves this cell population entirely dependent upon the related protein BRM, encoded by the SMARCA2 gene. In this context, a selective BRM inhibitor would be lethal to the ca. 10% of non-small cell lung cancers (NSCLC) where the SMARCA4 gene is lost or mutated, but well tolerated in healthy tissue due to their unaltered expression. However, due to the high sequence conservation between the two targets and despite extensive efforts, selective BRM inhibitors (for either the bromodomain or ATPase domain), remain elusive.
Here, PRT3789 from Prelude Therapeutics offers clear differentiation from these small molecule efforts [176]. Biochemically, the heterobifunctional molecule binds at equipotent nanomolar concentrations to both BRG and BRM1, yet in cells delivers 19-fold selectivity in BRM/BRG1 HiBiT assays. More interestingly, the compound delivers a 720 pM DC50 and 94% Dmax vs BRM, yet spares BRG1 (14 nM DC50 and 76% Dmax), delivering selective cell killing in mtSMARCA4/SMARCA4-del cells, but not in SMARCA4-wt cells, with this selectivity translating into matched in vivo xenograft studies. The precise mechanisms underlying this enhanced specificity have not been described but this outcome reflects those observed in the conversion of non-selective kinase inhibitors to selective kinase heterobifunctional degraders [38]. This approach offers the tantalising prospect of selectively degrading tumour specific protein homologs, or members of closely related protein families, in a way which is not possible with small molecule inhibitors. Whilst in its infancy, this paradigm may offer a real differentiator for heterobifunctional molecules, extending the remit beyond those proteins where high quality and effective small molecule therapies already exist.
5.1.2. Molecular Glue Degraders in Oncology
The molecular glue degrader landscape in oncology is represented by two predominant molecular classes – the dominant class of MGDs is derived from the phthalimide CRBN recruiters, and the smaller group are those derived from other chemotypes. Thalidomide, and related derivatives, had long been known to be effective in multiple myeloma, but through unknown mechanisms. Significant work by the Tokyo Institute of Technology and Celgene unraveled this mechanism [70,144] and led to the explosion of interest in CRBN-derived molecular glues. As such, thalidomide was the first approved CRBN-molecular glue, well before it was known to act in this manner. Approvals for lenalidomide and pomalidomide have since been granted.
As of the time of writing, a further 16 molecular glues have now entered the clinic (Table 2). Of these, for those where a structure has been disclosed, only three glues (Indisulam, CQS and E7820) do not display the structural motifs common to CRBN molecular glues, instead recruiting DCAF15 to effect protein degradation of RBM39. This overwhelming focus upon CRBN molecular glues raises several challenges, including the race to discover (and patent) novel chemical space. From a clinical standpoint, it also implies that many of the dozens of molecular glues now in pre-clinical development are likely to be competing for the same disease segment, and thus the same clinical trial populations, potentially limiting recruitment and delaying evaluation. Clearly, exploitation of a wider range of E3 ligases in the pursuit of MGDs is commercially attractive and likely to deliver wider patient benefit.
Given their smaller molecular weight and potentially improved physicochemical properties, oral bioavailability remains a key attractive feature of molecular glues, and all the compounds in the clinic which recruit CRBN as the E3 ligase are dosed orally, except for CC-90009 which is dosed via an iv infusion. The reasoning behind this outlier route of administration does not appear to have been publicly disclosed at this time. It is interesting to note that, in addition, all clinical examples of DCAF15 molecular glues are also dosed via an iv infusion, suggesting that oral bioavailability is not necessarily guaranteed for molecular glues outwith the CRBN IMiD-derived agents.
5.2.1. Heterobifunctional Degraders for Inflammatory Indications
TPD is emerging as a promising approach for the treatment of inflammatory diseases, offering a novel therapeutic strategy with potential advantages. Inflammatory diseases, characterized by abnormal immune responses, may involve dysregulated protein expression contributing to pathogenesis, and a TPD approach is suited to address this by selectively removing disease-associated proteins. Another potential benefit lies in the ability to modulate inflammatory signalling pathways by degrading previously un-druggable proteins.
An example of these strategies is the development of heterobifunctional degraders for the protein RIPK2. Acting downstream of pattern recognition receptors, RIPK2 activates NF-κB, leading to the production of inflammatory cytokines. Dysregulation of RIPK2 is implicated in various inflammatory diseases, such as Crohn’s Disease and Ulcerative Colitis. Degraders for RIPK2 have been explored pre-clinically as a strategy to improve kinase selectivity and pharmacokinetic properties of inhibitors, and have shown promising anti-inflammatory properties [20,177]. Further examples of targets in inflammatory pathways where degraders have been developed include TYK2 [178] and BTK [179], and bifunctional compounds for both are in pre-clinical development for inflammatory indications by Kymera Therapeutics and Nurix Therapeutics, respectively.
Currently, the only target where a bifunctional degrader has reached the clinic for an inflammatory indication is IRAK4. As a serine/threonine kinase, IRAK4 functions as a key mediator in the activation of signalling in response to inflammatory stimulation. IRAK4’s central role in inflammatory signalling makes it an attractive target in conditions associated with dysregulated immune activation, and several inhibitors of IRAK4′s kinase activity have entered clinical evaluation [180].
Multiple pre-clinical studies of IRAK4 degraders have highlighted that potent degraders for this kinase target can be generated [180], and KT-474 by Kymera Therapeutics is now in phase 2 for treatment of hidradenitis suppurativa (NCT06028230) and atopic dermatitis (NCT06058156). The phase 1 clinical results have demonstrated that single dosing of 600 mg-1600 mg KT-474 led to a rapid drop in IRAK4 levels in peripheral blood mononuclear cells (PBMCs), reaching nadir by 48 h. The degradation is sustained for at least 14 days, as IRAK4 levels did not return to baseline for these doses at that time. Multiple dosing over 14 days (once daily) showed that a dose as low as 25 mg led to robust IRAK4 degradation (92% at nadir). It’s worth noting that at the 100 mg dose, IRAK4 levels were still significantly below baseline at day 28, some 14 days post dosing. For both hidradenitis suppurativa and atopic dermatitis, there was an improvement in clinical symptoms after treatment with 75 mg KT-474 daily for 28 days. These clinical responses were either maintained or continued to improve in the two weeks that were evaluated after dosing was halted. Overall, the phase 1 results are promising, and indicate that longer term treatment can be achieved with a relatively low dose of the compound, and still achieve a strong degradation of IRAK4. In addition to Kymera Therapeutics, Nurix Therapeutics are in the IND enabling pre-clinical phase for their IRAK4 degrader for rheumatoid arthritis and other inflammatory conditions.
5.2.2. Molecular Glue Degraders for Inflammatory Indications
At this time, there are no clinical examples of rationally developed molecular glue degraders for treatment of inflammatory disease. However, in the pre-clinical pipeline is noteworthy that Monte Rosa Therapeutics is in the IND enabling phase for an MGD, MRT-6160, that targets VAV1, a protein implicated in T- and B-cell receptor signalling. Both Monte Rosa and Captor Therapeutics are also in the discovery phases for molecular glue degraders directed to NEK7, an activator of the NLRP3 inflammasome.
5.3.1. Heterobifunctional degraders for CNS disorders
Whereas targeted protein degradation in oncology has largely exploited targets with matched small molecule therapeutics, CNS disorders present opportunities for significant differentiation from both small- and large-molecule therapeutic approaches. Misfolded protein aggregates, a hallmark of neurodegenerative disorders, have been considered undruggable using conventional inhibitor approaches, potentially accounting for the failure of compounds in clinical trials targeting protein aggregates in the CNS [181].
At this time, a large number of CNS disorders lack effective treatment and heterobifunctional degraders represent an attractive therapeutic instrument due to their ability to remove proteins via proteasomal degradation. Despite their potential for degrading a POI in vitro, a major challenge for heterobifunctional degraders is their ability to reach the brain and treat the disease in vivo. Due to their high molecular weight and a large polar surface area, achieving blood brain barrier (BBB) permeability of heterobifunctional degraders is particularly challenging. In this context, degrader-antibody conjugates or encapsulated nanoparticles that can cross the BBB through receptor-mediated transcytosis, have been investigated and may provide an alternative route to access the CNS [182,183].
Ubiquitin ligases may have tissue-specific expression, such as RNF182, expressed preferentially in the brain; CNS restricted ligase expression enables the potential for a more specific targeting of the POI within the CNS [117,184], with the possible advantage of limiting unwanted effects in tissues not directly involved in disease pathology.
Unlike the oncology exemplars described above, PROTACs for neurodegenerative disorders remain in pre-clinical development, largely given the challenges of targeting the brain. Nevertheless, steps have been taken to advance PROTAC molecules in the neuroscience field, with a particular focus on Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS).
5.3.1.1. AD
The accumulation of mutated tau species into neurofibrillary tangles, both in the intracellular and extracellular space, leads to cellular toxicity and neuronal cell death [185]. Different peptide-based and small molecule-based PROTACs have been developed for tau degradation.
TH006, a peptide-based PROTAC with an ALAPYIP sequence as a VHL ligand, was shown to induce tau degradation in N2a cells overexpressing tau. Whilst the effect of degradation was observed at high concentrations (200 μM), TH006 administration in a mouse model of AD resulted in a reduction of tau levels in the cerebral cortex and hippocampus. TH006 has been administered both intranasally and intravenously, probably due to low BBB permeability, a common feature of these peptides [186]. Similarly, a Keap1-based peptide PROTAC was observed to recruit CUL3KEAP1 ubiquitin ligase, inducing the degradation of tau through the ubiquitin-proteasome system [187].
QC-01-175, a small-molecule tau-degrading PROTAC based on the PET tracer T807, with a CRBN ligand, was shown to induce Tau degradation in FTD patient-derived neuronal models including the tau-A152T and the tau-P301L variants [188]. Following linker optimization, second-generation CRBN-recruiting degraders (FMF-06-series) delivered a tenfold improvement in degradation potency of total tau and phospho-tau S396 with a tau reduction by 50% at 10 nM concentration [189]. Minimal E3-ligase occupancy was observed in in vitro cellular target engagement assays, indicating low cell permeability of the optimized analogues [189].
C004019, a VHL-based degrader, was demonstrated to lead to tau degradation through the proteasome, both in vitro and in vivo. C004019 has shown a DC50 of 7.9 nM in a HEK293-hTau overexpressed cell line. Furthermore, intracerebroventricular or subcutaneous administration of C004019 promoted a sustained tau clearance in vivo [190]. Another example is compound I3, a CRBN-based degrader with a THK5105 derivative as a tau ligand, where tau degradation has been demonstrated in vitro using PC12 cells [191].
In the clinical development pipeline, Arvinas claims to have achieved BBB penetration and removal of 95% of pathological tau in vivo after parenteral administration in animal models; the structure of this PROTAC has not been disclosed at the time of writing.
5.3.1.2. PD
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene and in the α-synuclein gene (SNCA) are linked to the onset of PD. The dysfunction of LRRK2 may contribute to the accumulation of α-synuclein, consequent microglia activation, neuroinflammation and ultimately neuronal cell death [192]. Selective degraders targeting LRRK2 have been recently disclosed in a patent by Arvinas, claiming the discovery of potent and selective CRBN-based LRRK2 PROTACs able to degrade LRRK2 in the cerebral cortex after single oral administration in a fPD mouse model with G2019S LRRK2 mutation [193,194]. In parallel, the Ciulli lab discovered the VHL-based degrader XL01126 that has been shown to be a potent degrader of LRRK2 in multiple cell lines and to pass through the BBB after oral or parenteral administration in mice, exhibiting an oral bioavailability of 15% [195]. These attractive in vitro and in vivo properties of XL01126 represent an intriguing starting point for further drug development.
