Submitted:
26 January 2024
Posted:
26 January 2024
You are already at the latest version
Abstract
The architecture, organization and functioning of biocatalytic reaction networks, which are coded in the cell specific genome and which work together in the small space of biological cells, are a fascinating feature of life evolved over more than 3 billion years. The knowledge about the diversity of biocatalytic functions and metabolic pathways sustaining life on our planet is highly important, especially as the currently occurring loss of biodiversity is considered a planetary boundary which is at high risk, and knowledge about the life of current biological organisms should be gained before they become extinct. In addition to the well-known enzymatic reactions involved in biochemical pathways, the enzyme universe offers numerous opportunities for discovering novel functions and pathways. Maintaining thousands of molecules and reactions functioning properly within biological cells, which may be exposed to various kinds of external hazards, environmental stress, enzymatic side reactions or non-enzymatic chemical reactions, is key for keeping cellular life healthy. This review aims at outlining advances in assigning enzyme functions to protein sequences and the discovery of novel biocatalytic functions and pathways.
Keywords:
Enzyme functions
; domains of unknown functions
; central metabolic pathways
; biosynthesis
; biosynthetic gene clusters
; cryptic pathways
; orphan pathways
; silent pathways
; metabolite repair enzymes
1. Introduction
The fundamental question of how life emerged on planet earth, which is up to now the only one among the thousands of exoplanets discovered, has been accompanying mankind in belief and scientific reflection systems for millenia and remains unsolved. Even though it is unknown how on early earth life originated and there are many conflicting models [1,2], the concept of a primordial soup by Alex Oparin [3] has provided inspirations to the question what type of components would have been needed in such a prebiotic soup [4]. Key features of living biological cells such as metabolism and self-organization, which are characterized by a complex organization in space and time of biocatalytic reaction networks coded in the cell-specific genome, have evolved over more than 3 billion years, with evidence for the oldest bacterial life forms near ancient submarine hydrothermal vents [5]. Whatever origin of life model is favoured, metabolism is a key part and criteria have been proposed which must satisfied by chemical reaction systems to be a simple metabolism able to support protocell growth and division [6]. It is of much interest what catalysts and non-enzymatic reaction networks of ancient metabolism have made use of simple starting materials and energy sources [7,8,9], prior to enzymatic reactions catalyzed by proteins and RNA enzymes. The known enzyme functions and pathways, which have been discovered over more than hundred years up to now, are already catalyzing a tremendous diversity of reactions, not only in central metabolism but also in more remote and specialized metabolism, such as natural product biosynthesis [10]. The universe of enzyme functions and pathways is however much bigger and is expanding with the investigations of enzymes and biosynthetic pathways in a growing number of microbes [11], plants [12] and animals [13].
The great advances of molecular biology and genetics as well as the impressive scientific and technological developments and improvements in methods for a) analyzing and sequencing DNA [14] and RNA [15], and b) synthesizing DNA [16] and RNA [17,18], have also revitalized the old interest in the influence of non-genetic factors such as nutrition, environment, competition, symbiosis, on health and disease of living organisms. The structural protein data collections, such as UniProt with more than 227 million protein sequences [19], PDB with more than 180'000 three-dimensional structures of proteins [20] and the more than 200 million predicted protein structures [21], provide a tremendous structural knowledge base for protein science and are advancing at a fast pace. Keeping up the pace of correct and experimentally verified functional annotations of protein sequences is essential [22], and the discovery of novel biocatalytic functions is supported by a variety of methodologies and tools linking genomics and enzymology [23]. Advances in the analysis of the whole small molecular domain (metabolome) of biological cells by MS and NMR methods have significantly enlarged the tools for identifying protein functions [24]. The simultaneous analysis of known protein functions in a biological cell, which has been shown for 132 enzyme catalytic rates in Escherichia coli by extracting from omics data the maximum of the catalytic rate which has been observed for an enzyme inside cells [25], is attractive for accelerating and extending the collection of enzyme kinetics data. For elucidation in vivo effects on enzyme kinetics, for experimentally verifying the discovery of a novel enzyme function or confirming an already known protein function, the synthesis of the respective protein and the small molecules predicted as substrate are essential.
Fundamental discoveries, new approaches and instrumental advances have enabled classical molecular biology and classical metabolism to meeting in a new integrated molecular view of life, where the molecular biology central dogma [26] regarding the sequential information transfer and the utilized alphabet is still key and the entire small molecular world of a biological organism is now also considered as an important part of a truly molecular biology [27,28,29,30,31]. The more than 55 natural metabolite-sensing riboswitches, which have been discovered to control transcription termination, translation initiation and alternative splicing, demonstrate the importance of linking the monitoring of biologically relevant elements, electrons and ions in the form of ligands to riboswitches with the gene expression control and the complex metabolic pathway regulation. The way how riboswitches typically achieve this sensing and control is by forming a mRNA domain in the 5′ untranslated region which is able to bind small molecules as ligands and is partly overlapping with an expression platform. Expression control is achieved by having the riboswitch in a genetic on mode when no ligand is bound, and a genetic off mode when the ligand is bound [32]. Although more than 55 classes of riboswitches for common cofactors and nucleotide-based signalling molecules have already been discovered, many opportunities still exist for discovering novel riboswitch classes sensing important metabolites, such as fatty acids, lipids, terpenoids, or unmodified sugars [33].
The opportunity to switch on or off metabolic pathways and the involved enzymes according to whether biologically relevant small molecules are presenr or absent in the nutrition or the environment connects metabolism with molecular biology and can be realized by different mechanisms. Metabolic pathway and network regulation can be achieved at multiple levels, such as genetic regulation [34], activation of silent pathways [35], or metabolite-protein interactions [36]. Metabolic efficiency can be achieved by separating highly reactive metabolites from the intracellular space in a protein-bound form, such as γ-glutamyl phosphate bound in L-glutamine synthase [37]. The hypothesis of metabolite-enzyme coevolution is attractive for integrating the avoidance of reaction losses through toxic and inhibitory metabolites, recruitment of enzymes and the evolution of metabolic pathways and networks [38].
The proper functioning of metabolic reactions needs also damage control and repair systems for the components of living biological cells which may be exposed to various kinds of damaging influences, either by external hazards and environmental stress, or by internal factors such as the working life span of an enzyme [39], side reactions due to additional non-native catalytic functions or additional non-native substrate acceptance of enzymes [40] or chemical reactions within biological cells, which can not only damage macromolecules like nucleic acids and proteins but also metabolites [41]. Repair systems for damaged metabolites, which may be just useless when having no biological effects but in more severe cases may have negative biological effects, are important for maintaining healthy life, and biocatalytic functions and pathways converting damaged metabolites back to useful and non-toxic pathway metabolites contribute to robustness and stability [42]. The analysis of a minimized Mycoplasma JCVI-Syn3A genome has shown that metabolite damage repair systems are still in place and cannot therefore be separated from life itself [43].
Metabolites play also key roles in the regulatory processes by the enzymatic modifications of essential biopolymers of molecular biology, such as DNA, RNA and proteins. The enzymatic modification of histones and DNA, which involves metabolites from nutrients and the microbiome, connects metabolism and epigenetics in a complex interplay [44]. Other important interfaces between metabolism and molecular biology appear in gene silencing [45], post-transcriptional RNA modification [46] and post-translational modifications of proteins [47].
Tremendous advances have been achieved in assigning enzyme functions to protein sequences and the discovery of novel biocatalytic functions and pathways. More than 6000 enzymes, which have been recognized and categorized according to the reaction they catalyze by a four-digit EC number within the seven EC classes [48], have been shown to have already a large variety of enzyme activities. As the universe of enzymes is however much larger, moving the frontiers of enzyme function knowledge is therefore highly important. This is clearly evident from the growing number of sequences of structures of known proteins, for which the corresponding enzyme function is unknown, or has not been experimentally verified. The main aim of the work is to bring back the attention to metabolism as a key research area for the study of life and as a valuable resource for applications in biocatalysis. The great progress in genomic enzymology tools and methodologies, discovery of unknown biocatalytic functions and pathways as well as experimental technologies for analysis and synthesis enlarge the power and opportunities of biocatalysis [49].
In addition to biocatalysts having a single enzyme function, biocatalysts have been discovered which can catalyze more than one reaction. Biocatalysts have been discovered which can switch their enzyme function depending on the pH, such as the terpene cyclases AaTPS and FgGS can act at basic pH as aromatic prenyltransferase for generating prenylindoles [50]. Biocatalysts containing multiple enzyme functions in the same protein are well known and of much interest for stabilizing labile intermediates and directing cascade reactions selectively towards rapid generation of molecular complexity [51]. Multifunctional enzymes, which are among the largest and most complex enzyme machineries, are involved as megasynthases in catalyzing the biosynthesis of numerous product groups, such as fatty acids, non-ribosomal peptides, polyketides, or terpenes, from simple natural building blocks. The biosynthesis of a large diversity of complex polyketides is catalyzed by multifunctional polyketide synthases, which can operate by the programmed use of the same enzyme functions repeatedly in an iterative mode, or by a linear channeling of the intermediates from one function to the subsequent function in an assembly-line mode [52,53,54].
2. Discovery and Characterization of Proteins with Unknown Biocatalytic Functions
Life on planet earth at this timepoint can be categorized into ancient living organisms which have become extinct now, the biological organisms living now, and the biological organisms evolving in the future. As the currently occurring loss of biodiversity his of major concern [55], it is of much interest to gain knowledge about the life of current biological organisms before they become extinct. From genes to proteins to metabolites the characterization of hidden biocatalytic functions and pathways, whether cryptic or silent [56], is an exciting research area. One key area for understanding the metabolism of a biological organism in healthy and diseased conditions is knowledge about sequence, structure and function of its constituting proteins. The knowledge of gene sequences coding for proteins is growing much faster than the corresponding experimental identification and verification of its corresponding protein functions. This widens the gap which represents a major challenge as the assignment and experimental characterization of biocatalytic functions to the corresponding gene products requires substantial efforts. The deep learning model DeepECtransformer has been developed to predict known enzyme functions at the level of EC numbers in order to reduce the number of unannotated genes [57]. From 464 un-annotated E. coli genes enzyme functions at the level of EC numbers were predicted for the corresponding proteins by DeepECtransformer and from these three proteins, the predicted glucose 1-dehydrogenase for YgfF, L-threonylcarbamoyladenylate synthase for YciO, and phosphonoacetate hydrolase for YjdM, have been randomly selected [57]. This facilitated experimental validation of the enzyme functions, which has been performed by in vitro enzyme assays of the overexpressed and affinity-purified proteins YgfF, YciO, and YjdM [57]. For special enzyme reactions or completely novel enzyme functions without EC numbers, a significant effort and time may be needed for the development of suitable analytical and preparative methods [58,59,60]. These include the expression and purification of proteins, synthesis of substrates, analysis of substrates and products, as the catalytic function of a protein needs to be demonstrated by its incubation with a potential substrate and the identification of the nature of the product formed from the substrate in a protein- and time-dependent way. Further experimental characterization is of much interest and includes the identification of the optimum reaction conditions and the measurement of catalytic performance parameters such kcat and KM. The reporting of these functional datasets according to the STRENDA guidelines, which are recommended by an increasing number of journals, and its deposition in the STRENDA database provide a modular framework in the workflow of processing, storing and retrieving an increasing number of enzyme function data [61]. The growing Pfam database of protein sequence families [62], which have been generated according to a significant degree of sequence similarity of a protein domain, is of much interest for connecting to enzyme functions and evolutionary history of proteins, and for guiding experiments. The activity of a known natural enzyme has also been a common starting point for engineering and evolving the properties of the enzyme towards optimum performance of a desired biocatalytic reaction under defined reaction conditions with respect to catalytic efficiency, selectivity stability, or substrate scope [63].
