Submitted:
29 December 2023
Posted:
03 January 2024
You are already at the latest version
Abstract
This review provides insight into the role that obesity, but especially mediators of the adipose tissue, have in both renal disease and transplantation. In particular, we focus on the functions of leptin and adiponectin, inflammatory mediators with widely known clinical implications, as well as on the role of arachidonic acid-derived eicosanoid metabolites. These latter compounds, epoxyeicosatrienoic (EETs) and hydroxyeicosatetraenoic (HETEs) acids, which modulate the liberation of cytokines in the adipose tissue, are generated in the epoxygenase pathway of arachidonic biotransformation via CYP450 enzymes. There is accumulating evidence suggesting that EETs and HETEs may have a critical role in the onset and outcome of chronic kidney disease and renal transplantation. Finally, we also discuss the reported effect that the presence of genetic variants in leptin, adiponectin, EETs and HETEs-related genes may cause in renal patients and recipients. The levels of cytokines of the adipose tissue and related compounds play a remarkable role in both renal disease and transplant. Accordingly, novel therapeutic strategies are being developed based on the modulation of their concentrations in patients with various diseases.
Keywords:
chronic kidney disease
; renal transplant
; arachidonic acid
; adipose tissue
Chronic kidney disease (CKD) is a global health problem whose incidence is around 8-16% (1). It consists of damage at the renal level that is usually characterized by a significant decrease in glomerular filtration rate (GFR), below 60 mL/min/1.73 m2, for more than three months (2). Among the susceptibility factors that increase the possibility of kidney damage, we can find advanced age, obesity, diabetes or arterial hypertension. Factors that initiate renal damage include autoimmune diseases, diabetes or systemic infections, whilst factors that worsen renal damage and accelerate renal function deterioration include smoking, dyslipidemia or obesity (3). The most advanced manifestation of chronic kidney disease is end-stage kidney disease (ESKD). Once this stage of the disease has been reached, the need to initiate renal replacement therapy for renal function by means of dialysis or renal transplantation is considered, the latter being the treatment of choice, even though it is not free of short- and long-term complications.
There is a myriad of mediators of kidney damage and post-transplant complications. The objective of this review was specifically to present information supporting the concept that adipose tissue mediators, mainly adiponectin and leptin and related substances such as eicosanoids derived from arachidonic acid, can also have an important role in the genesis and/or aggravation of kidney disease, as well as being associated with the clinical evolution of the graft after kidney transplantation.
Obesity in Chronic Kidney Disease
Obesity has been identified for years as a major cause of kidney disease, including CKD (4-6), with evidence of causality in several studies (4,7-10). Directly, obesity can lead to structural and inflammatory changes at the renal level (11), whilst it can indirectly influence the onset and worsening of diabetes mellitus (12). Obesity-related kidney injury may present a circulatory component, either by injury to blood vessels or compression by adipose tissue (13); an inflammatory component, by activation of inflammatory cytokines (14); or a hormonal component, through the effect on the renin-angiotensin system (15). In addition, obesity may produce metabolic and biochemical alterations that predispose to kidney disease, even in the presence of normal renal function (6,16).
Overweight and obesity are considered important risk factors for the onset of CKD, largely due to their close relationship with hypertension, diabetes mellitus (17) and cardiovascular risk (18), thus predisposing to pathologies such as diabetic nephropathy, hypertensive nephrosclerosis or focal and segmental glomerulosclerosis (6,7). However, the pathogenic mechanisms involved in the association of obesity with kidney disease are not yet fully elucidated (19). From a pathophysiological point of view, obesity triggers an increase in tubular sodium reabsorption, with altered natriuresis and consequent volume expansion due to activation of the sympathetic nervous system and the renin-angiotensin-aldosterone axis (17). From this point on, a series of pathophysiological mechanisms occur that trigger the development of arterial hypertension (HT), with increased renal flow and glomerular filtration rate. This increase in glomerular filtration rate, together with other metabolic alterations such as insulin resistance and diabetes mellitus, eventually lead to renal damage (4,6,7). The accumulation of adipose tissue in obesity, especially in the abdominal region, causes renal compression, and consequently an increase in intrarenal pressure, which leads to a reduction in tubular flow and subsequent increases in sodium reabsorption (4,6,7). In addition, obesity is associated with inflammation, which increases the production of inflammatory cytokines, being in itself a risk factor for the loss of renal function (20,21).
Reverse Epidemiology of Renal Transplantation
The concept of reverse epidemiology was born a few years ago, posing as an anodyne situation whose cause is still unknown, mainly present in the context of heart failure or hemodialysis, and which forces us to rethink certain medical axioms related to obesity. In view of this, it is inevitable to wonder whether renal transplant patients also present this phenomenon. Contrary to what happens in hemodialysis, the study of the effect of obesity in renal transplantation is controversial (22). On the one hand, a BMI > 30 kg/m2 has been identified as the most important risk factor for surgical wound infection in the renal transplant recipient (23), with consequent complications for both graft and patient. The relationship between a high BMI and a significant increase in surgical time and postoperative stay has also been described (24), as well as a higher incidence of early graft loss caused by vascular complications, such as renal artery thrombosis (25). Therefore, it seems logical to associate a poor post-transplant prognosis with obesity. However, on the other hand, in a meta-analysis conducted by Nicoletto et al. on the effects of obesity on the development of renal transplantation, the researchers conclude that the relationship between obesity and renal graft loss, death from cardiovascular causes or from any cause, differs according to the era of the study, such that in studies prior to the year 2000, obesity was a risk factor for the appearance of these variables, while in those after this year, obesity did not seem to influence them (22). There is another issue, pretansplant obesity, that makes the relationship between obesity and renal transplantation even more complex. Post-transplant obesity goes hand in hand with metabolic disorders such as HT, dyslipidemia or DM (26), classically associated with obesity. However, it has been paradoxically observed that a high BMI is associated with better survival in hemodialysis patients, including those on the list for renal transplantation (27-30). Since a large percentage of transplant patients come from hemodialysis, the effect of obesity in hemodialysis patients who are going to undergo renal transplantation would not be well established (31). According to the results of the meta-analysis performed by Ahmadi and co-workers (32), extreme pre-transplant BMI figures in adults are associated with an increase in transplant patient mortality and graft failure, and no paradoxical obesity effect was observed in their population. The authors argue that the paradoxical effect observed in hemodialysis patients who are candidates for kidney transplantation could be due to the contrast between the short- and long-term consequences of obesity, i.e., while obesity increases long-term cardiovascular mortality, it could at the same time attenuate short-term mortality associated with malnutrition, inflammation and protein energy wasting syndrome (33). A return to normal phenomenon, which would be in place after renal transplantation (33), has also been proposed.
The question thus arises regarding the recommendation of weight loss prior to transplantation. As previously discussed, there are several studies evidencing the negative effect of obesity on graft and renal transplant recipient (34). A calculator has even been developed that evaluates graft survival including BMI among its variables (35). This is why several authors recommend weight loss in potential renal transplant recipients (35,36), while other authors consider that BMI interventions should not be performed in patients on the transplant list, given the observed benefit of obesity on hemodialysis patients (32).
Adipose Tissue
Until a few years ago, adipose tissue was considered only a passive energy storage site. However, after the discovery of leptin, a protein secreted by adipose tissue, this concept changed, and it is now considered an active endocrine organ, secreting numerous adipokines, cytokines, growth factors and exosomal microRNAs with important systemic functions (37,38). Likewise, adipose tissue has a prominent role in the regulation of energy metabolism (39) and lipid homeostasis (40). It is now also known that adipose tissue is distributed diffusely throughout the body, varying the endocrine and metabolic functions it performs depending on its location (39).
There is increasing evidence of the involvement of adipose tissue in the physiology and pathophysiology of multiple diseases (41). Histologically, the adipose tissue is composed of two distinct types of elements, adipocytes and interadipocyte-vascular stroma, consisting of extracellular matrix with scattered fibroblasts, preadipocytes (immature adipocyte precursors), endothelial cells and immune cells (42,43). The presence of immune system cells in adipose tissue is not surprising if we consider the chronological evolution of the organs that comprise it. Inflammation, framed in the context of the immune response, consists of a series of humoral and cellular reactions aimed at defending the organism from aggressions, including infection and tissue damage, finally reaching the restoration of the functional and morphological integrity of the affected tissues (44,45). In this context, obesity is considered a state of chronic low-grade inflammation, characterized by an increase in proinflammatory markers, without obvious clinical signs, which is why it has been termed a subclinical inflammatory state (34). Even though this situation might seem to be a reaction of the organism of lesser intensity than that activated in the face of an acute aggression, it could also represent the ideal environment for the larval development of various diseases of later onset.
In a normal situation, the cells of the immune system residing in the adipose tissue actively participate in the maintenance of this tissue, eliminating detritus or apoptotic cells, and maintaining the homeostatic balance (46); however, in a situation of chronic inflammation as obesity represents, the existing balance can be altered. Adipose tissue undergoes marked hypertrophy and hyperplasia in response to an insult, leading to hypoxia, dysregulation of adipokines and, consequently, low-grade inflammation characterized by increased infiltration and activation of innate and adaptive immunity of immune cells (47). Chronic excess food intake leads to pathological expansion of adipose tissue, with hypertrophic adipocytes that fail to store energy, resulting in dysfunction, dyslipidemia and insulin resistance (48). This altered, inflamed, adipose tissue causes the release of various proinflammatory factors from adipocytes, thus amplifying the inflammatory situation (48). There are many questions about the in-situ activation of these inflammatory cells. It is now known that obesity leads to a qualitative alteration of the resident macrophages in adipose tissue, as well as the generation of oxygen free radicals, all of which leads to insulin resistance (49). The body tends to maintain the equilibrium situation at all times, so this inflammatory reaction of the adipose tissue may be necessary to maintain the homeostasis in the rest of the body. Moreover, the changes carried out in the adipose tissue also occur in other organs of the body, such as the liver, pancreas or muscle tissue, which gives us an idea of the universal nature of the metabolic dysfunction that takes place in the context of obesity (50).
