
Submitted:
01 February 2024
Posted:
01 February 2024
You are already at the latest version
Abstract
Amylin, also known as islet amyloid polypeptide (IAPP), is a metabolic homeostasis-related hormone that is produced and released by the β cells of the pancreas, the same cells that produce insulin. Insulin-degrading enzyme (IDE) is a protease enzyme that plays an essential role in the breakdown and degradation of various peptides, including insulin and amylin. Direct binding & interaction between amylin and IDE is inextricably linked to the degradation of amylin, and research and development effort in this area is crucial to understand the potential therapeutic implications of disrupting the IDE-amylin interaction in the context of conditions where metabolic homeostasis needs to be regulated, such as diabetes and obesity. Here, this article incorporates currently available experimental complex structure of amylin and IDE, and delves deep into the interstructural biophysics underlying the binding interface of the two interacting partners. With a set of comprehensive structural biophysical analysis, this article identified an intriguing region of high electrostatic potential indicative of strong binding sites between the first N-terminal lysine (Lys1, K1) residue of amylin and Glu341 (E341) of IDE. This unique electrostatic hotspot presented herein paves the way for the rational design of drug-like small molecules that can selectively disrupt this interaction, offering a targeted therapeutic strategy for improved metabolic homeostasis, particularly for patients with diabetes and obesity.
Keywords:
Amylin
; Insulin-degrading enzyme
; Electrostatic hotspots
; Salt bridge
; Hydrogen bond
1. Introduction
Amylin is a 37-amino-acid pancreatic hormone acting to control energy homeostasis and body weight [1,2,3,4]. Physiologically, amylin regulates glucose homeostasis by inhibiting insulin and glucagon secretion [5,6,7]. Furthermore, amylin modulates satiety and inhibits gastric emptying via the central nervous system [8,9,10,11]. Produced and released by pancreatic cells, amylin shares a common secretion pathway with insulin, collectively orchestrating the postprandial control of glucose homeostasis [12,13,14,15]. The intricate regulatory network governing blood sugar levels involves a delicate interplay between hormones and enzymes, among which amylin (islet amyloid polypeptide, IAPP) and insulin-degrading enzyme (IDE) stand as key players [12,16,17,18,19,20,21], where IDE plays a crucial role in the degradation and clearance of amylin from the bloodstream [22,23,24]. The cooperation between amylin and IDE, therefore, is pivotal in maintaining the delicate balance of glucose homeostasis and averting the detrimental consequences of amyloid deposition [25,26,27,28,29]. For instance, IDE defects are linked to the development of type 2 diabetes mellitus (T2DM) and Alzheimer’s disease (AD) [13,18,30].
2. Motivation
Thanks to the continued development of experimental structural biology and the half-a-century old Protein Data Bank (PDB) [31,32], a comprehensive structural biophysical (CSB) analysis becomes possible [33,34,35] for specific ligand-receptor [36,37,38], antigen-antibody [39] or enzyme-substrate [40,41,42] complex structures deposited in PDB, expanding our understanding of the structural and biophysical basis of their interfacial structural stability, and facilitating the design of drug analogues with improved affinity to their interacting partners [43,44].
As a matter of fact, structural and biochemical analyses [45,46,47,48] have already revealed the binding mode and pattern for the formation of the IDE-amylin complex structure, and this experimental information is useful but insufficient for the development of promising inhibitors (e.g., small molecules) of IDE-amylin interaction to improve glucose homeostasis. This manuscript, therefore, seeks to delve into the structural and biophysical aspects of the interaction between amylin and IDE, with a specific focus on identifying electrostatic hotspots at their binding interface. In case these hotspots are able to act as potential binding sites for drug-like small molecules, the aim of this article is to provide a precise and targeted approach for the development of small molecules to disrupt the amylin-IDE interaction.
3. Materials and Methods
As of February 1, 2024, a total of 101 experimental structures have been deposited in Protein Data Bank (PDB) [31] as listed in Table 1, according to a text query: QUERY: Full Text = "amylin" of the Protein Data Bank (PDB) [31]. Among them, only two experimental structures represent the amylin-IDE complex, with PDB IDs: 2G48 [45,46] and 3HGZ [47,48], respectively, providing an accurate structural basis of the IDE-amylin interaction specificity for subsequent comprehensive structural biophysical (CSB) analysis of the two structural models (two yellow rows in Table 1).
First, after the atomic coordinates file for PDB IDs: 2G48 [45,46] and 3HGZ [47,48] were downloaded from the PDB [31] website, Chimera [49] was employed to manually add hydrogen atoms to the structural model of the two structural models representing IDE-amylin complex. Afterwards, the two hydrogen-added structural models were subject to a set of comprehensive structural biophysical (CSB) analysis as described in [33] to identify key residue-specific interactions at the amylin-IDE binding interface and uncover the interstructural biophysics underlying the IDE-amylin complex structure.
Specifically, the CSB analysis here [33] consists of the structural identification of salt bridges and side chain hydrogen bonds at the binding interface of amylin and IDE. Given the fact that native proteins are in dynamic equilibrium with their less-structured, partially folded and/or unfolded states [50], and that, according to an NMR structure of human amylin bound to model membranes, the helix structure of amylin itself is also dynamic, this article uses two sets of screening criteria for the structural identification of potential hotspots at the IDE-amylin binding interface in the two structural models i.e., PDB IDs: 2G48 [45,46] and 3HGZ [47,48].
First, the same set of criteria as in [33] was used, i.e., the interfacial salt bridge analysis was conducted with an in-house python script only for titrateable residues (Asp, Glu, Lys, Arg and His), 4.0 Å was used as the cutoff distance for the two oppositely charged groups [33,51]. The hydrogen bond analysis was also conducted for only side chain nuclei with an in-house python script, and employed two geometric criteria: (a) a cutoff value of the angle formed by acceptor (A), donor (D) and hydrogen (H) () of 30; (b) a cutoff value of donor-acceptor distance at 3.0 Å. That is, a hydrogen bond is only considered to be formed if is not larger than 30 and the donor-acceptor distance is not larger than 3.0 Å [33,51].
Afterwards, a new set of criteria was used to account for the dynamic -helix structure of amylin itself, i.e., the interfacial salt bridge analysis was conducted with an in-house python script only for titrateable residues (Asp, Glu, Lys, Arg and His), 6.0 Å was used as the cutoff distance for the two oppositely charged groups [33,51]. The hydrogen bond analysis was also conducted for only side chain nuclei with an in-house python script, and employed two geometric criteria: (a) a cutoff value of the angle formed by acceptor (A), donor (D) and hydrogen (H) () of 50; (b) a cutoff value of donor-acceptor distance at 5.0 Å. That is, a hydrogen bond is only considered to be formed if is not larger than 30 and the donor-acceptor distance is not larger than 5.0 Å [33,51].
Here, the in-house python scripts essentially are the same as those used in [52], except for the differences in three key parameters related to the screening criteria, i.e., the salt bridge distance cutoff in Å, cutoff angle in for hydrogen bonding, and the cutoff distance (in Å) of donor-acceptor for hydrogen bonding.
4. Results
4.1. Characterization of residue-specific electrostatic interactions at the amylin-IDE binding interface
As of February 1, 2024, there are only two experimental structures representing the amylin-IDE complex, with PDB IDs: 2G48 [45,46] and 3HGZ [47,48], respectively (Table 1). As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively. For the two structural models (2G48 [45,46] and 3HGZ [47,48]), this article reports a set of comprehensive structural biophysical (CSB) analysis as described in [33], which lead to a set of residue-specific electrostatic interactions at the binding interface of amylin-IDE, as listed in Table 2, Table 3, Table 4, Table 5, Table 6 and Table 7. Specifically,
As discussed above, to account for the dynamic helix structure and less-structured or random coil region of amylin, this article uses two sets of screening criteria for structural identification of potential electrostatic hotspots at the IDE-amylin binding interface in the two structural models i.e., PDB IDs: 2G48 [45,46] and 3HGZ [47,48]. Specifically,
- among the 22, only 3 interfacial salt bridges (Table 5) were structurally identified between Lys1 and Glu341 (Figure 2 and Figure 3) at the binding interface of IDE and amylin for the amylin-IDE complex structure PDB ID 2G48 [45,46], according to a new set of criteria [33] as defined in the section of Materials and Methods.
- among the 16, only 4 interfacial salt bridges (Table 5) were structurally identified between Lys1 and Glu341 (Figure 2 and Figure 3) at the binding interface of IDE and amylin for the amylin-IDE complex structure PDB ID 3HGZ [47,48], according to a new set of criteria [33] as defined in the section of Materials and Methods.
4.2. Structural identification of an electrostatic hotspot at amylin-IDE binding interface
Among the residue-specific electrostatic interactions at the amylin-IDE binding interface described above, one extraordinary pair of oppositely charged residues appear rather outstanding: lysine (Lys1, K1) residue of amylin and Glu341 (E341) of IDE, as this residue pair is the only one where interfacial electrostatic interactions were structurally identified at the amylin-IDE binding interface for two experimental structures representing the amylin-IDE complex, with PDB IDs: 2G48 [45,46] and 3HGZ [47,48] as listed in Table 1.
Take 2G48 [45,46] for example, one 3.514 Å interfacial salt bridge (Figurer̃efpymol2) was found to be formed between the oppositely charged side chains of Lys1 of amylin and Glu341 of IDE, while two interfacial salt bridges (2.441 and 3.179 Å) were also found to be formed between the oppositely charged side chains of Lys1 of amylin and Glu341 of IDE for 3HGZ [47,48]. Moreover, while no further salt bridges were found to be formed according to the old set of criteria as defined in [33], they are still quite close to each other for the side chains of Lys1 of amylin and Glu341 of IDE. Take 2G48 [45,46] for example, apart from the salt bridge, other positively charged side chain atoms of Lys1 of amylin are only 4.876 and 5.297 Å away from the negatively charged side chain oxygens of Glu341 of IDE, as shown in Figuresr̃efpymol2 and Figure 3.
In addition to interfacial salt bridges, the one extraordinary pair, i.e., Lys1 of amylin and Glu341 of IDE, were also found to be involved in a set of interfacial side chain hydrogen bonds, as shown by two yellow rows in Table 7 and also two yellow rows in Table 6, according to a new set of criteria [33] as defined in the section of Materials and Methods. Finally, what even more interesting is one interfacial side chain bridges formed between Lys1 of amylin and Glu341 of IDE, as shown in Figuresr̃efpymol2 and Figure 3, the main chain amide nitrogen (positively charged) of Lys1 of amylin is only 3.4 Å away from the negatively charged side chain oxygen of Glu341 of IDE, making the residue pair close to each other enough to form a strong main chain-side chain interfacial salt bridge, further strengthening the binding between amylin and IDE.
