
Submitted:
02 January 2024
Posted:
03 January 2024
You are already at the latest version
Abstract
Adult-onset Still's disease (AOSD) is a rare systemic inflammatory disorder. Diagnosis can take a long time, especially in the presence of confounding factors, and it is, at least in part, of exclusion. AOSD is generally mild, however a small percentage of patients can develop a fearsome as well potentially fatal complication, the macrophage activation syndrome (MAS), which is also referred to as hemophagocytic lymphohistocytosis (HLH). This condition is correlated with a cytokine storm production and monocyte/macrophage overactivation, and typically occurs with rash, pyrexia, pancytopenia, hepatosplenomegaly and systemic involvement. Exitus occurs in approximately 10% of cases. The Histiocyte Society, which is a nonprofit organization committed to improving the lives of patients with histiocytic disorders, currently suggests etoposide in combination with dexamethasone for the treatment of HLH/MAS, although a multidisciplinary collaboration using the resources and expertise of several specialists (e.g. rheumatologist, infectiologist, critical care medicine specialist) is always recommended. Hereby we propose the detailed description of the clinical case of a previously healthy young woman in which MAS was the dramatic onset manifestation of AOSD and whose diagnosis posed a real clinical challenge.
Keywords:
adult-onset Still's disease
; macrophage activation syndrome
; hemophagocytic lymphohistiocytosis
; autoimmunity
; autoinflammatory diseases
; immune system
; inflammation
; steroids
; fever of unknown origin
; monocytes/macrophages
1. Introduction
Adult-onset Still’s disease (AOSD) is a complex autoimmune inflammatory disease of unknown etiology. It is a rare condition (annual incidence of 0.16 per 1,000,000 people), with a variable spectrum of signs/symptoms at onset which can mimic other inflammatory or infectious conditions [1]. AODS usually affects young people, without gender preference, and presents with two peaks at 20 and 40 years of age, even if cases have been described in elderly subjects. In any case, a presentation of the disease in subjects over 70 years of age does not exclude the diagnosis, but makes it highly unlikely [2]. Diagnosis is not always easy, and involves a process of exclusion: diagnostic delays typically depend on when it is considered in the differential diagnosis. During this process, the clinician can be supported by various sets of diagnostic criteria, amongst which Yamaguchi’s are currently the most widely used, as detailed in Table 1 [3].
The text continues here.
Looking more closely at the aforementioned classification, it can be seen that the clinical elements of AOSD include signs/symptoms (such as fever, arthralgia, rash, pharyngodynia, splenomegaly, lymphadenopathy) and alterations in laboratory tests (such as leukocytosis, neutrophilia, increased hepatic cytolysis indices) that are shared by numerous other rheumatological, infectious or neoplastic conditions. As a matter of fact, the classification criteria require the exclusion of such conditions. In other words, this algorithm guides and supports the diagnostic process only when it has already been considered by the treating physician.
The disease generally presents in a mild and self-limiting way, but sometimes it is more tumultuous from the very beginning or it rapidly evolves into fearsome complications. A possible uncommon but severe complication of AOSD is the macrophage activation syndrome (MAS), which is a form of hemophagocytic lymphohistiocytosis (HLH) occurring in the course of an immunorheumatological disease. As reported from some retrospective studies, between 6 and 19% of AOSD patients evolve into MAS [4,5]. MAS tends to arise after a certain period of illness and it is very rare that it can present at the onset of Still’s disease. The decisive diagnostic element is biopsy, with special reference to the presence of differentiated histiocytes engaged in the phagocytosis of hematopoietic elements of the red series. From a clinical point of view, HLH suspicion should arise on the basis of constitutional symptoms with or without high fever and some not-so-specific laboratory alterations, such as anemia associated with thrombocytopenia/leukopenia, a gap between ferritin (very high) and erythrocyte sedimentation rate (not so high), and elevated triglyceride levels with normal or low fibrinogen in the blood (Table 2) [6,7,8,9].
The text continues here.