With respect to α-synuclein, a synthetic peptide-based degrader has been shown to degrade intracellular α-synuclein in a recombinant expression system. The degradation of α-synuclein through the proteasome has been shown to rescue mitochondrial defects caused by aberrant α-synuclein accumulation [196]. Moreover, in a recent patent, Arvinas claims the discovery of a series of small-molecule PROTACs, containing a VHL, CRBN, IAP or MDM2 ligand, showing a 65% degradation of α-synuclein at 1 μM in different cell lines [197,198].
5.3.1.3. HD
The aggregation of mutant huntingtin in HD leads to progressive degeneration of neurons in the striatum and cerebral cortex. Small molecule-based bifunctional IAP-based degraders have been developed showing ubiquitination and degradation of mutant huntingtin in fibroblasts derived from HD patients [199]. Further studies are needed to fully exploit this approach to treat HD, as the degradation activity has been observed to also involve the wild-type huntingtin [200].
5.3.1.4. FTD and ALS
The TAR DNA-binding protein 43 (TDP-43) is an example of a misfolded protein implicated in FTD and ALS. The successful removal of the toxic C-terminal form using a heterobifunctional degrader has recently been reported [201].
Given the challenges of developing new drugs for targeting mutated proteins in the brain, further in vitro and in vivo validation in independent laboratories is required to further explore the viability of heterobifunctional as an effective therapeutic tool to treat neurodegenerative disorders.
5.3.2. Molecular Glue Degraders for CNS disorders
In principle, an MGD approach has distinct advantages for the CNS space as compared to heterobifunctional degraders, largely due to the generally much more attractive physicochemical properties of glues.
At this time, no MGDs have been reported for therapeutically relevant CNS targets. Discovery of MGDs here may favour non-aggregating targets, such as LRRK2 described above, however glues for aggregating proteins may also be possible to discover, since it has been demonstrated that heterobifunctional compounds can function in this area, e.g., for tau.
6. Concluding Remarks and Future Perspectives
Heterobifunctional degraders and MGDs are both groundbreaking modalities in drug discovery and therapeutics, where both options have gained attention for their ability to selectively degrade disease-causing proteins. A TPD approach offers advantages over traditional inhibition, potentially overcoming drug resistance and providing a more profound and sustained therapeutic effect.
Moreover, degraders offer a quicker avenue for investigating the molecular mechanisms underlying essential cellular processes in both physiology and pathology, bypassing the need for time-consuming gene silencing approaches.
The further development of these technologies may pave the way for highly specific and efficient therapeutics, with the potential to address previously undruggable targets and improve the overall efficacy and safety of treatments for different therapeutic areas.
This review has captured developments in oncology, neurodegenerative disorders, and inflammation. However, there is clear potential outside of these areas, e.g., for fibrotic disease, anti-infectives, metabolic disorders, etc. As research activities into new discovery approaches, DMPK strategies and novel applications progress, degraders are likely to play a pivotal role in shaping the future landscape of medicine, helping us break bad proteins to alleviate human disease burden.
Funding
We thank the COST Action CA20113 ‘PROTEOCURE’ supported by COST (European Cooperation in Science and Technology). MSR is supported by the Telethon grant number 23115, the JANUS (ANR-23-CE13-0013) and CONACyT-SRE (Mexico) grant 0280365. CB received support from the ARC foundation (ARCDOC42023010005996) and La ligue contre le Cancer (TAJV22303). WX received support from Consejo Nacional de Ciencia y Tecnología (CONACYT) grant CB-2017-2018-A1-S-22895.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1–Cullin–F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Słabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.-D.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK Inhibitor CR8 Acts as a Molecular Glue Degrader That Depletes Cyclin K. Nature 2020, 585, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Chen, P.; Cao, L.; Li, Y.; Zeng, Z.; Cui, Y.; Wu, Q.; Li, J.; Wang, J.-H.; Dong, M.-Q.; et al. Discovery of a Molecular Glue Promoting CDK12-DDB1 Interaction to Trigger Cyclin K Degradation. eLife 2020, 9, e59994. [Google Scholar] [CrossRef] [PubMed]
- Mayor-Ruiz, C.; Bauer, S.; Brand, M.; Kozicka, Z.; Siklos, M.; Imrichova, H.; Kaltheuner, I.H.; Hahn, E.; Seiler, K.; Koren, A.; et al. Rational Discovery of Molecular Glue Degraders via Scalable Chemical Profiling. Nat Chem Biol 2020, 16, 1199–1207. [Google Scholar] [CrossRef]
- Thomas, K.L.; Bouguenina, H.; Miller, D.S.J.; Sialana, F.J.; Hayhow, T.G.; Choudhary, J.S.; Rossanese, O.W.; Bellenie, B.R. Degradation by Design: New Cyclin K Degraders from Old CDK Inhibitors. ACS Chem. Biol. 2024, acschembio.3c00616. [CrossRef]
- Hsia, O.; Hinterndorfer, M.; Cowan, A.D.; Iso, K.; Ishida, T.; Sundaramoorthy, R.; Nakasone, M.A.; Imrichova, H.; Schätz, C.; Rukavina, A.; et al. Targeted Protein Degradation via Intramolecular Bivalent Glues; Biochemistry, 2023.
- Hassan, M.M.; Li, Y.-D.; Ma, M.W.; Teng, M.; Byun, W.S.; Puvar, K.; Lumpkin, R.; Sandoval, B.; Rutter, J.C.; Jin, C.Y.; et al. Exploration of the Tunability of BRD4 Degradation by DCAF16 Trans -Labelling Covalent Glues; Pharmacology and Toxicology, 2023.
- Shergalis, A.G.; Marin, V.L.; Rhee, D.Y.; Senaweera, S.; McCloud, R.L.; Ronau, J.A.; Hutchins, C.W.; McLoughlin, S.; Woller, K.R.; Warder, S.E.; et al. CRISPR Screen Reveals BRD2/4 Molecular Glue-like Degrader via Recruitment of DCAF16. ACS Chem. Biol. 2023, 18, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Advanced Drug Delivery Reviews 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat Chem Biol 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Belcher, B.P.; Ward, C.C.; Nomura, D.K. Ligandability of E3 Ligases for Targeted Protein Degradation Applications. Biochemistry 2023, 62, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Jiang, J.; Wu, Q.; Henning, N.J.; Donovan, K.A.; Yue, H.; Che, J.; Lu, W.; Fischer, E.S.; Bardeesy, N.; et al. Discovery of a Potent Degrader for Fibroblast Growth Factor Receptor 1/2. Angew Chem Int Ed 2021, 60, 15905–15911. [Google Scholar] [CrossRef]
- Shah, R.R.; De Vita, E.; Conole, D.; Sathyamurthi, P.S.; Zhang, X.; Fellows, E.; Fleites, C.M.; Queisser, M.A.; Harling, J.D.; Tate, E.W. Structure-Guided Design and Optimization of Covalent VHL-Targeted Sulfonyl Fluoride PROTACs; Molecular Biology, 2023.
- Nowak, R.P.; Ragosta, L.; Huerta, F.; Liu, H.; Ficarro, S.B.; Cruite, J.T.; Metivier, R.J.; Donovan, K.A.; Marto, J.A.; Fischer, E.S.; et al. Development of a Covalent Cereblon-Based PROTAC Employing a Fluorosulfate Warhead. RSC Chem. Biol. 2023, 4, 906–912. [Google Scholar] [CrossRef]
- Schiemer, J.; Maxwell, A.; Horst, R.; Liu, S.; Uccello, D.P.; Borzilleri, K.; Rajamohan, N.; Brown, M.F.; Calabrese, M.F. A Covalent BTK Ternary Complex Compatible with Targeted Protein Degradation. Nat Commun 2023, 14, 1189. [Google Scholar] [CrossRef]
- Gabizon, R.; Shraga, A.; Gehrtz, P.; Livnah, E.; Shorer, Y.; Gurwicz, N.; Avram, L.; Unger, T.; Aharoni, H.; Albeck, S.; et al. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J. Am. Chem. Soc. 2020, 142, 11734–11742. [Google Scholar] [CrossRef]
- Xue, G.; Chen, J.; Liu, L.; Zhou, D.; Zuo, Y.; Fu, T.; Pan, Z. Protein Degradation through Covalent Inhibitor-Based PROTACs. Chem. Commun. 2020, 56, 1521–1524. [Google Scholar] [CrossRef]
- Zeng, M.; Xiong, Y.; Safaee, N.; Nowak, R.P.; Donovan, K.A.; Yuan, C.J.; Nabet, B.; Gero, T.W.; Feru, F.; Li, L.; et al. Exploring Targeted Degradation Strategy for Oncogenic KRASG12C. Cell Chemical Biology 2020, 27, 19–31.e6. [Google Scholar] [CrossRef]
- Bond, M.J.; Chu, L.; Nalawansha, D.A.; Li, K.; Crews, C.M. Targeted Degradation of Oncogenic KRAS G12C by VHL-Recruiting PROTACs. ACS Cent. Sci. 2020, 6, 1367–1375. [Google Scholar] [CrossRef]
- Mares, A.; Miah, A.H.; Smith, I.E.D.; Rackham, M.; Thawani, A.R.; Cryan, J.; Haile, P.A.; Votta, B.J.; Beal, A.M.; Capriotti, C.; et al. Extended Pharmacodynamic Responses Observed upon PROTAC-Mediated Degradation of RIPK2. Commun Biol 2020, 3, 140. [Google Scholar] [CrossRef] [PubMed]
- Riching, K.M.; Caine, E.A.; Urh, M.; Daniels, D.L. The Importance of Cellular Degradation Kinetics for Understanding Mechanisms in Targeted Protein Degradation. Chem. Soc. Rev. 2022, 51, 6210–6221. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Schulman, B.A. An Expanded Lexicon for the Ubiquitin Code. Nat Rev Mol Cell Biol 2023, 24, 273–287. [Google Scholar] [CrossRef]
- Ishida, T.; Ciulli, A. E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discovery 2021, 26, 484–502. [Google Scholar] [CrossRef] [PubMed]
- Henning, N.J.; Manford, A.G.; Spradlin, J.N.; Brittain, S.M.; Zhang, E.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Rape, M.; Nomura, D.K. Discovery of a Covalent FEM1B Recruiter for Targeted Protein Degradation Applications. J. Am. Chem. Soc. 2022, 144, 701–708. [Google Scholar] [CrossRef]
- Hickey, C.M.; Digianantonio, K.M.; Zimmermann, K.; Harbin, A.; Quinn, C.; Patel, A.; Gareiss, P.; Chapman, A.; Tiberi, B.; Dobrodziej, J.; et al. Co-Opting the E3 Ligase KLHDC2 for Targeted Protein Degradation by Small Molecules. Nat Struct Mol Biol 2024. [CrossRef]
- Zhang, X.; Luukkonen, L.M.; Eissler, C.L.; Crowley, V.M.; Yamashita, Y.; Schafroth, M.A.; Kikuchi, S.; Weinstein, D.S.; Symons, K.T.; Nordin, B.E.; et al. DCAF11 Supports Targeted Protein Degradation by Electrophilic Proteolysis-Targeting Chimeras. J. Am. Chem. Soc. 2021, 143, 5141–5149. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Renatus, M.; Liang, X.; Meili, F.; Zoller, T.; Ferrand, S.; Gauter, F.; Li, X.; Sigoillot, F.; Gleim, S.; et al. DCAF1-Based PROTACs with Activity against Clinically Validated Targets Overcoming Intrinsic- and Acquired-Degrader Resistance. Nat Commun 2024, 15, 275. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, J.; Wang, T.; Luo, M.; Chen, Y.; Chen, C.; Ronai, Z.; Zhou, Y.; Ruppin, E.; Han, L. Expanding PROTACtable Genome Universe of E3 Ligases. Nat Commun 2023, 14, 6509. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Pillow, T.H.; Blake, R.A.; Sadowsky, J.D.; Adaligil, E.; Adhikari, P.; Bhakta, S.; Blaquiere, N.; Chen, J.; Dela Cruz-Chuh, J.; et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 1: Exploration of Antibody Linker, Payload Loading, and Payload Molecular Properties. J. Med. Chem. 2021, 64, 2534–2575. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A Selective BCL-XL PROTAC Degrader Achieves Safe and Potent Antitumor Activity. Nat Med 2019, 25, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.M.; Glennie, L.; Brewer, A.; Zhao, J.-F.; Crooks, J.; Shpiro, N.; Sapkota, G.P. Target Protein Localization and Its Impact on PROTAC-Mediated Degradation. Cell Chemical Biology 2022, 29, 1482–1504.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Crowley, V.M.; Wucherpfennig, T.G.; Dix, M.M.; Cravatt, B.F. Electrophilic PROTACs That Degrade Nuclear Proteins by Engaging DCAF16. Nat Chem Biol 2019, 15, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.; Tsai, W.-T.K.; Kee, Y.-S.; Ruiz, K.; He, J.; Cox, C.; Sun, T.; Penikalapati, S.; Dwivedi, P.; Choi, M.; et al. Antibody Targeting of E3 Ubiquitin Ligases for Receptor Degradation. Nature 2022, 610, 182–189. [Google Scholar] [CrossRef]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef]