Taken together, these three sets of interfacial electrostatic interactions between Lys1 of amylin and Glu341 of IDE, i.e., side chain-side chain interfacial salt bridges, side chain-side chain interfacial hydrogen bonds, main chain-side chain interfacial salt bridges, highlights an extraordinary electrostatic hotspot at the amylin-IDE binding interface, making it an attractive precise target for small molecule inhibitor to reach inside IDE (the cage, Figurer̃efpymol1) and disrupt the amylin-IDE interaction for improved metabolic homeostasis.
5. Conclusion
Starting from two experimental structures representing the amylin-IDE complex, with PDB IDs: 2G48 [45,46] and 3HGZ [47,48] as listed in Table 1, this article puts forward a set of structural characterization for residue-specific electrostatic interactions at the amylin-IDE binding interface, as listed in Table 2, Table 3, Table 4, Table 5, Table 6 and Table 7. Moreover, this article also highlights one intriguing electrostatic hotspot (Figuresr̃efpymol2 and Figure 3) between the first N-terminal lysine (Lys1, K1) residue of amylin and Glu341 (E341) of IDE, with both interfacial salt bridges and side chain hydrogen bonds formed between the two oppositely charged residues sitting at the binding interface of amylin-IDE.
To sum up,
- This finding also contributes to the growing body of knowledge aimed at unraveling the intricacies of protein–protein interactions and provides a foundation for future research endeavors in the development of targeted therapeutics for metabolic disorders, particularly diabetes (T2DM) and obesity [57,58].
- The rationale for targeting electrostatic hotspots in the amylin-IDE interaction lies in the role of such sites as preferred binding locations for drug-like small molecules. By pinpointing these hotspots, we aim to provide a precise target for the development of small molecules capable of disrupting the amylin-IDE interaction. The potential therapeutic implications of such disruptors extend to modulating glucose homeostasis and mitigating the risk of amyloid formation, which is particularly relevant in the context of type 2 diabetes.
6. Discussion
6.1. Disrupting the amylin-IDE interaction: a drug discovery and design perspective
In drug discovery and design, targeting the interaction between amylin and IDE could be a potential strategy for therapeutic intervention. As amylin is involved in the regulation of blood glucose levels, manipulating its interaction with IDE might be explored as a way to modulate glucose homeostasis. While this article identified an intriguing electrostatic hotspots at the amylin-IDE binding interface, offering precise targets for therapeutic intervention and the development of small molecule inhibitors targeting the amylin-IDE interaction, the inhibition of the interaction between amylin and IDE can also lead to side effects such as glucose intolerance [59], as the intricate interplay between amylin and IDE constitutes a pivotal aspect of glucose homeostasis, with implications for the prevention of amyloid formation and maintenance of metabolic health [60,61,62]. Moreover, disrupting the interaction between amylin and IDE could have unintended consequences, as IDE is involved in the degradation of various peptides, and altering its interaction with amylin might affect the levels of other important regulatory molecules, selectivity is a key challenge in the design of small molecule inhibitor or even other molecular modalities such as small peptide of peptide-small molecule conjugate. Finally, still essential is a thorough understanding of the biological consequences of disrupting the amylin-IDE interaction, while disrupting the interaction between amylin and IDE could be a potential avenue for drug development, especially in the context of diabetes, obesity or even neurodegenerative diseases.
6.2. High-throughput comprehensive structural biophysical analysis: a methological perspective
In 2017, a comprehensive structural biophysical (CSB) approach was for the first time used in the analysis of experimental complex structures deposited in PDB [31] to address this question: how do SMA-linked mutations of SMN1 lead to structural/functional deficiency of the SMA protein i.e., the survivor motor neuron protein [33]? Here, the same structural biophysical approach was used here for the analysis of experimental complex structures of amylin and IDE, allowing for the precise identification of electrostatic hotspots at the binding interface of amylin and insulin-degrading enzyme. This level of precision aids in targeting specific regions for the development of small molecules. Moreover, By exploring the structural details and biophysical characteristics of the interaction, this approach provides valuable insights into the molecular interactions between amylin and insulin-degrading enzyme. Understanding these interactions is crucial for rational design of effective small molecule disruptors with high specificity, i.e., without affecting other biological processes. This specificity is crucial for minimizing off-target effects and enhancing the safety profile of potential therapeutic agents.
In addition, the information obtained through this approach contributes to rational drug design by providing a solid foundation for the development of small molecules. The identified hotspots serve as rational targets for disrupting the interaction, potentially enhancing the success rate of drug development efforts. Furthermore, in light of the increasingly large size of the Protein Data Bank, this CSB approach technically is applicable for a exhaustive analysis of the entire PDB for high-throughput extraction of structural and biophysical features [63,64] and continued generation and accumulation of synthetic structural and biophysical data with reasonable accuracy to support the development of machine learning-models such as GIBAC [65]. With this respect, however, in original PDB-format data, the experimentally determined atomic coordinates are presented in the ATOM records, and chances are that they do not exactly match the sequence (consisting of nucleic and/or amino acid residues) of the experimental sample per se, be it protein, DNA, RNA or their complexes with drugs and/or other small molecules [66,67].
Take amylin and IDE for example, the fasta format sequence of amylin in PDB ID 2G48 [45,46] is as below:
KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY
KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY
As listed in Table 8, in PDB ID 2G48 [45,46], there are a total of 37 residues for amylin in the experimental sample, with atomic coordinate information of 19 missing for chain C (amylin in PDB ID 2G48) and 20 missing for chain D (amylin in PDB ID 2G48), while in PDB ID 3HGZ [47,48], there are also a total of 37 residues for amylin in the experimental sample, with atomic coordinate information of 30 missing for chain D (amylin in PDB ID 3HGZ) and 27 missing for chain D (amylin in PDB ID 3HGZ).
As previously described in [68] in 2017, the past 53 years of experimental structure deposition in PDB also saw continued accumulation of experimentally uncharted territories (EUTs) inside it, reaching a point already where it is increasingly pressing for biomolecular structures (especially membrane proteins like Ca2+ channel [69,70]) to be experimentally determined in an EUT-less manner, as exemplified here again by the experimentally uncharted territories (EUTs) in two structural models representing IDE-amylin complex (PDB IDs: 2G48 [45,46] and 3HGZ [47,48]).
7. Ethical statement
No ethical approval is required.
8. Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work, the author used OpenAI’s ChatGPT in order to improve the readability of the manuscript, and to make it as concise and short as possible. After using this tool, the author reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Author Contributions
Conceptualization, W.L.; methodology, W.L.; software, W.L.; validation, W.L.; formal analysis, W.L.; investigation, W.L.; resources, W.L.; data duration, W.L.; writing–original draft preparation, W.L.; writing–review and editing, W.L.; visualization, W.L.; supervision, W.L.; project administration, W.L.; funding acquisition, not applicable.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Mathiesen, D.S.; Bagger, J.I.; Knop, F.K. Long-acting amylin analogues for the management of obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2022, 29, 183–190. [Google Scholar] [CrossRef]
- Hay, D.L.; Chen, S.; Lutz, T.A.; Parkes, D.G.; Roth, J.D. Amylin: Pharmacology, Physiology, and Clinical Potential. Pharmacol. Rev. 2015, 67, 564–600. [Google Scholar] [CrossRef] [PubMed]
- Alghrably, M.; Czaban, I.; Jaremko, L.; Jaremko, M. Interaction of amylin species with transition metals and membranes. J. Inorg. Biochem. 2019, 191, 69–76. [Google Scholar] [CrossRef]
- Moracci, L.; Crotti, S.; Traldi, P.; Agostini, M.; Cosma, C.; Lapolla, A. Role of mass spectrometry in the study of interactions between amylin and metal ions. Mass Spectrom. Rev. 2021, 42, 984–1007. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.G.; Duckworth, W.C.; Hamel, F.G. Degradation of Amylin by Insulin-degrading Enzyme. J. Biol. Chem. 2000, 275, 36621–36625. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.J. Targeting Insulin-Degrading Enzyme to Treat Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2016, 27, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Adamek, R.N.; Suire, C.N.; Stokes, R.W.; Brizuela, M.K.; Cohen, S.M.; Leissring, M.A. Hydroxypyridinethione Inhibitors of Human Insulin-Degrading Enzyme. ChemMedChem 2021, 16, 1776–1788. [Google Scholar] [CrossRef] [PubMed]
- Ling, W.; Huang, Y.M.; Qiao, Y.C.; Zhang, X.X.; Zhao, H.L. Human Amylin: From Pathology to Physiology and Pharmacology. Curr. Protein Pept. Sci. 2019, 20, 944–957. [Google Scholar] [CrossRef]
- Yang, Y.; Song, W. Molecular links between Alzheimer’s disease and diabetes mellitus. Neuroscience 2013, 250, 140–150. [Google Scholar] [CrossRef]
