Submitted:
06 February 2024
Posted:
07 February 2024
You are already at the latest version
Abstract
Phosphatase and tensin homolog (PTEN) is a negative regulator of the phosphoinositide 3-kinases/protein kinase B (PI3K/AKT) signaling pathway. Notably, its active site harbors a cysteine residue that is susceptible to oxidation by hydrogen peroxide (H2O2). This oxidation inhibits the phosphatase function of PTEN, critically contributing to the activation of the PI3K/AKT pathway. Upon stimulation of cell surface receptors, the activity of NADPH oxidase 2 (NOX2) generates a transient amount of H2O2, serving as a mediator in this pathway by oxidizing PTEN. The mechanism underlying this oxidation, occurring despite the presence of highly efficient and abundant cellular oxidant-protecting and reducing systems, continues to pose a perplexing conundrum. Here, we demonstrate that the presence of bicarbonate (HCO3-) promoted the rate of H2O2-mediated PTEN oxidation, probably through the formation of peroxymonocarbonate (HCO4-), consequently potentiated the phosphorylation of AKT. In essence, our findings consolidate the crucial role of HCO3- in the redox regulation of PTEN by H2O2, lead to the presumption regarding HCO4- as a signaling molecule during cellular physiological processes.
Keywords:
PTEN
; redox regulation
; H2O2
; bicarbonate
; peroxymonocarbonate.
; cell signaling
1. Introduction
When cells are stimulated by growth factors and cytokines, such as platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin, granulocyte-macrophage colony-stimulating factor (GM-CSF), tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and interleukin-3 (IL-3), hydrogen peroxide (H2O2) is produced through the activity of NADPH oxidases (NOXs) [1]. The activation of NOXs results in the production of superoxide (O2•−), which is subsequently transformed to H2O2 by the activity of superoxide dismutase (SOD) [2]. This physiological H2O2 influences various intracellular signaling pathways. Its capacity to oxidize the cysteine residues of some proteins, such as protein tyrosine phosphatases (PTPs), results in a functional modification that notably impairs their activities [3]. PTPs play pivotal roles in cellular processes involving cell growth, proliferation, and differentiation [4]. Within their structural framework, all PTPs feature a cysteine residue in their active site [5]. When exposed to H2O2, the cysteine residue undergoes oxidation, transforming into cysteine-sulfenic acid (Cys-SOH) and leading to the oxidative inhibition of PTP’s enzymatic activity [6].
Phosphatase and tensin homolog (PTEN), a member of the PTP family, structurally comprises five functional domains: a short N-terminal phosphatidylinositol (PtdIns)(4,5)P2-binding domain (PBD), a catalytic phosphatase domain, a C2 lipid/membrane-binding domain, a C-terminal tail containing Pro, Glu, Ser, and Thr (PEST) sequences, and a class I PDZ-binding (PDZ-BD) motif. PTEN interacts with phospholipid membranes through the C2 domain located at the C-terminal end. The tight linkage between the C2 and phosphatase domains implies that the C2 domain not only aids in recruiting PTEN to the membrane but also optimizes the orientation of the catalytic domain associated with its membrane-bound substrate. Moreover, the phosphatase domain of PTEN has an enlarged active site to accommodate its phosphoinositide substrate, membrane-bound phosphatidylinositol-3,4,5-triphosphate (PIP3) [7,8]. Hence, PTEN is considered a tumor suppressor owing to its ability to dephosphorylate PIP3 and, consequently, negatively regulate the PI3K/AKT signaling pathway, which is pivotal in controlling cell survival, growth, and proliferation [9,10]. Besides cancer studies, substantial insights into PTEN’s role in physiological processes are available. Notably, PTEN inhibition has been demonstrated as a promising therapeutic intervention for neurodegenerative diseases, ischemia, infection, and insulin-resistant metabolic disorders [11].
Similar to other members of the PTP family, PTEN contains a cysteine residue in the active site of its phosphatase domain, rendering it susceptible to oxidative inhibition by ROS, particularly H2O2 [5]. Lee et al. were the first to demonstrate the reversible inactivation of PTEN by H2O2 through the oxidation of the Cys124 catalytic residue in the active site and the formation of an intramolecular disulfide bond with Cys71. This inactivation is reversible because oxidized PTEN can be converted back to the functional reduced form by cellular reducing agents, such as the Thioredoxin/Thioredoxin Reductase (Trx/TrxR) system [12]. Additionally, in the cell, Peroxiredoxins (Prx), thiol-specific antioxidants of the peroxidase family, can function as modulators of H2O2-induced phosphorylation signaling due to their capacity to sense and eliminate peroxides [13,14,15]. Thus, the mechanism of PTEN oxidation by transient H2O2, a signaling molecule produced in response to growth factor receptor stimulation, raises questions about how this small amount of physiological H2O2 can function to oxidize PTEN in a cellular environment rich in H2O2 scavengers such as catalase, glutathione peroxidase, and Prx, alongside the presence of ubiquitous Trx/TrxR reducing systems.
It has been known since 1980s that H2O2 can react with bicarbonate/carbon dioxide (HCO3-/CO2) to form peroxymonocarbonate (HCO4-)-: H2O2 + HCO3-/CO2 ⇌ HCO4- + H2O/H+. In neutral pH aqueous solution, HCO4- formation process occurs rapidly at 25 ℃ with half-life t1/2 of 10 min or below. Carbonic anhydrase and a Zinc model complex can accelerate this reaction [16,17]. HCO4- is a robust oxidant with a higher catalytic potency comparable to that of H2O2. Its interaction with target molecules proceeds at velocities ranging from 100 to 1000 times faster than those observed with H2O2 [18]. Sulfide oxidation caused by HCO4- surpasses that caused by H2O2 by approximately 300-fold [19]. Notably, HCO4- also serves as a potent two-electron oxidant primarily accountable for biothiol peroxidation [20]. Hence, the presence of HCO3- is demonstrated to be a pivotal factor in promoting the oxidative reactivity of H2O2 towards PTPs, such as PTP1B, SHP-2, and PTPN22, during signal transduction processes [21,22,23]. When cells are stimulated by growth factors, HCO3- is produced through the activation of a transmembrane enzyme, carbonic anhydrase IX, which catalyzes the hydration of CO2 (CO2 + H2O → HCO3- + H+) [24]. Dagnell et al. demonstrated that HCO3- not only facilitates but also plays an essential role in H2O2-mediated inactivation of PTP1B, surpassing the protection provided by the Trx/TrxR reducing systems [22]. Given that PTEN belongs to the PTP family, it is worth exploring whether HCO3- may somehow affect the H2O2-mediated oxidative inactivation of PTEN. Based on the available pieces of information, we anticipated that the presence of HCO3- in combination with H2O2, through the formation of HCO4-, would augment oxidation of PTEN.