- Squair, D.R.; Virdee, S. A New Dawn beyond Lysine Ubiquitination. Nat Chem Biol 2022, 18, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Hornberger, K.R.; Araujo, E.M.V. Physicochemical Property Determinants of Oral Absorption for PROTAC Protein Degraders. J. Med. Chem. 2023, 66, 8281–8287. [Google Scholar] [CrossRef] [PubMed]
- Donovan, K.A.; Ferguson, F.M.; Bushman, J.W.; Eleuteri, N.A.; Bhunia, D.; Ryu, S.; Tan, L.; Shi, K.; Yue, H.; Liu, X.; et al. Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell 2020, 183, 1714–1731.e10. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chemical Biology 2018, 25, 78–87.e5. [Google Scholar] [CrossRef]
- Roy, M.J.; Winkler, S.; Hughes, S.J.; Whitworth, C.; Galant, M.; Farnaby, W.; Rumpel, K.; Ciulli, A. SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 2019, 14, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.-H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat Chem Biol 2017, 13, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.M.; Walz, M.; Meyners, C.; Kuehn, A.; Dreizler, J.K.; Sugiarto, W.O.; Maciel, E.V.S.; Zheng, M.; Lermyte, F.; Hausch, F. Discovery of a Potent Proteolysis Targeting Chimera Enables Targeting the Scaffolding Functions of FK506-Binding Protein 51 (FKBP51). Angew Chem Int Ed 2023, e202309706. [Google Scholar] [CrossRef]
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. BAF Complex Vulnerabilities in Cancer Demonstrated via Structure-Based PROTAC Design. Nat Chem Biol 2019, 15, 672–680. [Google Scholar] [CrossRef]
- Kaneshige, A.; Bai, L.; Wang, M.; McEachern, D.; Meagher, J.L.; Xu, R.; Wang, Y.; Jiang, W.; Metwally, H.; Kirchhoff, P.D.; et al. A Selective Small-Molecule STAT5 PROTAC Degrader Capable of Achieving Tumor Regression in Vivo. Nat Chem Biol 2023, 19, 703–711. [Google Scholar] [CrossRef]
- Wurz, R.P.; Rui, H.; Dellamaggiore, K.; Ghimire-Rijal, S.; Choi, K.; Smither, K.; Amegadzie, A.; Chen, N.; Li, X.; Banerjee, A.; et al. Affinity and Cooperativity Modulate Ternary Complex Formation to Drive Targeted Protein Degradation. Nat Commun 2023, 14, 4177. [Google Scholar] [CrossRef]
- Chung, C.; Dai, H.; Fernandez, E.; Tinworth, C.P.; Churcher, I.; Cryan, J.; Denyer, J.; Harling, J.D.; Konopacka, A.; Queisser, M.A.; et al. Structural Insights into PROTAC-Mediated Degradation of Bcl-XL. ACS Chem. Biol. 2020, 15, 2316–2323. [Google Scholar] [CrossRef]
- Zorba, A.; Nguyen, C.; Xu, Y.; Starr, J.; Borzilleri, K.; Smith, J.; Zhu, H.; Farley, K.A.; Ding, W.; Schiemer, J.; et al. Delineating the Role of Cooperativity in the Design of Potent PROTACs for BTK. Proc. Natl. Acad. Sci. U.S.A. 2018, 115. [Google Scholar] [CrossRef]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING Finger Families of E3 Ubiquitin Ligases at a Glance. Journal of Cell Science 2012, 125, 531–537. [Google Scholar] [CrossRef]
- Budhidarmo, R.; Nakatani, Y.; Day, C.L. RINGs Hold the Key to Ubiquitin Transfer. Trends in Biochemical Sciences 2012, 37, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Rotin, D.; Kumar, S. Physiological Functions of the HECT Family of Ubiquitin Ligases. Nat Rev Mol Cell Biol 2009, 10, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Wolberger, C. New Insights into Ubiquitin E3 Ligase Mechanism. Nat Struct Mol Biol 2014, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 Ubiquitin Ligases: New Structures, New Insights, New Questions. Biochemical Journal 2014, 458, 421–437. [Google Scholar] [CrossRef]
- Cotton, T.R.; Lechtenberg, B.C. Chain Reactions: Molecular Mechanisms of RBR Ubiquitin Ligases. Biochemical Society Transactions 2020, 48, 1737–1750. [Google Scholar] [CrossRef]
- Zimmerman, E.S.; Schulman, B.A.; Zheng, N. Structural Assembly of Cullin-RING Ubiquitin Ligase Complexes. Current Opinion in Structural Biology 2010, 20, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Sarikas, A.; Hartmann, T.; Pan, Z.-Q. The Cullin Protein Family. Genome Biol 2011, 12, 220. [Google Scholar] [CrossRef]
- Zheng, N.; Schulman, B.A.; Song, L.; Miller, J.J.; Jeffrey, P.D.; Wang, P.; Chu, C.; Koepp, D.M.; Elledge, S.J.; Pagano, M.; et al. Structure of the Cul1–Rbx1–Skp1–F BoxSkp2 SCF Ubiquitin Ligase Complex. Nature 2002, 416, 703–709. [Google Scholar] [CrossRef]
- Kamura, T.; Maenaka, K.; Kotoshiba, S.; Matsumoto, M.; Kohda, D.; Conaway, R.C.; Conaway, J.W.; Nakayama, K.I. VHL-Box and SOCS-Box Domains Determine Binding Specificity for Cul2-Rbx1 and Cul5-Rbx2 Modules of Ubiquitin Ligases. Genes Dev. 2004, 18, 3055–3065. [Google Scholar] [CrossRef]
- Mahrour, N.; Redwine, W.B.; Florens, L.; Swanson, S.K.; Martin-Brown, S.; Bradford, W.D.; Staehling-Hampton, K.; Washburn, M.P.; Conaway, R.C.; Conaway, J.W. Characterization of Cullin-Box Sequences That Direct Recruitment of Cul2-Rbx1 and Cul5-Rbx2 Modules to Elongin BC-Based Ubiquitin Ligases. Journal of Biological Chemistry 2008, 283, 8005–8013. [Google Scholar] [CrossRef]
- Xu, L.; Wei, Y.; Reboul, J.; Vaglio, P.; Shin, T.-H.; Vidal, M.; Elledge, S.J.; Harper, J.W. BTB Proteins Are Substrate-Specific Adaptors in an SCF-like Modular Ubiquitin Ligase Containing CUL-3. Nature 2003, 425, 316–321. [Google Scholar] [CrossRef]
- Geyer, R.; Wee, S.; Anderson, S.; Yates, J.; Wolf, D.A. BTB/POZ Domain Proteins Are Putative Substrate Adaptors for Cullin 3 Ubiquitin Ligases. Molecular Cell 2003, 12, 783–790. [Google Scholar] [CrossRef]
- Hannah, J.; Zhou, P. Distinct and Overlapping Functions of the Cullin E3 Ligase Scaffolding Proteins CUL4A and CUL4B. Gene 2015, 573, 33–45. [Google Scholar] [CrossRef]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular Architecture and Assembly of the DDB1–CUL4A Ubiquitin Ligase Machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.S.; Scrima, A.; Böhm, K.; Matsumoto, S.; Lingaraju, G.M.; Faty, M.; Yasuda, T.; Cavadini, S.; Wakasugi, M.; Hanaoka, F.; et al. The Molecular Basis of CRL4DDB2/CSA Ubiquitin Ligase Architecture, Targeting, and Activation. Cell 2011, 147, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Skaar, J.R.; Pagan, J.K.; Pagano, M. Mechanisms and Function of Substrate Recruitment by F-Box Proteins. Nat Rev Mol Cell Biol 2013, 14, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Reitsma, J.M.; Mamrosh, J.L.; Zhang, Y.; Straube, R.; Deshaies, R.J. Cand1-Mediated Adaptive Exchange Mechanism Enables Variation in F-Box Protein Expression. Molecular Cell 2018, 69, 773–786.e6. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-Type E3 Ligases: Master Manipulators of E2 Ubiquitin-Conjugating Enzymes and Ubiquitination. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2014, 1843, 47–60. [Google Scholar] [CrossRef]
- Lucas, X.; Ciulli, A. Recognition of Substrate Degrons by E3 Ubiquitin Ligases and Modulation by Small-Molecule Mimicry Strategies. Current Opinion in Structural Biology 2017, 44, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, J.S.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.; Crews, C.M. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef] [PubMed]
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57, 8657–8663. [Google Scholar] [CrossRef] [PubMed]
- Soares, P.; Gadd, M.S.; Frost, J.; Galdeano, C.; Ellis, L.; Epemolu, O.; Rocha, S.; Read, K.D.; Ciulli, A. Group-Based Optimization of Potent and Cell-Active Inhibitors of the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase: Structure–Activity Relationships Leading to the Chemical Probe (2 S,4 R )-1-(( S )-2-(1-Cyanocyclopropanecarboxamido)-3,3-Dimethylbutanoyl)-4-Hydroxy- N -(4-(4-Methylthiazol-5-Yl)Benzyl)Pyrrolidine-2-Carboxamide (VH298). J. Med. Chem. 2018, 61, 599–618. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, P.P.; Lopez-Girona, A.; Miller, K.; Carmel, G.; Pagarigan, B.; Chie-Leon, B.; Rychak, E.; Corral, L.G.; Ren, Y.J.; Wang, M.; et al. Structure of the Human Cereblon–DDB1–Lenalidomide Complex Reveals Basis for Responsiveness to Thalidomide Analogs. Nat Struct Mol Biol 2014, 21, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide Induces Ubiquitination and Degradation of CK1α in Del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Słabicki, M.; Yoon, H.; Koeppel, J.; Nitsch, L.; Roy Burman, S.S.; Di Genua, C.; Donovan, K.A.; Sperling, A.S.; Hunkeler, M.; Tsai, J.M.; et al. Small-Molecule-Induced Polymerization Triggers Degradation of BCL6. Nature 2020, 588, 164–168. [Google Scholar] [CrossRef]
- House, C.M.; Frew, I.J.; Huang, H.-L.; Wiche, G.; Traficante, N.; Nice, E.; Catimel, B.; Bowtell, D.D.L. A Binding Motif for Siah Ubiquitin Ligase. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3101–3106. [Google Scholar] [CrossRef] [PubMed]
- McCoull, W.; Cheung, T.; Anderson, E.; Barton, P.; Burgess, J.; Byth, K.; Cao, Q.; Castaldi, M.P.; Chen, H.; Chiarparin, E.; et al. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6. ACS Chem. Biol. 2018, 13, 3131–3141. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemistry & Biology 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Wu, Y.; Xing, D. Developments of CRBN-Based PROTACs as Potential Therapeutic Agents. European Journal of Medicinal Chemistry 2021, 225, 113749. [Google Scholar] [CrossRef]
- Cieślak, M.; Słowianek, M. Cereblon-Recruiting PROTACs: Will New Drugs Have to Face Old Challenges? Pharmaceutics 2023, 15, 812. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Robert, E.I.; Van Breugel, P.C.; Strubin, M.; Zheng, N. A Promiscuous α-Helical Motif Anchors Viral Hijackers and Substrate Receptors to the CUL4–DDB1 Ubiquitin Ligase Machinery. Nat Struct Mol Biol 2010, 17, 105–111. [Google Scholar] [CrossRef]
- Meyers, M.; Cismoski, S.; Panidapu, A.; Chie-Leon, B.; Nomura, D.K. Targeted Protein Degradation through Recruitment of the CUL4 Complex Adaptor Protein DDB1. ACS Chem. Biol. 2024, 19, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Divakaran, A.; Osa, A.; Huang, O.W.; Wertz, I.E.; Nomura, D.K. Exploiting the Cullin E3 Ligase Adaptor Protein SKP1 for Targeted Protein Degradation. ACS Chem. Biol. 2024, acschembio.3c00642. [CrossRef]
- King, E.A.; Cho, Y.; Hsu, N.S.; Dovala, D.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Nomura, D.K. Chemoproteomics-Enabled Discovery of a Covalent Molecular Glue Degrader Targeting NF-ΚB. Cell Chemical Biology 2023, 30, 394-402.e9. [CrossRef]
- Forte, N.; Dovala, D.; Hesse, M.J.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Nomura, D.K. Targeted Protein Degradation through E2 Recruitment. ACS Chem. Biol. 2023, 18, 897–904. [Google Scholar] [CrossRef]
- Bashore, C.; Prakash, S.; Johnson, M.C.; Conrad, R.J.; Kekessie, I.A.; Scales, S.J.; Ishisoko, N.; Kleinheinz, T.; Liu, P.S.; Popovych, N.; et al. Targeted Degradation via Direct 26S Proteasome Recruitment. Nat Chem Biol 2023, 19, 55–63. [Google Scholar] [CrossRef]
- Alessio CIULLI, Andrea TESTA, Scott Hughes, Steven Peter Butcher Bifunctional Molecules for Targeting Uchl5.