- Sousa, L.; Guarda, M.; Meneses, M.J.; Macedo, M.P.; Vicente Miranda, H. Insulin-degrading enzyme: An ally against metabolic and neurodegenerative diseases. J. Pathol. 2021, 255, 346–361. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Grasso, G.; Gioia, M.; Coletta, A.; Polticelli, F.; Di Pierro, D.; Milardi, D.; Van Endert, P.; Marini, S.; Coletta, M. Multiple functions of insulin-degrading enzyme: A metabolic crosslight? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 554–582. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chorell, E.; Wittung-Stafshede, P. Insulin-degrading enzyme is activated by the C-terminus of -synuclein. Biochem. Biophys. Res. Commun. 2015, 466, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Elseweidy, M.M.; Amin, R.S.; Atteia, H.H.; Ali, M.A. Vitamin D3 intake as regulator of insulin degrading enzyme and insulin receptor phosphorylation in diabetic rats. Biomed. Pharmacother. 2017, 85, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Maianti, J.P.; McFedries, A.; Foda, Z.H.; Kleiner, R.E.; Du, X.Q.; Leissring, M.A.; Tang, W.J.; Charron, M.J.; Seeliger, M.A.; Saghatelian, A.; Liu, D.R. Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones. Nature 2014, 511, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Manolopoulou, M.; Bian, Y.; Schilling, A.B.; Tang, W.J. Molecular Basis for the Recognition and Cleavages of IGF-II, TGF-α, and Amylin by Human Insulin-Degrading Enzyme. J. Mol. Biol. 2010, 395, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.A. Unraveling Alzheimer’s: Making Sense of the Relationship between Diabetes and Alzheimer’s Disease1. J. Alzheimer’s Dis. 2016, 51, 961–977. [Google Scholar] [CrossRef]
- Bennett, R.G.; Hamel, F.G.; Duckworth, W.C. An Insulin-Degrading Enzyme Inhibitor Decreases Amylin Degradation, Increases Amylin-Induced Cytotoxicity, and Increases Amyloid Formation in Insulinoma Cell Cultures. Diabetes 2003, 52, 2315–2320. [Google Scholar] [CrossRef] [PubMed]
- QIU, W.; FOLSTEIN, M. Insulin, insulin-degrading enzyme and amyloid- peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- Durham, T.B.; Toth, J.L.; Klimkowski, V.J.; Cao, J.X.; Siesky, A.M.; Alexander-Chacko, J.; Wu, G.Y.; Dixon, J.T.; McGee, J.E.; Wang, Y.; Guo, S.Y.; Cavitt, R.N.; Schindler, J.; Thibodeaux, S.J.; Calvert, N.A.; Coghlan, M.J.; Sindelar, D.K.; Christe, M.; Kiselyov, V.V.; Michael, M.D.; Sloop, K.W. Dual Exosite-binding Inhibitors of Insulin-degrading Enzyme Challenge Its Role as the Primary Mediator of Insulin Clearance in Vivo. J. Biol. Chem. 2015, 290, 20044–20059. [Google Scholar] [CrossRef]
- Song, E.S.; Ozbil, M.; Zhang, T.; Sheetz, M.; Lee, D.; Tran, D.; Li, S.; Prabhakar, R.; Hersh, L.B.; Rodgers, D.W. An Extended Polyanion Activation Surface in Insulin Degrading Enzyme. PLoS ONE 2015, 10, e0133114. [Google Scholar] [CrossRef]
- Costes, S.; Butler, P. Insulin-Degrading Enzyme Inhibition, a Novel Therapy for Type 2 Diabetes? Cell Metab. 2014, 20, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Bellia, F.; Grasso, G. The role of copper(II) and zinc(II) in the degradation of human and murine IAPP by insulin-degrading enzyme: Metal ion modulation of IAPP degradation by IDE. J. Mass Spectrom. 2014, 49, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yin, F.; Liu, J.; Wang, Y. Geniposide protects pancreatic INS-1E cells from hIAPP-induced cell damage: Potential involvement of insulin degrading-enzyme. Cell Biol. Int. 2014, 39, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Suire, C.N.; Brizuela, M.K.; Leissring, M.A. Quantitative, High-Throughput Assays for Proteolytic Degradation of Amylin. Methods Protoc. 2020, 3, 81. [Google Scholar] [CrossRef] [PubMed]
- Hogan, M.F.; Meier, D.T.; Zraika, S.; Templin, A.T.; Mellati, M.; Hull, R.L.; Leissring, M.A.; Kahn, S.E. Inhibition of Insulin-Degrading Enzyme Does Not Increase Islet Amyloid Deposition in Vitro. Endocrinology 2016, 157, 3462–3468. [Google Scholar] [CrossRef]
- Song, E.S.; Jang, H.; Guo, H.F.; Juliano, M.A.; Juliano, L.; Morris, A.J.; Galperin, E.; Rodgers, D.W.; Hersh, L.B. Inositol phosphates and phosphoinositides activate insulin-degrading enzyme, while phosphoinositides also mediate binding to endosomes. Proc. Natl. Acad. Sci. 2017, 114. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, H.; Fang, G.G.; Walsh, K.; Mwamburi, M.; Wolozin, B.; Abdul-Hay, S.O.; Ikezu, T.; Leissring, M.A.; Qiu, W.Q. Characterization of Insulin Degrading Enzyme and Other Amyloid-β Degrading Proteases in Human Serum: A Role in Alzheimer’s Disease? J. Alzheimer’s Dis. 2012, 29, 329–340. [Google Scholar] [CrossRef]
- Shen, Y.; Joachimiak, A.; Rich Rosner, M.; Tang, W.J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006, 443, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J. Drugs on the horizon for diabesity. Curr. Diabetes Rep. 2005, 5, 353–359. [Google Scholar] [CrossRef]
- Fernandez-Gamba, A.; Leal, M.; Morelli, L.; Castano, E. Insulin-Degrading Enzyme: Structure-Function Relationship and its Possible Roles in Health and Disease. Curr. Pharm. Des. 2009, 15, 3644–3655. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. & Mol. Biol. 2003, 10, 980–980. [Google Scholar]
- Li, W. Half-a-century Burial of ρ, and φ in PDB. Preprint 2021. [Google Scholar]
- Li, W. How do SMA-linked mutations of SMN1 lead to structural/functional deficiency of the SMA protein? PLOS ONE 2017, 12, e0178519. [Google Scholar]
- Li, W. Delving deep into the structural aspects of a furin cleavage site inserted into the spike protein of SARS-CoV-2: A structural biophysical perspective. Biophys. Chem. 2020, 264, 106420. [Google Scholar] [CrossRef] [PubMed]
- Kortemme, T.; Kim, D.E.; Baker, D. Computational Alanine Scanning of Protein-Protein Interfaces. Sci. Signal. 2004, 2004, pl2–pl2. [Google Scholar] [CrossRef]
- Li, W. A: Structural Modifications of Insulin Icodec Contributes to Its Prolonged Duration of Action: A Structural and Biophysical Perspective 2023, 2023. [CrossRef]
- Li, Y.; Liang, Z.; Tian, Y.; Cai, W.; Weng, Z.; Chen, L.; Zhang, H.; Bao, Y.; Zheng, H.; Zeng, S.; Bei, C.; Li, Y. High-affinity PD-1 molecules deliver improved interaction with PD-L1 and PD-L2. Cancer Sci. 2018, 109, 2435–2445. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Delving Deep into the Structural Aspects of the BPro28-BLys29 Exchange in Insulin Lispro: A Structural Biophysical Lesson 2020.
- Li, W. Extracting the Interfacial Electrostatic Features from Experimentally Determined Antigen and/or Antibody-Related Structures inside Protein Data Bank for Machine Learning-Based Antibody Design. 2020. [Google Scholar] [CrossRef]
- Wong, S.K.; Li, W.; Moore, M.J.; Choe, H.; Farzan, M. A 193-Amino Acid Fragment of the SARS Coronavirus S Protein Efficiently Binds Angiotensin-converting Enzyme 2. J. Biol. Chem. 2003, 279, 3197–3201. [Google Scholar] [CrossRef]
- Ho, M.W.; O’Brien, J.S. Gaucher’s Disease: Deficiency of CID β-Glucosidase and Reconstitution of Enzyme Activity In Vitro. Proc. Natl. Acad. Sci. USA 1971, 68, 2810–2813. [Google Scholar] [CrossRef]
- Abraham, E.P.; Chain, E. An enzyme from bacteria able to destroy penicillin. Nature 1940, 146, 837–837. [Google Scholar] [CrossRef]
- Li, W. Strengthening Semaglutide-GLP-1R Binding Affinity Via a Val27-Arg28 Exchange in the Peptide Backbone of Semaglutide: A Computational Structural Approach. Journal of Computational Biophysics and Chemistry.
- Li, W. Designing rt-PA Analogs to Release its Trapped Thrombolytic Activity. J. Comput. Biophys. Chem. 2021, 20, 719–727. [Google Scholar] [CrossRef]
- Shen, Y.; Joachimiak, A.; Rich Rosner, M.; Tang, W.J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006, 443, 870–874. [Google Scholar] [CrossRef]
- Shen, Y.; Tang, W.J. crystal structure of human insulin-degrading enzyme in complex with amylin. 2006. [Google Scholar] [CrossRef]
- Guo, Q.; Bian, Y.; Tang, W. Crystal structure of human insulin-degrading enzyme in complex with amylin. 2009. [Google Scholar] [CrossRef]
- Guo, Q.; Manolopoulou, M.; Bian, Y.; Schilling, A.B.; Tang, W.J. Molecular Basis for the Recognition and Cleavages of IGF-II, TGF-α, and Amylin by Human Insulin-Degrading Enzyme. J. Mol. Biol. 2010, 395, 430–443. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; Zhang, X.; Gao, G.F. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Structural and Functional Consequences of the SMA-Linked Missense Mutations of the Survival Motor Neuron Protein: A Brief Update. In Novel Aspects on Motor Neuron Disease; IntechOpen, 2019.
- Deciphering critical amino acid residues to modify and enhance the binding affinity of ankyrin scaffold specific to capsid protein of human immunodeficiency virus type 1. Asian Pacific Journal of Allergy and Immunology 2017.
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Marmentini, C.; Guimarães, D.S.P.; de Lima, T.I.; Teófilo, F.B.S.; da Silva, N.S.; Soares, G.M.; Boschero, A.C.; Kurauti, M.A. Rosiglitazone protects INS-1E cells from human islet amyloid polypeptide toxicity. Eur. J. Pharmacol. 2022, 928, 175122. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Zhu, H. Amylin and its analogs: A friend or foe for the treatment of Alzheimer’s disease? Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Kuo, W.L.; Yousef, M.; Rosner, M.R.; Tang, W.J. The C-terminal domain of human insulin degrading enzyme is required for dimerization and substrate recognition. Biochem. Biophys. Res. Commun. 2006, 343, 1032–1037. [Google Scholar] [CrossRef]
- Lam, V.K.L.; Ma, R.C.W.; Lee, H.M.; Hu, C.; Park, K.S.; Furuta, H.; Wang, Y.; Tam, C.H.T.; Sim, X.; Ng, D.P.K.; Liu, J.; Wong, T.Y.; Tai, E.S.; Morris, A.P.; Tang, N.L.S.; Woo, J.; Leung, P.C.; Kong, A.P.S.; Ozaki, R.; Jia, W.P.; Lee, H.K.; Nanjo, K.; Xu, G.; Ng, M.C.Y.; So, W.Y.; Chan, J.C.N. Genetic Associations of Type 2 Diabetes with Islet Amyloid Polypeptide Processing and Degrading Pathways in Asian Populations. PLoS ONE 2013, 8, e62378. [Google Scholar] [CrossRef]
- Aston-Mourney, K.; Zraika, S.; Udayasankar, J.; Subramanian, S.L.; Green, P.S.; Kahn, S.E.; Hull, R.L. Matrix Metalloproteinase-9 Reduces Islet Amyloid Formation by Degrading Islet Amyloid Polypeptide. J. Biol. Chem. 2013, 288, 3553–3559. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.J. Targeting Insulin-Degrading Enzyme to Treat Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2016, 27, 24–34. [Google Scholar] [CrossRef]
- Sebokova, E.; Christ, A.; Boehringer, M.; Mizrahi, J. Dipeptidyl Peptidase IV Inhibitors: The Next Generation of New Promising Therapies for the Management of Type 2 Diabetes. Curr. Top. Med. Chem. 2007, 7, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Alafuzoff, I.; Aho, L.; Helisalmi, S.; Mannermaa, A.; Soininen, H. -Amyloid deposition in brains of subjects with diabetes. Neuropathol. Appl. Neurobiol. 2009, 35, 60–68. [Google Scholar] [CrossRef]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Clark, A.; Utzschneider, K.M.; Tong, J.; Gerchman, F.; Kahn, S.E. Identification of the Amyloid-Degrading Enzyme Neprilysin in Mouse Islets and Potential Role in Islet Amyloidogenesis. Diabetes 2007, 56, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Li, W. A Local Spherical Coordinate System Approach to Protein 3D Structure Description 2020.
