Submitted:
17 January 2024
Posted:
17 January 2024
You are already at the latest version
Abstract
Neurocutaneous disorders, also known as phakomatoses, are congenital and acquired syndromes resulting in simultaneous neurologic and cutaneous involvement. In several of these conditions, the genetic phenomenon is understood, providing a pivotal role in the development of therapeutic options. This review encompasses the discussion of the genetic and clinical involvement of neurocutaneous disorders and examines clinical management and treatment options. With the current advances in genetics, the role of precision medicine and targeted therapy plays a substantial role in addressing management of these conditions. The interconnectedness between therapeutic options highlights the importance of precision medicine in treating each disorder’s unique molecular pathway. This review provides an extensive synthesis of ongoing and current therapeutics in the management of such clinically unique and challenging conditions.
Keywords:
neurocutaneous disorders
; cutaneous
; neurologic
; phakomatoses
; genetic advances
; precision medicine
1. Introduction
Neurocutaneous disorders are a group of conditions which result in long term involvement of the nervous system and skin. Often, they are genetically inherited, and symptoms emerge throughout adolescents [1]. Many of these conditions involve an understood genetic phenomenon, although spontaneous mutations can occur. These conditions also exist on a wide spectrum of phenotypes and often have multi-system involvement [1]. Given the various systems involved and the varying difference in presentation, treatment is often difficult and focuses on palliative efforts. However, given the advances in genetics, targeted therapies offer a promising future for the development of treatments in the management of neurocutaneous disorders, offering patients a tailored therapeutic option.
2. Neurofibromatosis 1 & 2
Neurofibromatosis 1, also known as von Recklinghausen disease, makes up 96% of all neurofibromatosis cases and is an autosomal dominant neurocutaneous disorder caused by a germline mutation in the NF1 gene [2,3]. The NF1 gene is a tumor suppressor gene on chromosome 17 that promotes the production of neurofibromin, a protein that helps regulate the cell cycle by inhibiting RAS/MAPK and PI3K-AKT-mTOR signaling pathways [4,5]. Mutation of the NF1 gene leads to the production of nonfunctional or inadequate amounts of neurofibromin which is associated with an increased risk of various tumors [6].
Figure 1.
Pathogenesis of Neurofibromatosis 1. Mutations in the NF1 gene may result in a nonfunctional neurofibromin protein. Neurofibromin is a tumor suppressor that downregulates the RAS/MAPK and PI3K-AKT-mTOR signaling pathways which play a major role in cell proliferation. When these pathways go unchecked, the risk for tumor formation increases, resulting in the development of tumors in patients with neurofibromatosis 1.
Figure 1.
Pathogenesis of Neurofibromatosis 1. Mutations in the NF1 gene may result in a nonfunctional neurofibromin protein. Neurofibromin is a tumor suppressor that downregulates the RAS/MAPK and PI3K-AKT-mTOR signaling pathways which play a major role in cell proliferation. When these pathways go unchecked, the risk for tumor formation increases, resulting in the development of tumors in patients with neurofibromatosis 1.
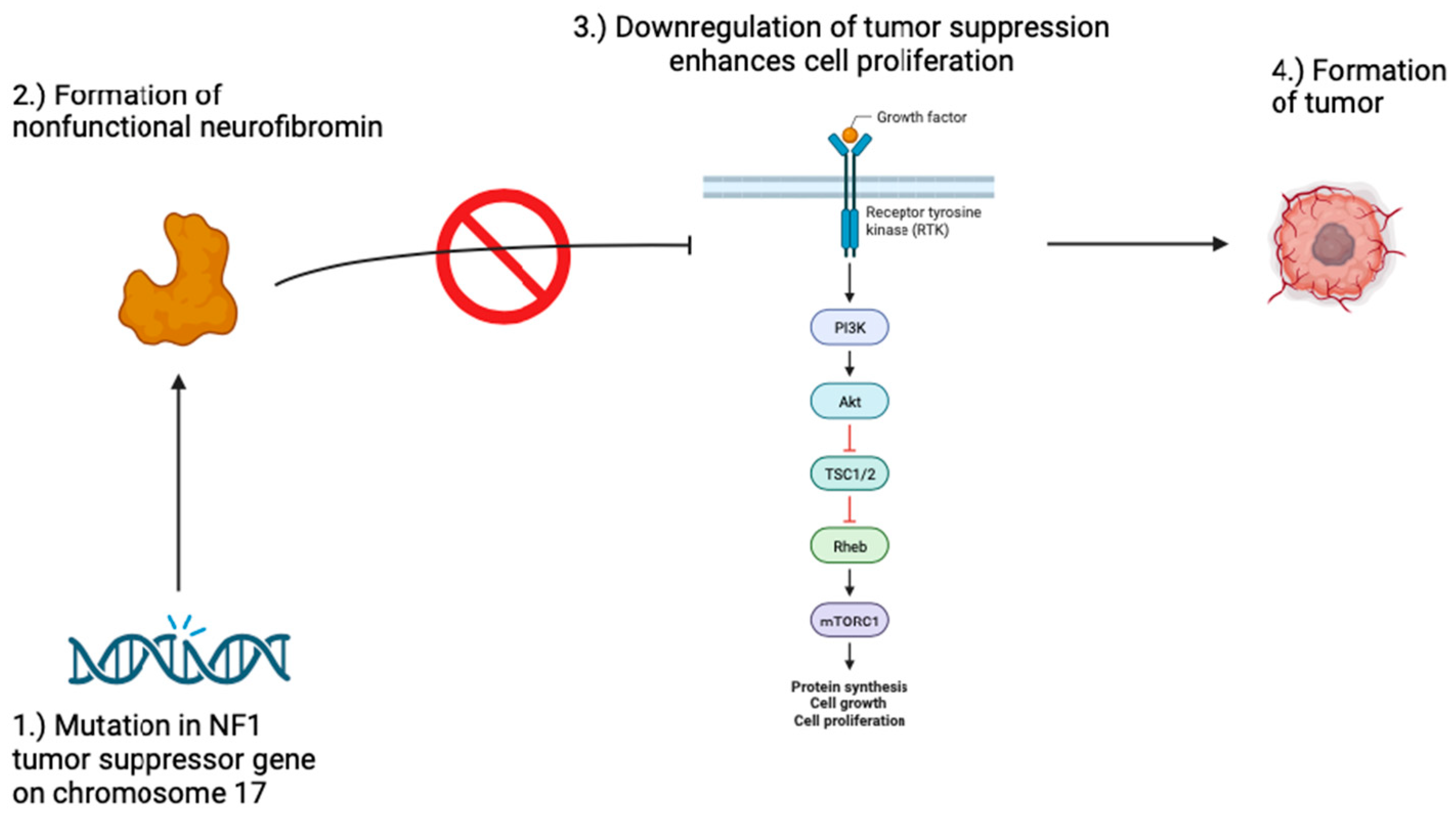
Neurofibromatosis 1 presents clinically with neurofibromas, café-au-lait spots, Lisch nodules, freckles, bone abnormalities like bowing of the legs or curvature of the spine, learning disabilities, and increased risk for tumor formation [7,8]. Additionally, Neurofibromatosis 1 can present with mosaicism and cutaneous manifestations may present only in affected body regions [9,10].
Diagnostic criteria according to National Institutes of Health for neurofibromatosis 1 should include more than two of the following: cafe’- au-lait spots, plexiform neurofibroma, Lisch nodules, optic glioma, freckling, first-degree relative with neurofibromatosis, or dysplasia of cortical bones [11,12,13]. Genetic testing has been gaining significance as a tool to distinguish neurofibromatosis 1 from other conditions and identify mosaicism in patients [6].
There is currently no cure for neurofibromatosis 1 and current treatment aims to prevent complications and improve patient quality of life. Regular monitoring is recommended for tracking the progression of the patient’s condition and to detect any complications early. Surgical management of painful or uncomfortable neurofibromas may not prevent the recurrence of the neurofibromas but may provide the patient with pain relief and improve quality of life. Selumetinib is the first FDA-approved treatment for inoperable plexiform neurofibroma in patients with neurofibromatosis 1 that seeks to induce antitumor activity by inhibiting the MEK pathway [14,15,16]. Selumetinib shows promise, but more research is needed.
Neurofibromatosis 2 is an autosomal dominant neurocutaneous disorder caused by mutations in the NF2 gene on chromosome 22q12. The NF2 gene codes for the formation of the protein, Merlin (schwannomin), a tumor suppressor that is associated with the formation of central nervous system tumors upon NF2 gene mutation [17,18]. Approximately half of neurofibromatosis 2 cases occur because of spontaneous mutations despite being an autosomal dominant disorder [19]. Like neurofibromatosis 1, neurofibromatosis 2 can exhibit mosaicism and symptoms can localize in regions of affected cells. The severity of the disorder is variable and can present early on in life with multiple tumors, or later in life with a milder presentation [20].
Cutaneous manifestations of neurofibromatosis 2 are less prominent compared to neurofibromatosis 1 and do not commonly include the dermatological findings normally seen in neurofibromatosis 1. Distinct features of neurofibromatosis 2 mainly include the development of tumors that arise from Schwann cells like vestibular schwannomas which can be associated with tinnitus, hearing loss, and balance dysfunction. Meningiomas and ependymomas are also common [21]. Additionally, neurofibromatosis 2 may present with cataracts, retinal abnormalities, and rarely with cutaneous schwannomas under the skin [2].
The less prominent cutaneous manifestations of neurofibromatosis 2 help distinguish it from neurofibromatosis 1 clinically. Diagnosis of neurofibromatosis 2 mainly involves clinical presentation of the associated tumors, family history of NF2, and genetic testing [22,23].
While neurofibromatosis 2 associated tumors are largely benign, lack of treatment can lead to auditory, facial, and vestibular functional deficiencies and extreme cases can lead to brainstem compression, obstructive hydrocephalus, and death. Surgery and radiation are treatment options that pose major risks like hearing loss, intracranial bleeding, and stroke. Additionally, surgical removal is not possible in multiple occasions where tumors commonly arise in patients with neurofibromatosis 2, and radiation treatment may transform or accelerate the growth of schwannomas. As a result, neurofibromatosis 2 does not have a curative or long-term therapy [24,25].
3. Tuberous Sclerosis
Tuberous sclerosis complex (TSC) represents a neurocutaneous genetic disorder characterized by the development of benign tumors known as hamartomas [28,29]. The clinical expression of TSC arises from disruptions in various cellular functions. Genomically, TSC results from autosomal dominant or sporadic mutations in the TSC1 or TSC2 genes, encoding the hamartin and tuberin proteins, respectively [29,30,31,32,33]. These proteins jointly regulate cell proliferation through the mammalian target of the rapamycin pathway (mTOR). Mutations in either gene lead to dysregulation of the TSC1:TSC2 complex, consequently affecting the mTOR pathway and causing tissue overgrowth [34].