- Alessio CIULLI, Andrea TESTA, Scott Hughes, Steven Peter Butcher Bifunctional Molecules for Targeting Rpn11.
- Andrea TESTA, Scott Hughes, Steven Peter Butcher, Alessio CIULLI Bifunctional Molecules for Targeting Usp14.
- Ali, E.M.H.; Loy, C.A.; Trader, D.J. ByeTAC: Bypassing an E3 Ligase for Targeted Protein Degradation; Biochemistry, 2024.
- Miyamoto-Sato, E.; Imanishi, S.; Huang, L.; Itakura, S.; Iwasaki, Y.; Ishizaka, M. A First-Class Degrader Candidate Targeting Both KRAS G12D and G12V Mediated by CANDDY Technology Independent of Ubiquitination. Molecules 2023, 28, 5600. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.-Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498-511.e17. [CrossRef]
- Tong, B.; Spradlin, J.N.; Novaes, L.F.T.; Zhang, E.; Hu, X.; Moeller, M.; Brittain, S.M.; McGregor, L.M.; McKenna, J.M.; Tallarico, J.A.; et al. A Nimbolide-Based Kinase Degrader Preferentially Degrades Oncogenic BCR-ABL. ACS Chem. Biol. 2020, 15, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Spradlin, J.N.; Boike, L.; Tong, B.; Brittain, S.M.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Maimone, T.J.; Nomura, D.K. Chemoproteomics-Enabled Discovery of Covalent RNF114-Based Degraders That Mimic Natural Product Function. Cell Chemical Biology 2021, 28, 559-566.e15. [CrossRef]
- De Nardo, D.; Balka, K.R.; Cardona Gloria, Y.; Rao, V.R.; Latz, E.; Masters, S.L. Interleukin-1 Receptor–Associated Kinase 4 (IRAK4) Plays a Dual Role in Myddosome Formation and Toll-like Receptor Signaling. Journal of Biological Chemistry 2018, 293, 15195–15207. [Google Scholar] [CrossRef]
- Li, W.; Sano, R.; Apatira, M.; DeAnda, F.; Gururaja, T.; Yang, M.; Lundgaard, G.; Pan, C.; Liu, J.; Zhai, Y.; et al. Bruton’s Tyrosine Kinase Inhibitors with Distinct Binding Modes Reveal Differential Functional Impact on B-Cell Receptor Signaling. Molecular Cancer Therapeutics 2024, 23, 35–46. [Google Scholar] [CrossRef]
- Montoya, S.; Bourcier, J.; Noviski, M.; Lu, H.; Thompson, M.C.; Chirino, A.; Jahn, J.; Sondhi, A.K.; Gajewski, S.; Tan, Y.S.; et al. Kinase-Impaired BTK Mutations Are Susceptible to Clinical-Stage BTK and IKZF1/3 Degrader NX-2127. Science 2024, 383, eadi5798. [CrossRef]
- Yuan, H.; Zhu, Y.; Cheng, Y.; Hou, J.; Jin, F.; Li, M.; Jia, W.; Cheng, Z.; Xing, H.; Liu, M.; et al. BTK Kinase Activity Is Dispensable for the Survival of Diffuse Large B-Cell Lymphoma. Journal of Biological Chemistry 2022, 298, 102555. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, L.; Acloque, G.; Bacchelli, S.; Schwartz, H.; Feinstein, B.J.; La Stella, P.; Alavi, A.; Gollerkeri, A.; Davis, J.; Campbell, V.; et al. IRAK4 Degrader in Hidradenitis Suppurativa and Atopic Dermatitis: A Phase 1 Trial. Nat Med 2023, 29, 3127–3136. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Sig Transduct Target Ther 2020, 5, 213. [Google Scholar] [CrossRef]
- Wang, Z.; He, N.; Guo, Z.; Niu, C.; Song, T.; Guo, Y.; Cao, K.; Wang, A.; Zhu, J.; Zhang, X.; et al. Proteolysis Targeting Chimeras for the Selective Degradation of Mcl-1/Bcl-2 Derived from Nonselective Target Binding Ligands. J. Med. Chem. 2019, 62, 8152–8163. [Google Scholar] [CrossRef]
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [Google Scholar] [CrossRef]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a Selective Inhibitor of the Abl Tyrosine Kinase on the Growth of Bcr–Abl Positive Cells. Nat Med 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed 2016, 55, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ding, X.; Liu, L.; Mi, Q.; Zhao, Q.; Shao, Y.; Ren, C.; Chen, J.; Kong, Y.; Qiu, X.; et al. Discovery of Novel BCR-ABL PROTACs Based on the Cereblon E3 Ligase Design, Synthesis, and Biological Evaluation. European Journal of Medicinal Chemistry 2021, 223, 113645. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Schultz, A.R.; Bondeson, D.P.; Eide, C.A.; Savage Stevens, S.L.; Druker, B.J.; Crews, C.M. Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Research 2019, 79, 4744–4753. [Google Scholar] [CrossRef]
- Lyczek, A.; Berger, B.-T.; Rangwala, A.M.; Paung, Y.; Tom, J.; Philipose, H.; Guo, J.; Albanese, S.K.; Robers, M.B.; Knapp, S.; et al. Mutation in Abl Kinase with Altered Drug-Binding Kinetics Indicates a Novel Mechanism of Imatinib Resistance. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2111451118. [Google Scholar] [CrossRef]
- Heinlein, C.A.; Chang, C. Androgen Receptor in Prostate Cancer. Endocrine Reviews 2004, 25, 276–308. [Google Scholar] [CrossRef]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; Watson, P.A.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L. Overcoming Mutation-Based Resistance to Antiandrogens with Rational Drug Design. eLife 2013, 2, e00499. [Google Scholar] [CrossRef]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen Receptor Degradation by the Proteolysis-Targeting Chimera ARCC-4 Outperforms Enzalutamide in Cellular Models of Prostate Cancer Drug Resistance. Commun Biol 2018, 1, 100. [Google Scholar] [CrossRef]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen Receptor Degraders Overcome Common Resistance Mechanisms Developed during Prostate Cancer Treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef]
- Lee, G.T.; Nagaya, N.; Desantis, J.; Madura, K.; Sabaawy, H.E.; Kim, W.-J.; Vaz, R.J.; Cruciani, G.; Kim, I.Y. Effects of MTX-23, a Novel PROTAC of Androgen Receptor Splice Variant-7 and Androgen Receptor, on CRPC Resistant to Second-Line Antiandrogen Therapy. Molecular Cancer Therapeutics 2021, 20, 490–499. [Google Scholar] [CrossRef]
- Shirasaki, R.; Matthews, G.M.; Gandolfi, S.; De Matos Simoes, R.; Buckley, D.L.; Raja Vora, J.; Sievers, Q.L.; Brüggenthies, J.B.; Dashevsky, O.; Poarch, H.; et al. Functional Genomics Identify Distinct and Overlapping Genes Mediating Resistance to Different Classes of Heterobifunctional Degraders of Oncoproteins. Cell Reports 2021, 34, 108532. [Google Scholar] [CrossRef]
- Zhang, L.; Riley-Gillis, B.; Vijay, P.; Shen, Y. Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Molecular Cancer Therapeutics 2019, 18, 1302–1311. [Google Scholar] [CrossRef]
- Ottis, P.; Palladino, C.; Thienger, P.; Britschgi, A.; Heichinger, C.; Berrera, M.; Julien-Laferriere, A.; Roudnicky, F.; Kam-Thong, T.; Bischoff, J.R.; et al. Cellular Resistance Mechanisms to Targeted Protein Degradation Converge Toward Impairment of the Engaged Ubiquitin Transfer Pathway. ACS Chem. Biol. 2019, acschembio.9b00525. [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat Rev Drug Discov 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 Degrader via Conjugation of Ineffectual Bromodomain and VHL Ligands. Nat Chem Biol 2018, 14, 405–412. [Google Scholar] [CrossRef]
- Prudent, R.; Annis, D.A.; Dandliker, P.J.; Ortholand, J.-Y.; Roche, D. Exploring New Targets and Chemical Space with Affinity Selection-Mass Spectrometry. Nat Rev Chem 2020, 5, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Disch, J.S.; Duffy, J.M.; Lee, E.C.Y.; Gikunju, D.; Chan, B.; Levin, B.; Monteiro, M.I.; Talcott, S.A.; Lau, A.C.; Zhou, F.; et al. Bispecific Estrogen Receptor α Degraders Incorporating Novel Binders Identified Using DNA-Encoded Chemical Library Screening. J. Med. Chem. 2021, 64, 5049–5066. [Google Scholar] [CrossRef] [PubMed]
- Chana, C.K.; Maisonneuve, P.; Posternak, G.; Grinberg, N.G.A.; Poirson, J.; Ona, S.M.; Ceccarelli, D.F.; Mader, P.; St-Cyr, D.J.; Pau, V.; et al. Discovery and Structural Characterization of Small Molecule Binders of the Human CTLH E3 Ligase Subunit GID4. J. Med. Chem. 2022, 65, 12725–12746. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.K.; Perveen, S.; Song, X.; Dong, A.; Szewczyk, M.M.; Calabrese, M.F.; Casimiro-Garcia, A.; Chakrapani, S.; Dowling, M.S.; Ficici, E.; et al. Chemical Tools for the Gid4 Subunit of the Human E3 Ligase C-Terminal to LisH (CTLH) Degradation Complex; Biophysics, 2023.