Whole genome sequencing of biological organisms has yielded a large dataset of genes which code for proteins whose function is completely unknown. The description of domains of unknown functions (DUF) for uncharacterized protein families started in 1998 with the first two members DUF1 and DUF2 [64]. Since then, the DUFs, both in absolute numbers and as a percentage of all protein families, have been continuously increasing to more than 20% of all protein families over the years [65], reaching DUF 6807 in release 35.0 of the Pfam database, which has now been integrated into the InterPro database [66]. Genomic enzymology web tools, such as sequence similarity networks, enzyme similarity tools, genome neighbourhood tools and taxonomy tools, enable the exploration of databases towards the in vitro characterization of enzyme activities to uncharacterized proteins [67,68,69,70].
Screening of transport system proteins which bind solutes and applying sequence similarity networks and genome neighbourhood networks has enabled the identification of novel kinases, which are ATP-dependent and are acting on four-carbon sugar acids, from the DUF1537 protein family [71]. Thereby the novel DUF1537 enzymes D-threonate kinase DtnK and D-erythronate kinase DenK (see Figure 1) have been identified and characterized [71]. A strategy for finding enzyme activities within protein families of unknown function has been based on defining a generic conserved reaction in the protein family, high-throughput screening, analysis of genomic and metabolic context [71].
The protein Cj1418 from Campylobacter jejuni, which has been recombinantly expressed and affinity-purified, has been discovered as first enzyme to directly phosphorylate the amide nitrogen [72]. Cj1418 has been clearly demonstrated to act as ATP-dependent L-glutamine kinase (see Figure 1), which corrected its former annotation as putative phosphoenolpyruvate synthase or pyruvate phosphate dikinase [72]. The application of this approach to the DUF849 Pfam family enabled the discovery of various novel β-keto acid cleavage enzymes [73].
For functional annotation of the proteins Ms0025 from Mycoplasma synoviae and Mag6390 from Mycoplasma agalactiae (see Figure 1) as novel lactonases a combination of approaches was needed, from the consideration of genetic context, computational, empirical and structural screening, to the comparison of sequences and addition of newly synthesized substrates to the original libraries [74]. Both lactonases have been demonstrated to catalyze the hydrolysis of D-xylono-1,4-lactone-5-phosphate with a kcat/Km value of 5.7 × 104 M−1s−1 for Mag6390 and 4.7 × 104 M−1s−1 for Ms0025, and the hydrolysis of L-arabino-1,4-lactone-5-phosphate with a kcat/Km value of 2.2 × 104 M−1s−1 for Mag6390 and 1.3 × 104 M−1s−1 for Ms0025 [74].
3. Discovery and Characterization of Unknown Metabolic Pathways
Central metabolic cycles and pathways, such as glycolysis, mevalonate and methyl-erythritol phosphate pathways, the Calvin cycle, citric acid cycle, or urea cycle, have been discovered through significant scientific efforts and fundamental investigations, which have been honoured by many Nobel Prizes and have become standard biochemistry knowledge. In addition to the central metabolic pathways for sustaining healthy life in the large diversity of biological species, specialized pathways for preparing bioactive small molecules from the nutrients available may be connected with special living conditions, diseases or tasks. From microbes to plants, animals and humans, the identification of the relevant biochemical pathways and missing enzymes continues to be highly important not only for natural and synthetic pathways to known bioactive metabolites and natural products, but also for orphan, cryptic or silent pathways to unknown metabolites, salvage and repair pathways. Therefore methods for identifying functional genes, such as gene expression profiling in real time, knockouts or heterologous expression of all the target genes of a complete biosynthetic pathway [75], combined analysis of genome and transcriptome data, as well as metabolome and enzymatic analysis are essential for elucidating biochemical pathways [76]. This requires also outlining the organic chemistry of the biochemical pathways and connecting the metabolites with the genes that encode their biosynthesis [10,77]. Computational methods and tools using the databases of biochemical compounds and principles of biochemical reactions [78] are of much interest for potential unknown metabolic pathways towards shining light on the dark matter of metabolism. The identification of all enzymes and their functions along a biocatalytic pathway, as well as the metabolic intermediates is key for a molecular understanding of the natural pathway and for designing synthetic pathways.
The identification of missing enzymatic reaction steps in metabolic pathways has been a classical area in the discovery of now well established metabolic pathways. Newly developed experimental tools and methods have however enabled fresh and straightforward approaches to identify missing enzymatic reaction steps often leading to the discovery of entirely novel enzyme functions. The question of how nature catalyzes the synthesis of altemicidin by the gene products of a recently identified biosynthetic gene cluster has been addressed by a smart combination of various experimental techni-ques [79]. This has led to the discovery of a fascinating novel pathway from β-nicotin-amide adenine dinucleotide to altemicidin in eight enzymatic reaction steps (see Figure 2), whereby a novel enzymatic [3+2]-annulation between β-nicotinamide adenine dinuc-leotide and S-adenosyl-L-methionine has been discovered [79]. From the separately expressed genes of the Streptomyces lividans biosynthetic gene cluster sbz and functional anaylsis by untargeted metabolomics analysis, SpzP has been found as the gatekeeping enzyme in the generation of the 6-azatetrahydroindane backbone [79].
The identification of all missing enzymatic reaction steps for completing the whole bio-synthetic pathway to metabolites traditionally extracted in low yields from biological species is not only of fundamental interest but provides also a starting point for develo-ping sustainable multi-step enzyme-catalyzed processes for their production. The identi-fication of all missing enzymes in the complex 31-step vinblastin biosynthetic pathway of Catharanthus roseus has demonstrated how these enzymes catalyze the resource-effi-cient generation of chemical complexity from the simple metabolites tryptophan and ge-ranylpyrohosphate by a combination of divergent and convergent synthesis strategies [80,81,82,83]. The question of how stemmadenine acetate, which is formed from strictosidine [81], is converted by divergent biocatalytic reactions to the two metabolite building blocks cataranthine and tabersonine has been addressed by chemical investigations, sequence data, gene silencing, synthesis of metabolite standards, NMR and mass spectrometry [80]. These methods and the validation of the biocatalytic reaction steps in vitro with expressed and purified proteins enabled the discovery of two novel redox enzymes which have been named precondylocarpine acetate synthase (PAS) and dihydroprecondylocarpine acetate synthase (DPAS), and the characterization of the two hydrolases tabersonine synthase (TS) and catharanthine synthase (CS) [80]. The two enzymes PAS and DPAS have been shown to catalyze the conversion of stemmadenine acetate into the unstable intermediates precondylorapine acetate and dihydroprecondy-locarpine acetate, which is converted by TS- or CS-catalyzed desacetoxylation to dehy-drosecodine [80], to subsequently generate through Diels-Alder cyclizations, either the TS-catalyzed reaction to tabersonine or the CS-catalyzed reaction to catharanthine [80]. After the biotransformation of tabersonine to vindoline, which is catalyzed by seven enzymes [82], the convergent synthesis of the anticancer natural product vinblastine in the biosynthetic pathway is completed by the condensation of catharanthine and vindo-line [83]. The benefit of identifying all missing enzymes in a pathway has been demon-strated by the impressive achievement of engineering the thirty enzymes catalyzing the reactions to vindoline and catharanthine into yeast, using a chemical coupling reaction in the final step to vinblastine [84].
It is also of much interest how the diversity of biological cells and their environments, from which the uptake of nutrients and energy is needed, is also reflected in diverse core metabolic pathways for the biosynthesis of the central molecules of life. Investigating different domains of life for central metabolic pathways can provide not only insights into fundamental reactions, unusual pathways and evolution, but also valuable novel biocatalytic functions. A reversible reductive tricarboxylic acid cycle, which has been discovered in the chemolithotrophic thermophile Thermosulfidibacter takaii required a combination of genomic, metabolomic and enzymatic analysis [85]. The biodiversity and biosynthetic potential of the human gut microbiome has been demonstrated by the identification of 19890 primary metabolic gene clusters in 4240 genomes, which represents an important milestone for advancing the understanding of its role in human physiology [86]. While the biosynthetic pathway to the essential cofactor coenzyme A has been well established in bacteria and eukarya, it was only recently that the entire coenzyme biosynthetic pathway in archaea has been experimentally validated and demonstrated to be different from bacteria and eukarya [87].
The fast growth of genome sequences in all domains of life has revealed the extent of unknown metabolic and biosynthetic capabilities of living organisms [88,89]. An impressive 231534 biosynthetic gene cluster regions have been selected from archaeal, bacterial and fungal genomes for the antiSMASH database version 4 [90]. Specialized metabolic pathways to complex and uniquely functionalized natural products remain therefore a very promising and vast area for discovering novel biosynthetic logic and biocatalytic functions. The biosynthetic pathway for enediyne aromatic polyketides has been investigated utilizing the coexpression in E. coli of combinations of genes which code for a polyketide synthase and a thioesterase from the enediyne biosynthetic gene clusters have been expressed in recombinant E. coli strains to complement mutant strains able to produce anthraquinone-fused enediynes but lacking the corresponding polyketide synthase [91]. A combination of synthetic biology, chemical complementation and 13C stable isotope labeling experiments enabled the identification of the common linear polyene intermediate 1,3,5,7,9,11,13-pentadecaheptaene and the proposal of a unifying pathway (see Figure 3) for enediyne aromatic polyketides [91]. This provides an excellent groundwork for the exploration of the intriguing biocatalytic reactions by which the pathways are diverging from 1,3,5,7,9,11,13-pentadecaheptaene, whereby one molecule is transformed to the enediyne core, while the anthraquinone moiety is formed from a second one [91].
The search for various types of completely unknown or hidden biosynthetic pathways, such as cryptic, silent or orphan pathways, leading to still unknown metabolites/natural products may not only provide novel biocatalytic functions and exciting new chemistry but also attractive novel scaffolds for biologically active small molecules [35,56]. A range of approaches have been developed for discovering novel structures of biologically active small molecules and for uncovering the links with the respective genes coding for the enzymes which catalyze the reactions leading to their biosynthesis [92]. Specialized biologically active small molecules may be only needed at specific times or certain conditions of life and it is therefore not surprising that their biosynthesis is dependent on cultivation conditions, environmental signals, stress, presence of elicitors, inducers, or exogenous metabolites from co-cultivation [92]. These empirical approaches may however be challenging and impractical for larger numbers of samples. With the advances in genomics and the findings that often the genes which code for the enzymes used in a specific pathway are organized in biosynthetic gene clusters [93], genome-guided methods have attracted much interest [92]. Bioinformatics analysis tools for discovering biosynthetic pathways and for identifying biosynthetic gene clusters are very promising [94,95,96]. While genome sequences are important, much more is needed for connecting genes and the functions coded by them, and for deciphering the biosynthetic logic of the corresponding metabolic pathways. Important complementary information can be derived from the combination of chemical and biochemical knowledge with genomic context [97], identification of co-expressed genes by RNA-sequencing and transcriptome-wide analysis of differential gene expression [98], metabolomic analysis and their correlation with absent or present expression of a biosynthetic gene cluster [99]. Finally, the integration of powerful analytical technologies with high information content is key for establishing the sequence of the biocatalytic reactions along the pathway, and the molecular structure of the metabolites and natural products [100,101].
Bacterial and fungal genomes have also numerous silent biosynthetic gene clusters [88,89,90], which are poorly and not at all expressed under standard laboratory conditions. The question how cryptic, orphan and silent biosynthetic gene clusters, which outnumber the active ones, can be expressed is of fundamental importance for the discovery of unknown metabolic pathways. Therefore much attention has been paid to general approaches and methods for activating and characterizing natural product biosynthetic routes as well as the corresponding genes encoding all the enzymes catalyzing the biosynthetic reactions. Various methods have been shown to be valuable, such as perturbation of epigenetic regulation, promoter exchange, control of the translation machinery by ribosome engineering, activator gene overexpression, or repressor gene inactivation [92]. The novel urea natural product class gaburedins has been discovered via the derepression of its silent gbnABC biosynthetic gene cluster, which has been achieved by deleting the putative regulatory gene gbnR, which is pathway-specific for the transcription repressor in Streptomyces venezuelae [102]. Structure determination of the metabolites which were present in the derepressed gbnR mutant but lacking in the silent wild type, feeding of possible precursors and the demonstration of no gaburedin biosynthesis when the gbnB gene was deleted in the gbnR mutant have led to the proposed roles of the enzymes GbnA and GbnB in gaburedin pathways [102]. The expression of silent biosynthetic genes was derepressed in a Streptomyces host by CRISPR/Cas9-mediated genome editing, after the capture of a specific Streptomyces sclerotialus biosynthetic gene cluster, which was cryptic and silent, and he transfer into the Streptomyces host [103]. A novel natural product class has been discovered by this approach, as demonstrated by (2-(benzoyloxy)acetyl)-L-proline, named scleric acid, and its proposed biosynthesis [103].
