Submitted:
25 January 2024
Posted:
25 January 2024
You are already at the latest version
Abstract
The emergence of bispecific antibodies has transformed cancer immunotherapy, highlighting increased clinical efficacy, especially in hematological malignancies. These innovative molecules uniquely target two distinct tumor antigens or separate epitopes simultaneously, demonstrating potent antitumor activity across various cancers. Despite their promise, challenges like rapid drug clearance, off-target effects, and cytokine release syndrome hinder their widespread therapeutic application. Recent engineering advancements in bispecific antibody systems aim to overcome these challenges, broadening therapeutic coverage. This review offers insights into the latest clinical and preclinical progress in bispecific immunotherapy, outlining key challenges faced by the technique, and exploring emerging strategies to address these obstacles.
Keywords:
Bispecific
; immunotherapy
; cancer
; cytokine
; antibodies
Introduction
Before the introduction of bispecific antibodies (BsAbs), traditional cancer immunotherapy relied on monoclonal antibody (MoAb) molecules targeting a single tumor antigen [1]. However, the complex nature of some cancers, with their ability to switch signaling pathways and evade immune responses, posed challenges for this approach [2]. A prime example is the interaction between Programmed Cell Death Protein 1 (PD-1) and Programmed Cell Death Ligand 1 (PD-L1), where tumor cells exploit this interaction to attenuate the immune response [3]. This manipulation involves inducing apoptosis in antigen-specific T cells and inhibiting the apoptosis of regulatory T cells, affecting the efficacy of single antibody-targeted immunotherapy [3]. The arrival of bispecific antibodies marks a significant shift in addressing these challenges, offering a promising avenue for more effective cancer treatment.
BsAbs are a promising type of therapy that can target two different tumor antigens simultaneously [4]. These antibodies typically consist of two single-chain variable fragment (scFv) antigen-binding parts linked by a flexible amino acid linker, offering a more refined approach against cancer cells [5]. In the case of the PD-1/PD-L1 axis, bispecific antibodies can be designed to bind both PD-1 and a tumor-specific antigen at the same time. This disrupts immune evasion mechanisms and strengthens the immune response [6]. Over 100 bispecific antibodies have been evaluated across various cancer types, with many receiving marketing approvals (Table 1) [7,8]. A significant achievement occurred in 2022 when the FDA approved a Bispecific T cell Engager (BiTE) product targeting CD3/BCMA for treating relapsed or refractory multiple myeloma [9]. Subsequently, talquetamab and elranatamab, both CD3 T-cell engagers, received FDA approval in 2023 for multiple myeloma treatment (Table 1) [10,11]. These approvals mark substantial progress in treating adult patients with relapsed or refractory multiple myeloma.
While most bispecific antibodies focus on cancer treatment, some are directed at chronic inflammatory, autoimmune, neurodegenerative diseases, and infections. Examples include emacizumab and faricimab, both developed for hemophilia A and retinal vascular disease treatment, respectively [12,13]. These diverse applications highlight the expanding role of bispecific antibodies in transformative therapeutic interventions. While bispecific antibodies have shown success in cancer treatment, they still face challenges like a short in vivo half-life, on-target off-tumor effects, cytokine release syndrome, and issues in manufacturing [14,15,16]. These challenges hinder their broad application. However, recent advancements have led to innovative approaches addressing these challenges, paving the way for improved clinical practices. In this review, we shed light on the evolving field of bispecific antibodies, providing insights into their present status in clinical development. Additionally, we delve into the challenges associated with bispecific antibodies and explore recent modifications aimed at enhancing their therapeutic efficacy.
Bispecific T Cell Engager
The concept of bispecific antibodies (BsAbs) has evolved significantly since their initial description by Nisonoff in 1960, resulting in the development of several hundred formats categorized into six diverse mechanisms of action: (1) bridging cells, (2) receptor inhibition, (3) receptor activation, (4) co-factor mimetic, (5) piggybacking I, and (6) piggybacking II [8,17]. These diverse BsAb formats have been engineered to target various components such as tumor signaling pathways, immune checkpoint inhibitors (ICIs), inflammatory cytokines, and more [18,19,20]. Among these formats, the bridging cell or Bispecific T-cell Engager stands out as the most common BsAb employed for the treatment of both liquid and solid tumors [21]. A crucial aspect of BiTEs is their ability to redirect naïve T cells to target tumor cells, leading to T-cell activation, clonal expansion, and subsequent tumor cytotoxicity [21,22]. First-generation BiTE constructs were typically designed with two monoclonal antibody (mAb) moieties tandemly fused, with one moiety targeting a specific tumor antigen and the other binding to CD3 antigen on T-cell surfaces. This design ensures that T cells engaged by BiTE molecules become activated and effectively eliminate malignant cells [23]. More than six decades, seven BiTEs are approved for cancer treatment (see Table 1), and several more are undergoing clinical testing [24]. Despite their efficacy, the use of bispecific T-cell engagers has faced challenges associated with 'on-target, off-tumor' toxicities [25,26]. BiTE therapy primarily involves identifying suitable tumor-associated antigens (TAAs) on target cells that differ from those on normal cells, aiming to prevent on-target/off-tumor toxicity [25]. However, the identification of antigenic targets exclusive to tumor cells presents challenges, as many target antigens are expressed on both normal and tumor cells [27]. Even minimal antigen expression on normal cells can result in adverse on-target off-tumor toxicities leading to cytokine release syndrome (CRS). CRS, characterized by an excessive immune response leading to the release of proinflammatory cytokines, can potentially result in organ failure and, in severe cases, death [28]. Currently, the primary clinical interventions to manage CRS in TCE therapies involve dose reduction or the administration of anti-interleukin antibodies and corticosteroids [28]. While these interventions have proven effective in certain scenarios, they do not provide a complete prevention of CRS. Accordingly, increasing reports have highlighted the occurrences of off-target, on-target toxicity associated with bispecific antibody molecules, especially BiTE therapeutics [23,29,30].
To overcome the significant challenge of on-target, off-tumor adverse effects including CRS, and enhanced the therapeutic index of BsAbs particularly in the context of solid malignancies, researchers have been exploring several modification strategies. One such strategy focuses on employing avidity-mediated specificity or the 2 + 1 architecture [31,32]. In this novel approach, a bivalent antibody with low affinity for the tumor antigen is combined with a monovalent anti-CD3 molecule [32]. This unique design enables the BiTE to selectively bind to tumor cells that overexpress the target tumor-associated antigen (TAA), facilitating the specific killing of tumor cells while sparing normal cells expressing the target antigen at lower densities.
A study conducted by Bacac et al. exemplifies this approach, utilizing a bivalent anti-CEA scFv domain linked with a monovalent anti-CD3 domain for the treatment of solid tumors expressing carcinoembryonic antigen (CEA) [33]. CEA, also known as carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5), is associated with glycosylphosphatidylinositol and is overexpressed in various cancers, playing a role in adhesion and invasion [34]. The resulting CEA T cell bispecific (TCB) demonstrated sustained antitumor activity in a preclinical model, exhibiting a notable increase in T-cell longevity [33]. Moreover, the CEA+CD3 TCB transformed PD-L1-negative tumors into PD-L1-positive, creating a highly inflamed tumor microenvironment. This promising development has advanced to phase 1 clinical investigation (NCT02324257), showcasing pronounced efficacy and manageable safety profiles [33].
In line with these advancements, another group used an anti-HER2/CD3 T cell-dependent bispecific (TDB) antibody to redirect T cells to eliminate HER2-overexpressing cells, demonstrating potent antitumor activity [31]. This suggests that avidity-mediated selection holds promise for treating solid tumors, as its potentially addresses one of the major challenges associated with TCE therapies, offering a more targeted and controlled immune response. However, since high expression levels of the TAA are crucial for avidity specificity and bispecific antibody-mediated tumor lysis, this strategy is applicable primarily to cancer cells expressing very high levels of the target antigen. The challenge arises when dealing with solid tumors expressing variable densities of the target antigen. To address this challenge and enhance the versatility of the approach, future studies are needed to develop a dual bispecific antibody with a 2+1+1 architecture, where one target incorporates avidity-mediated specificity and the other features high-affinity binding. This approach would offer a comprehensive solution to rapidly target and eliminate solid tumors expressing differential levels of the target antigen.
Generally, there is currently no FDA-approved BiTE molecule for treating solid malignancies. However, catumaxomab, the first bispecific T-cell engager approved by the EMA in 2009 to treat malignant ascites of epithelial cancers, was later withdrawn from the market due to severe adverse events, including CRS and dose-dependent liver toxicity [35]. Ongoing research and development aim to address these challenges and further enhance the clinical applicability of BiTEs, emphasizing their significance in advancing cancer immunotherapy.
Figure 1.
