
Submitted:
12 January 2024
Posted:
15 January 2024
You are already at the latest version
Abstract
How Nature discovered genetic coding is a largely ignored question, yet the answer is key to explaining the transition from biochemical building blocks to life. Other, related puzzles also fall inside the aegis enclosing the codes themselves. The peptide bond is unstable with respect to hydrolysis. So, it requires some form of chemical free energy to drive it. Amino acid activation and acyl-transfer are also slow and call for catalysis. All living things must thus also convert free energy and synchronize cellular chemistry. Most importantly, functional proteins occupy only small, isolated regions of sequence space. Nature evolved heritable symbolic data processing to seek out and use those sequences. That system has three parts: a memory of how amino acids behave in solution and inside proteins, a set of code-keys to access that memory, and a scoring function. The code-keys themselves are the genes for cognate pairs of tRNA and aminoacyl-tRNA synthetases, AARS. I outline the surprising links between all these questions and the structural duality of the base-pairing that holds genes together and how those links arise from the experiments.
Keywords:
aminoacyl-tRNA synthetase•tRNA cognate pairs
; bidirectional genetic coding
; protein folding and gating
; origin of catalysis
; origin of free energy transduction
; genome propagation into the proteome
1. Introduction
Watson and Crick’s model for DNA structure [1,2] may be the most decisive dividing line in the history of human awareness. Nucleotide base-pairing revealed the molecular basis of inheritance, redirecting biological and human health research in a single stroke, from inspired—but blind—guesswork to lucid inevitability. Inheritance, with variation, is only one of several pillars underpinning living matter. Sentience as we know it also requires molecular processes that store and manipulate both information and free energy. Biology is a singularly self-constructing form of matter.
Interpreting information stored in genes required the symbolic transformation embodied in the universal genetic coding table. That table pairs amino acids to one or more of the 64 triplet “codons” possible using the four nucleotide bases found in nucleic acid genes. That such a code existed [3,4,5], what the assignments were [6,7], and the Nature of the assignment catalysts [8,9] all emerged rapidly after Watson and Crick described the DNA double helix. It was roughly 30 more years until the first clues [10,11,12,13] began to emerge that would lead, ultimately, to the surprising conclusion that base-pairing might also propagate into the proteome itself, and be central to the emergence of both the coding table and its molecular implementation [14].
How Nature discovered genetic coding is a largely ignored question, yet the answer is key to explaining the transition from biochemical building blocks to life. Genetic coding also conceals a deeper, rarely stated question. The molecular assignment catalysts, aminoacyl-tRNA synthetases (AARS), must implement the very language in which their own genes are written. That means that the AARS are reflexive [15,16,17]. Reflexivity sets living matter apart from all other forms of active matter. Its roots lie deep in evolutionary molecular biology, which must have resulted from a complex historical progression. Each step in that historical progression exploited only what was available at the time and must have enabled the next step. Solving the puzzle of genetic coding means charting that process.
The proteome amplifies the chemical engineering diversity embedded in genes by perhaps a billion-fold [15]. That amplification introduced computational control enabling life to emerge and flourish on earth. Nature enabled it by evolving the genetic code. That reagent-to-token assignment is done by nanomachines—(AARS)•tRNA cognate pairs. As their name suggests, AARS use ATP to activate and transfer the α-carboxyl group of amino acids covalently to cognate tRNAs, thus enforcing the code.
Nature built these self-describing machines within an as yet unknown historical context. Pre-existing conditions made each successive step possible. Each step, in turn, enabled those that followed. Creating rudimentary assignment catalysts, in turn, enabled the explosive transition to living organisms by a process we have compared to booting a computer’s operating system [15]. The code was almost entirely completed before the Last Universal Common Ancestor, LUCA [18]. Hence, most aspects of the process must have been highly cooperative. Cooperativity, in turn, meant that many unlikely processes had to promote one another, increasing their joint probability.
The dashed curves in Figure 1a suggest four separate areas in which evolutionary advances must promoted one another as the coding alphabet grew. We imagine that the coding alphabet was initially modest, perhaps only a single bit. Acquiring new bits required the AARS to speciate, in order to better discriminate between amino acid and tRNA substrates. That meant that mutant sequences folded into structures with more sophisticated cognition (Figure 1a). The coding alphabet size, specificity, folding, and function (Figure 1a) all have experimentally accessible signatures (Figure 1b). These can help us track development of the code, using experimental models of evolutionary intermediate AARS•tRNA cognate pairs.
The interdependence evident in Figure 1a also orders events in time. Models for the origin of coding must thus identify plausible pathways between successive steps. Processes that favor one another confer selective advantage and tend to survive. Our studies suggest that catalysis itself was prominent even in the earliest ancestral models [2]. Moreover, peptide bond formation is an unfavorable reaction that must be driven by coupling it to a source of free energy. So, the earliest catalysts must also have coupled chemical free energy into biological reactions, as we have observed [2]. Specificity, however, required lengthy refining of binding specificities to enhance precision. It came only later.
How did Nature solve the “chicken-and-egg” questions by converting random chemistry into symbolic coding to interpret genes? This question is especially vexing when we consider those whose translated products that enforced the coding rules. How too did Nature learn to exploit sources of chemical free energy necessary to sustain itself far from equilibrium? Nearly 4 billion years later, these questions remain deeply mysterious to us—the only products of that explosive transformation who are able to ask them.
My colleague, Peter Wills, and I re-focused attention [15] to a specialized subset of questions. We considered only a narrow range of phenomena with two characteristics. First, we examined only areas where the relevant fields still have useful signals. Second, we looked for the elements of reflexivity—specific recognition of amino acid and tRNA substrates—in models for the primordial AARS assignment catalysts.
Coherent shards from evolutionary molecular biology, structural biology, bioinformatics, and biochemistry are beginning to suggest useful answers. These come from a wide range of experimental and conceptual disciplines. Discussion will proceed as follows. §II summarizes the key model for experimental study of the origin of the genetic coding table and its implementation. §III asks what can be expected from analysis of the historical record. §IV describes the constraint surface—the scoring function that probably shaped Nature’s self-organization prior to the advent of Darwinian selection. §V surveys challenges that remain to be addressed and the experimental models with which to address them. §VI considers whether or not understanding the emergence of animate matter required new physics.
2. Creating the first bit of genetic information
At the boundary between non-random chemistry and biology the earliest AARS•tRNA cognate pairs began to embed two distinct kinds of symbolic information into nucleic acids. Transfer RNAs are a like a computer programming language connecting amino acid physical chemistry to symbols, i.e., the 64 codons. Messenger RNAs are blueprints for making functional proteins written using that language [20]. Together, they separated phenotype from genotype. Unlike the Morse code, which was assembled to represent a pre-existing alphabet, Nature created alphabet, symbols, and programs simultaneously and from scratch.
2.1. AARS/tRNA cognate pairs function as mutually exclusive molecular AND gates.
The elemental barrier to creating the code was creating the first bit of information. AARS “assignment catalysis”—selecting and combining two substrates from closely-related homologs—resembles AND gating in computer hardware. The first genetic coding “bit” likely was defined by two, mutually exclusive AARS•tRNA cognate pairs (Figure 2). Both AARSs and tRNAs use separate domains for catalysis and anticodon recognition. AARS still recognize the acceptor stem in “minihelices” lacking the anticodon-binding domain derived from tRNAs for several amino acids [12]. Thus, an “operational” code likely drove recognition in single-domain AARS•tRNA complexes prior to the advent of the anticodon stem-loop.
2.2. Bidirectional genetic coding projected duality into the proteome,
The molecular basis of this rudimentary substrate differentiation surfaced with the recognition that AARS have two distinct versions, Class I and II [21,22,23]. The two Classes have different architectures. Class I active-site domains are modified Rossman dinucleotide-binding folds with parallel β-strands interspersed with α-helices. Class II AARS have multi-stranded antiparallel β-sheets [24,25,26]. The Class distinction also rationalized earlier studies showing that, with the notable exception of aromatic amino acids, those AARS now belonging to Class I acylated the 3’terminal ribose 2’ OH; those belonging to Class II acylated the 3’ OH. Ribas and Schimmel [11] later summarized subclass-specific pairwise interactions matching Class I AARS with the minor groove and Class II AARS with the major groove of cognate tRNAs (Figure 2a).
Despite their contrasting architectures, Class I and II AARS appear to share a common origin. Rodin and Ohno [13] observed unusually high base pairing between the antiparallel aligned coding sequences of the highly conserved PxxxxHIGH and KMSKS signatures of Class I AARS with Motifs 2 and 1, respectively, in Class II AARS. They proposed that the original Class I and Class II ancestors were coded by opposite strands of the same bidirectional gene. Their proposal received little attention for another decade. Subsequent work, however, [2,27,28,29,30,31,32] provided substantial supporting evidence.
The base-paired segments represent only ~130 residues, about 40% of the smallest Class I AARS (TrpRS). Remarkably, those segments include the active sites of both Class I and II AARS. When purified, they retain ~60% of their activity in both key reactions [2,31,33,34,35]. We called them AARS “urzymes”. Improbably elevated middle-base pairing between signature sequences extends throughout >75 % of the paired, antiparallel coding sequences [27]. Moreover, the base-pairing between sequences independently reconstructed for Class I and II urzymes increases toward the oldest ancestral node [36].
AARS urzymes contain several structural modules. One module, the protozyme, appears to be older than the others [19]. Protozymes contain the ATP binding sites in both AARS Classes. The protozymes appear to have arisen simultaneously on a single bidirectional gene [2], as outlined further below. The protozyme is at the amino terminus of Class I and at the carboxy terminus of the Class II AARS urzymes. Class I AARS have three modules in addition to the protozyme (A)—the connecting peptide insertion within the catalytic domain, the second half of the urzyme, C, and the anticodon-binding domain, D. We settled on a nomenclature for AARS urzymes using these modular designations. Class I urzyme modules begin with the protozyme and so are designated with the three letter amino acid plus AC. Class II urzymes begin with the bidirectional partner or module C and end with the protozyme, so are designated (Aa)CA.
We have described urzymes derived from two Class I (TrpAC [27,28] and LeuAC [37,38]) and two Class II (HisCA [29] and GlyCA [39]) AARS. They all speed amino acid activation up by ~ 109-fold over the uncatalyzed rates. They speed up cognate tRNA acylation by ~ 103-fold [30,38,39]. Further, the LeuAC urzyme actually acylates TΨC minihelixLeu about an order of magnitude faster than tRNALeu [40].