Other general approaches involve the insertion of promoters which are constitutively active using CRISPR-Cas9, identifying small molecules as inducers by high-throughput elicitor screening, and creating overproducing strains by reporter-guided mutant selection [104,105].
Biological cells may have various provisioning paths to more complex metabolites and natural products in between their uptake from the environment and their complete biosynthesis from essential and simple low-molecular weight biochemicals by natural or synthetic pathways, also termed de novo pathways. Nutrient supply issues may thereby be experiences at different levels, from intermediates and precursors to the more com-plex metabolites and natural products, in the uptake from the environment or in the bio-chemical degradation of biopolymers. Therefore the recycling and utilization of such intermediates and precursors by pathways to the more complex metabolites and natural products, also termed salvage pathways, is not only avoiding the accumulation of waste and ensuring resource efficiency but also supporting the life, resilience and stability of biological cells, for example by keeping adequate cofactor levels. Major coenzymes have been shown to be remarkably stable in vivo in Escherichia coli, Bacillus subtilis and Saccharomyces cerevisiae [106].
The cofactor nicotinamide adenine dinucleotide (NAD+) with its de novo biosynthe-tic pathway from L-tryptophan via kynurenine is important for health and mitochon-drial function [107]. The maintenance of adequate NAD+-levels is critical for a multitude of enzymatic and cellular functions, and several additional biosynthetic pathways to NAD+ have been discovered. NAD+ biosynthesis can be achieved in 3 enzymatic reaction steps (Preiss-Handler pathway) from niacin (nicotinic acid), whereby the last two steps converge with the kynurenine pathway [108,109,110]. Other biosynthetic path-ways to NAD+ have been discovered which start from the intermediates nicotinamide [111], nicotinamide riboside [112], nicotinic acid riboside [113] and its reduced form [114,115], and nicotinamide mononucleotide [116]. As these pathways are requiring less reac-tion steps to NAD+, due to their use of metabolites containing the pyridine structure, and are recycling these to the production of NAD+, a more inclusive use of the term salvage pathway seems reasonable [117,118].
The recycling and re-utilization of materials in salvage pathways provides biolo-gical cells with additional flexibility for maintaining the levels of key cellular compo-nents, such as cofactors, under changing living conditions, while benefitting from shor-ter biosynthetic routes to complex cellular components. This re-utilization blueprint from nature is also of much interest for synthetic applications, for example in the re-cycling of cofactors. Further benefits of salvaging precursors have been demonstrated in populations of engineered Escherichia coli strains, where salvagers able to use the precur-sors cobinamide and 5,6-dimethylbenzimidazole for the biosynthesis of the complete cobamide vitamin B12 make this cofactor available to nonproducing consumer strains, are not overexploited and remove nonfunctional and inhibiting precursors [119]. For the cofactor S-adenosyl-L-methionine (SAM) several salvage pathways for L-methionine and SAM byproducts are known and two novel oxygen-independent salvage pathways for the SAM byproducts 5ʹ-deoxy-adenosine and 5ʹ-methylthioadenosine have been discovered (see Figure 4) in Rhodospirillum rubrum and pathogenic Escherichia coli [120]. Salvage Pathways leading back to SAM, either from nature or by design, can provide important tools for advancing and broadening the synthetic applications of different classes of SAM-dependent enzymes [121].
Genetically encoded biocatalytic systems for preventing the formation of damaged metabolites, or for repairing damaged metabolites and transforming them back into valuable metabolic intermediates, which cells can utilize again, are of key importance for maintaining the health of living cells. Biocatalytic damage control systems may be as important for cellular life over the course of time as biosynthetic pathways, as they can systematically counteract the negative effects of damaged metabolites, which can be for-med from normal physiological metabolites by enzymatic side reactions or spontaneous chemical reactions to toxic products [41,42,43]. The enzymatic formation of damaged meta-bolites can occur under a variety of condtions, such as an unintended enzymatic trans-formation of a normal physiological metabolite, or an enzymatic transformation of an unintended substrate in addition to the normal physiological substrate. Spontaneous chemical reactions occurring under physiological conditions have been assembled in the database CD-MINE, which is the abbreviation for Chemical Damage - Metabolic In Silico Network Expansion [122].
The activities of metabolite repair and clearance enzymes, which have been found to eliminate damaged metabolites and side-products in glycolysis [123], citric acid cycle [124], photosynthesis [125], and other major pathways are essential for the proper funcioning of metabolic pathways. Damaged metabolites and side products which are toxic to biological cells have been a good starting point in the search for enzymes cataly-zing its conversion to non-toxic natural metabolites which can be utilized in the corres-ponding cell metabolism. The repair of the intermediate L-4-hydroxy-threonine, which is toxic, by its phosphorylation to L-4-hydroxythreonine-5-phosphate (see figure 1), an essential metabolite of the pyridoxal-5-posphate pathway, has been demonstrated to be catalyzed by kinase STM0162 (DUF1537) [126]. The unnatural metabolite L-glyceralde-hyde 3-phosphate, which can be taken up by E. coli cells through different transport systems, or can be formed by glycerolkinase-catalyzed phosphorylation of L-glyceralde-hyde, or by a very slow non-enzymatic racemization of D-glyceraldehyde 3-phosphate [127], is toxic to biological cells due to its action as bactericidal agent and enzyme inhibitor [128]. L-glyceraldehyde-3-phosphate reductase YghZ from E. coli has been discovered as enzyme for catalyzing the removal of toxic L-glyceraldehyde 3-phosphate by its conversion to L-glycerol-3-phosphate (see Figure 5), a natural non-toxic metabolite for use in phospholipid biosynthesis or for bypassing the triosephosphate isomerase-catalyzed reaction [128,129].
4. Discussion
The inventories of gene sequence information acquired from living organisms in all kingdoms of life, from genomes of uncultivated microorganisms as well as from metagenomes collected from different habitats, provide important resources [130,131,132] and document the genetic parts of life on planet earth even when biologically endangered species become extinct due to the ongoing loss of biodiversity. Whole genome sequencing and the growth of gene sequence datasets are however much faster than the assignment of enzyme functions to the corresponding protein sequences. Therefore, the percentage of annotations with experimental evidence of even the ten most annotated genomes has been found to vary from 2.5 to 21% in the year 2022 [133]. This sequence-function gap regarding genetically encoded biocatalytic functions is thus an exciting and fertile field for the discovery and characterization of novel sequence-function relationships. Great advances and novel approaches, methods and tools [67,68,69,70] are very promising for reducing the number of un-annotated genes and for facilitating the significant effort and time connected with the experimental validation of enzyme functions in annotated genes. Biological organisms have also been shown to possess numerous metabolic and biosynthetic capabilities, coded by clustered and non-clustered biosynthetic genes, for which the identification of all enzyme functions in a pathway is essential. The combination of various approaches for identifying the missing enzymatic reaction steps in a biosynthetic pathway can create a fertile research environment for discovering entirely novel biocatalytic functions. Not only is this highly important for delineating all the reaction types and the complete reaction architecture to known bioactive small molecules, but also for deciphering orphan, cryptic or silent pathways to unknown metabolites. As keeping cellular life healthy over the course of time, biocatalytic housekeeping and maintenance systems for repairing damage, clearing toxic side products and removing waste represent another highly important field for discovering novel biocatalytic functions and pathways. Elucidating the machinery to systematically prevent, repair and overcome damage from enzymatic side reactions or spontaneous chemical reactions is not only of fundamental interest for life, but have important implications in the context of diseases with the corresponding damage profile and therapeutic approaches in molecular medicine.
5. Outlook
Interactions between synthetic organic chemistry and synthetic blueprint of natural biosynthesis are of much interest in comparing reaction mechanisms, selectivities and controlling sensitive intermediates, for example in the comparison of the biosynthesis [76] and the recent total synthesis of altemicidin [134]. While many compounds, such as altemicidin, have been discovered in nature before their total synthesis in the laboratory, the laboratory synthesis of a compound inspiring the discovery of this structure in nature, a coincidence of synthetic work and independent isolation from nature, or the synthesis of an anticipated natural product not yet confirmed by isolation, have been outlined as very interesting explorative area for the collaboration of synthetic organic chemistry and natural biosynthesis [135]. This could not only be instrumental in the synthesis of potential substrates for as yet unknown enzyme functions, but for the discovery of completely novel reactions and whole biocatalytic pathways to these anticipated natural products.
The use of omics technologies and the integration of genome mining and metabolomics analysis [136], designated as metabologenomics, looks very promising for directing the discovery of the correct links between the biosynthetic gene clusters, the encoded enzymes and enzyme-catalyzed reactions to the corresponding natural products, as recently shown by the identification of 21 natural products from Ascomycetes to their biosynthetic gene clusters [137].
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Smoukov, S.K.; Seckbach, J.; Gordon, R. (Eds.) Conflicting Models for the Origin of Life. John Wiley & Sons, Inc., Hoboken, NJ 07030, and Scrivener Publishing LLC, Beverly, MA 01915, USA, 2023.
- Preiner, M.; Asche, S.; Becker, S.; Betts, H.C.; Boniface, A.; Camprubi, E.; Chandru, K.; Erastova, V.; Garg, S.G.; Khawaja, N.; Kostyrka, G.; Machné, R.; Moggioli, G.; Muchowska, K.B.; Neukirchen, S.; Peter, B.; Pichlhöfer, E.; Radványi, A.; Rossetto, D.; Salditt, A.; Schmelling, N.M.; Sousa, F.L.; Tria, F.D.K.; Vörös, D.; Xavier, J.C. The Future of Origin of Life Research: Bridging Decades-Old Divisions. Life 2020, 10, 20. [Google Scholar] [CrossRef]
- Oparin, A.I. 1957. The origin of life on the earth, 3rd ed.; Oliver & Boyd, Edinburgh & London, Country, UK, 1957.
- Vincent, L.; Colón-Santos, S.; Cleaves, H.J.; Baum, D.A.; Maurer, S.E. The prebiotic kitchen: A guide to composing prebiotic soup recipes to test origins of life hypotheses. Life, 2021, 11(11), 1221.
- Dodd, M.S.; Papineau, D.; Grenne, T.; Slack, J.F.; Rittner, M.; Pirajno, F.; O’Neil, J.; Little, C.T.S. Evidence for early life in Earth’s oldest hydrothermal vent precipitates. Nature 2017, 543, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G. When Is a Reaction Network a Metabolism? Criteria for Simple Metabolisms That Support Growth and Division of Protocells. Life 2021, 11(9), 966. [Google Scholar] [CrossRef] [PubMed]
- Muchowska, K.B.; Varma, S.J.; Moran, J. Nonenzymatic Metabolic Reactions and Life’s Origins. Chem. Rev. 2020, 120, 15, 7708–7744. [Google Scholar] [CrossRef] [PubMed]
- Akbaria, A.; Palsson, B.O. Metabolic homeostasis and growth in abiotic cells. Proc. Natl. Acad. Sci. 2023, 120(19), e2300687120. [Google Scholar] [CrossRef] [PubMed]
- Aitken, H.R.M.; Wright, T.H.; Radakovic, A.; Szostak, J.W. Small-Molecule Organocatalysis Facilitates In Situ Nucleotide Activation and RNA Copying. J. Am. Chem. Soc, 2023; Articles ASAP. [Google Scholar] [CrossRef]
- Walsh, C.T.; Tang, Y. Natural Product Biosynthesis: Chemical Logic and Enzymatic Machinery, Royal Society of Chemistry, London, 2017.