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorganic & Medicinal Chemistry Letters 2008, 18, 5904–5908. [CrossRef]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The DTAG System for Immediate and Target-Specific Protein Degradation. Nat Chem Biol 2018, 14, 431–441. [Google Scholar] [CrossRef]
- Bensimon, A.; Pizzagalli, M.D.; Kartnig, F.; Dvorak, V.; Essletzbichler, P.; Winter, G.E.; Superti-Furga, G. Targeted Degradation of SLC Transporters Reveals Amenability of Multi-Pass Transmembrane Proteins to Ligand-Induced Proteolysis. Cell Chemical Biology 2020, 27, 728-739.e9. [CrossRef]
- Ege, N.; Bouguenina, H.; Tatari, M.; Chopra, R. Phenotypic Screening with Target Identification and Validation in the Discovery and Development of E3 Ligase Modulators. Cell Chemical Biology 2021, 28, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; He, M.; Wang, L.; He, Y.; Rao, Y. Chemistries of Bifunctional PROTAC Degraders. Chem. Soc. Rev. 2022, 51, 7066–7114. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Drummond, M.L.; Williams, C.I. In Silico Modeling of PROTAC-Mediated Ternary Complexes: Validation and Application. J. Chem. Inf. Model. 2019, 59, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Drummond, M.L.; Henry, A.; Li, H.; Williams, C.I. Improved Accuracy for Modeling PROTAC-Mediated Ternary Complex Formation and Targeted Protein Degradation via New In Silico Methodologies. J. Chem. Inf. Model. 2020, 60, 5234–5254. [Google Scholar] [CrossRef] [PubMed]
- Zaidman, D.; Prilusky, J.; London, N. PRosettaC: Rosetta Based Modeling of PROTAC Mediated Ternary Complexes. J. Chem. Inf. Model. 2020, 60, 4894–4903. [Google Scholar] [CrossRef] [PubMed]
- Bai, N.; Miller, S.A.; Andrianov, G.V.; Yates, M.; Kirubakaran, P.; Karanicolas, J. Rationalizing PROTAC-Mediated Ternary Complex Formation Using Rosetta. J. Chem. Inf. Model. 2021, 61, 1368–1382. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Li, D.; Kang, Y.; Hou, T. Integrative Modeling of PROTAC-Mediated Ternary Complexes. J. Med. Chem. 2021, 64, 16271–16281. [Google Scholar] [CrossRef]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current Strategies for the Design of PROTAC Linkers: A Critical Review.Exploration of Targeted Anti-tumor Therapy. 2020; 1. [Google Scholar] [CrossRef]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great Opportunities for Academia and Industry. Sig Transduct Target Ther 2019, 4, 64. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Small-Molecule Modulation of Protein Homeostasis. Chem. Rev. 2017, 117, 11269–11301. [Google Scholar] [CrossRef]
- Maple, H.J.; Clayden, N.; Baron, A.; Stacey, C.; Felix, R. Developing Degraders: Principles and Perspectives on Design and Chemical Space. Med. Chem. Commun. 2019, 10, 1755–1764. [Google Scholar] [CrossRef]
- Borsari, C.; Trader, D.J.; Tait, A.; Costi, M.P. Designing Chimeric Molecules for Drug Discovery by Leveraging Chemical Biology. J. Med. Chem. 2020, 63, 1908–1928. [Google Scholar] [CrossRef]
- Hendrick, C.E.; Jorgensen, J.R.; Chaudhry, C.; Strambeanu, I.I.; Brazeau, J.-F.; Schiffer, J.; Shi, Z.; Venable, J.D.; Wolkenberg, S.E. Direct-to-Biology Accelerates PROTAC Synthesis and the Evaluation of Linker Effects on Permeability and Degradation. ACS Med. Chem. Lett. 2022, 13, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Merritt, A.; Skerratt, S.; Swarbrick, M.E. Recent Trends in Medicinal Chemistry and Enabling Technologies. Highlights from the Society for Medicines Research Conference. London - December 8, 2022. Drugs of the Future 2023, 48, 211–219. [CrossRef]
- Plesniak, M.P.; Taylor, E.K.; Eisele, F.; Kourra, C.M.B.K.; Michaelides, I.N.; Oram, A.; Wernevik, J.; Valencia, Z.S.; Rowbottom, H.; Mann, N.; et al. Rapid PROTAC Discovery Platform: Nanomole-Scale Array Synthesis and Direct Screening of Reaction Mixtures. ACS Med. Chem. Lett. 2023, 14, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon Is a Direct Protein Target for Immunomodulatory and Antiproliferative Activities of Lenalidomide and Pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Owa, T.; Yoshino, H.; Okauchi, T.; Yoshimatsu, K.; Ozawa, Y.; Sugi, N.H.; Nagasu, T.; Koyanagi, N.; Kitoh, K. Discovery of Novel Antitumor Sulfonamides Targeting G1 Phase of the Cell Cycle. J. Med. Chem. 1999, 42, 3789–3799. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer Sulfonamides Target Splicing by Inducing RBM39 Degradation via Recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef] [PubMed]
- Bussiere, D.E.; Xie, L.; Srinivas, H.; Shu, W.; Burke, A.; Be, C.; Zhao, J.; Godbole, A.; King, D.; Karki, R.G.; et al. Structural Basis of Indisulam-Mediated RBM39 Recruitment to DCAF15 E3 Ligase Complex. Nat Chem Biol 2020, 16, 15–23. [Google Scholar] [CrossRef]
- Kozicka, Z.; Suchyta, D.J.; Focht, V.; Kempf, G.; Petzold, G.; Jentzsch, M.; Zou, C.; Di Genua, C.; Donovan, K.A.; Coomar, S.; et al. Design Principles for Cyclin K Molecular Glue Degraders. Nat Chem Biol 2024, 20, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.-A.; Singh, S.; Han, H.; Davis, C.T.; Borgeson, B.; Hartland, C.; Kost-Alimova, M.; Gustafsdottir, S.M.; Gibson, C.C.; Carpenter, A.E. Cell Painting, a High-Content Image-Based Assay for Morphological Profiling Using Multiplexed Fluorescent Dyes. Nat Protoc 2016, 11, 1757–1774. [Google Scholar] [CrossRef]
- Ng, A.; Offensperger, F.; Cisneros, J.A.; Scholes, N.S.; Malik, M.; Villanti, L.; Rukavina, A.; Ferrada, E.; Hannich, J.T.; Koren, A.; et al. Discovery of Molecular Glue Degraders via Isogenic Morphological Profiling. ACS Chem. Biol. 2023, 18, 2464–2473. [Google Scholar] [CrossRef]
- Bonazzi, S.; d’Hennezel, E.; Beckwith, R.E.J.; Xu, L.; Fazal, A.; Magracheva, A.; Ramesh, R.; Cernijenko, A.; Antonakos, B.; Bhang, H.C.; et al. Discovery and Characterization of a Selective IKZF2 Glue Degrader for Cancer Immunotherapy. Cell Chemical Biology 2023, 30, 235-247.e12. [CrossRef]
- Tutter, A.; Buckley, D.; Golosov, A.A.; Ma, X.; Shu, W.; McKay, D.J.J.; Darsigny, V.; Dovala, D.; Beckwith, R.; Solomon, J.; et al. A Small Molecule VHL Molecular Glue Degrader for Cysteine Dioxygenase 1; Biochemistry, 2024.
- Simonetta, K.R.; Taygerly, J.; Boyle, K.; Basham, S.E.; Padovani, C.; Lou, Y.; Cummins, T.J.; Yung, S.L.; Von Soly, S.K.; Kayser, F.; et al. Prospective Discovery of Small Molecule Enhancers of an E3 Ligase-Substrate Interaction. Nat Commun 2019, 10, 1402. [Google Scholar] [CrossRef]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat Commun 2019, 10, 131. [Google Scholar] [CrossRef]
- Daniels, D.L.; Riching, K.M.; Urh, M. Monitoring and Deciphering Protein Degradation Pathways inside Cells. Drug Discovery Today: Technologies 2019, 31, 61–68. [CrossRef]
- Riching, K.M.; Mahan, S.; Corona, C.R.; McDougall, M.; Vasta, J.D.; Robers, M.B.; Urh, M.; Daniels, D.L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13, 2758–2770. [Google Scholar] [CrossRef] [PubMed]
- Hyer, M.L.; Milhollen, M.A.; Ciavarri, J.; Fleming, P.; Traore, T.; Sappal, D.; Huck, J.; Shi, J.; Gavin, J.; Brownell, J.; et al. A Small-Molecule Inhibitor of the Ubiquitin Activating Enzyme for Cancer Treatment. Nat Med 2018, 24, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An Inhibitor of NEDD8-Activating Enzyme as a New Approach to Treat Cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Nowak, R.P.; DeAngelo, S.L.; Buckley, D.; He, Z.; Donovan, K.A.; An, J.; Safaee, N.; Jedrychowski, M.P.; Ponthier, C.M.; Ishoey, M.; et al. Plasticity in Binding Confers Selectivity in Ligand-Induced Protein Degradation. Nat Chem Biol 2018, 14, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef]
- Hjerpe, R.; Aillet, F.; Lopitz-Otsoa, F.; Lang, V.; England, P.; Rodriguez, M.S. Efficient Protection and Isolation of Ubiquitylated Proteins Using Tandem Ubiquitin-binding Entities. EMBO Reports 2009, 10, 1250–1258. [Google Scholar] [CrossRef]
- Serna, S.; Xolalpa, W.; Lang, V.; Aillet, F.; England, P.; Reichardt, N.; Rodriguez, M.S. Efficient Monitoring of Protein Ubiquitylation Levels Using TUBE S-based Microarrays. FEBS Letters 2016, 590, 2748–2756. [Google Scholar] [CrossRef]
- Huang, H.-T.; Dobrovolsky, D.; Paulk, J.; Yang, G.; Weisberg, E.L.; Doctor, Z.M.; Buckley, D.L.; Cho, J.-H.; Ko, E.; Jang, J.; et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-Kinase Degrader. Cell Chemical Biology 2018, 25, 88-99.e6. [CrossRef]
- Zeng, S.; Ye, Y.; Xia, H.; Min, J.; Xu, J.; Wang, Z.; Pan, Y.; Zhou, X.; Huang, W. Current Advances and Development Strategies of Orally Bioavailable PROTACs. European Journal of Medicinal Chemistry 2023, 261, 115793. [Google Scholar] [CrossRef]
- Schott, A.F.; Hurvitz, S.; Ma, C.; Hamilton, E.; Nanda, R.; Zahrah, G.; Hunter, N.; Tan, A.R.; Telli, M.; Mesias, J.A.; et al. Abstract GS3-03: GS3-03 ARV-471, a PROTAC® Estrogen Receptor (ER) Degrader in Advanced ER-Positive/Human Epidermal Growth Factor Receptor 2 (HER2)-Negative Breast Cancer: Phase 2 Expansion (VERITAC) of a Phase 1/2 Study. Cancer Research 2023, 83, GS3-03-GS3-03. [CrossRef]
- Gao, X.; Burris Iii, H.A.; Vuky, J.; Dreicer, R.; Sartor, A.O.; Sternberg, C.N.; Percent, I.J.; Hussain, M.H.A.; Rezazadeh Kalebasty, A.; Shen, J.; et al. Phase 1/2 Study of ARV-110, an Androgen Receptor (AR) PROTAC Degrader, in Metastatic Castration-Resistant Prostate Cancer (MCRPC). JCO 2022, 40, 17–17. [Google Scholar] [CrossRef]