2.3. AARS protozymes are amino-acid activating catalysts that can be coded by a bidirectional gene.
A bidirectional gene encodes functional proteins from both of its strands. We made a computationally designed bidirectional gene to code 46-residue, ATP-binding subsites from Class I and II AARS on opposite strands. Both excerpts, called protozymes, are active. In fact, catalytic proficiencies, kcat/KM, of bidirectionally coded Class I and II protozymes are the same, within experimental error, as those of wild type TrpRS and HisRS protozyme sequences [2]. Tamura and co-workers verified our conclusions [32].
The bidirectionally coded protozymes increase in the rate of amino acid activation by more than ~106-fold. That overcomes the rate-limiting step in protein synthesis [31,34]. Thus, it strongly supports participation of such genes early in the genesis of genetic coding. Moreover, the AARS protozymes were very likely to be the earliest catalytic polypeptides with direct, ancestral phylogenetic relationships to the contemporary proteome.
2.4. The inverse complementarity of nucleic acid base-pairing duality projects deeply into the proteome.
The coding table is exquisitely well-designed to promote bidirectional coding [41,42,43]. Thirty codons—those with middle base U or A—occur as complementary codon:anticodon pairs one of which encodes a core, the other a surface amino acid. The amino acids related in this way define the insides and outsides of folded proteins. Water-to-cyclohexane transfer free energies, ΔGw>c, for Class I and II protozymes show high reflection symmetry in antiparallel alignment (Figure 3a). The tertiary structures that result from each strand are consequently anticorrelated. Surfaces that form tertiary structures (amber stripes) in one Class lie opposite amino acids that form the solvent-accessible surface (green stripes) of the other Class. Proteins translated from opposite strands of bidirectional genes thus fold up inside-out from one another!
2.5. The projected duality creates rudimentary nanomachinery for chemical free energy transduction.
This inversion also has a strong impact on Class-dependent AARS ligand binding (Fig 3b). Remarkably, both protozymes furnish binding determinants for both ATP and amino acid substrates. They bind ATP with higher affinity than full-length enyzmes [2]. Indeed, Class I protozymes appear to be ancestral forms for NTP binding sites in a broad variety of ATP- and GTPAses [44]. NMR studies by Mildvan of ATP-binding peptides excerpted from DNA polymerase I [45], F1 ATPase [46,47], and adenylate kinase [48,49] suggest that ATP induces folding from a largely disordered form to something resembling the structures of these peptides in X-ray crystal structures of the intact proteins. Thus, the two AARS protozymes appear to underlie the origin not only of amino acid activation, but also of NTP-linked free-energy coupling to biochemistry in general.
2.6. The projected duality constrains substrate recognition by AARS urzymes, dividing amino acids and tRNA acceptor stems into parallel groups.
These studies reinforce the extensive homologies of the protozymes from across the ten families of both Class I and II AARS [19]. As is the case with the three proteins Mildvan studied, Class I and II secondary structures appear in a similar order, β–α–β. Crystal structures of AARS complexed with analogs of their activated amino acids [50,51] (Figure 3b) reveal patterns that account for their preference for large (Class I) or small (Class II) side chains. Contacts from the central α-helix specify ATP (blue) in Class I and amino acid (salmon) in Class II protozymes. Conversely, those from the β-strands specify amino acid in Class I and ATP in Class II protozymes.
The inversion also differentiates both amino acid and tRNA acceptor stem groove recognition by AARS urzymes [52,53]. The ATP and amino acid binding sites put the prochiral α-phosphate into diastereroisomeric environments in Class I and II AARS because the amino acid binding determinants arise from opposite sides of the adenine ring plane. Consequently, there is less room for side chains in Class II AARS [54], consistent with the uniformly smaller size of Class II amino acids.
Most Class I and II AARS approach the tRNA acceptor stem from opposite grooves (Figure 4) [14,52,53]. Cognate tRNA binding by s differentiated decisively because the enzymes Class I tRNA substrates, which approach via the minor groove, must make a sharp hairpin to enter the cognate synthetase active site. Class I urzymes promote hairpin formation via specific interactions between the amino terminus of the specificity-determining helix and the phosphate group and ribose of A76. Class II tRNA substrates approach the major groove and do not require a hairpin. Rather, the extended polypeptide hairpin at the C-terminus of the Class II protozyme fits across the 1-72 base pair, fortifying the 3’-DCCA helical extension.
Both Class I and II urzymes are therefore replete with determinants not only for catalytic rate acceleration of both amino acid activation and acyl-transfer to tRNA, but also for differential recognition of both amino acid and tRNA substrates. Notably, with the exception of the conserved aromatic residue – A76 ribose interaction in Class I (here mediated by Y198, Figure 4A), these determinants appear to be rooted in side-chain independent secondary structure. Recent combinatorial mutagenesis of full-length Class I Leucyl-tRNA synthetase and its urzyme confirmed this by showing that the native HIGH and KMSKS catalytic sequences are either non-functional or inhibitory in the LeuAC urzyme, whereas they favor catalysis of tRNA acylation synergistically in the full-length enzyme [37]. Bidirectional coding thus appears to account for the requisite primordial differentiation of both amino acids and tRNAs, hence for creating the first informational bit of the eventual genetic code.
3. Phylogenetics
Any account of the origin of translation must seek consistency with the historical record embedded into the proteome structural (sequence and tertiary) databases. Readout from those databases is challenging because Nature took highly convoluted pathways—assimilation of hard-to-define modular bits of genetic information, horizontal gene transfer, mutation reversals—to elaborate the proteome. It is far easier to interpret the contemporary primary and 3D structural databases and work backward. However, the most challenging questions posed by the origins of translation, including the order in which amino acids entered the coding table, concern behavior near the oldest nodes which remain inherently ambiguous. These require supplementary assumptions.
Caetano-Anollés and colleagues [55,56,57,58,59,60,61] clarify many difficult issues posed by these databases. Their models capture salient points other investigators [62,63,64,65,66,67] fail to address. Foremost among these are the requirement for co-evolution of multiple functions [58], especially of catalysis with the interpretive machinery; the widespread sharing of modular components as the proteome diverged [56,57,58,68]; and the historical separation between architectural diversification and the adaptive radiation of genes within species [68], which parallels that between self-organization [69,70,71,72] and natural selection.
Models drawn from phylogenetics must be validated by excerpting experimental prototypes for putative nodes in phylogenetic trees and testing them for both catalytic proficiency and specificity [27]. The catalytic proficiency of the bidirectional 46-residue protozyme gene products [2,32] implies remarkable functionality in much smaller structural motifs than those examined by Caetano-Anollés. The functional granularity of the proteome’s structural patchwork has substantially higher resolution than that inferred from the SCOP database of intact protein domains [73]—which have an average length of nearly 190 residues [74].
That high-resolution mosaicity likely has crucial details about the birth of the proteome [26]. The Class I urzyme is structurally isomorphous with the TOPRIM domain [35]. Phylogenetic metrics suggest that it, too, has significant mosaic substructure, including evidence that the protozyme [19] is its oldest module. The linear dependence of the transition-state stabilization and Michaelis constant free energies on sequence length of the protozyme, urzyme, catalytic domains and intact Class I and II AARS (see Figure 6 in [33]) constitutes pivotal experimental evidence for the hypothesis that these represent meaningful states along the evolutionary pathway to mature AARS.
A key goal is to define sequence probability distributions for nodes at which ancestral AARS bifurcated to form two mutually exclusive new forms. Despite the considerable promise of ancestral sequence reconstruction [75,76,77,78], however, unambiguous phylogenetic trees for both AARS superfamilies remain elusive for at least two reasons. First, specific substrate recognition required the advent of sophisticated allosteric phenomena [2,37,79,80,81,82,83]. So, the earliest genes and biological peptides were doubtless quasispecies until the proteome was almost complete. Second, the functionally-relevant sequence space at the root was substantially more comprehensive than expected, in the sense that very diverse sequences had similar activity [2,37]. Third, each AARS family bifurcation to form two exclusive new letters in the coding alphabet forced Nature to decide which of the two, new amino acids worked best in every extant context, and re-equilibrate the entire extant proteome to the enlarged coding alphabet [84].
Thus, phylogenetics algorithms must be revised not only to incorporate asymmetric transition matrices [85], but also to dynamically recognize and equilibrate increases in the coding alphabet. Recent developments may improve prospects for realizing this goal [86].
Experimental testing of reconstructed ancestral sequences highlights many of the challenges of other higher-order combinatorial problems. Artificial intelligence tools to aid the design of protein sequences, given a backbone scaffolding constraint have recently transformed our ability to evaluate and improve upon reconstructed ancestral sequences [87]. This capability, combined with other bioinformatic tools [88,89,90,91] affords a path for selecting sequences likely to have useful properties, simplifying a large combinatorial array to a much smaller representative sample.
As the coding specificity of the current 20 amino acid table decreases with successive ancestral nodes, the resulting ancestral sequences become increasingly like quasispecies. It becomes important to be able to characterize populations. An appropriate way to meet that challenge is to express recombinant libraries, which implies assaying populations, rather than individual species. Douglas, anticipating this requirement, has created a Bayesian estimation for the mean value and variance of the two Michaelis-Menten parameters, kcat and KM [92]. A key purpose of that software is to estimate the enzymatic heterogeneity of the population.
4. Constraints: impedance matching and reciprocally-coupled gating
The most significant obstacles Nature overcame to produce animate matter were explosive combinatorial optimization problems. Emergence of symbolic coding from random chemical processes was unlikely without mechanisms for selecting improbable combinations of multiple processes. Such problems preoccupied the protein folding community [93,94,95]. In keeping with the metaphor of bootstrapping nature’s OS, artificial intelligence neural network algorithms called constraint programming [96] provide a general approach to solving them. Such methods recently succeeded for both protein folding [88] and its inverse, protein design [87].
Aspects of the constraint surface Nature likely navigated as it built the coding table are illustrated in Figure 5. Wills developed a rigorous equivalence between the energetic cost of errors in information transfer and the physical concept of impedance [97]. He then showed that the most efficient pathway to the coding table matched the error rates in translation to those in replication. Thus, the gradual sharpening of AARS specificity and correspondingly decreased redundancies in coding assignments should be correlated with error rates in nucleic acid replication [15,97,98]. More coding letters lead to increasingly specific AARS in analogy to successive derailleur gears on a bicycle, so the most probable path from a binary to the contemporary 20-letter alphabet also dissipated the least chemical free energy.