2. Materials and Methods
2.1. Materials
Dulbecco’s modified Eagle’s medium (DMEM) D5648 and N-Ethylmaleimide (NEM) were sourced from Sigma-Aldrich, 1 M HEPES from Enzynomics, 100X Penicillin/Streptomycin from Capricorn Scientific, fetal bovine serum (FBS) from Welgene, 3% hydrogen peroxide from Samchun, sodium bicarbonate from Amresco, Pro-prep Protein Extraction Solution from iNtRON Biotechnology, PageRuler Prestained Protein Ladder No. 26616 and BCA Protein Assay Kit from Thermo Scientific. The antibodies used for western blotting comprised primary PTEN antibody, β-actin antibody, phosphorylated-Serine-473 AKT, total AKT and anti-rabbit immunoglobulin G horseradish peroxidase-linked secondary antibodies. All other chemicals and reagents were of analytical grade.
2.2. Cell Culture
HepG2 cells were cultured in DMEM supplemented with 5% FBS and 1X Penicillin/Streptomycin.
2.3. Immunoblot Analysis of H2O2-Induced Oxidation of PTEN
To assess the redox state of PTEN, we performed mobility shift assays in cells, employing N-Ethylmaleimide (NEM) as an alkylating agent, as described in our previous publication [25]. HepG2 cells were cultured until they reached 90% confluency. Subsequently, they were washed with PBS and switched to media containing 0.1% FBS and 25 mM HEPES, with or without sodium bicarbonate, and then pre-incubated at 37 °C with 0.1% CO2. Serum-free stimulation media containing 0.5-1 mM H2O2, with various bicarbonate conditions, were prepared before applying to the cells. After pre-incubation for 4 h, the media were removed, and cells were incubated with the prepared stimulation media. Subsequently, the stimulation media were removed at varying time points, and the cells were washed twice using cold PBS. The reaction was stopped by adding Pro-prep lysis buffer containing 10 mM NEM. Cell lysates were collected, and the protein concentrations were quantified using the BCA method. Subsequently, the samples were subjected to non-reducing SDS-PAGE, followed by western blotting using PTEN, β-Actin, pAKT and total AKT specific antibodies. (Figure 1).
2.4. Statistical Analysis
PTEN oxidation levels in the samples were quantified by analyzing the oxidized/reduced PTEN states using ImageJ. Data significances were evaluated using unpair Student’s t test. Differences were significant when P < 0.05. Graphical values are presented as the mean ± standard error (SE). The trendlines for PTEN oxidation represent linear regression.
3. Results
The Presence of HCO3- Potentiates the PTEN Oxidation by H2O2
HepG2 cells were pre-incubated in HCO3--free media at 37 °C with 0.1% CO2 for 4 h. Various stimulation media were made before administering to the cells: 44 mM HCO3- alone, 1 mM H2O2 alone, a combination of 1 mM H2O2 and 22 mM HCO3-, and a combination of 1 mM H2O2 and 44 mM HCO3-. The percentages of oxidized PTEN were assessed after 10 min treatment. The result showed that with the presence of 22 mM or 44 mM bicarbonate in the stimulation media, the oxidations of PTEN in 10 min were significantly higher (P < 0.05), compared to the cells which were treated with H2O2 alone. The oxidation did not exhibit an increase magnitude when adding more HCO3-. In addition, stimulation media with only 44 mM HCO3- exhibited no oxidation effect. (Figure 2)
The Presence of HCO3- Accelerates the Redox Regulation of PTEN by H2O2
To investigate the change in PTEN oxidation, we observed the percentages of oxidized PTEN in different time period. Treatments of HepG2 cells by H2O2, with the presence and absence of 44 mM HCO3-, in 5, 10, 15, 30 60, and 120 min, resulted in PTEN oxidation in varying time-dependent manners. In the first group, cells were pre-incubated and treated in stimulation media containing 44 mM HCO3-. In the second group, the condition was similar except that the media was HCO3--free. Our data indicated that in the HCO3--containing group, the PTEN oxidation rate soared to its peak at 10 min and then decreased gradually. At 60 min, almost all oxidized PTEN were reduced back and the recovery was completed by 120 min. In the HCO3--free group, the oxidation rate increased at a slower speed, reaching its peak at 30 min. At 60 min, there was a considerable amount of oxidized PTEN that was not reduced back, and the amount was much higher than that observed in the HCO3--containing group. In addition, the maximum oxidation level was higher in the HCO3--containing group. The linear regression trendlines illustrate that PTEN oxidation was faster in the HCO3--containing group than in the HCO3--free group. Comparing the slopes between the trendlines (2.87 and 1.16), the difference in oxidation speed was nearly 2.5 times. During the recovery period, the absence of HCO3- remarkably impaired the reduction of oxidized PTEN. A comparison of the slopes revealed that the difference in reduction speed was also approximately equivalent (2.3-fold). These results clearly demonstrate that HCO3- accelerates the H2O2-mediated redox regulation of PTEN. (Figure 3)
The Activation of PI3K/AKT Pathway via PTEN Oxidation by H2O2 and HCO3-
PTEN oxidation inhibits its phosphatase function and subsequently leads to the activation of PI3K/AKT signaling pathway [10]. We have shown that PTEN can be oxidized at faster speed by the combination of HCO3- and H2O2 than by H2O2 alone. To investigate how this PTEN oxidative inhibition affect the phosphorylation of AKT, we conducted the same protocol, cell lysates were subjected to SDS-PAGE with antibodies specific for pAKT and total AKT. With the addition of 44 mM HCO3-, AKT phosphorylation started to rise significantly following 10 min exposure to H2O2, reaching maximum at 15 min before gradually declining. Meanwhile, the absence of HCO3- delayed this process with a slower increase to its peak at 30 min. Furthermore, the HCO3--containing group also exhibited a higher proportion of pAKT across all timepoints. Comparing the slopes between the trendlines (0.0704 and 0.0263), the disparity in phosphorylation velocity was approximately 2.7-fold. Overall, under stimulating by H2O2, the presence of HCO3- potentiates the phosphorylation of AKT in a time-dependent manner. (Figure 4)
4. Discussion
The activation of receptor tyrosine kinases (RTK) is a pivotal event in transmitting phosphorylation signals upon stimulation by growth factors and, therefore, holds substantial importance in cell physiology [26]. In this situation, a transient amount of H2O2 is generated by membrane NOXs [27]. Subsequently, H2O2 causes reversible oxidative inhibition of PTEN, leading to an increase in PI3K/AKT signaling pathway and, consequently, eliciting cellular substantial effects [28,29]. The activation of AKT and its downstream cascades are proved to be related to variety of cancers [30]. Besides, studies also indicate that the elevated AKT activity through oxidative inactivation of PTEN can yield benefits in particular physiological processes that require cell growth such as cardiac remodeling following ischemia [31], neuronal regeneration [32], immune response [33], insulin-related metabolism [34], and myogenesis [35]. Within the cellular environment, H2O2 can be eliminated by thiol proteins from the Prx family. Furthermore, oxidized PTEN and Prx can be rapidly converted back to their active reduced forms via the Trx/TrxR/NADPH system [36,37]. Thus, PTEN phosphatase catalytic activity is conserved by Trx/TrxR and Prx systems [14,15,36,38]. However, PTEN is inhibited during PI3K/AKT signaling pathway activation. Through our experiment result, we presume the factor that facilitates PTEN oxidation by H2O2 in the presence of HCO3- is the formation of HCO4- (Figure 5).