- Shaffer, J.P.; Nothias, LF.; Thompson, L.R.; Sanders, J.G.; Salido, R.A.; Couvillion, S.P.; Brejnrod, A.D.; Lejzerowicz, F.; Haiminen, N.; Huang, S.; Lutz, H.L.; Zhu, Q.; Martino, C.; Morton, J.T.; Karthikeyan, S.; Nothias-Esposito, M.; Dührkop, K.; Böcker, S.; Kim, H.W.; Aksenov, A.A.; Bittremieux, W.; Minich, J.J.; Marotz, C.; Bryant, M.M.; Sanders, K.; Schwartz, T.; Humphrey, G.; Vásquez-Baeza, Y.; Tripathi, A.; Parida, L.; Carrieri, A.P.; Beck, K.L.; Das, P.; González, A.; McDonald, D.; Ladau, J.; Karst, S.M.; Albertsen, M.; Ackermann, G.; DeReus, J.; Thomas, T.; Petras, D.; Shade, A.; Stegen, J.; Song, S.J.; Metz, T.O.; Swafford, A.D.; Dorrestein, P.C.; Jansson, J.K.; Gilbert, J.A.; Knight, R.; Earth Microbiome Project 500 (EMP500) Consortium. Standardized multi-omics of Earth’s microbiomes reveals microbial and metabolite diversity. Nat. Microbiol. 2022, 7, 2128–2150. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.K.; Lynch, J.H.; Matos, J.O.; Dudareva, N. Adaptive mechanisms of plant specialized metabolism connecting chemistry to function. Nat. Chem. Biol. 2021, 17 (10), 1037-1045. [CrossRef]
- Torres, J.P.; Eric, W. Schmidt, E.W. The biosynthetic diversity of the animal world. J. Biol. Chem 2019, 294(46), 17684–17692. [Google Scholar]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: past, present and future. Nature 2017, 550(7676), 345–353. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: the teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Hoose, A.; Vellacott, R.; Storch, M.; Freemont, P.S.; Ryadnov, M.G. DNA synthesis technologies to close the gene writing gap. Nat. Rev. Chem. 2023, 7, 144–161. [Google Scholar] [CrossRef]
- Nelissen, F.H.T.; Leunissen, E.H.P.; van de Laar, L.; Tessari, M.; Heus, H.A.; Wijmenga, S.S. Fast production of homogeneous recombinant RNA—towards large-scale production of RNA. Nucleic Acids Res. 2012, 40(13), e102. [Google Scholar] [CrossRef]
- Flamme, M.; McKenzie, L.K.; Sarac, I.; Hollenstein, M. Chemical methods for the modification of RNA. Methods 2019, 161, 64–82. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium, UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; Žídek, A.; Green, T.; Tunyasuvunakool, K.; Petersen, S.; Jumper, J.; Clancy, F.; Green, R.; Vora, A.; Lutfi, M.; Figurnov, M.; Cowie, A.; Hobbs, N.; Kohli, P.; Kleywegt, G.; Birney, E.; Hassabis, D.; Velankar, S. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50(D1), D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Hu, Z., Rachlin, J.; Anton, B.P.; Kasif, S.; Roberts, R.J.; Steffen, M. COMBREX-DB: an experiment centered database of protein function: knowledge, predictions and knowledge gaps. Nucleic Acids Res. 2016, 44, D330–D335. [CrossRef]
- Oberg, N.; Zallot, R.; Gerlt, J.A. 2023. EFI-EST, EFI-GNT, and EFI-CGFP: Enzyme Function Initiative (EFI) Web Resource for Genomic Enzymology Tools. J. Mol. Biol. 2023, 435(14), 168018. [Google Scholar] [CrossRef] [PubMed]
- Sévin, D.C.; Fuhrer, T.; Zamboni, N.; Sauer, U. Nontargeted in vitro metabolomics for high-throughput identification of novel enzymes in Escherichia coli. Nat. Methods 2017, 14(2), 187–194. [Google Scholar] [CrossRef] [PubMed]
- Davidi, D.; Noor, E.; Liebermeister, W.; Bar-Even, A.; Flamholz, A.; Tummler, K.; Barenholz, U.; Goldenfeld, M.; Shlomi, T.; Milo, R. Global characterization of in vivo enzyme catalytic rates and their correspondence to in vitro kcat measurements. Proc. Natl. Acad. Sci. 2016, 113(12), 3401–3406. [Google Scholar] [CrossRef]
- Crick, F. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef]
- Schreiber, S. L. Small molecules: The missing link in the central dogma. Nat. Chem. Biol. 2005, 1(2), 64–66. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324(5930), 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- McKnight, S.L. Back to the future: molecular biology meets metabolism. In: Cold Spring Harbor symposia on quantitative biology 2011, 76, 403-411. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY 11724, USA.
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Alam, M.T.; Keller, M.A.; Breitenbach, M.; Brindle, K.M.; Rabinowitz, J.D.; Ralser, M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metabolism 2022, 34(3), 355–377. [Google Scholar] [CrossRef] [PubMed]
- Kavita, K.; Breaker, R.R. Discovering riboswitches: the past and the future. Trends Biochem. Sci. 2023, 48(2), P119–141. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R. The Biochemical Landscape of Riboswitch Ligands. Biochemistry 2022, 61, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3(3), 318–356. [Google Scholar] [CrossRef]
- Rutledge, P.; Challis, G. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol, 2015, 13, 509–523. [Google Scholar] [CrossRef]
- Diether, M.; Nikolaev, Y.; Allain, F.H.; Sauer, U. Systematic mapping of protein-metabolite interactions in central metabolism of Escherichia coli. Mol. Syst. Biol. 2019, 15(8), e9008. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Gutierrez-Rus, L.I.; Manssour-Triedo, F.; Newton, M.S.; Gamiz-Arco, G.; Mota, A.J.; Reiné, P.; Cuerva, J.M.; Ortega-Muñoz, M.; Andrés-León, E.; Ortega-Roldan, J.L.; Seelig, B.; Ibarra-Molero, B.; Sanchez-Ruiz, J.M. Cell Survival Enabled by Leakage of a Labile Metabolic Intermediate. Mol. Biol. Evol. 2023, 40(3), msad032. [Google Scholar] [CrossRef]
- Noda-Garcia, L.; Liebermeister, W.; Tawfik, D.S. Metabolite–Enzyme Coevolution: From Single Enzymes to Metabolic Pathways and Networks. Annu. Rev. Biochem. 2018, 87, 187–216. [Google Scholar] [CrossRef] [PubMed]
- Bathe, U.; Leong, B.J.; McCarty, D.R.; Henry, C.S.; Abraham, P.E.; Wilson, M.A.; Hanson, A.D. The moderately (d) efficient enzyme: catalysis-related damage in vivo and its repair. Biochemistry 2021, 60(47), 3555–3565. [Google Scholar] [CrossRef] [PubMed]
- Hult, K.; Berglund, P. Enzyme promiscuity: mechanism and applications. Trends Biotechnol. 2007, 25(5), 231–238. [Google Scholar] [CrossRef] [PubMed]
- Linster, C.L.; Van Schaftingen, E.; Hanson; A.D. Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 2013, 9, 72–80. [CrossRef] [PubMed]
- Griffith, C.M.; Walvekar, A.S.; Linster, C.L. Approaches for completing metabolic networks through metabolite damage and repair discovery. Curr. Opin. Syst. Biol. 2021, 28, 100379. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.; Thamm, A.M.; Sun, J.; Huang, L.; Sun, L.; Beaudoin, G.A.; Wise, K.S.; Lerma-Ortiz, C.; Bruner, S.D.; Breuer, M.; Luthey-Schulten, Z.; Lin, J.; Wilson, M.A.; Brown, G.; Yakunin, A.F.; Kurilyak, I.; Folz, J.; Fiehn, O.; Glass, J.I.; Hanson, A.D.; Henry, C.S.; de Crécy-Lagard; V. Metabolite damage and damage control in a minimal genome. Mbio 2022, 13(4), e01630-22. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Ramesh, V.; Locasale, J.W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 2020, 21(12), 737–753. [Google Scholar] [CrossRef]
- Goga, A.; Stoffel, M. Therapeutic RNA-silencing oligonucleotides in metabolic diseases. Nat. Rev. Drug Discov. 2022, 21(6), 417–439. [Google Scholar] [CrossRef]
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; Avşar, G.; Romitelli, A.; Pir, P.; Dassi, E.; Conticello, S.G.; Aguilo, F.; Bujnicki, J.M. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022, 50(D1), D231–D235. [Google Scholar] [CrossRef]
- Tarazona, O.A.; Pourquie, O. Exploring the influence of cell metabolism on cell fate through protein post-translational modifications. Dev. Cell 2020, 54(2), 282–292. [Google Scholar] [CrossRef]
- McDonald, A.G.; Tipton, K.F. Enzyme nomenclature and classification: The state of the art. FEBS J. 2023, 290(9), 2214–2231. [Google Scholar] [CrossRef] [PubMed]
- Alcántara, A.R.; Dominguez de Maria, P.; Littlechild, J.A.; Schürmann, M.; Sheldon, R.A.; Wohlgemuth, R. Biocatalysis as key to sustainable industrial chemistry. ChemSusChem 2022, 15(9), e202102709. [Google Scholar] [CrossRef] [PubMed]
- He, H., Bian, G., Herbst-Gervasoni, C.J., Mori, T., Shinsky, S.A., Hou, A., Mu, X., Huang, M., Cheng, S., Deng, Z.; Christianson, D.W.; Abe, I.; Liu, T. Discovery of the cryptic function of terpene cyclases as aromatic prenyltransferases. Nat. Commun. 2020, 11(1), 3958. [CrossRef]
- Walsh, C.T.; Moore, B.S. Enzymatic cascade reactions in biosynthesis. Angew. Chem. Int. Ed. 2019, 58(21), 6846–6879. [Google Scholar] [CrossRef]
- Hertweck, C. The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. 2009, 48, 4688–4716. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Du, L. Iterative polyketide biosynthesis by modular polyketide synthases in bacteria. Appl. Microbiol. Biotechnol. 2016, 100, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Nivina, A.; Yuet, K.P.; Hsu, J.; Khosla, C. Evolution and diversity of assembly-line polyketide synthases: focus review. Chem. Rev. 2019, 119(24), 12524–12547. [Google Scholar] [CrossRef] [PubMed]
- Steffen, W.; Richardson, K.; Rockström, J.; Cornell, S.E.; Fetzer, I.; Bennett, E.M.; Biggs, R.; Carpenter, S.R.; De Vries, W.; De Wit, C.A.; Folke, C. Planetary boundaries: Guiding human development on a changing planet. Science 2015, 347(6223), 1259855. [Google Scholar] [CrossRef] [PubMed]
- Hoskisson, P.A.; Seipke, R.F. Cryptic or silent? The known unknowns, unknown knowns, and unknown unknowns of secondary metabolism. MBio 2020, 11(5), 10–1128. [Google Scholar] [CrossRef]
- Kim, G.B.; Kim, J.Y.; Lee, J.A.; Charles, J. Norsigian, C.J.; Palsson, B.O.; Lee, S.Y. Functional annotation of enzyme-encoding genes using deep learning with transformer layers. Nat. Commun. 2023, 14, 7370. [Google Scholar] [CrossRef]
- Chang, Y.C.; Hu, Z., Rachlin, J.; Anton, B.P.; Kasif, S.; Roberts, R.J.; Steffen, M. COMBREX-DB: an experiment centered database of protein function: knowledge, predictions and knowledge gaps. Nucleic Acids Res. 2016, 44(D1), D330-D335. [CrossRef]
- Price, M.N.; Wetmore, K.M.; Waters, R.J.; Callaghan, M.; Ray, J.; Liu, H.; Kuehl, J.V.; Melnyk, R.A.; Lamson, J.S.; Suh, Y.; Carlson, H.K.; Esquivel, Z.; Sadeeshkumar, H.; Chakraborty, R.; Zane, G.M.; Rubin, B.E.; Wall, J.D.; Visel, A.; Bristow, J.; Blow, M.J.; Arkin, A.P.; Deutschbauer, A.M. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature 2018, 557(7706), 503–509. [Google Scholar] [CrossRef]
- Rhee, K.Y.; Jansen, R.S.; Grundner, C. Activity-based annotation: the emergence of systems biochemistry. Trends Biochem. Sci. 2022, 47(9), 785–794. [Google Scholar] [CrossRef] [PubMed]