- Li, W. Calcium Channel Trafficking Blocker Gabapentin Bound to the -2–1 Subunit of Voltage-Gated Calcium Channel: A Computational Structural Investigation 2020.
- Li, W.; Vottevor, G. Towards a Truly General Intermolecular Binding Affinity Calculator for Drug Discovery & Design. 2023. [Google Scholar] [CrossRef]
- Crunkhorn, S. Role of the Protein Data Bank. Nat. Rev. Drug Discov. 2019, 18, 98–98. [Google Scholar] [CrossRef]
- Westbrook, J.D.; Burley, S.K. How Structural Biologists and the Protein Data Bank Contributed to Recent FDA New Drug Approvals. Structure 2019, 27, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Visualising the Experimentally Uncharted Territories of Membrane Protein Structures inside Protein Data Bank 2020.
- Wu, J.; Yan, Z.; Li, Z.; Qian, X.; Lu, S.; Dong, M.; Zhou, Q.; Yan, N. Structure of the voltage-gated calcium channel CaV1.1 at 3.6 Å resolution. Nature 2016, 537, 191–196. [Google Scholar] [CrossRef]
- Li, W.; Shi, G. How CaV1.2-bound verapamil blocks Ca2+ influx into cardiomyocyte: Atomic level views. Pharmacol. Res. 2019, 139, 153–157. [Google Scholar] [CrossRef]
Figure 1.
The overall structure of human insulin-degrading enzyme in complex with amylin. This figure is prepared by PyMol [53] with PDB ID 2G48 [45,46]. In this figure, IDE as an amylin-degrading enzyme is like a cage for amylin as its substrate, making it a preferable choice for the potential development of small molecule inhibitor(s) to reach inside IDE (the cage) and disrupt the amylin-IDE interaction for improved metabolic homeostasis.
Figure 1.
The overall structure of human insulin-degrading enzyme in complex with amylin. This figure is prepared by PyMol [53] with PDB ID 2G48 [45,46]. In this figure, IDE as an amylin-degrading enzyme is like a cage for amylin as its substrate, making it a preferable choice for the potential development of small molecule inhibitor(s) to reach inside IDE (the cage) and disrupt the amylin-IDE interaction for improved metabolic homeostasis.
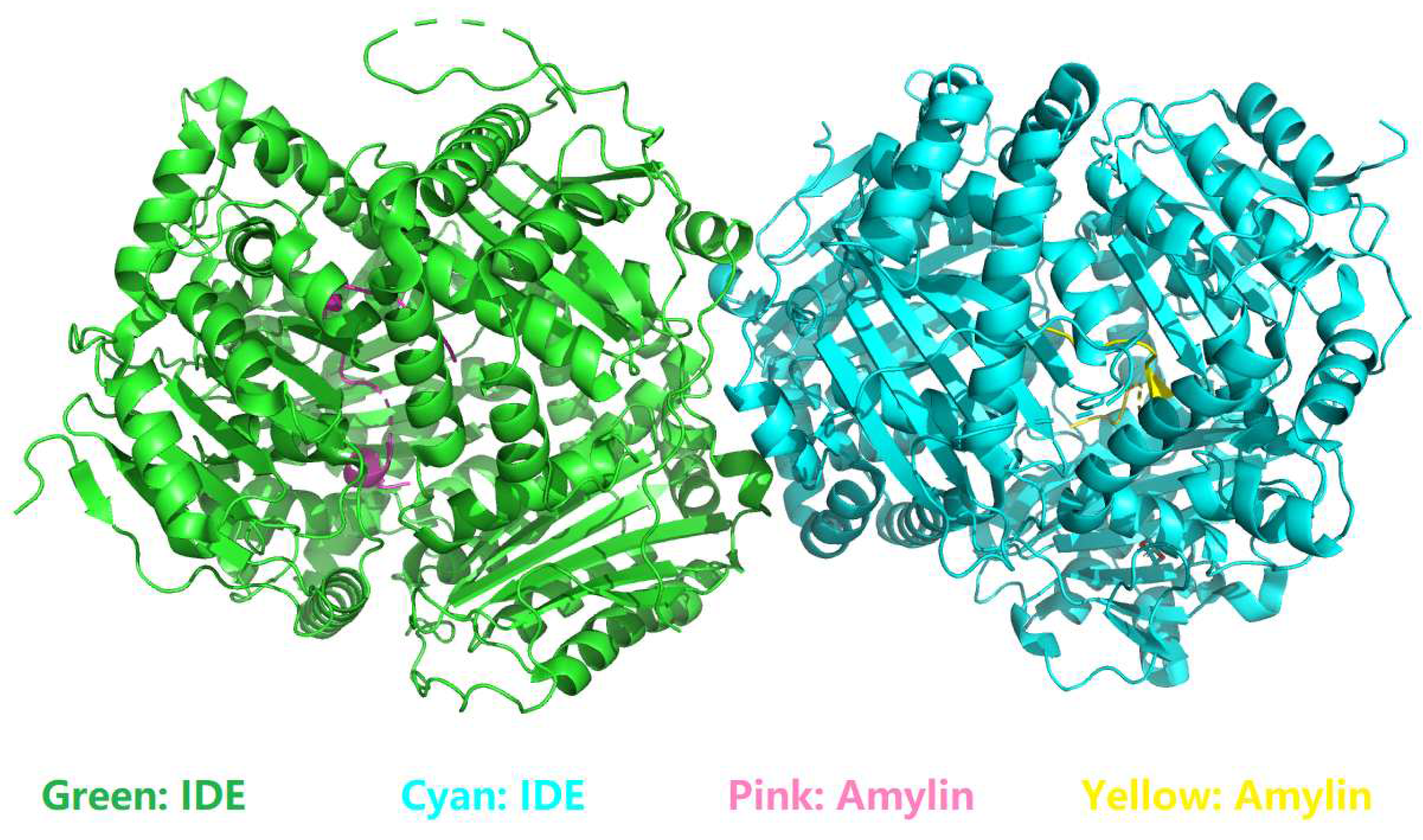
Figure 2.
Two sets of interfacial salt bridges (dotted yellow sticks) between Lys1 (K1) of amylin and Glu341 (E341) of IDE. This figure is prepared by PyMol [53] with PDB ID 2G48 [45,46]. In this figure, the color scheme is the same as in Figure 1.
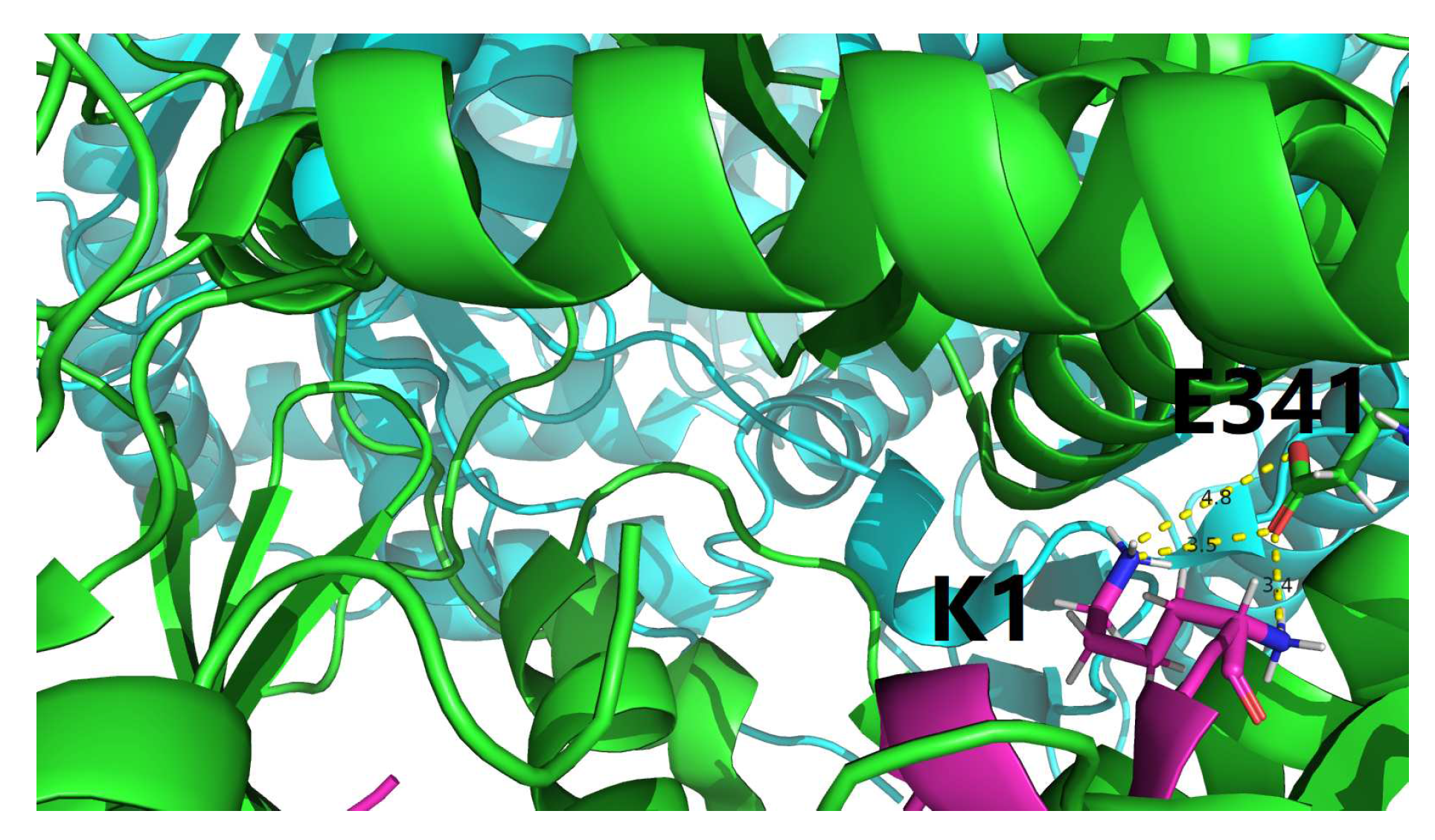
Figure 3.
A closer (than Figurer̃efpymol2) view of two sets of interfacial salt bridges (dotted yellow sticks) between Lys1 (K1) of amylin and Glu341 (E341) of IDE. This figure is prepared by PyMol [53] with PDB ID 2G48 [45,46]. In this figure, the color scheme is the same as in Figure 1.