The addition of 44 mM sodium bicarbonate to DMEM D5648, which lacks HCO3-, replicates the common condition of culture media. Previous investigations on NIH 3T3 cells exposed to H2O2 in standard DMEM revealed reversible PTEN oxidation in a time-dependent manner. Incubation with 0.5 mM H2O2 resulted in increased levels of oxidized PTEN, peaking at 10 min before declining gradually, suggesting that PTEN was initially inactivated by H2O2 and then reactivated by cellular reductants as the H2O2 declined. This reversible PTEN inactivation was also observed in HeLa cells exposed to H2O2 [12]. In our recent studies, cells were treated with H2O2 for specific durations, extending the time points to 120 min to ensure the completion of the reduction process. Similarly, oxidized PTEN exhibited a time-dependent increase followed by a decline. The PTEN oxidation rate reached its peak after exposure for 10 min, and the oxidized PTEN was completely converted to the reduced form within 120 min of exposure. Based on these results, it is evident that treatments with H2O2 in standard HCO3--containing media yield maximum PTEN oxidation after 10 min of exposure, followed by the conversion of oxidized PTEN to its reduced state by the cellular Trx/TrxR system within approximately 60 min after exposure [25,38]. Therefore, we initially tested the role of HCO3- in PTEN oxidation by H2O2 through evaluating the proportions of oxidized PTEN following 10 min exposure to stimulation media. The pre-incubation period aimed to diminish the HCO3- in both extracellular and intracellular environments. Our findings indicated HCO3- alone did not cause any PTEN oxidation. Meanwhile, H2O2 alone has significant lower percentage than H2O2-HCO3- combination. Additionally, there was no notable difference between HCO3- concentration of 22 mM and 44 mM. This implies that the greater percentages observed in the H2O2-HCO3- combination groups were not merely the cumulative effect of each H2O2 and HCO3-. The plausible explanation is that HCO3- can empower the catalytic activity of H2O2, supporting the hypothesis there might be a formation of HCO4-, a more reactive oxidant.
Given that HCO4- was demonstrated to exhibit a greater oxidation rate towards sulfide when compared to H2O2 [19], we suppose that it may also influence PTEN oxidation. This implies that the peak of PTEN oxidation in each group may not be solely at the 10 min timepoint. Therefore, we extended experiments to additional time points to investigate the redox regulation of HCO3- and H2O2. Additionally, we reduced the concentration of H2O2 to 0.5 mM and expect it could make the difference more visible. A similar pattern to results of our previous studies was observed this time on HepG2 cells in 44 mM HCO3--supplemented media. PTEN was predominantly oxidized within 5–15 min, with the highest oxidation rate observed at 10 min, then reduced back to active form. Thus, the PTEN oxidation observed in the HCO3--containing group in the present study follows a trend consistent with our former studies.
The exclusion of HCO3- from the experimental conditions considerably prolonged the PTEN oxidation period, requiring up to 30 min to reach its peak. However, the H2O2-mediated oxidative inactivation of intracellular PTPs in signaling events typically happens swiftly within the timeframe of 5–15 minutes, typically peaking at 10 min [12,39,40]. This disparity suggests that the absence of HCO3- adversely affects the PTEN oxidation speed. Our result aligns with the findings of Dagnell et al., indicating that HCO3- can potentiate the H2O2-dependent oxidative inactivation of PTP [22]. Illustrative data analysis suggests that the combination of HCO3- and H2O2 accelerated the rate of PTEN oxidation in the cellular environment by approximately 2.5-fold in a time-dependent manner. Notably, this was also strongly correlated to the similar decrease in the PTEN reduction speed in the HCO3--free group. Based on growing evidence about the role of HCO3- in H2O2-mediated oxidation under cell signaling processes, we suppose the formed HCO4- promotes the PTEN redox regulation, therefore exerts function as a signaling molecule.
PTEN acts as a negative regulator of PI3K/AKT signaling pathway, which induces various cellular processes including protein synthesis, cell survival, proliferation, and migration. This mechanism plays important role in human physiological and pathological conditions [7,11,41]. Following the result that H2O2-mediated oxidative inhibition of PTEN is potentiated and accelerated by the HCO3-, we examined its subsequent impact on phosphorylation of AKT. The elevating trends of pAKT/AKT reflect similar patterns observed in PTEN oxidation, both in bicarbonate-containing and bicarbonate-free status. Our findings are consistent with the previous evidence suggesting that H2O2 can activate PI3K/AKT pathway by oxidizing PTEN [10]. Additionally, the time-dependent increase of pAKT/AKT is equivalent to the rise of PTEN oxidation in the presence of bicarbonate, 2.7-fold and 2.5-fold, correspondingly. In another words, without bicarbonate, PTEN oxidation is impaired, leading to the delay in AKT phosphorylation. Thus, in this context, the increase in AKT activation definitely arises from the surge in PTEN oxidation. Considering HCO4- is formed through the reaction between H2O2 and HCO3-, we propose that HCO4- can positively regulate PI3K/AKT signaling pathway via oxidative inactivation of PTEN.