A broader constraint is associated with recognizing that self-referential “strange loops” [99] evident in coding, catalysis, and bioenergetics can be formulated generally as reciprocally coupled AND gates [100,101] that explicitly filter large numbers of inputs. Coupling the antecedent of a second AND gate to the consequent of the first compounds the strength of the first. As an example, efficient coupling of ATP consumption to useful work requires both transition-state stabilization and a conformational change, while at the same time conformational changes require both transition-state stabilization and ATP consumption (i.e., product release) [100,101]. That counterintuitive coupling illustrates how reciprocally-coupled gating reduces many possibilities to few, efficiently bypassing Darwinian natural selection. Iterative application may contribute significantly to the emergence of order from chaos.
5. Experimental Challenges
Coherent experimental and conceptual results described here amount to a plausible scenario by which Nature implemented genetic coding by elaborating two Classes of AARS (Figure 6). It rests on a substantial, though partial experimental base. Deconstructing the patchwork of contemporary AARS genes, we characterized a nested hierarchy of functional modules from both AARS classes as experimental models [28,29,30,35,102] for evolutionary intermediates Nature used to create the genetic coding language, and by implication the necessary genetic messages. Remarkably, these intermediates also trace the emergence of catalytic proficiency and the capture of chemical free energy from ATP.
The hypothesis of Rodin and Ohno that the AARS Classes originated from opposite strands of the same ancestral gene provides a plausible model for the creation of the first bit of coding information by providing a structural rationale for the amino acid and tRNA specificities of the earliest AARS•tRNA cognate pairs. In a qualitative sense, the structural data in Figures 3,4 thus enhance the likelihood and Bayesian posterior probability of the bidirectional coding hypothesis substantially beyond that provided by direct experimental evidence presented in §II. Many details, however, remain obscure.
5.1. Validating the role of bidirectional coding
The designed 46-residue bidirectional protozyme gene [2,32] and four Class I/II urzymes are fledgling experimental outposts in the remote terrain from which genetic coding emerged. Were such bidirectional ancestral genes and their cognate tRNAs necessary and sufficient to elaborate the full coding table? In other words, how close were they to a functional boot-block for Nature’s operating system? Despite promises captured in Figures 3,4 and evidence for their remarkable catalytic properties, there remain fundamental questions about whether and, if so how, protozyme and urzyme specificity for the two substrates became sufficient to launch and refine coded protein synthesis.
These questions fall into three categories: (a) What functionalities are possible with alphabets of a given size and composition? (b) When and how did tRNA recognition arise? (c) What factors, besides the growth of the coding alphabet induced successive specificity improvements? These questions and the experimental tools to answer them, especially if supplemented by enhanced phylogenetics, are considered briefly here.
- How many bits (pairs of coding letters) were necessary to make bidirectional gene products sufficiently specific to achieve reflexivity? Experimental validation of reflexivity refers to designing bidirectional genes for Class I and II AARS precursors capable of experimentally discriminating between appropriate subsets of amino acids and tRNAs well enough to implement the corresponding alphabet, forming a self-consistent alphabet and set of implementing genes.
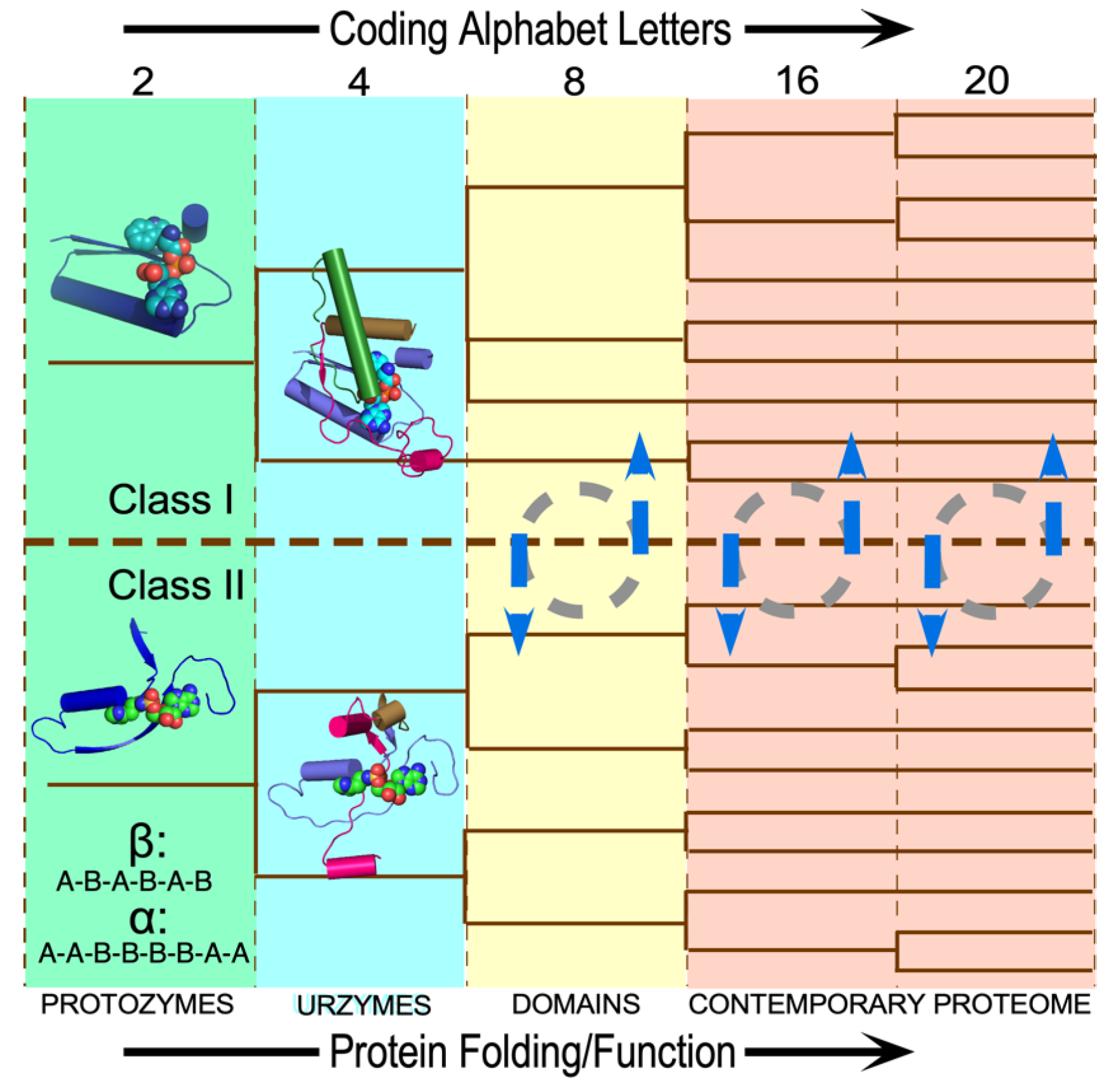
Experimental LeuAC, HisCA, and GlyCA urzyme specificity spectra (see Figure 5 in [39]) discriminate against amino acids from the opposite class on average four times out of five, and all have a within-Class preference for about five amino acids. Reinforcement from tRNA groove recognition might strengthen preferences to nine times out of ten. These probabilities are consistent with urzymes administering a two-bit, four letter alphabet as suggested in Figure 6. Considerable phylogenetic work to establish likely sequence probability distributions and amino acid specificities at the two-bit nodes, remains before bidirectional genes can be designed to confirm such hypotheses experimentally.
Tamura’s studies [32] revealed that protozymes may discriminate poorly between different amino acid side chains, and no one has yet tested their ability to transfer the aminoacyl group to tRNA (see §V.2). Thus, even more challenging work will be required to demonstrate reflexive enforcement by protozymes of a 1-bit alphabet with two kinds of letters.
- 2.
- Is a bidirectional urzyme gene feasible? Naïve analysis of the modular patchwork of Class I and II urzymes (see Figure 4A in [27]) has not resulted in an antiparallel alignment Class I and II urzyme sequences compatible with continuous bidirectional coding of their respective three-dimensional structures; likely that analysis cannot be used to constrain protein design programs as hoped. Recently, a more suitable, alternatively threaded antiparallel alignment emerged. That alignment, also consistent with the high resolution modularity [19], may provide a template for bidirectionally coded urzymes. However, we have yet to test it.
- 3.
- What limitations of bidirectional coding forced its breakdown by providing new functionality? The CP1 insertion at the C-terminus of the protozyme interrupted all extant Class I urzymes, definitively ending bidirectional coding. Eventually, CP1 significantly enhanced amino acid specificity, but only when complemented by the anticodon-binding domain [81,82]. Simultaneous acquisition of both domains seems unlikely, so one might expect a more decisive selective advantage for so significant a modular acquisition. The LeuAC urzyme converts substantial amounts of ATP to ADP in single turnover experiments [38]. If comparable analysis of the intact catalytic domain reduced ADP production, that would suggest that CP1 initially increased the efficiency of free energy transduction. Modular deconstruction of Class II AARS should also shed new light.
- 4.
- Can AARS urzymes acylation of TΨC-minihelices confirm details of the operational code? Acylation of minihelices partially substantiated the “single-domain” model for the origin of coding [12]. Evidence that AARS urzymes catalyze acylation of full-length tRNAs [30] further strengthened that model. Recently we showed that minihelixLeu is an even better substrate for LeuAC than tRNALeu [40]. That can now enable a detailed test of the operational code.
- 5.
- Can AARS protozymes catalyze tRNA acylation? Polypeptide catalysis of aminoacylation must have appeared sometime between the ancestral bidirectional protozyme gene and the emergence of urzymes. Structures illustrated in Figure 4 are quite sophisticated, even though far simpler than contemporary AARS. As AARS protozymes likely exemplify earlier ancestral catalytic polymers, it may be notable that the motif 2 loop Class II protozymes contains much of the tRNA binding site [53] whereas the Class I tRNA binding site is formed largely by a helix present only in the urzyme. That asymmetry, suggesting that Class II AARS preceded Class I AARS, raises the profound objection that functional polypeptides must have depended minimally at least on a binary code. Ribozymes similar to the flexizyme family [103,104] might have accelerated acyl transfer from aminoacyl-5’AMP produced by Class I protozymes to proto-tRNAs, assuring provision of aminoacylated RNAs for templated protein synthesis. That would have required an ad hoc mechanism to discriminate between two types of tRNA.
5.2. Beyond genetic coding.
The bidirectional gene construct suggests experimental, computational, and theoretical approaches to other questions implicit in Figure 1.