Clinically, TSC impacts multiple organ systems (figure #), with a predilection for the central nervous system (CNS), skin, and kidneys [28]. Most common CNS symptoms encompass seizures, subependymal nodules (SENs), and subependymal giant cell astrocytomas (SEGA) [35]. Renal complications arise from renal angiomyolipomas, which are benign tumors forming in the kidneys. Lastly, cutaneous manifestations involve hypomelanotic macules, adenoma sebaceum, shagreen patches, and ungual fibromas [36].
Figure 2.
Multisystem manifestations of TSC. Multisystemic manifestations of TSC include the nervous system, skin, lungs, heart, kidneys, and eyes, presenting a variable phenotype.
Figure 2.
Multisystem manifestations of TSC. Multisystemic manifestations of TSC include the nervous system, skin, lungs, heart, kidneys, and eyes, presenting a variable phenotype.
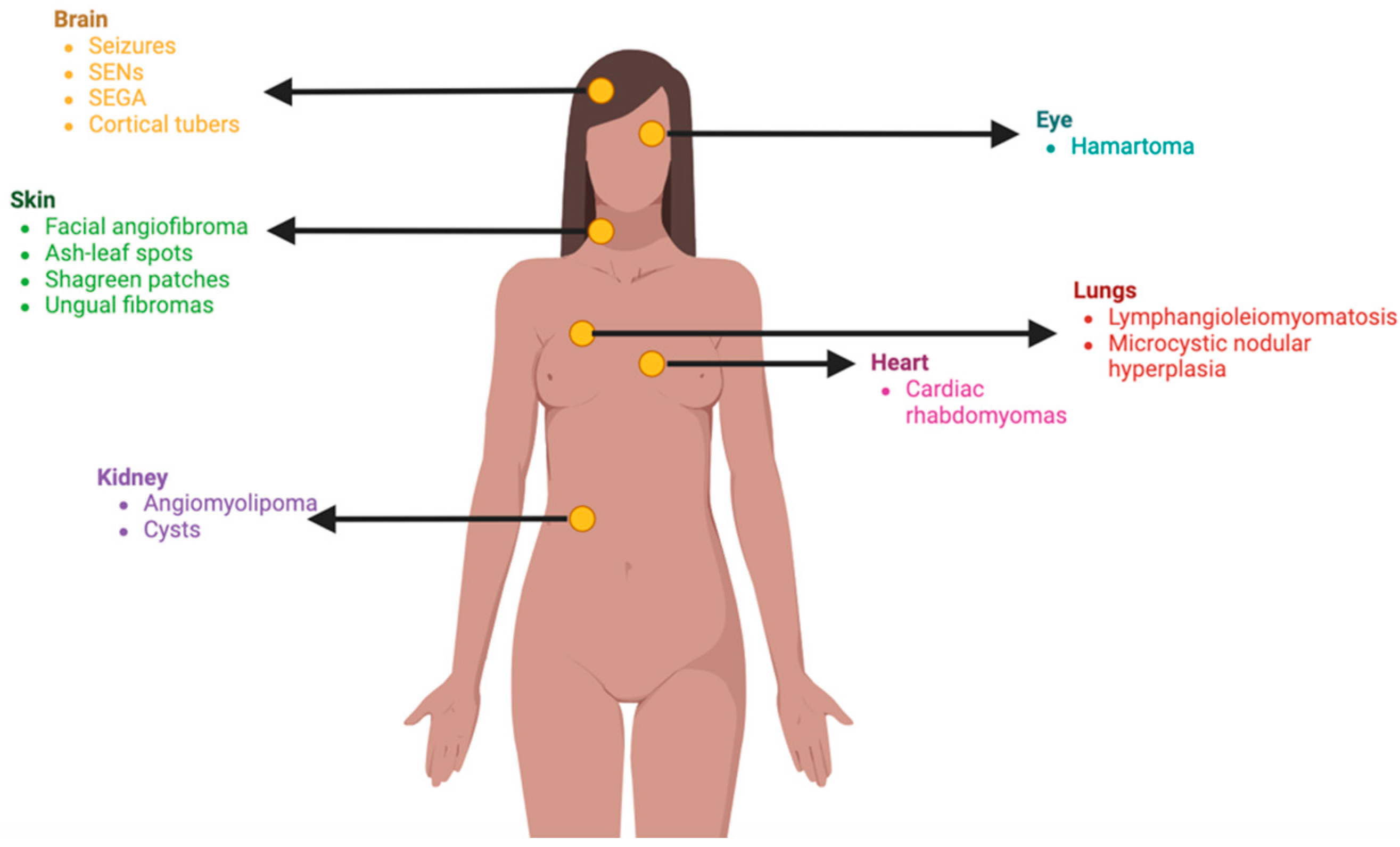
Regarding dermatologic abnormalities, approximately 90% of patients develop hypomelanotic macules, also known as ash leaf spots [28,37,38]. These are oval-shaped characteristic that are usually present at birth, and a Wood’s lamp examination improves its detection [39]. Adenoma sebaceum, formerly known as facial angiofibroma, manifests as small hyperpigmented macules in a butterfly pattern on the bilateral cheeks [39]. Shagreen patches are leather plaques with an orange-peel appearance due to slightly depressed hair follicles [39]. Ungual fibromas are commonly observed periungually and subungually, particularly in toenails, emerging later in puberty [39].
Diagnosing TSC relies on a constellation of features rather than a singular symptom. Definitive diagnostic criteria necessitate two major features or one major and two or more minor features. Major features, observed more frequently in TSC patients, include 11 clinical findings uncommon in the general population [40]. Minor features, also more prevalent in TSC patients, are common in the general population [40]. Additionally, imaging studies, such as CT and MRI, are crucial for assessing CNS manifestations. A genetic testing is also conducted to identify mutations in the TSC1 or TSC2 genes.
Treatment options for TSC-associated skin lesions encompass both non-pharmacological and pharmacological approaches. Systemic mTOR inhibitor treatment is indicated in patients with multisystemic manifestations, addressing the underlying TSC pathophysiology and potentially improving dermatologic symptoms [40]. Alternatively, topical mTOR inhibitors or surgical procedures may be considered in patients not receiving systemic treatment [40].
Notably, in the year 2022, the Food and Drug Administration (FDA) granted approval for HYFTOR (sirolimus) gel, the first FDA-approved topical intervention designed for individuals with TSC-presenting facial angiofibroma [41]. Lastly, diverse surgical modalities are also available, including laser-based procedures. For elevated angiofibromas, ablative lasers such as carbon dioxide (CO2) or erbium: YAG laser are frequently used [42]. Additionally, a vascular laser, capable of selectively obliterating blood vessels with minimal scarring risk, can be employed for the treatment of flat red spots [42].
4. Sturge-Weber-Syndrome
Sturge-Weber Syndrome (SWS) is a rare neurocutaneous disorder, distinctively marked by port-wine vascular macules and café-au-lait spots on the skin and eyes [43,44,45]. Unlike many genetic conditions, SWS is not hereditary but stems from a sporadic mutation in the GNAQ gene, leading to a range of abnormalities in the eyes, skin, and brain with varied clinical presentations, from asymptomatic to severe [46,47].
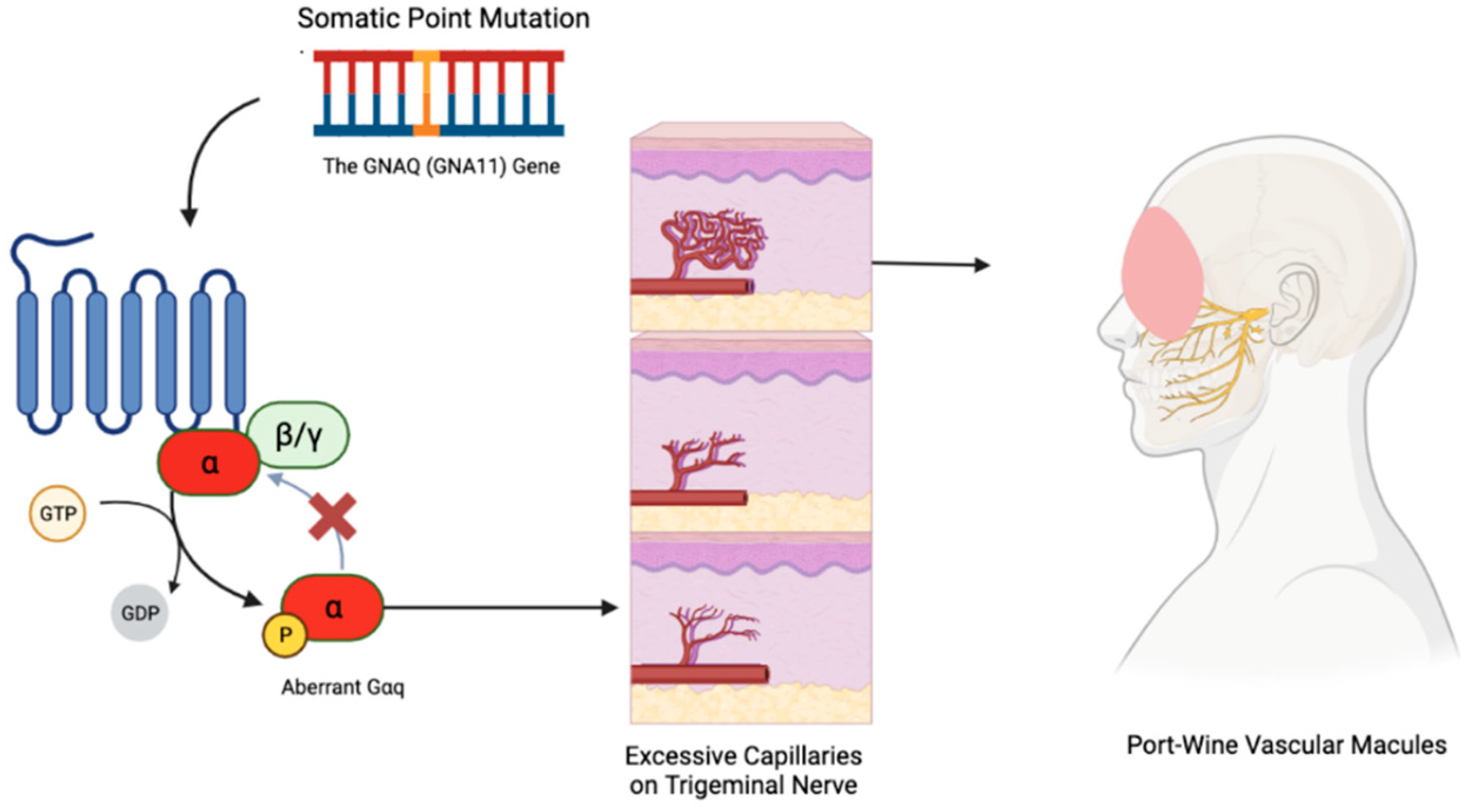
The underlying mechanism involves a postzygotic somatic mutation in GNAQ, sometimes also implicating the homogenous GNA11 gene [46]. These mutations create a defect in the Gαq protein that doesn’t allow it to properly shut-off, contributing to conditions like phakomatosis pigmentovascularis through mosaic expression [48,49,50]. SWS’s port-wine lesions result from excessive capillaries around the trigeminal nerve in the face, refer to Figure 1, and abnormal brain vessels that can lead to tissue atrophy and tram-line calcification [51,52,53,54]. Diagnosis primarily relies on clinical observation and imaging studies like MRI and CT scans, which reveal the extent of cerebral involvement [53].