BiTE and its mechanism of action. a. BiTE antibody construct comprises two single-chain variable fragments of monoclonal antibodies linked together through a flexible linker. b. One arm of the BiTE molecule is designed to bind to CD3, an antigen located on the surface of T cells. Simultaneously, the other arm is engineered to bind to a tumor-associated antigen (TAA). Upon successful binding of both arms to their specific targets, a synapse is formed between the T cell and the cancer cell. Subsequently, the T cells undergo expansion and release perforin, creating a pore in the cancer cell's membrane. This pore allows toxic molecules called granzymes to flow through, ultimately inducing the death of the cancer cell.
Figure 1.
BiTE and its mechanism of action. a. BiTE antibody construct comprises two single-chain variable fragments of monoclonal antibodies linked together through a flexible linker. b. One arm of the BiTE molecule is designed to bind to CD3, an antigen located on the surface of T cells. Simultaneously, the other arm is engineered to bind to a tumor-associated antigen (TAA). Upon successful binding of both arms to their specific targets, a synapse is formed between the T cell and the cancer cell. Subsequently, the T cells undergo expansion and release perforin, creating a pore in the cancer cell's membrane. This pore allows toxic molecules called granzymes to flow through, ultimately inducing the death of the cancer cell.
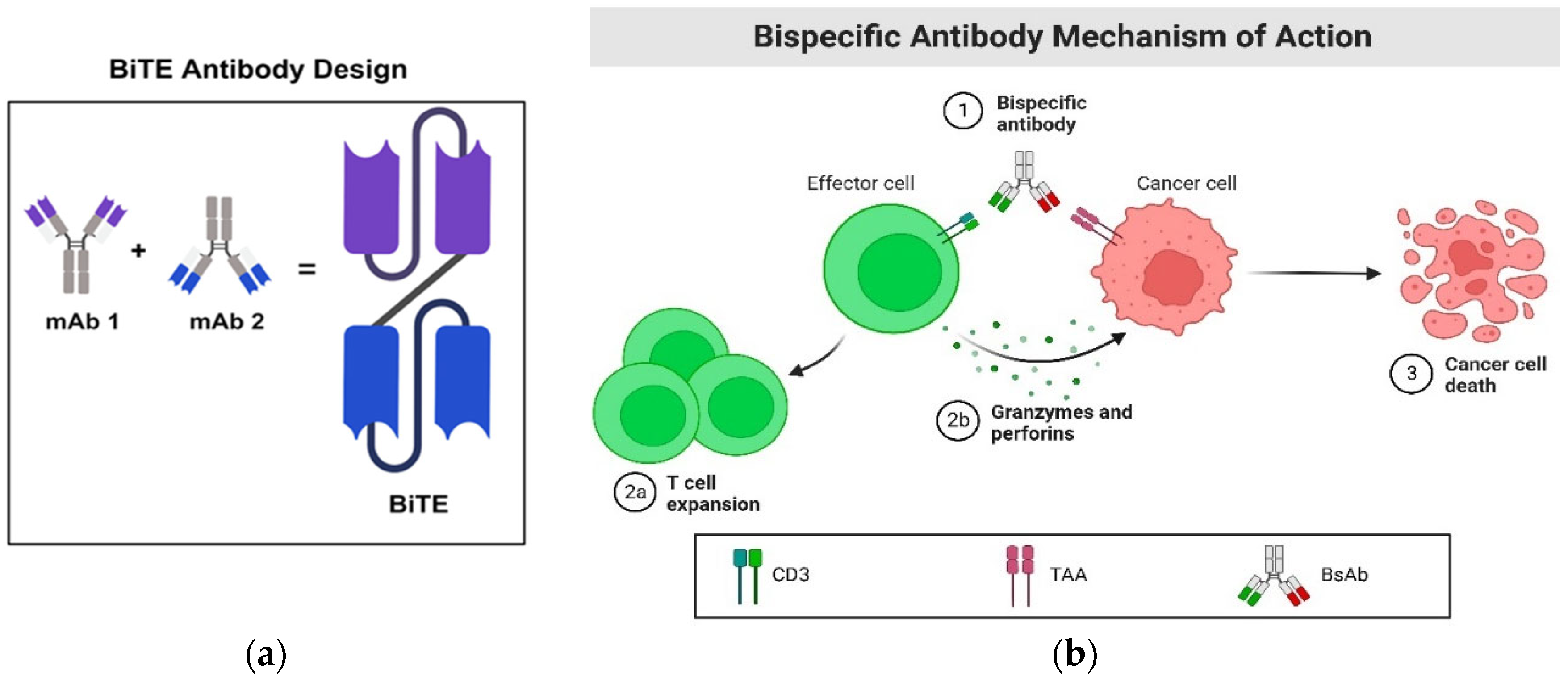
While BiTEs encounter challenges in battling solid tumors, a promising alternative, immune-mobilizing monoclonal T-cell receptors against cancer (ImmTACs), has emerged [36]. Like BiTEs, ImmTACs facilitate the interaction between cancer cells and T cells by simultaneously engaging their proteins. However, ImmTACs take a different approach by employing a T-cell receptor instead of an antibody fragment to recognize proteins on cancer cells [36]. This unique strategy allows ImmTACs to bind to intracellular proteins processed and presented externally, expanding their target range beyond cell surface proteins. This characteristic makes ImmTACs more effective in addressing solid tumors, where many cancer-specific proteins are primarily expressed inside the cell. Tebentafusp (Kimmtrak), an ImmTAC therapeutic, has already gained approval for treating uveal melanoma [37]. Considering the risks associated with BiTEs in solid tumors, especially CRS, ImmTACs emerge as a promising class of therapeutics, offering cancer-fighting immune cells a distinct advantage.
Blinatumomab, the First FDA Approved BiTE Construct
Blinatumomab stands out as a significant success in BiTE therapy, marking the first FDA-approved BiTE molecule to treat B-cell acute lymphoblastic leukemia (ALL) [38]. This therapy combines anti-CD19 and anti-CD3 single-chain variable fragment (scFv) domains, demonstrating notable clinical efficacy. Many patients experienced complete tumor regression, contributing to improved overall survival rates [39]. In a study with 54 relapsed or refractory (R/R) patients, 91% (49/54) achieved a complete response with blinatumomab treatment, highlighting its clinical effectiveness in challenging R/R settings [40]. These outcomes emphasize Blinatumomab's therapeutic potential and its crucial role in advancing treatment options for B-cell ALL patients. Importantly, Blinatumomab's activity is independent of major histocompatibility complex activation, ensuring rapid activation of T cells and the destruction of tumor cells [41].
Figure 2.
The mechanism of action for blinatumomab, the first-in-class bispecific T cell engager (BiTE), involves one arm binding to CD3 and the other to CD19. This interaction activates unstimulated T cells, initiating their attack on CD19+ cells.
Figure 2.
The mechanism of action for blinatumomab, the first-in-class bispecific T cell engager (BiTE), involves one arm binding to CD3 and the other to CD19. This interaction activates unstimulated T cells, initiating their attack on CD19+ cells.
