Submitted:
24 January 2024
Posted:
24 January 2024
You are already at the latest version
Abstract
Considering the increasing interest in understanding the biotic component of methane removal from our atmosphere, it becomes essential to study the physiological characteristics and genomic potential of methanotroph isolates, especially their traits allowing them to adapt to elevated growth temperatures. The genetic signatures of Methylocaldum species have been detected in many terrestrial and aquatic ecosystems. A small set of representatives of this genus has been isolated and maintained in culture. The genus is commonly described as moderately thermophilic, with the growth optimum reaching 50oC for some strains. Here, we present a comparative analysis of genomes of three Methylocaldum strains—two terrestrial M. szegediense strains (O-12 and Norfolk) and one marine strain Methylocaldum marinum (S8). The examination of the core genome inventory of this genus uncovers significant redundancy in primary metabolic pathways, including the machinery for methane oxidation (numerous copies of pmo genes) and methanol oxidation (duplications of mxaF, xoxF1-5 genes), three pathways for one-carbon (C1) assimilation, and two methods of carbon storage (glycogen and polyhydroxyalkanoates). We also investigate the genetics of melanin production pathways, as a key feature of the genus.
Keywords:
thermophilic methanotrophic bacteria
; Methylocaldum
; methane monooxygenase
; pyomelanin
; methanol dehydrogenase
; extremophiles
1. Introduction
Microbial methane oxidation is a key process in the carbon cycle at local and global scales. Methane-utilizing bacteria (methanotrophs) inhabiting high-temperature ecosystems include members of the genera Methylococcus, Methylothermus and Methylocaldum from the phylum Pseudomonadota (Proteobacteria) and the genus Methylacidiphilum in the phylum Verrucomicrobia [1,2,3]. Thermophilic and thermotolerant species of the Methylocaldum genus (Gammaproteobacteria class in the family Methylococcales) are commonly identified as important members of the bacterial communities in soils from a range of geographical locations [4] including hot springs [5], landfill cover soils [6,7], tin-mining ponds [8], oil sands [9], flooded rice fields [10], and marine sediments [11]. The genus Methylocaldum was introduced by Bodrossy et al. (1997). The genus currently includes four species, M. szegediense, M. tepidum, M. gracile [5] and M. marinum [11]. The isolate M. szegediense O-12, was obtained from a sample of cow manure from a farm near Pushchino in the Moscow region in Russia. The pure culture was obtained from an initial enrichment, followed by a serial dilution to extinction technique [12]. M. szegediense O-12 grows at a temperature range of 37–59°C, with optimal growth at 53–55°C. M. szegediense Norfolk was isolated from 50°C biofilter soil enrichments from a landfill in Norfolk County Council, UK, and grows between 37°C and 62°C. Here we conduct a comparative analysis between thermotolerant methanotrophs to identify key genes potentially involved in their capability to grow at elevated temperatures. The genomic features of two terrestrial moderate thermophilic M. szegediense O-12 and Norfolk strains are compared with the thermotolerant, M. marinus S8 (= NBRC 109686T = DSM 27392T) isolated from marine sediments in Kagoshima Bay, Japan which grows optimally at 36°C and a salinity of up to 5% NaCl [11].
2. Materials and Methods
Genome sequencing, assembly, and annotation. M. szegediense O-12 genome sequencing and assembly was conducted in collaboration with U.S. Department of Energy Joint Genome Institute (taxon ID: 2508501066 (IMG)/ 675524 (NCBI)). The genome and assembly of M. szegediense Norfolk was generated by Dr. David Pearce in the laboratory of Dr. J. Colin Murrell, at University of East Anglia, Norwich, UK, using the MicrobesNG (Birmingham UK) enhanced genome service (taxon ID: 8069803063 (IMG)/ 73780 (NCBI)). The genome sequence of M. marinum S8 was generated by National Institute of Advanced Industrial Science and Technology (AIST), Japan (taxon ID: 2832923104 (IMG)/ 1432792 (NCBI)). In addition to being available in NCBI, the three genomes were uploaded and annotated in IMG/MER [14,15].
Methylocaldum phylogenic analysis was carried out using all (20) complete sequences available for the 16S rRNA gene assigned to Methylocaldum strains on NCBI at the time of the analysis (2022). The alignment was generated using the multiple alignment program for amino acid or nucleotide sequences MAFFT (https://mafft.cbrc.jp/alignment/software/) choosing the E-INS-i method [16,17,18]. The phylogenetic relationship was inferred using the MEGA X software using the Maximum Likelihood method and the model with the lowest Bayesian Information Criterion (BIC) scores for the set, which corresponded to Tamura 3-parameter (T92) with a discrete Gamma distribution (+G) with 5 rate categories and by assuming that a certain fraction of sites is evolutionarily invariable (+I). This analysis involved 32 nucleotide sequences and all positions with less than 95% site coverage were eliminated [19,20,21].
pMMO synteny, functional genomic comparison and ANI x AF. The synteny and functional genomic comparison of the genomes was based on the information obtained using Anvi’o v7 software [22]. The visual representation of the pMMO operon syntheny was generated combining the outputs of circos plot utilizing the corresponding nucleotidic sequence coordinate data and combining them with the gene cluster visualization output from Geneious software. The generated data included the list of all Coding Sequences (CDS) from the detected open reading frames identified by prodigal [23]. The CDS predicted functionality was annotated using the NCBI’s Clusters of Orthologous Groups (COG) database [24,25,26] and the KEGG KOfamKOALA database [27]. The pangenome analysis [28] generated orthologous groups (OG) of genes, based on the homology of their amino acid sequences. A >70% of combined homogeneity index (derived from geometrical and functional homology) was the cut off to consider a gene as part of an OG and each of them contained at least one gene. Additionally, the OG were organized by presence/absence through euclidean distance and ward linkage, which was used to categorize them as shared by the three genomes or unique for each of them. Then within each of the shared and unique subsets, all CDS with COG assigned to them were used to determine which COG categories were the most represented (>5%). Additionally, a whole genome comparison was generated using the anvi-compute-genome-similarity to obtain the average nucleotide identity multiplied by the aligned fraction (ANI x AF). Genomes of Methylocaldum, Methylobacter, and Methylococcus available on IMG/MER were used to search for the range of copies of pmoC. The search was based on IMG annotations using the different available identifiers for pmoC: TIGR03078, KO10946, and gene product name. A table containing the Anvi’o v7 results for synteny and functional genomic comparison of the genomes can be found in Appendix Table A1.
xoxF phylogeny. The alignment was generated using the multiple alignment program for amino acid or nucleotide sequences MAFFT (https://mafft.cbrc.jp/alignment/software/) choosing the E-INS-i method [16,17,18]. The tree topology was obtained after 100 bootstrap replications using the Maximum Likelihood method and General Time Reversible model [20], using MEGA X [19,21]. The tree with the highest log likelihood (-175992.24) [29] was used to generate the summarized version of it.
Fdh phylogeny. The alignment was generated using the multiple alignment program for amino acid or nucleotide sequences MAFFT (https://mafft.cbrc.jp/alignment/software/) choosing the E-INS-i method [16,17,18]. The tree topology was obtained after 100 bootstrap replications using the Maximum Likelihood method and the Whelan and Goldman + Freq. model [30], The tree with the highest log likelihood (-50331.00) (supplementary Figure S1) was used to generate the cartoon version of it (Figure 5). The percentage of trees in which the associated taxa clustered together is shown next to the branches. This analysis involved 193 amino acid sequences. There were a total of 1638 positions in the final dataset. Evolutionary analyses were conducted in MEGA X [19,21].
3. Results
3.1. Genome Overview
The M. szegediense O-12 genome (taxon ID: 2508501066 (IMG)/ 675524 (NCBI)) includes two scaffolds of 2.37Mb and 2.65Mb, resulting in a genome of 5.02 Mb with an average GC content of 57.03 %. It includes two rRNA operons, 22 types of tRNAs (47 total) and 4693 predicted protein-coding genes (3448 predicted functions). The genome of M. szegediense sp. Norfolk (taxon ID: 8069803063 (IMG)/ 73780 (NCBI)) includes a chromosome of 4.87Mb and a 25.7kb plasmid, resulting in a genome of 4.89 Mb with an average GC content of 57.17 %. It contains two rRNA operons and 24 types of tRNAs (53 total) and 4491 predicted protein coding genes (3378 predicted functions). Methylocaldum marinum S8 (taxon ID: 2832923104 (IMG) / 1432792 (NCBI)) includes one scaffold, having a genome of 6.08 Mb with a 58.75% GC content, including two rRNA operons, 24 types of tRNAs (51 total) and 5596 predicted protein coding genes (3846 predicted functions).
3.2. Methylocaldum Phylogeny
The phylogeny of Methylocaldum strains based on available 16S rRNA sequences shows five clades (Figure 1A). Four of these clades correspond to currently characterized species within the Methylocaldum genera (Methylocaldum tepidum LK6T, Methylocaldum szegediense OR2T, Methylocaldum gracile VKM 14LT, and Methylocaldum marinum S8T), while the fifth clade formed by strains BFH1, dr65 and r6f does not have a type strain. A comparison based on the presence or absence of orthologous genes between the three strains analyzed in this study revealed that the M. szegediense O-12 and Norfolk strains share 90% and 93%, respectively, of their predicted protein sequences as orthologous, while only 47% of the predicted protein sequences in M. marinum S8 were orthologous to genes in the O-12 and Norfolk strains (Figure 1B). The average nucleotide identity times the aligned fraction (ANI x AF) percentage between the three strains confirmed that M. szegediense O-12 and Norfolk are members of the same species (≥90%), while M. marinum S8 shared only a 38% with the M. szegediense strains (Figure 1B).