Neurologically, seizures are a significant concern in SWS, often correlating with the size of leptomeningeal angiomas [55]. The onset age of seizures is crucial for prognosis, with early-onset seizures linked to more significant neurological challenges [56]. Glaucoma, another critical aspect of SWS, tends to emerge in early childhood and can lead to vision loss if not managed promptly. The variable nature of glaucoma’s progression and visual outcomes underscores the importance of early intervention [57,58].
The prevalence of autism spectrum disorders and communication difficulties in SWS patients highlights the need for comprehensive neurodevelopmental assessment [59]. Furthermore, the disorder can lead to a range of developmental and neuropsychiatric comorbidities, significantly affecting psychological functioning, especially in younger patients [60].
SWS management requires a personalized, interdisciplinary approach. Laser therapy, particularly pulsed dye laser, is widely recognized for treating port-wine stains [61]. In terms of neurological complications, anticonvulsant therapy is crucial for seizure management, tailored to the seizure type and patient’s age [62]. For refractory cases, surgical options like hemispherectomy might be necessary, though they carry significant risks [58,63]. Glaucoma management often involves a combination of medical and surgical interventions, with regular ophthalmological assessments being essential for early detection and treatment [59]. Additionally, addressing neurodevelopmental and psychological aspects through early intervention programs and neuropsychological evaluations is vital for managing developmental delays, learning disabilities, and neuropsychiatric issues [43,60].
Recent discoveries, particularly the identification of the GNAQ gene mutation, have improved our understanding of SWS and opened possibilities for targeted molecular therapies [43,64]. Advances in MRI technology have enhanced the visualization of leptomeningeal angiomas, aiding in early diagnosis and informing treatment decisions, especially concerning epilepsy management [65]. However, treatment strategies, especially regarding epilepsy and cutaneous symptoms, remain subjects of debate. Discussions are ongoing about the timing, aggressiveness of seizure management, and the balance between medication side effects and seizure control [66]. Surgical interventions in intractable cases and the optimal timing and type of laser therapy for port-wine stains are also debated, considering the risks of skin damage and the psychological impact [67,68].
Figure 3.
How the Vascular Macules Manifest in Sturge-Weber Syndrome. Sturge-Weber Syndrome results from a sporadic mutation in the GNAQ gene. This mutation leads to impaired function of the Gαq protein, leading to excess capillary proliferation in a V1/V2 distribution on the face. This results in the classic port-wine stain presentation.
Figure 3.
How the Vascular Macules Manifest in Sturge-Weber Syndrome. Sturge-Weber Syndrome results from a sporadic mutation in the GNAQ gene. This mutation leads to impaired function of the Gαq protein, leading to excess capillary proliferation in a V1/V2 distribution on the face. This results in the classic port-wine stain presentation.
5. Von-Hippel-Lindau Disease
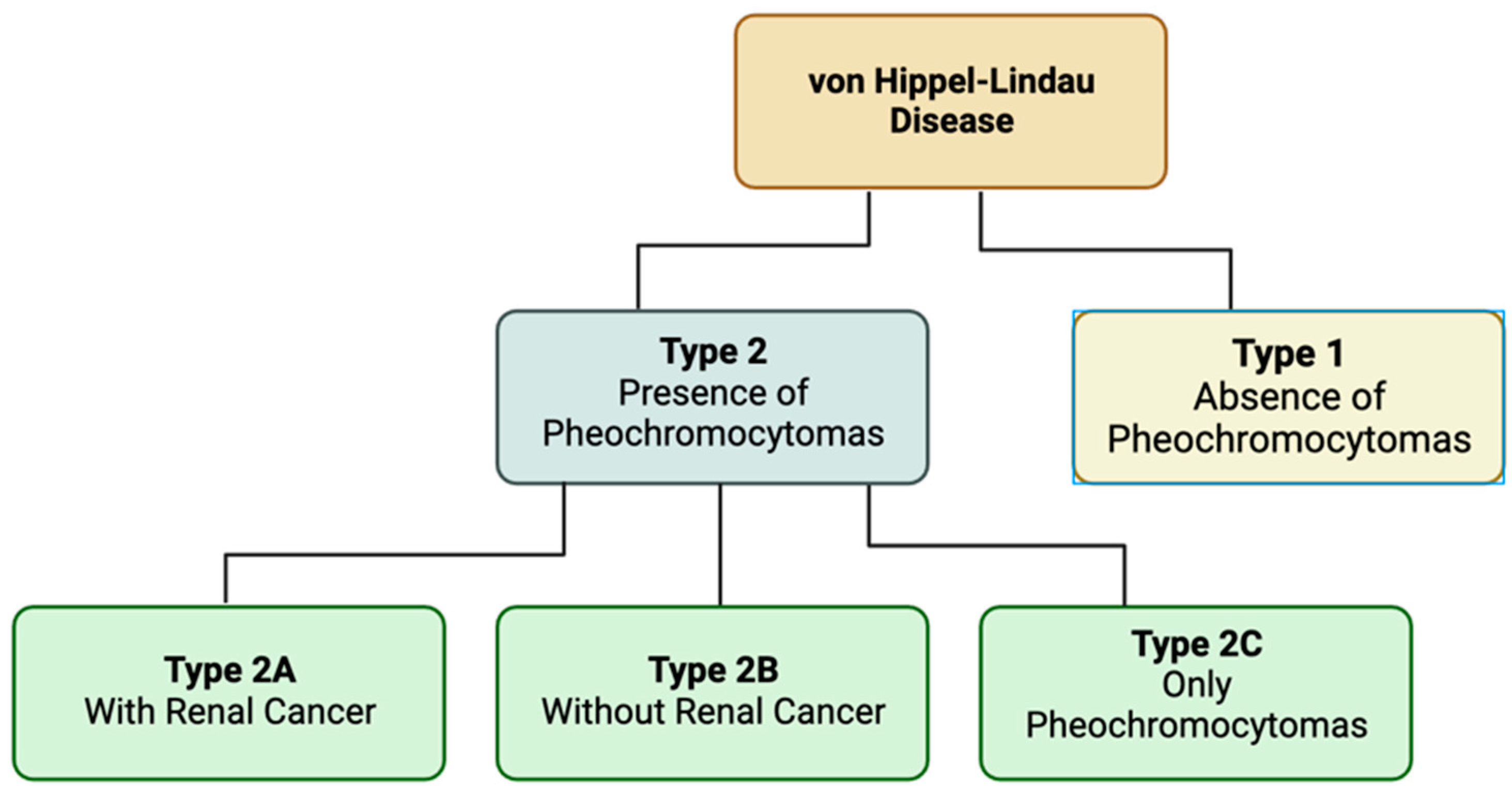
Von Hippel-Lindau (VHL) disease is a rare genetic disorder that causes a variety of tumors and cysts to manifest throughout various organ systems [69]. These tumors can manifest as benign or malignant and show commonly in the central nervous system, kidneys, adrenal glands, and pancreas. Most notably, Von Hippel-Lindau was found to be the first renal cancer disorder with a defined genetic basis [70]. Specifically, this relates to inherited renal cell carcinoma. Another common manifestation of the disease are hemangioblastomas especially those in the eye [71]. There are two different types of VHL dependent on the presence of pheochromocytoma and the additional presence of renal cancers. Pheochromocytomas are benign tumors typically found in the chromaffin cells of the adrenal medulla or paraganglion [72]. Type 1 VHL are classified via the absence of pheochromocytoma. VHL type 2 are characterized by the presence of pheochromocytomas and further break down into three subtypes. Type 2A is with renal cancer. Type 2B is without renal cancer. Type 2C is individuals who only develop pheochromocytomas [73].
Von Hippel-Lindau is an autosomal dominant disease. VHL is rare and found in about 1 in 36,000 people, and it has a de novo manifestation rate of about 20% in new cases [74,75]. It is an inherited germline mutation of the VHL gene on chromosome 3p25 [76]. This leads to a mutated end product of the VHL gene, the VHL protein (pVHL). This mutation causes the degradation of the of the hypoxia-inducible factor (HIF) which is responsible for oxygen regulation in cells [69]. When this protein is degraded HIF is uncontrolled and upregulated along with other growth factors promoting tumor formation. The platelet derived growth factor (PDGF) and vascular endothelial growth factors (VEGF) contribute significantly to tumor formation [74]. The HIFs encourage an angiogenic state of continuous mitogenic signaling [77]. Overall, this combination of growth factors leads to an environment where tumors can grow unchecked and cause a variety of lesions throughout the body.
It is important to note that Von Hippel-Lindau has a variety of cutaneous manifestations. While most of the effects of the disease are internal, about five percent of cases do show significant cutaneous features [78]. However, VHL is a phakomatoses with very few and sporadic cutaneous findings. These are seen as melanocytic nevi, café au lait spots, and capillary malformations [79]. Melanocytic nevi (non-cancerous moles) are a common benign neoplasm of the skin. They appear on the skin as darkened noncancerous moles, and they affect the melanocytes responsible for skin pigment synthesis [80]. This is important to consider in patients showing this cutaneous manifestation of the disease as melanomas arise from nevi about 25-33% of the time [81]. Café au lait spots are hyperpigmented spots that are dark to light brown and often appear at birth or early in life [82]. Typically, they are flat and vary in size. The presence of multiple spots is often linked to genetic syndromes as seen in the association with VHL. While these are uncommon findings in patients with Von Hippel-Lindau disease, they are a key feature that put them in the category of neurocutaneous diseases and can help in the diagnosis and treatment of patients. It is unclear whether the type of VHL determines the likelihood of developing cutaneous manifestations.
The diagnosis of Von Hippel-Lindau uses a mix of clinical criteria involving manifestations and genetics/family history of the disease along with genetic testing to confirm a diagnosis. There are three main clinical diagnostic criteria used in proband: Dutch, Danish, and International [83]. Each vary slightly in what they utilize for family history of VHL or a related tumor and individual diagnosis of a VHL related manifestation [84]. The Dutch and International criteria require one VHL-related tumor and a first or second degree relative diagnosed with VHL or with more than one VHL related tumor. The Danish criteria is stricter in that it only requires a relative to the first degree to be positive for VHL [84]. If no known family history is available, then the Dutch criteria requires two or more VHL related manifestations to be present. The Danish and International criteria require either two or more hemangioblastomas or one hemangioblastoma and one other VHL related manifestation to be present in order to be considered for this disease [84]. Molecular diagnosis or genetic testing is used to find a heterozygous pathogenic variant of pVHL and is a conclusive way to obtain a diagnosis [69]. The two types of genetic testing are gene targeted testing and comprehensive genomic testing. This includes single and multigene panel tests. The comprehensive genomic testing consists of either exome or genome sequencing. These criteria are the standard combination for assessing an individual’s likelihood of having the disease and follows through with genetic testing to confirm a diagnosis.