The prognosis of MAS is variable, and strictly depends on the diagnostic delay: usually, when recognized early or presenting with a mild severity, it resolves rather quickly. However, cases with poor prognosis leading to death have also been reported. In any case, therefore, for all the above considerations it should always be considered as a serious manifestation that must be treated aggressively. The Histiocyte Society has recommended the use of etoposide (VP-16) in combination with dexamethasone for the treatment of various forms of HLH, including HLH secondary to autoimmunity, since 1994 [6,10,11,12]. A valid alternative is represented by anakinra (a recombinant human interleukin-1 receptor antagonist), especially by the intravenous route (4 times a day) interspersed with boluses of high-dose methylprednisolone [13].
Here, a case is reported of MAS secondary to AODS (hereafter referred to as AOSD-MAS) in a previously healthy patient which responded to the aforementioned therapies with corticosteroids (CS) and etoposide. In our opinion, this is a paradigmatic example of how coexisting medical diagnoses may condition the judgment of the treating physicians, possibly leading to potentially very dangerous underestimations of problems. This case is, therefore, suitable for educational purposes for doctors who, often, do not have a specialization or at least a specific interest in the care of rheumatological patients.
2. Case presentation
The patient was a 34-year-old woman with a history of cigarette smoking and bronchial asthma who presented to the emergency department for persistent fever over 39°C lasting for one week, associated with general malaise, pharyngodynia and cervical lymphadenopathy. Four days before hospital admission the patient started a home therapy with amoxicillin 1 g three times a day (t.i.d) and non-steroidal anti-inflammatory drugs (NSAIDs), but obtained an inadequate control of body temperature, which never fell below 38.5°C. She also reported headaches and skin rashes which tended to be migratory and transient. The complete timeline of her clinical case is presented in Figure 1.
The text continues here.
The patient was admitted to the Infectious Diseases Department. At clinical examination, she was a severely ill febrile subject with physical weakness and anorexia as the main associated constitutional symptoms; she complained of headache and diffuse arthralgias to knees, wrists and ankles. Salmon-pink nonpruritic skin rashes were evident on the trunk and extremities during fever episodes; cervical lymph nodes, liver and spleen were palpable. Her main initial blood tests, showing a severe inflammatory state, are represented in Table 3.
The text continues here.
Serial blood cultures were obtained, of which only one was found to be positive for meticillin-sensitive Streptococcus Thermophilus. Targeted antibiotic therapy with ceftriaxone 2 g quaque die (q.d.) and teicoplanin (maintenance dose: 12 mg/kg q.d.) was then started and discontinued after five days both due to lack of clinical response and the development of diarrhea. A transesophageal echocardiography was performed and showed no endocardial vegetations. As stool samples were positive for Clostridium difficile, the patient also received targeted antibiotic therapy with oral vancomycin 500 mg q.d.
After her hospital admission, the patient’s clinical condition continued to deteriorate with hemodynamic instability (systolic pressure of 75 mmHg during crystalloid fluid replacement), so that after two weeks she was transferred to the Intensive Care Unit (ICU) with a working diagnosis of "fever of unknown origin associated with severe hypotension". In the ICU, vasopressin and norepinephrine were administered to support organ perfusion. Due to the persistence of fever, an additional course of empiric broad-spectrum antibiotic therapy with meropenem 2 g t.i.d. was started, and a myeloid activation test at flow cytometry on peripheral blood was performed, which showed a picture of marked granulocytic activation (i.e. granulocyte to lymphocyte ratio of CD64 intensity expression: 13.8; normal value < 2.6) consistent with a possible infection. Therefore, an antifungal therapy (caspofungin 70 mg on day 1 and 50 mg q.d. for 3 days) was added to meropenem, again without any improvement. Because of all these treatment failures, after a multidisciplinary discussion, it was finally decided to start a steroid therapy with methylprednisolone 125 mg t.i.d., suspecting an autoimmune disease. For the differential diagnosis between infectious and autoimmune diseases, the patient in the meantime underwent all of the additional investigations reported in Table 4.
The text continues here.