3.3. Synteny of Methane Monooxygenase (MMO) Gene Clusters
The identification of key genes necessary for methane oxidation in bacteria revealed the presence of several copies of the pmoC gene, which is usually part of the pmoCAB operon for the particulate methane monooxygenase (pMMO). Five copies were found in M. marinum S8, six in M. szegediense O-12 and five in M. szegediense Norfolk (Figure 2). In the M. marinum S8 and M. szegediense Norfolk strains, two pmoC genes formed part of the complete gene cluster for the synthesis of the pMMO, whereas only one pmoC gene formed part of the complete gene cluster in M. szegediense O-12 (Figure 2). All pmoC genes forming part of pMMO operons had high sequence identity to each other and were denominated as type 1 (purple ribbon, Figure 2). Furthermore, the three Methylocaldum genomes contained paralogs of pmoC as independent genetic components in different loci of their genomes, which have been previously named as stand-alone copies of pmoC [31]. Except for a single stand-alone pmoC in M. szegediense O-12 assigned to type 1, the rest of the stand-alone copies of pmoC have different nucleotide sequences than the one forming the pMMO cluster (type 1) and form two separate groups of paralogs, denominated as type 2 and 5 (pink and magenta ribbons, respectively, Figure 2). Furthermore, both M. szegediense strains exhibit two stand-alone pmoC type 2 relatively next to each other. Additionally, two stand-alone pmoC without high homology to any other pmoC sequences within the three genomes were found in M. marinum S8 (type 4) and M. szegediense O-12 (type 3), shown in black-edged triangles, Figure 2. Only M. marinum S8 exhibits a complete gene cluster for the soluble methane monooxygenase (sMMO). When compared with other methanotrophs with genomes annotated on IMG/MER, Methylobacter (16) and Methylococcus (10), and Methylocaldum strains have between five and six coding sequences (CDS) assigned to pmoC, while Methylobacter have between one and two and Methylococcus have between two and three pmoC in the available genomes.
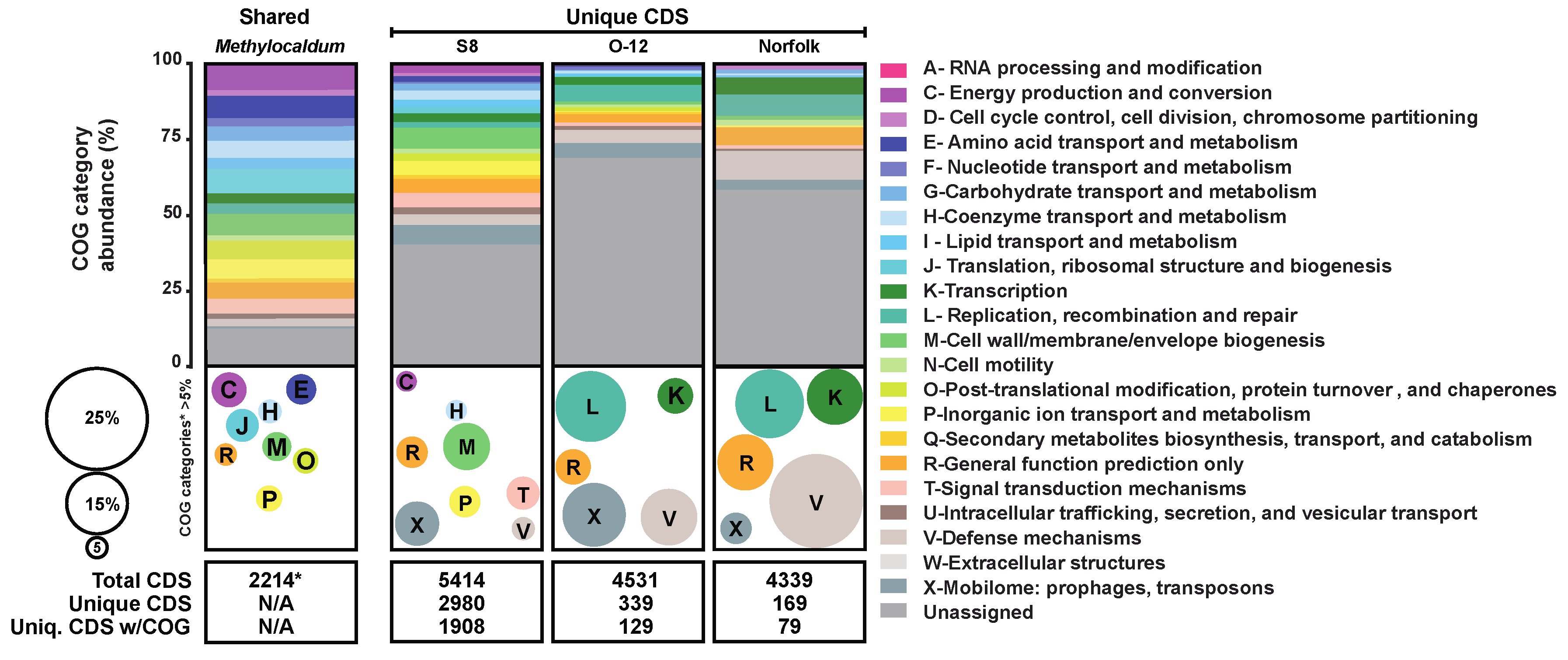
3.4. Comparative Abundance CDS Assigned to COG Categories
According to the pangenome analysis of the three Methylocaldum strains, from a total of 14,284 CDS predicted for the three genomes, 2,214 CDS formed orthologous groups (OG) of amino acid sequences, based on a ≥70% threshold of combined homogeneity index (derived from geometrical and functional homology; Figure 3). Each genome had at least one gene copy as part of each OG assigned as shared among the three strains. Based on their relative abundances, the main shared COG categories (comprising >5% of the total gene calls that could be categorized) were genes related to categories C (8.58%) involved in energy production and conversion; E (7.36%) for amino acid transport and metabolism; H (5.74%) encoding coenzyme transport and metabolism; J (8.04%) for translation, ribosomal structure and biogenesis; M (7.09%) cell wall/membrane biogenesis; O (6.23%) post translational modification; P (6.32%) inorganic ion transport and metabolism; and R (5.42%) classified only as general function.
On the contrary, the CDS found to be unique to each genome, based on a low combined homogeneity index of their amino acidic sequences when compared to their counterparts in the other genomes, were mainly assigned to COG categories M, cell wall/membrane biogenesis (11.57%); X, mobilome including prophages and transposons (10.9%); T, signal transduction mechanisms (8.15%); P, inorganic ion transport and metabolism (7.70%); R, general function (7.47%); V, defense mechanisms (5.67%); H, coenzyme transport and metabolism (5.17%), and C, energy production and conversion (5%) for M. marinum S8. While the pattern was similar between M. szegediense O-12 and Norfolk with V (13.91% & 23.07%), X (15.65% & 7.69%), L, replication, recombination, and repair (17.39% & 16.92%), R (8.7% & 13.84), and K, transcription (8.70% & 13.84%), respectively (Figure 3).
Figure 3.
Comparative abundance of COG categories for Methylocaldum genomes. The relative percentage of each COG category is depicted by colored horizontal bars and bubbles according to the color-coded legend. Horizontal bars represent the percentage of CoDing Sequences (CDS) assigned to each COG category among the three strains (Shared), and the unique for each strain (S8, O-12, and Norfolk). The total number of CDS considered for this comparison are at the bottom of each column. Bubbles size and colors depict COG categories that represented> 5% of the total when unassigned CDS were not considered.
Figure 3.
Comparative abundance of COG categories for Methylocaldum genomes. The relative percentage of each COG category is depicted by colored horizontal bars and bubbles according to the color-coded legend. Horizontal bars represent the percentage of CoDing Sequences (CDS) assigned to each COG category among the three strains (Shared), and the unique for each strain (S8, O-12, and Norfolk). The total number of CDS considered for this comparison are at the bottom of each column. Bubbles size and colors depict COG categories that represented> 5% of the total when unassigned CDS were not considered.
3.5. Conjugation
Several predicted functions were only found in the two moderate thermophilic M. szegediense strains including K12056 corresponding to the conjugal transfer mating pair stabilization protein TraG, which is one of the pilus assembly proteins in bacterial type IV secretion systems (T4SS) [32]. While Norfolk has only two conjugal transfer proteins, TraG and TraN, and O-12 genome contains two TraG, and additionally TraW, TraF, TraE, TraL which participate in pilus assembly; TraA (pilin); TraN and TraU which are responsible for mating pair stabilization; TraV, TraK, and TraB which form the core complex; TraD, and TraC which are ATPase proteins reported to be complexed in the cytosol and inner membrane, and also TrbL, which are a subset of protein described as F-like T4SS [32,33].
3.6. O-Antigen Biosynthesis
Interestingly, two homologs of O-antigen biosynthesis protein RfbC (K20444; COG0438|COG1216) were found only in the M. szegediense strains. The RfbC is predicted to catalyze the incorporation of sugars and their products to form the O-antigen polysaccharides in the lipopolysaccharides (LPS) [34] and the enzyme specificity toward rare sugars enables production of unusual LPS, thus providing remarkable diversity of the cell envelop recognition patterns observed among bacteria [35,36,37,38,39]. RfbC is a member of the glycosyltransferase family 2 (GT2). A search for other glycosyltransferases belonging to that family of which RfbC is part, glycosyltransferase family 2 (GT2), revealed the presence of six other OGs shared between the M. szegediense strains and absent in M. marinum S8. Additionally, the three Methylocaldum strains share three OGs of GT2, and M. marinum S8 has four different GT2, not shared with M. szegediense strains. Differences between the genetic toolkit for the biosynthesis of the O-antigen among Methylocaldum offers a starting point for future studies to elucidate the mechanisms of specialization in their different niches, and how they can tolerate a wide range of temperatures. It has been shown that O-antigen structural differences can confer resistance to different types of stress, such as oxidative [40], mechanical stress [39] and osmotic stress [41]. Additionally, antigen molecular diversity is a current target of study to understand interdomain symbiosis [42].