Both protozyme genes have similar distributions of conformational angles, φ and ψ, consistent with β-α-β secondary structures. Class I superfamily tertiary structures are based on the Rossmann dinucleotide binding fold [105] and parallel β strands interspersed with α-helices. Class II structures are based on antiparallel β strands. Crystal structures suggest this difference is nascent in the respective protozymes. Do sequence differences between Class I and II protozymes dictate their ultimate tertiary structures. If so, how? Do they emanate from the bidirectional coding inversion (Figure 3a)? The experimental models we created to study AARS evolution may serve in answering these questions:
- (i)
- We can infer sequence/structure relationships from variations in both naturally occurring and designed sequence databases.
- (ii)
- Bioinformatic tools reducing tertiary structures to lower dimensions—conformational angles [φ, ψ]; residue transfer free energies (ΔGvapor>chx, ΔGwater>chx) [20,106,107]; TetraDA one-dimensional strings derived from Delaunay tesselation [108]; SNAPP scoring [89,90]—provide alternative multidimensional windows into structural and evolutionary determinants.
- (iii)
- The highly sensitive Malachite Green assay for phosphates generated on amino acid activation [32] affords a five-fold increase in the rate at which assays can be performed on variants of this gene.
- (iv)
- Artificial intelligence has improved both protein design [87] and structure prediction[88], creating a dynamic virtual feedback loop capable of sampling substantially larger regions of the protein sequence space expected for early nodes in AARS speciation. Pruning those sequence distributions virtually, before committing to experimental construction, expression, and testing will greatly enhance the experimental tools described above.
6. Did creating the genetic code require new physics?
String theorist Edward Witten has written that “physics—like history—does not precisely repeat itself, (but) it does rhyme” [109]. This work has uncovered significant rhymes. Constraint surfaces in Figure 5 point to analogies with physical laws, notably the equivalence of the energetic cost of errors and the physical concept of impedance [97] (Figure 5a). Figure 5b draws on less obvious potential analogies that nonetheless likely conform to known physics [100]. Putting it the opposite way, impedance matching and reciprocally coupled gating provide possible mechanistic heuristics for universal extremum principles, including the minimum action principle. In that sense, investigating the genesis of animate matter may have opened new ways to view physical laws, rather than identifying a need for new physics. Biology’s secrets lie in structural and symbolic coincidences that solve otherwise improbably difficult problems, rather than requiring new laws.
Funding
This work was funded by the Alfred P. Sloan Foundation Matter-to-Life program Grant number G-2021-16944.
Competing interest statement
The author declares no competing interests.
Data availability
All data cited have been published and are available via public databases (The Protein Databank) or upon request from the author.
Acknowledgments
Peter Wills participated in many aspects of this work and is equally responsible for the directions it has taken. Comments from Jordan Douglas and Laurie Betts improved the manuscript.
References
- Watson, J.D.; Crick, F.H.C. A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Rodriguez, L.; Jimenez-Rodriguez, M.; Gonzalez-Rivera, K.; Williams, T.; Li, L.; Weinreb, V.; Chandrasekaran, S.N.; Collier, M.; Ambroggio, X.; Kuhlman, B.; et al. Functional Class I and II Amino Acid Activating Enzymes Can Be Coded by Opposite Strands of the Same Gene. J. Biol. Chem. 2015, 290, 19710–19725. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H.C. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H.C. The Origin of the Genetic Code. J. Mol. Biol. 1968, 38, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H.C. Codon-Anticodon Pairing: The Wobble Hypothesis. J. Mol. Biol. 1966, 19, 548–555. [Google Scholar] [CrossRef]
- Jones, O.W., Jr.; Nirenberg, M.W. Degeneracy in the amino acid code. Biochimica et Biophysica Acta (BBA) - Nucleic Acids and Protein Synthesis 1966, 119, 400–406. [Google Scholar] [CrossRef]
- Trupin, J.S.; Rottman, F.M.; Brimacome, R.; Leder, P.; Bernfield, M.R.; Nirenberg, M. RNA Codewords and Protein Synthesis, VI. On the Nucleotide Sequences of Degenerate Codeword Sets for Isoleucine, Tyrosine, Asparagine, and Lysine. Proc. Natl. Acad. Sci. U.S.A. 1965; 53, 807–811. [Google Scholar]
- Berg, P.; Ofengand, E.J. An Enzymatic Mechanism for Linking Amino Acids to RNA. Proc. Nat. Acad. Sci. USA 1958, 44, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, M.B.; E. B. Keller; Zamecnik., P.C. Enzymatic Carboxyl Activation of Amino Acids. J. Biol. Chem. 1956, 21, 345–358.
- Eriani, G.; Delarue, M.; Poch, O.; Gangloff, J.; Moras, D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 1990, 347, 203–206. [Google Scholar] [CrossRef]
- Ribas de Pouplana, L.; Schimmel, P. Two Classes of tRNA Synthetases Suggested by Sterically Compatible Dockings on tRNA Acceptor Stem. Cell 2001, 104, 191–193. [Google Scholar] [CrossRef]
- Schimmel, P.; Giegé, R.; Moras, D.; Yokoyama, S. An operational RNA code for amino acids and possible relationship to genetic code. Proc. Nat. Acad. Sci. USA 1993, 90, 8763–8768. [Google Scholar] [CrossRef] [PubMed]
- Rodin, S.N.; Ohno, S. Two Types of Aminoacyl-tRNA Synthetases Could be Originally Encoded by Complementary Strands of the Same Nucleic Acid. Orig. Life Evol. Biosph. 1995, 25, 565–589. [Google Scholar] [CrossRef]
- Carter, C.W., Jr.; Wills, P.R. The Roots of Genetic Coding in Aminoacyl-tRNA Synthetase Duality. Annual Review of Biochemistry 2021, 90, 349–373. [CrossRef]
- Carter, C.W., Jr; Wills, P.R. Interdependence, Reflexivity, Fidelity, and Impedance Matching, and the Evolution of Genetic Coding. Molecular Biology and Evolution 2018, 35, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Wills, P.R. Autocatalysis, information, and coding. BioSystems 2001, 50, 49–57. [Google Scholar] [CrossRef]
- Nieselt-Struwe, K.; Wills, P.R. The Emergence of Genetic Coding in Physical Systems. J. theor. Biol. 1997, 187, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fournier, G.P.; Andam, C.P.; Alm, E.J.; Gogarten, J.P. Molecular Evolution of Aminoacyl tRNA Synthetase Proteins in the Early History of Life. Orig Life Evol Biosph 2011, 41, 621–632. [Google Scholar] [CrossRef]
- Carter, C.W., Jr.; Popinga, A.; Bouckaert, R.; Wills, P.R. Multidimensional Phylogenetic Metrics Identify Class I Aminoacyl-tRNA Synthetase Evolutionary Mosaicity and Inter-modular Coupling. International Journal of Molecular Sciences 2022, 23, 1520. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W., Jr.; Wolfenden, R. Acceptor-stem and anticodon bases embed amino acid chemistry into tRNA. RNA Biology 2016, 13, 145–151. [Google Scholar] [CrossRef]
- Eriani, G.; Delarue, M.; Poch, O.; Gangloff, J.; Moras, D. Partition of tRNA Synthetases into Two Classes Based on Mutually Exclusive Sets of Sequence Motifs. Nature 1990, 347, 203–206. [Google Scholar] [CrossRef]
- Cusack, S.; Berthet-Colominas, C.; Hartlein, M.; Nassar, N.; Leberman, R. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 A. Nature 1990, 347, 249–255. [Google Scholar] [CrossRef]
- Ruff, M.; Krishnaswamy, S.; Boeglin, M.; Poterszman, A.; Mitschler, A.; Podjarny, A.; Rees, B.; Thierry, J.C.; Moras, D. Class II Aminoacyl Transfer RNA Synthetases: Crystal Structure of Yeast Aspartyl-tRNA Synthetase Complexed with tRNAAsp. Science 1991, 252, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W., Jr. Cognition Mechanism and Evolutionary Relationships in Aminoacyl-tRNA Synthetases. Annual Review of Biochemistry 1993, 62, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Cusack, S. Evolutionary Implications. Nat. Struct. Mol. Biol. 1994, 1, 760. [Google Scholar] [CrossRef]
- Douglas, J.; Bouckaert, R.; Carter, C.W., Jr.; Wills, P. Enzymic recognition of amino acids drove the evolution of primordial genetic codes. Nucleic Acids Research 2023. [Google Scholar] [CrossRef] [PubMed]
- Pham, Y.; Li, L.; Kim, A.; Erdogan, O.; Weinreb, V.; Butterfoss, G.; Kuhlman, B.; Carter, C.W., Jr. A Minimal TrpRS Catalytic Domain Supports Sense/Antisense Ancestry of Class I and II Aminoacyl-tRNA Synthetases. Mol Cell 2007, 25, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Pham, Y.; Kuhlman, B.; Butterfoss, G.L.; Hu, H.; Weinreb, V.; Carter, C.W., Jr. Tryptophanyl-tRNA synthetase Urzyme: a model to recapitulate molecular evolution and investigate intramolecular complementation. J. Biol. Chem. 2010, 285, 38590–38601. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Weinreb, V.; Francklyn, C.; Carter, C.W., Jr. Histidyl-tRNA Synthetase Urzymes: Class I and II Aminoacyl-tRNA Synthetase Urzymes have Comparable Catalytic Activities for Cognate Amino Acid Activation. J. Biol. Chem. 2011, 286, 10387–10395. [Google Scholar] [CrossRef]
- Li, L.; Francklyn, C.; Carter, C.W., Jr. Aminoacylating Urzymes Challenge the RNA World Hypothesis. J. Biol. Chem. 2013, 288, 26856–26863. [Google Scholar] [CrossRef]
- Carter, C.W., Jr.; Li, L.; Weinreb, V.; Collier, M.; Gonzales-Rivera, K.; Jimenez-Rodriguez, M.; Erdogan, O.; Chandrasekharan, S.N. The Rodin-Ohno Hypothesis That Two Enzyme Superfamilies Descended from One Ancestral Gene: An Unlikely Scenario for the Origins of Translation That Will Not Be Dismissed. Biology Direct 2014, 9, 11. [Google Scholar] [CrossRef]
- Onodera, K.; Suganuma, N.; Takano, H.; Sugita, Y.; Shoji, T.; Minobe, A.; Yamaki, N.; Otsuka, R.; Mutsuro-Aoki, H.; Umehara, T. , et al. Amino acid activation analysis of primitive aminoacyl-tRNA synthetases encoded by both strands of a single gene using the malachite green assay. BioSystems 2021, 208, 104481. [Google Scholar] [CrossRef]
- Carter, C.W., Jr. Coding of Class I and II aminoacyl-tRNA synthetases. Advances in Experimental Medicine and Biology: Protein Reviews 2017, 18, 103–148. [Google Scholar] [CrossRef]
- Carter, C.W., Jr. What RNA World? Why a Peptide/RNA Partnership Merits Renewed Experimental Attention. Life 2015, 5, 294–320. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W., Jr. Urzymology: Experimental Access to a Key Transition in the Appearance of Enzymes. J. Biol. Chem. 2014, 289, 30213–30220. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.N.; Yardimci, G.; Erdogan, O.; Roach, J.M.; Carter, C.W., Jr. Statistical Evaluation of the Rodin-Ohno Hypothesis: Sense/Antisense Coding of Ancestral Class I and II Aminoacyl-tRNA Synthetases. Molecular Biology and Evolution 2013, 30, 1588–1604. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.Q.; Hobson, J.J.; Carter, C.W.J. Domain Acquisition by Class I Aminoacyl-tRNA Synthetase Urzymes Coordinated the Catalytic Functions of HVGH and KMSKS Motifs. TBD 2023. [CrossRef] [PubMed]
- Hobson, J.J.; Li, Z.; Carter, C.W., Jr. A leucyl-tRNA synthetase urzyme: authenticity of tRNA Synthetase urzyme catalytic activities and production of a non-canonical product. International Journal of Molecular Sciences 2022, 23, 4229. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.K.; Betts, L.; Tang, G.Q.; Douglas, J.; Wills, P.R.; Bouckeart, R.; Carter, C.W., Jr. . Genomic databases furnish a spontaneous example of a functional Class II Glycyl-tRNA synthetase urzyme. In Preparation 2024.