A total body computed tomography was also performed, showing a 22-mm pericardial effusion, bilateral pleural effusions, and a pelvic intraperitoneal fluid flap; both liver and spleen were enlarged; a small amount of free fluid was reported within the peritoneal space, the paracolic gutters and around the liver and gallbladder (Figure 2).
The text continues here.
Due to the occurrence of anemia and thrombocytopenia requiring red blood cell/plasma transfusions and human fibrinogen concentrate administrations, a diagnosis of acute disseminated intravascular coagulation was made; prothrombin time was moderately prolonged, while activated partial thromboplastin time, ADAMTS13 enzyme activity levels and schistocyte count at peripheral blood smear were within normal ranges; autoantibodies directed against ADAMTS13 were absent and Coombs test (direct and indirect) was negative. Flow cytometry analysis of bone marrow/peripheral blood did not show any clonal B-cell or abnormal T-cell populations, while confirmed an inflammatory circulating neutrophilic leukocytosis. A bone marrow biopsy was finally performed, revealing the presence hemophagocytosis at bone marrow smear and a histological pattern of hypercellular marrow with mature myeloid hyperplasia, marked reduction/absence of erythroblastic series, and diffuse infiltration of histocytes, some of which had a hemophagocytic pattern (Figure 3).
The text continues here.
After the procedure, due to the appearance of desaturation (arterial partial pressure of oxygen in room air: 44.0 mmHg) and orthopnea it was necessary to put the patient on a course of continuous positive airway pressure (CPAP), which was later discontinued due to the improvement of the respiratory failure.
Both the aforementioned biopsy findings and the appearance of a weak clinical response only after the initiation of steroid therapy guided at this stage the diagnostic suspicion toward the presence of a HLH, possibly secondary to an autoimmune disease. In this respect, the onset of an AOSD appeared to be plausible, so Yamaguchi’s classification criteria were applied (Table 1) [3]. A positivity for all 4 major and 5 minor criteria was found. This despite the possible concomitant infectious process, which -in itself- would be an exclusion criterion; in any case, infections alone ─even had they been present─ could not have accounted for the patient’s clinical and histological patterns. Therefore, a final diagnosis of MAS as a complication of a new onset AOSD was finally made.
Given the patient’s progressive and rapid deterioration, treatment with an intensive therapy with immunosuppressive and cytotoxic agents was administered with the aim to induce remission of the disease activity, following the HLH-94 treatment protocol [10]. More in detail, this initial therapy included etoposide -which is proapoptotic in HLH- 150 mg/m2 IV twice weekly during the first 2 weeks and then weekly, in combination with daily dexamethasone (initially 10 mg/m2 IV for 2 weeks, followed by 5 mg/m2 IV for 2 weeks, and 2.5 mg/m2 IV for 2 weeks). Filgrastim was administered as needed, while trimethoprim/sulfamethoxazole (180/800 mg/q.d. orally) and acyclovir (800 mg/q.d. intravenously) therapy were started as secondary prophylaxis.
After the first two doses of chemotherapy administered in the ICU, the patient became hemodynamically stable again and could be transferred to an Internal Medicine Division, where she experienced a progressive rapid clinical and laboratory improvement. More in detail, the fever completely resolved and the performance status significantly recovered, although some remaining tendency to fatigue. From a musculoskeletal perspective, arthralgias gradually reduced until they disappeared, but a marked sarcopenia, requiring an in-hospital rehabilitation program, persisted as a result of the prolonged intrahospital bed rest. With regard to the main laboratory issues, inflammatory indices gradually decreased to normalization, platelet and hemoglobin levels stabilized, and transaminase and ferritin levels dramatically reduced (Table 1).
After two months from the initial hospitalization the patient was finally discharged in a stable clinical condition (switching dexamethasone to oral prednisone at an initial dosage of 25 mg q.d.) and referred to our rheumatology outpatient clinic. During the first eight follow-up months she remained apyretic and symptom-free at home expect for a residual easy fatigability. From a biochemical point of view, the only relevant data consisted in a slight transient increase in lactate dehydrogenase and transaminase levels within the first month, so that methotrexate (MTX) 12.5 mg SC quaque week was associated, while the patient continued her slow tapering of the steroid therapy (current prednisone dosage: 2.5 mg q.d.) (Table 3).