3.7. Catalase
Among the distinct functions that were found to be unique for M. marinum S8, is catalase KatE (COG 0753, KEGG K03781) which can convert toxic hydrogen peroxide (H2O2) into water and oxygen. H2O2 is one of the three products of methanethiol (CH3SH) oxidation (HCOH, H2S, H2O2), for which all three genomes have a homolog to the gene mtoX, a copper-dependent methanethiol oxidase (MTO) gene characterized in Hyphomicrobium sp. VS [43] and annotated in our analysis as K17285. CH3SH is a volatile organic sulfur compound that can co-occur with methane. It can be consumed by the verrucomicrobial methanotroph Methylacidiphilum fumariolicum SolV which produces H2S that is subsequently oxidized for energy [44]. It seems likely that this gene would be beneficial for methanotrophs, since it could allow them to flourish in niches that release methane and CH3SH, an inhibitor of methane oxidation [45].
3.8. Cobalamin
Another unique function on M. marinum S8 among the three genomes, was a nearly complete de novo cobalamin (B12) biosynthetic pathway. The only missing gene corresponds to cbiJ-cobK whose product, precorrin-6X reductase, converts (Co)Precorrin-6A to (Co)Precorrin-6B. This gene had been previously described as missing in almost all characterized cobalamin-producing archaea [46,47,48]. This suggests independency of an external source of the essential cofactor, which would be an advantage, given that most predicted cobalamin producers in the ocean belong to photoautotrophic Cyanobacteria, chemoautotrophic Nitrososphaerota (Thaumarcheota), and a selected group of Pseudomonadota (Proteobacteria), including Rhodobacterales, Rhodospirillales, Deltaproteobacteria, Oceanospirillales, and Pseudomonadales [49]. Both M. szegediense strains lacked any recognizable genetic elements required for the corrinoid ring biosynthesis [50]. Nonetheless, all three genomes contain the genes for 5,6-dimethylbenzimidazole (DMB) synthesis, 5,6-dimethylbenzimidazole synthase, bluB (COG0778, K04719) which is the lower ligand of coenzyme B12; all the genes for the final synthesis and repair (K19222 (CobA), K00798 (CobO), K02232 (CobQ), K02225 (CobC), K02227 (CobD), K02231 (CobP/CobU), and K02233 (CobS), and for B12 transport K16092 (BtuB), which match the previous classification of M. szegediense O-12 as potentially unable to do de novo synthesis, but instead to salvage cobamide precursors, assemble the nucleotide loop, and attach a lower ligand, based on its in silico analysis of 11,000 strains [51].
3.9. M. marinum S8 Unique Predicted Functions
Additionally, only M. marinum S8 has several hydrogenases subunits, including Fe-S-cluster-containing hydrogenase component 2 (HycB) COG1142, hydrogenase-4 membrane subunit HyfE COG4237 (K12140), Ni,Fe-hydrogenase III small subunit (HycG) (PDB:6CFW) COG3260, and the Ni,Fe-hydrogenase III large subunit HycE2|Ni,Fe-hydrogenase III component G (HycE1) COG3261|COG3262, which suggests the metabolic potential to obtain energy through membrane-associated H2 involving hydrogenases. These hydrogenases are in the immediate vicinity of homologs to the formate hydrogenlyase subunit HyfC (COG0650).
Other functions found only in the genome of M. marinum S8 are homologs for molybdopterin biosynthesis (COG0303|1910, K07219), peroxiredoxin DsrE/DsrF-like family (COG2044), putative NADH-Flavin reductase YWNB (COG2910), RecA superfamily ATPase KaiC/GvpD/Rad55 (COG0467, K0882), Fe3+ permease component FepD, (COG0609|0614) Fe2+ transporter FeoB|FeoA (COG0370|1918, K04759), periplasmic components for ABC-type tungstate transport system, TupA (COG4662, K05773) and TupB (COG2998, K05772), heavy metal-binding TRASH/YHS Cu/Ag metallochaperone (COG3350, K16157), nitrous oxide reductase accessory protein NosL (COG4314, K19342), and cobalt transporter membrane subunit CgtA (COG5446).
3.10. C1-Oxidation Pathways Are Highly Redundant
As described above, in addition to having two pmoCAB operons in the Norfolk and S8 genomes and the sMMO operon in S8, the three genomes also contain multiple stand-alone pmoCs with unknown functions. They also have multiple methanol dehydrogenases. The mxaFJGIRACKLD gene clusters in the three Methylocaldum strains encode the two subunits of methanol dehydrogenase mxaF (K14028) and mxaI (K14029) and the natural electron acceptor, cytochrome cL (mxaG). Moreover, the three strains share two OGs of additional methanol dehydrogenases, lanthanide-dependent MDHs: xoxF corresponding to xoxF types 5 and 3 according to the phylogenetic reconstructions (Figure 4) following previously xoxF type assignation [52,53,54] . M. marinum has a third xoxF with no homology to genes in the M. szegediense strains, and which also belongs to xoxF type3.
The three Methylocaldum have the set of genes for the tetrahydromethanopterin (HMPT)-dependent pathway required for the oxidation of formaldehyde to formate. Gene redundancy was also found in this pathway. Based on the OG analysis, three different OGs for fae (K10713) which encodes the 5,6,7,8-tetrahydromethanopterin hydro-lyase were found among Methylocaldum. One fae OG included three CDS from M. marinum S8, and two for M. szegediense O-12 and Norfolk. The other two OGs had only one CDS in each strain. Each strain had two methylene-tetrahydromethanopterin dehydrogenases mtdB (K10714) in separate OG. Only one of the following genes were found in each of the strains: methylenetetrahydrofolate dehydrogenase mtdA (K00300), methenyltetrahydromethanopterin cyclohydrolase mch (K01499), formylmethanofuran--tetrahydromethanopterin N-formyltransferase ftr (K00672), formylmethanofuran dehydrogenase fwdABC (K00200, K00201, K00202), glycine hydroxymethyltransferase glyA (K00600), and coenzyme F420 hydrogenase subunit beta frhB (K00441) for which only M. marinum S8 had two CDS.
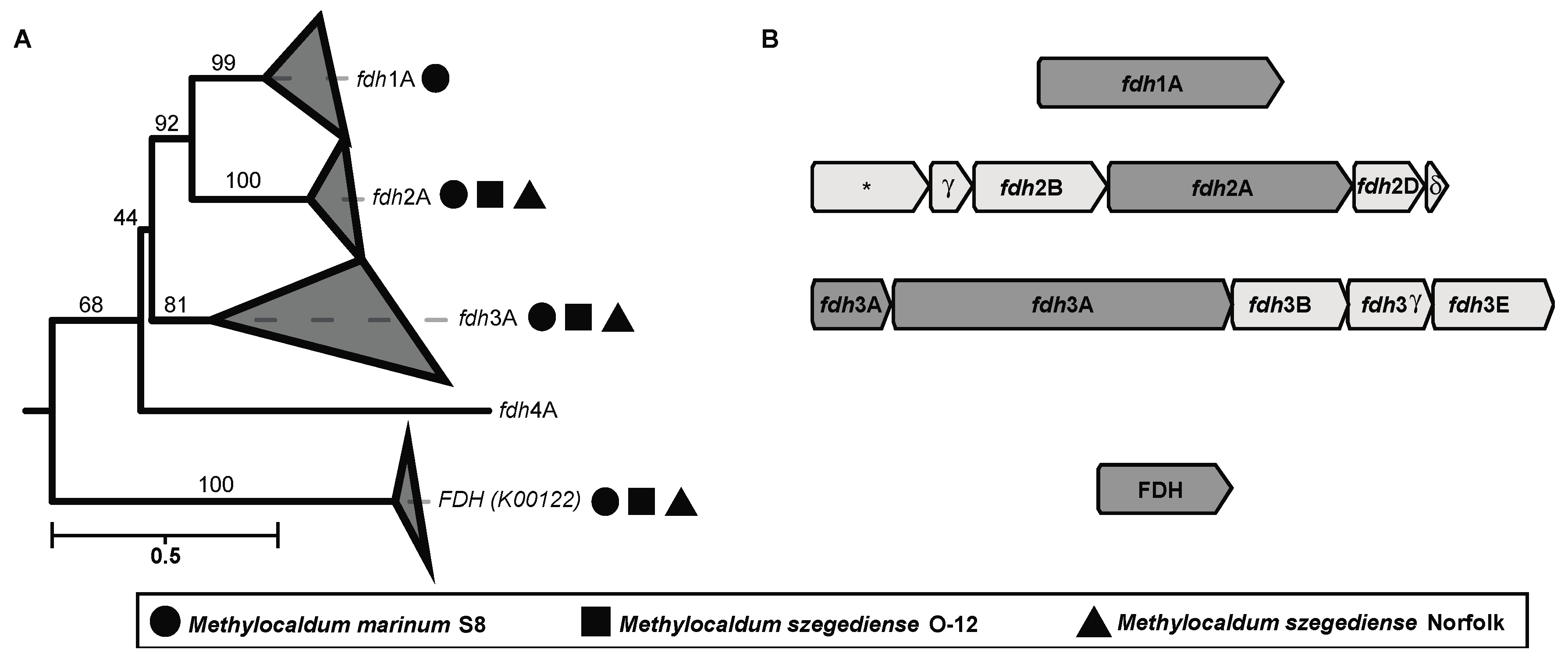
Interestingly, several formate dehydrogenases encoding genes (fdh) were found in the three strains. These enzymes catalyze the oxidation of formate to CO2, donating electrons to NAD+ or cytochromes. The phylogenetic reconstruction based on the amino acid sequence of the major subunits (K00123) of the different formate dehydrogenases found in Methylocaldum revealed that they have four types of fdh (Figure 5 and supplementary Figure S1). According to the previously described types of fdh in Methylobacterium extorquens AM1 [55,56], we detected the presence of three different fdh in the three Methylocaldum strains, corresponding to fdh type 2 and 3 including all their subunits. Additionally, FDH (K00122) was found in the three genomes as well. Moreover, M. marinum S8 has the major subunit for fdh type 1, although it was previously described to require the subunit B for functioning in the M. extorquens AM1 model [56,57].