- Tang, G.Q.; Carter, C.W., Jr. The LeuAC Urzyme Catalyzes Aminoacylation of tRNALeu Minihelix Using Leucine and ATP. In preparation 2023.
- Zull, J.E.; Smith, S.K. Is genetic code redundancy related to retention of structural information in both DNA strands? TIBS 1990, 15, 257–261. [Google Scholar] [CrossRef]
- Opuu, V.; Silvert, M.; Simonson, T. Computational design of fully overlapping coding schemes for protein pairs and triplets. Scientific REPORTS 2017, 7, 15873. [Google Scholar] [CrossRef]
- Carter, C.W., Jr. Simultaneous codon usage, the origin of the proteome, and the emergence of de-novo proteins. Current Opinion in Structural Biology 2021, 68, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W., Jr. How did the proteome emerge from pre-biotic chemistry? In Pre-Biotic Chemistry and Life's Origin, Fiore, M., Ed. The Royal Society of Chemistry: London, UK, 2022; pp. 317-346.
- Mullen, G.P.; Vaughn, J.B., Jr.; Mildvan, A.S. Sequential Proton NMR Resonance Assignments, Circular Dichroism, and Structural Properties of a 50-Residue Substrate-Binding Peptide from DNA Polymerase I. Arch. Biochem. Biophys. 1993, 301, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Chuang, W.-J.; Abeygunawardana, C.; Pedersen, P.L.; Mildvan, A.S. Two-Dimensional NMR, Circular Dichroism, and Fluorescence Studies of PP-50, a Synthetic ATP-Binding Peptide from the b-Subunit of Mitochondrial ATP Synthase. Biochem. 1992, 31, 7915–7921. [Google Scholar] [CrossRef]
- Chuang, W.-J.; Abeygunawardana, C.; Gittis, A.G.; Pedersen, P.L.; Mildvan, A.S. Solution Structure and Function in Trifluoroethanol of PP-50, an ATP-Binding Peptide from F1ATPase. Arch. Biochem. Biophys. 1992, 319, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.C.; Byler, D.M.; Sisu, H.; Brown, E.M.; Kuby, S., A.; Mildvan, A.S. Solution Structure of the 45-Residue MgATP-Binding Peptide of Adenylate Kinase As Examined by 2-D NMR, FTIR, and CD Spectroscopy. Biochem. 1988, 27, 3588–3598. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.C.; Kuby, S., A.; Mildvan, A.S. NMR Studies of the MgATP Binding Site of Adenylate Kinase and of a 45-Residue Peptide Fragment of the Enzyme. Biochem. 1985, 24, 4680–4694. [Google Scholar] [CrossRef]
- Kaiser, F.; Krautwurst, S.; Salentin, S.; Haupt, V.J.; Leberecht, C.; Bittrich, S.; Labudde, D.; Schroeder, M. The structural basis of the genetic code: amino acid recognition by aminoacyl-tRNA synthetases. Sci Rep 2020, 10, 12647. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, F.; Bittrich, S.; Salentin, S.; Leberecht, C.; Haupt, V.J.; Krautwurst, S.; Schroeder, M.; Labudde, D. Backbone Brackets and Arginine Tweezers delineate Class I and Class II aminoacyl tRNA synthetases. PLoS Comput Biol 2018, 14, e1006101. [Google Scholar] [CrossRef]
- Carter, C.W., Jr; Wills, P.R. Class I and II aminoacyl-tRNA synthetase tRNA groove discrimination created the first synthetase•tRNA cognate pairs and was therefore essential to the origin of genetic coding. IUBMB Life 2019, 71, 1088–1098. [Google Scholar] [CrossRef]
- Carter, C.W., Jr.; Wills, P.R. Hierarchical groove discrimination by Class I and II aminoacyl-tRNA synthetases reveals a palimpsest of the operational RNA code in the tRNA acceptor-stem bases. Nucleic Acids Research 2018, 46, 9667–9683. [Google Scholar] [CrossRef]
- Carter, C.W., Jr.; Wills, P.R. Experimental Solutions to Problems Defining the Origin of Codon-Directed Protein Synthesis. BioSystems 2019, 183, 103979. [Google Scholar] [CrossRef]
- Fizza MughalI; Caetano-Anollés, G. MANET 3.0: Hierarchy and modularity in evolving metabolic networks. PLOS ONE 2019, 14, e0224201. [CrossRef]
- Caetano-Anollés, G.; Aziz, M.F.; Mughal, F.M.; Gräter, F.; Koç, I.; Caetano-Anollés, K.; Caetano-Anollés, D. Emergence of Hierarchical Modularity in Evolving Networks Uncovered by Phylogenomic Analysis. Evolutionary Bioinformatics 2019, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Anollés, G.; Nasir, A.; Kim, K.M.; Caetano-Anollés, D. Rooting Phylogenies and the Tree of Life While Minimizing Ad Hoc and Auxiliary Assumptions. Evolutionary Bioinformatics 2018, 14, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Koç, I.; Caetano-Anollés, G. The natural history of molecular functions inferred from an extensive phylogenomic analysis of gene ontology data. PLoS ONE 2017, 12, e0176129. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Anollés, D.; Caetano-Anollés, G. Piecemeal Buildup of the Genetic Code, Ribosomes, and Genomes from Primordial tRNA Building Blocks. Life 2016, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.F.; Caetano-Anollés, K.; Caetano-Anollés, G. The early history and emergence of molecular functions and modular scale-free network behavior. Scientific Reports | 2016, 6, 25058. [Google Scholar] [CrossRef]
- Caetano-Anollés, G.; Wang, M.; Caetano-Anollés, D. Structural Phylogenomics Retrodicts the Origin of the Genetic Code and Uncovers the Evolutionary Impact of Protein Flexibility. Plos One 2013, 8, e72225. [Google Scholar] [CrossRef]
- Koonin, E.V.; Novozhilov, A.S. Origin and Evolution of the Universal Genetic Code. Annu. Rev. Genet. 2017, 51, 45–62. [Google Scholar] [CrossRef]
- O’Donoghue, P.; Luthey-Schulten, Z. On the Evolution of Structure in Aminoacyl-tRNA Synthetases. Microbiol. Mol. Biol. Rev. 2003, 67, 550–573. [Google Scholar] [CrossRef]
- Hohn, M.J.; Park, H.-S.; O’Donoghue, P.; Schnitzbauer, M.; Söll, D. Emergence of the universal genetic code imprinted in an RNA record. Proc. Nat. Acad. Sci. USA 2006, 103, 18095–18100. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Burton, Z.F. Evolution of Life on Earth: tRNA, Aminoacyl-tRNA Synthetases and the Genetic Code. Life 2020, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Ibba, M.; Soll, D. Aminoacyl-tRNAs: setting the limits of the genetic code. Genes and Development 2004, 18, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Olsen, G.J.; Ibba, M.; Soll, D. Aminoacyl-tRNA Synthetases, the Genetic Code, and the Evolutionary Process. Microbiol. Mol. Biol. Rev. 2000, 64, 202–236. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Anollés, G.; Sun, F.-J. The natural history of transfer RNA and its interactions with the ribosome. Fronteirs in Genetics 2014, 5, 127. [Google Scholar]
- Johnson, B.R.; Lam, S.K. Self-organization, Natural Selection,and Evolution: Cellular Hardware and Genetic Software. BioScience 2010, 60, 879–885. [Google Scholar] [CrossRef]
- Füchslin, R.M.; McCaskill, J.S. Evolutionary self-organization of cell-free genetic coding. Proc Natl Acad Sci USA 2001, 98, 9185–9190. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Selforganization of Matter and the Evolution of Biological Macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef]
- Johnson, K.A. New standards for collecting and fitting steady state kinetic data. Beilstein J. Org. Chem. 2019, 15, 16–29. [Google Scholar] [CrossRef]
- Andreeva, A.; Howorth, D.; Chandonia, J.-M.; Brenner, S.E.; Hubbard, T.J.P.; Chothia, C.; Murzin, A.G. Data growth and its impact on the SCOP database: new developments. Nucl. Acids Res. 2008, 36, D419–D425. [Google Scholar] [CrossRef]
- Zhang, Y.; Chandonia, J.-M.; Ding, C.; Holbrook, S.R. Comparative mapping of sequence-based and structure-based protein domains. BMC Bioinformatics 2005, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Hanson-Smith, V.; Kolaczkowski, B.; Thornton, J.W. Robustness of Ancestral Sequence Reconstruction to Phylogenetic Uncertainty. Mol. Biol. Evol. 2010, 27, 1988–1999. [Google Scholar] [CrossRef] [PubMed]
- Liberles, D.A. Ancestral Sequence Reconstruction; Oxford University Press: Oxford, 2007. [Google Scholar]
- Benner, S.A.; Sassi, S.O.; Gaucher, E.A. Molecular Paleoscience: Systems Biology from the Past. Advances in Enzymology and Related Areas of Molecular Biology 2007, 75, 9–140. [Google Scholar]
- Stackhouse, J.; Presnell, S.R.; McGeehan, G.M.; Nambiar, K.P.; Benner, S.A. The Ribonuclease from an extinct bovid ruminant. FEBS Letters 1990, 262, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Praetorius-Ibba, M.; Stange-Thomann, N.; Kitabatake, M.; Ali, K.; Söll, I.; Carter, C.W., Jr.; Ibba, M.; Söll, D. Ancient Adaptation of the Active Site of Tryptophanyl-tRNA Synthetase for Tryptophan Binding. Biochemistry 2000, 39, 13136–13143. [Google Scholar] [CrossRef] [PubMed]
- Bullock, T.; Uter, N.; Nissan, T.A.; Perona, J.J. Amino Acid Discrimination by a class I aminoacyl-tRNA synthetase specified by negative determinants. J. Mol. Biol. 2003, 328, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Carter, C.W., Jr. Full Implementation of the Genetic Code by Tryptophanyl-tRNA Synthetase Requires Intermodular Coupling. J. Biol. Chem. 2013, 288, 34736–34745. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, V.; Li, L.; Chandrasekaran, S.N.; Koehl, P.; Delarue, M.; Carter, C.W., Jr Enhanced Amino Acid Selection in Fully-Evolved Tryptophanyl-tRNA Synthetase, Relative to its Urzyme, Requires Domain Movement Sensed by the D1 Switch, a Remote, Dynamic Packing Motif J Biol Chem 2014, 289, 4367-4376. [CrossRef]
- Perona, J.J.; Gruic-Sovulj, I. Synthetic and Editing Mechanisms of Aminoacyl-tRNA Synthetases. Topics in Current Chemistry 2014, 344, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Shore, J.; Holland, B.R.; Sumner, J.G.; Nieselt, K.; Wills, P.R. The Ancient Operational Code is Embedded in the Amino Acid Substitution Matrix and aaRS Phylogenies. J. Mol. Evol. 2019, 88, 136–150. [Google Scholar] [CrossRef]
- Dang, C.C.; Minh, B.Q.; McShea, H.; Masel, J.; James, J.E.; Vinh, L.S. Dang, C.C.; Minh, B.Q.; McShea, H.; Masel, J.; James, J.E.; Vinh, L.S.; Robert Lanfear. nQMaker: Estimating Time Nonreversible Amino Acid Substitution Models. Syst. Biol. 2022, 71, 1110–1123. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Sottani, J.B.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N., et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput Biol 2019, 15, e1006650.