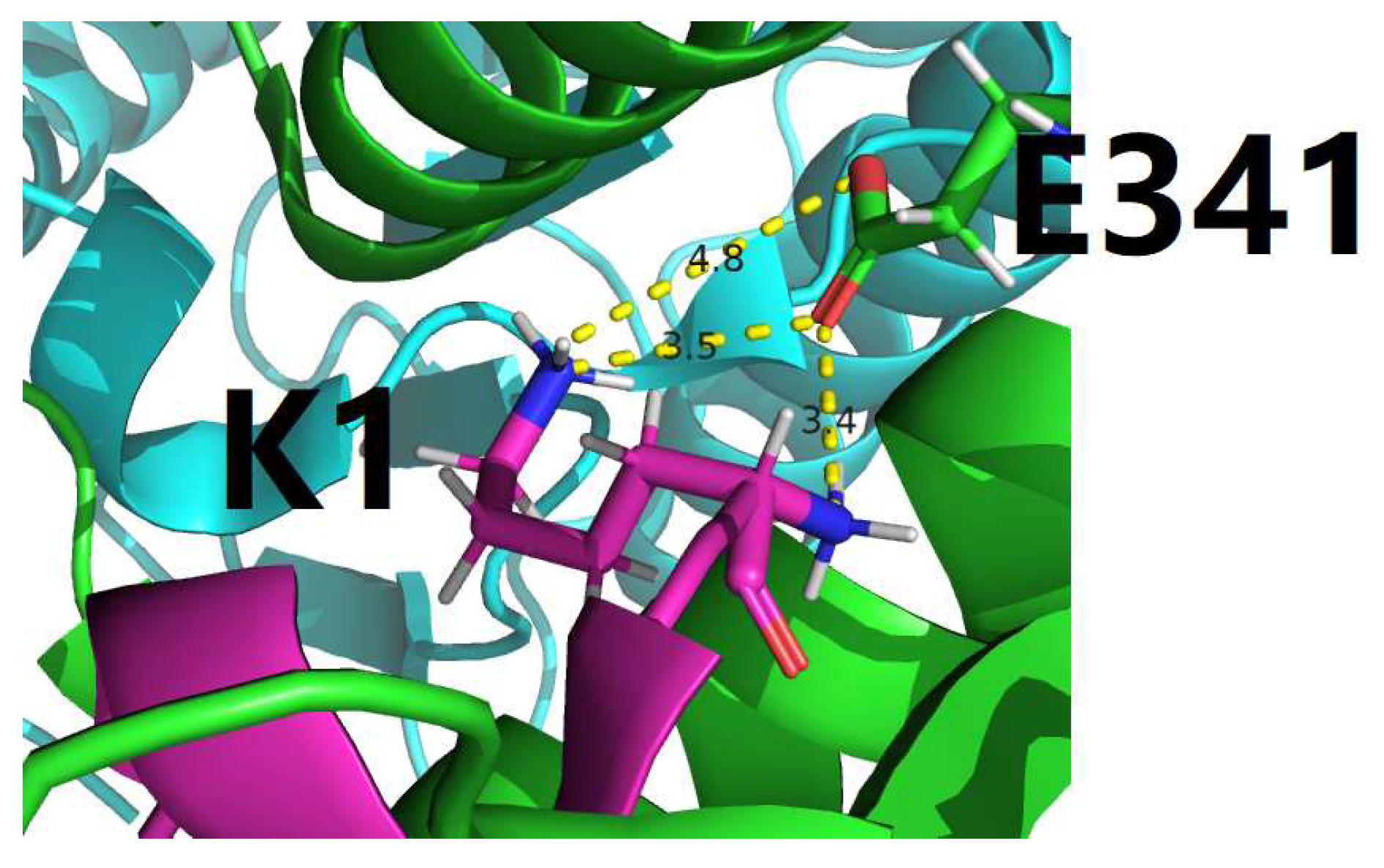

Table 1.
Experimentally determined amylin-related structures in the Protein Data Bank (PDB [31]) as of February 1, 2024. QUERY: Full Text = "amylin". In this table, the two structural models representing IDE-amylin complex are highlighted in two yellow rows, i.e., PDB IDs: 2G48 [45,46] and 3HGZ [47,48].
Table 1.
Experimentally determined amylin-related structures in the Protein Data Bank (PDB [31]) as of February 1, 2024. QUERY: Full Text = "amylin". In this table, the two structural models representing IDE-amylin complex are highlighted in two yellow rows, i.e., PDB IDs: 2G48 [45,46] and 3HGZ [47,48].
| PDB ID | Structure Title |
|---|---|
| 8AZ7 | IAPP S20G plateau-phase fibril polymorph 4PF-LJ |
| 8AZ6 | IAPP S20G plateau-phase fibril polymorph 4PF-LU |
| 8AZ5 | IAPP S20G plateau-phase fibril polymorph 4PF-CU |
| 8AZ4 | IAPP S20G plateau-phase fibril polymorph 2PF-L |
| 8AZ3 | IAPP S20G growth-phase fibril polymorph 4PF-CU |
| 8AZ2 | IAPP S20G growth-phase fibril polymorph 3PF-CU |
| 8AZ1 | IAPP S20G growth-phase fibril polymorph 2PF-C |
| 8AZ0 | IAPP S20G growth-phase fibril polymorph 2PF-L |
| 8AWT | IAPP S20G lag-phase fibril polymorph 2PF-P |
| 8T89 | Racemic mixture of amyloid beta segment 16-KLVFFA-21 forms heterochiral rippled beta-sheet |
| 8T86 | Racemic mixture of amylin segment 25-AILSS-29 forms heterochiral rippled beta-sheet |
| 8T84 | Racemic mixture of amyloid beta segment 35-MVGGVV-40 forms heterochiral rippled beta-sheet, includes hexafluoroisopropanol |
| 8T82 | Racemic mixture of amyloid beta segment 35-MVGGVV-40 forms heterochiral rippled beta-sheet, includes pentafluoropropionic acid |
| 8F2B | Amylin 3 Receptor in complex with Gs and Pramlintide analogue peptide San45 |
| 8F2A | Human Amylin3 Receptor in complex with Gs and Pramlintide analogue peptide San385 (Cluster 5 conformation) |
| 8F0K | Human Amylin3 Receptor in complex with Gs and Pramlintide analogue peptide San385 |
| 8F0J | Calcitonin Receptor in complex with Gs and Pramlintide analogue peptide San45 |
| 7YKW | Structure of hIAPP fibril at 3.6 Angstroms resolution |
| 7YL7 | Structure of hIAPP-TF-type3 |
| 7YL3 | Structure of hIAPP-TF-type1 |
| 7YL0 | Structure of hIAPP-TF-type2 |
| 8AX7 | Crystal structure of a CGRP receptor ectodomain heterodimer bound to macrocyclic inhibitor HTL0031448 |
| 8AX6 | Crystal structure of a CGRP receptor ectodomain heterodimer bound to macrocyclic inhibitor HTL0029882 |
| 8AX5 | Crystal structure of a CGRP receptor ectodomain heterodimer bound to macrocyclic inhibitor HTL0029881 |
| 7P0I | Crystal structure of a CGRP receptor ectodomain heterodimer bound to macrocyclic inhibitor Compound 13 |
| 7P0F | Crystal structure of a CGRP receptor ectodomain heterodimer bound to macrocyclic inhibitor HTL0028125 |
| 7TYX | Human Amylin2 Receptor in complex with Gs and rat amylin peptide |
| 7TYN | Calcitonin Receptor in complex with Gs and salmon calcitonin peptide |
| 7TYI | Calcitonin Receptor in complex with Gs and rat amylin peptide, CT-like state |
| 7TZF | Human Amylin3 Receptor in complex with Gs and rat amylin peptide |
| 7TYY | Human Amylin2 Receptor in complex with Gs and salmon calcitonin peptide |
| 7TYW | Human Amylin1 Receptor in complex with Gs and salmon calcitonin peptide |
| 7TYO | Calcitonin receptor in complex with Gs and human calcitonin peptide |
| 7TYL | Calcitonin Receptor in complex with Gs and rat amylin peptide, bypass motif |
| 7TYH | Human Amylin2 Receptor in complex with Gs and human calcitonin peptide |
| 7TYF | Human Amylin1 Receptor in complex with Gs and rat amylin peptide |
| 7VV0 | Cryo-EM structure of pseudoallergen receptor MRGPRX2 complex with PAMP-12, local |
| 7M65 | Cryo-EM structure of human islet amyloid polypeptide (hIAPP, or amylin) fibrils seeded by patient extracted fibrils, polymorph 4 |
| 7M64 | Cryo-EM structure of human islet amyloid polypeptide (hIAPP, or amylin) fibrils seeded by patient extracted fibrils, polymorph 3 |
| 7M62 | Cryo-EM structure of human islet amyloid polypeptide (hIAPP, or amylin) fibrils seeded by patient extracted fibrils, polymorph 2 |
| 7M61 | Cryo-EM structure of human islet amyloid polypeptide (hIAPP, or amylin) fibrils seeded by patient extracted fibrils, polymorph 1 |
| 7BG0 | Fusion of MBP and the backbone of the long-acting amylin analog AM833. |
| 7KNU | CryoEM structure of the CGRP receptor with bound CGRP peptide in a detergent micelle |
| 7KNT | CryoEM structure of the apo-CGRP receptor in a detergent micelle |
| 6ZRR | three-protofilament amyloid structure of S20G variant of human amylin (IAPP - Islet Amyloid Polypeptide) |
| 6ZRQ | two-protofilament amyloid structure of S20G variant of human amylin (IAPP - islet amyloid polypeptide) |
| 6ZRF | amyloid structure of amylin (IAPP - islet amyloid polypeptide) |
| 6V2E | Crystal structure of the human CLR:RAMP2 extracellular domain heterodimer with bound high-affinity adrenomedullin S45R/K46L/S48G/Q50W variant |
| 6ZIS | Crystal structure of a CGRP receptor ectodomain heterodimer with bound high affinity inhibitor |
| 6ZHO | Crystal structure of a CGRP receptor ectodomain heterodimer with bound high affinity inhibitor |
| 6VW2 | Cryo-EM structure of human islet amyloid polypeptide (hIAPP, or amylin) fibrils |
| 6UVA | CryoEM Structure of the active Adrenomedullin 2 receptor G protein complex with adrenomedullin 2 peptide |
| 6UUS | CryoEM Structure of the active Adrenomedullin 2 receptor G protein complex with adrenomedullin peptide |
| 6UUN | CryoEM Structure of the active Adrenomedullin 1 receptor G protein complex with adrenomedullin peptide |
| 6Y1A | Amyloid fibril structure of islet amyloid polypeptide |
| 6UCK | proIAPP in DPC Micelles - Two-Conformer Ensemble Refinement, Bent Conformer |
| 6UCJ | proIAPP in DPC Micelles - Two-Conformer Ensemble Refinement, Open Conformer |
| 6UMG | Crystal structure of erenumab Fab bound to the extracellular domain of CGRP receptor |
| 6PGQ | Crystal structure of N-glycosylated human calcitonin receptor extracellular domain in complex with salmon calcitonin (22-32) |
| 6PFO | Crystal structure of N-glycosylated human calcitonin receptor extracellular domain in complex with salmon calcitonin (16-32) |
| 6NIY | A high-resolution cryo-electron microscopy structure of a calcitonin receptor-heterotrimeric Gs protein complex |
| 6E3Y | Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor |
| 6D1U | Crystal structure of the human CLR:RAMP1 extracellular domain heterodimer in complex with adrenomedullin 2/intermedin |
| 5V6Y | Crystal structure of the human CLR:RAMP1 extracellular domain heterodimer with bound high-affinity and altered selectivity adrenomedullin variant |
| 5UZ7 | Volta phase plate cryo-electron microscopy structure of a calcitonin receptor-heterotrimeric Gs protein complex |
| 5MGQ | Solution structure of oxidized and amidated human IAPP (1-37), the diabetes II peptide. |
| 5KO0 | Human Islet Amyloid Polypeptide Segment 15-FLVHSSNNFGA-25 Determined by MicroED |
| 5KNZ | Human Islet Amyloid Polypeptide Segment 19-SGNNFGAILSS-29 with Early Onset S20G Mutation Determined by MicroED |
| 5K5G | Structure of human islet amyloid polypeptide in complex with an engineered binding protein |
| 5II0 | Crystal structure of the human calcitonin receptor ectodomain in complex with a truncated salmon calcitonin analogue |