Renal transplantation is a real challenge for the balance of the immune system, making the nephrologist to keep a challenging balance between the complex mechanism perfected over the years that allows us to survive pathological processes, and to avoid rejection of an organ understood by the organism as foreign. The immune system is the fundamental axis around which transplantation revolves and there are continuous advances in this field, controlling the body’s immune response by means of immunosuppression. As a fundamental organ of the immune system, adipose tissue plays an important role in this balance, interrelating the rest of the organs through inflammatory mediators.
Inflammatory Mediators of Adipose Tissue
Leptin
Leptin is a 16 kDa protein with 167 amino acids (51) that belongs to the interleukin-6 family, a group of inflammatory cytokines (52,53). Associated since its inception with adipose tissue, numerous pleiotropic effects of this protein are now known (54). Blood leptin concentrations follow a pulsatile and circadian pattern, with lower levels from early to mid-afternoon and higher levels between midnight and early morning (55). Leptin concentrations are influenced by factors such as sex, hormones or drugs. Thus, glucocorticoids increase leptin levels, as do insulin or estrogens (56), while fasting, metabolic acidosis, androgens, cold or beta-adrenergic agonists inhibit its production (57,58). Leptin values are higher in overweight and obese individuals (59), and also in women compared with men, after adjusting for BMI (60). This sexual dimorphism, independent of BMI, is attributed to differences in fat mass, body fat distribution and sex hormones (61).
Leptin in CKD
There is some controversy surrounding the relationship of leptin levels in CKD patients. Leptin is filtered in the renal glomerulus and catabolized in the renal tubules (62) via megalin (63). It has been observed how leptin levels are elevated in patients with moderate CKD (64-67), presumably due to lack of renal clearance of the protein (57,68). However, there may be other pathophysiological mechanisms contributing to this phenomenon. For instance, hyperleptinemia in patients with CKD could be produced by an increase in the production of this protein by the adipocytes of these patients. Thus, in two different studies, the authors demonstrated that adipocytes in a uremic environment produced overproduction of leptin (69,70). In addition, factors such as increased fat mass, hyperinsulinemia or inflammation (54), could contribute to the higher figures observed in patients with CKD. In this line, several studies have evidenced the relationship between this adipokine and renal damage, presumably through its participation in the development and progression of vascular damage, increased collagen production and increased mesangial cellularity (71-73). In addition, taking into account the close relationship between leptin and the immune system, and considering that CKD is characterized by a chronic inflammatory state, several in vitro studies have evidenced how leptin secretion was regulated by proinflammatory cytokines such as TNF-α (69,74,75) or IL-6 (67), resulting in higher levels in these individuals. In contrast, Silva et al. observed that leptin levels in patients with CKD and normal BMI were similar to those of healthy subjects, but lower than those of overweight or obese patients (76), which attributes the effect on leptin levels to the impact of BMI rather than to CKD (76). Likewise, another study showed that plasma leptin levels were similar in HD patients and healthy subjects with the same BMI (BMI of 25.0 ± 4.2 kg/m2) (77). All this suggests that the hyperleptinemia that accompanies CKD has a complex and multifactorial origin, including a possible increase in leptin production by the adipose tissue of these patients, a consequence of decreased renal clearance, or the hyperinsulinemia and the inflammatory state present in many renal patients (54).
Leptin in Renal Transplant
With regard to renal transplant, leptin levels are elevated in ESKD, drop dramatically after grafting (78) but increase again with time after transplant (79,80), reaching similar levels to the pre-transplantation period in a few years (81). The possible relationship of leptin with the elevated cardiovascular risk in renal transplant recipients is very interesting. As it is known, the main cause of death in these patients is cardiovascular disease, and leptin, which increases its levels post-transplantation, is a peptide closely related to hypertension and arteriosclerosis. In addition, the germ of cardiovascular risk is calcification and vascular stiffness, whose manifestations throughout the body will mark the evolution of a transplant patient. In this regard, Lee et al. have shown that leptin levels presented a clear correlation with peripheral vessel stiffness in renal transplant recipients (45), indicating a putative implication of leptin in the evolution and cardiovascular events of renal transplant patients. In the same manner, an association could be established, from the endothelial point of view, between leptin and delayed graft function (DGF), given the inflammatory effects associated with leptin and knowing its relationship with vascular proliferation in the glomerulus, vascular damage, increased collagen and cell hypertrophy (71,72). In short, leptin has a proinflammatory action, with a close relationship with angiogenesis and hematopoiesis, which could explain its importance in the innate and adaptive immunity so relevant in renal transplantation (82,83).
Implications of Genetic Variability in Leptin Genes
Among the genes in the leptin pathway, the one encoding its receptor, LEPR (ENSG00000116678, HGNC:6554), has attracted most of the attention. It consists of 24 exons and is located on chromosome 1 between positions 65,420,652 and 65,641,559 (84). In recent years, it has been observed that single nucleotide polymorphisms (SNPs) in the LEPR gene encoding the protein receptor could influence susceptibility to certain pathologies, such as obesity (85), diabetes mellitus (86), hypertension (87) or cardiovascular mortality (88). However, in the field of renal transplantation, published articles are scarce. One study demonstrated the influence of the LEPR Gln223Arg variant on the development of post-transplant Diabetes mellitus (PTDM) (89), which has been confirmed in a later work by our group that analyzed more SNPs in this gene locus (86). In addition, renal recipients who were carriers of the SNP rs1805094 showed longer graft survival than non-carriers did (90). Furthermore, three SNPs in the LEPR gene, namely rs1137101, rs1805094 and rs1137100 have been associated with delayed graft function, acute rejection and renal function one year post-transplant in renal recipients (91).
The mechanism by which variants in the LEPR gene might translate into clinical events is unknown. Daghestani et al. (92) observed how carriers of the rs1137101 SNP presented higher leptin levels, but this association was only described in women with obesity. In other studies, this association between LEPR SNPs and leptin levels has not been confirmed (93,94). A different explanation for clinical repercussions of these genetic variants would be that the effect on the leptin receptor might alter the subsequent intracellular metabolic cascade that occurs after its activation.
Adiponectin
Adiponectin, a 30-kDa protein linked since its discovery to adipose tissue, has subsequently been found to be also produced, in smaller quantities, from cardiac myocytes and skeletal muscle cells (95,96). In plasma, adiponectin can be found in various multimeric forms: globular adiponectin, full-length and low (LMW), medium and high molecular weight (HMW) adiponectin (97). The HMW multimer appears to be the most active form of adiponectin, with the plasma concentration of the HMR multimer being related to insulin sensitivity, so that a failure in multimerization is associated with type 2 diabetes mellitus (98).
Adiponectin in CKD
The main route of elimination of adiponectin is hepatic and, secondarily, the renal route (99). Thus, it has been observed how adiponectin monomers and dimers are small enough to cross the glomerular filtration barrier, so these substances are identified in urine; however, the HMW multimer has also been observed to be excreted in the urine of patients with proteinuria, which may reflect a defect in the filtration barrier (100). The relationship between adiponectin and the kidney rise numerous questions. In patients diagnosed with CKD, adiponectin values could be related to the progression of kidney disease, even reaching ESKD, as evidenced by several studies (101-103). However, the specific reason is not well established. Even though in CKD there is increased resistance to insulin action (104), higher risk of cardiovascular disease and dyslipidemia (105), paradoxically, adiponectin levels are elevated in this situation. It would be logical to attribute this to the decline in renal function. However, other factors could be related, namely a possible malfunction of adiponectin receptors leading to resistance to the action of the protein, or an increase in its production, secondary to a state of generalized inflammation of the organism (105). The increase in plasma adiponectin levels observed in CKD is accompanied by an increase in adiponectin protein and mRNA expression in subcutaneous and visceral adipose tissue, suggesting that there is a stimulus that produces an increase in adiponectin, despite elevated plasma levels (105). In parallel, it has been shown that in the muscle tissue of patients with CKD, the expression of adiponectin receptors increase (106), suggesting that uremia could confer resistance to adiponectin (107). In any case, and similar to the case of leptin, the relationship between adiponectin and renal function is not yet fully understood.
Adiponectin in Renal Transplant
There are very few studies on adiponectin carried out in renal transplant recipients, where the restoration of renal function does not necessarily imply a return to a better cardiovascular situation. Chudek et al. showed that pre-transplant adiponectin levels were significantly higher than those of healthy subjects. After the transplant, concentrations decreased but did not reach the levels seen in the control population (108). Moreover, Idorn et al. have argued that, despite the decrease in protein levels, its relationship with various cardiovascular and metabolic variables will not be reestablished until several months after transplantation, with a period of clinical and metabolic instability (109). It is again worth asking whether the persistence of high adiponectin concentrations is due to a larval inflammatory state, or perhaps to the existence of alterations in the metabolic cascades triggered at the intra- and extracellular level by adiponectin (110). In relation to the high cardiovascular risk of this population group, one could also wonder whether patients with higher adiponectin levels post-transplantation are also those with better cardiovascular profile and longer survival. According to a study, renal recipients with lower baseline adiponectin levels also had a worse metabolic profile, with lower HDL cholesterol levels, higher CRP (C-Reactive Protein) levels, total cholesterol and higher BMI. In contrast, patients with higher levels had worse survival, similar to what happens in CKD (111). Based on the above, it is evident that the relationship between adiponectin and renal transplantation is complex. The levels of this hormone decrease after grafting, which evidences the restoration of certain metabolic feed-back and, perhaps, a decrease in the inflammatory state of the individual. Conversely, the figures remain higher than in the general population. To make matters more complicated, and similar to what is observed in hemodialysis, patients with higher levels present worse survival instead of the expected better cardiovascular and metabolic profile. In this complex relationship, it is worth highlighting the possible impact of various post-transplant events, such as weight gain, improved renal function, the use of drugs that alter the lipid profile, diabetes or the use of immunosuppressants (109).
Regarding to post-transplant graft evolution, a study showed that rejection was observed more rapidly in heart transplanted mice which were deficient in adiponectin than in healthy mice (112), which could indicate a lower T-cell response in the presence of adiponectin (113). Furthermore, adiponectin-deficient heart transplanted mice show more incidence of ischemia-reperfusion, myocardial infarction and myocardial apoptosis rate (114).