3. Discussion
This case is emblematic in terms of the difficulty of managing a young and previously healthy subject who undergoes a rapid deterioration of clinical conditions and develops life-threatening diseases. Initially the patient experienced mild and non-worrisome symptoms, which were interpreted as a common form of bacterial upper respiratory tract infection. Subsequently the clinical course changed and the following impression was that of being faced with an infection of unknown origin in the septic evolution phase. And it is on this front that the doctors’ attention was focused during the first part of the hospitalization, also because of the aforementioned misleading elements that supported the initial working hypothesis. Probably, one of the main struggles in this clinical case was represented by the classic diagnostic dilemma of infection versus autoimmunity, incidentally always bearing in mind that not only one thing does not exclude the other, but also that an infection can trigger an autoimmunity flare or, in some cases, even its onset. Moreover, immune activation from an infection is also a common trigger for HLH/MAS both in patients with a genetic predisposition and in sporadic cases with no underlying genetic cause identified.
Our patient ultimately resulted affected by AOSD. This is a rare systemic autoinflammatory disorder characterized by recurrent high fever, a fading salmon-pink rash, and arthritis. These signs/symptoms are often associated with pharyngodynia, myalgias, lymphadenopathy, splenomegaly, and neutrophilic leukocytosis (Table 5) [14,15,16,17,18,19,20,21,22,23,24,25,26].
The text continues here.
In younger individuals, the counterpart of AOSD is called systemic juvenile idiopathic arthritis (s-JIA), which shares some of its features, including recurrent fever, salmon-pink rash, and polyarthritis; in other words, these two conditions are considered to represent the same disease continuum with different ages of onset [1]. This type of symptoms, as is known, is shared by various pathologies much more common than AOSD; in fact, in our case, they were initially interpreted as of bacterial or fungal origin.
The etiology of the disease is unknown, but it is generally believed that various infectious agents can play an important role in its pathogenesis, acting -as mentioned above- like a trigger in some predisposed individuals [27]. Known causative agents may include a variety of viruses (such as Rubivirus, Morbillivirus, Echovirus 7, Coxsackievirus B4, Cytomegalovirus, and Epstein-Barr Virus) and some bacteria (including Mycoplasma Pneumoniae, Chlamydia Pneumoniae, Yersinia Enterocolitica, Brucella Abortus and Borrelia Burgdorferi). It should be noted, however, that no single pathogenic trigger has ever been clearly identified, indicating the likely involvement of multiple concurrent factors [28]. In any case, in the specific case of our young patient no definite elements emerged that could guide us in this direction.
Being a rare disease, AOSD is notoriously difficult to cure, but it is even more difficult to diagnose. Patients typically face a journey of confusing symptoms, misdiagnoses or delayed diagnoses, and a series of ineffective treatments before a proper diagnosis and an effective treatment plan can be provided. Delayed diagnosis can lead to longer hospital stays and higher financial costs, and it can also hasten the onset of the rare and potentially fatal AOSD complications which we will discuss below. In any case, diagnosis is generally by exclusion, as clinicians do not currently have a tool to make it in a definite way, but can only rely on a few specific sets of diagnostic criteria, among which the most sensitive and widely used are Yamaguchi ones, as shown in Table 1 [3]. A diagnosis can be made when ≧ 5 criteria are present, with at least two being major diagnostic criteria, preferably in the absence of any exclusion criterion. Despite their widespread use and proven clinical utility, these criteria only consider the patient clinical presentation, which is sometimes nuanced and, in rare cases, overlapping with a transient acute infectious pattern that may facilitate the onset of AOSD itself as reported above [29]. Moreover, it goes without saying that these criteria are applicable since the disease is suspected on the basis of the clinical history and the lack of response to therapies considered effective for the previously diagnosed conditions, as in the case currently described.