| 4RWG | Crystal structure of the CLR:RAMP1 extracellular domain heterodimer with bound high affinity CGRP analog |
| 4RWF | Crystal structure of the CLR:RAMP2 extracellular domain heterodimer with bound adrenomedullin |
| 3AQE | Crystal structure of the extracellular domain of human RAMP2 |
| 3AQF | Crystal structure of the human CRLR/RAMP2 extracellular complex |
| 2L7S | Determination of the three-dimensional structure of adrenomedullin, a first step towards the analysis of its interactions with receptors and small molecules |
| 2L86 | Solution NMR structure of human amylin in SDS micelles at pH 7.3 |
| 2XVT | Structure of the extracellular domain of human RAMP2 |
| 3N7S | Crystal structure of the ectodomain complex of the CGRP receptor, a Class-B GPCR, reveals the site of drug antagonism |
| 3N7R | Crystal structure of the ectodomain complex of the CGRP receptor, a Class-B GPCR, reveals the site of drug antagonism |
| 3N7P | Crystal structure of the ectodomain complex of the CGRP receptor, a Class-B GPCR, reveals the site of drug antagonism |
| 3HGZ | Crystal structure of human insulin-degrading enzyme in complex with amylin |
| 2WK3 | Crystal structure of human insulin-degrading enzyme in complex with amyloid-beta (1-42) |
| 2KIB | Protein Fibril |
| 3E50 | Crystal structure of human insulin degrading enzyme in complex with transforming growth factor-alpha |
| 3E4Z | Crystal structure of human insulin degrading enzyme in complex with insulin-like growth factor II |
| 3FTR | Structure of an amyloid forming peptide SSTNVG from IAPP (alternate polymorph) |
| 3FTL | NVGSNTY segment from Islet Amyloid Polypeptide (IAPP or Amylin), dehydrated crystal form |
| 3FTK | NVGSNTY segment from Islet Amyloid Polypeptide (IAPP or Amylin), hydrated crystal form |
| 3FTH | NFLVHSS segment from Islet Amyloid Polypeptide (IAPP or Amylin) |
| 3FR1 | NFLVHS segment from Islet Amyloid Polypeptide (IAPP or Amylin) |
| 3FPO | HSSNNF segment from Islet Amyloid Polypeptide (IAPP or Amylin) |
| 3G7W | Islet Amyloid Polypeptide (IAPP or Amylin) Residues 1 to 22 fused to Maltose Binding Protein |
| 3G7V | Islet Amyloid Polypeptide (IAPP or Amylin) fused to Maltose Binding Protein |
| 2KJ7 | Three-Dimensional NMR Structure of Rat Islet Amyloid Polypeptide in DPC micelles |
| 2KB8 | The dynamic alpha-helix structure of micelle-bound human amylin. |
| 3DGJ | NNFGAIL segment from Islet Amyloid Polypeptide (IAPP or amylin) |
| 3DG1 | Segment SSTNVG derived from IAPP |
| 2YX8 | Crystal structure of the extracellular domain of human RAMP1 |
| 2G48 | crystal structure of human insulin-degrading enzyme in complex with amylin |
| 2FLY | Proadrenomedullin N-Terminal 20 Peptide |
| 1KUW | High-Resolution Structure and Localization of Amylin Nucleation Site in Detergent Micelle |
Table 2.
Interfacial salt bridging network analysis of the two structural models of IDE-amylin complex (Table 1), i.e., PDB IDs 2G48 [45,46] and 3HGZ [47,48] according to the old set of criteria as in [33]. In this table, the residue naming scheme is Chain ID_residue name_residue number. As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
Table 2.
Interfacial salt bridging network analysis of the two structural models of IDE-amylin complex (Table 1), i.e., PDB IDs 2G48 [45,46] and 3HGZ [47,48] according to the old set of criteria as in [33]. In this table, the residue naming scheme is Chain ID_residue name_residue number. As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
| PDB ID | Residue A | Atom A | Residue B | Atom B | Distance (Å) |
|---|---|---|---|---|---|
| 2G48 | A_ARG_722 | NH1 | B_ASP_706 | OD1 | 2.987 |
| 2G48 | A_ARG_722 | NH1 | B_ASP_706 | OD2 | 2.560 |
| 2G48 | A_ARG_722 | NH2 | B_GLU_702 | OE2 | 3.255 |
| 2G48 | A_LYS_756 | NZ | B_ASP_706 | OD1 | 3.852 |
| 2G48 | B_ARG_722 | NH1 | A_ASP_706 | OD1 | 2.728 |
| 2G48 | B_ARG_722 | NH1 | A_ASP_706 | OD2 | 3.524 |
| 2G48 | B_ARG_722 | NH2 | A_ASP_706 | OD1 | 3.653 |
| 2G48 | B_LYS_756 | NZ | A_ASP_706 | OD1 | 3.253 |
| 2G48 | B_LYS_756 | NZ | A_ASP_706 | OD2 | 2.575 |
| 2G48 | C_LYS_1 | NZ | A_GLU_341 | OE1 | 3.514 |
| 3HGZ | A_ARG_164 | NH1 | B_GLU_408 | OE1 | 3.083 |
| 3HGZ | A_ARG_164 | NH1 | B_GLU_408 | OE2 | 3.662 |
| 3HGZ | A_ARG_164 | NH2 | B_GLU_408 | OE1 | 3.032 |
| 3HGZ | B_LYS_327 | NZ | A_GLU_880 | OE1 | 3.476 |
| 3HGZ | D_LYS_1 | NZ | A_GLU_341 | OE1 | 2.441 |
| 3HGZ | E_LYS_1 | NZ | B_GLU_341 | OE1 | 3.179 |
Table 3.
2G48-specific interfacial side chain and main chain hydrogen bonding analysis according to the old set of criteria as in [33]. In this table, the residue naming scheme is Chain ID_residue name_residue number, represents the angle formed by acceptor (A), donor (D) and hydrogen (H) . As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
Table 3.
2G48-specific interfacial side chain and main chain hydrogen bonding analysis according to the old set of criteria as in [33]. In this table, the residue naming scheme is Chain ID_residue name_residue number, represents the angle formed by acceptor (A), donor (D) and hydrogen (H) . As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
| PDB ID | Acceptor (A) | Donor (D) | Hydrogen (H) | D-A (Å) | H-A (Å) | |
|---|---|---|---|---|---|---|
| 2G48 | O, C_LYS_1 | N, A_GLY_361 | H, A_GLY_361 | 2.72 | 1.72 | 5.66 |
| 2G48 | OE2, B_GLU_699 | OG, A_SER_761 | HG, A_SER_761 | 2.52 | 1.67 | 22.41 |
| 2G48 | O, C_LEU_16 | NH2, A_ARG_824 | HH21, A_ARG_824 | 2.98 | 2.09 | 22.98 |
| 2G48 | O, D_ASN_14 | N, B_THR_142 | H, B_THR_142 | 2.89 | 1.99 | 21.69 |
| 2G48 | O, D_LYS_1 | N, B_GLY_361 | H, B_GLY_361 | 2.89 | 1.89 | 5.77 |
| 2G48 | OD1, A_ASP_706 | NH1, B_ARG_722 | HH12, B_ARG_722 | 2.73 | 1.91 | 29.32 |
| 2G48 | OD2, A_ASP_706 | NZ, B_LYS_756 | HZ1, B_LYS_756 | 2.57 | 1.59 | 10.38 |
| 2G48 | OE2, A_GLU_699 | OG, B_SER_761 | HG, B_SER_761 | 2.51 | 1.64 | 19.94 |
| 2G48 | O, A_GLY_361 | N, C_ASN_3 | H, C_ASN_3 | 2.65 | 1.76 | 22.83 |
| 2G48 | O, A_ALA_140 | N, C_LEU_16 | H, C_LEU_16 | 2.97 | 2.01 | 15.18 |
| 2G48 | OE1, B_GLU_341 | N, D_LYS_1 | H2, D_LYS_1 | 2.85 | 1.85 | 6.72 |
| 2G48 | O, B_GLY_361 | N, D_ASN_3 | H, D_ASN_3 | 2.67 | 1.76 | 20.40 |
| 2G48 | O, B_GLN_363 | ND2, D_ASN_3 | HD22, D_ASN_3 | 2.73 | 1.89 | 26.92 |
| 2G48 | O, B_ALA_140 | N, D_LEU_16 | H, D_LEU_16 | 2.83 | 1.95 | 23.79 |
Table 4.
2G48-specific interfacial side chain hydrogen bonding analysis according to the old set of criteria as in [33]. In this table, the residue naming scheme is Chain ID_residue name_residue number, represents the angle formed by acceptor (A), donor (D) and hydrogen (H) . As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
Table 4.
2G48-specific interfacial side chain hydrogen bonding analysis according to the old set of criteria as in [33]. In this table, the residue naming scheme is Chain ID_residue name_residue number, represents the angle formed by acceptor (A), donor (D) and hydrogen (H) . As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
| PDB ID | Acceptor (A) | Donor (D) | Hydrogen (H) | D-A (Å) | H-A (Å) | |
|---|---|---|---|---|---|---|
| 2G48 | OE2, B_GLU_699 | OG, A_SER_761 | HG, A_SER_761 | 2.52 | 1.67 | 22.41 |
| 2G48 | OD1, A_ASP_706 | NH1, B_ARG_722 | HH12, B_ARG_722 | 2.73 | 1.91 | 29.32 |
| 2G48 | OD2, A_ASP_706 | NZ, B_LYS_756 | HZ1, B_LYS_756 | 2.57 | 1.59 | 10.38 |
| 2G48 | OE2, A_GLU_699 | OG, B_SER_761 | HG, B_SER_761 | 2.51 | 1.64 | 19.94 |
Table 5.