Implications of Genetic Variability in Adiponectin Genes
The adiponectin, ADIPOQ, gene (ENSG00000181092, HGNC:13633), consists of three exons and is located on chromosome 3 between positions 186,842,704 and 186,858,463.
Several genetic variants have been described linked to the plasma levels of this adipokine, which directly or indirectly play an important role in the susceptibility to certain pathologies such as obesity (115), insulin resistance, type 2 diabetes mellitus (116) or metabolic syndrome (117). With regard to renal transplantation, there is a marked paucity of data. Only Kang et al., who studied the association of PTDM with eight polymorphisms in ADIPOQ and also in the gene receptor ADIPOR1 in a sample of 575 renal transplant recipients, obtained an association between PTDM and the homozygous ADIPOQ rs1501299 TT genotype in males carriers (118). Our group studied the influence of ADIPOQ SNPs on graft survival and found that carriers of the rs1501299 variant showed worse survival compared to non-carriers (90).
Arachidonic-Derived Vasoactive Eicosanoids
The secretion of leptin and adiponectin in the adipose tissue is affected by the eicosanoid metabolites of arachidonic acid. Heme oxygenases are responsible for maintaining normal metabolic cellular functions and the production of potent antioxidant and anti-inflammatory molecules (119,120). In turn, the expression of these enzymes is up-regulated by arachidonic-derived epoxyeicosatrienoic acids (EETs). Burgess et al. (121,122) have reported that EETs levels have a direct influence on the regulation of adipogenesis and adipocyte function, by decreasing the production of circulating inflammatory cytokines, e.g. leptin, while increasing adiponectin levels (Figure 1). Indeed, Dai et al. have described the beneficial properties of these metabolites in inflammatory diseases related with obesity (123).
Arachidonic acid (AA) main function is to work as a precursor of proinflammatory bioactive mediators in at least three known metabolic pathways related to the onset, development, and regression of renal inflammation (Figure 2).
First, the cyclooxygenase (COX) pathway, through which AA is metabolized to prostaglandins and thromboxanes; second, the lipooxygenase (LOX) pathway, which leads to the formation of leukotrienes and lipoxins, and the third pathway, the epoxygenase pathway, in which cytochrome P450 (CYP450)-mediated metabolism produce different vasoactive eicosanoids, namely EETs and hydroxyeicosatetraenoic acids (HETEs) (124). The latter is the main pathway of AA metabolism in the kidney (125,126).
Several isomers of EETs are produced by this epoxygenase metabolic route, being CYP2 is the main CYP450 involved (124). These metabolites are very rapidly metabolized by the soluble epoxide hydrolase (sEH) enzyme, encoded by the EPHX2 gene, to dihydroxyeicosatrienoic acids (DHETs), with considerably weaker biological activity (127). In addition, a number of HETE isomers are also formed, of which 20-HETE is the main product and whose formation in the human kidney is mediated by CYP4F2 and CYP4A11 (124).
Eicosanoids in CKD
Renal physiological function is clearly influenced by both the vascular and tubular actions of these AA-derived metabolites, which contribute to the kidney’s ability to maintain electrolyte and body fluid homeostasis. Among other functions, EETs stand out for their renoprotective role. A decrease in the concentration of EETs can significantly influence the occurrence of vascular and tubular abnormalities of renovascular disease (128). These eicosanoids protect the kidney against inflammatory processes and renal injury by helping to increase renal blood flow, GFR and sodium excretion. Thus, EET analogs lower blood pressure, decrease kidney inflammation, improve vascular endothelial function, and decrease kidney fibrosis and apoptosis (129).
On the other hand, HETEs, especially 20-HETE, are vasoconstrictor metabolites implicated in the development of acute and chronic kidney disease, as well as polycystic kidney disease, in addition to increasing renal cell vasoconstriction and inducing hypertension (130). Likewise, they stimulate renal tubular cell hypertrophy and podocyte destruction. These effects have been confirmed by using 20-HETE antagonists, whose function was protective, reducing vascular inflammation, tubular injury, and loss of renal function (131).
Eicosanoids in Renal Transplant
Chronic graft nephropathy represents the major cause of long-term dysfunction in renal transplantation (132-134), which is determined by ischemia-reperfusion events, innate and adaptive immune response and the effect of drugs such as anticalcineurinics, which produce endothelial dysfunction (135-137). These factors, along with the fact that cardiovascular disease continues to be one of the leading causes of graft loss (138) and that the AA-epoxygenase pathway is key in the cardiovascular function, have led to an increase in the number of studies on the link between this route and renal transplantation. Indeed, Duflot et al. have highlighted the importance of preserving the bioavailability of EETs for their short- and long-term benefits in renal transplantation (139). There is also available literature on the role of HETEs in renal transplant outcomes. Dołegowska et al. showed that the dynamics of 20-HETE changes, which occurs during early phase of allograft reperfusion, is associated with early post-transplant graft function. The authors regarded 20-HETE as a novel clinical marker of post-transplant allograft function (140).
Implications of Genetic Variability in Eicosanoid Genes
The most important genes involved in the metabolism of AA to EETs are CYP2J2 and CYP2C8., with two widely studied variants, CYP2C8*3 R139K/K399R (rs10509681) and CYP2J2*7 G-50T (rs890293), strongly related to decreased enzymatic activity or lower transcription rate (141,142). In particular CYP2C8*3 appears to be associated with altered metabolism of CYP2C8 substrates (143,144), and CYP2J2*7 causes a loss of the Sp1 transcription factor binding site resulting in decreased expression of the enzyme (145). Interestingly, these SNPs have shown to produce a significant decrease in EETs levels (146-148).
CYP2C8*3 has been observed mostly in Caucasian population and has been associated with an increased risk of delayed renal function and reduced creatinine clearance in renal transplant recipients (149) as well as with increased susceptibility of diabetic kidney disease (150). As for CYP2J2*7, considering that the enzyme synthesizes renoprotective EETs, carriers could present a decrease in this protective activity. In addition, a lower transcription rate of CYP2J2 caused by the SNP could contribute to arterial hypertension because of the reduced levels of vasodilator EETs (147). Indeed, we studied the effect of this variant on cardiovascular event-free survival after kidney transplantation and reported a significantly lower survival for carriers of the CYP2J2*7 allele.
The EPHX2 gene, coding for the enzyme responsible for EETs degradation, can also present different SNPs that have been associated with enzymatic and kinetic activity, being the most relevant EPHX2 3’UTR A>G (rs1042032), R287Q (rs751141) and K55R (rs41507953) (151). The GG genotype of the EPHX2 3’UTR A>G variant has been related to lower GFR and higher serum creatine values in renal transplant recipients as well as with an increased risk of acute rejection and worse graft function. In this case, both the donor’s and the recipient’s genotype showed this association with rejection (152). In contrast, Lee et al. reported an increased risk of graft dysfunction for the AA genotype (153). EPHX2 R287Q results in decreased sEH activity and has been associated with coronary artery calcification (153) and hypertension (154). Finally, carriers of the EPHX2 K55R show increased sEH activity and therefore higher EETs degradation (155). The variant produces a decreased endothelium-dependent vasodilation, which in turn implies reduced blood flow (151).
The formation of 20-HETE, the most relevant HETE, is mediated by the CYP4F2 and CYP4A11 genes. Initially, the CYP4F2 V433M (rs2108622) polymorphism was reported to reduce the production of 20-HETE by 50% (156). However, subsequent studies suggest this SNP to be associated with increased urinary excretion of 20-HETE (157,158). We have analyzed associations of the CYP4F2 V433M SNP with the risk of diabetic kidney disease and with clinical outcomes in these patients. We observed that carriers of the 433M variant allele had lower risk of diabetic kidney disease, in addition to lower urinary 20-HETE levels. We hypothesized that 433M carriers would produce less 20-HETE, resulting in reduced vasoconstrictor activity in kidney tissue. In turn, this would alleviate glomerular capillary pressure causing the observed reduction in filtration (150).
In renal transplantation, the V433M polymorphism has been related to acute rejection, delayed graft function (159) and PTDM (160). With regard to CYP4A11, the F434S substitution has been shown to lead to a decrease in enzymatic activity and has been related to hypertension and increased vasoconstriction (161,162). Consistent with these findings, renal recipients whose donors carried the CYP4A11 434S variant showed impaired creatinine clearance compared to wild-type carriers (163).
Perspectives and Conclusions
In this review, we have focused on the role that obesity can play in CKD and in the clinical evolution of renal transplant. In particular, we have discussed in depth the mediators of the adipose tissue, which are gaining increasing attention as significant actors in processes leading to renal injury as well as in renoprotective mechanisms.
Experimental and clinical evidence implicates the two major adipose tissue cytokines, adiponectin and leptin, in renal damage. There is a growing body of evidence indicating various potential therapeutic strategies based on this. For instance, the development of leptin signaling modulators, namely peptide-based receptor antagonists, leptin mutants, antibodies, and nanobodies (164) represents a promising strategy for the treatment of diseases where leptin is involved. Indeed, a very recent study shows in an animal model of renal ischemia how local, intrarenal postischemic treatment at reperfusion with a leptin antagonist prevented apoptosis and inflammation and was renoprotective (165). In the same line, leptin antagonists have also been shown to ameliorate CKD-associated cachexia in mice (166). Common drugs have also been used to antagonize leptin damaging effects. For instance, a clinical trial showed how treatment with lovastatin can reduce leptin serum levels in patients with type 2 diabetic nephropathy (167).