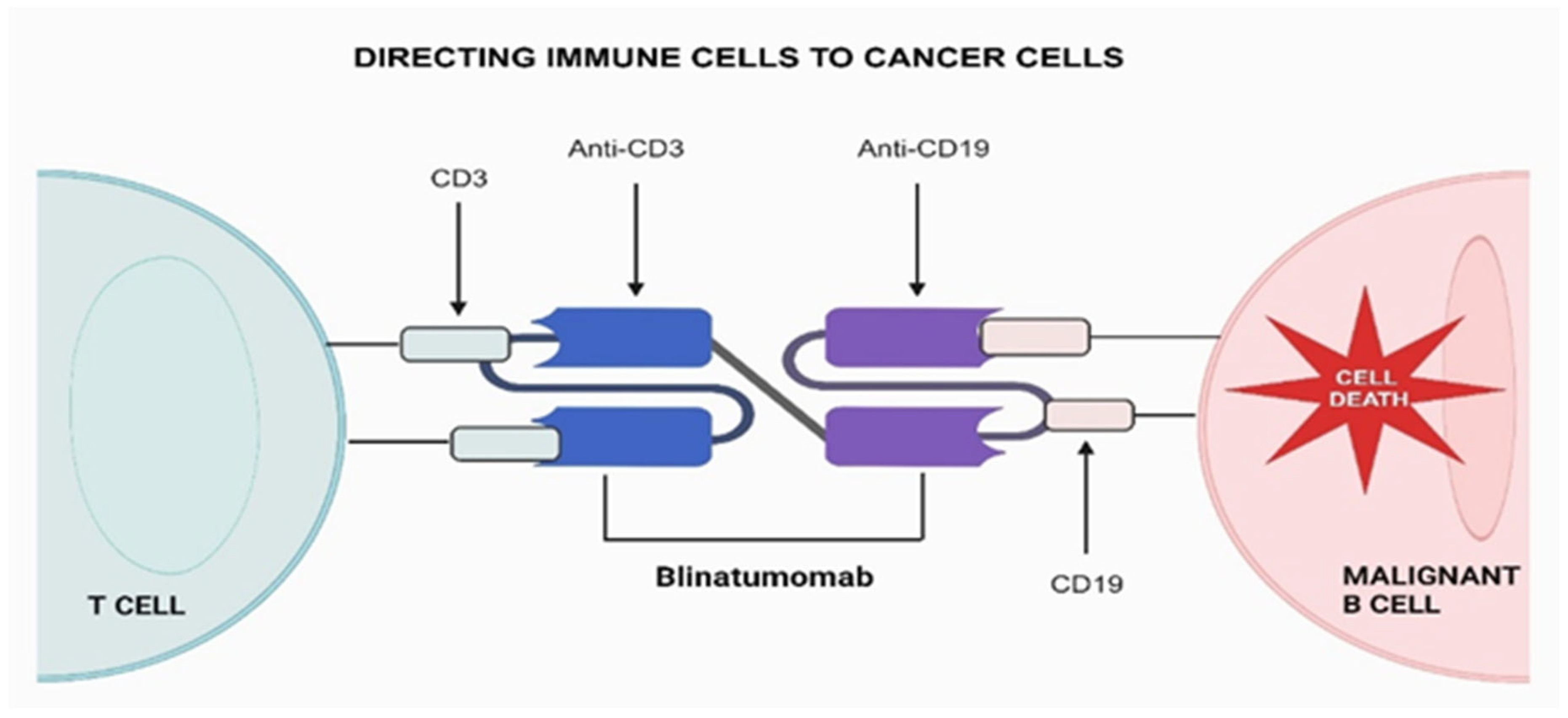
Although Blinatumomab has demonstrated significant success, crucial challenges persist. Factors such as rapid drug clearance, on-target/off-tumor adverse effects, cytokine release syndrome, and activation of peripheral immune cells may potentially limit therapeutic efficacy in both hematological malignancies [42]. Recent reports indicate instances of relapse among patients following Blinatumomab treatment, with the phenomenon associated not only with the loss of CD19 but also CD58, as proposed by Jabbour et al. [43]. Previous research has explored mechanisms contributing to CD19 escape, including CD19 mutations, CD19-mutant allele-specific expression, low CD19 RNA expression, and mutations in CD19 signaling member CD81 [44]. However, limited attention has been given to CD58 loss and its mechanism in the context of Blinatumomab treatment.
A recent study by Yizhen et al. has identified a crucial intrinsic factor, PAX5 mutation, significantly downregulating CD58. This downregulation has been linked to reduction in Blinatumomab activity, particularly observed in patients with acute lymphoblastic leukemia (ALL) [45]. Further research is needed to address the PAX5 mutation in ALL models under Blinatumomab treatment, providing a more comprehensive understanding of the role of PAX5 in CD58 loss. Moreover, additional studies have suggested regulatory T cells (Tregs) as potential regulators in the resistance process against Blinatumomab, indicating that multiple factors may contribute to resistance and a reduced response rate to this therapeutic approach [44]. These findings indicate the complexity of the mechanisms behind resistance to Blinatumomab, emphasizing the necessity for ongoing research to unravel these intricacies and ultimately pave the way for more effective and personalized treatment strategies.
Furthermore, the phenomenon of lineage switch represents a significant challenge associated with blinatumomab treatment, wherein refractory B lymphoblastic leukemia (B-ALL) can undergo a transition to acute myeloid leukemia (AML) [46,47,48]. This shift in lineage was initially documented by Stass and colleagues following standard chemotherapy for acute leukemia [49]. The occurrence of lineage switching has been observed not only in blinatumomab therapy but also in other immunotherapies, including CD19-specific chimeric antigen receptor (CAR) T cells [50]. It is particularly noteworthy that this switch occurs when CD19 B-cells acquire a distinct phenotype after the loss of CD19 [51,52,53]. While other several theories have been proposed to explain the mechanisms leading to lineage switch [54,55], the prevailing view suggests that the selective pressure resulting from CD19-directed therapy plays a crucial role in this phenotypic transition [56,57,58]. Studies on lineage switching highlight various rearrangements of the gene encoding histone–lysine N-methyl-transferase 2A (KMT2A, also known as mixed-lineage leukemia, MLL) as a key regulator of this switch [59,60,61]. The development of this immunophenotype is recognized as a critical factor contributing to relapses and resistance to several antibody-targeted therapies.
In the case of blinatumomab, five chromosomal rearrangements linked to lineage switch have been identified: KMT2A-AFF1 [62,63], KMT2A/AFF4 [58], BCR-ABL1 [64], hyperdiploidy [65], and KMT2A/EPS15 [66]. The t(4;11) (q21;q23) rearrangement with the KMT2A/AFF1 fusion protein is particularly common, especially in infants with ALL [67,68,69]. Lineage conversion has been observed in pediatric patients with ALL, impacting blinatumomab treatment monitoring. A switch from CD19-positive B-precursor ALL to CD19-negative AML has been documented following blinatumomab therapy [47]. Efforts to overcome this challenge include incorporating blinatumomab into the Interfant-06 backbone regimen. In an analysis of 30 infants with acute leukemia treated with standard chemotherapy and post-induction blinatumomab, no lineage switches were observed [citation needed]. Similarly, promising outcomes have been reported in infants with KMT2A-rearranged ALL, where the addition of blinatumomab to the Interfant-06 chemotherapy trial significantly improved the 2-year overall survival compared to the Interfant-06 alone [70]. It is essential to note that the follow-up time in these studies was relatively short, and longer-term monitoring is required to comprehensively evaluate the safety and efficacy of this combined therapy. Furthermore, blinatumomab has shown promise as an effective salvage therapy following anti-CD19-CAR-T failure, surpassing chemotherapy options. In R/R B-ALL patients, blinatumomab showed an improved complete remission rate, even in those expressing low CD19 levels [71]. However, inconsistent findings warrant further comparable studies to validate its potency as a rescue or pretreatment therapy, as some reports suggest prior blinatumomab treatment can maintain anti-CD19-CAR-T efficacy [72].
Immune Checkpoint Bispecific Antibodies
In cancer immunotherapy, the use of immune checkpoint inhibitors (ICIs) has been a major breakthrough, particularly when used as monotherapies [73,74]. These inhibitors tap into the potential of natural T cells that infiltrate tumors. Cancer cells often exploit immune checkpoints to avoid immune responses, and ICIs counteract this by blocking specific checkpoints [75,76]. Approvals of drugs like ipilimumab, pembrolizumab, and nivolumab signify significant strides in ICI development [73]. However, the effectiveness of single antibody targets against immune checkpoints and their ligands has shown limited impact, especially in treating "cold tumors" – tumors that hinder immune responses by preventing the infiltration of immune cells into the tumor [77,78]. Consequently, only a minimal fraction of the patient population has experienced significant benefits from ICI monotherapies.
Recent advancements in bispecific antibodies have addressed this limitation by focusing on the dual targeting of immune checkpoints, encompassing both receptors and ligands [79]. Notably, programmed death protein 1 (PD-1) and programmed cell death ligand 1 (PD-L1) checkpoint inhibitors have gained attention for their ability to restore T cells exhausted due to tumor-induced suppression [79]. PD-L1 and PD-L2, widely expressed ligands across various cancer types, have been a focus of study. PD-L2, known to bind PD-1 more strongly than PD-L1, presents an opportunity for more impactful outcomes when targeted [80,81]. In contrast to monospecific PD-1 and PD-L1 antibodies, bispecific antibodies targeting both PD-1 and PD-L1 have demonstrated powerful antitumor responses. LY3434172, a bispecific antibody co-targeting PD-1 and PD-L1, exhibited significant in vivo antitumor potency even at lower doses in preclinical studies, suggesting a synergistic effect and a distinctive pathway interaction in modulating immune responses [82].
Approximately 60% of cancers express both PD-L1 and PD-L2, while around 30% express either PD-L1 or PD-L2, expanding the binding effect and reducing off-target toxicities of bispecific antibody constructs [83]. Ongoing studies are exploring dual-specific antibodies to co-target stromal cells, T regulatory cells (Tregs), and myofibroblasts in the tumor microenvironment, facilitating the influx of T cells into poorly infiltrated tumors [84]. Emerging strategies aim to target specific surface proteins, including PD-L1/PD-L2, CD25/CTLA-4, PD-L1/ICOS, PD-1/CD47, and PD-L1/T cell immunoreceptor with Ig and ITIM domains (TIGIT) (Table 1) [85,86]. For instance, dual-specific monoclonal antibodies designed to bind PD-L1 and PD-L2 have demonstrated enhanced immune-driven anti-tumor activity [87]. In the context of treating HER2-positive solid tumors, a bispecific combination of PD1 and HER2 exhibited high effectiveness in killing HER2-positive tumor cells through antibody-dependent cellular cytotoxicity [88].
Undoubtedly, bispecific antibodies tailored against PD-L1 and PD-L2 play a pivotal role in facilitating the migration of host immune responses to tumor cells, thereby enhancing antitumor responses. The targeting of PD-L1 in dual antibody regimens has demonstrated effectiveness in various settings of human tumors, as evidenced by the numerous ongoing clinical trials exploring PD1/PDL1 combination regimens [89].