It is important to note that AOSD is a complex and multifaceted condition. It is generally a mild disease, but approximately 20% of patients develop a potentially fatal complication, the most frequent being MAS (also called reactive hemophagocytic syndrome), a secondary form of HLH. The latter one manifests with rash, pyrexia, pancytopenia, hepatosplenomegaly and systemic involvement, leading to death in approximately 10% of cases (Table 2). This syndrome is correlated with a cytokine storm expression and monocyte/macrophage overactivation, leading to multi-organ dysfunction [30,31]. When HLH occurs as a familial disorder it is called familial hemophagocytic lymphohistiocytosis (FHL), a condition where gene mutations map to loci that code for elements of the cytotoxic granule formation and release pathway.
For milder forms of AOSD, NSAIDs alone may be sufficient to treat symptoms, but an initial CS therapy is usually required for moderate and severe cases which may include serositis, debilitating arthritis, fever refractory to NSAIDs or internal organ involvement. Due to the significant toxicities associated with continuous CS therapy, CS-sparing medications are typically needed to achieve steroid-free remissions and treat refractory cases: MTX, cyclosporine A and leflunomide are the most commonly used conventional synthetic disease-modifying antirheumatic drugs (csDMARDs). In addition, recent developments in biologic drugs have greatly improved the quality of life and coping ability of AOSD patients: for the most severe and refractory subjects, anti-cytokine drugs are an effective and reliable alternative to csDMARDs as well as the only remaining treatment option. Currently, a safe and effective strategy is the specific suppression of interleukin (IL)-6 (with tocilizumab) or IL-1 (with anakinra, canakinumab or rilonacept); tumor necrosis factor (TNF) inhibitors may be used in refractory arthritis forms [32].
For patients in whom incipient HLH/MAS remains a concern, a combination of high dose anakinra, preferably IV, with pulse-dose IV CS therapy is generally suggested, rather than using a single agent. Concern for MAS is particularly high in patients with elevation in ferritin levels out of proportion to other inflammatory markers, transaminase elevation, marked elevation in D-dimer, thrombocytopenia, and/or a decreasing erythrocyte sedimentation rate despite continued elevation of C-reactive protein [33].
However, in the present case, HLH/MAS diagnosis was not only suspected but also confirmed at a given point in time. As a matter of fact, our patient fulfilled 7 out of 9 HLH-2004 criteria, in addition to having increased liver enzymes (Table 2) [6,8,9]. Moreover, her Hscore ─a scoring system developed to estimate the probability of HLH incorporating points for immunosuppression, fever, organomegaly, levels of serum analytes (triglycerides, ferritin, alanine aminotransferase and fibrinogen), degree of cytopenias, and presence of hemophagocytosis on the bone marrow aspirate─ was 215 which confers a >95% probability of HLH [34]. When MAS diagnosis is confirmed or strongly suspected, current guidelines suggest an initial empiric treatment for the presumed underlying pathology, deferring HLH-specific therapy, only for patients without deteriorating cardiac, pulmonary, hepatic, renal, or neurological function; nonetheless, these subjects should be monitored carefully and treatment should be intensified in case of clinical worsening. Examples may include antimicrobials for triggering infections, CS for rheumatologic conditions, or antineoplastics for cancers; for the rare adults with FHL, an allogeneic hematopoietic cell transplantation (HCT) may be proposed, using a graft from an unaffected donor.