The management and treatment of Von Hippel-Lindau disease varies in each case as the manifestations of the disease vary. There is no standard or universal treatment for VHL, but options exist for the varying symptoms that may arise. Targeted therapies for advanced renal cell carcinoma include the oral treatment Pazopanib that inhibits VEGF, PEGF, and the stem-cell factor receptor c-kit [85]. Similarly, Belzutifan is a newly FDA-approved, oral drug that is used for the treatment of renal cell carcinoma in VHL. It is an inhibitor of HIF-2α and has been very effective in treatment [86]. Renal cell carcinoma can also be treated using radiofrequency ablation or cryoablation over surgical resections [87]. Most tumors in VHL are safe to be removed. Central nervous system hemangioblastomas and selected spinal hemangioblastomas are safe and allow for full tumor resection [88,89]. Retinal hemangioblastomas are treated early as they can lead to vision loss. Treatment is safe and does not cause damage to eyesight. Treatment of the retinal angiomas include diathermy, xenon, laser, cryocoagulation, and external beam radiotherapy [69]. Pheochromocytomas should be removed laparoscopically and it is recommended to watch pheochromocytomas that are less than two centimeters in size as they are not considered harmful [83]. Surveillance is recommended for the common manifestations of VHL as preventative care and early finding of tumors can help to improve patient outcome. Lastly, it is important to consider the genetic counseling in VHL patients who wish to have children. The disease is autosomal dominant meaning an affected individual has a 50% chance of passing the mutation on to offspring [83].
Figure 4.
Types of Von-Hippel-Lindau Disease. VHL can be broken into two main types based on the presence of pheochromocytomas. Type 2 has three subtypes based on the presence of renal cancers.
Figure 4.
Types of Von-Hippel-Lindau Disease. VHL can be broken into two main types based on the presence of pheochromocytomas. Type 2 has three subtypes based on the presence of renal cancers.
6. Ataxia-Telangiectasia
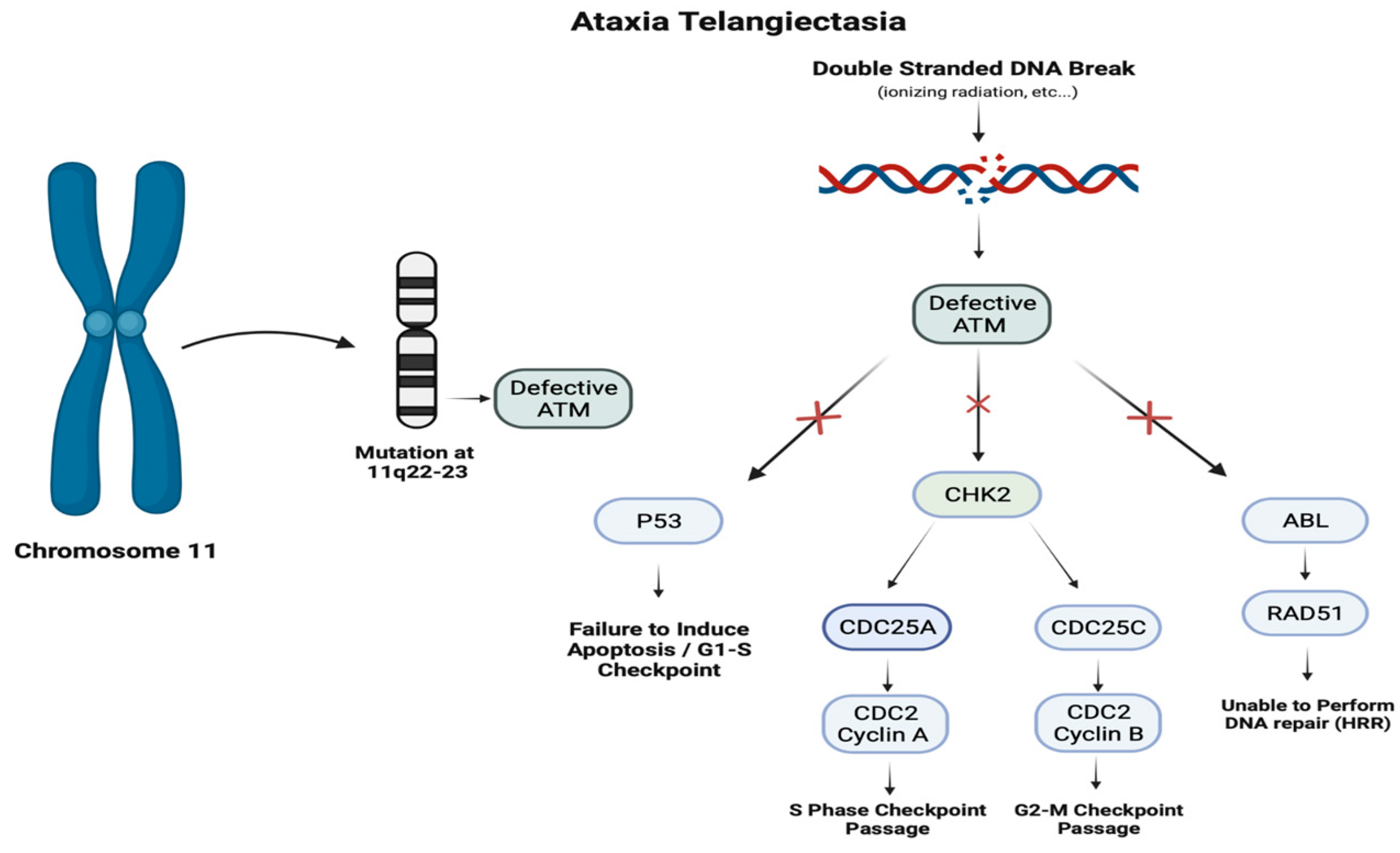
Mutations in the ATM gene result in a defective ataxia telangiectasia mutated serine/threonine kinase. Functioning ATM protein acts as a tumor suppressor with vital roles in the progression of the cell cycle, induction of apoptosis, and repair of double-stranded DNA breaks through paramount interactions with p53, CHK2, and ABL. Downstream effects of the deficient ATM result in the inability to correct damage in the genome, hindering the production of mature T and B cells, leading to immunodeficiency and increased susceptibility to the development of certain malignancies.
In the discourse of inherited neurological disorders, Ataxia Telangiectasia (AT), also recognized as Louis-Bar syndrome, remains a staple of discussion with its characteristic cutaneous manifestations and etiology [90]. Fortunately, as a neurodegenerative disorder, AT is seldom diagnosed, occurring in 1 out of 100,000 births in the United States, with some populations having an increased incidence of up to 1 out of every 40,000 births [91]. Ataxia Telangiectasia remains one of the most debilitating Primary Immunodeficiencies with patients generally dying prior to the third decade of life [92]. Although, AT’s prognosis and comprehension have undergone significant expansion with the advent of genetic testing paired with an increased understanding of its clinical presentation and pathophysiology.
This autosomal recessive disorder’s morbidity is directly linked to its deleterious involvement in multiple organ systems within the body [93]. Arising from mutations in the ATM gene, its viral roles in the repair of damaged DNA can lead to a variety of manifestations such as the gradual degeneration of the cerebellum leading to progressive ataxia, the presence of cutaneous telangiectasias, elevated risk of malignancies (especially lymphoid malignancies), compromised immune function, and frequent sinopulmonary infections [94]. Specifically, a mutation located at 11q22-23, which encodes for the ATM gene, results in a defective Ataxia Telangiectasia Mutated Serine Threonine Kinase (ATM) protein that insufficiently addresses double-stranded breaks in DNA [95]. This impaired response is vital in the presence of external stressors such as ionizing radiation or chemicals as well as the development of mature T cells [96]. The subsequent faulty ATM protein is unable to maintain critical interactions with p53, and ABL, leading to inability to induce apoptosis, and performing DNA repair by homologous recombination repair [97]. Additionally, CHK2′s interactions with CDC25A and CDC25C contribute to the bypassing of the cell cycle checkpoints at S and G2-M, further exacerbating the loss of p53 bypassing the G1-S checkpoint [98].
Figure 5.
Pathogenesis of Ataxia Telangiectasia. Ataxia telangiectasia is an autosomal recessive disorder due to mutations on the ATM gene on chromosome 11q22. The ATM gene encodes an ATM kinase involved in detecting DNA damage and regulating the cell cycle. Mutations result in failure to induce apoptosis during the G1-S transition and an inability to repair DNA, resulting in cell cycle progression.
Figure 5.
Pathogenesis of Ataxia Telangiectasia. Ataxia telangiectasia is an autosomal recessive disorder due to mutations on the ATM gene on chromosome 11q22. The ATM gene encodes an ATM kinase involved in detecting DNA damage and regulating the cell cycle. Mutations result in failure to induce apoptosis during the G1-S transition and an inability to repair DNA, resulting in cell cycle progression.
One of the distinctive features prompting clinicians to diagnose AT is its characteristic cutaneous features, most notably the telangiectasias [99]. Telangiectasias are identified by the presence of smaller, vasodilated blood vessels on the skin, frequently associated with dilation or broken blood vessels located near the surface of the skin or mucous membranes [100]. In the progression of AT, telangiectasias are observed on the whites of the eyes and various areas on the skin, particularly the bulbar conjunctiva, ears, neck, and cubital fossa [101]. The exact cause of telangiectasia remains unknown; however, researchers propose that development of these manifestations include weakening of the blood vessel structure and function, stemming from genetic, environmental, or an interplay of these influences [94]. It is widely accepted that there is a consensus in acknowledging AT patients exhibit a variability in the cutaneous manifestation of the disease [102]. The degree of telangiectasias can vary influenced by the aforementioned factors, patients may present with extensive telangiectasias while others may not manifest them at all [103].
Diagnosing AT presents a multifaceted challenge from the absence of a consensus on clinical diagnostic criteria, requiring a comprehensive evaluation of clinical features to confirm [104]. Arising from the diverse array of affected organs as well as the varying severity observed in patients, the complexity of diagnosing patients accounts includes the combining neurologic clinical features in tandem with one or more cutaneous or laboratory findings [99]. Diminished control and coordination of muscles (ataxia), development of clusters of dilated blood vessels (telangiectasias) on mucous membranes, and frequent infections are indicative symptoms that lead towards AT diagnosis [105]. The diverse range of symptomatology underscores the importance of a comprehensive personal and family history, which can lead a practitioner towards genetic testing to identify the presence of the 11q22-23 mutation in an individual suffering from cerebellar effects [106]. Moreover, analysis of the patient’s blood may provide key insights in diagnosing AT, revealing the presence of either decreased immunoglobulins or increased alpha-fetoprotein (AFP) levels in adults [107]. A significant reduction in immunoglobulins in AT results from the lack of mature T cells and the impaired interaction activating B cells [108]. While elevated AFP levels in infants are commonplace, their decline is associated with age; however, markedly higher levels in adults are characteristic of AT, making the recovery of AFP a hallmark diagnostic tool [109].