Table 2.
Studies investigating the efficacy of PD1/PDL1 combination regimens in patients with advanced solid tumors (Clinical trials are registered at clinicaltrials.gov).
Table 2.
Studies investigating the efficacy of PD1/PDL1 combination regimens in patients with advanced solid tumors (Clinical trials are registered at clinicaltrials.gov).
| Target | Name | Condition | Status | Phase | NCT ID |
| PD-L1 and TGF-β | SHR-1701 | Advanced solid tumors | Unknown | Phase I | NCT03710265 |
| CTLA-4×PD-L1 | KN064 | Advanced Solid Tumors | Completed | Phase 1 | NCT03733951 |
| PD-1 and CTLA-4 | MEDI5752 | Advanced solid tumors | Recruiting | Phase I | NCT03530397 |
| MGD019 | Advanced solid tumors | Active, not recruiting | Phase 1 | NCT03761017 | |
| AK104 | Hepatocellular carcinoma | Recruiting | Phase I/II | NCT04444167 | |
| COMPASSION-03 | Advanced solid tumors | Active, not recruiting | Phase I/II | NCT03852251 | |
| LAG-3 × PD-L1 | ABL501 | Advanced solid tumors | Recruiting | Phase I | NCT05101109 |
| FS118 | Advanced solid tumors | Active, not recruiting | Phase I/II | NCT03440437 | |
| AK104 | NSCLC | Active, not recruiting | Phase I/II | NCT04646330 | |
| LAG-3 × PD-1 | MGD013 | Advanced liver cancer | Terminated | Phase I/II | NCT04212221 |
| RG6139 | Advanced solid tumors | Recruiting | Phase I/II | NCT04140500 | |
| Not Given | Advanced solid tumors | Recruiting | Phase I | NCT05577182 | |
| TIM-3 × PD-L1 | LY3415244 | Advanced solid tumors | Terminated | Phase I | NCT03752177 |
| ABL501 | Advanced solid tumors | Recruiting | Phase I | NCT05101109 | |
| TIGIT×PD-L1 | HLX301 | Advanced solid tumors | Recruiting | Phase I/II | NCT05102214 |
| TIGIT×PD-1 | ARTEMIDE-01 | Advanced NSCLC | Recruiting | Phase I/II | NCT04995523 |
| LB1410 | Advanced Solid Tumor | Recruiting | Phase I | NCT05357651 | |
| TIM-3 × PD-1 | AZD7789 | Lymphoma | Recruiting | Phase I/II | NCT04931654 |
| RG7769 | Advanced Solid Cancer | Recruiting | Phase I | NCT03708328 | |
| Lomvastomig | Advanced Solid Cancer | Active, not recruiting | Phase II | NCT04785820 | |
| Tobemstomig | Non-small Cell Lung Cancer | Recruiting | Phase II | NCT05775289 | |
| 4-1BB×PD-L1 | ABL503 | Advanced Solid Cancer | Recruiting | Phase I | NCT04762641 |
| PRS-344 | Advanced Solid Cancer | Recruiting | Phase I/II | NCT05159388 | |
| GEN1046 | Advanced Solid Cancer | Recruiting | Phase I/II | NCT03917381 | |
| CD27×PD-L1 | CDX-527 | Advanced Solid Cancer | Completed | Phase I | NCT04440943 |
| PD-L1 and CD137 | MCLA-145 | Advanced Solid Cancer | Recruiting | Phase I | NCT03922204 |
| AP203 | Advanced Solid Cancer | Not yet recruiting | Phase I/II | NCT05473156 | |
| FS222 | Advanced Solid Cancer | Recruiting | Phase I | NCT04740424 | |
| PD-L1 and VEGF | PM8002 | Advanced Solid Cancer | Recruiting | Phase II | NCT05879055 |
| HB0025 | Advanced Solid Cancer | Recruiting | Phase I | NCT04678908 | |
| IMM2510 | Advanced Solid Cancer | Recruiting | Phase I | NCT05972460 | |
| PD-1/ VEGF | AK112 | NSCLC | Recruiting | Phase II | NCT04736823 |
Future Directions
Looking ahead, the success of Bispecific Antibodies (BsAbs) in effectively treating hematological malignancies is evident with FDA approvals. However, it's noteworthy that there is currently no FDA-approved BiTE molecule for addressing solid malignancies. Ongoing initiatives are exploring innovative approaches, such as incorporating masks linked through protease-cleavable linkers into first-generation TCEs, including Conditional Bispecific Redirected Activation, Probody TCB, and precision-activated TCEs. These attempts aim to enhance the therapeutic efficacy of bispecific T-cell engagers in treating solid tumors.
In addressing complications like cytokine release syndrome (CRS) associated with BsAbs therapy, future research is focused on optimizing design to trigger immunological responses exclusively towards tumors. Unlike previous designs involving a single BsAb agent, emerging strategies adopt a unique approach by employing two Bispecific Antibodies (BsAbs) components. Each component features a split anti-CD3 paratope and a binding moiety for a tumor antigen. These advancements signify a promising direction in the evolution of bispecific T-cell engagers for more effective and targeted treatments of solid tumors.
Conclusions
The field of bispecific antibodies (BsAbs), particularly exemplified by bispecific T cell–engaging therapies, has witnessed remarkable strides in cancer immunotherapy, and appears superior to conventional chemotherapy, in at least hematological malignancy settings. The clinical success of over 100 evaluated bsAbs, with seven BiTE approved for market use, highlights their remarkable achievements. However, challenges such as rapid drug clearance, off-target effects, and cytokine release syndrome persist, limiting their widespread application. Despite, innovative modifications, including avidity-mediated specificity, paratope masking and two BsAbs system hold promise in addressing on-target/off-tumor adverse effects. Moreover, immune checkpoint bispecific antibodies, co-targeting receptors and ligands like PD-1 and PD-L1, present a paradigm shift in cancer immunotherapy, offering enhanced antitumor responses. The evolving landscape of bispecific immunotherapeutic holds great potential in advancing personalized and effective cancer treatments, emphasizing the need for ongoing research and development to overcome existing challenges and broaden therapeutic applications.
References
- Salvaris, R., J. Ong, and G. P. Gregory. 2021. "Bispecific Antibodies: A Review of Development, Clinical Efficacy and Toxicity in B-Cell Lymphomas." J Pers Med 11 (5). [CrossRef]
- Henricks, L. M., J. H. Schellens, A. D. Huitema, and J. H. Beijnen. 2015. "The use of combinations of monoclonal antibodies in clinical oncology." Cancer Treat Rev 41 (10): 859-67. [CrossRef]
- Han, Y., D. Liu, and L. Li. 2020a. "PD-1/PD-L1 pathway: current researches in cancer." Am J Cancer Res 10 (3): 727-742.