Moreover, leptin and adiponectin, being as they are associated with cardiovascular disease, have also been proposed to be used as a combined biomarker (leptin-to-adiponectin ratio) of adverse events in patients on kidney replacement therapy (168). Nevertheless, it should be mentioned that information regarding adiponectin in chronic kidney disease (CKD) is not consistently uniform, likely owing to its intricate and varied actions. Moreover, the existence of various adiponectin isoforms, each with distinct roles, the initiation of diverse signaling pathways, and the varied expression of its receptors in different tissues, could further contribute to the controversial nature of the data (169). In any case, it has been proposed that enhancing the synthesis of adiponectin in CKD patients may have therapeutic potential, as inferred from the results of studies on rodent models of CKD, where adiponectin administration was able to attenuate kidney injury and fibrosis (170). For instance, PPAR agonist tesaglitazar has shown promising results in clinical trials for the treatment of type-2 diabetes trough increasing adiponectin circulating concentrations (171).
As previously explained, inhibiting soluble epoxide hydrolase (sEH) proves effective in maintaining endogenous epoxyeicosatrienoic acids (EETs) levels while reducing dihydroxyeicosatrienoic acids (DHETs) levels. This has therapeutic potential for cardiovascular, central nervous system, and metabolic diseases (172). Consequently, the development of sEH inhibitors has been a prominent research focus since the early 21st century. Indeed, sEH inhibitors have demonstrated a protective role in animal models of renal damage and experimental arterial hypertension (173,174). Moreover, mice deficient in sEH exhibit decreased blood pressure, attenuated renal inflammation, and reduced glomerular injury (175). Among the different sEH inhibitors, urea-based compounds are particularly notable due to their general high selectivity for sEH. Various potent sEH inhibitors, whether synthesized chemically or isolated from natural sources, include compounds like AR9281, EC5026, or GSK2256294. These are currently undergoing clinical trials to address conditions such as heart failure, insulin resistance, glucose intolerance, hypertension, endothelial dysfunction, and pain (176).
Additionally, combining sEH inhibitors with other agents, such as COX-2 inhibitors like celecoxib, has proven to enhance efficiency in reducing pain and hypotension (177), which has led to the development of sEH/COX2 dual inhibitors (178). Having said that, further research in areas like CKD is necessary to fully understand the clinical applications of these promising compounds in the future.
In addition to the therapeutic possibilities that targeting these mediators open for renal patients, we should not underestimate the role of genetics. Whilst genetic-based therapies are still far from being developed in this field, there are numerous studies indicating that genetic variants may regulate the levels of adiponectin, leptin, EETs and HETEs, and hence hold the potential to be clinically relevant in the renal setting (150,179,180).
Chronic kidney disease is expected to be one of the global leading causes of death by 2040, and the need for new biomarkers of early diagnosis and progression to ESKD has been repeatedly stressed. In the same manner, renal transplant recipients might benefit from novel markers of clinical outcomes, particularly cardiovascular-related, as this is the main cause of death in this population. These mediators of the adipose tissue, together with the arachidonic metabolites that regulate their secretion, hold the potential to be useful clinical tools in this context.
Funding
This research was funded by grant PI22/00181 and RD21/0005/0031 from Instituto de Salud Carlos III, Madrid (Spain), financed by the European Union – NextGeneration UE, Recovery and Resilience Mechanism, and grant GR21026 from Junta de Extremadura, Mérida (Spain) and Fondo Europeo de Desarrollo Regional (FEDER) “Una manera de hacer Europa”. Funding sources did not have any involvement in the study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication..
Conflicts of Interest
All authors declare that they have no conflict of interest.
References
- Chen TK, Knicely DH, Grams ME. Chronic Kidney Disease Diagnosis and Management: A Review. Jama. 2019;322(13):1294-304. [CrossRef]
- Drawz P, Rahman M. Chronic kidney disease. Annals of internal medicine. 2015;162(11):ITC1-16. [CrossRef]
- Martinez-Castelao A, Gorriz JL, Segura-de la Morena J, Cebollada J, Escalada J, Esmatjes E, et al. Consensus document for the detection and management of chronic kidney disease. Nefrologia : publicacion oficial de la Sociedad Espanola Nefrologia. 2014;34(2):243-62. [CrossRef]
- Hall JE, Henegar JR, Dwyer TM, Liu J, Da Silva AA, Kuo JJ, et al. Is obesity a major cause of chronic kidney disease? Advances in renal replacement therapy. 2004;11(1):41-54. [CrossRef]
- Taylor EN, Stampfer MJ, Curhan GC. Obesity, weight gain, and the risk of kidney stones. Jama. 2005;293(4):455-62. [CrossRef]
- Kopple JD, Feroze U. The effect of obesity on chronic kidney disease. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation. 2011;21(1):66-71. [CrossRef]
- Kopple JD. Obesity and chronic kidney disease. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation. 2010;20(5 Suppl):S29-30. [CrossRef]
- Chang A, Van Horn L, Jacobs DR, Jr., Liu K, Muntner P, Newsome B, et al. Lifestyle-related factors, obesity, and incident microalbuminuria: the CARDIA (Coronary Artery Risk Development in Young Adults) study. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2013;62(2):267-75. [CrossRef]
- Song YM, Sung J, Lee K. Longitudinal relationships of metabolic syndrome and obesity with kidney function: Healthy Twin Study. Clinical and experimental nephrology. 2015;19(5):887-94. [CrossRef]
- Lee HS, Lee KB, Hyun YY, Chang Y, Ryu S, Choi Y. DASH dietary pattern and chronic kidney disease in elderly Korean adults. European journal of clinical nutrition. 2017;71(6):755-61. [CrossRef]
- Wickman C, Kramer H. Obesity and kidney disease: potential mechanisms. Seminars in nephrology. 2013;33(1):14-22. [CrossRef]
- Pinto KRD, Feckinghaus CM, Hirakata VN. Obesity as a predictive factor for chronic kidney disease in adults: systematic review and meta-analysis. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas. 2021;54(4):e10022. [CrossRef]
- Ritz E, Koleganova N. Obesity and chronic kidney disease. Seminars in nephrology. 2009;29(5):504-11. [CrossRef]
- Arner P. Introduction: the inflammation orchestra in adipose tissue. Journal of internal medicine. 2007;262(4):404-7.
- Zoccali C. Overweight, obesity and metabolic alterations in chronic kidney disease. Prilozi. 2009;30(2):17-31.
- Decleves AE, Sharma K. Obesity and kidney disease: differential effects of obesity on adipose tissue and kidney inflammation and fibrosis. Current opinion in nephrology and hypertension. 2015;24(1):28-36. [CrossRef]
- Silva Junior GB, Bentes AC, Daher EF, Matos SM. Obesity and kidney disease. Jornal brasileiro de nefrologia. 2017;39(1):65-9. [CrossRef]
- Kalaitzidis RG, Siamopoulos KC. The role of obesity in kidney disease: recent findings and potential mechanisms. International urology and nephrology. 2011;43(3):771-84. [CrossRef]
- Navarro-Diaz M, Serra A, Lopez D, Granada M, Bayes B, Romero R. Obesity, inflammation, and kidney disease. Kidney international Supplement. 2008(111):S15-8. [CrossRef]
- Bash LD, Erlinger TP, Coresh J, Marsh-Manzi J, Folsom AR, Astor BC. Inflammation, hemostasis, and the risk of kidney function decline in the Atherosclerosis Risk in Communities (ARIC) Study. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2009;53(4):596-605. [CrossRef]
- Lin J, Hu FB, Mantzoros C, Curhan GC. Lipid and inflammatory biomarkers and kidney function decline in type 2 diabetes. Diabetologia. 2010;53(2):263-7. [CrossRef]
- Nicoletto BB, Fonseca NK, Manfro RC, Goncalves LF, Leitao CB, Souza GC. Effects of obesity on kidney transplantation outcomes: a systematic review and meta-analysis. Transplantation. 2014;98(2):167-76. [CrossRef]
- Humar A, Ramcharan T, Denny R, Gillingham KJ, Payne WD, Matas AJ. Are wound complications after a kidney transplant more common with modern immunosuppression? Transplantation. 2001;72(12):1920-3. ttps://doi.org/10.1097/00007890-200112270-00009.
- Singh D, Lawen J, Alkhudair W. Does pretransplant obesity affect the outcome in kidney transplant recipients? Transplantation proceedings. 2005;37(2):717-20. [CrossRef]
- Halme L, Eklund B, Kyllonen L, Salmela K. Is obesity still a risk factor in renal transplantation? Transplant international : official journal of the European Society for Organ Transplantation. 1997;10(4):284-8. [CrossRef]
- Chan W, Bosch JA, Jones D, McTernan PG, Phillips AC, Borrows R. Obesity in kidney transplantation. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation. 2014;24(1):1-12. [CrossRef]
- Kalantar-Zadeh K, Abbott KC, Salahudeen AK, Kilpatrick RD, Horwich TB. Survival advantages of obesity in dialysis patients. The American journal of clinical nutrition. 2005;81(3):543-54. [CrossRef]
- Herselman M, Esau N, Kruger JM, Labadarios D, Moosa MR. Relationship between serum protein and mortality in adults on long-term hemodialysis: exhaustive review and meta-analysis. Nutrition. 2010;26(1):10-32. [CrossRef]
- Park J, Jin DC, Molnar MZ, Dukkipati R, Kim YL, Jing J, et al. Mortality predictability of body size and muscle mass surrogates in Asian vs white and African American hemodialysis patients. Mayo Clinic proceedings. 2013;88(5):479-86. [CrossRef]
- Park J, Ahmadi SF, Streja E, Molnar MZ, Flegal KM, Gillen D, et al. Obesity paradox in end-stage kidney disease patients. Progress in cardiovascular diseases. 2014;56(4):415-25. [CrossRef]
- Ahmadi F, Mohebi-Nejad A. Re: Lovastatin for reduction of leptin in nondialysis patients with type 2 diabetic nephropathy. Iranian journal of kidney diseases. 2014;8(4):344-5.