- Dauparas, J.; Anishchenko, I.; Bennett, N.; Bai, H.; Ragotte, R.J.; Milles, L.F.; Wicky, B.I.M.; Courbet, A.; de Haas, R.J.; Bethel, N., et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science 2022, 378, , 49–56 (2022).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–592. [Google Scholar] [CrossRef]
- Tropsha, A.; Carter, C.W.J.; Cammer, S.; Vaisman, I.I. Simplicial Neighborhood Analysis of Protein Packing (SNAPP): A Computational Geometry Approach to Studying Proteins. Methods in Enzymology 2003, 374, 509–544. [Google Scholar]
- Carter, C.W., Jr.; LeFebvre, B.; Cammer, S.A.; Tropsha, A.; Edgell, M.H. Four-body potentials reveal protein-specific correlations to stability changes caused by hydrophobic core mutations. Journal of Molecular Biology 2001, 311, 625–638. [Google Scholar] [CrossRef]
- Wang, C.; Zou, Q. Prediction of protein solubility based on sequence physicochemical patterns and distributed representation information with DeepSoluE. BMC Biology 2023, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.; Carter, C.W., Jr; Peter, R. Wills. HetMM: A Michaelis-Menten model for non-homogeneous enzyme mixtures. BioRxiv, 1101. [Google Scholar]
- Ivankov, D.N.; Finkelstein, A.V. Solution of Levinthal’s Paradox and a Physical Theory of Protein Folding Times. Biomolecules 2020, 10, 250. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.; Chan, H.S. From Levinthal to pathways to funnels. Nat. Str. Biol. 1997, 4, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Levinthal, C. ARE THERE PATHWAYS FOR PROTEIN FOLDING ? Journal de Chimie Physique 1968, 65, 44. [Google Scholar] [CrossRef]
- Mattenet, A.L.; Davidson, I.; Nijssen, S.; Schaus, P. Constraint Programming for an Efficient and Flexible Block Modeling Solver. AAAI Conference on Artificial Intelligence 2020, 34, 13685–13688. [Google Scholar] [CrossRef]
- Wills, P.R.; Carter, C.W., Jr. Impedance matching and the choice between alternative pathways for the origin of genetic coding. International Journal of Molecular Sciences 2020, 21, 7392. [Google Scholar] [CrossRef]
- San Andrés, L. Impedance Matching. Availabe online: https://oaktrust.library.tamu.edu/handle/1969.1/188313 (accessed on.
- Hofstadter, D.R. Gödel, Escher, Bach: an eternal golden braid; Basic Books, Inc: New York, 1979; p. 777. [Google Scholar]
- Carter, C.W., Jr.; Wills, P.R. Reciprocally-coupled Gating: Strange Loops in Bioenergetics, Genetics, and Catalysis. Biomolecules 2021, 11, 265. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W., Jr. Escapement mechanisms: efficient free energy transduction by reciprocally-coupled gating. Proteins: Structure, Function, and Bioinformatics 2019, 88, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Pham, Y.; Li, L.; Kim, A.; Weinreb, V.; Butterfoss, G.; Kuhlman, B.; Carter, C.W., Jr. A Minimal TrpRS Catalytic Domain Supports Sense/Antisense Ancestry of Class I and II Aminoacyl-tRNA Synthetases. Mol. Cell 2007, 25, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Niwa, N.; Yamagishi, Y.; Murakami, H.; Suga, H. A flexizyme that selectively charges amino acids activated by a water-friendly leaving group. Bioorg. Med. Chem. Lett. 2009, 19, 3892–3894. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Murakami, H.; Suga, H.; Ferre-D’Amare, A.R. Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature 2008, 454, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Buehner, M.; Ford, G.C.; Moras, D.; Olsen, K.W.; Rossmann, M.G. D-Glyceraldehyde 3-Phosphate Dehydrogenase: Three Dimensional Structure and Evolutionary Significance. Proc. Nat. Acad. Sci. USA 1973, 70, 3052–3054. [Google Scholar] [CrossRef]
- Carter, C.W., Jr.; Wolfenden, R. tRNA Acceptor-Stem and Anticodon Bases Form Independent Codes Related to Protein Folding. Proc. Nat. Acad. Sci. USA 2015, 112 7489-7494. [CrossRef]
- Wolfenden, R.; Lewis, C.A.; Yuan, Y.; Carter, C.W., Jr. Temperature dependence of amino acid hydrophobicities. Proc. Nat. Acad. Sci. USA 2015, 112, 7484–7488. [Google Scholar] [CrossRef] [PubMed]
- Roach, J.M.; Sharma, S.; Kapustina, M.; Carter, C.W., Jr. Structure alignment via Delaunay tetrahedralization. PROTEINS: Structure, Function and Bioinformatics 2005, 60, 66–81. [Google Scholar] [CrossRef]
- Witten, E. What every physicist should know about String Theory. Physics Today 2015, November, 38–43. [Google Scholar] [CrossRef]
Figure 1.
Coupled evolutionary advances favoring emergence and fostering the specialization of genetic coding. a. Interdependences involved in embedding specific recognition into the sequence space of folded proteins [19]. b. Experimentally measurable metrics corresponding to a. Although experimental models exist for each process, confirming details requires further work.
Figure 1.
Coupled evolutionary advances favoring emergence and fostering the specialization of genetic coding. a. Interdependences involved in embedding specific recognition into the sequence space of folded proteins [19]. b. Experimentally measurable metrics corresponding to a. Although experimental models exist for each process, confirming details requires further work.
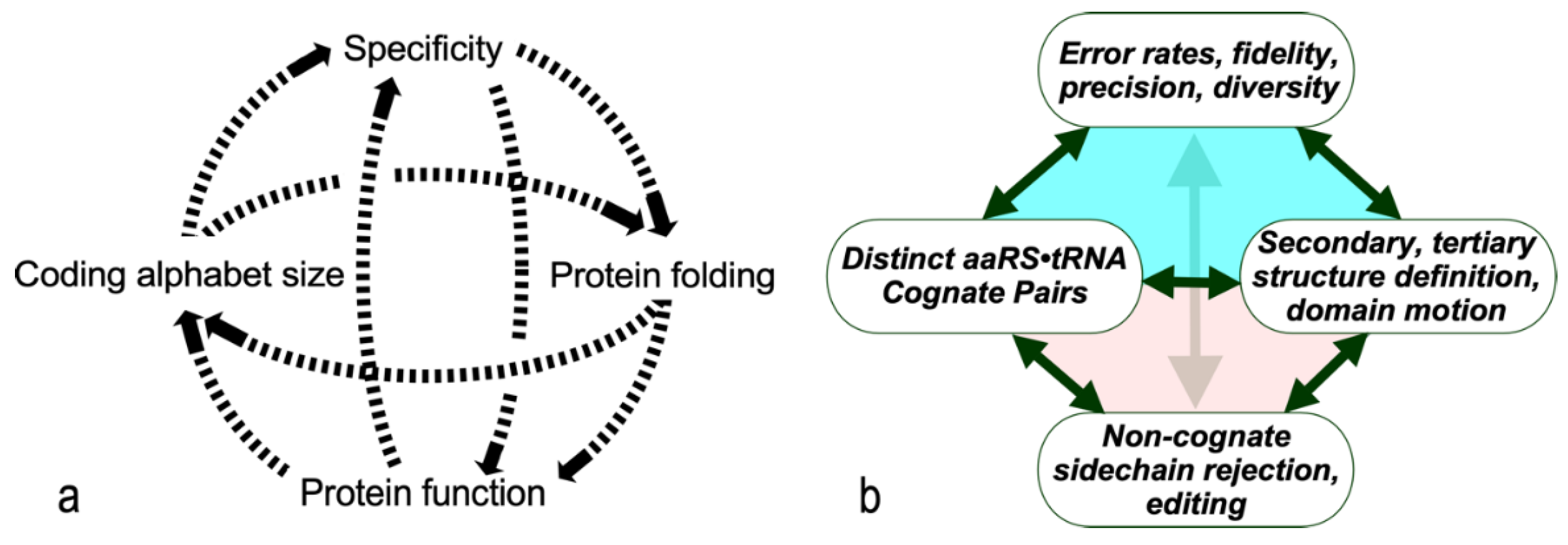
Figure 2.