- Ma, J., Y. Mo, M. Tang, J. Shen, Y. Qi, W. Zhao, Y. Huang, Y. Xu, and C. Qian. 2021. "Bispecific Antibodies: From Research to Clinical Application." Front Immunol 12: 626616. [CrossRef]
- Acheampong, D. O., C. K. Adokoh, P. Ampomah, D. S. Agyirifor, I. Dadzie, F. A. Ackah, and E. A. Asiamah. 2017. "Bispecific Antibodies (bsAbs): Promising Immunotherapeutic Agents for Cancer Therapy." Protein Pept Lett 24 (5): 456-465. [CrossRef]
- Ma, Y., J. Xue, Y. Zhao, Y. Zhang, Y. Huang, Y. Yang, W. Fang, Y. Guo, Q. Li, X. Ge, J. Sun, B. Zhang, J. Xiao, L. Zhang, and H. Zhao. 2023. "Phase I trial of KN046, a novel bispecific antibody targeting PD-L1 and CTLA-4 in patients with advanced solid tumors." J Immunother Cancer 11 (6). [CrossRef]
- Wu, Y., M. Yi, S. Zhu, H. Wang, and K. Wu. 2021. "Recent advances and challenges of bispecific antibodies in solid tumors." Exp Hematol Oncol 10 (1): 56. [CrossRef]
- Brinkmann, U., and R. E. Kontermann. 2021. "Bispecific antibodies." Science 372 (6545): 916-917. [CrossRef]
- Moreau, P., A. L. Garfall, N. W. C.J van de Donk, H. Nahi, J. F. San-Miguel, A. Oriol, A. K. Nooka, T. Martin, L. Rosinol, A. Chari, L. Karlin, L. Benboubker, M. V. Mateos, N. Bahlis, R. Popat, B. Besemer, J. Martínez-López, S. Sidana, M. Delforge, L. Pei, D. Trancucci, R. Verona, S. Girgis, S. X. W. Lin, Y. Olyslager, M. Jaffe, C. Uhlar, T. Stephenson, R. Van Rampelbergh, A. Banerjee, J. D. Goldberg, R. Kobos, A. Krishnan, and S. Z. Usmani. 2022. "Teclistamab in Relapsed or Refractory Multiple Myeloma." N Engl J Med 387 (6): 495-505. [CrossRef]
- Lesokhin, A. M., M. H. Tomasson, B. Arnulf, N. J. Bahlis, H. Miles Prince, R. Niesvizky, P. Rodrίguez-Otero, J. Martinez-Lopez, G. Koehne, C. Touzeau, Y. Jethava, H. Quach, J. Depaus, H. Yokoyama, A. E. Gabayan, D. A. Stevens, A. K. Nooka, S. Manier, N. Raje, S. Iida, M. S. Raab, E. Searle, E. Leip, S. T. Sullivan, U. Conte, M. Elmeliegy, A. Czibere, A. Viqueira, and M. Mohty. 2023. "Elranatamab in relapsed or refractory multiple myeloma: phase 2 MagnetisMM-3 trial results." Nat Med 29 (9): 2259-2267. [CrossRef]
- Keam, S. J. 2023. "Talquetamab: First Approval." Drugs 83 (15): 1439-1445. [CrossRef]
- Scott, L. J., and E. S. Kim. 2018. "Emicizumab-kxwh: First Global Approval." Drugs 78 (2): 269-274. [CrossRef]
- Shirley, M. 2022. "Faricimab: First Approval." Drugs 82 (7): 825-830. [CrossRef]
- Wu, B., R. Jug, C. Luedke, P. Su, C. Rehder, C. McCall, A. S. Lagoo, and E. Wang. 2017. "Lineage Switch Between B-Lymphoblastic Leukemia and Acute Myeloid Leukemia Intermediated by "Occult" Myelodysplastic Neoplasm: Two Cases of Adult Patients With Evidence of Genomic Instability and Clonal Selection by Chemotherapy." Am J Clin Pathol 148 (2): 136-147. [CrossRef]
- Li, H., P. Er Saw, and E. Song. 2020. "Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics." Cell Mol Immunol 17 (5): 451-461. [CrossRef]
- Underwood, D. J., J. Bettencourt, and Z. Jawad. 2022. "The manufacturing considerations of bispecific antibodies." Expert Opin Biol Ther 22 (8): 1043-1065. [CrossRef]
- Zhang, T., Y. Lin, and Q. Gao. 2023. "Bispecific antibodies targeting immunomodulatory checkpoints for cancer therapy." Cancer Biol Med 20 (3): 181-95. [CrossRef]
- Yu, G. H., A. M. Li, X. Li, Z. Yang, and H. Peng. 2017. "Bispecific antibody suppresses osteosarcoma aggressiveness through regulation of NF-κB signaling pathway." Tumour Biol 39 (6): 1010428317705572. [CrossRef]
- Zhong, Z., M. Zhang, Y. Ning, G. Mao, X. Li, Q. Deng, X. Chen, D. Zuo, X. Zhao, E. Xie, H. Wang, L. Guo, B. Li, K. Xiao, and X. He. 2022. "Development of a bispecific antibody targeting PD-L1 and TIGIT with optimal cytotoxicity." Sci Rep 12 (1): 18011. [CrossRef]
- Esfandiari, A., S. Cassidy, and R. M. Webster. 2022. "Bispecific antibodies in oncology." Nat Rev Drug Discov 21 (6): 411-412. [CrossRef]
- Löffler, A., P. Kufer, R. Lutterbüse, F. Zettl, P. T. Daniel, J. M. Schwenkenbecher, G. Riethmüller, B. Dörken, and R. C. Bargou. 2000. "A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes." Blood 95 (6): 2098-103.
- Emmanuel Owusu Ansah, Andy Baah, Emmanuel Boateng Agyenim, "Vaccine Boosting CAR-T Cell Therapy: Current and Future Strategies", Advances in Cell and Gene Therapy, vol. 2023, Article ID 8030440, 9 pages, 2023. [CrossRef]
- Viardot, A., M. E. Goebeler, G. Hess, S. Neumann, M. Pfreundschuh, N. Adrian, F. Zettl, M. Libicher, C. Sayehli, J. Stieglmaier, A. Zhang, D. Nagorsen, and R. C. Bargou. 2016. "Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma." Blood 127 (11): 1410-6. https://www.ncbi.nlm.nih.gov/pubmed/26755709. [CrossRef]
- Simão, D. C., K. K. Zarrabi, J. L. Mendes, R. Luz, J. A. Garcia, W. K. Kelly, and P. C. Barata. 2023. "Bispecific T-Cell Engagers Therapies in Solid Tumors: Focusing on Prostate Cancer." Cancers (Basel) 15 (5). [CrossRef]
- Subklewe, M. 2021. "BiTEs better than CAR T cells." Blood Adv 5 (2): 607-612. [CrossRef]
- Buzzetti, M., and M. Gerlinger. 2023. "Assessing the toxicity of bispecific antibodies." Nat Biomed Eng. [CrossRef]
- Wang, D. R., X. L. Wu, and Y. L. Sun. 2022. "Therapeutic targets and biomarkers of tumor immunotherapy: response versus non-response." Signal Transduct Target Ther 7 (1): 331. https://www.ncbi.nlm.nih.gov/pubmed/36123348.
- Li, J., R. Piskol, R. Ybarra, Y. J. Chen, D. Slaga, M. Hristopoulos, R. Clark, Z. Modrusan, K. Totpal, M. R. Junttila, and T. T. Junttila. 2019. "CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity." Sci Transl Med 11 (508). [CrossRef]
- Wang, K., G. Wei, and D. Liu. 2012. "CD19: a biomarker for B cell development, lymphoma diagnosis and therapy." Exp Hematol Oncol 1 (1): 36. [CrossRef]
- Shimabukuro-Vornhagen, A., P. Gödel, M. Subklewe, H. J. Stemmler, H. A. Schlößer, M. Schlaak, M. Kochanek, B. Böll, and M. S. von Bergwelt-Baildon. 2018. "Cytokine release syndrome." J Immunother Cancer 6 (1): 56. [CrossRef]
- Slaga, D., D. Ellerman, T. N. Lombana, R. Vij, J. Li, M. Hristopoulos, R. Clark, J. Johnston, A. Shelton, E. Mai, K. Gadkar, A. A. Lo, J. T. Koerber, K. Totpal, R. Prell, G. Lee, C. Spiess, and T. T. Junttila. 2018. "Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3." Sci Transl Med 10 (463). [CrossRef]
- Singh, A., S. Dees, and I. S. Grewal. 2021. "Overcoming the challenges associated with CD3+ T-cell redirection in cancer." Br J Cancer 124 (6): 1037-1048. [CrossRef]
- Bacac, M., T. Fauti, J. Sam, S. Colombetti, T. Weinzierl, D. Ouaret, W. Bodmer, S. Lehmann, T. Hofer, R. J. Hosse, E. Moessner, O. Ast, P. Bruenker, S. Grau-Richards, T. Schaller, A. Seidl, C. Gerdes, M. Perro, V. Nicolini, N. Steinhoff, S. Dudal, S. Neumann, T. von Hirschheydt, C. Jaeger, J. Saro, V. Karanikas, C. Klein, and P. Umaña. 2016a. "A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors." Clin Cancer Res 22 (13): 3286-97. [CrossRef]
- Zhu, X. Y., Q. X. Li, Y. Kong, K. K. Huang, G. Wang, Y. J. Wang, J. Lu, G. Q. Hua, Y. L. Wu, and T. L. Ying. 2023. "A novel human single-domain antibody-drug conjugate targeting CEACAM5 exhibits potent in vitro and in vivo antitumor activity." Acta Pharmacol Sin. [CrossRef]