- Ahmadi SF, Zahmatkesh G, Streja E, Molnar MZ, Rhee CM, Kovesdy CP, et al. Body mass index and mortality in kidney transplant recipients: a systematic review and meta-analysis. American journal of nephrology. 2014;40(4):315-24. [CrossRef]
- Kalantar-Zadeh K, Kovesdy CP, Derose SF, Horwich TB, Fonarow GC. Racial and survival paradoxes in chronic kidney disease. Nature clinical practice Nephrology. 2007;3(9):493-506. [CrossRef]
- Pommer W. Preventive Nephrology: The Role of Obesity in Different Stages of Chronic Kidney Disease. Kidney Dis (Basel). 2018;4(4):199-204. [CrossRef]
- Ashby VB, Leichtman AB, Rees MA, Song PX, Bray M, Wang W, et al. A Kidney Graft Survival Calculator that Accounts for Mismatches in Age, Sex, HLA, and Body Size. Clinical journal of the American Society of Nephrology : CJASN. 2017;12(7):1148-60. [CrossRef]
- Hossain M, Woywodt A, Augustine T, Sharma V. Obesity and listing for renal transplantation: weighing the evidence for a growing problem. Clinical kidney journal. 2017;10(5):703-8. [CrossRef]
- Fantuzzi G. Adipose tissue, adipokines, and inflammation. The Journal of allergy and clinical immunology. 2005;115(5):911-9; quiz 20. [CrossRef]
- Thomou T, Mori MA, Dreyfuss JM, Konishi M, Sakaguchi M, Wolfrum C, et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature. 2017;542(7642):450-5. [CrossRef]
- Proenca AR, Sertie RA, Oliveira AC, Campana AB, Caminhotto RO, Chimin P, et al. New concepts in white adipose tissue physiology. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas. 2014;47(3):192-205. [CrossRef]
- Roman S, Agil A, Peran M, Alvaro-Galue E, Ruiz-Ojeda FJ, Fernandez-Vazquez G, et al. Brown adipose tissue and novel therapeutic approaches to treat metabolic disorders. Translational research : the journal of laboratory and clinical medicine. 2015;165(4):464-79. [CrossRef]
- Bates R, Huang W, Cao L. Adipose Tissue: An Emerging Target for Adeno-associated Viral Vectors. Molecular therapy Methods & clinical development. 2020;19:236-49. [CrossRef]
- Curat CA, Miranville A, Sengenes C, Diehl M, Tonus C, Busse R, et al. From blood monocytes to adipose tissue-resident macrophages: induction of diapedesis by human mature adipocytes. Diabetes. 2004;53(5):1285-92. [CrossRef]
- Jung UJ, Choi MS. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. International journal of molecular sciences. 2014;15(4):6184-223. [CrossRef]
- Cildir G, Akincilar SC, Tergaonkar V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends in molecular medicine. 2013;19(8):487-500. [CrossRef]
- Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochimica et biophysica acta. 2014;1842(3):446-62. [CrossRef]
- Schipper HS, Prakken B, Kalkhoven E, Boes M. Adipose tissue-resident immune cells: key players in immunometabolism. Trends in endocrinology and metabolism: TEM. 2012;23(8):407-15. [CrossRef]
- AlZaim I, Hammoud SH, Al-Koussa H, Ghazi A, Eid AH, El-Yazbi AF. Adipose Tissue Immunomodulation: A Novel Therapeutic Approach in Cardiovascular and Metabolic Diseases. Frontiers in cardiovascular medicine. 2020;7:602088. [CrossRef]
- Exley MA, Hand L, O'Shea D, Lynch L. Interplay between the immune system and adipose tissue in obesity. The Journal of endocrinology. 2014;223(2):R41-8. [CrossRef]
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of clinical investigation. 2007;117(1):175-84. [CrossRef]
- Cao H. Adipocytokines in obesity and metabolic disease. The Journal of endocrinology. 2014;220(2):T47-59. [CrossRef]
- Faggioni R, Feingold KR, Grunfeld C. Leptin regulation of the immune response and the immunodeficiency of malnutrition. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001;15(14):2565-71. [CrossRef]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425-32. [CrossRef]
- Zhang F, Chen Y, Heiman M, Dimarchi R. Leptin: structure, function and biology. Vitamins and hormones. 2005;71:345-72. [CrossRef]
- Alix PM, Guebre-Egziabher F, Soulage CO. Leptin as an uremic toxin: Deleterious role of leptin in chronic kidney disease. Biochimie. 2014;105:12-21. [CrossRef]
- Minocci A, Savia G, Lucantoni R, Berselli ME, Tagliaferri M, Calo G, et al. Leptin plasma concentrations are dependent on body fat distribution in obese patients. International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity. 2000;24(9):1139-44. [CrossRef]
- Briley LP, Szczech LA. Leptin and renal disease. Seminars in dialysis. 2006;19(1):54-9. [CrossRef]
- Sharma K, Considine RV. The Ob protein (leptin) and the kidney. Kidney international. 1998;53(6):1483-7. [CrossRef]
- Teta D, Bevington A, Brown J, Pawluczyk I, Harris K, Walls J. Acidosis downregulates leptin production from cultured adipocytes through a glucose transport-dependent post-transcriptional mechanism. Journal of the American Society of Nephrology : JASN. 2003;14(9):2248-54. [CrossRef]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. The New England journal of medicine. 1996;334(5):292-5. [CrossRef]
- Schrauwen P, van Marken Lichtenbelt WD, Westerterp KR, Saris WH. Effect of diet composition on leptin concentration in lean subjects. Metabolism: clinical and experimental. 1997;46(4):420-4. [CrossRef]
- Leshan RL, Bjornholm M, Munzberg H, Myers MG, Jr. Leptin receptor signaling and action in the central nervous system. Obesity (Silver Spring). 2006;14 Suppl 5:208S-12S. [CrossRef]
- Zhang J, Wang N. Leptin in chronic kidney disease: a link between hematopoiesis, bone metabolism, and nutrition. International urology and nephrology. 2014;46(6):1169-74. [CrossRef]
- Hama H, Saito A, Takeda T, Tanuma A, Xie Y, Sato K, et al. Evidence indicating that renal tubular metabolism of leptin is mediated by megalin but not by the leptin receptors. Endocrinology. 2004;145(8):3935-40. [CrossRef]
- Heimburger O, Lonnqvist F, Danielsson A, Nordenstrom J, Stenvinkel P. Serum immunoreactive leptin concentration and its relation to the body fat content in chronic renal failure. Journal of the American Society of Nephrology : JASN. 1997;8(9):1423-30. [CrossRef]
- Merabet E, Dagogo-Jack S, Coyne DW, Klein S, Santiago JV, Hmiel SP, et al. Increased plasma leptin concentration in end-stage renal disease. The Journal of clinical endocrinology and metabolism. 1997;82(3):847-50. [CrossRef]
- Nordfors L, Lonnqvist F, Heimburger O, Danielsson A, Schalling M, Stenvinkel P. Low leptin gene expression and hyperleptinemia in chronic renal failure. Kidney international. 1998;54(4):1267-75. [CrossRef]
- Pecoits-Filho R, Nordfors L, Heimburger O, Lindholm B, Anderstam B, Marchlewska A, et al. Soluble leptin receptors and serum leptin in end-stage renal disease: relationship with inflammation and body composition. European journal of clinical investigation. 2002;32(11):811-7. [CrossRef]
- Shankar A, Syamala S, Xiao J, Muntner P. Relationship between Plasma Leptin Level and Chronic Kidney Disease. International journal of nephrology. 2012;2012:269532. [CrossRef]
- Aminzadeh MA, Pahl MV, Barton CH, Doctor NS, Vaziri ND. Human uraemic plasma stimulates release of leptin and uptake of tumour necrosis factor-alpha in visceral adipocytes. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2009;24(12):3626-31. [CrossRef]
- Kalbacher E, Koppe L, Zarrouki B, Pillon NJ, Fouque D, Soulage CO. Human uremic plasma and not urea induces exuberant secretion of leptin in 3T3-L1 adipocytes. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation. 2011;21(1):72-5. [CrossRef]
- Ambarkar M, Pemmaraju SV, Gouroju S, Manohar SM, Bitla AR, Yajamanam N, et al. Adipokines and their Relation to Endothelial Dysfunction in Patients with Chronic Kidney Disease. Journal of clinical and diagnostic research : JCDR. 2016;10(1):BC04-8. [CrossRef]
- Ding N, Liu B, Song J, Bao S, Zhen J, Lv Z, et al. Leptin promotes endothelial dysfunction in chronic kidney disease through AKT/GSK3beta and beta-catenin signals. Biochemical and biophysical research communications. 2016;480(4):544-51. [CrossRef]
- D'Elia L, Manfredi M, Perna L, Iacone R, Russo O, Strazzullo P, et al. Circulating leptin levels predict the decline in renal function with age in a sample of adult men (The Olivetti Heart Study). Internal and emergency medicine. 2019;14(4):507-13. [CrossRef]
- Grunfeld C, Zhao C, Fuller J, Pollack A, Moser A, Friedman J, et al. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. The Journal of clinical investigation. 1996;97(9):2152-7. [CrossRef]
- Kirchgessner TG, Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Tumor necrosis factor-alpha contributes to obesity-related hyperleptinemia by regulating leptin release from adipocytes. The Journal of clinical investigation. 1997;100(11):2777-82. [CrossRef]
- Silva MI, Vale BS, Lemos CC, Torres MR, Bregman R. Body adiposity index assess body fat with high accuracy in nondialyzed chronic kidney disease patients. Obesity (Silver Spring). 2013;21(3):546-52. [CrossRef]
- Leal VO, Lobo JC, Stockler-Pinto MB, Farage NE, Calixto A, Geloneze B, et al. Apelin: a peptide involved in cardiovascular risk in hemodialysis patients? Renal failure. 2012;34(5):577-81. [CrossRef]
- Souza GC, Costa C, Scalco R, Goncalves LF, Manfro RC. Serum leptin, insulin resistance, and body fat after renal transplantation. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation. 2008;18(6):479-88. [CrossRef]
- Kayacan SM, Yildiz A, Kazancioglu R, Sahin S, Sever MS, Ark E. The changes in serum leptin, body fat mass and insulin resistance after renal transplantation. Clinical transplantation. 2003;17(1):63-8. [CrossRef]
- El Haggan W, Chauveau P, Barthe N, Merville P, Potaux L, Aparicio M. Serum leptin, body fat, and nutritional markers during the six months post-kidney transplantation. Metabolism: clinical and experimental. 2004;53(5):614-9. [CrossRef]
- Nicoletto BB, Souza GC, Goncalves LF, Costa C, Perry IS, Manfro RC. Leptin, insulin resistance, and metabolic changes 5 years after renal transplantation. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation. 2012;22(4):440-9. [CrossRef]
- Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394(6696):897-901. [CrossRef]
- Sanchez-Margalet V, Martin-Romero C, Santos-Alvarez J, Goberna R, Najib S, Gonzalez-Yanes C. Role of leptin as an immunomodulator of blood mononuclear cells: mechanisms of action. Clinical and experimental immunology. 2003;133(1):11-9. [CrossRef]
- Garavito P, Mosquera-Heredia MI, Fang L, Payares F, Ruiz M, Arias I, et al. Polymorphisms of leptin-melanocortin system genes associated with obesity in an adult population from Barranquilla. Biomedica : revista del Instituto Nacional de Salud. 2020;40(2):257-69. Polimorfismos de los genes del sistema leptina-melanocortina asociados con la obesidad en la poblacion adulta de Barranquilla. [CrossRef]
- Mendez-Hernandez A, Gallegos-Arreola MP, Moreno-Macias H, Espinosa Fematt J, Perez-Morales R. LEP rs7799039, LEPR rs1137101, and ADIPOQ rs2241766 and 1501299 Polymorphisms Are Associated With Obesity and Chemotherapy Response in Mexican Women With Breast Cancer. Clinical breast cancer. 2017;17(6):453-62. [CrossRef]
- Mota-Zamorano S, Luna E, Garcia-Pino G, Gonzalez LM, Gervasini G. Variability in the leptin receptor gene and other risk factors for post-transplant diabetes mellitus in renal transplant recipients. Annals of medicine. 2019;51(2):164-73. [CrossRef]
- Lian Y, Tang Z, Xie Y, Chen Z. Leptin receptor gene polymorphisms and risk of hypertension: a meta-analysis. International journal of clinical and experimental medicine. 2015;8(8):14277-82.