Figure 5.
(A) Maximum likelihood tree reconstructing the phylogenetic relationship between the amino acidic sequences of four different types of formate dehydrogenases major subunits (K00123). Geometric figures next to each collapsed clade indicates the presence of a sequence from one of the three Methylocaldum strains analyzed in this study. Circles represent M. marinum S8, squares M. szegediense O-12, and triangles M. szegediense Norfolk. Formate dehydrogenase (FDH) K00122 was used as outgroup. (B) Gene cluster genomic arrangement found on the three Methylocaldum strains analyzed in this study. The asterisk indicates the presence of a predicted protein with unknown function.
Figure 5.
(A) Maximum likelihood tree reconstructing the phylogenetic relationship between the amino acidic sequences of four different types of formate dehydrogenases major subunits (K00123). Geometric figures next to each collapsed clade indicates the presence of a sequence from one of the three Methylocaldum strains analyzed in this study. Circles represent M. marinum S8, squares M. szegediense O-12, and triangles M. szegediense Norfolk. Formate dehydrogenase (FDH) K00122 was used as outgroup. (B) Gene cluster genomic arrangement found on the three Methylocaldum strains analyzed in this study. The asterisk indicates the presence of a predicted protein with unknown function.
3.11. Methylocaldum Potential Single-Carbon Assimilation Pathways
These Methylocaldum genomes encode three single-carbon assimilation pathways—the ribulose monophosphate (RuMP), serine, and Calvin Benson Bassham (CBB) cycle. The genes present in the RuMP pathway include two CDS for 3-hexulose-6-phosphate synthase in each genome, hxlA (K08093) and hps-pi (K13831), catalyzing condensation of formaldehyde with ribulose-5-phosphate to D-arabino-hex-3-ulose 6-phosphate, which is converted to D-fructose 6-phosphate by the 6-phospho-3-hexuloisomerase, hxlB (K08094) present in the three genomes. D-fructose 6-P can then enter the pentose phosphate pathway.
The large rbcL/cbbL (K01601) and small rbcS/cbbS (K01602) subunits of the ribulose 1,5-bisphosphate carboxylase/oxygenase (RuBisCO) were found in the three strains, having a single gene difference between M. szegediense and M. marinum. This includes the presence of norQ and norD next to RuBisCO genes of the three Methylocaldum. M. marinum S8 also has pimeloyl-ACP methyl ester carboxylesterase between the RuBisCO and nor genes. The presence of rcb/cbb genes had been reported in other methanotrophs; however, its activity had not been demonstrated yet [58,59,60].
Similarly to other gammaproteobacterial methanotrophs, the regeneration of pentose phosphates from hexosephosphates requires fructose-bisphosphate aldolase, for which class I, ALDO (K01623), is present in all three Methylocaldum genomes.
As in other gammaproteobacterial methanotrophs, the Methylocaldum genomes contain some key genes for the serine pathway of formaldehyde assimilation. These include serine-glyoxylate transaminase, AGXT (K00830) which converts glyoxylate to hydroxypyruvate; and glycine hydroxymethyltransferase, glyA (K00600) which uses 5,10-methylenetetrahydrofolate and glycine as substrates to produce tetrahydrofolate and L-serine, which can continue to the carbon fixation pathway or sulfur or serine metabolism, respectively. Also, as part of the serine pathway, the three genomes encode malyl-CoA/(S)-citramalyl-CoA lyase mcl (K08691) which produces acetyl-CoA and glyoxylate from L-malyl-CoA as a substrate. However, all strains lack phosphoenolpyruvate carboxylase, ppc (K01595), which produces oxaloacetate from phosphoenolpyruvate, necessary for the serine pathway. Anaplerotic function of the serine pathways has been previously proposed in Methylococcus capsulatus Bath. Like the strain Bath, all three genomes have the necessary genes to convert malyl-CoA (mcl, K08691) to glyoxylate, and then to glycine (AGXT, K00830), and subsequently to serine (glyA, K00600) as part of the serine pathway. In the same pathway, they also have the genes to convert from D-glycerate to glycerate-2P (gck, K11529) to phosphoenolpyruvate (eno, K01689).
3.12. Nitrogen Metabolism
All three strains of Methylocaldum have the structural genes for nitrogenase (nifH (K02588), nifD (K02586), and nifK (K02591)) in an operon with nifE, nifN, nifX, and nifQ genes, and also a nif-specific ferredoxin III (TIGR02936) (Figure 6B). Two additional homologs of nifH and nifK genes were also found in the three strains, although their genetic context was different from each other and may not be related to nitrogen fixation. The genetic elements required for nitrate assimilation were found in the three strains, having as a difference the additional presence of the nrtABC gene cluster (K15576, K15577, K15578) for the nitrate/nitrite transport system in M. marinum S8 only (Figure 6A). The three strains have the norCB gene cluster (K02305, K04561) for the reduction of nitric oxide (NO) to nitrous oxide (N2O), however only O-12 and Norfolk had the nirK (K00368) for dissimilatory nitrite reductase for denitrification, and none of them have alternative nirS (K15864) nitrite reductase. Similarly, to other methanotrophic species, homologs of hydroxylamine dehydrogenase (K10535) responsible for NH2OH oxidation to NO2 as well as hydroxylamine reductase reducing NH2OH to form NH4+ (K05601) are present in the three genomes. Additionally, the three strains have the genetic potential to assimilate NH4+ through the glutamate cycle, with their genes for glutamine synthetase (GS) glnA (K01915) and glutamate synthase (GOGAT) gltBD (K00265, K00266) [58]. The three strains have the genetic potential for the reversible conversion of glutamate to 2-oxoglutarate/α-ketoglutarate (which is an intermediate of Krebs cycle) and ammonia through their glutamate dehydrogenase GDH2 (K15371). Additionally, the three strains also have the gene for alanine dehydrogenase ald (K00259), which had been described to participate in the reductive amination of pyruvate in other methanotrophs, under high NH4+ environments [58]. The two M. szegediense strains also have the gene for another glutamate dehydrogenase gdhA (K00262), which had been demonstrated to be required for Streptococcus pneumoniae for adaptation to high temperature (40°C)[61].
3.13. Carbon Storage
The carbon storage inventory includes glycogen biosynthesis glgABC as well as for the genes for polyhydroxybutyrate biosynthesis (Supplementary Figure S2). The key gene for polyhydroxyalkanoate synthase phaC (K03821), as well as acetyl-CoA C-acetyltransferase phaA (K00626), and 3-oxoacyl-[acyl-carrier-protein] reductase phaB (K00023) were present in the three Methylocaldum strains. Furthermore, additional phaC homologs were found in the three strains, indicating possible PHB co-polymer biosynthesis. The ability to produce polyhydroxyalkanoates has been recently observed for axenic cultures of Methylocaldum, supporting the genetic observations [62]. In addition to genes for polymer biosynthesis, all three strains contain genes encoding pathways of sucrose biosynthesis.