- Simão, D. C., K. K. Zarrabi, J. L. Mendes, R. Luz, J. A. Garcia, W. K. Kelly, and P. C. Barata. 2023a. "Bispecific T-Cell Engagers Therapies in Solid Tumors: Focusing on Prostate Cancer." Cancers (Basel) 15 (5). [CrossRef]
- Oates, J., N. J. Hassan, and B. K. Jakobsen. 2015. "ImmTACs for targeted cancer therapy: Why, what, how, and which." Mol Immunol 67 (2 Pt A): 67-74. [CrossRef]
- Howlett, S., T. J. Carter, H. M. Shaw, and P. D. Nathan. 2023. "Tebentafusp: a first-in-class treatment for metastatic uveal melanoma." Ther Adv Med Oncol 15: 17588359231160140. [CrossRef]
- Pulte, E. D., J. Vallejo, D. Przepiorka, L. Nie, A. T. Farrell, K. B. Goldberg, A. E. McKee, and R. Pazdur. 2018a. "FDA Supplemental Approval: Blinatumomab for Treatment of Relapsed and Refractory Precursor B-Cell Acute Lymphoblastic Leukemia." Oncologist 23 (11): 1366-1371. https://www.ncbi.nlm.nih.gov/pubmed/30018129. [CrossRef]
- Mocquot, P., Y. Mossazadeh, L. Lapierre, F. Pineau, and F. Despas. 2022. "The pharmacology of blinatumomab: state of the art on pharmacodynamics, pharmacokinetics, adverse drug reactions and evaluation in clinical trials." J Clin Pharm Ther 47 (9): 1337-1351. [CrossRef]
- Boissel, N., S. Chiaretti, C. Papayannidis, J. M. Ribera, R. Bassan, A. N. Sokolov, N. Alam, A. Brescianini, I. Pezzani, G. Kreuzbauer, G. Zugmaier, R. Foà, and A. Rambaldi. 2023. "Real-world use of blinatumomab in adult patients with B-cell acute lymphoblastic leukemia in clinical practice: results from the NEUF study." Blood Cancer J 13 (1): 2. [CrossRef]
- Burt, R., D. Warcel, and A. K. Fielding. 2019. "Blinatumomab, a bispecific B-cell and T-cell engaging antibody, in the treatment of B-cell malignancies." Hum Vaccin Immunother 15 (3): 594-602. [CrossRef]
- Zhou, S., M. Liu, F. Ren, X. Meng, and J. Yu. 2021. "The landscape of bispecific T cell engager in cancer treatment." Biomark Res 9 (1): 38. [CrossRef]
- Jabbour, E., and H. Kantarjian. 2016. "Chemoimmunotherapy as a new standard of care for Burkitt leukaemia/lymphoma." Lancet 387 (10036): 2360-1. [CrossRef]
- Zhao, Y., I. Aldoss, C. Qu, J. C. Crawford, Z. Gu, E. K. Allen, A. E. Zamora, T. B. Alexander, J. Wang, H. Goto, T. Imamura, K. Akahane, G. Marcucci, A. S. Stein, R. Bhatia, P. G. Thomas, S. J. Forman, C. G. Mullighan, and K. G. Roberts. 2021a. "Tumor-intrinsic and -extrinsic determinants of response to blinatumomab in adults with B-ALL." Blood 137 (4): 471-484. https://www.ncbi.nlm.nih.gov/pubmed/32881995. [CrossRef]
- Li, Y., T. Moriyama, S. Yoshimura, X. Zhao, Z. Li, X. Yang, E. Paietta, M. R. Litzow, M. Konopleva, J. Yu, H. Inaba, R. C. Ribeiro, C. H. Pui, and J. J. Yang. 2022. "PAX5 epigenetically orchestrates CD58 transcription and modulates blinatumomab response in acute lymphoblastic leukemia." Sci Adv 8 (50): eadd6403. [CrossRef]
- Haddox, C. L., A. A. Mangaonkar, D. Chen, M. Shi, R. He, J. L. Oliveira, M. R. Litzow, A. Al-Kali, W. J. Hogan, and M. A. Elliott. 2017. "Blinatumomab-induced lineage switch of B-ALL with t(4:11)(q21;q23) KMT2A/AFF1 into an aggressive AML: pre- and post-switch phenotypic, cytogenetic and molecular analysis." Blood Cancer J 7 (9): e607. [CrossRef]
- Rayes, A., R. L. McMasters, and M. M. O'Brien. 2016. "Lineage Switch in MLL-Rearranged Infant Leukemia Following CD19-Directed Therapy." Pediatr Blood Cancer 63 (6): 1113-5. [CrossRef]
- Jacoby, E., S. M. Nguyen, T. J. Fountaine, K. Welp, B. Gryder, H. Qin, Y. Yang, C. D. Chien, A. E. Seif, H. Lei, Y. K. Song, J. Khan, D. W. Lee, C. L. Mackall, R. A. Gardner, M. C. Jensen, J. F. Shern, and T. J. Fry. 2016. "CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity." Nat Commun 7: 12320. [CrossRef]
- Stass, S., J. Mirro, S. Melvin, C. H. Pui, S. B. Murphy, and D. Williams. 1984. "Lineage switch in acute leukemia." Blood 64 (3): 701-6.
- Perna, F., and M. Sadelain. 2016. "Myeloid leukemia switch as immune escape from CD19 chimeric antigen receptor (CAR) therapy." Transl Cancer Res 5 (Suppl 2): S221-S225. [CrossRef]
- Aldulescu, M., K. Leuer, L. J. Jennings, K. L. Yap, and S. Gong. 2023. "Lineage switch from acute myeloid leukemia to B-lymphoblastic lymphoma with an acquired PIK3R1 loss-of-function mutation." Am J Hematol 98 (1): E1-E3. [CrossRef]
- Dorantes-Acosta, E., and R. Pelayo. 2012a. "Lineage switching in acute leukemias: a consequence of stem cell plasticity?" Bone Marrow Res 2012: 406796. [CrossRef]
- Yang, W., S. Xie, Y. Li, J. Wang, J. Xiao, K. Huang, X. Wang, Y. Wu, L. Ma, and D. Nie. 2022. "Lineage switch from lymphoma to myeloid neoplasms: First case series from a single institution." Open Med (Wars) 17 (1): 1466-1472. [CrossRef]
- Pui, C. H., S. C. Raimondi, F. G. Behm, J. Ochs, W. L. Furman, N. J. Bunin, R. C. Ribeiro, P. A. Tinsley, and J. Mirro. 1986. "Shifts in blast cell phenotype and karyotype at relapse of childhood lymphoblastic leukemia." Blood 68 (6): 1306-10.
- Zuna, J., H. Cavé, C. Eckert, T. Szczepanski, C. Meyer, E. Mejstrikova, E. Fronkova, K. Muzikova, E. Clappier, D. Mendelova, P. Boutard, A. Schrauder, J. Sterba, R. Marschalek, J. J. van Dongen, O. Hrusak, J. Stary, and J. Trka. 2007. "Childhood secondary ALL after ALL treatment." Leukemia 21 (7): 1431-5. [CrossRef]
- Li, L. Z., Q. Sun, Y. Fang, L. J. Yang, Z. Y. Xu, J. H. Hu, L. Cao, J. Y. Huang, M. Hong, J. Y. Li, and S. X. Qian. 2020. "A report on Lineage switch at relapse of CD19 CAR-T therapy for Philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia." Chin Med J (Engl) 133 (16): 2001-2003. [CrossRef]
- Rayes, A., R. L. McMasters, and M. M. O'Brien. 2016. "Lineage Switch in MLL-Rearranged Infant Leukemia Following CD19-Directed Therapy." Pediatr Blood Cancer 63 (6): 1113-5. [CrossRef]
- Wölfl, M., M. Rasche, M. Eyrich, R. Schmid, D. Reinhardt, and P. G. Schlegel. 2018. "Spontaneous reversion of a lineage switch following an initial blinatumomab-induced ALL-to-AML switch in." Blood Adv 2 (12): 1382-1385. [CrossRef]
- Iacobucci, I., and C. G. Mullighan. 2022. "KMT2A-rearranged leukemia: the shapeshifter." Blood 140 (17): 1833-1835. [CrossRef]
- Shimony, S., and M. R. Luskin. 2023. "Unraveling KMT2A-rearranged ALL." Blood 142 (21): 1764-1766. [CrossRef]
- Meyer, C., P. Larghero, B. Almeida Lopes, T. Burmeister, D. Gröger, R. Sutton, N. C. Venn, G. Cazzaniga, L. Corral Abascal, G. Tsaur, L. Fechina, M. Emerenciano, M. S. Pombo-de-Oliveira, T. Lund-Aho, T. Lundán, M. Montonen, V. Juvonen, J. Zuna, J. Trka, P. Ballerini, H. Lapillonne, V. H. J. Van der Velden, E. Sonneveld, E. Delabesse, R. R. C. de Matos, M. L. M. Silva, S. Bomken, K. Katsibardi, M. Keernik, N. Grardel, J. Mason, R. Price, J. Kim, C. Eckert, L. Lo Nigro, C. Bueno, P. Menendez, U. Zur Stadt, P. Gameiro, L. Sedék, T. Szczepański, A. Bidet, V. Marcu, K. Shichrur, S. Izraeli, H. O. Madsen, B. W. Schäfer, S. Kubetzko, R. Kim, E. Clappier, H. Trautmann, M. Brüggemann, P. Archer, J. Hancock, J. Alten, A. Möricke, M. Stanulla, J. Lentes, A. K. Bergmann, S. Strehl, S. Köhrer, K. Nebral, M. N. Dworzak, O. A. Haas, C. Arfeuille, A. Caye-Eude, H. Cavé, and R. Marschalek. 2023. "The KMT2A recombinome of acute leukemias in 2023." Leukemia 37 (5): 988-1005. [CrossRef]