In summary, the results of our experiment support the hypothesis that the presence of HCO3- promotes the H2O2-mediated inactivation of PTEN. This subsequently promotes the activation of PI3K/AKT signaling pathway. In addition, the reduction of oxidized PTEN by antioxidant systems is also be equivalently affected. The plausible explanation can be the formation of HCO4-. In the future, further experiments would be conducted to consolidate the role of HCO4- in the redox regulation of PTEN.
Author Contributions
Conceptualization, S.-R.L.; methodology, V.H.T.; validation, S.-R.L.; writing—original draft preparation, V.H.T.; writing—review and editing, V.H.T., T.N.H., D.K.S., J.M.C., H.J.Y., S.C.P. and J.Y.S.; visualization, D.K.S.; supervision, S.-R.L.; funding acquisition, S.-R.L. and S.C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Basic Research Program (NRF-2022M3A9E4017151 to S.-RL) through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT, and Technology, and by the KBRI basic research program through the Korea Brain Research Institute (23-BR-03-05 to S.-RL and S.C.P.). This research was also supported by the National Research Foundation of Korea (2018R1D1A1B06051438), Republic of Korea. Thang Nguyen Huu is supported in part by the Center for Global Future Biomedical Scientists at Chonnam National University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
Illustrating figures were made using Biorender.com
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rhee, S.G.; Bae, Y.S.; Lee, S.-R.; Kwon, J. Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Science’s STKE 2000, 2000, pe1-pe1. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- van der Geer, P.; Hunter, T.; Lindberg, R.A. Receptor protein-tyrosine kinases and their signal transduction pathways. Annual review of cell biology 1994, 10, 251–337. [Google Scholar] [CrossRef]
- Denu, J.M.; Dixon, J.E. Protein tyrosine phosphatases: mechanisms of catalysis and regulation. Curr Opin Chem Biol 1998, 2, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Tanner, K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nature reviews Molecular cell biology 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Lee, J.-O.; Yang, H.; Georgescu, M.-M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef]
- Stambolic, V.; Suzuki, A.; De La Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. The EMBO journal 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- Pulido, R. PTEN inhibition in human disease therapy. Molecules 2018, 23, 285. [Google Scholar] [CrossRef]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. Journal of Biological Chemistry 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free radical biology and Medicine 2005, 38, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. Journal of Biological Chemistry 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends in biochemical sciences 2015, 40, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Flangan, J.; Jones, D.P.; Griffith, W.P.; Skapski, A.C.; West, A.P. On the existence of peroxocarbonates in aqueous solution. Journal of the Chemical Society, Chemical Communications 1986, 20–21. [Google Scholar] [CrossRef]
- Bakhmutova-Albert, E.V.; Yao, H.; Denevan, D.E.; Richardson, D.E. Kinetics and mechanism of peroxymonocarbonate formation. Inorganic chemistry 2010, 49, 11287–11296. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Interplay of carbon dioxide and peroxide metabolism in mammalian cells. Journal of Biological Chemistry 2022, 298. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.E.; Yao, H.; Frank, K.M.; Bennett, D.A. Equilibria, kinetics, and mechanism in the bicarbonate activation of hydrogen peroxide: oxidation of sulfides by peroxymonocarbonate. Journal of the American Chemical Society 2000, 122, 1729–1739. [Google Scholar] [CrossRef]
- Trindade, D.F.; Cerchiaro, G.; Augusto, O. A role for peroxymonocarbonate in the stimulation of biothiol peroxidation by the bicarbonate/carbon dioxide pair. Chem Res Toxicol 2006, 19, 1475–1482. [Google Scholar] [CrossRef]
- Zhou, H.; Singh, H.; Parsons, Z.D.; Lewis, S.M.; Bhattacharya, S.; Seiner, D.R.; LaButti, J.N.; Reilly, T.J.; Tanner, J.J.; Gates, K.S. The biological buffer bicarbonate/CO2 potentiates H2O2-mediated inactivation of protein tyrosine phosphatases. Journal of the American Chemical Society 2011, 133, 15803–15805. [Google Scholar] [CrossRef]
- Dagnell, M.; Cheng, Q.; Rizvi, S.H.M.; Pace, P.E.; Boivin, B.; Winterbourn, C.C.; Arnér, E.S. Bicarbonate is essential for protein-tyrosine phosphatase 1B (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades. Journal of Biological Chemistry 2019, 294, 12330–12338. [Google Scholar] [CrossRef]