- Aijala M, Santaniemi M, Bloigu R, Kesaniemi YA, Ukkola O. Leptin receptor Arg109 homozygotes display decreased total mortality as well as lower incidence of cardiovascular disease and related death. Gene. 2014;534(1):88-92. [CrossRef]
- Romanowski M, Dziedziejko V, Maciejewska-Karlowska A, Sawczuk M, Safranow K, Domanski L, et al. Adiponectin and leptin gene polymorphisms in patients with post-transplant diabetes mellitus. Pharmacogenomics. 2015;16(11):1243-51. [CrossRef]
- Gervasini G, Garcia-Pino G, Mota-Zamorano S, Luna E, Garcia-Cerrada M, Tormo MA, et al. Association of polymorphisms in leptin and adiponectin genes with long-term outcomes in renal transplant recipients. The pharmacogenomics journal. 2020;20(3):388-97. [CrossRef]
- Guadalup García-Pino,, Enrique Luna,, Sonia Mota-Zamorano,, et al. Effect of leptin concentrations and leptin receptor gene polymorphisms on the outcome of renal transplantation. Archives of medical science. 2020. [CrossRef]
- Daghestani M, Purohit R, Daghistani M, Warsy A. Molecular dynamic (MD) studies on Gln233Arg (rs1137101) polymorphism of leptin receptor gene and associated variations in the anthropometric and metabolic profiles of Saudi women. PloS one. 2019;14(2):e0211381. [CrossRef]
- Lopez-Quintero A, Garcia-Zapien AG, Flores-Martinez SE, Diaz-Burke Y, Gonzalez-Sandoval CE, Lopez-Roa RI, et al. Contribution of polymorphisms in the LEP, LEPR and RETN genes on serum leptin and resistin levels in young adults from Mexico. Cell Mol Biol (Noisy-le-grand). 2017;63(8):10-8. [CrossRef]
- Foucan L, Bassien-Capsa V, Rambhojan C, Lacorte JM, Larifla L. Influence of K656N Polymorphism of the Leptin Receptor Gene on Obesity-Related Traits in Nondiabetic Afro-Caribbean Individuals. Metabolic syndrome and related disorders. 2019;17(4):197-203. [CrossRef]
- Brochu-Gaudreau K, Rehfeldt C, Blouin R, Bordignon V, Murphy BD, Palin MF. Adiponectin action from head to toe. Endocrine. 2010;37(1):11-32. [CrossRef]
- Sharma K. The link between obesity and albuminuria: adiponectin and podocyte dysfunction. Kidney international. 2009;76(2):145-8. [CrossRef]
- Neumann E, Frommer KW, Vasile M, Muller-Ladner U. Adipocytokines as driving forces in rheumatoid arthritis and related inflammatory diseases? Arthritis and rheumatism. 2011;63(5):1159-69. [CrossRef]
- Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, et al. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. The Journal of biological chemistry. 2003;278(41):40352-63. [CrossRef]
- Achari AE, Jain SK. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. International journal of molecular sciences. 2017;18(6). [CrossRef]
- Kim Y, Park CW. Mechanisms of Adiponectin Action: Implication of Adiponectin Receptor Agonism in Diabetic Kidney Disease. International journal of molecular sciences. 2019;20(7). [CrossRef]
- Jorsal A, Tarnow L, Frystyk J, Lajer M, Flyvbjerg A, Parving HH, et al. Serum adiponectin predicts all-cause mortality and end stage renal disease in patients with type I diabetes and diabetic nephropathy. Kidney international. 2008;74(5):649-54. [CrossRef]
- Heidari M, Nasri P, Nasri H. Adiponectin and chronic kidney disease; a review on recent findings. Journal of nephropharmacology. 2015;4(2):63-8.
- Kuo IC, Wu PH, Lin HY, Niu SW, Huang JC, Hung CC, et al. The association of adiponectin with metabolic syndrome and clinical outcome in patients with non-diabetic chronic kidney disease. PloS one. 2019;14(7):e0220158. [CrossRef]
- DeFronzo RA, Alvestrand A, Smith D, Hendler R, Hendler E, Wahren J. Insulin resistance in uremia. The Journal of clinical investigation. 1981;67(2):563-8. [CrossRef]
- Martinez Cantarin MP, Waldman SA, Doria C, Frank AM, Maley WR, Ramirez CB, et al. The adipose tissue production of adiponectin is increased in end-stage renal disease. Kidney international. 2013;83(3):487-94. [CrossRef]
- Martinez Cantarin MP, Keith SW, Waldman SA, Falkner B. Adiponectin receptor and adiponectin signaling in human tissue among patients with end-stage renal disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2014;29(12):2268-77. [CrossRef]
- Markaki A, Psylinakis E, Spyridaki A. Adiponectin and end-stage renal disease. Hormones (Athens). 2016;15(3):345-54. [CrossRef]
- Chudek J, Adamczak M, Karkoszka H, Budzinski G, Ignacy W, Funahashi T, et al. Plasma adiponectin concentration before and after successful kidney transplantation. Transplantation proceedings. 2003;35(6):2186-9. [CrossRef]
- Idorn T, Hornum M, Bjerre M, Jorgensen KA, Nielsen FT, Hansen JM, et al. Plasma adiponectin before and after kidney transplantation. Transplant international : official journal of the European Society for Organ Transplantation. 2012;25(11):1194-203. [CrossRef]
- Cortazar F, Molnar MZ, Isakova T, Czira ME, Kovesdy CP, Roth D, et al. Clinical outcomes in kidney transplant recipients receiving long-term therapy with inhibitors of the mammalian target of rapamycin. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2012;12(2):379-87. [CrossRef]
- Alam A, Molnar MZ, Czira ME, Rudas A, Ujszaszi A, Kalantar-Zadeh K, et al. Serum adiponectin levels and mortality after kidney transplantation. Clinical journal of the American Society of Nephrology : CJASN. 2013;8(3):460-7. [CrossRef]
- Okamoto Y, Christen T, Shimizu K, Asano K, Kihara S, Mitchell RN, et al. Adiponectin inhibits allograft rejection in murine cardiac transplantation. Transplantation. 2009;88(7):879-83. [CrossRef]
- Wolf AM, Wolf D, Rumpold H, Enrich B, Tilg H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochemical and biophysical research communications. 2004;323(2):630-5. [CrossRef]
- Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nature medicine. 2005;11(10):1096-103. [CrossRef]
- Wu J, Liu Z, Meng K, Zhang L. Association of adiponectin gene (ADIPOQ) rs2241766 polymorphism with obesity in adults: a meta-analysis. PloS one. 2014;9(4):e95270. [CrossRef]
- Dastani Z, Hivert MF, Timpson N, Perry JR, Yuan X, Scott RA, et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS genetics. 2012;8(3):e1002607. [CrossRef]
- Canale MP, Manca di Villahermosa S, Martino G, Rovella V, Noce A, De Lorenzo A, et al. Obesity-related metabolic syndrome: mechanisms of sympathetic overactivity. International journal of endocrinology. 2013;2013:865965. [CrossRef]
- Kang ES, Magkos F, Kim BS, Zhai R, Su L, Kim YS, et al. Variants of the adiponectin and adiponectin receptor-1 genes and posttransplantation diabetes mellitus in renal allograft recipients. The Journal of clinical endocrinology and metabolism. 2012;97(1):E129-35. [CrossRef]
- Abraham NG, Drummond G. CD163-Mediated hemoglobin-heme uptake activates macrophage HO-1, providing an antiinflammatory function. Circulation research. 2006;99(9):911-4. [CrossRef]
- Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiological reviews. 2006;86(2):583-650. [CrossRef]
- Burgess A, Vanella L, Bellner L, Schwartzman ML, Abraham NG. Epoxyeicosatrienoic acids and heme oxygenase-1 interaction attenuates diabetes and metabolic syndrome complications. Prostaglandins & other lipid mediators. 2012;97(1-2):1-16. [CrossRef]
- Burgess AP, Vanella L, Bellner L, Gotlinger K, Falck JR, Abraham NG, et al. Heme oxygenase (HO-1) rescue of adipocyte dysfunction in HO-2 deficient mice via recruitment of epoxyeicosatrienoic acids (EETs) and adiponectin. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2012;29(1-2):99-110. [CrossRef]
- Dai M, Wu L, Wang P, Wen Z, Xu X, Wang DW. CYP2J2 and Its Metabolites EETs Attenuate Insulin Resistance via Regulating Macrophage Polarization in Adipose Tissue. Scientific reports. 2017;7:46743. [CrossRef]
- Wang T, Fu X, Chen Q, Patra JK, Wang D, Wang Z, et al. Arachidonic Acid Metabolism and Kidney Inflammation. International journal of molecular sciences. 2019;20(15). [CrossRef]
- Capdevila J, Chacos N, Werringloer J, Prough RA, Estabrook RW. Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(9):5362-6. [CrossRef]
- Lasker JM, Chen WB, Wolf I, Bloswick BP, Wilson PD, Powell PK. Formation of 20-hydroxyeicosatetraenoic acid, a vasoactive and natriuretic eicosanoid, in human kidney. Role of Cyp4F2 and Cyp4A11. The Journal of biological chemistry. 2000;275(6):4118-26. [CrossRef]
- Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circulation research. 2000;87(11):992-8. [CrossRef]
- Sporkova A, Kopkan L, Varcabova S, Huskova Z, Hwang SH, Hammock BD, et al. Role of cytochrome P-450 metabolites in the regulation of renal function and blood pressure in 2-kidney 1-clip hypertensive rats. American journal of physiology Regulatory, integrative and comparative physiology. 2011;300(6):R1468-75. [CrossRef]