Interfacial salt bridging network analysis within the PDB entries (2G48 [45,46] and 3HGZ [47,48]) according to a new set of criteria [33] as defined in the section of Materials and Methods. In this table, the residue naming scheme is Chain ID_residue name_residue number. As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
Table 5.
Interfacial salt bridging network analysis within the PDB entries (2G48 [45,46] and 3HGZ [47,48]) according to a new set of criteria [33] as defined in the section of Materials and Methods. In this table, the residue naming scheme is Chain ID_residue name_residue number. As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
| PDB ID | Residue A | Atom A | Residue B | Atom B | Distance (Å) |
|---|---|---|---|---|---|
| 2G48 | A_ARG_722 | NH1 | B_GLU_702 | OE2 | 4.700 |
| 2G48 | A_ARG_722 | NH1 | B_ASP_706 | OD1 | 2.987 |
| 2G48 | A_ARG_722 | NH1 | B_ASP_706 | OD2 | 2.560 |
| 2G48 | A_ARG_722 | NH2 | B_GLU_702 | OE1 | 5.134 |
| 2G48 | A_ARG_722 | NH2 | B_GLU_702 | OE2 | 3.255 |
| 2G48 | A_ARG_722 | NH2 | B_ASP_706 | OD1 | 4.711 |
| 2G48 | A_ARG_722 | NH2 | B_ASP_706 | OD2 | 4.433 |
| 2G48 | A_LYS_756 | NZ | B_GLU_702 | OE2 | 5.525 |
| 2G48 | A_LYS_756 | NZ | B_ASP_706 | OD1 | 3.852 |
| 2G48 | A_LYS_756 | NZ | B_ASP_706 | OD2 | 5.449 |
| 2G48 | A_LYS_1009 | NZ | B_GLU_990 | OE1 | 5.840 |
| 2G48 | B_ARG_722 | NH1 | A_ASP_706 | OD1 | 2.728 |
| 2G48 | B_ARG_722 | NH1 | A_ASP_706 | OD2 | 3.524 |
| 2G48 | B_ARG_722 | NH2 | A_GLU_702 | OE1 | 5.354 |
| 2G48 | B_ARG_722 | NH2 | A_ASP_706 | OD1 | 3.653 |
| 2G48 | B_ARG_722 | NH2 | A_ASP_706 | OD2 | 5.078 |
| 2G48 | B_LYS_756 | NZ | A_ASP_706 | OD1 | 3.253 |
| 2G48 | B_LYS_756 | NZ | A_ASP_706 | OD2 | 2.575 |
| 2G48 | B_LYS_1009 | NZ | A_GLU_997 | OE1 | 5.095 |
| 2G48 | C_LYS_1 | NZ | A_GLU_341 | OE1 | 3.514 |
| 2G48 | C_LYS_1 | NZ | A_GLU_341 | OE2 | 4.876 |
| 2G48 | C_LYS_1 | NZ | A_GLU_612 | OE2 | 5.297 |
| 3HGZ | A_LYS_123 | NZ | B_ASP_416 | OD1 | 4.754 |
| 3HGZ | A_ARG_164 | NH1 | B_GLU_408 | OE1 | 3.083 |
| 3HGZ | A_ARG_164 | NH1 | B_GLU_408 | OE2 | 3.662 |
| 3HGZ | A_ARG_164 | NH2 | B_GLU_408 | OE1 | 3.032 |
| 3HGZ | A_ARG_164 | NH2 | B_GLU_408 | OE2 | 4.613 |
| 3HGZ | A_LYS_884 | NZ | B_GLU_457 | OE1 | 4.680 |
| 3HGZ | B_HIS_53 | ND1 | A_GLU_875 | OE1 | 4.690 |
| 3HGZ | B_HIS_53 | NE2 | A_GLU_875 | OE1 | 4.274 |
| 3HGZ | B_LYS_327 | NZ | A_GLU_880 | OE1 | 3.476 |
| 3HGZ | B_LYS_327 | NZ | A_GLU_880 | OE2 | 5.083 |
| 3HGZ | B_LYS_415 | NZ | A_GLU_133 | OE1 | 5.186 |
| 3HGZ | D_LYS_1 | NZ | A_GLU_341 | OE1 | 2.441 |
| 3HGZ | D_LYS_1 | NZ | A_GLU_341 | OE2 | 4.468 |
| 3HGZ | E_LYS_1 | NZ | B_GLU_341 | OE1 | 3.179 |
| 3HGZ | E_LYS_1 | NZ | B_GLU_341 | OE2 | 5.147 |
Table 6.
2G48 [45,46]-specific interfacial side chain hydrogen bonding analysis according to a new set of criteria [33] as defined in the section of Materials and Methods. In this table, the residue naming scheme is Chain ID_residue name_residue number, represents the angle formed by acceptor (A), donor (D) and hydrogen (H) . As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
Table 6.
2G48 [45,46]-specific interfacial side chain hydrogen bonding analysis according to a new set of criteria [33] as defined in the section of Materials and Methods. In this table, the residue naming scheme is Chain ID_residue name_residue number, represents the angle formed by acceptor (A), donor (D) and hydrogen (H) . As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
| PDB ID | Acceptor (A) | Donor (D) | Hydrogen (H) | D-A (Å) | H-A (Å) | |
|---|---|---|---|---|---|---|
| 2G48 | NE2, C_HIS_18 | NZ, A_LYS_192 | HZ3, A_LYS_192 | 4.89 | 4.23 | 44.36 |
| 2G48 | OD1, C_ASN_14 | OG1, A_THR_220 | HG1, A_THR_220 | 3.22 | 2.28 | 9.48 |
| 2G48 | ND2, C_ASN_14 | OG1, A_THR_220 | HG1, A_THR_220 | 3.21 | 2.65 | 47.15 |
| 2G48 | OE1, B_GLN_718 | NE2, A_HIS_589 | HE2, A_HIS_589 | 4.14 | 3.18 | 16.40 |
| 2G48 | NE2, B_GLN_718 | NE2, A_HIS_589 | HE2, A_HIS_589 | 4.55 | 3.58 | 13.52 |
| 2G48 | OG, B_SER_721 | NH1, A_ARG_711 | HH12, A_ARG_711 | 4.90 | 3.97 | 20.43 |
| 2G48 | NE2, B_GLN_718 | NH2, A_ARG_711 | HH21, A_ARG_711 | 3.92 | 3.14 | 33.89 |
| 2G48 | OG, B_SER_721 | NH2, A_ARG_711 | HH21, A_ARG_711 | 4.86 | 3.92 | 19.33 |
| 2G48 | NE2, B_HIS_589 | NE2, A_GLN_718 | HE22, A_GLN_718 | 3.40 | 2.61 | 32.75 |
| 2G48 | OD1, B_ASP_706 | NH1, A_ARG_722 | HH12, A_ARG_722 | 2.99 | 2.41 | 46.66 |
| 2G48 | OE2, B_GLU_702 | NH2, A_ARG_722 | HH22, A_ARG_722 | 3.25 | 2.43 | 29.82 |
| 2G48 | OE1, B_GLU_699 | OG, A_SER_761 | HG, A_SER_761 | 3.84 | 2.92 | 13.96 |
| 2G48 | OE2, B_GLU_699 | OG, A_SER_761 | HG, A_SER_761 | 2.52 | 1.67 | 22.41 |
| 2G48 | OD2, B_ASP_586 | NE2, A_GLN_762 | HE22, A_GLN_762 | 3.64 | 3.02 | 45.51 |
| 2G48 | NE2, B_GLN_770 | NE2, A_GLN_770 | HE21, A_GLN_770 | 4.30 | 3.39 | 22.59 |
| 2G48 | OG, D_SER_19 | ND2, B_ASN_139 | HD22, B_ASN_139 | 4.49 | 3.91 | 48.99 |
| 2G48 | OD1, D_ASN_14 | OG1, B_THR_220 | HG1, B_THR_220 | 3.09 | 2.14 | 8.07 |
| 2G48 | ND2, D_ASN_14 | OG1, B_THR_220 | HG1, B_THR_220 | 3.42 | 2.85 | 46.81 |
| 2G48 | OE1, A_GLN_718 | NE, B_ARG_711 | HE, B_ARG_711 | 4.89 | 3.96 | 20.15 |
| 2G48 | OG, A_SER_721 | NE, B_ARG_711 | HE, B_ARG_711 | 4.94 | 4.15 | 34.49 |
| 2G48 | OG, A_SER_721 | NH1, B_ARG_711 | HH12, B_ARG_711 | 4.28 | 3.36 | 20.95 |
| 2G48 | OD1, A_ASP_706 | NH1, B_ARG_722 | HH12, B_ARG_722 | 2.73 | 1.91 | 29.32 |
| 2G48 | OD2, A_ASP_706 | NH1, B_ARG_722 | HH12, B_ARG_722 | 3.52 | 2.69 | 29.77 |
| 2G48 | OD1, A_ASP_706 | NH2, B_ARG_722 | HH21, B_ARG_722 | 3.65 | 3.09 | 49.48 |
| 2G48 | OD2, A_ASP_706 | NZ, B_LYS_756 | HZ1, B_LYS_756 | 2.57 | 1.59 | 10.38 |
| 2G48 | OE1, A_GLU_699 | OG, B_SER_761 | HG, B_SER_761 | 4.00 | 3.07 | 11.68 |
| 2G48 | OE2, A_GLU_699 | OG, B_SER_761 | HG, B_SER_761 | 2.51 | 1.64 | 19.94 |
| 2G48 | OD2, A_ASP_586 | NE2, B_GLN_762 | HE22, B_GLN_762 | 4.17 | 3.60 | 49.58 |
| 2G48 | NE2, A_GLN_770 | NE2, B_GLN_770 | HE21, B_GLN_770 | 4.30 | 3.46 | 30.40 |
| 2G48 | OD1, D_ASN_22 | NH2, B_ARG_847 | HH22, B_ARG_847 | 4.10 | 3.14 | 17.00 |
| 2G48 | ND2, D_ASN_22 | NH2, B_ARG_847 | HH22, B_ARG_847 | 4.95 | 3.97 | 13.60 |
| 2G48 | OE1, A_GLU_341 | NZ, C_LYS_1 | HZ3, C_LYS_1 | 3.51 | 2.72 | 32.95 |
| 2G48 | OE2, A_GLU_341 | NZ, C_LYS_1 | HZ3, C_LYS_1 | 4.88 | 4.20 | 43.27 |
| 2G48 | ND1, A_HIS_332 | OG1, C_THR_4 | HG1, C_THR_4 | 3.10 | 2.41 | 37.40 |
| 2G48 | OG1, A_THR_220 | ND2, C_ASN_14 | HD22, C_ASN_14 | 3.21 | 2.37 | 28.58 |
| 2G48 | NE2, A_HIS_679 | NE2, C_HIS_18 | HE2, C_HIS_18 | 4.27 | 3.60 | 43.28 |
| 2G48 | OG1, B_THR_220 | ND2, D_ASN_14 | HD22, D_ASN_14 | 3.42 | 2.67 | 36.00 |
| 2G48 | NE2, B_HIS_679 | NE2, D_HIS_18 | HE2, D_HIS_18 | 4.32 | 3.75 | 49.86 |
Table 7.