- Lee, D.K.; Chang, C. Expression and Degradation of Androgen Receptor: Mechanism and Clinical Implication. The Journal of Clinical Endocrinology & Metabolism 2003, 88, 4043–4054. [CrossRef]
- Lung, D.K.; Warrick, J.W.; Hematti, P.; Callander, N.S.; Mark, C.J.; Miyamoto, S.; Alarid, E.T. Bone Marrow Stromal Cells Transcriptionally Repress ESR1 but Cannot Overcome Constitutive ESR1 Mutant Activity. Endocrinology 2019, 160, 2427–2440. [Google Scholar] [CrossRef]
- Chirnomas, D.; Hornberger, K.R.; Crews, C.M. Protein Degraders Enter the Clinic — a New Approach to Cancer Therapy. Nat Rev Clin Oncol 2023, 20, 265–278. [Google Scholar] [CrossRef]
- Muller, F.L.; Aquilanti, E.A.; DePinho, R.A. Collateral Lethality: A New Therapeutic Strategy in Oncology. Trends in Cancer 2015, 1, 161–173. [Google Scholar] [CrossRef]
- Hoffman, G.R.; Rahal, R.; Buxton, F.; Xiang, K.; McAllister, G.; Frias, E.; Bagdasarian, L.; Huber, J.; Lindeman, A.; Chen, D.; et al. Functional Epigenetics Approach Identifies BRM/SMARCA2 as a Critical Synthetic Lethal Target in BRG1-Deficient Cancers. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 3128–3133. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.; March, N.; Thomson, G.; Fairweather, E.; Fritzl, S.; Hitchin, J.; Hamilton, N.; Jordan, A.; Waddell, I.; Ogilvie, D. Abstract 5429: Inhibition of SMARCA2: A Novel Target for SMARCA4-Deficient Lung Adenocarcinoma. Cancer Research 2015, 75, 5429–5429. [Google Scholar] [CrossRef]
- Hulse, M.; Agarwal, A.; Wang, M.; Carter, J.; Sivakumar, M.; Vidal, B.; Brown, J.; Moore, A.; Grego, A.; Bhagwat, N.; et al. Abstract 3263: Preclinical Characterization of PRT3789, a Potent and Selective SMARCA2 Targeted Degrader. Cancer Research 2022, 82, 3263–3263. [Google Scholar] [CrossRef]
- Miah, A.H.; Smith, I.E.D.; Rackham, M.; Mares, A.; Thawani, A.R.; Nagilla, R.; Haile, P.A.; Votta, B.J.; Gordon, L.J.; Watt, G.; et al. Optimization of a Series of RIPK2 PROTACs. J. Med. Chem. 2021, 64, 12978–13003. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Korenaga, S.; Iwakura, M. Discovery of a Potent and Subtype-Selective TYK2 Degrader Based on an Allosteric TYK2 Inhibitor. Bioorganic & Medicinal Chemistry Letters 2023, 79, 129083. [CrossRef]
- Huang, J.; Ma, Z.; Yang, Z.; He, Z.; Bao, J.; Peng, X.; Liu, Y.; Chen, T.; Cai, S.; Chen, J.; et al. Discovery of Ibrutinib-Based BTK PROTACs with in Vivo Anti-Inflammatory Efficacy by Inhibiting NF-ΚB Activation. European Journal of Medicinal Chemistry 2023, 259, 115664. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.-R.; Yang, W.-G.; Hou, X.-H.; Shen, D.-D.; Zhang, S.-N.; Li, Y.; Qiao, Y.-Y.; Wang, S.-Q.; Yuan, S.; Liu, H.-M. The Recent Advance of Interleukin-1 Receptor Associated Kinase 4 Inhibitors for the Treatment of Inflammation and Related Diseases. European Journal of Medicinal Chemistry 2023, 258, 115606. [Google Scholar] [CrossRef]
- Gribkoff, V.K.; Kaczmarek, L.K. The Need for New Approaches in CNS Drug Discovery: Why Drugs Have Failed, and What Can Be Done to Improve Outcomes. Neuropharmacology 2017, 120, 11–19. [Google Scholar] [CrossRef]
- Tashima, T. Proteolysis-Targeting Chimera (PROTAC) Delivery into the Brain across the Blood-Brain Barrier. Antibodies (Basel) 2023, 12, 43. [Google Scholar] [CrossRef]
- Tashima, T. Delivery of Intravenously Administered Antibodies Targeting Alzheimer’s Disease-Relevant Tau Species into the Brain Based on Receptor-Mediated Transcytosis. Pharmaceutics 2022, 14, 411. [Google Scholar] [CrossRef] [PubMed]
- Potjewyd, F.M.; Axtman, A.D. Exploration of Aberrant E3 Ligases Implicated in Alzheimer’s Disease and Development of Chemical Tools to Modulate Their Function. Front. Cell. Neurosci. 2021, 15, 768655. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.A.I.; Lewis, H.L.; Jones, D.H.; Ward, S.E. Central Nervous System Targeted Protein Degraders. Biomolecules 2023, 13, 1164. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.-T.; Gao, N.; Li, Q.-Q.; Chen, P.-G.; Yang, X.-F.; Chen, Y.-X.; Zhao, Y.-F.; Li, Y.-M. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chemical Biology 2016, 23, 453–461. [Google Scholar] [CrossRef]
- Lu, M.; Liu, T.; Jiao, Q.; Ji, J.; Tao, M.; Liu, Y.; You, Q.; Jiang, Z. Discovery of a Keap1-Dependent Peptide PROTAC to Knockdown Tau by Ubiquitination-Proteasome Degradation Pathway. European Journal of Medicinal Chemistry 2018, 146, 251–259. [Google Scholar] [CrossRef]
- Silva, M.C.; Ferguson, F.M.; Cai, Q.; Donovan, K.A.; Nandi, G.; Patnaik, D.; Zhang, T.; Huang, H.-T.; Lucente, D.E.; Dickerson, B.C.; et al. Targeted Degradation of Aberrant Tau in Frontotemporal Dementia Patient-Derived Neuronal Cell Models. eLife 2019, 8, e45457. [Google Scholar] [CrossRef]
- Silva, M.C.; Nandi, G.; Donovan, K.A.; Cai, Q.; Berry, B.C.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Ferguson, F.M.; Haggarty, S.J. Discovery and Optimization of Tau Targeted Protein Degraders Enabled by Patient Induced Pluripotent Stem Cells-Derived Neuronal Models of Tauopathy. Front. Cell. Neurosci. 2022, 16, 801179. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, Q.; Jiang, T.; Li, S.; Ye, J.; Zheng, J.; Wang, X.; Liu, Y.; Deng, M.; Ke, D.; et al. A Novel Small-Molecule PROTAC Selectively Promotes Tau Clearance to Improve Cognitive Functions in Alzheimer-like Models. Theranostics 2021, 11, 5279–5295. [Google Scholar] [CrossRef]
- Liang, M.; Gu, L.; Zhang, H.; Min, J.; Wang, Z.; Ma, Z.; Zhang, C.; Zeng, S.; Pan, Y.; Yan, D.; et al. Design, Synthesis, and Bioactivity of Novel Bifunctional Small Molecules for Alzheimer’s Disease. ACS Omega 2022, 7, 26308–26315. [Google Scholar] [CrossRef]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. CN 2018, 16, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Steven, M. Sparks, Erika Araujo, Michael Berlin, Wei Zhang, Jing Wang Selective Modulators of Mutant Lrrk2 Proteolysis and Associated Methods of Use.
- Erika Araujo, Steven M. Sparks, Michael Berlin, Wei Zhang, Jing Wang Indazole Based Compounds and Associated Methods of Use.
- Liu, X.; Kalogeropulou, A.F.; Domingos, S.; Makukhin, N.; Nirujogi, R.S.; Singh, F.; Shpiro, N.; Saalfrank, A.; Sammler, E.; Ganley, I.G.; et al. Discovery of XL01126: A Potent, Fast, Cooperative, Selective, Orally Bioavailable, and Blood–Brain Barrier Penetrant PROTAC Degrader of Leucine-Rich Repeat Kinase 2. J. Am. Chem. Soc. 2022, 144, 16930–16952. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Ren, X.; Xue, F.; He, Y.; Zhang, R.; Zheng, Y.; Huang, H.; Wang, W.; Zhang, J. Specific Knockdown of α-Synuclein by Peptide-Directed Proteasome Degradation Rescued Its Associated Neurotoxicity. Cell Chemical Biology 2020, 27, 751-762.e4. [CrossRef]
- Andrew P CREW, Hanqing Dong, Michael Berlin, Steven M. Sparks Proteolysis Targeting Chimeric (Protac) Compound with E3 Ubiquitin Ligase Binding Activity and Targeting Alpha-Synuclein Protein for Treating Neurodegenerative Diseases.
- Kargbo, R.B. PROTAC Compounds Targeting α-Synuclein Protein for Treating Neurogenerative Disorders: Alzheimer’s and Parkinson’s Diseases. ACS Med. Chem. Lett. 2020, 11, 1086–1087. [Google Scholar] [CrossRef]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Discovery of Small Molecules That Induce the Degradation of Huntingtin. Angew Chem Int Ed 2017, 56, 11530–11533. [Google Scholar] [CrossRef] [PubMed]
- Tomoshige, S.; Ishikawa, M. PROTACs and Other Chemical Protein Degradation Technologies for the Treatment of Neurodegenerative Disorders. Angew Chem Int Ed 2021, 60, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-L.; Lu, P.-C.; Lee, C.-C.; He, R.-Y.; Huang, Y.-A.; Tseng, Y.-C.; Cheng, T.-J.R.; Huang, J.J.-T.; Fang, J.-M. Degradation of Neurodegenerative Disease-Associated TDP-43 Aggregates and Oligomers via a Proteolysis-Targeting Chimera. J Biomed Sci 2023, 30, 27. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the mechanisms of proteolysis-targeting chimeras (PROTACs) and molecular glue degraders (MGDs) (A) The UPS consists of a cascade involving three enzymes that sequentially attach ubiquitin molecules to target proteins, marking them for proteasomal degradation. Degraders exploit this system by inducing proximity between a target protein (POI) and an E3 ligase, facilitating ubiquitination and subsequent degradation of the target protein. PROTACs are heterobifunctional molecules comprising a POI warhead and an E3 connected by a linker while molecular glue degraders are monomeric molecules. (B) Representation of the hook effect illustrating that at high concentrations, bifunctional degraders form binary complexes that reduce target degradation. This figure was created with BioRender.com.
Figure 1.
Schematic representation of the mechanisms of proteolysis-targeting chimeras (PROTACs) and molecular glue degraders (MGDs) (A) The UPS consists of a cascade involving three enzymes that sequentially attach ubiquitin molecules to target proteins, marking them for proteasomal degradation. Degraders exploit this system by inducing proximity between a target protein (POI) and an E3 ligase, facilitating ubiquitination and subsequent degradation of the target protein. PROTACs are heterobifunctional molecules comprising a POI warhead and an E3 connected by a linker while molecular glue degraders are monomeric molecules. (B) Representation of the hook effect illustrating that at high concentrations, bifunctional degraders form binary complexes that reduce target degradation. This figure was created with BioRender.com.