- Imig JD. Orally active epoxyeicosatrienoic acid analogs in hypertension and renal injury. Adv Pharmacol. 2022;94:27-55. [CrossRef]
- Fan F, Ge Y, Lv W, Elliott MR, Muroya Y, Hirata T, et al. Molecular mechanisms and cell signaling of 20-hydroxyeicosatetraenoic acid in vascular pathophysiology. Front Biosci (Landmark Ed). 2016;21(7):1427-63. [CrossRef]
- Hoopes SL, Garcia V, Edin ML, Schwartzman ML, Zeldin DC. Vascular actions of 20-HETE. Prostaglandins & other lipid mediators. 2015;120:9-16. [CrossRef]
- Stegall MD, Gaston RS, Cosio FG, Matas A. Through a glass darkly: seeking clarity in preventing late kidney transplant failure. Journal of the American Society of Nephrology : JASN. 2015;26(1):20-9. [CrossRef]
- Chapman JR. Progress in Transplantation: Will It Be Achieved in Big Steps or by Marginal Gains? American journal of kidney diseases : the official journal of the National Kidney Foundation. 2017;69(2):287-95. [CrossRef]
- Hart A, Smith JM, Skeans MA, Gustafson SK, Wilk AR, Castro S, et al. OPTN/SRTR 2018 Annual Data Report: Kidney. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2020;20 Suppl s1:20-130. [CrossRef]
- Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nature medicine. 2011;17(11):1391-401. [CrossRef]
- Rodrigues-Diez R, Gonzalez-Guerrero C, Ocana-Salceda C, Rodrigues-Diez RR, Egido J, Ortiz A, et al. Calcineurin inhibitors cyclosporine A and tacrolimus induce vascular inflammation and endothelial activation through TLR4 signaling. Scientific reports. 2016;6:27915. [CrossRef]
- Jourde-Chiche N, Fakhouri F, Dou L, Bellien J, Burtey S, Frimat M, et al. Endothelium structure and function in kidney health and disease. Nature reviews Nephrology. 2019;15(2):87-108. [CrossRef]
- Rangaswami J, Mathew RO, Parasuraman R, Tantisattamo E, Lubetzky M, Rao S, et al. Cardiovascular disease in the kidney transplant recipient: epidemiology, diagnosis and management strategies. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2019;34(5):760-73. [CrossRef]
- Duflot T, Laurent C, Soudey A, Fonrose X, Hamzaoui M, Iacob M, et al. Preservation of epoxyeicosatrienoic acid bioavailability prevents renal allograft dysfunction and cardiovascular alterations in kidney transplant recipients. Scientific reports. 2021;11(1):3739. [CrossRef]
- Dolegowska B, Blogowski W, Domanski L. Is it possible to predict the early post-transplant allograft function using 20-HETE measurements? A preliminary report. Transplant international : official journal of the European Society for Organ Transplantation. 2009;22(5):546-53. [CrossRef]
- King LM, Ma J, Srettabunjong S, Graves J, Bradbury JA, Li L, et al. Cloning of CYP2J2 gene and identification of functional polymorphisms. Molecular pharmacology. 2002;61(4):840-52. [CrossRef]
- Smith HE, Jones JP, 3rd, Kalhorn TF, Farin FM, Stapleton PL, Davis CL, et al. Role of cytochrome P450 2C8 and 2J2 genotypes in calcineurin inhibitor-induced chronic kidney disease. Pharmacogenetics and genomics. 2008;18(11):943-53. [CrossRef]
- Bahadur N, Leathart JB, Mutch E, Steimel-Crespi D, Dunn SA, Gilissen R, et al. CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha-hydroxylase activity in human liver microsomes. Biochemical pharmacology. 2002;64(11):1579-89. [CrossRef]
- Yu L, Shi D, Ma L, Zhou Q, Zeng S. Influence of CYP2C8 polymorphisms on the hydroxylation metabolism of paclitaxel, repaglinide and ibuprofen enantiomers in vitro. Biopharmaceutics & drug disposition. 2013;34(5):278-87. [CrossRef]
- Spiecker M, Liao J. Cytochrome P450 epoxygenase CYP2J2 and the risk of coronary artery disease. Trends in cardiovascular medicine. 2006;16(6):204-8. [CrossRef]
- Dai D, Zeldin DC, Blaisdell JA, Chanas B, Coulter SJ, Ghanayem BI, et al. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics. 2001;11(7):597-607. [CrossRef]
- Spiecker M, Darius H, Hankeln T, Soufi M, Sattler AM, Schaefer JR, et al. Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation. 2004;110(15):2132-6. [CrossRef]
- Liu PY, Li YH, Chao TH, Wu HL, Lin LJ, Tsai LM, et al. Synergistic effect of cytochrome P450 epoxygenase CYP2J2*7 polymorphism with smoking on the onset of premature myocardial infarction. Atherosclerosis. 2007;195(1):199-206. [CrossRef]
- Genvigir FDV, Campos-Salazar AB, Felipe CR, Tedesco-Silva H, Jr., Medina-Pestana JO, Doi SQ, et al. CYP3A5*3 and CYP2C8*3 variants influence exposure and clinical outcomes of tacrolimus-based therapy. Pharmacogenomics. 2020;21(1):7-21. [CrossRef]
- Mota-Zamorano S, Robles NR, Gonzalez LM, Valdivielso JM, Lopez-Gomez J, Cancho B, et al. Genetics Variants in the Epoxygenase Pathway of Arachidonic Metabolism Are Associated with Eicosanoids Levels and the Risk of Diabetic Nephropathy. Journal of clinical medicine. 2021;10(17). [CrossRef]
- Lee CR, Pretorius M, Schuck RN, Burch LH, Bartlett J, Williams SM, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) is associated with forearm vasodilator responses in humans. Hypertension. 2011;57(1):116-22. [CrossRef]
- Gervasini G, Garcia-Cerrada M, Coto E, Vergara E, Garcia-Pino G, Alvarado R, et al. A 3'-UTR Polymorphism in Soluble Epoxide Hydrolase Gene Is Associated with Acute Rejection in Renal Transplant Recipients. PloS one. 2015;10(7):e0133563. [CrossRef]
- Lee SH, Lee J, Cha R, Park MH, Ha JW, Kim S, et al. Genetic variations in soluble epoxide hydrolase and graft function in kidney transplantation. Transplantation proceedings. 2008;40(5):1353-6. [CrossRef]
- Ma L, Zhao H, Yu M, Wen Y, Zhao T, Yan M, et al. Association of Epoxide Hydrolase 2 Gene Arg287Gln with the Risk for Primary Hypertension in Chinese. International journal of hypertension. 2020;2020:2351547. [CrossRef]
- Shuey MM, Billings FTt, Wei S, Milne GL, Nian H, Yu C, et al. Association of gain-of-function EPHX2 polymorphism Lys55Arg with acute kidney injury following cardiac surgery. PloS one. 2017;12(5):e0175292. [CrossRef]
- Stec DE, Roman RJ, Flasch A, Rieder MJ. Functional polymorphism in human CYP4F2 decreases 20-HETE production. Physiological genomics. 2007;30(1):74-81. [CrossRef]
- Hu BC, Li Y, Li FH, Zhang Y, Sheng CS, Fan HQ, et al. Peripheral and central augmentation indexes in relation to the CYP4F2 polymorphisms in Chinese. Journal of hypertension. 2011;29(3):501-8. [CrossRef]
- Ward NC, Croft KD, Puddey IB, Phillips M, van Bockxmeer F, Beilin LJ, et al. The effect of a single nucleotide polymorphism of the CYP4F2 gene on blood pressure and 20-hydroxyeicosatetraenoic acid excretion after weight loss. Journal of hypertension. 2014;32(7):1495-502; discussion 502. [CrossRef]
- Mota-Zamorano S, Gonzalez LM, Luna E, Fernandez JJ, Gomez A, Nieto-Fernandez A, et al. Polymorphisms in vasoactive eicosanoid genes of kidney donors affect biopsy scores and clinical outcomes in renal transplantation. PloS one. 2019;14(10):e0224129. [CrossRef]
- Gervasini G, Luna E, Garcia-Cerrada M, Garcia-Pino G, Cubero JJ. Risk factors for post-transplant diabetes mellitus in renal transplant: Role of genetic variability in the CYP450-mediated arachidonic acid metabolism. Molecular and cellular endocrinology. 2016;419:158-64. [CrossRef]
- Fava C, Montagnana M, Almgren P, Rosberg L, Lippi G, Hedblad B, et al. The V433M variant of the CYP4F2 is associated with ischemic stroke in male Swedes beyond its effect on blood pressure. Hypertension. 2008;52(2):373-80. [CrossRef]
- Hermann M, Hellermann JP, Quitzau K, Hoffmann MM, Gasser T, Meinertz T, et al. CYP4A11 polymorphism correlates with coronary endothelial dysfunction in patients with coronary artery disease--the ENCORE Trials. Atherosclerosis. 2009;207(2):476-9. [CrossRef]
- Gervasini G, Garcia-Cerrada M, Vergara E, Garcia-Pino G, Alvarado R, Fernandez-Cavada MJ, et al. Polymorphisms in CYP-mediated arachidonic acid routes affect the outcome of renal transplantation. European journal of clinical investigation. 2015;45(10):1060-8. [CrossRef]
- Greco M, De Santo M, Comande A, Belsito EL, Ando S, Liguori A, et al. Leptin-Activity Modulators and Their Potential Pharmaceutical Applications. Biomolecules. 2021;11(7). [CrossRef]
- Jonas M, Simon AJ, Gilburd B, Schneiderman J. Intrarenal Anti-Leptin Treatment Attenuates Ischemia and Reperfusion Injury. American journal of nephrology. 2023;54(7-8):337-48. [CrossRef]
- Cheung WW, Ding W, Gunta SS, Gu Y, Tabakman R, Klapper LN, et al. A pegylated leptin antagonist ameliorates CKD-associated cachexia in mice. Journal of the American Society of Nephrology : JASN. 2014;25(1):119-28. [CrossRef]
- Gholamin S, Razavi SM, Taghavi-Garmestani SM, Ghorbanihaghjo A, Rashtchizadeh N, Safa J, et al. Lovastatin for reduction of leptin in nondialysis patients with type 2 diabetic nephropathy. Iranian journal of kidney diseases. 2014;8(3):201-6.