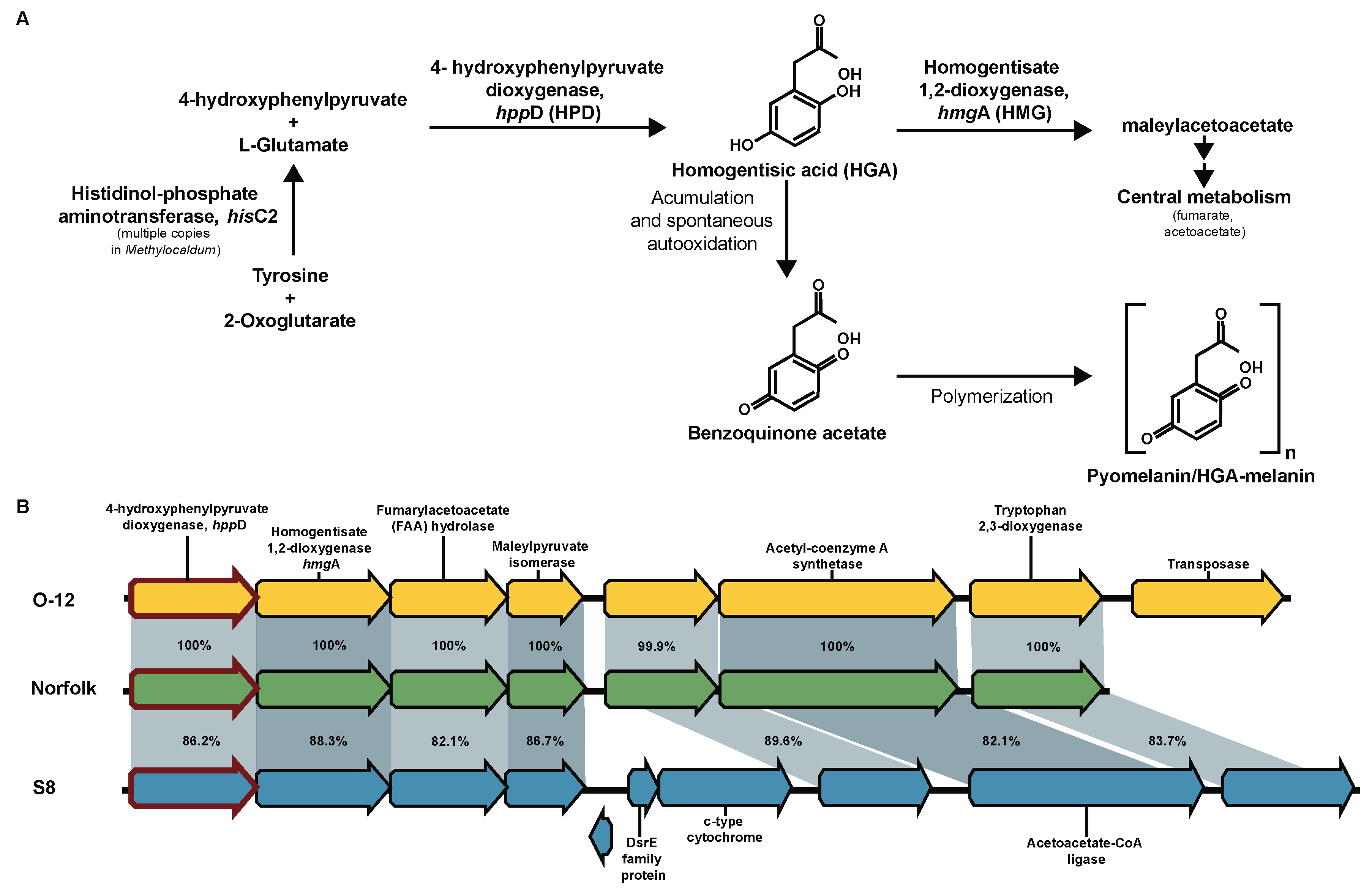
3.14. Pyomelanin/HGA-Melanin Proposed Production
A signature characteristic of Methylocaldum strains is the development of light to dark brown colored colonies/culture [63]. Previous analysis shown that M. szegediense O-12 synthesizes a tyrosine-derived melanin-like pigment upon decrease of growth temperature (at suboptimal temperature 42 °C)[64]. Because of the lack of the canonical melC1 for tyrosinase (K00505) for the production eumelanin in Methylocaldum, here it is proposed that this color corresponds to a type of melanin, known as pyomelanin or HGA-melanin [65]. The three Methylocaldum strains have genes encoding tyrosine degradation pathway via homogentisic acid (HGA), hppD (K00457) and hmgA (K00451). In this pathway, the HGA intermediate can continue to be reincorporated into central metabolism as fumarate and acetoacetate [66] or accumulated and, through an spontaneous autoxidation process, converted to benzoquinone, which polymerizes to form pyomelanin [67,68] (Figure 7). The gene encoding histidinol-phosphateaminotransferase hisC (K00817), which produces the HGA precursor 4-hydroxyphenylpyruvate, has four CDS assigned to it in M. marinum S8, three CDS in M. szegediense O-12 and two CDS in Norfolk.
4. Conclusions
Here we analyze three genomes of thermophilic/thermotolerant methanotrophs of the genus Methylocaldum. Most methanotrophs display metabolic plasticity in response to availability of key nutrients (nitrogen, phosphates) or metals (copper, tungsten, lanthanides). However, all three members of the Methylocaldum genus are superior to other microbes in the number of paralogs for key enzymes as well as the number of metabolic pathways for C1-carbon conversions and storage. The significance of preservation of orthologs in organisms inhabiting very different ecological niches remains to be elucidated. However, it is tempting to propose that the evolution of C1-genes was driven as an adaptation to tolerate dramatic changes in temperature.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Maximum likelihood tree showing the phylogenetic relationship between the aminoacidic sequences of four different types of formate dehydrogenases major subunits.
Author Contributions
ND, DP and SB contributed to genome sequencing, assembly and annotation. ND carried out most of the genomic comparisons presented in this study. MGK, MCJ and VNK provided conceptual framework of the study. Original Draft Preparation, ND, MGK and SB. Review & Editing, ND, SB, DP, MGK, MCJ and VNK. All authors have read and agreed to the published version of the manuscript.
Funding
Genome sequencing of Methylocaldum Strain Norfork was performed by MicrobesNG (http://www.microbesng.com). M. szegediense O-12 genome sequencing and assembly was carried out by the Joint Genome Institute (JGI) under the Office of Science of the U.S. Department of Energy contract DE-AC02-05CH11231. This work was funded by the EnvEast DTP (UKRI grant NE/L002582/1) and Norfolk County Council and by the U.S. Department of Energy under the DOE Office of Science (SC) RENEW contract DE-SC0024289.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
We would like to thank Dr. Mio Takeuchi, from Biomedical Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Midorigaoka, Ikeda, Osaka, Japan, for providing the genome of M. marinum S8 and for her insightful comments on the manuscript. We also thank Alvaro Munoz Plominsky, for his bioinformatic support and feedback.
Conflicts of Interest
Declare conflicts of interest or state “The authors declare no conflicts of interest.” Authors must identify and declare any personal circumstances or interest that may be perceived as inappropriately influencing the representation or interpretation of reported research results. Any role of the funders in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; or in the decision to publish the results must be declared in this section. If there is no role, please state “The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results”.
Appendix A
Table containing Anvi’o v7 outputs for synteny and functional genomic comparison of the Methylocaldum genomes.
References
- Dunfield, P.F.; Yuryev, A.; Senin, P.; Smirnova, A.V.; Stott, M.B.; Hou, S.; Ly, B.; Saw, J.H.; Zhou, Z.; Ren, Y. Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 2007, 450, 879–882. [Google Scholar] [CrossRef]
- Islam, T.; Jensen, S.; Reigstad, L.J.; Larsen, Ø.; Birkeland, N.-K. Methane oxidation at 55 C and pH 2 by a thermoacidophilic bacterium belonging to the Verrucomicrobia phylum. Proc. Natl. Acad. Sci. 2008, 105, 300–304. [Google Scholar] [CrossRef]
- Pol, A.; Heijmans, K.; Harhangi, H.R.; Tedesco, D.; Jetten, M.S.; Op den Camp, H.J. Methanotrophy below pH 1 by a new Verrucomicrobia species. Nature 2007, 450, 874–878. [Google Scholar] [CrossRef]
- Yang, Y.; Shan, J.; Zhang, J.; Zhang, X.; Xie, S.; Liu, Y. Ammonia-and methane-oxidizing microorganisms in high-altitude wetland sediments and adjacent agricultural soils. Appl. Microbiol. Biotechnol. 2014, 98, 10197–10209. [Google Scholar] [CrossRef] [PubMed]
- Bodrossy, L.; Holmes, E.M.; Holmes, A.J.; Kovács, K.L.; Murrell, J.C. Analysis of 16S rRNA and methane monooxygenase gene sequences reveals a novel group of thermotolerant and thermophilic methanotrophs, Methylocaldum gen. nov. Arch. Microbiol. 1997, 168, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.Y.; Su, Y.; Zhang, Q.Q.; Bai, Y.; Xia, F.F.; Fang, C.R.; He, R. Vertical profiles of community and activity of methanotrophs in landfill cover soils of different age. J. Appl. Microbiol. 2013, 115, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kong, J.-Y.; Xia, F.-F.; Su, Y.; He, R. Effects of ammonium on the activity and community of methanotrophs in landfill biocover soils. Syst. Appl. Microbiol. 2014, 37, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Sow, S.; Khoo, G.; Chong, L.; Smith, T.; Harrison, P.; Ong, H. Molecular diversity of the methanotrophic bacteria communities associated with disused tin-mining ponds in Kampar, Perak, Malaysia. World J. Microbiol. Biotechnol. 2014, 30, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Saidi-Mehrabad, A.; He, Z.; Tamas, I.; Sharp, C.E.; Brady, A.L.; Rochman, F.F.; Bodrossy, L.; Abell, G.C.; Penner, T.; Dong, X. Methanotrophic bacteria in oilsands tailings ponds of northern Alberta. ISME J. 2013, 7, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, S.Y.; Kim, P.J.; Madsen, E.L.; Jeon, C.O. Methane emission and dynamics of methanotrophic and methanogenic communities in a flooded rice field ecosystem. FEMS Microbiol. Ecol. 2014, 88, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Kamagata, Y.; Oshima, K.; Hanada, S.; Tamaki, H.; Marumo, K.; Maeda, H.; Nedachi, M.; Hattori, M.; Iwasaki, W. Methylocaldum marinum sp. nov., a thermotolerant, methane-oxidizing bacterium isolated from marine sediments, and emended description of the genus Methylocaldum. Int. J. Syst. Evol. Microbiol. 2014, 64, 3240–3246. [Google Scholar] [CrossRef]
- Eshinimaev, B.T.; Medvedkova, K.; Khmelenina, V.; Suzina, N.; Osipov, G.; Lysenko, A.; Trotsenko, Y.A. New thermophilic methanotrophs of the genus Methylocaldum. Microbiology 2004, 73, 448–456. [Google Scholar] [CrossRef]
- Ward, N.; Larsen, Ø.; Sakwa, J.; Bruseth, L.; Khouri, H.; Durkin, A.S.; Dimitrov, G.; Jiang, L.; Scanlan, D.; Kang, K.H. Genomic insights into methanotrophy: the complete genome sequence of Methylococcus capsulatus (Bath). PLoS Biol. 2004, 2, e303. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.; Varghese, N.; Seshadri, R.; et al. The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res. 2021, 49, D751–D763. [Google Scholar] [CrossRef]
- Mukherjee, S.; Stamatis, D.; Bertsch, J.; Ovchinnikova, G.; Sundaramurthi, Jagadish C.; Lee, J.; Kandimalla, M.; Chen, I.M.A.; Kyrpides, N.C.; Reddy, T.B.K. Genomes OnLine Database (GOLD) v.8: overview and updates. Nucleic Acids Res. 2021, 49, D723–D733. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.-i.; Toh, H.; Miyata, T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular evolution and phylogenetics; Oxford University Press, USA: 2000.