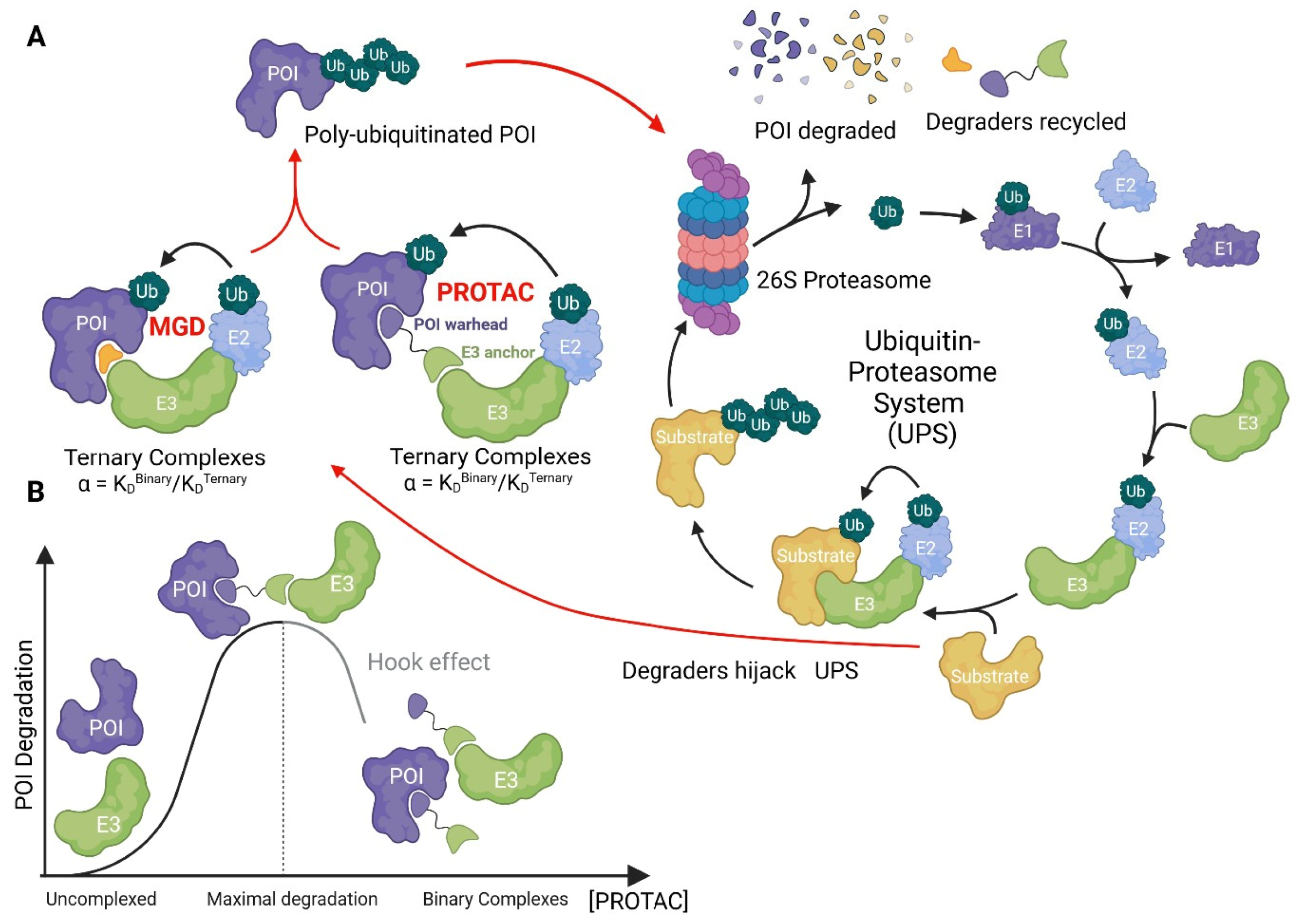
Figure 2.
Ubiquitin ligase families and examples of degraders directly recruiting E3 ligases or alternative UPS components. (A) The homologous to E6-AP C terminus (HECT) E3 family forms a thioester ubiquitin intermediate with a HECT domain cysteine, before transferring it to the substrate protein. Really interesting new gene (RING) finger E3 ligases utilize a RING domain to facilitate direct transfer of ubiquitin from E2 enzymes to substrate proteins. RING-in-between-RING (RBR) E3 ligases combine features of both RING and HECT ligases; the RING1 domain first recruits the E2-Ub complex before transferring ubiquitin onto a RING2 catalytic cysteine residue, ultimately facilitating its transfer onto the substrate protein. (B) The Cullin-RING ligases (CRL) are modular RING-family E3 ligases that utilize exchangeable adaptors (adpt) and receptors (recept) organized around a CUL domain to recruit specific substrates. Exemplified are the CUL2-RBX1-ElonginB-ElonginC-VHL (CRL2VHL) and the CUL4–RBX1–DDB1–CRBN (CRL4CRBN) complexes, with PROTAC and MGD recruiters as indicated. (C) MGDs can induce ubiquitination by triggering oligomerization of a protein. For example, MGD BI-3802 induces the dimerization of BCL6, facilitating its recognition by the E3 ligase SIAH1, which then mediates polyubiquitination of BCL6, ultimately resulting in its degradation. (D) Degraders can also recruit alternative UPS components to induce POI degradation such as E3 adaptors, E2 enzymes or directly the 26S proteasome. For example, the adaptor DDB1 can be recruited by MGD dCeMM3, forming a ternary complex with CDK12-Cyclin K conjugate, leading to Cyclin K ubiquitination and degradation. The E2 enzyme UBE2D can form a ternary complex with the MGD EN450 and NFKB1, leading to NFKB1 ubiquitination and subsequent degradation. MG EN67 can also be converted into a PROTAC. The proteasome can be directly recruited with compounds such as chemical inducers of degradation (CIDE). These degraders can be used to target a protein of interest (POI), such as BRD4, to the 26S proteasome for ubiquitin-independent proteasomal degradation. This figure was created with BioRender.com.
Figure 2.
Ubiquitin ligase families and examples of degraders directly recruiting E3 ligases or alternative UPS components. (A) The homologous to E6-AP C terminus (HECT) E3 family forms a thioester ubiquitin intermediate with a HECT domain cysteine, before transferring it to the substrate protein. Really interesting new gene (RING) finger E3 ligases utilize a RING domain to facilitate direct transfer of ubiquitin from E2 enzymes to substrate proteins. RING-in-between-RING (RBR) E3 ligases combine features of both RING and HECT ligases; the RING1 domain first recruits the E2-Ub complex before transferring ubiquitin onto a RING2 catalytic cysteine residue, ultimately facilitating its transfer onto the substrate protein. (B) The Cullin-RING ligases (CRL) are modular RING-family E3 ligases that utilize exchangeable adaptors (adpt) and receptors (recept) organized around a CUL domain to recruit specific substrates. Exemplified are the CUL2-RBX1-ElonginB-ElonginC-VHL (CRL2VHL) and the CUL4–RBX1–DDB1–CRBN (CRL4CRBN) complexes, with PROTAC and MGD recruiters as indicated. (C) MGDs can induce ubiquitination by triggering oligomerization of a protein. For example, MGD BI-3802 induces the dimerization of BCL6, facilitating its recognition by the E3 ligase SIAH1, which then mediates polyubiquitination of BCL6, ultimately resulting in its degradation. (D) Degraders can also recruit alternative UPS components to induce POI degradation such as E3 adaptors, E2 enzymes or directly the 26S proteasome. For example, the adaptor DDB1 can be recruited by MGD dCeMM3, forming a ternary complex with CDK12-Cyclin K conjugate, leading to Cyclin K ubiquitination and degradation. The E2 enzyme UBE2D can form a ternary complex with the MGD EN450 and NFKB1, leading to NFKB1 ubiquitination and subsequent degradation. MG EN67 can also be converted into a PROTAC. The proteasome can be directly recruited with compounds such as chemical inducers of degradation (CIDE). These degraders can be used to target a protein of interest (POI), such as BRD4, to the 26S proteasome for ubiquitin-independent proteasomal degradation. This figure was created with BioRender.com.
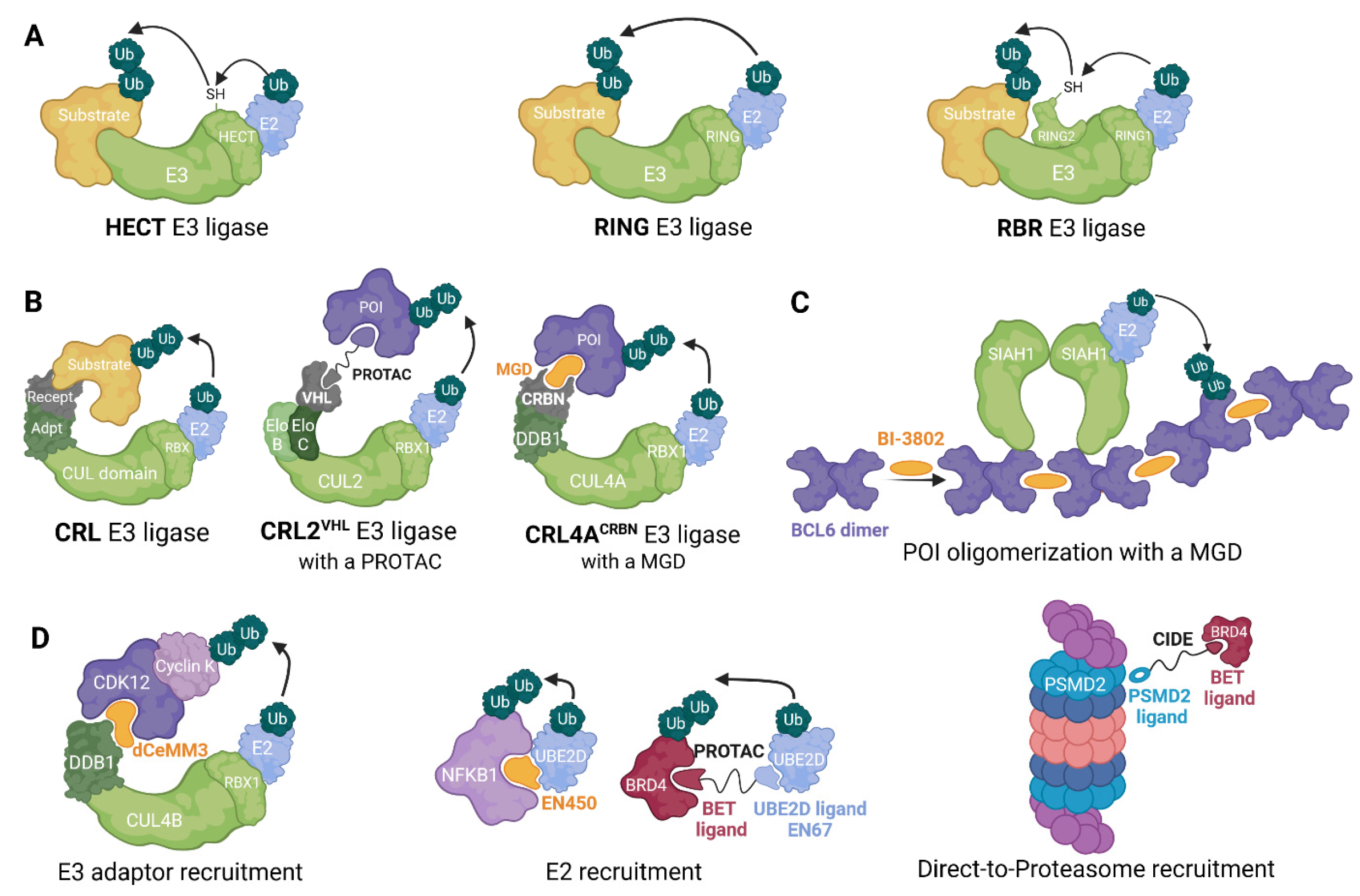
Figure 3.