- Swainston, N.; Baici, A.; Bakker, B.M.; Cornish-Bowden, A.; Fitzpatrick, P.F.; Halling, P.; Leyh, T.S.; O’Donovan, C.; Raushel, F.M.; Reschel, U.; Rohwer, J.M.; Schnell, S.; Schomburg, D.; Tipton, K.F.; Tsai, M.D.; Westerhoff, H.V.; Wittig, U.; Wohlgemuth, R.; Kettner, K. STRENDA DB: enabling the validation and sharing of enzyme kinetics data. FEBS J. 2018, 1–12. [Google Scholar] [CrossRef]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; Heger, A.; Holm, L.; Sonnhammer, E.L.; Eddy, S.R.; Bateman, A.; Finn, R.D. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–301. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, D.L.; Tawfik, D.S. Protein engineers turned evolutionists—the quest for the optimal starting point. Curr. Opin. Biotechnol. 2019, 60, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. 1998, 95(11), 5857–5864. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Penny Coggill, P.; Robert D. Finn, R.D. DUFs: families in search of function. Acta Cryst. 2010, F66, 1148–1152. [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Lázaro Pinto, B.; Salazar, G.A.; Bileschi, M.I.; Bork, P.; Bridge, A.; Colwell, L.; Gough, J.; Haft, D.H.; Letunić, I.; Marchler-Bauer, A.; Mi, H.; Natale, D.A.; Orengo, C.A.; Pandurangan, A.P.; Rivoire, C.; Sigrist, C.A.; Sillitoe, I.; Thanki, N.; Thomas, P.D.; Tosatto, S.C.E.; Wu, C.H.; Bateman, A. InterPro in 2022, Nucleic Acids Res. , 51(D1), Pages D418–D427. [CrossRef]
- Gerlt, J.A.; Allen, K.N.; Almo, S.C.; Armstrong, R.N.; Babbitt, P.C.; Cronan, J.E.; Dunaway-Mariano, D.; Imker, H.J.; Jacobson, M.P.; Minor, W.; Poulter, C.D. The enzyme function initiative. Biochemistry 2011, 50(46), 9950–9962. [Google Scholar] [CrossRef] [PubMed]
- Gerlt, J.A.; Bouvier, J.T.; Davidson, D.B.; Imker, H.J.; Sadkhin, B.; Slater, D.R.; Whalen, K.L. Enzyme function initiative-enzyme similarity tool (EFI-EST): a web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 2015, 1854(8), 1019–1037. [Google Scholar] [CrossRef] [PubMed]
- Zallot, R.; Oberg, N.; Gerlt, J.A. The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58(41), 4169–4182. [Google Scholar] [CrossRef] [PubMed]
- Oberg, N.; Zallot, R.; Gerlt, J.A. EFI-EST, EFI-GNT, and EFI-CGFP: Enzyme Function Initiative (EFI) Web Resource for Genomic Enzymology Tools. J. Mol. Biol. 2023, 435, 168018. [Google Scholar] [CrossRef]
- Zhang, X.; Carter, M.S.; Vetting, M.W.; San Francisco, B.; Zhao, S.; Al-Obaidi, N.F.; Solbiati, J.O.; Thiaville, J.J.; de Crécy-Lagard, V.; Jacobson, M.P.; Almo, S.C. Assignment of function to a domain of unknown function: DUF1537 is a new kinase family in catabolic pathways for acid sugars. Proc. Natl. Acad. Sci. 2016, 113(29), E4161–E4169. [Google Scholar] [CrossRef]
- Taylor, Z.W.; Brown, H.A.; Narindoshvili, T.; Wenzel, C.Q.; Szymanski, C.M.; Holden, H.M.; Raushel, F.M. Discovery of a glutamine kinase required for the biosynthesis of the O-methyl phosphoramidate modifications found in the capsular polysaccharides of Campylobacter jejuni. J. Am. Chem. Soc. 2017, 139(28), 9463–9466. [Google Scholar] [CrossRef]
- Bastard, K.; Smith, A.A.T.; Vergne-Vaxelaire, C.; Perret, A.; Zaparucha, A.; De Melo-Minardi, R.; Mariage, A.; Boutard, M.; Debard, A.; Lechaplais, C.; Pelle, C. Revealing the hidden functional diversity of an enzyme family. Nat. Chem. Biol. 2014, 10(1), 42–49. [Google Scholar] [CrossRef] [PubMed]
- Korczynska, M.; Xiang, D.F.; Zhang, Z.; Xu, C.; Narindoshvili, T.; Kamat, S.S.; Williams, H.J.; Chang, S.S.; Kolb, P.; Hillerich, B.; Sauder, J.M.; Burley, S.K.; Almo, S.C.; Swaminathan, S.; Shoichet, B.K.; Raushel, F.M. Functional annotation and structural characterization of a novel lactonase hydrolyzing D-xylono-1, 4-lactone-5-phosphate and L-arabino-1, 4-lactone-5-phosphate. Biochemistry, 2014, 53(28), 4727-4738. [CrossRef]
- Malpartida, F.; Hopwood, D.A. Molecular cloning of the whole biosynthetic pathway of a Streptomyces antibiotic and its expression in a heterologous host. Nature 1984, 309(5967), 462–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Huo, Y.X. Using genome and transcriptome analysis to elucidate biosynthetic pathways. Curr. Opin. Biotechnol. 2022, 75, 102708. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Fischbach, M.A. Natural Products Version 2.0: Connecting Genes to Molecules. J. Am. Chem. Soc. 2010, 132, 2469–249392469. [Google Scholar] [CrossRef] [PubMed]
- MohammadiPeyhani, H.; Hafner, J.; Sveshnikova, A.; Viterbo, V.; Hatzimanikatis, V. Expanding biochemical knowledge and illuminating metabolic dark matter with ATLASx. Nat. Commun. 2022, 13, 1560. [Google Scholar] [CrossRef] [PubMed]
- Barra, L.; Awakawa, T.; Shirai, K.; Hu, Z.; Bashiri, G.; Abe, I. β-NAD as a building block in natural product biosynthesis. Nature 2021, 600(7890), 754–758. [Google Scholar] [CrossRef] [PubMed]
- Caputi, L.; Franke, J.; Farrow, S.C.; Chung, K.; Payne, R.M.; Nguyen, T.D.; Dang, T.T.T.; Soares Teto Carqueijeiro, I.; Koudounas, K.; Dugé de Bernonville, T.; Ameyaw, B. , Jones, D.M.; Curcino Vieira, I.J.; Courdavault, V.; O’Connor, S.E. Missing enzymes in the biosynthesis of the anticancer drug vinblastine in Madagascar periwinkle. Science 2018, 360(6394), 1235-1239.
- Qu, Y.; Easson, M.E.A.M.; Simionescu, R.; Hajicek, J.; Thamm, A.M.K.; Salim, V.; De Luca, V. Solution of the multistep pathway for assembly of corynanthean, strychnos, iboga, and aspidosperma monoterpenoid indole alkaloids from 19E-geissoschizine. Proc. Natl. Acad. Sci. 2018, 115(12), 3180–3185. [Google Scholar] [CrossRef]
- Qu, Y.; Easson, M.L.; Froese, J.; Simionescu, R.; Hudlicky, T.; De Luca, V. Completion of the seven-step pathway from taber-sonine to the anticancer drug precursor vindoline and its assembly in yeast. Proc. Natl. Acad. Sci. 2015, 112(19), 6224–6229. [Google Scholar] [CrossRef]
- Qu, Y.; Safonova, O.; De Luca, V. Completion of the canonical pathway for assembly of anticancer drugs vincristine/vinblastine in Catharanthus roseus. Plant J. 2019, 97(2), 257–266. [Google Scholar] [CrossRef]
- Zhang, J.; Hansen, L.G.; Gudich, O.; Viehrig, K.; Lassen, L.M.M.; Schrübbers, L.; Adhikari, K.B.; Rubaszka, P.; Carrasquer-Alvarez, E.; Chen, L.; D’Ambrosio, V.; Lehka, B.; Haidar, A.K.; Nallapareddy, S.; Giannakou, K.; Laloux, M.; Arsovska, D.; Jørgensen, M.A.K.; Chan, L.J.G.; Kristensen, M.; Christensen, H.B.; Sudarsan, S.; Stander, E.A.; Baidoo, E.; Petzold, C.J.; Wulff, T.; O’Connor, S.E.; Courdavault, V.; Jensen; M.K.; Keasling, J.D. A microbial supply chain for production of the anti-cancer drug vinblastine. Nature 2022, 609, 341–347. [CrossRef]
- Nunoura, T.; Chikaraishi, Y.; Izaki, R.; Suwa, T.; Sato, T.; Harada, T.; Mori, K.; Kato, Y.; Miyazaki, M.; Shimamura, S.; Yanagawa, K.; Shuto, A.; Ohkouchi, N.; Fujita, N.; Takaki, Y.; Atomi, H.; Takai, K. A primordial and reversible TCA cycle in a facultatively chemolithoautotrophic thermophile. Science 2018, 359, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Pascal Andreu, V.; Augustijn, H.E.; Chen, L.; Zhernakova, A.; Fu, J.; Fischbach, M.A.; Dodd, D.; Medema, M.H. gutSMASH predicts specialized primary metabolic pathways from the human gut microbiota. Nat. Biotechnol. 2023, 41, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Shimosaka, T.; Makarova, K.S.; Koonin, E.V.; Atomi, H. Identification of dephospho-coenzyme A (dephospho-CoA) kinase in Thermococcus kodakarensis and elucidation of the entire CoA biosynthesis pathway in Archaea. Mbio 2019, 10(4), 10–1128. [Google Scholar] [CrossRef]
- Medema, M.H.; de Rond, T.; Moore, B.S. Mining genomes to illuminate the specialized chemistry of life. Nat. Rev. Genet. 2021, 22(9), 553–571. [Google Scholar] [CrossRef] [PubMed]
- Robey, M.T.; Caesar, L.K.; Drott, M.T.; Keller, N.P.; Kelleher, N.L. An interpreted atlas of biosynthetic gene clusters from 1,000 fungal genomes. Proc. Natl. Acad. Sci. 2021, 118(19), e2020230118. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Medema, M.H.; Weber, T. The antiSMASH database version 4: additional genomes and BGCs, new sequence-based searches and more. Nucleic Acids Res. 2024, 52, D586–D589. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, M.; Cui, Z.; Hankore, E.D.; Moonschi, F.H.; Esfahani, H.S.; Kalkreuter, E.; Gui, C.; Yang, D.; Phillips Jr., G. N.; Thorson, J.S.; Shen, B.; Van Lanen, S.G. A discrete intermediate for the biosynthesis of both the enediyne core and the anthraquinone moiety of enediyne natural products. Proc. Natl. Acad. Sci. 2023, 120(9), e2220468120. [Google Scholar] [CrossRef]
- Scherlach, K.; Hertweck, C. Mining and unearthing hidden biosynthetic potential. Nat. Commun. 2021, 12, 3864. [Google Scholar] [CrossRef]
- Terlouw, B.R.; Blin, K.; Navarro-Muñoz, J.C.; Avalon, N.E.; Chevrette, M.G.; Egbert, S.; Lee, S.; Meijer, D.; Recchia, M.J.J.; Reitz, Z.L.; van Santen, J.A.; Selem-Mojica, N., Tørring, T.; Zaroubi, L.; Alanjary, M.; Aleti, G.; Aguilar, C.; Al-Salihi, S.A.A.; Augustijn, H.E.; Avelar-Rivas, J.A.; Avitia-Domínguez, L.A.; Barona-Gómez, F.; Bernaldo-Agüero, J.; Bielinski, V.A.; Biermann, F.; Booth, T.J.; Carrion Bravo, V.J.; Castelo-Branco, R.; Chagas, F.O.; Cruz-Morales, P.; Du, C.; Duncan, K.R.; Gavriilidou, A.; Gayrard, D.; Gutiérrez-García, K.; Haslinger, K.; Helfrich, E.J.N.; van der Hooft, J.J.J.; Jati, A.P.; Kalkreuter, E.; Kalyvas, N.; Kang, K.B.; Kautsar, S.; Kim, W.; Kunjapur, A.M.; Li, Y.X.; Lin, G.M.; Loureiro, C.; Louwen, J.J.R.; Louwen, N.L.L.; Lund, G.; Parra, J.; Philmus, B.; Pourmohsenin, B.; Pronk, L.J.U.; Rego, A.; Rex, D.A.B.; Robinson, S.; Rosas-Becerra, L.R.; Roxborough, E.T.; Schorn, M.A.; Scobie, D.J:, Singh, K.S.; Sokolova, N.; Tang, X.; Udwary, D.; Vigneshwari, A.; Vind, K.; Vromans, S.P.J.M.; Waschulin, V.; Williams, S.E.; Winter, J.M.; Witte, T.E.; Xie, H.; Yang, D.; Yu, J.; Zdouc, M.; Zhong, Z.; Collemare, J.; Linington, R.G.; Weber, T.; Medema, M.H. MIBiG 3.0: a community-driven effort to annotate experimentally validated biosynthetic gene clusters, Nucleic Acids Res. 2023, 51(D1), D603–D610. [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids oRes. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.; van Wezel, G.P., Medema, M.H.; Weber, T. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023, 51(W1), W46–W50. [CrossRef]
- Ren, H.; Shi, C.; Zhao, H. Computational tools for discovering and engineering natural product biosynthetic pathways. Iscience 2020, 23(1), 100795. [Google Scholar] [CrossRef]