- James, J.; Chen, Y.; Hernandez, C.M.; Forster, F.; Dagnell, M.; Cheng, Q.; Saei, A.A.; Gharibi, H.; Lahore, G.F.; Åstrand, A. Redox regulation of PTPN22 affects the severity of T-cell-dependent autoimmune inflammation. Elife 2022, 11, e74549. [Google Scholar] [CrossRef]
- Dorai, T.; Sawczuk, I.S.; Pastorek, J.; Wiernik, P.H.; Dutcher, J.P. The role of carbonic anhydrase IX overexpression in kidney cancer. European journal of cancer 2005, 41, 2935–2947. [Google Scholar] [CrossRef]
- Han, S.-J.; Ahn, Y.; Park, I.; Zhang, Y.; Kim, I.; Kim, H.W.; Ku, C.-S.; Chay, K.-O.; Yang, S.Y.; Ahn, B.W. Assay of the redox state of the tumor suppressor PTEN by mobility shift. Methods 2015, 77, 58–62. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases: from genes, to function, to disease. Nature reviews Molecular cell biology 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nature Reviews Immunology 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R. The redox regulation of PI 3-kinase-dependent signaling. Antioxid Redox Signal 2006, 8, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Downes, C.P.; Ross, S.; Maccario, H.; Perera, N.; Davidson, L.; Leslie, N.R. Stimulation of PI 3-kinase signaling via inhibition of the tumor suppressor phosphatase, PTEN. Adv Enzyme Regul 2007, 47, 184–194. [Google Scholar] [CrossRef]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nature Reviews Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Semenza, G.L. PTEN activity is modulated during ischemia and reperfusion: involvement in the induction and decay of preconditioning. Circulation research 2005, 97, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Hervera, A.; De Virgiliis, F.; Palmisano, I.; Zhou, L.; Tantardini, E.; Kong, G.; Hutson, T.; Danzi, M.C.; Perry, R.B.; Santos, C.X.C.; et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat Cell Biol 2018, 20, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.-J.; Liu, P.; Bajrami, B.; Xu, Y.; Park, S.-Y.; Nombela-Arrieta, C.; Mondal, S.; Sun, Y.; Zhu, H.; Chai, L. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity 2015, 42, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M. Reactive oxygen species enhance insulin sensitivity. Cell metabolism 2009, 10, 260–272. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, T.G.; Park, S.; Yun, H.R.; Nguyen, N.N.Y.; Jo, Y.H.; Jang, M.; Kim, J.; Kim, J.; Kang, I.; et al. Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ 2018, 25, 1921–1937. [Google Scholar] [CrossRef]
- Nguyen Huu, T.; Park, J.; Zhang, Y.; Park, I.; Yoon, H.J.; Woo, H.A.; Lee, S.R. Redox Regulation of PTEN by Peroxiredoxins. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. Embo j 2009, 28, 1505–1517. [Google Scholar] [CrossRef]
- Zhang, Y.; Park, J.; Han, S.-J.; Yang, S.Y.; Yoon, H.J.; Park, I.; Woo, H.A.; Lee, S.-R. Redox regulation of tumor suppressor PTEN in cell signaling. Redox biology 2020, 34, 101553. [Google Scholar] [CrossRef]
- Mahadev, K.; Zilbering, A.; Zhu, L.; Goldstein, B.J. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. Journal of Biological Chemistry 2001, 276, 21938–21942. [Google Scholar] [CrossRef]
- Meng, T.-C.; Buckley, D.A.; Galic, S.; Tiganis, T.; Tonks, N.K. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. Journal of Biological Chemistry 2004, 279, 37716–37725. [Google Scholar] [CrossRef]
- Boosani, C.S.; Gunasekar, P.; Agrawal, D.K. An update on PTEN modulators–a patent review. Expert opinion on therapeutic patents 2019, 29, 881–889. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Experimental scheme for detecting redox status of PTEN by mobility shift. Upon HCO4- exposure, a proportion of PTEN can be oxidized in the Cystein124 residue of their active site through creating a reversible intracellular disulfide bond with Cystein71. Subsequently, the supplement of NEM akylates PTEN reduced form into PTEN-NEM complex, increasing the molecular weight, thereby result in a lower shift on non-reducing SDS-PAGE.
Figure 1.
Experimental scheme for detecting redox status of PTEN by mobility shift. Upon HCO4- exposure, a proportion of PTEN can be oxidized in the Cystein124 residue of their active site through creating a reversible intracellular disulfide bond with Cystein71. Subsequently, the supplement of NEM akylates PTEN reduced form into PTEN-NEM complex, increasing the molecular weight, thereby result in a lower shift on non-reducing SDS-PAGE.
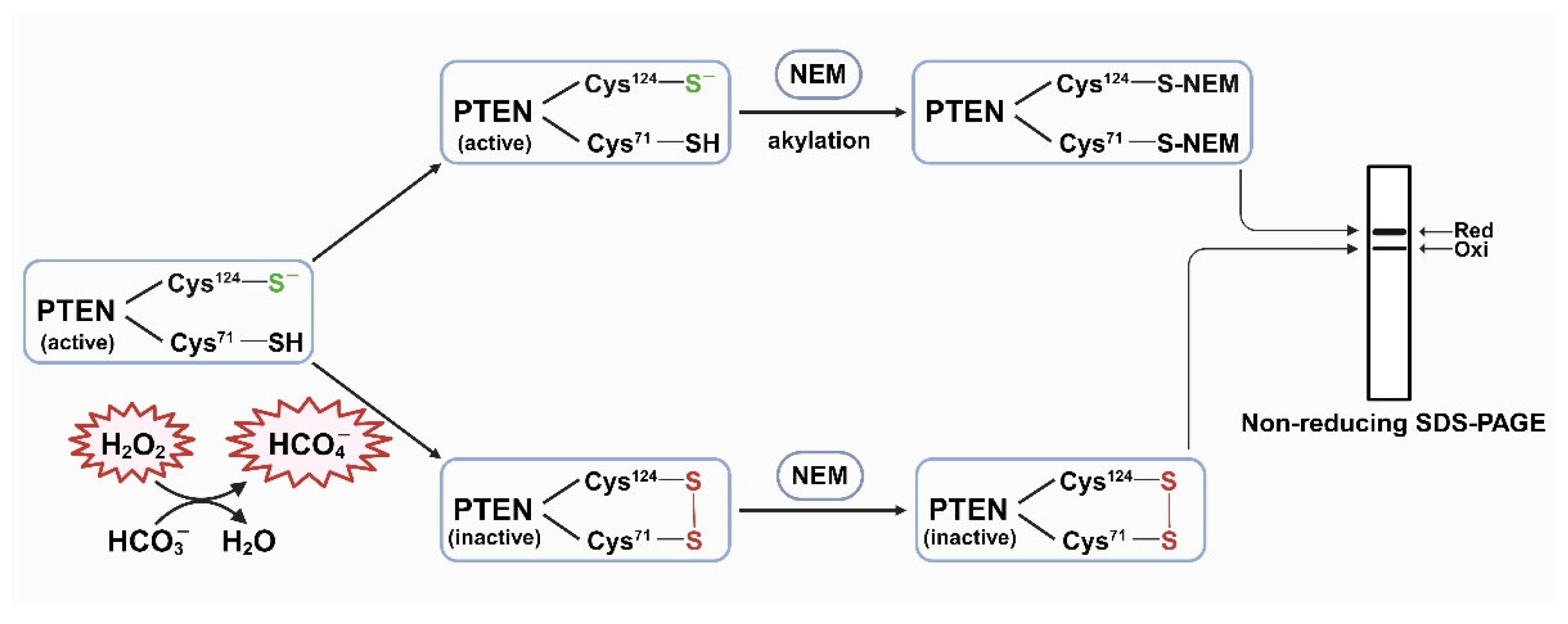
Figure 2.