Unfortunately, as of now, there is no cure to AT, the extent of treatment is supportive care aiming to address neurological dysfunction and slow deterioration [110]. The challenge in treating AT stems from the inherited gene mutation on chromosome 11, the lack of gene editing capabilities to correct such a mutation leaves the population in a persistent state risk, while being minimal [111]. In terms of supportive care, immunoglobulin supplementation and antibiotics are routinely utilized to proactively prevent and halt opportunistic infection from the immunodeficiency seen in patients with AT [112]. Moreover, the utilization of chest physiotherapy and airway clearance techniques in children with recurrent sinopulmonary disease have demonstrated reduced development of chronic lung disease in AT [113]. Lastly, administration of glucocorticoids in a prospective cohort study has demonstrated promise in relieving the ataxia symptoms in AT, however the long-term effectiveness has yet to be established [114,115]. Recent advancements in genetics and immunology have yielded transformative work igniting a diverse range of novel treatments that hold considerable potential as innovative therapies in the treatment of AT. In an effort to correct the hematologic abnormalities seen in AT, bone marrow transplantation is becoming an increasing option of therapy to promote immune competence while preventing leukemia [116]. Lastly, recent publications have shed light on the use of splice switching antisense oligonucleotides (ASOs) which are able to modify cellular pre-mRNA to alter and correct splice altering genetic variants that cause AT; leading practitioners in the future to potentially use ASOs enabling correction of splice altering genetic variants that cause disease [117].
7. Osler-Weber-Rendu
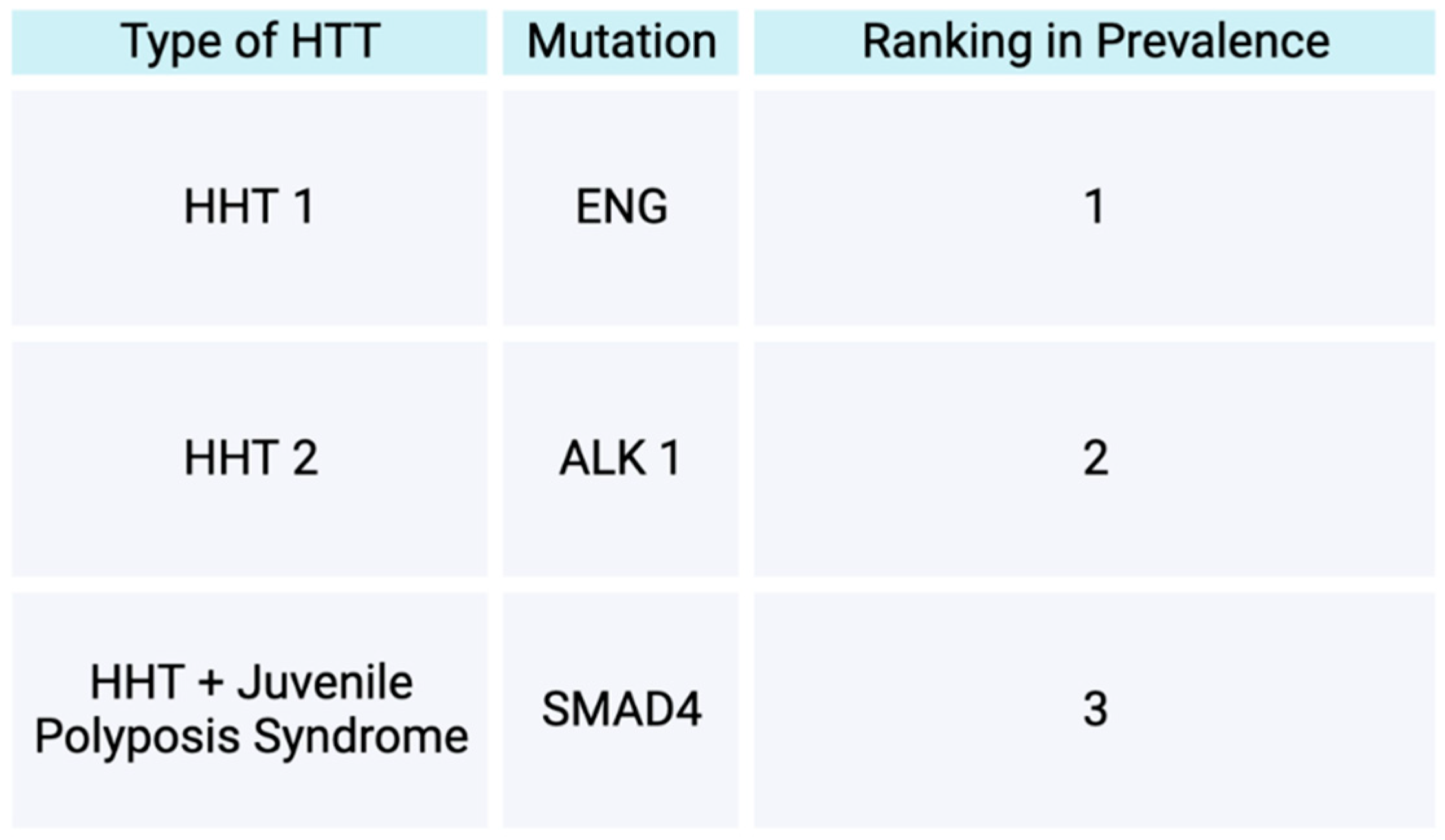
Osler Weber Rendu disease also known as Hereditary Hemorrhagic Telangiectasias (HTT) is a disease inherited an autosomal manner with a later onset of symptoms [118,119]. Prevalence of HHT in the general population is reported to range from 1.5-2 out of 10,000. In terms of the types of HHT, HHT is divided into two main forms, HHT 1 and HHT 2. HHT 1 form is reported to be more prevalent than HHT 2 form [118]. The two forms are initiated by heterozygous mutations [118]. HHT 1 is initiated by a mutation in endoglin, while HHT 2 is initiated by a mutation in Activin A receptor-like type 1 [118]. Both of these mutations lead to interruption of the typical TGF beta mediated pathways [118]. Consequently, the arteriovenous malformations develop and affect the way blood vessels form, initiating atypical connections between veins and arteries and subsequently avoiding passing through capillaries [118]. AVMs of the pulmonary system, brain, spine, or liver can occur in patients with HHT [118]. Visceral AVMs, such as brain AVMs, can lead to complications such as a hemorrhagic strokes and seizures [119]. Brain AVMs may also lead to motor weakness and aphasia [120]. A less common form of HHT, is one with mutations within SMAD4, in which a patient subsequently exhibits traits of both HHT and juvenile polyposis syndrome [121].
A common manifestation of Osler Weber Rendu disease is the formation of telangiectasias [121]. About three fourths of the HHT patient populations display telangiectatic lesions [121]. Telangiectasias are dilated blood vessels formed in various parts of the body, both visceral and cutaneous [119]. The cutaneous telangiectasias are often described as “spider-web-like” in appearance, affecting several external surfaces of the body, including commonly over the lips, tongue, and face [118]. In an article by Hyldahl et al. (2022), a more thorough description and evaluation of telangiectasias of HHT patients was performed [121]. Per the Hyldahl et al. article, most commonly the lesions of HHT patients were described as round, flat, or minimally raised/elevated [121]. More specifically, telangiectatic lesions can be categorized as confluent round, radiating round, lacunar round, tortuous round, hemangioma-like round, dilated, and arborized [121]. Overall, per the Hyldahl et al. article, HHT1 type patients are more likely to develop telangiectatic lesions, both mucosal and cutaneous, when compared to HHT2 type patients. Additionally, as patients age, HHT patients are more likely to form telangiectatic lesions and acquire dilated blood vessels [121]. Additionally, over 90% of HHT patients from the Hyldahl article experience epistaxis. Less commonly, patients may also experience bleeding from the gums, lips, ears, cheeks, and digits [121].
Early screening is often recommended if HHT is suspected in patients due to the severe health threat visceral AVMs may pose [119]. Some criterion to consider when screening for HHT includes family history/first degree relative with HHT, recurrent epistaxis, visceral lesions, and formation of telangiectasias [122]. Genetic testing for mutations in Activin A receptor-like type 1 (ALK1) and endoglin (ENG) is recommended for diagnosis of HHT [118]. In terms of the epistaxis and subsequent bleeding disorders, such as anemia, that HHT patients often develop, patients may often be treated with antiangiogenics [119,123]. More commonly in the older HHT patient population, GI bleeding may be developed and subsequently need to be addressed with antiangiogenic modalities and iron transfusions [119]. As a result, clinical recommendations for HHT patients also include iron level and anemia testing [124]. Treatments patients are provided in regards to nasal cavity telangiectasias include bevacizumab, laser therapies, cauterizations, nasal implants, and surgical procedures to close nasal cavities [121]. In terms of treatment of visceral AVMs, depending on the location of lesion, patients may undergo embolization, resection, or focal radiation [119]. Specifically for hepatic AVMs, liver transplants or bevacizumab are the mainstays of treatment, since hepatic lesion embolization leads to subsequent liver issues in the future [119]. However, the full consequences of long-term use of bevacizumab have not been fully determined at this time, as increased thrombotic risk occurs with use [124]. More research is currently inquiring about the efficacy of targeting VEGF in medications for HHT patients in order to inhibit angiogenesis, given HHT patients often have a pro-angiogenesis process active [124].
Figure 6.
Types of Hereditary Hemorrhagic Telangiectasias. This figure demonstrates the types of hereditary hemorrhagic telangiectasias based off gene mutation types and their prevalence in the HHT patient population.
Figure 6.
Types of Hereditary Hemorrhagic Telangiectasias. This figure demonstrates the types of hereditary hemorrhagic telangiectasias based off gene mutation types and their prevalence in the HHT patient population.
8. Conclusions
Although the clinical characteristics and genetic basis of neurocutaneous disorders differ, the role of precision medicine and targeted therapy provides a shared theme. Emerging therapeutics for NF-1 includes selumetinib, am MEK inhibitor. Antiangiogenic agents are being used for the management of NF-2 along with Osler-Weber-Rendu to manage symptoms. mTOR inhibitors are gaining prevalence for the management of TSC. Regarding VLH, treatments focus on managing renal cell carcinoma. Overall, the role of precision medicine and targeted therapies is increasing, elucidating the genetic phenomenon behind neurocutaneous disorders. With an improved understanding, modern medicine can develop interventions to mitigate symptoms and improve patient outcomes.