- Kountz, D.J.; Balskus, E.P. Leveraging Microbial Genomes and Genomic Context for Chemical Discovery. Acc. Chem. Res. 2021, 54, 2788–2797. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: the teenage years. Nat. Rev. Genetics 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Doroghazi, J.R.; Albright, J.C.; Goering, A.W.; Ju, K.S.; Haines, R.R.; Tchalukov, K.A.; Labeda, D.P.; Kelleher, N.L.; Metcalf, W.W. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol. 2014, 10(11), 963–968. [Google Scholar] [CrossRef] [PubMed]
- Avalon, N.E.; Murray, A.E.; Baker, B.J. Integrated Metabolomic–Genomic Workflows Accelerate Microbial Natural Product Discovery. Anal. Chem. 2022, 94, 35, 11959–11966. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, B.; Timári, I.; Somogyi, Á.; Li, D.W.; Adcox, H.E.; Gunn, J.S.; Bruschweiler-Li, L.; Brüschweiler, R. Accurate and efficient determination of unknown metabolites in metabolomics by NMR-based molecular motif identification. Anal. Chem. 2019, 91(24), 15686–15693. [Google Scholar] [CrossRef] [PubMed]
- Sidda, J.D.; Song, L.; Poon, V.; Al-Bassam, M.; Lazos, O.; Buttner, M.J.; Challis, G.L.; Corre, C. Discovery of a family of γ-aminobutyrate ureas via rational derepression of a silent bacterial gene cluster. Chem. Sci. 2014, 5, 86–89. [Google Scholar] [CrossRef]
- Alberti, F.; Leng, D.J.; Wilkening, I.; Song, L.; Tosin, M.; Corre, C. Triggering the expression of a silent gene cluster from genetically intractable bacteria results in scleric acid discovery. Chem. Sci. 2019, 10, 453–463. [Google Scholar] [CrossRef]
- Mao, D.; Okada, B.K.; Wu, Y.; Xu, F.; Seyedsayamdost, M.R. Recent advances in activating silent biosynthetic gene clusters in bacteria. Curr. Opin. Microbiol. 2018, 45, 156–163. [Google Scholar] [CrossRef]
- Covington, B.C.; Xu, F.; Seyedsayamdost, M.R. A natural product chemist's guide to unlocking silent biosynthetic gene clusters. Ann. Rev. Biochem. 2021, 90, 763–788. [Google Scholar] [CrossRef]
- Hartl, J.; Kiefer, P.; Meyer, F.; Vorholt; J. A. Longevity of major coenzymes allows minimal de novo synthesis in microorganisms. Nat. Microbiol. 2017, 2, 17073. [Google Scholar] [CrossRef]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; Giacchè, N.; Stokar-Regenscheit, N.; Legouis, D.; de Seigneux, S.; Ivanisevic, J.; Raffaelli, N.; Schoonjans, K.; Pellicciari, R.; Auwerx, J. . De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 2018, 563(7731), 354–359. Nature 2018, 563(7731), 354–359. [Google Scholar] [CrossRef] [PubMed]
- Preiss, J.; Handler, P. Biosynthesis of Diphosphopyridine Nucleotide. I. Identification of Intermediates. J. Biol. Chem. 1958, 233(2), 488–492. [Google Scholar] [CrossRef] [PubMed]
- Preiss, J.; Handler, P. Biosynthesis of Diphosphopyridine Nucleotide. II. Enzymatic Aspects. J. Biol. Chem. 1958, 233(2), 493–500. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.A.; Lee, E.; Li, W.; Fan, W.; Li, J.L.; Sokolsky, M.; Kabanov, A.V.; Li, L.; Migaud, M.E.; Locasale, J.W.; Li, X. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metabolism 2020, 31(3), 564–579. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117(4), 495–502. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Christensen, K.C.; Gazzaniga, F.; Pletnev, A.A.; Brenner, C. Nicotinamide riboside and nicotinic acid riboside salvage in fungi and mammals: Quantitative basis for Urh1 and purine nucleoside phosphorylase function in NAD+ metabolism. J. Biol. Chem. 2009, 284(1), 158–164. [Google Scholar] [CrossRef]
- Giroud-Gerbetant, J.; Joffraud, M.; Giner, M.P.; Cercillieux, A.; Bartova, S.; Makarov, M.V.; Zapata-Pérez, R.; Sánchez-García, J.L.; Houtkooper, R.H.; Migaud, M.E.; Moco, S. A reduced form of nicotinamide riboside defines a new path for NAD+ biosynthesis and acts as an orally bioavailable NAD+ precursor. Mol. Metab. 2019, 30, 192–202. [Google Scholar] [CrossRef]
- Yang, Y.; Mohammed, F.S.; Zhang, N.; Sauve, A.A. Dihydronicotinamide riboside is a potent NAD+ concentration enhancer in vitro and in vivo, J. Biol. Chem. 2019, 294(23), 9295–9307. [Google Scholar] [CrossRef]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018, 27(3), 513–528. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nature Metabolism 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22(2), 119–141. [Google Scholar] [CrossRef] [PubMed]
- Gude, S.; Pherribo, G.J.; Taga, M.A. A Salvaging Strategy Enables Stable Metabolite Provisioning among Free-Living Bacteria. mSystems 2022, 7(4), 1-14. [CrossRef]
- North, J.A.; Wildenthal, J.A.; Erb, T.J.; Evans, B.S.; Byerly, K.M. , Gerlt, J.A.; Fred R. Tabita, F.R. A bifunctional salvage pathway for two distinct S-adenosylmethionine by-products that is widespread in bacteria, including pathogenic Escherichia coli. Mol. Microbiol, 2020; 113, 923–937. [Google Scholar] [CrossRef]
- Gericke, L.; Mhaindarkar, D.; Karst, L.C.; Jahn, S.; Kuge, M.; Mohr, M.K.F.; Gagsteiger, J.; Cornelissen, N.V.; Wen, X.; Mordhorst, S.; Jessen, H.J.; Rentmeister, A.; Seebeck, F.P.; Layer, G.; Loenarz, C.; Andexer, J.A. Biomimetic S-Adenosylmethionine Regeneration Starting from Multiple Byproducts Enables Biocatalytic Alkylation with Radical SAM Enzymes. ChemBioChem 2023, 24, e202300133. [Google Scholar] [CrossRef] [PubMed]
- Jeffryes, J.G.; Lerma-Ortiz, C.; Liu, F.; Golubev, A.; Niehaus, T.D.; Elbadawi-Sidhu, M.; Fiehn, O.; Hanson, A.D.; Tyo, K.E.; Henry, C.S. Chemical-damage MINE: A database of curated and predicted spontaneous metabolic reactions. Metab. Eng. 2022, 69, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Bommer, G.T.; Van Schaftingen, E.; Veiga-da-Cunha, M. Metabolite repair enzymes control metabolic damage in glycolysis. Trends Biochem. Sci. 2020, 45(3), 228–243. [Google Scholar] [CrossRef] [PubMed]
- Niehaus, T.D.; Katie, B. Hillmann, K.B. Enzyme promiscuity, metabolite damage, and metabolite damage control systems of the tricarboxylic acid cycle. FEBS J. 2020, 287, 1343–1358. [Google Scholar] [CrossRef] [PubMed]
- Leister, D.; Sharma, A.; Kerber, N.; Nägele, T.; Reiter, B.; Pasch, V.; Beeh, S.; Jahns, P.; Barbato, R.; Pribil, M.; Rühle, T. An ancient metabolite damage-repair system sustains photosynthesis in plants. Nat. Commun. 2023, 14, 3023. [Google Scholar] [CrossRef]
- Thiaville, J.J.; Flood, J.; Yurgel, S.; Prunetti, L.; Elbadawi-Sidhu, M.; Hutinet, G.; Forouhar, F.; Zhang, X.; Ganesan, V.; Reddy, P.; Fiehn, O.; Gerlt, J.A.; Hunt, J.F.; Copley, S.D.; de Crécy-Lagard, V. Members of a novel kinase family (DUF1537) can recycle toxic intermediates into an essential metabolite. ACS Chem. Biol. 2016, 11(8), 2304–2311. [Google Scholar] [CrossRef]
- Hall, A.; Knowles, J.R. The Uncatalyzed Rates of Enolization of Dihydroxyacetone Phosphate and of Glyceraldehyde 3-Phosphate in Neutral Aqueous Solution. The Quantitative Assessment of the Effectiveness of an Enzyme Catalyst. Biochemistry 1975, 14(19), 4348–4351. [Google Scholar] [CrossRef]
- Kalyananda, M.K.G.S.; Engel, R.; Tropp, B.E. Metabolism of L-Glyceraldehyde 3-Phosphate in Escherichia coli. J. Bact. 1987, 169(6), 2488–2493. [Google Scholar] [CrossRef]
- Desai, K.D.; Miller, B.G. A Metabolic Bypass of the Triosephosphate Isomerase Reaction. Biochemistry 2008, 47, 7983–7985. [Google Scholar] [CrossRef] [PubMed]
- Lewin, H.A.; Richards, S.; Lieberman Aiden, E.; Allende, M.L.; Archibald, J.M.; Bálint, M.; Barker, K.B.; Baumgartner, B.; Belov, K.; Bertorelle, G.; Blaxter, M.L. The earth BioGenome project 2020: Starting the clock. Proc. Natl. Acad. Sci. 2022, 119(4), e2115635118. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, T.E.; Muigai, A.W.T.; Nouala, S.; Badaoui, B.; Blaxter, M.; Buddie, A. G.; Jarvis, E.D.; Korlach, J.; Kuja, J.O.; Lewin, H.A.; Majewska, R.; Mapholi, N.; Maslamoney, S.; Mbo’o-Tchouawou, M.; Osuji, J.O.; Seehausen, O.; Shorinola, O.; Tiambo, C.K.; Mulder, N.; Ziyomo, C.; Djikeng, A. Africa: sequence 100,000 species to safeguard biodiversity. Nature 2022, 603(7901), 388–392. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Roux, S.; Seshadri, R.; Udwary, D.; Varghese, N.; Schulz, F.; Wu, D.; Paez-Espino, D.; Chen, I.M.; Huntemann, M.; Palaniappan, K.; Ladau, J.; Mukherjee, S.; Reddy, T.B.K.; Nielsen, T.; Kirton, E.; Faria, J.P.; Edirisinghe, J.N.; Henry, C.S.; Jungbluth, S.P.; Chivian, D.; Dehal, P.; Wood-Charlson, E.M.; Arkin, A.P.; Tringe, S.G.; Visel, A.; IMG/M Data Consortium, Woyke, T.; Mouncey, N.J.; Ivanova, N.N.; Kyrpides, N.C.; Eloe-Fadrosh, E.A. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 2021, 39, 520. [CrossRef]
- de Crécy-Lagard, V.; Amorin de Hegedus, R.; Arighi, C.; Babor, J.; Bateman, A.; Blaby, I.; Blaby-Haas, C.; Bridge, A.J.; Burley, S.K.; Cleveland, S.; Colwell, L.J.; Conesa, A.; Dallago, C.; Danchin, A.; de Waard, A.; Deutschbauer, A.; Dias, R.; Ding, Y.; Fang, G.; Friedberg, I.; Gerlt, J.; Goldford, J.; Gorelik, M.; Gyori, B.M.; Henry, C.; Hutinet, G.; Jaroch, M.; Karp, P.D.; Kondratova, L.; Lu, Z.; Marchler-Bauer, A.; Martin, M.J.; McWhite, C.; Moghe, G.D.; Monaghan, P.; Morgat, A.; Mungall, C.J.; Natale, D.A.; Nelson, W.C.; O’Donoghue, S.; Orengo, C.; O’Toole, K.H.; Radivojac, P.; Reed, C.; Roberts, R.J.; Rodionov, D.; Rodionova, I.A.; Rudolf, J.D.; Saleh, L.; Sheynkman, G.; Thibaud-Nissen, F.; Thomas, P.D.; Uetz, P.; Vallenet, D.; Watson Carter, E.; Weigele, P.R.; Wood, V.; Wood-Charlson, E.M.; Xu, J. A roadmap for the functional annotation of protein families: a community perspective. Database 2022, 2022, baac062. [Google Scholar] [CrossRef] [PubMed]
- Harmange Magnani, C.S.; Hernández-Meléndez, J.R.; Tantillo, D.J.; Maimone, T.J. Total Synthesis of Altemicidin: A Surprise Ending for a Monoterpene Alkaloid. JACS Au 2023, 3, 2883–2893. [Google Scholar] [CrossRef] [PubMed]
- Hetzler, B. E.; Trauner, D.; Lawrence, A. L. Natural product anticipation through synthesis. Nat. Rev. Chem. 2022, 6, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Caesar, L.K.; Montaser, R.; Keller, N.P.; Kelleher, N.L. Metabolomics and Genomics in Natural Products Research: Complementary Tools for Targeting New Chemical Entities. Nat. Prod. Rep. 2021, 38, 2041–2065. [Google Scholar] [CrossRef]
- Caesar, L.K.; Butun, F.A.; Robey, M.T.; Ayon, N.J.; Gupta, R.; Dainko, D.; Bok, J.W.; Nickles, G.; Stankey, R.J.; Johnson, D.; Mead, D.; Cank, K.B.; Earp, C.E.; Raja, H.A.; Oberlies, N.H.; Keller, N.P.; Kelleher, N.L. Correlative metabologenomics of 110 fungi reveals metabolite–gene cluster pairs. Nat. Chem. Biol. 2023, 19(7), 846–854. [Google Scholar] [CrossRef]
Figure 1.