Assignment catalysis by AARS. a. Class I and II AARS•tRNA cognate pairs as envisioned by Ribas and Schimmel [11] with molecular cartoons of respective tRNA acceptor stems superimposed to highlight the opposite directions of their 3’ CCA extensions. Synthetases are shown as elliptical shapes to highlight the five distinct regions in their active-site pockets. Three are cavities (white) for amino acid, ATP, and 3’ terminal adenosine. Stars represent transition-state complementarity during amino acid activation (red) and acyl transfer to cognate tRNA (purple). b. AND gate pseudocode. Two inputs are detected and compared to select exclusive, complementary subsets of the two substrate groups, amino acids and tRNAs. tRNA substrates contain cognate anticodons used to read mRNA, hence are explicitly symbolic. Covalent bonds result if and only if both substrates are correct.
Figure 2.
Assignment catalysis by AARS. a. Class I and II AARS•tRNA cognate pairs as envisioned by Ribas and Schimmel [11] with molecular cartoons of respective tRNA acceptor stems superimposed to highlight the opposite directions of their 3’ CCA extensions. Synthetases are shown as elliptical shapes to highlight the five distinct regions in their active-site pockets. Three are cavities (white) for amino acid, ATP, and 3’ terminal adenosine. Stars represent transition-state complementarity during amino acid activation (red) and acyl transfer to cognate tRNA (purple). b. AND gate pseudocode. Two inputs are detected and compared to select exclusive, complementary subsets of the two substrate groups, amino acids and tRNAs. tRNA substrates contain cognate anticodons used to read mRNA, hence are explicitly symbolic. Covalent bonds result if and only if both substrates are correct.
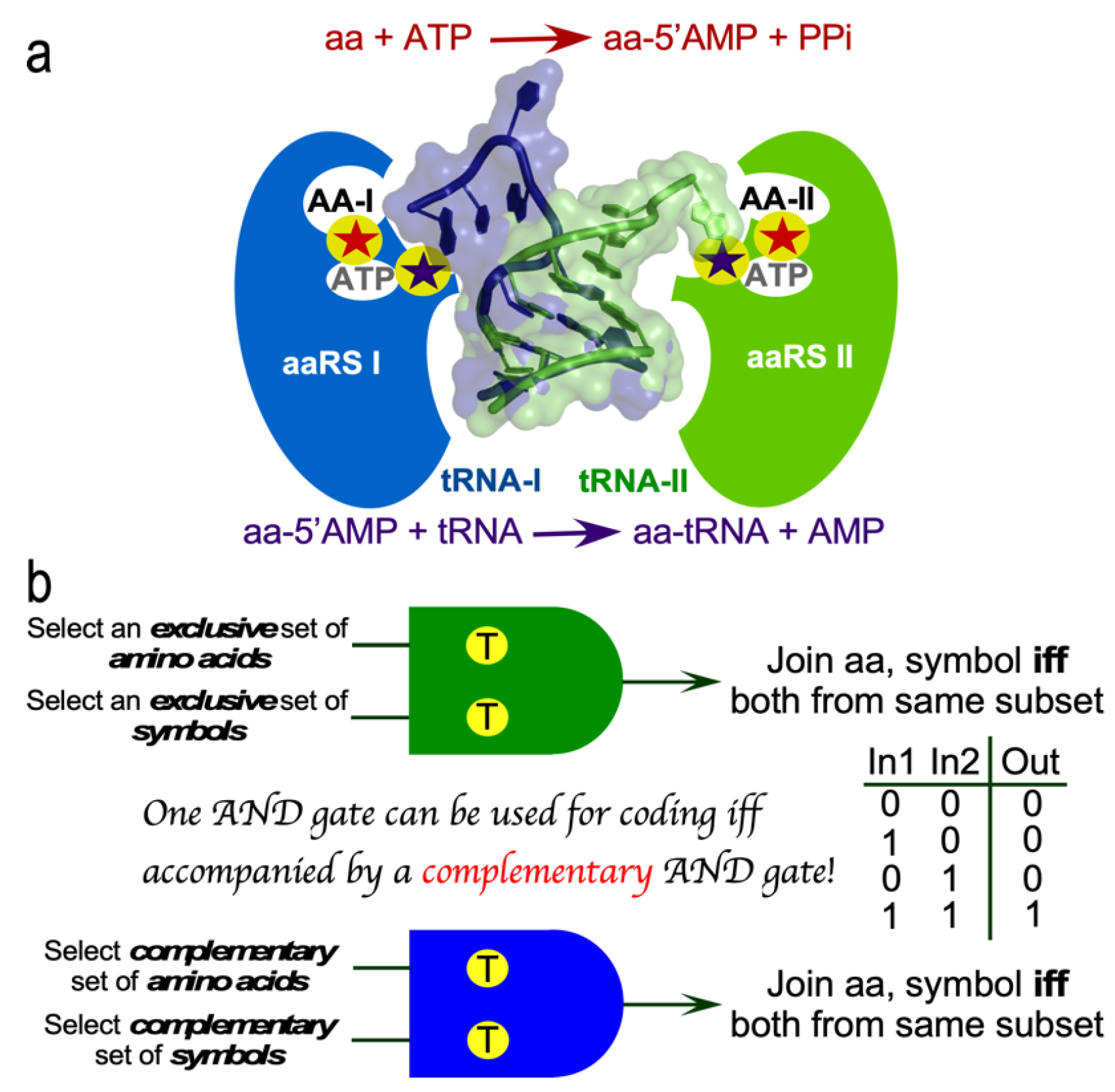
Figure 3.
Bidirectional coding induces inside-out folding and inverted substrate binding. a. Antiparallel alignment of Class I (red) and II (blue) protozymes described previously [2,32]. A significant majority (74 %) of amino acid side chains have inversely related transfer free energies, ΔGw>c, from water to cyclohexane, leading to highly symmetric hydrophobicity profiles for those residues. Solid dots denote residues from restricted subsets [Ile, Val, Leu; subclass IA] and [Asp, Lys, Asn; subclass IIB] and account for 46% of the sequence. Open dots denote residues defining the HIGH and Motif 2 signatures. b. Genetic complementarity propagates, via reflection symmetry in (a) into the resulting Class I and II protozyme and tertiary structures. This has functional consequences. Their succession of secondary structures is similar, but their inverted polarity means that the substrate-binding loci are inverted. ATP binds to light blue segments at the N-terminus of the α-helix in Class I (dark background) and to the C-terminus of the second β-strand in Class II (light background). Similarly, the amino acid substrate binds to salmon segments of both β-strands in Class I but to the N-terminus of the α-helix in Class II. Binding sites have the adenine ring (lower left) in approximately the same orientation to highlight the approximate stereroisomerism of the 5’ phosphate (circles). Class I amino acids point left, away from, while Class II amino acids point right, toward their protein binding determinants (arrows).
Figure 3.
Bidirectional coding induces inside-out folding and inverted substrate binding. a. Antiparallel alignment of Class I (red) and II (blue) protozymes described previously [2,32]. A significant majority (74 %) of amino acid side chains have inversely related transfer free energies, ΔGw>c, from water to cyclohexane, leading to highly symmetric hydrophobicity profiles for those residues. Solid dots denote residues from restricted subsets [Ile, Val, Leu; subclass IA] and [Asp, Lys, Asn; subclass IIB] and account for 46% of the sequence. Open dots denote residues defining the HIGH and Motif 2 signatures. b. Genetic complementarity propagates, via reflection symmetry in (a) into the resulting Class I and II protozyme and tertiary structures. This has functional consequences. Their succession of secondary structures is similar, but their inverted polarity means that the substrate-binding loci are inverted. ATP binds to light blue segments at the N-terminus of the α-helix in Class I (dark background) and to the C-terminus of the second β-strand in Class II (light background). Similarly, the amino acid substrate binds to salmon segments of both β-strands in Class I but to the N-terminus of the α-helix in Class II. Binding sites have the adenine ring (lower left) in approximately the same orientation to highlight the approximate stereroisomerism of the 5’ phosphate (circles). Class I amino acids point left, away from, while Class II amino acids point right, toward their protein binding determinants (arrows).
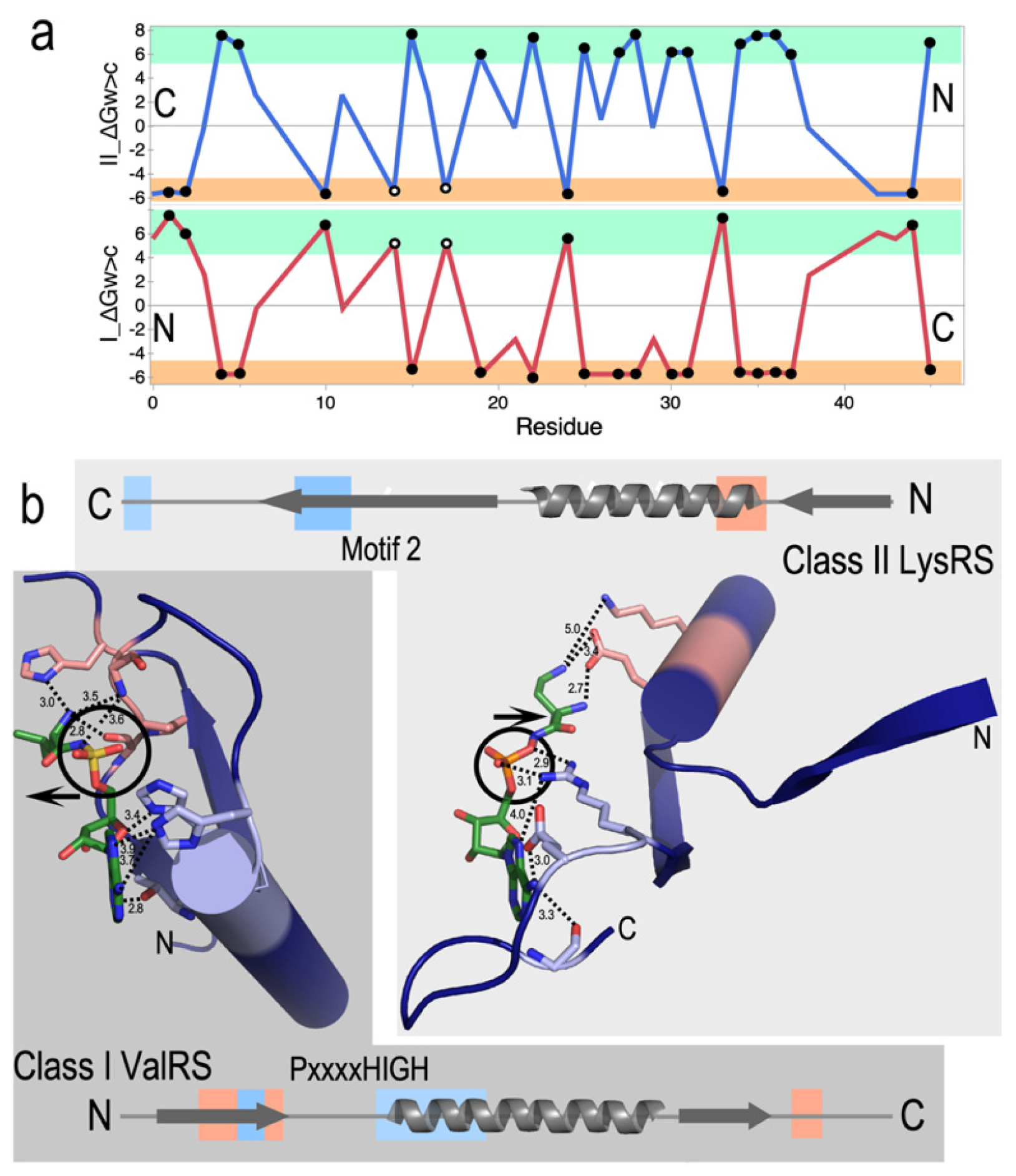
Figure 4.