Author Contributions
Conceptualization, I.K.; investigation, I.K. D.F., R.T., J.C., J.S., C.C., H.K.; writing—original draft preparation, I.K. D.F., R.T., J.C., J.S., C.C., H.K.; writing—review and editing, I.K. D.F., R.T., J.C., J.S., C.C., H.K; supervision, B.L.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Klar, N.; Cohen, B.; Lin, D.D.M. Neurocutaneous Syndromes. Handb. Clin. Neurol. 2016, 135, 565–589. [Google Scholar] [CrossRef] [PubMed]
- Le, C.; Bedocs, P.M. Neurofibromatosis. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023. [Google Scholar]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis Type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef] [PubMed]
- Giraud, J.-S.; Bièche, I.; Pasmant, É.; Tlemsani, C. NF1 Alterations in Cancers: Therapeutic Implications in Precision Medicine. Expert. Opin. Investig. Drugs 2023, 32, 941–957. [Google Scholar] [CrossRef]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef] [PubMed]
- Legius, E.; Brems, H. Genetic Basis of Neurofibromatosis Type 1 and Related Conditions, Including Mosaicism. Childs Nerv. Syst. 2020, 36, 2285–2295. [Google Scholar] [CrossRef] [PubMed]
- Bikowska-Opalach, B.; Jackowska, T. [Neurofibromatosis type 1 - description of clinical features and molecular mechanism of the disease]. Med. Wieku Rozwoj 2013, 17, 334–340. [Google Scholar] [PubMed]
- Nasi, L.; Alexopoulos, A.; Kokkinou, E.; Roka, K.; Tzetis, M.; Tsipi, M.; Kakourou, T.; Kanaka-Gantenbein, C.; Chrousos, G.; Kattamis, A.; et al. Characteristics of Café-Au-Lait Macules and Their Association with the Neurofibromatosis Type I Genotype in a Cohort of Greek Children. Acta Derm. Venereol. 2023, 103, adv5758. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y. Neurofibromatosis 1 (von Recklinghausen Disease). Keio J. Med. 2023. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Idogawa, M.; Okura, M.; Sugita, S.; Sugawara, T.; Sasaki, Y.; Tokino, T.; Yamashita, T.; Uhara, H. Genetic Analyses of Mosaic Neurofibromatosis Type 1 with Giant Café-Au-Lait Macule, Plexiform Neurofibroma and Multiple Melanocytic Nevi. J. Dermatol. 2020, 47, 658–662. [Google Scholar] [CrossRef]
- Walaszek, Z.; Hanausek-Walaszek, M.; Webb, T.E. Dietary Glucarate-Mediated Reduction of Sensitivity of Murine Strains to Chemical Carcinogenesis. Cancer Lett. 1986, 33, 25–32. [Google Scholar] [CrossRef]
- Taal, W.; van Dijk, S.A.; Noordhoek, C.; Broen, M.P.G.; Gijtenbeek, J.M.M.A.; Oostenbrink, R. [Symptomatic tumors in neurofibromatosis type 1: a diagnostic challenge]. Ned. Tijdschr. Geneeskd. 2023, 167, D7864. [Google Scholar] [PubMed]
- Khatri, N.; Raza, M.L.; Aijaz, A.; Ramesh, R.; Gianchand, N.; Khan, F.A.A. Neurofibroma: Case Series with Clinical Features and Recommendations. Acta Neurol. Taiwan. 2024, 33(3), 112–121. [Google Scholar]
- Magwood, G.S.; Ellis, C.; Buie, J.N.J.; Slan, S.; Bonilha, L.; Adams, R.J. High Tech and High Touch: Recruitment Strategies for Enrolling African American Stroke Survivors in Community Based Intervention under Nurse Guidance after Stroke (CINGS) Trial. Contemp. Clin. Trials Commun. 2021, 24, 100844. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Espírito Santo, V.; Passos, J.; Nzwalo, H.; Carvalho, I.; Santos, F.; Martins, C.; Salgado, L.; Silva, C.E.; Vinhais, S.; Vilares, M.; et al. Selumetinib for Plexiform Neurofibromas in Neurofibromatosis Type 1: A Single-Institution Experience. J. Neurooncol 2020, 147, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Ardern-Holmes, S.; Fisher, G.; North, K. Neurofibromatosis Type 2. J. Child. Neurol. 2017, 32, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 Inactivation in Tumor Biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Freeman, S.; Gokhale, C.; Wallace, A.; Lloyd, S.K.; Axon, P.; Ward, C.L.; Rutherford, S.; King, A.; Huson, S.M.; et al. Bilateral Vestibular Schwannomas in Older Patients: NF2 or Chance? J. Med. Genet. 2015, 52, 422–424. [Google Scholar] [CrossRef]
- Halliday, D.; Emmanouil, B.; Pretorius, P.; MacKeith, S.; Painter, S.; Tomkins, H.; Evans, D.G.; Parry, A. Genetic Severity Score Predicts Clinical Phenotype in NF2. J. Med. Genet. 2017, 54, 657–664. [Google Scholar] [CrossRef]
- Gregory, G.E.; Jones, A.P.; Haley, M.J.; Hoyle, C.; Zeef, L.A.H.; Lin, I.-H.; Coope, D.J.; King, A.T.; Evans, D.G.; Paszek, P.; et al. The Comparable Tumour Microenvironment in Sporadic and NF2-Related Schwannomatosis Vestibular Schwannoma. Brain Commun. 2023, 5, fcad197. [Google Scholar] [CrossRef]
- Lloyd, S.K.W.; Evans, D.G.R. Neurofibromatosis Type 2 (NF2): Diagnosis and Management. Handb. Clin. Neurol. 2013, 115, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.R.; Messiaen, L.; Legius, E.; Pancza, P.; Avery, R.A.; Blakeley, J.O.; Babovic-Vuksanovic, D.; Ferner, R.; Fisher, M.J.; Friedman, J.M.; et al. Updated Diagnostic Criteria and Nomenclature for Neurofibromatosis Type 2 and Schwannomatosis: An International Consensus Recommendation. Genet. Med. 2022, 24, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Cumpston, E.C.; Rhodes, S.D.; Yates, C.W. Advances in Targeted Therapy for Neurofibromatosis Type 2 (NF2)-Associated Vestibular Schwannomas. Curr. Oncol. Rep. 2023, 25, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, S.; Akagami, R. Prospective Comparison of Quality of Life before and after Observation, Radiation, or Surgery for Vestibular Schwannomas. J. Neurosurg. 2009, 111, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Chari, D.A.; Vasilijic, S.; Welling, D.B.; Stankovic, K.M. New Developments in Neurofibromatosis Type 2 and Vestibular Schwannoma. Neurooncol Adv. 2021, 3, vdaa153. [Google Scholar] [CrossRef] [PubMed]
- Hiruta, R.; Saito, K.; Bakhit, M.; Fujii, M. Current Progress in Genomics and Targeted Therapies for Neurofibromatosis Type 2. Fukushima J. Med. Sci. 2023, 69, 95–103. [Google Scholar] [CrossRef]
- Northrup, H.; Krueger, D.A. International Tuberous Sclerosis Complex Consensus Group Tuberous Sclerosis Complex Diagnostic Criteria Update: Recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013, 49, 243–254. [Google Scholar] [CrossRef]
- Roach, E.S. Applying the Lessons of Tuberous Sclerosis: The 2015 Hower Award Lecture. Pediatr Neurol 2016, 63, 6–22. [Google Scholar] [CrossRef]
- Jones, A.C.; Shyamsundar, M.M.; Thomas, M.W.; Maynard, J.; Idziaszczyk, S.; Tomkins, S.; Sampson, J.R.; Cheadle, J.P. Comprehensive Mutation Analysis of TSC1 and TSC2-and Phenotypic Correlations in 150 Families with Tuberous Sclerosis. Am. J. Hum. Genet. 1999, 64, 1305–1315. [Google Scholar] [CrossRef]
- Au, K.S.; Williams, A.T.; Roach, E.S.; Batchelor, L.; Sparagana, S.P.; Delgado, M.R.; Wheless, J.W.; Baumgartner, J.E.; Roa, B.B.; Wilson, C.M.; et al. Genotype/Phenotype Correlation in 325 Individuals Referred for a Diagnosis of Tuberous Sclerosis Complex in the United States. Genet. Med. 2007, 9, 88–100. [Google Scholar] [CrossRef]
- van Slegtenhorst, M.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; van den Ouweland, A.; Halley, D.; Young, J.; et al. Identification of the Tuberous Sclerosis Gene TSC1 on Chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef] [PubMed]
- European Chromosome 16 Tuberous Sclerosis Consortium Identification and Characterization of the Tuberous Sclerosis Gene on Chromosome 16. Cell 1993, 75, 1305–1315. [CrossRef] [PubMed]
- Caban, C.; Khan, N.; Hasbani, D.M.; Crino, P.B. Genetics of Tuberous Sclerosis Complex: Implications for Clinical Practice. Appl. Clin. Genet. 2017, 10, 1–8. [Google Scholar] [CrossRef]
- Feliciano, D.M. The Neurodevelopmental Pathogenesis of Tuberous Sclerosis Complex (TSC). Front. Neuroanat. 2020, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.; Chakraborty, R.; Mahajan, Z.; Sharma, A.; Sethi, S.K.; Raina, R. Renal Manifestations of Tuberous Sclerosis Complex. J. Kidney Cancer VHL 2020, 7, 5–19. [Google Scholar] [CrossRef]
- Teng, J.M.C.; Cowen, E.W.; Wataya-Kaneda, M.; Gosnell, E.S.; Witman, P.M.; Hebert, A.A.; Mlynarczyk, G.; Soltani, K.; Darling, T.N. Dermatologic and Dental Aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol 2014, 150, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Jacks, S.K.; Witman, P.M. Tuberous Sclerosis Complex: An Update for Dermatologists. Pediatr. Dermatol. 2015, 32, 563–570. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Kanish, B.; Bhatia, A. Tuberous Sclerosis. Indian. Dermatol. Online J. 2015, 6, 142–143. [Google Scholar] [CrossRef]
- Cardis, M.A.; DeKlotz, C.M.C. Cutaneous Manifestations of Tuberous Sclerosis Complex and the Paediatrician’s Role. Arch. Dis. Child. 2017, 102, 858–863. [Google Scholar] [CrossRef]
- Dao, D.-P.D.; Pixley, J.N.; Akkurt, Z.M.; Feldman, S.R. A Review of Topical Sirolimus for the Treatment of Facial Angiofibromas in Tuberous Sclerosis Complex. Ann. Pharmacother. 2023, 10600280231182421. [Google Scholar] [CrossRef]