3HGZ [47,48]-specific interfacial side chain hydrogen bonding analysis according to a new set of criteria [33] as defined in the section of Materials and Methods. In this table, the residue naming scheme is Chain ID_residue name_residue number, represents the angle formed by acceptor (A), donor (D) and hydrogen (H) . As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
Table 7.
3HGZ [47,48]-specific interfacial side chain hydrogen bonding analysis according to a new set of criteria [33] as defined in the section of Materials and Methods. In this table, the residue naming scheme is Chain ID_residue name_residue number, represents the angle formed by acceptor (A), donor (D) and hydrogen (H) . As defined in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48], chains A and B represent IDE in 3HGZ, chains D and E represent amylin in 3HGZ, while chains A and B represent IDE in 2G48, and chains C and D represent amylin in 2G48, respectively.
| PDB ID | Acceptor (A) | Donor (D) | Hydrogen (H) | D-A (Å) | H-A (Å) | |
|---|---|---|---|---|---|---|
| 3HGZ | NE2, B_GLN_407 | NZ, A_LYS_120 | HZ3, A_LYS_120 | 4.83 | 4.00 | 31.04 |
| 3HGZ | OE1, B_GLU_408 | NH1, A_ARG_164 | HH11, A_ARG_164 | 3.08 | 2.20 | 24.18 |
| 3HGZ | OE2, B_GLU_408 | NH1, A_ARG_164 | HH11, A_ARG_164 | 3.66 | 2.70 | 14.52 |
| 3HGZ | OE1, B_GLU_408 | NH2, A_ARG_164 | HH21, A_ARG_164 | 3.03 | 2.13 | 21.99 |
| 3HGZ | OE2, B_GLU_408 | NH2, A_ARG_164 | HH21, A_ARG_164 | 4.61 | 3.89 | 39.58 |
| 3HGZ | OE1, B_GLN_412 | NH2, A_ARG_164 | HH22, A_ARG_164 | 3.72 | 2.94 | 34.36 |
| 3HGZ | OE1, B_GLU_457 | OG1, A_THR_878 | HG1, A_THR_878 | 2.79 | 2.15 | 40.71 |
| 3HGZ | OE2, B_GLU_457 | OG1, A_THR_878 | HG1, A_THR_878 | 3.07 | 2.47 | 43.72 |
| 3HGZ | OE1, B_GLU_457 | NZ, A_LYS_884 | HZ3, A_LYS_884 | 4.68 | 3.74 | 18.88 |
| 3HGZ | OG1, B_THR_55 | NZ, A_LYS_933 | HZ2, A_LYS_933 | 3.39 | 2.77 | 44.49 |
| 3HGZ | OE1, A_GLU_880 | NZ, B_LYS_327 | HZ1, B_LYS_327 | 3.48 | 2.53 | 17.30 |
| 3HGZ | OE1, A_GLU_341 | NZ, D_LYS_1 | HZ1, D_LYS_1 | 2.44 | 1.85 | 44.11 |
| 3HGZ | OH, A_TYR_609 | NZ, D_LYS_1 | HZ2, D_LYS_1 | 3.91 | 3.25 | 43.14 |
| 3HGZ | OE1, B_GLU_341 | NZ, E_LYS_1 | HZ2, E_LYS_1 | 3.18 | 2.42 | 34.86 |
| 3HGZ | OH, B_TYR_609 | NZ, E_LYS_1 | HZ3, E_LYS_1 | 3.97 | 3.23 | 37.37 |
Table 8.
Experimentally uncharted territories (EUTs) in two structural models representing IDE-amylin complex, as described by REMARK 465 in the PDB format content of PDB IDs: 2G48 [45,46] and 3HGZ [47,48].
| PDB ID | REMARK | REMARK ID | ResName | Chain ID | ResID |
|---|---|---|---|---|---|
| 3HGZ | REMARK | 465 | ASN | D | 3 |
| 3HGZ | REMARK | 465 | THR | D | 4 |
| 3HGZ | REMARK | 465 | ALA | D | 5 |
| 3HGZ | REMARK | 465 | THR | D | 6 |
| 3HGZ | REMARK | 465 | CYS | D | 7 |
| 3HGZ | REMARK | 465 | ALA | D | 8 |
| 3HGZ | REMARK | 465 | THR | D | 9 |
| 3HGZ | REMARK | 465 | GLN | D | 10 |
| 3HGZ | REMARK | 465 | ARG | D | 11 |
| 3HGZ | REMARK | 465 | VAL | D | 17 |
| 3HGZ | REMARK | 465 | HIS | D | 18 |
| 3HGZ | REMARK | 465 | SER | D | 19 |
| 3HGZ | REMARK | 465 | SER | D | 20 |
| 3HGZ | REMARK | 465 | ASN | D | 21 |
| 3HGZ | REMARK | 465 | ASN | D | 22 |
| 3HGZ | REMARK | 465 | PHE | D | 23 |
| 3HGZ | REMARK | 465 | GLY | D | 24 |
| 3HGZ | REMARK | 465 | ALA | D | 25 |
| 3HGZ | REMARK | 465 | ILE | D | 26 |
| 3HGZ | REMARK | 465 | LEU | D | 27 |
| 3HGZ | REMARK | 465 | SER | D | 28 |
| 3HGZ | REMARK | 465 | SER | D | 29 |
| 3HGZ | REMARK | 465 | THR | D | 30 |
| 3HGZ | REMARK | 465 | ASN | D | 31 |
| 3HGZ | REMARK | 465 | VAL | D | 32 |
| 3HGZ | REMARK | 465 | GLY | D | 33 |
| 3HGZ | REMARK | 465 | SER | D | 34 |
| 3HGZ | REMARK | 465 | ASN | D | 35 |
| 3HGZ | REMARK | 465 | THR | D | 36 |
| 3HGZ | REMARK | 465 | TYR | D | 37 |
| 3HGZ | REMARK | 465 | THR | E | 4 |
| 3HGZ | REMARK | 465 | ALA | E | 5 |
| 3HGZ | REMARK | 465 | THR | E | 6 |
| 3HGZ | REMARK | 465 | THR | E | 9 |
| 3HGZ | REMARK | 465 | GLN | E | 10 |
| 3HGZ | REMARK | 465 | ARG | E | 11 |
| 3HGZ | REMARK | 465 | VAL | E | 17 |
| 3HGZ | REMARK | 465 | HIS | E | 18 |
| 3HGZ | REMARK | 465 | SER | E | 19 |
| 3HGZ | REMARK | 465 | SER | E | 20 |
| 3HGZ | REMARK | 465 | ASN | E | 21 |
| 3HGZ | REMARK | 465 | ASN | E | 22 |
| 3HGZ | REMARK | 465 | PHE | E | 23 |
| 3HGZ | REMARK | 465 | GLY | E | 24 |
| 3HGZ | REMARK | 465 | ALA | E | 25 |
| 3HGZ | REMARK | 465 | ILE | E | 26 |
| 3HGZ | REMARK | 465 | LEU | E | 27 |
| 3HGZ | REMARK | 465 | SER | E | 28 |
| 3HGZ | REMARK | 465 | SER | E | 29 |
| 3HGZ | REMARK | 465 | THR | E | 30 |
| 3HGZ | REMARK | 465 | ASN | E | 31 |
| 3HGZ | REMARK | 465 | VAL | E | 32 |
| 3HGZ | REMARK | 465 | GLY | E | 33 |
| 3HGZ | REMARK | 465 | SER | E | 34 |
| 3HGZ | REMARK | 465 | ASN | E | 35 |
| 3HGZ | REMARK | 465 | THR | E | 36 |
| 3HGZ | REMARK | 465 | TYR | E | 37 |
| 2G48 | REMARK | 465 | CYS | C | 7 |
| 2G48 | REMARK | 465 | ALA | C | 8 |
| 2G48 | REMARK | 465 | THR | C | 9 |
| 2G48 | REMARK | 465 | ASN | C | 22 |
| 2G48 | REMARK | 465 | PHE | C | 23 |
| 2G48 | REMARK | 465 | GLY | C | 24 |
| 2G48 | REMARK | 465 | ALA | C | 25 |
| 2G48 | REMARK | 465 | ILE | C | 26 |
| 2G48 | REMARK | 465 | LEU | C | 27 |
| 2G48 | REMARK | 465 | SER | C | 28 |
| 2G48 | REMARK | 465 | SER | C | 29 |
| 2G48 | REMARK | 465 | THR | C | 30 |
| 2G48 | REMARK | 465 | ASN | C | 31 |
| 2G48 | REMARK | 465 | VAL | C | 32 |
| 2G48 | REMARK | 465 | GLY | C | 33 |
| 2G48 | REMARK | 465 | SER | C | 34 |
| 2G48 | REMARK | 465 | ASN | C | 35 |
| 2G48 | REMARK | 465 | THR | C | 36 |
| 2G48 | REMARK | 465 | TYR | C | 37 |
| 2G48 | REMARK | 465 | ALA | D | 5 |
| 2G48 | REMARK | 465 | THR | D | 6 |
| 2G48 | REMARK | 465 | CYS | D | 7 |
| 2G48 | REMARK | 465 | ALA | D | 8 |
| 2G48 | REMARK | 465 | THR | D | 9 |
| 2G48 | REMARK | 465 | GLN | D | 10 |
| 2G48 | REMARK | 465 | GLY | D | 24 |
| 2G48 | REMARK | 465 | ALA | D | 25 |
| 2G48 | REMARK | 465 | ILE | D | 26 |
| 2G48 | REMARK | 465 | LEU | D | 27 |
| 2G48 | REMARK | 465 | SER | D | 28 |
| 2G48 | REMARK | 465 | SER | D | 29 |
| 2G48 | REMARK | 465 | THR | D | 30 |
| 2G48 | REMARK | 465 | ASN | D | 31 |
| 2G48 | REMARK | 465 | VAL | D | 32 |
| 2G48 | REMARK | 465 | GLY | D | 33 |
| 2G48 | REMARK | 465 | SER | D | 34 |
| 2G48 | REMARK | 465 | ASN | D | 35 |
| 2G48 | REMARK | 465 | THR | D | 36 |
| 2G48 | REMARK | 465 | TYR | D | 37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