Assignment and characterization of enzyme functions for selected DUF proteins.
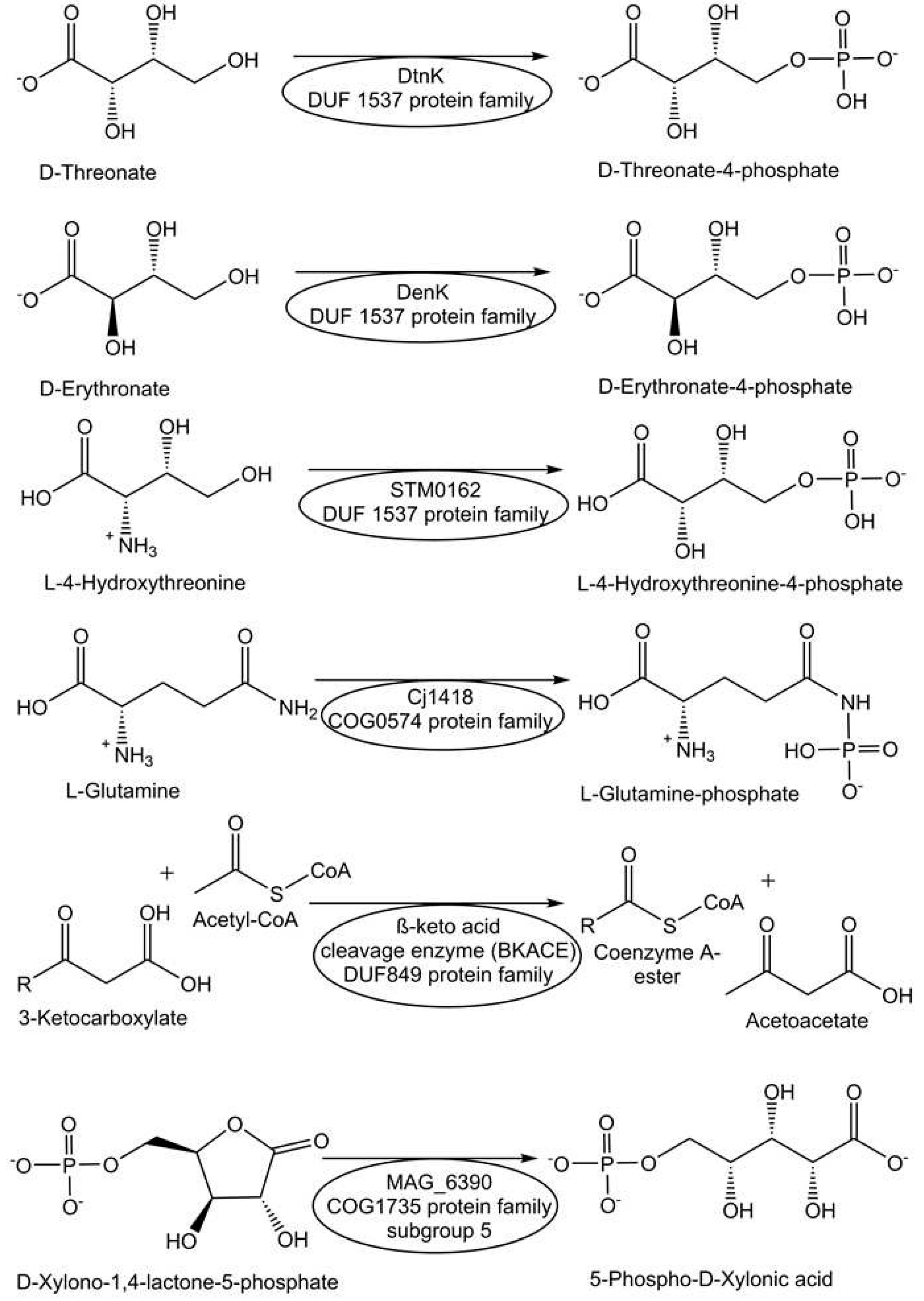
Figure 2.
Identification of a missing enzymatic reaction step in the biosynthesis of altemicidin.
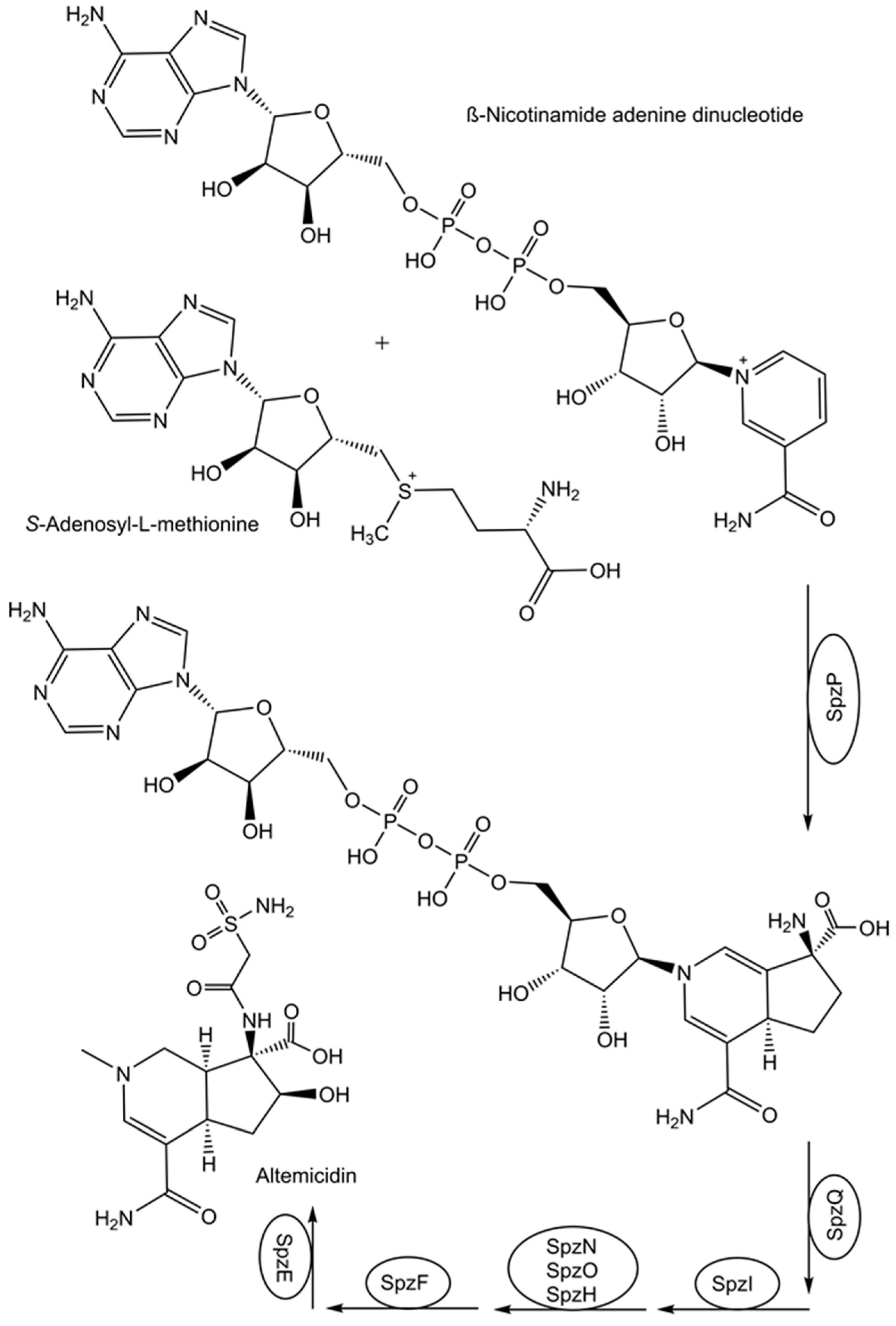
Figure 3.
Proposed unifying pathway in the deciphering of the biosynthesis of the enediyne aromatic polyketides.
Figure 3.
Proposed unifying pathway in the deciphering of the biosynthesis of the enediyne aromatic polyketides.