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen Ö, C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Vera Alvarez, R.; Landsman, D.; Koonin, E.V. COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef]
- Delmont, T.O.; Eren, A.M. Linking pangenomes and metagenomes: the Prochlorococcus metapangenome. PeerJ 2018, 6, e4320. [Google Scholar] [CrossRef]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+ C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef]
- Chen, L.-X.; Méheust, R.; Crits-Christoph, A.; McMahon, K.D.; Nelson, T.C.; Slater, G.F.; Warren, L.A.; Banfield, J.F. Large freshwater phages with the potential to augment aerobic methane oxidation. Nat. microbiol. 2020, 5, 1504–1515. [Google Scholar] [CrossRef]
- Bragagnolo, N.; Audette, G.F. Solution characterization of the dynamic conjugative entry exclusion protein TraG. Struct. Dyn. 2022, 9. [Google Scholar] [CrossRef]
- Lawley, T.; Klimke, W.; Gubbins, M.; Frost, L. F factor conjugation is a true type IV secretion system. FEMS Microbiol. Lett. 2003, 224, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bowden, M.G.; Pershad, R.; Kaplan, H.B. The Myxococcus xanthus rfbABC operon encodes an ATP-binding cassette transporter homolog required for O-antigen biosynthesis and multicellular development. J. Bacteriol. 1996, 178, 1631–1639. [Google Scholar] [CrossRef]
- Liu, B.; Furevi, A.; Perepelov, A.V.; Guo, X.; Cao, H.; Wang, Q.; Reeves, P.R.; Knirel, Y.A.; Wang, L.; Widmalm, G. Structure and genetics of Escherichia coli O antigens. FEMS Microbiol. Rev. 2020, 44, 655–683. [Google Scholar] [CrossRef] [PubMed]
- Merino, S.; Gonzalez, V.; Tomás, J.M. The first sugar of the repeat units is essential for the Wzy polymerase activity and elongation of the O-antigen lipopolysaccharide. Future Microbiol. 2016, 11, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Valvano, M.A. Export of O-specific lipopolysaccharide. Front. Biosci. (Landmark Ed.) 2003, 8, 452–471. [Google Scholar] [CrossRef]
- Balsanelli, E.; Serrato, R.V.; De Baura, V.A.; Sassaki, G.; Yates, M.G.; Rigo, L.U.; Pedrosa, F.O.; De Souza, E.M.; Monteiro, R.A. Herbaspirillum seropedicae rfbB and rfbC genes are required for maize colonization. Environ. Microbiol 2010, 12, 2233–2244. [Google Scholar] [CrossRef]
- Jefferies, D.; Shearer, J.; Khalid, S. Role of O-antigen in response to mechanical stress of the E. coli outer membrane: insights from coarse-grained MD simulations. J. Phys. Chem. B. 2019, 123, 3567–3575. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, H.; Huang, L.; Zhang, T.; Zong, B.; Ren, X.; Zhu, Y.; Song, F.; Wang, X.; Chen, H. Effect of O antigen ligase gene mutation on oxidative stress resistance and pathogenicity of NMEC strain RS218. Microb. Pathog. 2019, 136, 103656. [Google Scholar] [CrossRef]
- Manieri, F.Z.; Moreira, C.G. Salmonella Typhimurium O-antigen and VisP play an important role in swarming and osmotic stress response during intracellular conditions. Brazilian Journal of Microbiology 2022, 53, 557–564. [Google Scholar] [CrossRef]
- Lerouge, I.; Vanderleyden, J. O-antigen structural variation: mechanisms and possible roles in animal/plant–microbe interactions. FEMS Microbiol. Rev. 2002, 26, 17–47. [Google Scholar] [CrossRef] [PubMed]
- Eyice, Ö.; Myronova, N.; Pol, A.; Carrión, O.; Todd, J.D.; Smith, T.J.; Gurman, S.J.; Cuthbertson, A.; Mazard, S.; Mennink-Kersten, M.A. Bacterial SBP56 identified as a Cu-dependent methanethiol oxidase widely distributed in the biosphere. ISME J. 2018, 12, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.A.; Mohammadi, S.S.; Van Erven, T.; Berben, T.; Jetten, M.S.; Pol, A.; den Camp, H.J.O. Methanethiol consumption and hydrogen sulfide production by the thermoacidophilic methanotroph Methylacidiphilum fumariolicum SolV. Front. Microbiol. 2022, 13. [Google Scholar] [CrossRef]
- Börjesson, G. Inhibition of methane oxidation by volatile sulfur compounds (CH3SH and CS2) in landfill cover soils. Waste Manag. Res. 2001, 19, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Major, T.A.; Whitman, W.B. Role of the precorrin 6-X reductase gene in cobamide biosynthesis in Methanococcus maripaludis. Archaea 2005, 1, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, D.A.; Vitreschak, A.G.; Mironov, A.A.; Gelfand, M.S. Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J. Biol. Chem. 2003, 278, 41148–41159. [Google Scholar] [CrossRef]
- Santoro, A.E.; Dupont, C.L.; Richter, R.A.; Craig, M.T.; Carini, P.; McIlvin, M.R.; Yang, Y.; Orsi, W.D.; Moran, D.M.; Saito, M.A. Genomic and proteomic characterization of “Candidatus Nitrosopelagicus brevis”: an ammonia-oxidizing archaeon from the open ocean. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 1173–1178. [Google Scholar] [CrossRef]
- Heal, K.R.; Qin, W.; Ribalet, F.; Bertagnolli, A.D.; Coyote-Maestas, W.; Hmelo, L.R.; Moffett, J.W.; Devol, A.H.; Armbrust, E.V.; Stahl, D.A. Two distinct pools of B12 analogs reveal community interdependencies in the ocean. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 364–369. [Google Scholar] [CrossRef]
- Lu, X.; Heal, K.R.; Ingalls, A.E.; Doxey, A.C.; Neufeld, J.D. Metagenomic and chemical characterization of soil cobalamin production. ISME J. 2020, 14, 53–66. [Google Scholar] [CrossRef]
- Shelton, A.N.; Seth, E.C.; Mok, K.C.; Han, A.W.; Jackson, S.N.; Haft, D.R.; Taga, M.E. Uneven distribution of cobamide biosynthesis and dependence in bacteria predicted by comparative genomics. ISME J. 2019, 13, 789–804. [Google Scholar] [CrossRef]
- Chistoserdova, L.; Lidstrom, M.E. Molecular and mutational analysis of a DNA region separating two methylotrophy gene clusters in Methylobacterium extorquens AM1. Microbiology 1997, 143, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yu, Z.; Chistoserdova, L. Lanthanide-dependent methanol dehydrogenases of XoxF4 and XoxF5 clades are differentially distributed among methylotrophic bacteria and they reveal different biochemical properties. Front. Microbiol. 2018, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Keltjens, J.T.; Pol, A.; Reimann, J.; den Camp, H.J.O. PQQ-dependent methanol dehydrogenases: rare-earth elements make a difference. Appl. Microbiol. Biotechnol. 2014, 98, 6163–6183. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L.; Crowther, G.J.; Vorholt, J.A.; Skovran, E.; Portais, J.-C.; Lidstrom, M.E. Identification of a fourth formate dehydrogenase in Methylobacterium extorquens AM1 and confirmation of the essential role of formate oxidation in methylotrophy. J. Bacteriol. 2007, 189, 9076–9081. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L.; Laukel, M.; Portais, J.-C.; Vorholt, J.A.; Lidstrom, M.E. Multiple formate dehydrogenase enzymes in the facultative methylotroph Methylobacterium extorquens AM1 are dispensable for growth on methanol. J. Bacteriol. 2004, 186, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Laukel, M.; Chistoserdova, L.; Lidstrom, M.E.; Vorholt, J.A. The tungsten-containing formate dehydrogenase from Methylobacterium extorquens AM1: purification and properties. Eur. J. Biochem. 2003, 270, 325–333. [Google Scholar] [CrossRef]
- Khmelenina, V.N.; Murrell, J.C.; Smith, T.J.; Trotsenko, Y.A. Physiology and biochemistry of the aerobic methanotrophs. In Aerobic utilization of hydrocarbons, oils and lipids; Springer: 2018; pp. 1–25.
- Schmitz, R.A.; Peeters, S.H.; Versantvoort, W.; Picone, N.; Pol, A.; Jetten, M.S.; Op den Camp, H.J. Verrucomicrobial methanotrophs: ecophysiology of metabolically versatile acidophiles. FEMS Microbiol. Rev. 2021, 45, fuab007. [Google Scholar] [CrossRef]
- Stein, L.Y.; Roy, R.; Dunfield, P.F. Aerobic methanotrophy and nitrification: processes and connections. eLS 2012. [Google Scholar] [CrossRef]
- Gazioglu, O.; Kareem, B.O.; Afzal, M.; Shafeeq, S.; Kuipers, O.P.; Ulijasz, A.T.; Andrew, P.W.; Yesilkaya, H. Glutamate Dehydrogenase (GdhA) of Streptococcus pneumoniae is required for high temperature adaptation. Infect. Immun. 2021, 89. [Google Scholar] [CrossRef]
- Luangthongkam, P. Biosynthesis of Polyhydroxyalkanoates (PHAs) in methane-utilizing mixed cultures. Ph.D. thesis. St Lucia QLD: The University of Queensland. 2019.