- He, R. R., Z. Nayer, M. Hogan, R. S. Cuevo, K. Woodward, D. Heyer, C. A. Curtis, and J. F. Peterson. 2019. "Immunotherapy- (Blinatumomab-) Related Lineage Switch of." Case Rep Hematol 2019: 7394619. [CrossRef]
- Fournier, E., L. Inchiappa, C. Delattre, J. M. Pignon, F. Danicourt, M. Bemba, C. Roche-Lestienne, A. Daudignon, G. Decool, C. Roumier, F. Dumezy, L. Fournier, N. Grardel, C. Preudhomme, and N. Duployez. 2019. "Increased risk of adverse acute myeloid leukemia after anti-CD19-targeted immunotherapies in." Leuk Lymphoma 60 (7): 1827-1830. [CrossRef]
- Nagel, I., M. Bartels, J. Duell, H. H. Oberg, S. Ussat, H. Bruckmueller, O. Ottmann, H. Pfeifer, H. Trautmann, N. Gökbuget, A. Caliebe, D. Kabelitz, M. Kneba, H. A. Horst, D. Hoelzer, M. S. Topp, I. Cascorbi, R. Siebert, and M. Brüggemann. 2017. "Hematopoietic stem cell involvement in." Blood 130 (18): 2027-2031. [CrossRef]
- Zoghbi, A., U. Zur Stadt, B. Winkler, I. Müller, and G. Escherich. 2017. "Lineage switch under blinatumomab treatment of relapsed common acute lymphoblastic leukemia without MLL rearrangement." Pediatr Blood Cancer 64 (11). [CrossRef]
- Du, J., K. M. Chisholm, K. Tsuchiya, K. Leger, B. M. Lee, J. C. Rutledge, C. R. Paschal, C. Summers, and M. Xu. 2021. "Lineage Switch in an Infant B-Lymphoblastic Leukemia With t(1;11)(p32;q23);." Pediatr Dev Pathol 24 (4): 378-382. [CrossRef]
- Meyer, C., J. Hofmann, T. Burmeister, D. Gröger, T. S. Park, M. Emerenciano, M. Pombo de Oliveira, A. Renneville, P. Villarese, E. Macintyre, H. Cavé, E. Clappier, K. Mass-Malo, J. Zuna, J. Trka, E. De Braekeleer, M. De Braekeleer, S. H. Oh, G. Tsaur, L. Fechina, V. H. van der Velden, J. J. van Dongen, E. Delabesse, R. Binato, M. L. Silva, A. Kustanovich, O. Aleinikova, M. H. Harris, T. Lund-Aho, V. Juvonen, O. Heidenreich, J. Vormoor, W. W. Choi, M. Jarosova, A. Kolenova, C. Bueno, P. Menendez, S. Wehner, C. Eckert, P. Talmant, S. Tondeur, E. Lippert, E. Launay, C. Henry, P. Ballerini, H. Lapillone, M. B. Callanan, J. M. Cayuela, C. Herbaux, G. Cazzaniga, P. M. Kakadiya, S. Bohlander, M. Ahlmann, J. R. Choi, P. Gameiro, D. S. Lee, J. Krauter, P. Cornillet-Lefebvre, G. Te Kronnie, B. W. Schäfer, S. Kubetzko, C. N. Alonso, U. zur Stadt, R. Sutton, N. C. Venn, S. Izraeli, L. Trakhtenbrot, H. O. Madsen, P. Archer, J. Hancock, N. Cerveira, M. R. Teixeira, L. Lo Nigro, A. Möricke, M. Stanulla, M. Schrappe, L. Sedék, T. Szczepański, C. M. Zwaan, E. A. Coenen, M. M. van den Heuvel-Eibrink, S. Strehl, M. Dworzak, R. Panzer-Grümayer, T. Dingermann, T. Klingebiel, and R. Marschalek. 2013. "The MLL recombinome of acute leukemias in 2013." Leukemia 27 (11): 2165-76. [CrossRef]
- Piciocchi, A., M. Messina, L. Elia, A. Vitale, S. Soddu, A. M. Testi, S. Chiaretti, M. Mancini, F. Albano, A. Spadano, M. Krampera, M. Bonifacio, R. Cairoli, C. Vetro, F. Colella, F. Ferrara, G. Cimino, R. Bassan, P. Fazi, and M. Vignetti. 2021. "Prognostic impact of KMT2A-AFF1-positivity in 926 BCR-ABL1-negative B-lineage acute lymphoblastic leukemia patients treated in GIMEMA clinical trials since 1996." Am J Hematol 96 (9): E334-E338. [CrossRef]
- Richard-Carpentier, G., H. M. Kantarjian, G. Tang, C. C. Yin, J. D. Khoury, G. C. Issa, F. Haddad, N. Jain, F. Ravandi, N. J. Short, C. D. DiNardo, K. Takahashi, M. Y. Konopleva, N. G. Daver, T. Kadia, G. Garcia-Manero, R. Garris, S. O'Brien, and E. Jabbour. 2021. "Outcomes of acute lymphoblastic leukemia with KMT2A (MLL) rearrangement: the MD Anderson experience." Blood Adv 5 (23): 5415-5419. [CrossRef]
- van der Sluis, I. M., P. de Lorenzo, R. S. Kotecha, A. Attarbaschi, G. Escherich, K. Nysom, J. Stary, A. Ferster, B. Brethon, F. Locatelli, M. Schrappe, P. E. Scholte-van Houtem, M. G. Valsecchi, and R. Pieters. 2023. "Blinatumomab Added to Chemotherapy in Infant Lymphoblastic Leukemia." N Engl J Med 388 (17): 1572-1581. [CrossRef]
- Qi, Y., H. Liu, X. Li, Y. Shi, J. Mu, J. Li, Y. Wang, and Q. Deng. 2023. "Blinatumomab as salvage therapy in patients with relapsed/refractory B-ALL who have failed/progressed after anti-CD19-CAR T therapy." Ann Med 55 (1): 2230888. [CrossRef]
- Shah, B. D., M. R. Bishop, O. O. Oluwole, A. C. Logan, M. R. Baer, W. B. Donnellan, K. M. O'Dwyer, H. Holmes, M. L. Arellano, A. Ghobadi, J. M. Pagel, Y. Lin, R. D. Cassaday, J. H. Park, M. Abedi, J. E. Castro, D. J. DeAngelo, A. K. Malone, R. Mawad, G. J. Schiller, J. M. Rossi, A. Bot, T. Shen, L. Goyal, R. K. Jain, R. Vezan, and W. G. Wierda. 2021. "KTE-X19 anti-CD19 CAR T-cell therapy in adult relapsed/refractory acute lymphoblastic leukemia: ZUMA-3 phase 1 results." Blood 138 (1): 11-22. [CrossRef]
- Lee, J. B., H. R. Kim, and S. J. Ha. 2022. "Immune Checkpoint Inhibitors in 10 Years: Contribution of Basic Research and Clinical Application in Cancer Immunotherapy." Immune Netw 22 (1): e2. [CrossRef]
- Razaghi, A., M. Durand-Dubief, N. Brusselaers, and M. Björnstedt. 2023. "Combining PD-1/PD-L1 blockade with type I interferon in cancer therapy." Front Immunol 14: 1249330. [CrossRef]
- Meybodi, S. M., B. Farasati Far, A. Pourmolaei, F. Baradarbarjastehbaf, M. Safaei, N. Mohammadkhani, and A. A. Samadani. 2023. "Immune checkpoint inhibitors promising role in cancer therapy: clinical evidence and immune-related adverse events." Med Oncol 40 (8): 243. [CrossRef]
- Wang, Y., S. Yang, L. Wan, W. Ling, H. Chen, and J. Wang. 2023. "New developments in the mechanism and application of immune checkpoint inhibitors in cancer therapy (Review)." Int J Oncol 63 (1). [CrossRef]
- Bonaventura, P., T. Shekarian, V. Alcazer, J. Valladeau-Guilemond, S. Valsesia-Wittmann, S. Amigorena, C. Caux, and S. Depil. 2019. "Cold Tumors: A Therapeutic Challenge for Immunotherapy." Front Immunol 10: 168. [CrossRef]
- Wang, L., H. Geng, Y. Liu, L. Liu, Y. Chen, F. Wu, Z. Liu, S. Ling, Y. Wang, and L. Zhou. 2023. "Hot and cold tumors: Immunological features and the therapeutic strategies." MedComm (2020) 4 (5): e343. [CrossRef]
- Blanco, B., C. Domínguez-Alonso, and L. Alvarez-Vallina. 2021. "Bispecific Immunomodulatory Antibodies for Cancer Immunotherapy." Clin Cancer Res 27 (20): 5457-5464. [CrossRef]
- Wang, Y., J. Du, Z. Gao, H. Sun, M. Mei, Y. Ren, and X. Zhou. 2023. "Evolving landscape of PD-L2: bring new light to checkpoint immunotherapy." Br J Cancer 128 (7): 1196-1207. [CrossRef]
- Takehara, T., E. Wakamatsu, H. Machiyama, W. Nishi, K. Emoto, M. Azuma, K. Soejima, K. Fukunaga, and T. Yokosuka. 2021. "PD-L2 suppresses T cell signaling via coinhibitory microcluster formation and SHP2 phosphatase recruitment." Commun Biol 4 (1): 581. [CrossRef]