HCO3- potentiates the PTEN oxidation by H2O2. HepG2 cells were cultured until they reached 90% confluency, then washed with PBS and transferred to DMEM (L-glutamine, 1X penicillin-streptomycin, 0.1% FBS, and 25 mM HEPES) without 44 mM sodium HCO3-, and the pH was neutral. Then, the cells were incubated at 37 °C with 0.1% CO2 for 4 h and, subsequently, treated with prepared stimulation media containing 44 mM HCO3- alone, or 1 mM H2O2 alone, or combination of 1 mM H2O2 and 22 mM HCO3-, or combination of 1 mM H2O2 and 44 mM HCO3-. After 10 min, the reaction was stopped by adding lysis buffer containing protease inhibitors and 10 mM NEM. Then, the cell lysates were collected and used for SDS-PAGE and western blotting analyses. The results obtained using PTEN antibody are presented in (A). The quantification result (B) indicated that with the presence of 22 mM or 44 mM HCO3-, PTEN oxidation is significantly higher than treatment with only H2O2 (P < 0.05). 44 mM HCO3- alone showed no oxidation effect.
Figure 2.
HCO3- potentiates the PTEN oxidation by H2O2. HepG2 cells were cultured until they reached 90% confluency, then washed with PBS and transferred to DMEM (L-glutamine, 1X penicillin-streptomycin, 0.1% FBS, and 25 mM HEPES) without 44 mM sodium HCO3-, and the pH was neutral. Then, the cells were incubated at 37 °C with 0.1% CO2 for 4 h and, subsequently, treated with prepared stimulation media containing 44 mM HCO3- alone, or 1 mM H2O2 alone, or combination of 1 mM H2O2 and 22 mM HCO3-, or combination of 1 mM H2O2 and 44 mM HCO3-. After 10 min, the reaction was stopped by adding lysis buffer containing protease inhibitors and 10 mM NEM. Then, the cell lysates were collected and used for SDS-PAGE and western blotting analyses. The results obtained using PTEN antibody are presented in (A). The quantification result (B) indicated that with the presence of 22 mM or 44 mM HCO3-, PTEN oxidation is significantly higher than treatment with only H2O2 (P < 0.05). 44 mM HCO3- alone showed no oxidation effect.
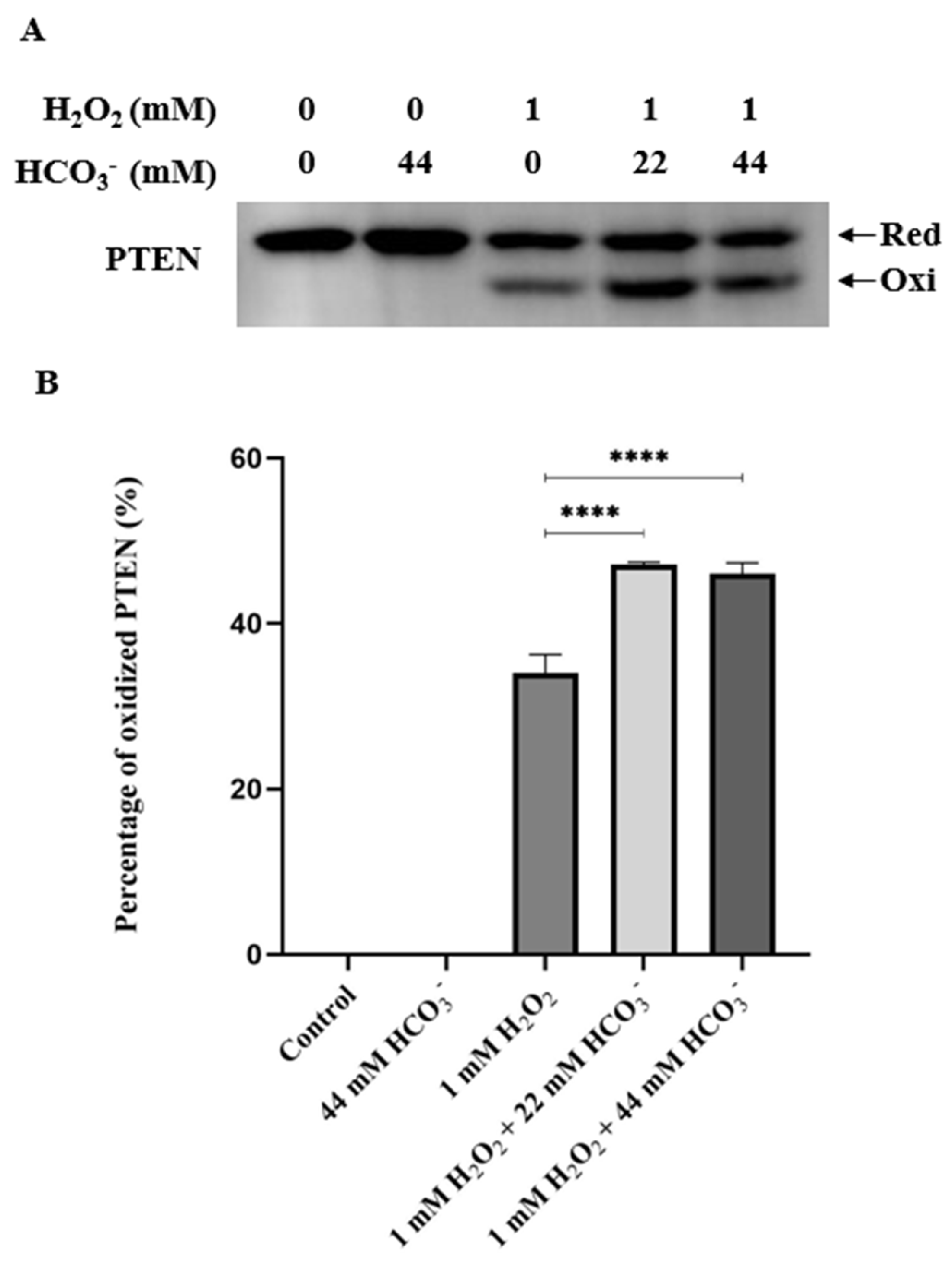
Figure 3.