- Takeuchi, M. Methylocaldum. Bergey’s Manual of Systematics of Archaea and Bacteria . 2016; 1–5. [Google Scholar] [CrossRef]
- Medvedkova, K.; Khmelenina, V.; Baskunov, B.; Trotsenko, Y.A. Synthesis of melanin by a moderately thermophilic methanotroph Methylocaldum szegediense depends on cultivation temperature. Microbiology 2008, 77, 112–114. [Google Scholar] [CrossRef]
- Singh, S.; Nimse, S.B.; Mathew, D.E.; Dhimmar, A.; Sahastrabudhe, H.; Gajjar, A.; Ghadge, V.A.; Kumar, P.; Shinde, P.B. Microbial melanin: Recent advances in biosynthesis, extraction, characterization, and applications. Biotechnol. Adv. 2021, 53, 107773. [Google Scholar] [CrossRef]
- Donoso, R.A.; Ruiz, D.; Gárate-Castro, C.; Villegas, P.; González-Pastor, J.E.; de Lorenzo, V.; Gonzalez, B.; Pérez-Pantoja, D. Identification of a self-sufficient cytochrome P450 monooxygenase from Cupriavidus pinatubonensis JMP134 involved in 2-hydroxyphenylacetic acid catabolism, via homogentisate pathway. Microb. Biotechnol. 2021, 14, 1944–1960. [Google Scholar] [CrossRef] [PubMed]
- Galeb, H.A.; Lamantia, A.; Robson, A.; König, K.; Eichhorn, J.; Baldock, S.J.; Ashton, M.D.; Baum, J.V.; Mort, R.L.; Robinson, B.J. The Polymerization of Homogentisic Acid In Vitro as a Model for Pyomelanin Formation. Macromol. Chem. Phys. 2022, 223, 2100489. [Google Scholar] [CrossRef]
- Singh, D.; Kumar, J.; Kumar, A. Isolation of pyomelanin from bacteria and evidences showing its synthesis by 4-hydroxyphenylpyruvate dioxygenase enzyme encoded by hppD gene. Int. J. Biol. Macromol. 2018, 119, 864–873. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A) Maximum likelihood tree representing the phylogenetic relationship of Methylocaldum based on 16S rRNA gene sequences. Leaf names with the strains analyzed in this article are in bold. Identifiers for each IMG gene ID are at the tip of each leaf. (B) Dendogram showing the association between cluster of groups of orthologous genes, represented by thin vertical black lines, which when grouped form the rectangular blocks depicted on each genome. The clustering is based on the presence or absence of the orthologous genes on each genome. Average nucleotide identity (ANI) times the aligned fraction (AF) percentage of each genome when compared with one of the other Methylocaldum strains are shown inside of blue or light blue squares.
Figure 1.
(A) Maximum likelihood tree representing the phylogenetic relationship of Methylocaldum based on 16S rRNA gene sequences. Leaf names with the strains analyzed in this article are in bold. Identifiers for each IMG gene ID are at the tip of each leaf. (B) Dendogram showing the association between cluster of groups of orthologous genes, represented by thin vertical black lines, which when grouped form the rectangular blocks depicted on each genome. The clustering is based on the presence or absence of the orthologous genes on each genome. Average nucleotide identity (ANI) times the aligned fraction (AF) percentage of each genome when compared with one of the other Methylocaldum strains are shown inside of blue or light blue squares.

Figure 2.
Synteny of methane monooxygenase (MMO) gene clusters in Methylocaldum genomes. Schematics for genomes of M. marinum S8, M. szegediense O-12 and M. szegediense Norfolk depicting the locus of MMO gene clusters. Ribbons connecting genomes show pmoC genes with high identity, as part of pMMO gene cluster (purple), and as stand-alone genetic component (pink and magenta). Black-edge triangles indicate stand-alone copies of pmoC without high identity to any other pmoC within the three genomes. The gene cluster for soluble methane monooxygenase, sMMO, only found on M. marinum S8 is also shown.
Figure 2.
Synteny of methane monooxygenase (MMO) gene clusters in Methylocaldum genomes. Schematics for genomes of M. marinum S8, M. szegediense O-12 and M. szegediense Norfolk depicting the locus of MMO gene clusters. Ribbons connecting genomes show pmoC genes with high identity, as part of pMMO gene cluster (purple), and as stand-alone genetic component (pink and magenta). Black-edge triangles indicate stand-alone copies of pmoC without high identity to any other pmoC within the three genomes. The gene cluster for soluble methane monooxygenase, sMMO, only found on M. marinum S8 is also shown.
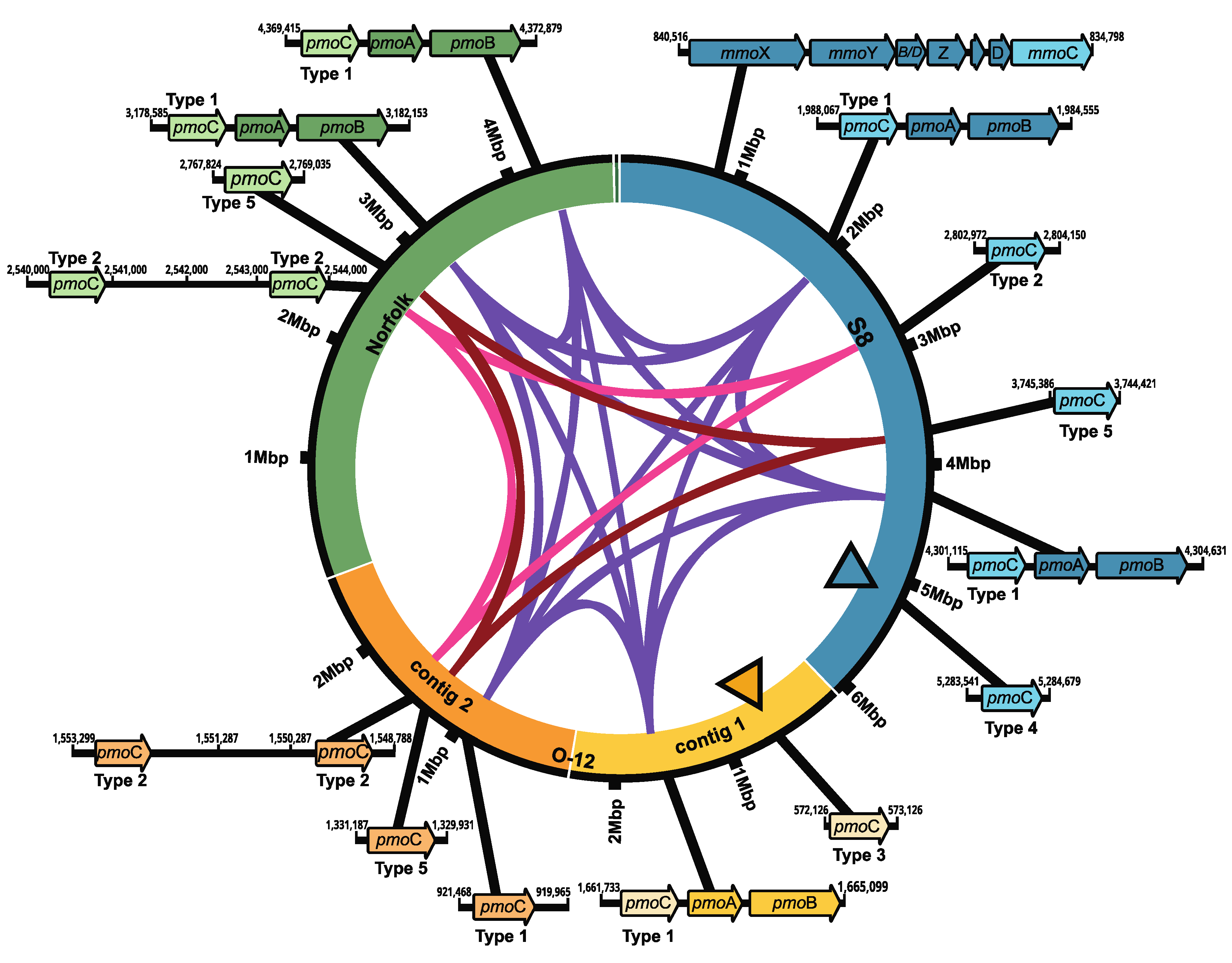
Figure 4.
Phylogenetic reconstruction based on all xoxF and mxaF genes found in the three Methylocaldum genomes analyzed (highlighted in bold). The corresponding IMG gene ID are at the tip of each leaf. Collapsed clades did not contain any gene from the three analyzed genomes.
Figure 4.
Phylogenetic reconstruction based on all xoxF and mxaF genes found in the three Methylocaldum genomes analyzed (highlighted in bold). The corresponding IMG gene ID are at the tip of each leaf. Collapsed clades did not contain any gene from the three analyzed genomes.