- Borzęcki, A.; Chyl-Surdacka, K.; Turska, M. Spectacular Effect of Massive Facial Angiofibromas Removal With a Carbon Dioxide Laser as a Manifestation of a Tuberous Sclerosis Complex. J. Lasers Med. Sci. 2021, 12, e24. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M.D.; Tang, H.; Gallione, C.J.; Baugher, J.D.; Frelin, L.P.; Cohen, B.; North, P.E.; Marchuk, D.A.; Comi, A.M.; Pevsner, J. Sturge-Weber Syndrome and Port-Wine Stains Caused by Somatic Mutation in GNAQ. N. Engl. J. Med. 2013, 368, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Happle, R. Phacomatosis Pigmentovascularis Revisited and Reclassified. Arch. Dermatol. 2005, 141, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Fois, A.; Calistri, L.; Balestri, P.; Vivarelli, R.; Bartalini, G.; Mancini, L.; Berardi, A.; Vanni, M. Relationship between Café-Au-Lait Spots as the Only Symptom and Peripheral Neurofibromatosis (NF1): A Follow-up Study. Eur. J. Pediatr. 1993, 152, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Espino, L.F.; Ivars, M.; Antoñanzas, J.; Baselga, E. Sturge-Weber Syndrome: A Review of Pathophysiology, Genetics, Clinical Features, and Current Management Approache. Appl. Clin. Genet. 2023, 16, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Poliner, A.; Fernandez Faith, E.; Blieden, L.; Kelly, K.M.; Metry, D. Port-Wine Birthmarks: Update on Diagnosis, Risk Assessment for Sturge-Weber Syndrome, and Management. Pediatr. Rev. 2022, 43, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Wetzel-Strong, S.E.; Galeffi, F.; Benavides, C.; Patrucco, M.; Bullock, J.L.; Gallione, C.J.; Lee, H.K.; Marchuk, D.A. Developmental Expression of the Sturge-Weber Syndrome-Associated Genetic Mutation in Gnaq: A Formal Test of Happle’s Paradominant Inheritance Hypothesis. Genetics 2023, 224, iyad077. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, J.; Frelin, L.P.; McCann, M.; Pardo, C.A.; Cohen, B.A.; Comi, A.M.; Pevsner, J. Identification of a Mosaic Activating Mutation in GNA11 in Atypical Sturge-Weber Syndrome. J. Invest. Dermatol. 2021, 141, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Zeng, Z.; Rivière, J.-B.; O’Shaughnessy, R.; Al-Olabi, L.; St-Onge, J.; Atherton, D.J.; Aubert, H.; Bagazgoitia, L.; Barbarot, S.; et al. Mosaic Activating Mutations in GNA11 and GNAQ Are Associated with Phakomatosis Pigmentovascularis and Extensive Dermal Melanocytosis. J. Invest. Dermatol. 2016, 136, 770–778. [Google Scholar] [CrossRef]
- Waelchli, R.; Aylett, S.E.; Robinson, K.; Chong, W.K.; Martinez, A.E.; Kinsler, V.A. New Vascular Classification of Port-Wine Stains: Improving Prediction of Sturge-Weber Risk. Br. J. Dermatol. 2014, 171, 861–867. [Google Scholar] [CrossRef]
- Dompmartin, A.; van der Vleuten, C.J.M.; Dekeuleneer, V.; Duprez, T.; Revencu, N.; Désir, J.; Te Loo, D.M.W.M.; Flucke, U.; Eijkelenboom, A.; Schultze Kool, L.; et al. GNA11-Mutated Sturge-Weber Syndrome Has Distinct Neurological and Dermatological Features. Eur. J. Neurol. 2022, 29, 3061–3070. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, J.; Mehta, S.; Mulye, S. Paroxysmal Vascular Events in Sturge-Weber Syndrome: Role of Aspirin. J. Pediatr. Neurosci. 2014, 9, 39–41. [Google Scholar] [CrossRef]
- Yeom, S.; Comi, A.M. Updates on Sturge-Weber Syndrome. Stroke 2022, 53, 3769–3779. [Google Scholar] [CrossRef] [PubMed]
- Comi, A.M. Sturge-Weber Syndrome and Epilepsy: An Argument for Aggressive Seizure Management in These Patients. Expert. Rev. Neurother. 2007, 7, 951–956. [Google Scholar] [CrossRef]
- Day, A.M.; McCulloch, C.E.; Hammill, A.M.; Juhász, C.; Lo, W.D.; Pinto, A.L.; Miles, D.K.; Fisher, B.J.; Ball, K.L.; Wilfong, A.A.; et al. Physical and Family History Variables Associated With Neurological and Cognitive Development in Sturge-Weber Syndrome. Pediatr. Neurol. 2019, 96, 30–36. [Google Scholar] [CrossRef]
- Koenraads, Y.; van Egmond-Ebbeling, M.B.; de Boer, J.H.; Imhof, S.M.; Braun, K.P.J.; Porro, G.L. SWS study group Visual Outcome in Sturge-Weber Syndrome: A Systematic Review and Dutch Multicentre Cohort. Acta Ophthalmol. 2016, 94, 638–645. [Google Scholar] [CrossRef]
- Sujansky, E.; Conradi, S. Sturge-Weber Syndrome: Age of Onset of Seizures and Glaucoma and the Prognosis for Affected Children. J. Child. Neurol. 1995, 10, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Gittins, S.; Steel, D.; Brunklaus, A.; Newsom-Davis, I.; Hawkins, C.; Aylett, S.E. Autism Spectrum Disorder, Social Communication Difficulties, and Developmental Comorbidities in Sturge-Weber Syndrome. Epilepsy Behav. 2018, 88, 1–4. [Google Scholar] [CrossRef]
- Chapieski, L.; Friedman, A.; Lachar, D. Psychological Functioning in Children and Adolescents with Sturge-Weber Syndrome. J. Child. Neurol. 2000, 15, 660–665. [Google Scholar] [CrossRef]
- Léauté-Labréze, C.; Boralevi, F.; Pedespan, J.-M.; Meymat, Y.; Taïeb, A. Pulsed Dye Laser for Sturge-Weber Syndrome. Arch. Dis. Child. 2002, 87, 434–435. [Google Scholar] [CrossRef]
- Oakes, W.J. The Natural History of Patients with the Sturge-Weber Syndrome. Pediatr. Neurosurg. 1992, 18, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Parsa, C.F. Sturge-Weber Syndrome: A Unified Pathophysiologic Mechanism. Curr. Treat. Options Neurol. 2008, 10, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Miyajima, M.; Sugano, H.; Iimura, Y.; Kato, M.; Tsurusaki, Y.; Miyake, N.; Saitsu, H.; Arai, H.; Matsumoto, N. The Somatic GNAQ Mutation c.548G>A (p.R183Q) Is Consistently Found in Sturge-Weber Syndrome. J. Hum. Genet. 2014, 59, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, S.; Ball, K.L.; Bhattacharya, S.K.; Bitrian, E.; Blieden, L.S.; Brandt, J.D.; Burkhart, C.; Chugani, H.T.; Falchek, S.J.; Jain, B.G.; et al. Consensus Statement for the Management and Treatment of Sturge-Weber Syndrome: Neurology, Neuroimaging, and Ophthalmology Recommendations. Pediatr. Neurol. 2021, 121, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Smegal, L.F.; Sebold, A.J.; Hammill, A.M.; Juhász, C.; Lo, W.D.; Miles, D.K.; Wilfong, A.A.; Levin, A.V.; Fisher, B.; Ball, K.L.; et al. Multicenter Research Data of Epilepsy Management in Patients With Sturge-Weber Syndrome. Pediatr. Neurol. 2021, 119, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, B.; Yang, J.; Bi, M.; Bi, L.; Fan, W. Efficacy of Hemoporfin-Mediated Photodynamic Therapy in Treating Sturge-Weber Syndrome Associated Port-Wine Stains: A Retrospective Study. Indian. J. Dermatol. Venereol. Leprol. 2023, 1–8. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, A.J.; Luat, A.F.; Juhász, C.; Ho, M.L.; Argersinger, D.P.; Cavuoto, K.M.; Enriquez-Algeciras, M.; Tikkanen, S.; North, P.; Burkhart, C.N.; et al. A Multidisciplinary Consensus for Clinical Care and Research Needs for Sturge-Weber Syndrome. Pediatr. Neurol. 2018, 84, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, M.I.; Singh, A.K. Von Hippel-Lindau Syndrome. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023. [Google Scholar]
- Schmidt, L.S.; Linehan, W.M. Genetic Predisposition to Kidney Cancer. Semin. Oncol. 2016, 43, 566–574. [Google Scholar] [CrossRef]
- Klingler, J.-H.; Gläsker, S.; Bausch, B.; Urbach, H.; Krauss, T.; Jilg, C.A.; Steiert, C.; Puzik, A.; Neumann-Haefelin, E.; Kotsis, F.; et al. Hemangioblastoma and von Hippel-Lindau Disease: Genetic Background, Spectrum of Disease, and Neurosurgical Treatment. Childs Nerv. Syst. 2020, 36, 2537–2552. [Google Scholar] [CrossRef]
- Reisch, N.; Peczkowska, M.; Januszewicz, A.; Neumann, H.P.H. Pheochromocytoma: Presentation, Diagnosis and Treatment. J. Hypertens. 2006, 24, 2331–2339. [Google Scholar] [CrossRef]
- Gläsker, S.; Neumann, H.P.H.; Koch, C.A.; Vortmeyer, A.; Von Hippel-Lindau, Disease. Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth (MA), 2000. [Google Scholar]
- Chittiboina, P.; Lonser, R.R. Von Hippel-Lindau Disease. Handb. Clin. Neurol. 2015, 132, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Sgambati, M.T.; Stolle, C.; Choyke, P.L.; Walther, M.M.; Zbar, B.; Linehan, W.M.; Glenn, G.M. Mosaicism in von Hippel-Lindau Disease: Lessons from Kindreds with Germline Mutations Identified in Offspring with Mosaic Parents. Am. J. Hum. Genet. 2000, 66, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. Molecular Basis of the VHL Hereditary Cancer Syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.C.; Payne, L.B.; Rathmell, W.K. Hypoxia, Angiogenesis, and Metabolism in the Hereditary Kidney Cancers. J. Clin. Invest. 2019, 129, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Ferzli, P.G.; Millett, C.R.; Newman, M.D.; Heymann, W.R. The Dermatologist’s Guide to Hereditary Syndromes with Renal Tumors. Cutis 2008, 81, 41–48. [Google Scholar] [PubMed]
- Singh, A.D.; Shields, C.L.; Shields, J.A. Von Hippel-Lindau Disease. Surv. Ophthalmol. 2001, 46, 117–142. [Google Scholar] [CrossRef]
- Ahmad, W.A.W.; Khanom, M.; Yaakob, Z.H. Heart Failure in Pregnancy: An Overview. Int. J. Clin. Pract. 2011, 65, 848–851. [Google Scholar] [CrossRef]
- Damsky, W.E.; Bosenberg, M. Melanocytic Nevi and Melanoma: Unraveling a Complex Relationship. Oncogene 2017, 36, 5771–5792. [Google Scholar] [CrossRef]
- Jha, S.K.; Mendez, M.D. Cafe Au Lait Macules. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023. [Google Scholar]
- van Leeuwaarde, R.S.; Ahmad, S.; van Nesselrooij, B.; Zandee, W.; Giles, R.H. Von Hippel-Lindau Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle (WA), 1993. [Google Scholar]