Bicarbonate accelerates the redox regulation of PTEN by H2O2. HepG2 cells were cultured until they reached 90% confluency, then washed with PBS and transferred to DMEM (L-glutamine, 1X penicillin-streptomycin, 0.1% FBS, and 25 mM HEPES) with or without 44 mM sodium bicarbonate, and the pH was neutral. Then, the cells were incubated at 37 °C with 0.1% CO2 for 4 h and, subsequently, treated with prepared stimulation media containing 0.5 mM H2O2. After 5, 10, 15, 30, 60, and 120 min, the reaction was stopped by adding lysis buffer containing protease inhibitors and 10 mM NEM. Then, the cell lysates were collected and used for SDS-PAGE and western blotting analyses. The results obtained using PTEN and β-actin antibodies are presented in (A). The quantification results (B) indicate that in the absence of HCO3-, the rate of PTEN oxidation diminished, and the reduction period was prolonged. Data are presented as the mean ± SEM of three independent experiments. The linear regression trendlines of PTEN oxidation and reduction rates presented in (C) show that the presence of HCO3- accelerates the H2O2-mediated redox regulation of PTEN.
Figure 3.
Bicarbonate accelerates the redox regulation of PTEN by H2O2. HepG2 cells were cultured until they reached 90% confluency, then washed with PBS and transferred to DMEM (L-glutamine, 1X penicillin-streptomycin, 0.1% FBS, and 25 mM HEPES) with or without 44 mM sodium bicarbonate, and the pH was neutral. Then, the cells were incubated at 37 °C with 0.1% CO2 for 4 h and, subsequently, treated with prepared stimulation media containing 0.5 mM H2O2. After 5, 10, 15, 30, 60, and 120 min, the reaction was stopped by adding lysis buffer containing protease inhibitors and 10 mM NEM. Then, the cell lysates were collected and used for SDS-PAGE and western blotting analyses. The results obtained using PTEN and β-actin antibodies are presented in (A). The quantification results (B) indicate that in the absence of HCO3-, the rate of PTEN oxidation diminished, and the reduction period was prolonged. Data are presented as the mean ± SEM of three independent experiments. The linear regression trendlines of PTEN oxidation and reduction rates presented in (C) show that the presence of HCO3- accelerates the H2O2-mediated redox regulation of PTEN.
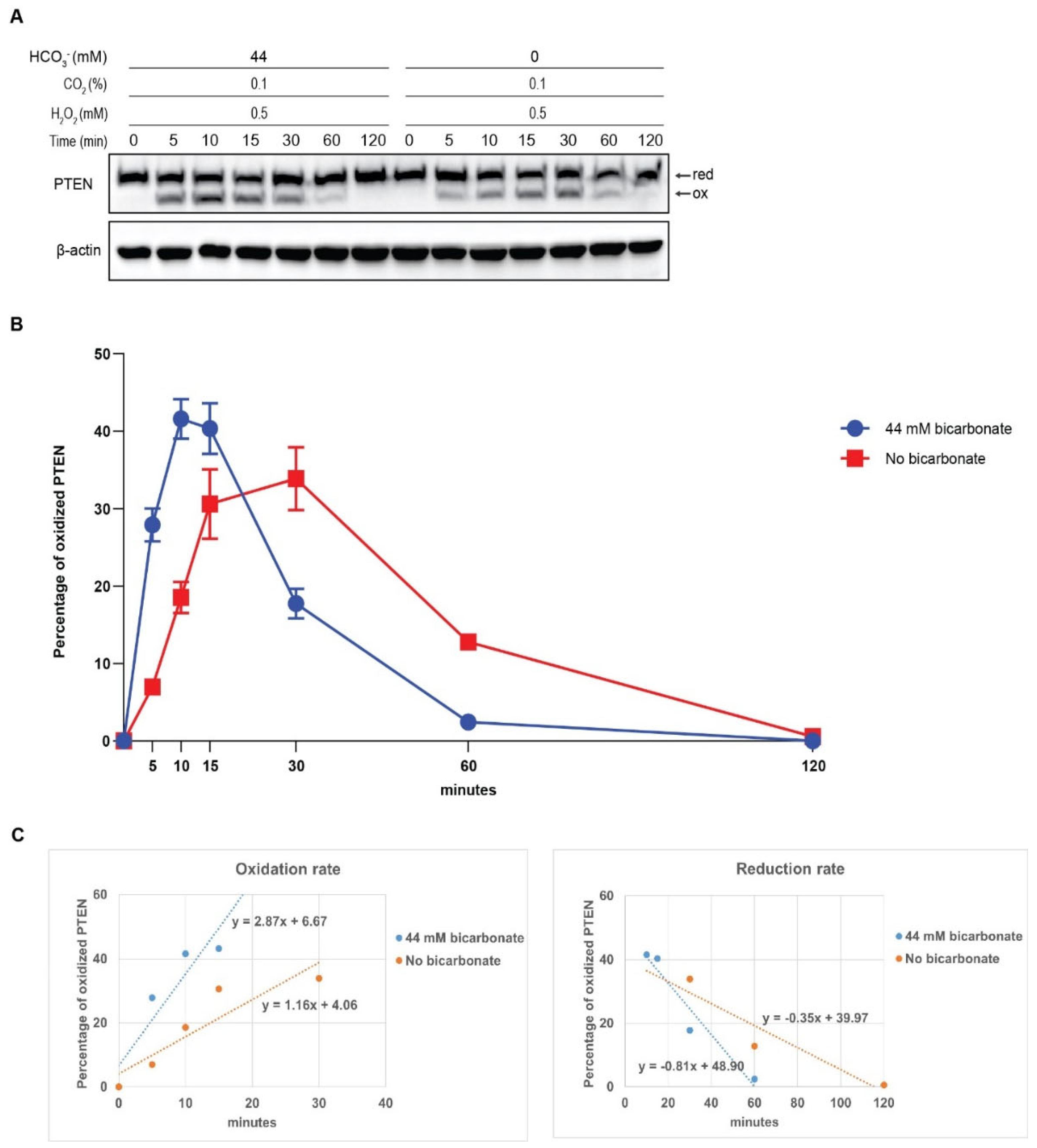
Figure 4.
HCO3- accelerates and potentiates the phosphorylation of AKT induced by H2O2. HepG2 cells were preincubated and exposed to 0.5 mM H2O2 in HCO3--supplemented or HCO3--free media. After 5, 10, 15, 30, 60 and 120 min, cell lysates were collected and subjected to Western blot using pAKT and total AKT antibody (A). Data are quantified by pAKT fold over total AKT and presented as mean ± SEM. The combination of HCO3- and H2O2 resulted in faster and higher AKT phosphorylation than H2O2 alone (B). The linear regression trendlines of pAKT/AKT presented in (C) show that the presence of HCO3- elevated the AKT phosphorylation rate.