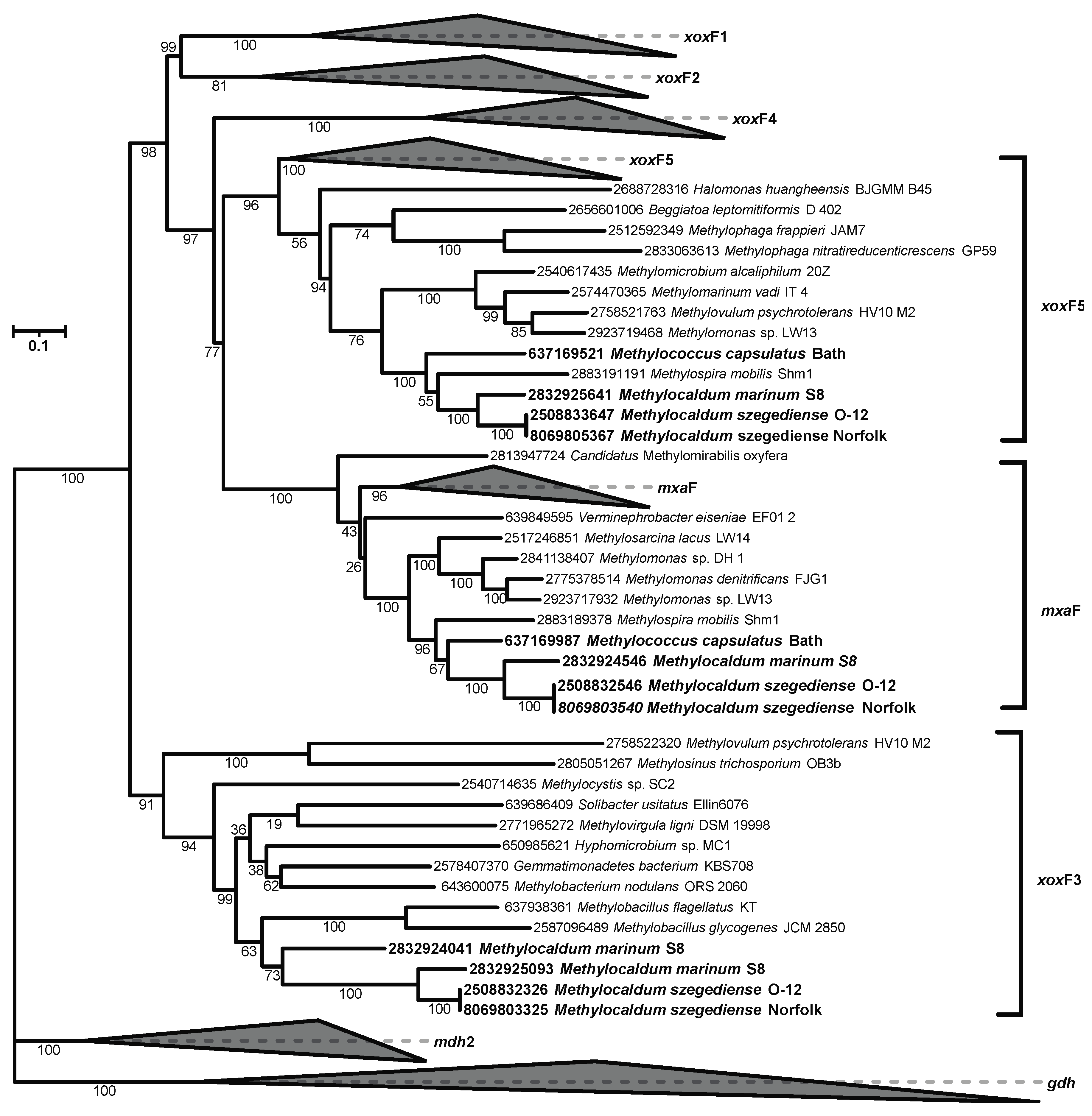
Figure 6.
Nitrogen metabolism genes in Methylocaldum strains. (A) Diagram representing the presence and absence of genes involved in nitrogen cycling. (B) Synteny of nitrogenase gene cluster present in the three Methylocaldum strains. Pairwise identity percentage is indicated inside ribbons connecting homologous genes.
Figure 6.
Nitrogen metabolism genes in Methylocaldum strains. (A) Diagram representing the presence and absence of genes involved in nitrogen cycling. (B) Synteny of nitrogenase gene cluster present in the three Methylocaldum strains. Pairwise identity percentage is indicated inside ribbons connecting homologous genes.
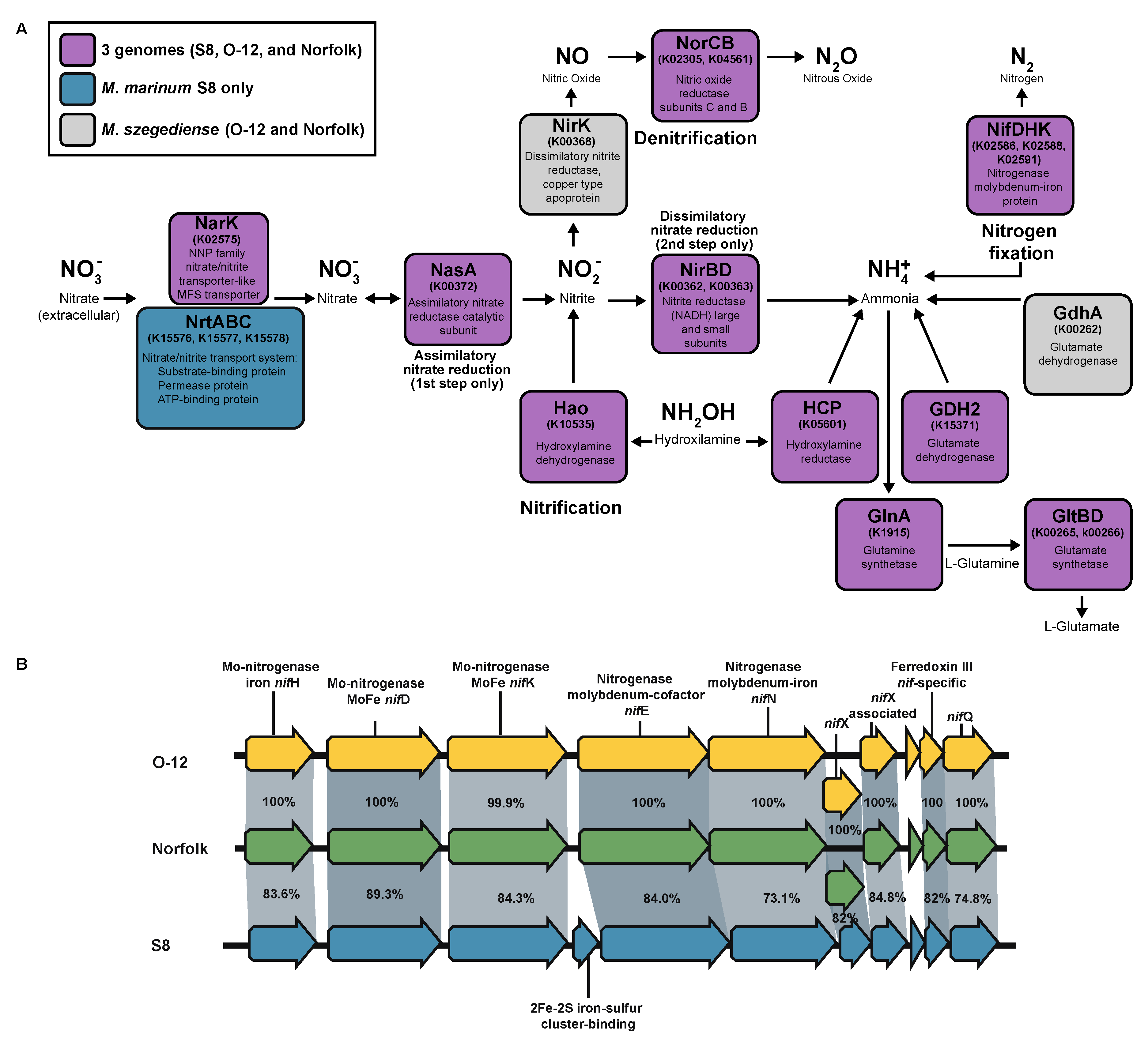
Figure 7.
Proposed pyomelanin/HGA-melanin synthesis pathway via accumulation of homogentisic acid (HGA) in Methylocaldum. (A) Schematic representation of tyrosine degradation pathway having HGA as intermediate synthetized by 4-hydroxyphenylpyruvate dioxygenase (hppD, HPD, K00457), which can be degraded via homogentisate 1,2-dioxygenase (hmgA, HMG, K00451) to be reincorporated into central metabolism as fumarate and acetoacetate or accumulated and converted to pyomelanin/HGA-melanin via polymerization of the monomer benzoquinone acetate. (B) Synteny of gene cluster where the hppD gene (red-edge arrows) was present in the three Methylocaldum strains. Pairwise identity percentage is indicated inside ribbons connecting homologous genes.
Figure 7.
Proposed pyomelanin/HGA-melanin synthesis pathway via accumulation of homogentisic acid (HGA) in Methylocaldum. (A) Schematic representation of tyrosine degradation pathway having HGA as intermediate synthetized by 4-hydroxyphenylpyruvate dioxygenase (hppD, HPD, K00457), which can be degraded via homogentisate 1,2-dioxygenase (hmgA, HMG, K00451) to be reincorporated into central metabolism as fumarate and acetoacetate or accumulated and converted to pyomelanin/HGA-melanin via polymerization of the monomer benzoquinone acetate. (B) Synteny of gene cluster where the hppD gene (red-edge arrows) was present in the three Methylocaldum strains. Pairwise identity percentage is indicated inside ribbons connecting homologous genes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