- Villasis-Keever MA, Zurita-Cruz JN, Serret-Montoya J, Barbosa-Cortes L, Zepeda-Martinez CDC, Alegria-Torres G, et al. The leptin/adiponectin ratio as prognostic marker for dyslipidemia during 1 year of follow-up in pediatric patients receiving kidney replacement therapy. Nutricion hospitalaria. 2022;39(5):977-87. Indice leptina/adiponectina como indicador pronostico de dislipidemia durante 1 ano de seguimiento en pacientes pediatricos que reciben terapia de reemplazo renal. [CrossRef]
- Coimbra S, Rocha S, Valente MJ, Catarino C, Bronze-da-Rocha E, Belo L, et al. New Insights into Adiponectin and Leptin Roles in Chronic Kidney Disease. Biomedicines. 2022;10(10). [CrossRef]
- Tian M, Tang L, Wu Y, Beddhu S, Huang Y. Adiponectin attenuates kidney injury and fibrosis in deoxycorticosterone acetate-salt and angiotensin II-induced CKD mice. American journal of physiology Renal physiology. 2018;315(3):F558-F71. [CrossRef]
- Zha D, Wu X, Gao P. Adiponectin and Its Receptors in Diabetic Kidney Disease: Molecular Mechanisms and Clinical Potential. Endocrinology. 2017;158(7):2022-34. [CrossRef]
- HC. Soluble epoxide hydrolase inhibitors: a patent review. Expert opinion on therapeutic patents. 2010;20(7):941-56. [CrossRef]
- Noh MR, Jang HS, Salem FE, Ferrer FA, Kim J, Padanilam BJ. Epoxyeicosatrienoic acid administration or soluble epoxide hydrolase inhibition attenuates renal fibrogenesis in obstructive nephropathy. American journal of physiology Renal physiology. 2023;324(2):F138-F51. [CrossRef]
- Papiashvili N, Gongadze N, Bakuridze A, Bakuridze K. Antihypertensive and Cardioprotective Effects of Epoxyeicosatrienoic Acid Analogs and Soluble Epoxide Hydrolase Inhibitors (Review). Georgian medical news. 2021(312):125-32.
- Chen G, Xu R, Wang Y, Wang P, Zhao G, Xu X, et al. Genetic disruption of soluble epoxide hydrolase is protective against streptozotocin-induced diabetic nephropathy. American journal of physiology Endocrinology and metabolism. 2012;303(5):E563-75. [CrossRef]
- Sun CP, Zhang XY, Morisseau C, Hwang SH, Zhang ZJ, Hammock BD, et al. Discovery of Soluble Epoxide Hydrolase Inhibitors from Chemical Synthesis 180and Natural Products. Journal of medicinal chemistry. 2021;64(1):184-215. [CrossRef]
- Burmistrov V, Morisseau C, Danilov D, Harris TR, Dalinger I, Vatsadze I, et al. 1,3-Disubstituted and 1,3,3-trisubstituted adamantyl-ureas with isoxazole as soluble epoxide hydrolase inhibitors. Bioorganic & medicinal chemistry letters. 2015;25(23):5514-9. [CrossRef]
- Zhang G, Panigrahy D, Hwang SH, Yang J, Mahakian LM, Wettersten HI, et al. Dual inhibition of cyclooxygenase-2 and soluble epoxide hydrolase synergistically suppresses primary tumor growth and metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(30):11127-32. [CrossRef]
- Oliveira CS, Giuffrida FM, Crispim F, Saddi-Rosa P, Reis AF. ADIPOQ and adiponectin: the common ground of hyperglycemia and coronary artery disease? Arquivos brasileiros de endocrinologia e metabologia. 2011;55(7):446-54. [CrossRef]
- Hao JQ, Zhang QK, Zhou YX, Chen LH, Wu PF. Association between circulating leptin concentration and G-2548A gene polymorphism in patients with breast cancer: a meta-analysis. Archives of medical science : AMS. 2019;15(2):275-83. [CrossRef]
Figure 1.
Interaction between arachidonic-acid derived epoxyeicosatrienoic acids (EETs) and cytokines secretion.
Figure 1.
Interaction between arachidonic-acid derived epoxyeicosatrienoic acids (EETs) and cytokines secretion.

Figure 2.
Arachidonic acid metabolism pathways.
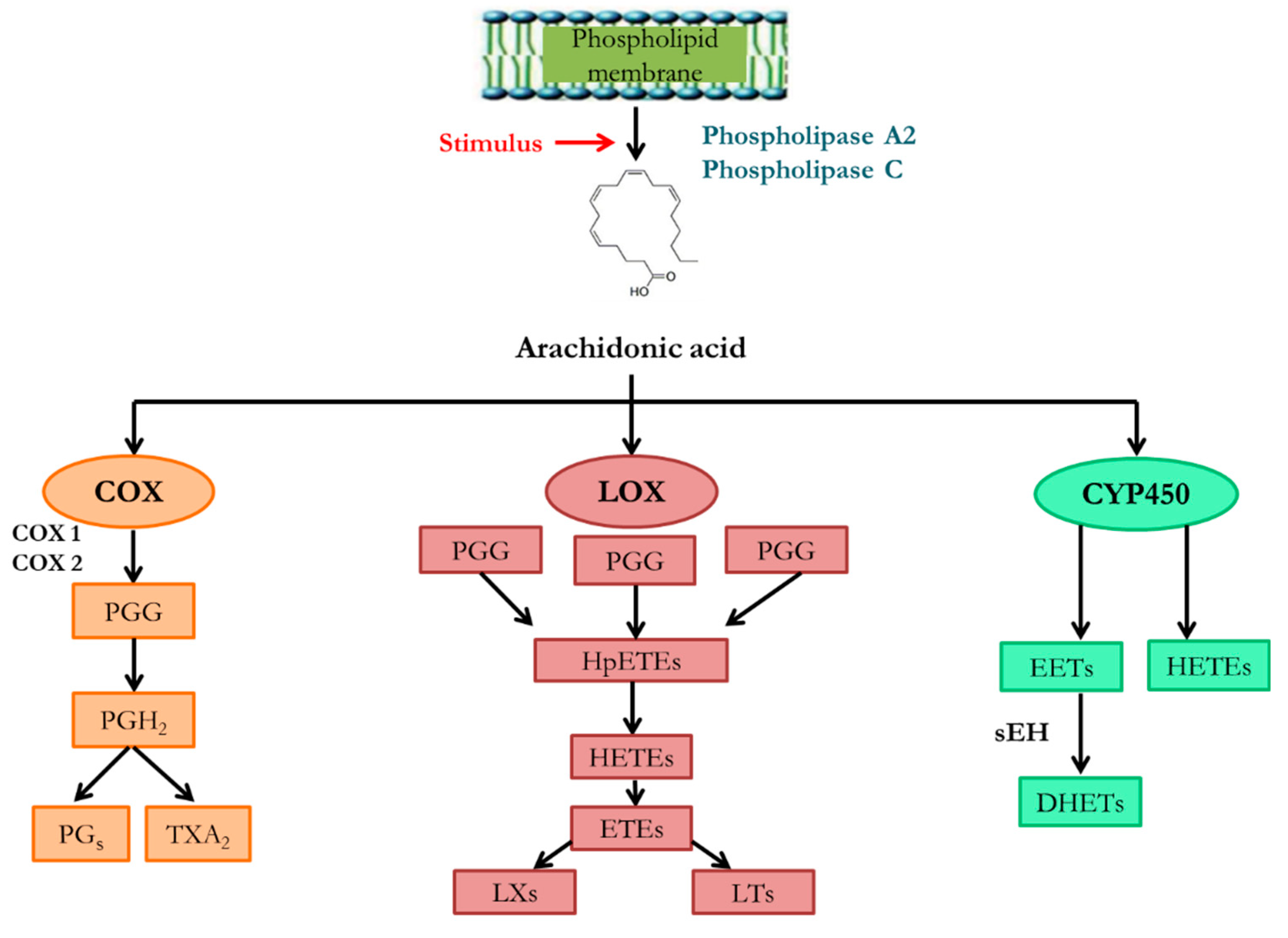
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