Figure 4.
HCO3- accelerates and potentiates the phosphorylation of AKT induced by H2O2. HepG2 cells were preincubated and exposed to 0.5 mM H2O2 in HCO3--supplemented or HCO3--free media. After 5, 10, 15, 30, 60 and 120 min, cell lysates were collected and subjected to Western blot using pAKT and total AKT antibody (A). Data are quantified by pAKT fold over total AKT and presented as mean ± SEM. The combination of HCO3- and H2O2 resulted in faster and higher AKT phosphorylation than H2O2 alone (B). The linear regression trendlines of pAKT/AKT presented in (C) show that the presence of HCO3- elevated the AKT phosphorylation rate.
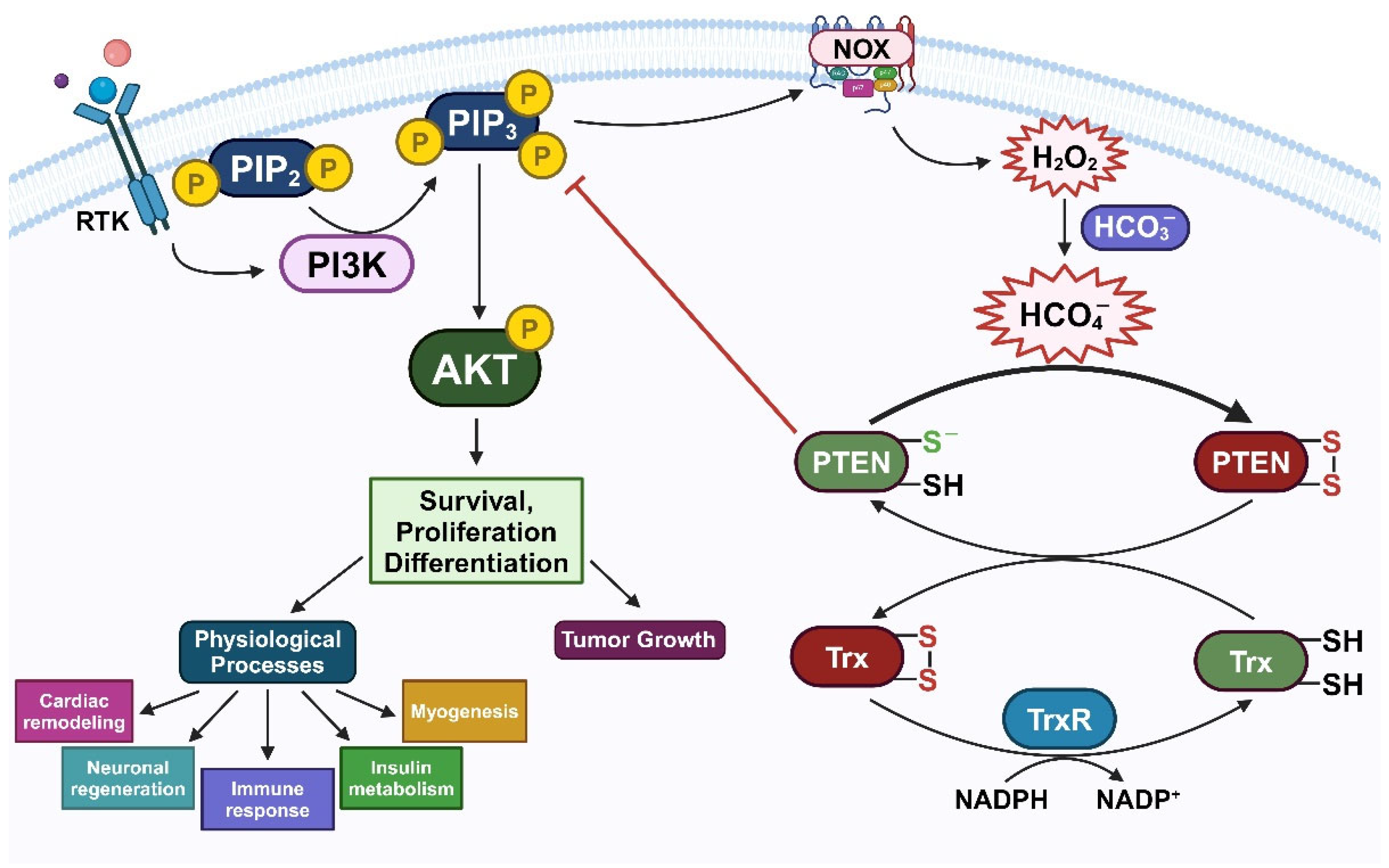
Figure 5.
Suggesting role of HCO4- in redox regulation of PTEN. Stimulation of RTK induces the assembly of PI3K to phosphorylate PIP2 to PIP3, subsequently activating AKT signaling cascade. PTEN can negatively modulate this pathway. During this condition, H2O2, produced through the activity of NOX2, oxidizes PTEN. In cellular environment, PTEN oxidation can be impaired and reversed by Trx/TrxR/NADPH reducing system. However, in the presence of HCO3- together with H2O2, a higher reactive oxidant HCO4- is formed. Hence, HCO4- is supposed to accelerate the oxidative inactivation of PTEN, consequently increase the PI3K/AKT signaling pathway, and as a result, promoting cell survival, proliferation, and differentiation in physiological processes or tumor growth.
Figure 5.
Suggesting role of HCO4- in redox regulation of PTEN. Stimulation of RTK induces the assembly of PI3K to phosphorylate PIP2 to PIP3, subsequently activating AKT signaling cascade. PTEN can negatively modulate this pathway. During this condition, H2O2, produced through the activity of NOX2, oxidizes PTEN. In cellular environment, PTEN oxidation can be impaired and reversed by Trx/TrxR/NADPH reducing system. However, in the presence of HCO3- together with H2O2, a higher reactive oxidant HCO4- is formed. Hence, HCO4- is supposed to accelerate the oxidative inactivation of PTEN, consequently increase the PI3K/AKT signaling pathway, and as a result, promoting cell survival, proliferation, and differentiation in physiological processes or tumor growth.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