- Wolters, W.P.G.; Dreijerink, K.M.A.; Giles, R.H.; van der Horst-Schrivers, A.N.A.; van Nesselrooij, B.; Zandee, W.T.; Timmers, H.J.L.M.; Seute, T.; de Herder, W.W.; Verrijn Stuart, A.A.; et al. Multidisciplinary Integrated Care Pathway for von Hippel-Lindau Disease. Cancer 2022, 128, 2871–2879. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kakutani, S.; Sato, Y.; Hanashi, A.; Kinoshita, Y.; Ishikawa, A. Drug Review: Pazopanib. Jpn. J. Clin. Oncol. 2018, 48, 503–513. [Google Scholar] [CrossRef]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N. Engl. J. Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.W.-S.; Lenton, J.; Smith, J.; Jagdev, S.; Ralph, C.; Vasudev, N.; Bhattarai, S.; Lewington, A.; Kimuli, M.; Cartledge, J.; et al. Multimodal Image-Guided Ablation on Management of Renal Cancer in Von-Hippel-Lindau Syndrome Patients from 2004 to 2021 at a Specialist Centre: A Longitudinal Observational Study. Eur. J. Surg. Oncol. 2022, 48, 672–679. [Google Scholar] [CrossRef]
- Gläsker, S.; Krüger, M.T.; Klingler, J.-H.; Wlodarski, M.; Klompen, J.; Schatlo, B.; Hippchen, B.; Neumann, H.P.H.; Van Velthoven, V. Hemangioblastomas and Neurogenic Polyglobulia. Neurosurgery 2013, 72, 930–935; discussion 935. [Google Scholar] [CrossRef]
- Krüger, M.T.; Steiert, C.; Gläsker, S.; Klingler, J.-H. Minimally Invasive Resection of Spinal Hemangioblastoma: Feasibility and Clinical Results in a Series of 18 Patients. J. Neurosurg. Spine 2019, 1–10. [Google Scholar] [CrossRef]
- Greenberger, S.; Berkun, Y.; Ben-Zeev, B.; Levi, Y.B.; Barziliai, A.; Nissenkorn, A. Dermatologic Manifestations of Ataxia-Telangiectasia Syndrome. J. Am. Acad. Dermatol. 2013, 68, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.Y.; Shaikh, A.G. Past and Present of Eye Movement Abnormalities in Ataxia-Telangiectasia. Cerebellum 2019, 18, 556–564. [Google Scholar] [CrossRef]
- van Os, N.J.H.; van Deuren, M.; Weemaes, C.M.R.; van Gaalen, J.; Hijdra, H.; Taylor, A.M.R.; van de Warrenburg, B.P.C.; Willemsen, M.A.A.P. Classic Ataxia-Telangiectasia: The Phenotype of Long-Term Survivors. J. Neurol. 2020, 267, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Sirajwala, A.A.; Khan, S.; Rathod, V.M.; Gevariya, V.C.; Jansari, J.R.; Patel, Y.M. Ataxia-Telangiectasia: A Case Report and a Brief Review. Cureus 2023, 15, e39346. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Samanta, D.; Frucht, S. Ataxia Telangiectasia. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023. [Google Scholar]
- Jiang, Y.; Chen, H.-C.; Su, X.; Thompson, P.A.; Liu, X.; Do, K.-A.; Wierda, W.; Keating, M.J.; Plunkett, W. ATM Function and Its Relationship with ATM Gene Mutations in Chronic Lymphocytic Leukemia with the Recurrent Deletion (11q22.3-23.2). Blood Cancer J. 2016, 6, e465. [Google Scholar] [CrossRef]
- Amirifar, P.; Ranjouri, M.R.; Yazdani, R.; Abolhassani, H.; Aghamohammadi, A. Ataxia-Telangiectasia: A Review of Clinical Features and Molecular Pathology. Pediatr. Allergy Immunol. 2019, 30, 277–288. [Google Scholar] [CrossRef]
- Yildiz, A.; Kaya, Y.; Tanriverdi, O. Effect of the Interaction Between Selenium and Zinc on DNA Repair in Association With Cancer Prevention. J. Cancer Prev. 2019, 24, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia Telangiectasia and Rad3-Related Inhibitors and Cancer Therapy: Where We Stand. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia Telangiectasia: A Review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Cavone, F.; Cappelli, S.; Bonuccelli, A.; D’Elios, S.; Costagliola, G.; Peroni, D.; Orsini, A.; Consolini, R. Ataxia Telangiectasia Arising as Immunodeficiency: The Intriguing Differential Diagnosis. J. Clin. Med. 2023, 12, 6041. [Google Scholar] [CrossRef]
- Kumar, N.; Aggarwal, P.; Dev, N.; Kumar, G. Ataxia Telangiectasia: Learning from Previous Mistakes. BMJ Case Rep. 2012, 2012, bcr2012007246. [Google Scholar] [CrossRef] [PubMed]
- Asadollahi, R.; Britschgi, C.; Joset, P.; Oneda, B.; Schindler, D.; Meier, U.R.; Rauch, A. Severe Reaction to Radiotherapy Provoked by Hypomorphic Germline Mutations in ATM (Ataxia-Telangiectasia Mutated Gene). Mol. Genet. Genomic Med. 2020, 8, e1409. [Google Scholar] [CrossRef]
- Moeini Shad, T.; Yazdani, R.; Amirifar, P.; Delavari, S.; Heidarzadeh Arani, M.; Mahdaviani, S.A.; Sadeghi-Shabestari, M.; Aghamohammadi, A.; Rezaei, N.; Abolhassani, H. Atypical Ataxia Presentation in Variant Ataxia Telangiectasia: Iranian Case-Series and Review of the Literature. Front. Immunol. 2021, 12, 779502. [Google Scholar] [CrossRef] [PubMed]
- Veenhuis, S.; van Os, N.; Weemaes, C.; Kamsteeg, E.-J.; Willemsen, M. Ataxia-Telangiectasia. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle (WA), 1993. [Google Scholar]
- Perlman, S.; Becker-Catania, S.; Gatti, R.A. Ataxia-Telangiectasia: Diagnosis and Treatment. Semin. Pediatr. Neurol. 2003, 10, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Gatti, R.A.; Berkel, I.; Boder, E.; Braedt, G.; Charmley, P.; Concannon, P.; Ersoy, F.; Foroud, T.; Jaspers, N.G.; Lange, K. Localization of an Ataxia-Telangiectasia Gene to Chromosome 11q22-23. Nature 1988, 336, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Paucar, M.; Taylor, A.M.R.; Hadjivassiliou, M.; Fogel, B.L.; Svenningsson, P. Progressive Ataxia with Elevated Alpha-Fetoprotein: Diagnostic Issues and Review of the Literature. Tremor Other Hyperkinet Mov. (N. Y) 2019, 9. [Google Scholar] [CrossRef]
- Chopra, C.; Davies, G.; Taylor, M.; Anderson, M.; Bainbridge, S.; Tighe, P.; McDermott, E.M. Immune Deficiency in Ataxia-Telangiectasia: A Longitudinal Study of 44 Patients. Clin. Exp. Immunol. 2014, 176, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Long, S.S. Parents Can Accurately Observe the Severity of Respiratory Illness in Their Infants Born Term and Preterm. J. Pediatr. 2019, 214, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Giardino, G.; Fusco, A.; Romano, R.; Gallo, V.; Maio, F.; Esposito, T.; Palamaro, L.; Parenti, G.; Salerno, M.C.; Vajro, P.; et al. Betamethasone Therapy in Ataxia Telangiectasia: Unraveling the Rationale of This Serendipitous Observation on the Basis of the Pathogenesis. Eur. J. Neurol. 2013, 20, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Hirch, T.; Brander, N.; Schenk, F.; Pöllmann, S.J.; Reichenbach, J.; Schubert, R.; Modlich, U. Expression of a Large Coding Sequence: Gene Therapy Vectors for Ataxia Telangiectasia. Sci. Rep. 2023, 13, 19386. [Google Scholar] [CrossRef] [PubMed]
- McGrath-Morrow, S.A.; Rothblum-Oviatt, C.C.; Wright, J.; Schlechter, H.; Lefton-Greif, M.A.; Natale, V.A.; Crawford, T.O.; Lederman, H.M. Multidisciplinary Management of Ataxia Telangiectasia: Current Perspectives. J. Multidiscip. Healthc. 2021, 14, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Unes, S.; Tuncdemir, M.; Eroglu-Ertugrul, N.G.; Kerem Gunel, M. Effectiveness of Physical Therapy on Ataxia-Telangiectasia: A Case Report. Pediatr. Phys. Ther. 2021, 33, E103–E107. [Google Scholar] [CrossRef] [PubMed]
- Zannolli, R.; Buoni, S.; Betti, G.; Salvucci, S.; Plebani, A.; Soresina, A.; Pietrogrande, M.C.; Martino, S.; Leuzzi, V.; Finocchi, A.; et al. A Randomized Trial of Oral Betamethasone to Reduce Ataxia Symptoms in Ataxia Telangiectasia. Mov. Disord. 2012, 27, 1312–1316. [Google Scholar] [CrossRef]
- Kwei, K.T.; Kuo, S.-H. An Overview of the Current State and the Future of Ataxia Treatments. Neurol. Clin. 2020, 38, 449–467. [Google Scholar] [CrossRef]
- Sabino Pinho de Oliveira, B.; Putti, S.; Naro, F.; Pellegrini, M. Bone Marrow Transplantation as Therapy for Ataxia-Telangiectasia: A Systematic Review. Cancers (Basel) 2020, 12, 3207. [Google Scholar] [CrossRef]
- Kim, J.; Woo, S.; de Gusmao, C.M.; Zhao, B.; Chin, D.H.; DiDonato, R.L.; Nguyen, M.A.; Nakayama, T.; Hu, C.A.; Soucy, A.; et al. A Framework for Individualized Splice-Switching Oligonucleotide Therapy. Nature 2023, 619, 828–836. [Google Scholar] [CrossRef]
- Macri, A.; Wilson, A.M.; Shafaat, O.; Sharma, S. Osler-Weber-Rendu Disease. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023. [Google Scholar]
- Hammill, A.M.; Wusik, K.; Kasthuri, R.S. Hereditary Hemorrhagic Telangiectasia (HHT): A Practical Guide to Management. Hematology Am. Soc. Hematol. Educ. Program. 2021, 2021, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.A.; Turcios, N.L. Pulmonary Manifestations of Skin Disorders in Children. Pediatr. Clin. North. Am. 2021, 68, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Hyldahl, S.J.; El-Jaji, M.Q.; Schuster, A.; Kjeldsen, A.D. Skin and Mucosal Telangiectatic Lesions in Hereditary Hemorrhagic Telangiectasia Patients. Int. J. Dermatol. 2022, 61, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Mylavarapu, C.; Lu, A.J.; Burns, E.A.; Samorajski, J.; Gotur, D.; Baker, K. Diffuse Cerebral Edema and Impending Herniation Complicating Hepatic Encephalopathy in Hereditary Hemorrhagic Telangiectasia. Case Rep. Med. 2022, 2022, 2612544. [Google Scholar] [CrossRef] [PubMed]
- Alkhalid, Y.; Darji, Z.; Shenkar, R.; Clancy, M.; Dyamenahalli, U.; Awad, I.A. the multidisciplinary faculty of the HHT Center of Excellence at University of Chicago Medicine Multidisciplinary Coordinated Care of Hereditary Hemorrhagic Telangiectasia (Osler-Weber-Rendu Disease). Vasc. Med. 2023, 28, 153–165. [Google Scholar] [CrossRef]
- Al-Samkari, H. Hereditary Hemorrhagic Telangiectasia: Systemic Therapies, Guidelines, and an Evolving Standard of Care. Blood 2021, 137, 888–895. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