Class-dependent AARS•tRNA cognate pair formation is dictated by synthetase tertiary structure. A. Class I complexes require the 3’CCA terminus to form a hairpin that is recognized by a combination of electrostatic (+, –) and aromatic/hydrophobic (green symbols) interactions with the 3’ terminal phosphate and ribose, respectively. The 3AKZ GluRS complex is typical of functionally relevant complexes of LeuRS, ArgRS, and GlnRS. B. The 5E6M GlyRS complex is typical of functionally relevant complexes of Class II ThrRS, AspRS, and AlaRS. Acceptor stems are indicated by dashed cylinders in both A and B. Aminoacylation sites are circled, with A76 nucleophile-to-aminoacyl-5’AMP distance shown with a bold dashed line. The direction of the incoming CCA is indicated by yellow arrows. Interactions with cognate AARS (sand) make very different interactions with the 1-72 base-pairs (spheres). The Class I α-helix with many side chains recognizing the amino acid forms key interactions with the 3’ phosphate and the ribose moieties of A76, enforcing the characteristic hairpin turn. Those interactions do not entail the terminal base pair. In Class II complexes, the antiparallel β-hairpin of Motif 2 (brown ribbon) covers the 1-72 base pair, enforcing interactions with the helical extension of the CCA terminus [52,53] (adapted from [14]).
Figure 4.
Class-dependent AARS•tRNA cognate pair formation is dictated by synthetase tertiary structure. A. Class I complexes require the 3’CCA terminus to form a hairpin that is recognized by a combination of electrostatic (+, –) and aromatic/hydrophobic (green symbols) interactions with the 3’ terminal phosphate and ribose, respectively. The 3AKZ GluRS complex is typical of functionally relevant complexes of LeuRS, ArgRS, and GlnRS. B. The 5E6M GlyRS complex is typical of functionally relevant complexes of Class II ThrRS, AspRS, and AlaRS. Acceptor stems are indicated by dashed cylinders in both A and B. Aminoacylation sites are circled, with A76 nucleophile-to-aminoacyl-5’AMP distance shown with a bold dashed line. The direction of the incoming CCA is indicated by yellow arrows. Interactions with cognate AARS (sand) make very different interactions with the 1-72 base-pairs (spheres). The Class I α-helix with many side chains recognizing the amino acid forms key interactions with the 3’ phosphate and the ribose moieties of A76, enforcing the characteristic hairpin turn. Those interactions do not entail the terminal base pair. In Class II complexes, the antiparallel β-hairpin of Motif 2 (brown ribbon) covers the 1-72 base pair, enforcing interactions with the helical extension of the CCA terminus [52,53] (adapted from [14]).
Figure 5.
Elements of the constraint surface over which Nature optimized the coding alphabet. a. Representative curve (blue) for coded information content Iσ (bits per codon or amino acid; left hand Y axis) versus alphabet size, n. Nominal rate of translation, μ, on log(kcat/KM) is indicated by the dashed proxy scale. Typical curves (dashed, color spectrum) for information transferred per unit energy expended per monomer concatenated, η, (bits per Joule per monomer, arbitrary dashed scale on right hand Y-axis) for AARS evolution through increasing amino alphabet size, n. Redundancy (red curve), I’red versus n, is averaged out over the codon and amino acid alphabets, making the most significant contribution to impedance-matching. b. Reciprocally coupled gating of two AND gates with the consequent of the first linked to the antecedent of the second introduces two kinds of forces. Gating efficiency is an attractive force because it concentrates inputs that satisfy both AND conditions. The coupled gates form a strange loop leading to incompleteness and a chemical potential-like reservoir.
Figure 5.
Elements of the constraint surface over which Nature optimized the coding alphabet. a. Representative curve (blue) for coded information content Iσ (bits per codon or amino acid; left hand Y axis) versus alphabet size, n. Nominal rate of translation, μ, on log(kcat/KM) is indicated by the dashed proxy scale. Typical curves (dashed, color spectrum) for information transferred per unit energy expended per monomer concatenated, η, (bits per Joule per monomer, arbitrary dashed scale on right hand Y-axis) for AARS evolution through increasing amino alphabet size, n. Redundancy (red curve), I’red versus n, is averaged out over the codon and amino acid alphabets, making the most significant contribution to impedance-matching. b. Reciprocally coupled gating of two AND gates with the consequent of the first linked to the antecedent of the second introduces two kinds of forces. Gating efficiency is an attractive force because it concentrates inputs that satisfy both AND conditions. The coupled gates form a strange loop leading to incompleteness and a chemical potential-like reservoir.
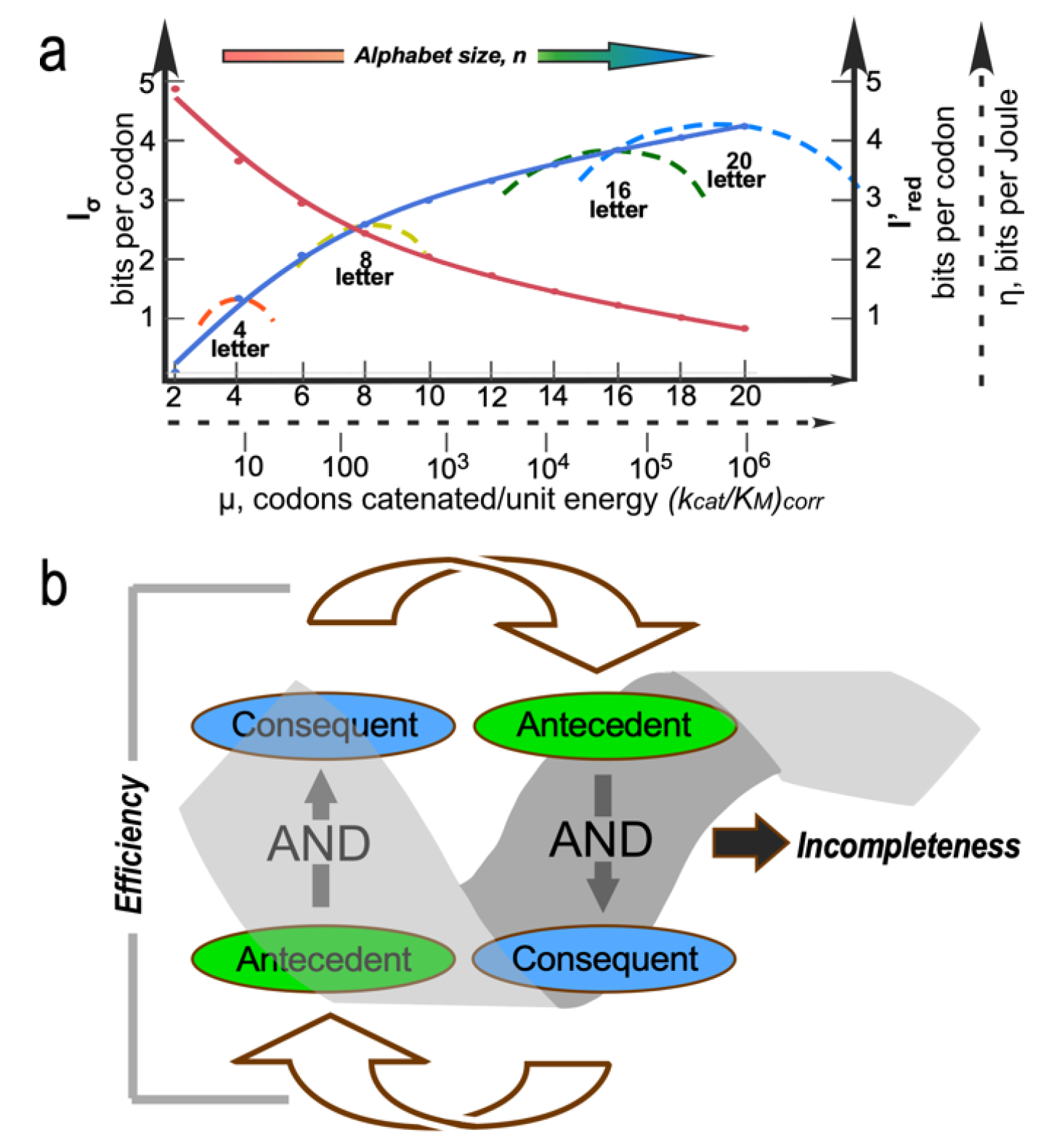
Figure 6.
Scenario for the origin and evolution of the 20 AARS. The size of the coding alphabet along the top defines the differently colored panels. Approximate functionality achieved for a given alphabet size is indicated along the bottom. The tree superimposed on the panels and the identities of coding letters at any stage are arbitrary. Dashed circular arrows denote the equilibration of all extant genes as AARS speciation defined more coding letters.
Figure 6.
Scenario for the origin and evolution of the 20 AARS. The size of the coding alphabet along the top defines the differently colored panels. Approximate functionality achieved for a given alphabet size is indicated along the bottom. The tree superimposed on the panels and the identities of coding letters at any stage are arbitrary. Dashed circular arrows denote the equilibration of all extant genes as AARS speciation defined more coding letters.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