Overview of concepts and considerations for degrader discovery, characterization, and validation. This figure was created with BioRender.com.
Figure 3.
Overview of concepts and considerations for degrader discovery, characterization, and validation. This figure was created with BioRender.com.
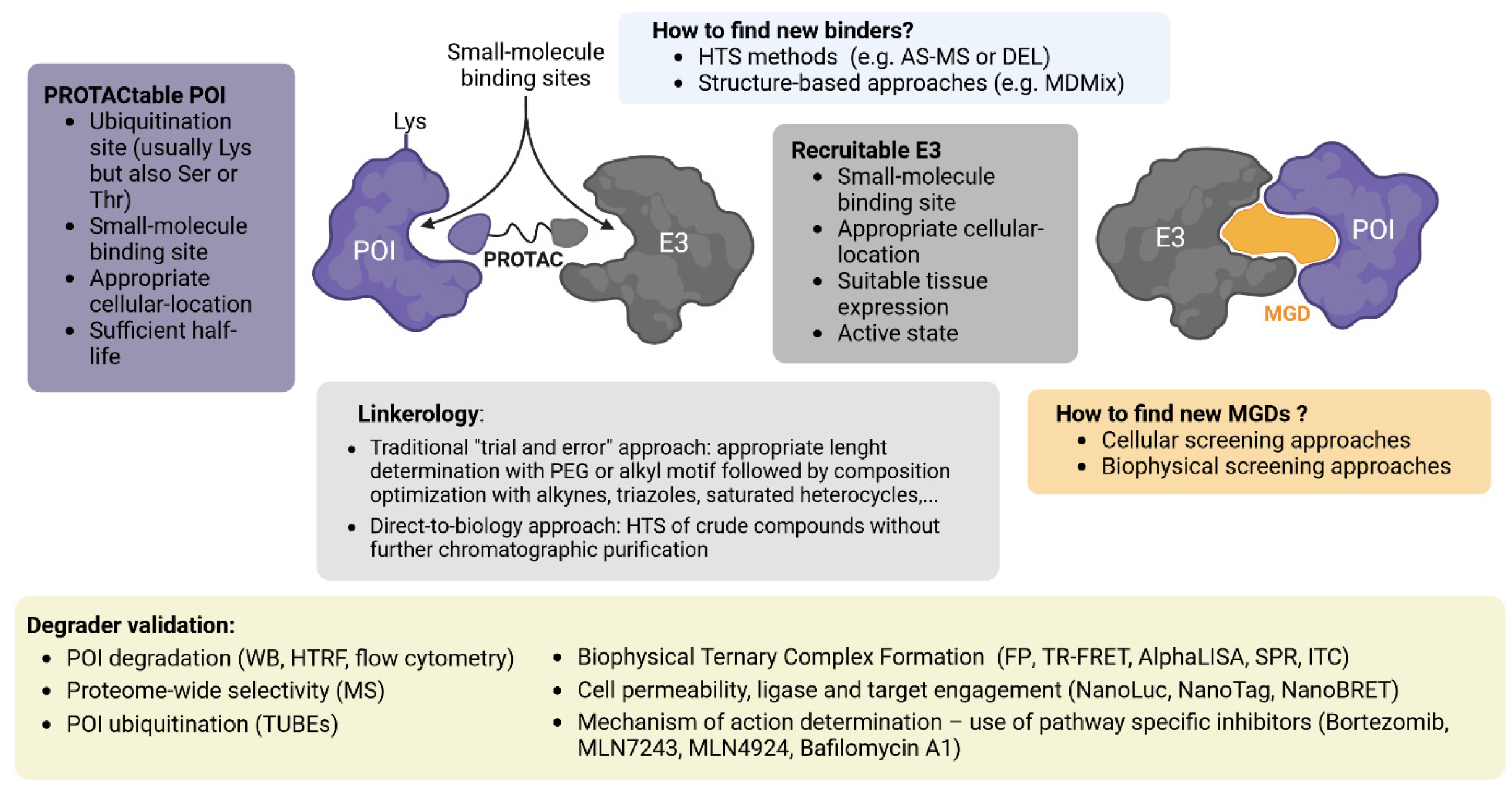

Table 1.
Clinically investigated heterobifunctional degraders for oncology indications. Source: https://clinicaltrials.gov, 4 December 2023. Clinical trial identifiers in green denote trials actively recruiting patients. Identifiers in orange are active trials not presently recruiting patients.
Table 1.
Clinically investigated heterobifunctional degraders for oncology indications. Source: https://clinicaltrials.gov, 4 December 2023. Clinical trial identifiers in green denote trials actively recruiting patients. Identifiers in orange are active trials not presently recruiting patients.
| Compound | Company | Protein Target | Disease Setting | Dose Route | Current Phase | Clinical Trial ID |
|---|---|---|---|---|---|---|
| ARV-471 | Arvinas | Estrogen Receptor | BC | Oral | Phase 3 | NCT05909397; NCT05654623 |
| ARV-110 | Arvinas | Androgen Receptor | mCRPC | Oral | Phase 2 | NCT03888612 |
| ARV-766 | Arvinas | Androgen Receptor | mCRPC | Oral | Phase 2 | NCT05067140 |
| RNK05047 | Ranok Therapeutics | BRD4 | DLBCL | iv | Phase 1/2 | NCT05487170 |
| BGB-16673 | Beigene | BTK | B-cell lymphomas | Oral | Phase 1/2 | NCT05294731; NCT05006716 |
| CFT1946 | C4 Therapeutics | mtBRAF V600 | NSCLC, mCRC, melanoma | Oral | Phase 1/2 | NCT05668585 |
| AC176 | Accutar Biotech | Androgen Receptor | mCRPC | Oral | Phase 1 | NCT05673109; NCT05241613 |
| ARD-LCC/CC-94676 | Celgene/BMS | Androgen Receptor | mCRPC | Oral | Phase 1 | NCT04428788 |
| RO7656594 | Gemicure/Genentech | Androgen Receptor | mCRPC | Oral | Phase 1 | NCT05800665 |
| HP518 | Hinova | Androgen Receptor | mCRPC | Oral | Phase 1 | NCT05252364 |
| DT-2216 | Dialectic Therapeutics | BCL-xL | Solid tumours/ Haematological tumours |
iv | Phase 1 | NCT04886622 |
| CFT8634 | C4 Therapeutics | BRD9 | Synovial Sarcoma | Oral | Phase 1 | NCT05355753 |
| FHD-609 | Foghorn Therapeutics | BRD9 | Synovial Sarcoma | iv | Phase 1 | NCT04965753 |
| AC676 | Accutar Biotech | BTK (wt/C481S) | B-cell lymphomas | Oral | Phase 1 | NCT05780034 |
| NX-2127 | Nurix | BTK | B-cell lymphomas | Oral | Phase 1 | NCT04830137 |
| NX-5948 | Nurix | BTK | B-cell lymphomas | Oral | Phase 1 | NCT05131022 |
| AC682 | Accutar Biotech | Estrogen Receptor | BC | Oral | Phase 1 | NCT05489679; NCT05080842 |
| AC699 | Accutar Biotech | Estrogen Receptor | BC | Oral | Phase 1 | NCT05654532 |
| KT-253 | Kymera | MDM2 | Various tumours | iv | Phase 1 | NCT05775406 |
| PRT3789 | Prelude | SMARCA2 | SMARCA4-del NSCLC | iv | Phase 1 | NCT05639751 |
| KT-333 | Kymera | STAT3 | Various lymphoma, leukaemia, solid tumours | iv | Phase 1 | NCT05225584 |
Table 2.
Clinically investigated molecular glue degrader for oncology indications. Source: https://clinicaltrials.gov, 4 December 2023. Clinical trial identifiers in green denote trials actively recruiting patients. Identifiers in orange are active trials not presently recruiting patients, those in purple indicate completed studies and those in red are terminated.
Table 2.
Clinically investigated molecular glue degrader for oncology indications. Source: https://clinicaltrials.gov, 4 December 2023. Clinical trial identifiers in green denote trials actively recruiting patients. Identifiers in orange are active trials not presently recruiting patients, those in purple indicate completed studies and those in red are terminated.
| Compound | Company | Protein Target | Disease Setting | Dose Route | Current Phase | Clinical Trial ID |
|---|---|---|---|---|---|---|
| Thalidomide (Thalomid) | Celgene/BMS | IKZF1/3 | Multiple myeloma | Oral | Approved | |
| Lenalidomide (Revlimid) | Celgene/BMS | IKZF1/3 | Multiple myeloma, Myleodysplastic syndrome | Oral | Approved | |
| Pomalidomide (Pomalyst) | Celgene/BMS | IKZF1/3 | Multiple myeloma | Oral | Approved | |
| CC-122 | Celgene/BMS | IKZF1/3 | Melanoma, Hepatocellular carcinoma, Chronic lymphocytic leukaemia | Oral | Phase II | NCT03834623, NCT02859324, NCT02406742 |
| CC-92480 (Mezigdomide) | Celgene/BMS | IKZF1/3 | Multiple myeloma | Oral | Phase II | NCT05372354 |
| CC-220 (Iberdomide) | Celgene/BMS | IKZF1/3 | Multiple myeloma | Oral | Phase II | NCT05199311 |
| ICP-490 | InnoCare Pharma | IKZF1/3 | Multiple myeloma | Oral | Phase II | NCT05719701 |
| CC-90009 | BMS/Celgene | GSPT1 | AML | iv | Phase II | NCT04336982 NCT02848001 |
| CQS | City of Hope Medical Center | RBM39 | Colorectal cancer, Small cell lung cancer | iv | Phase II | NCT00005864; NCT00008372 |
| E7070 (Indisulam) | Eisai | RBM39 | BC, AML, Melanoma, Renal cell carcinoma, Colorectal cancer, | iv | Phase II |
NCT00080197,NCT01692197; NCT00014625;NCT00059735; NCT00165867,NCT00165854 |
| E7820 | Eisai | RBM39 | AML, Colorectal | Oral | Phase II | NCT05024994; NCT00309179 |
| MRT-2359 | Monte Rosa | GSPT1 | Myc-driven tumours | Oral | Phase I/II | NCT05546268 |
| CC-99282 (Golcadomide) | BMS/Celgene | IKZF1/3 | Non-Hodgkin and B-Cell Lymphomas | Oral | Phase I/II | NCT03930953; NCT04884035 |
| CFT-7455 | C4 Therapeutics | IKZF1/3 | Multiple myeloma, Non-Hodgkin and B-Cell Lymphomas | Oral | Phase I/II | NCT04756726 |
| BTX-1188 | Biotheryx | IKZF1/3 | Non-Hodgkin lymphoma, AML, Solid tumours | Oral | Phase I | NCT05144334 |
| GT-919 | Gluetacs Therapeutics | IKZF1/3 | Haematological tumours | Oral | Phase I | Undefined |
| GT-929 | Gluetacs Therapeutics | IKZF1/3 | Haematological tumours | Oral | Phase I | Undefined |
| SP-3164 | Salarius | IKZF1/3 | Non-Hodgkin lymphoma | Oral | Phase I | NCT05979857 |
| DKY-709 | Novartis | IKZF2 | Advanced solid tumours | Oral | Phase I/Ib | NCT03891953 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