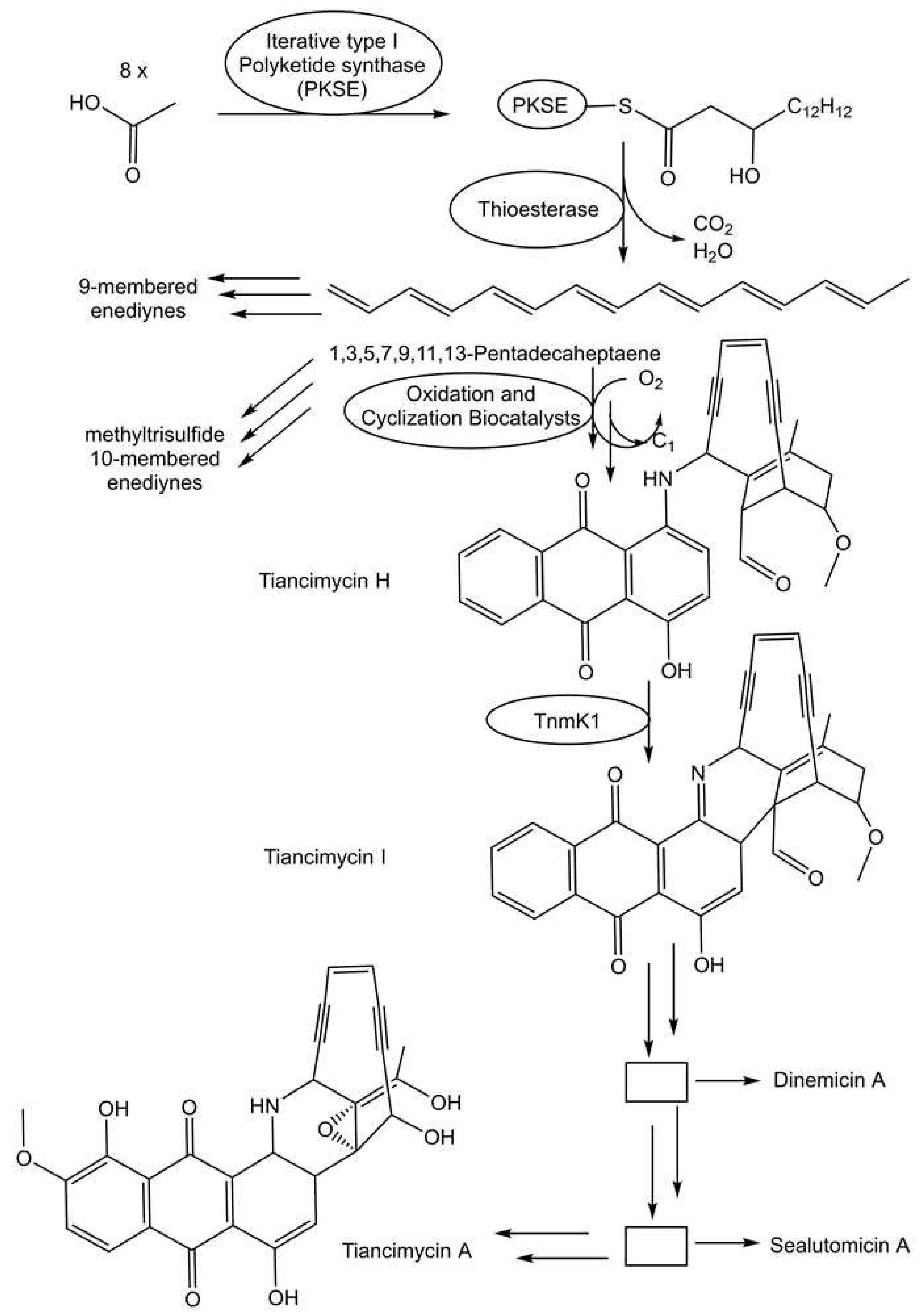
Figure 4.
Diverse natural and synthetic salvage pathways which have been discovered, proposed and designed for S-adenosyl-L-homocysteine, L-methionine, 5'-methylthioadenosine, 5'-deoxyadenosine.
Figure 4.
Diverse natural and synthetic salvage pathways which have been discovered, proposed and designed for S-adenosyl-L-homocysteine, L-methionine, 5'-methylthioadenosine, 5'-deoxyadenosine.
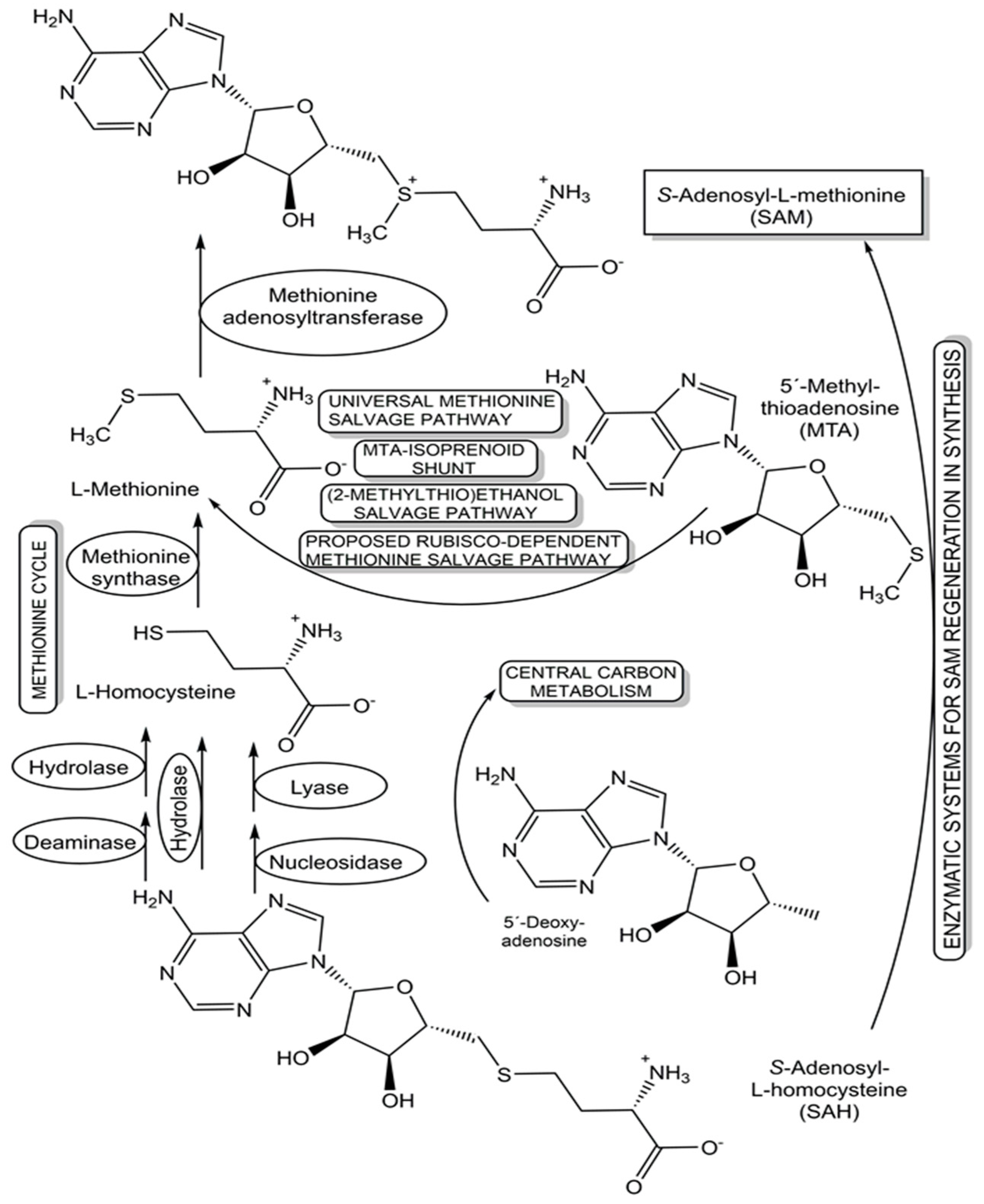
Figure 5.
Discovery of the metabolite repair enzyme L-glyceraldehyde-3-phosphate reductase catalyzing the stereospecific, NADPH-dependent reduction of L-glyceraldehyde-3-phosphate to the non-toxic natural metabolite L-glycerol-3-phosphate (sn-glycerol-3-phosphate) .
Figure 5.
Discovery of the metabolite repair enzyme L-glyceraldehyde-3-phosphate reductase catalyzing the stereospecific, NADPH-dependent reduction of L-glyceraldehyde-3-phosphate to the non-toxic natural metabolite L-glycerol-3-phosphate (sn-glycerol-3-phosphate) .
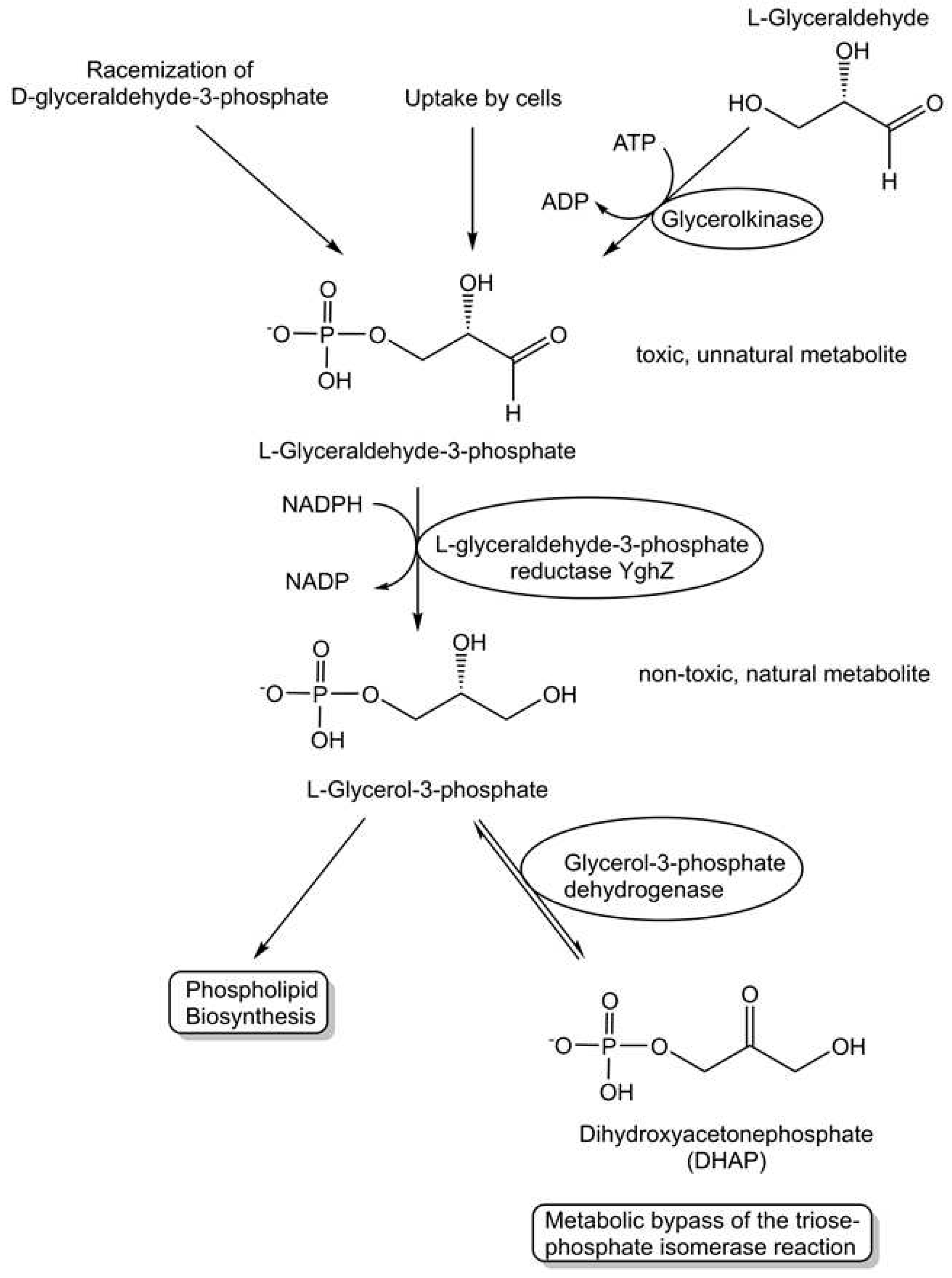
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