- Kotanides, H., Y. Li, M. Malabunga, C. Carpenito, S. W. Eastman, Y. Shen, G. Wang, I. Inigo, D. Surguladze, A. L. Pennello, K. Persaud, S. Hindi, M. Topper, X. Chen, Y. Zhang, D. K. Bulaon, T. Bailey, Y. Lao, B. Han, S. Torgerson, D. Chin, A. Sonyi, J. N. Haidar, R. D. Novosiadly, C. M. Moxham, G. D. Plowman, D. L. Ludwig, and M. Kalos. 2020. "Bispecific Targeting of PD-1 and PD-L1 Enhances T-cell Activation and Antitumor Immunity." Cancer Immunol Res 8 (10): 1300-1310. [CrossRef]
- Yearley, J. H., C. Gibson, N. Yu, C. Moon, E. Murphy, J. Juco, J. Lunceford, J. Cheng, L. Q. M. Chow, T. Y. Seiwert, M. Handa, J. E. Tomassini, and T. McClanahan. 2017. "PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer." Clin Cancer Res 23 (12): 3158-3167. [CrossRef]
- Wei, J., W. Montalvo-Ortiz, L. Yu, A. Krasco, K. Olson, S. Rizvi, N. Fiaschi, S. Coetzee, F. Wang, E. Ullman, H. S. Ahmed, E. Herlihy, K. Lee, L. Havel, T. Potocky, S. Ebstein, D. Frleta, A. Khatri, S. Godin, S. Hamon, J. Brouwer-Visser, T. Gorenc, D. MacDonald, A. Hermann, A. Chaudhry, A. Sirulnik, W. Olson, J. Lin, G. Thurston, I. Lowy, A. J. Murphy, E. Smith, V. Jankovic, M. A. Sleeman, and D. Skokos. 2022. "CD22-targeted CD28 bispecific antibody enhances antitumor efficacy of odronextamab in refractory diffuse large B cell lymphoma models." Sci Transl Med 14 (670): eabn1082. [CrossRef]
- Ke, H., F. Zhang, J. Wang, L. Xiong, X. An, X. Tu, C. Chen, Y. Wang, B. Mao, S. Guo, C. Ju, X. He, R. Sun, L. Zhang, O. A. O'Connor, and Q. X. Li. 2023. "HX009, a novel BsAb dual targeting PD1 x CD47, demonstrates potent anti-lymphoma activity in preclinical models." Sci Rep 13 (1): 5419. [CrossRef]
- Dovedi, S. J., M. J. Elder, C. Yang, S. I. Sitnikova, L. Irving, A. Hansen, J. Hair, D. C. Jones, S. Hasani, B. Wang, S. A. Im, B. Tran, D. S. Subramaniam, S. D. Gainer, K. Vashisht, A. Lewis, X. Jin, S. Kentner, K. Mulgrew, Y. Wang, M. G. Overstreet, J. Dodgson, Y. Wu, A. Palazon, M. Morrow, G. J. Rainey, G. J. Browne, F. Neal, T. V. Murray, A. D. Toloczko, W. Dall'Acqua, I. Achour, D. J. Freeman, R. W. Wilkinson, and Y. Mazor. 2021. "Design and Efficacy of a Monovalent Bispecific PD-1/CTLA4 Antibody That Enhances CTLA4 Blockade on PD-1." Cancer Discov 11 (5): 1100-1117. [CrossRef]
- Geuijen, C., P. Tacken, L. C. Wang, R. Klooster, P. F. van Loo, J. Zhou, A. Mondal, Y. B. Liu, A. Kramer, T. Condamine, A. Volgina, L. J. A. Hendriks, H. van der Maaden, E. Rovers, S. Engels, F. Fransen, R. den Blanken-Smit, V. Zondag-van der Zande, A. Basmeleh, W. Bartelink, A. Kulkarni, W. Marissen, C. Y. Huang, L. Hall, S. Harvey, S. Kim, M. Martinez, S. O'Brien, E. Moon, S. Albelda, C. Kanellopoulou, S. Stewart, H. Nastri, A. B. H. Bakker, P. Scherle, T. Logtenberg, G. Hollis, J. de Kruif, R. Huber, P. A. Mayes, and M. Throsby. 2021. "A human CD137×PD-L1 bispecific antibody promotes anti-tumor immunity via context-dependent T cell costimulation and checkpoint blockade." Nat Commun 12 (1): 4445. [CrossRef]
- Lin, W., Y. Zhang, Y. Yang, B. Lin, M. Zhu, J. Xu, Y. Chen, W. Wu, B. Chen, X. Chen, J. Liu, H. Wang, F. Teng, X. Yu, J. Lu, Q. Zhou, and L. Teng. 2023. "Anti-PD-1/Her2 Bispecific Antibody IBI315 Enhances the Treatment Effect of Her2-Positive Gastric Cancer through Gasdermin B-Cleavage Induced Pyroptosis." Adv Sci (Weinh) 10 (30): e2303908. [CrossRef]
- Perez-Santos, M. 2020a. "Bispecific anti-PD-1/CTLA-4 antibody for advanced solid tumors." Pharm Pat Anal 9 (5): 149-154. [CrossRef]
Table 1.
Summary of BsAbs approved for market worldwide for clinical use, as of 2014.
| Drug (Company) |
Trade name | Target antigen | Approved Countries | Year Approved | Approved indications |
| Blinatumomab (Amgen) | Blincyto | CD3/CD19 | FDA | 2014 | adults and children with B-cell precursor acute lymphoblastic leukemia (ALL) in first or second complete remission with minimal residual disease (MRD) greater than or equal to 0.1%. |
| Emacizumab-kxwh (Genentech) |
Hemlibra | FIXa/ FX | FDA | 2017 | the treatment is recommended for adult and pediatric patients, including newborns, with hemophilia A. This includes individuals with congenital factor VIII deficiency, whether or not they have developed factor VIII (FVIII) inhibitors |
| Amivantamab-vmjw(Janssen Biotech) | Rybrevant | EGFR/c-Met | FDA/EMA | 2021 | adult patients with locally advanced or metastatic non-small cell lung cancer who have EGFR exon 20 insertion mutations and have previously received platinum-based chemotherapy |
| Tebentafusp-tebn (Immunocore) |
Kimmtrak* | CD3/ gp100 | FDA | 2022 | for the treatment of adult patients with unresectable or metastatic uveal melanoma who are HLA-A*02:01-positive. |
| Faricimab-svoa (Roche) | Vabysmo | VEGF-A/Ang-2 | FDA | 2022 | To treat neovascular (wet) age-related macular degenerated and diabetic macular edema |
| Mosunetuzumab-axgb (Genentech) | Lunsumio | CD3/CD20 | EMA/FDA | 2022 | Patients with advanced non-small cell lung cancer (NSCLC), harboring EGFR exon 20 insertion mutations, facing disease progression after platinum-based chemotherapy, |
| Cadonilimab (Akeso) |
Kaitanni | PD-1/CTLA-4 | CFDA | 2022 | For patients with relapsed or metastatic cervical cancer (r/mCC) who have experienced disease progression following platinum-based chemotherapy |
| Teclistamab-cqyv (Janssen Biotech) |
Tecvavli | CD3/BCMA | EMA/FDA | 2022 | adult patients with relapsed or refractory multiple myeloma who have received at least four prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody |
| Epcoritamab-bysp (Genmab) |
Epkinly | CD3/CD20 | FDA/EMA | 2023 | adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), including cases arising from indolent lymphoma and high-grade B-cell lymphoma after two or more lines of systemic therapy |
| Glofitamab-gxbm (Genentech) | Columvi | CD3/CD20 | FDA | 2023 | For adult with relapsed or refractory diffuse large B-cell lymphoma (DLBCL, NOS) or large B-cell lymphoma (LBCL) arising from follicular lymphoma, after two or more lines of systemic therapy. |
| Talquetamab-tgvs (Janssen Biotech) | Talvey | GPRC5D/ CD3 | EMA/FDA | 2023 | adults with relapsed or refractory multiple myeloma who have undergone at least four prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody. |
| Elranatamab (Pfizer) | Elrexfio | BCMA/CD3 | FDA/EMA | 2023 | for adults with relapsed or refractory multiple myeloma who have received at least four prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody. |
| Odronextamab* | Regeneron | CD20/CD3 | FDA | FDA decision is on March 31, 2024. | adult patients with relapsed/refractory (R/R) follicular lymphoma (FL) or R/R diffuse large B-cell lymphoma (DLBCL) who have progressed after at least two prior systemic therapies |
*Kimmtrak is technically a bispecific molecule, not a bispecific antibody. Like some of the other bispecific antibodies used to treat some cancers, Kimmtrak has one arm using an antibody fragment to bring killer T cells to the tumor. Kimmtrak’s other arm is an analogous structure found on T cells, the T cell receptor, instead of an antibody fragment to target a tumor antigen.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