Acutely ill or clinically deteriorating patients, as in the case described here, should instead be promptly treated. The current more validated therapeutic regimen is the HLH-94 protocol developed by the Histiocyte Society [10,35], although alternative regimens have also been used to treat HLH, such as the similar HLH-2004 protocol, which shares the same regimen used in HLH-94 that will be described shortly, but incorporates cyclosporine as part of the initial therapy [6]. None of the latter ones, however, has been directly compared with HLH-94 or has shown clear superiority. Clinicians should also consider including patients in clinical trials, when available. HLH-94 protocol consists in an induction therapy with etoposide (150 mg/m2 IV twice/week for weeks 1-2 and then once/week for weeks 3-8) and dexamethasone at in initial dose of 10 mg/m2 q.d.), with intrathecal therapy (with MTX ± hydrocortisone) only for those with central nervous system involvement. Moreover, all patients should receive supportive care, such as blood product transfusions for cytopenias and antimicrobials for prevention/management of infections. Of course, patients receiving treatment will require daily clinical monitoring, laboratory studies including markers of inflammation, and evaluation of cerebrospinal fluid with each intrathecal treatment. It is important to note that HLH-94 protocol is widely used to treat several forms of HLH, including pregnancy-related/perinatal HLH, HLH following allogeneic HCT, and HLH secondary to viral infections or autoimmunity (i.e. MAS) [36,37,38]. Cumulatively, patients treated on the HLH-94 protocol have a median survival of 54 percent at 6.2 years [10].
Taking a closer look at the recommended treatment with etoposide, the latter has long been known as one of the most commonly prescribed anticancer drugs. Being inexpensive, it is particularly useful in poor countries for the management of advanced, refractory or relapsed malignancies. It works by inhibiting DNA topoisomerase II, an essential nuclear enzyme that manipulates DNA. This leads to the creation of permanent DNA breaks which, in turn, induce cell cycle inhibition and cell death if not repaired. Unluckily, etoposide-induced DNA damage can also be the cause of chromosome rearrangements, and its prolonged use has been correlated with therapy-induced secondary leukemia, particularly in children and young adults [39,40,41]. Etoposide has historically been used to treat aggressive solid malignancies and certain oncohematologic disorders but, in some cases, it is also utilized to cure other diseases. However, this non-tumor use of etoposide is so far largely unexplored. In particular, as previously mentioned, it is increasingly being used to treat immune-mediated inflammatory disorders associated with cytokine storm syndrome (CSS), such as precisely MAS. Etoposide dampens inflammation in patients with HLH through inhibition of the production of pro-inflammatory cytokine, such as IL-6, IL-10, IL-18, interferon (IFN)-γ, and TNF-α, and consequent deactivation of T cells and attenuation of the immune stimulation [42,43].
One consideration to keep in mind is that the aforementioned original HLH-94 and HLH-2004 protocols were initially developed for pediatric patients with primary HLH [11]. Although being very effective, these regimens may result in overtreatment and increased toxicity in adults, limiting their application in patients with AOSD-MAS [10,12]. While dose reduction and individualized tailoring of treatment are therefore recommended for adult patients with MAS at least in selected cases [12,44,45], there is still a shortage of reports guiding the proper application of such protocols. For instance, a recently published study showed that modifying the HLH-2004 scheme with low-dose, short-time courses of etoposide (100 mg twice weekly for 4 times) can be very effective in treating patients with AOSD-MAS, both in terms of survival rate and clinical and laboratory improvements, with also a better safety profile and a significantly less exposure to CS than the standard protocol [46]. Based on these considerations, in the present case the HLH -94 classical regimen was also modified, in that the patient’s initial clinical response was so dramatically good that the etoposide induction phase could be shortened to only 5 weeks instead of the 8 weeks described in the original protocol, while continuing slow CS tapering (Table 3).
4. Conclusions
AOSD is a rare autoinflammatory disease with heterogeneous clinical presentation and nonspecific hallmarks that can simulate many different diseases. MAS is a life-threatening complication that occurs in a significant proportion of patients with AOSD, with a short-term mortality rate as high as 10% [46]. In both conditions the diagnosis can take a long time, especially in the presence of confounding factors.
In the present case, the concomitance of a suspected systemic infection and the presence of an iatrogenic Clostridium Difficile colitis initially ruled out the hypothesis of AOSD, despite the fulfillment of Yamaguchi’s criteria. The diagnosis was taken into consideration only with the development of HLH. This allowed a complete re-evaluation of the clinical case, leading to the hypothesis of MAS as a complication of AOSD, probably triggered by an infectious/inflammatory background. Timely initiation of high-dose steroids and topoisomerase II inhibitors was crucial and effective.
Although our understanding of AOSD increased during the last decade, the disease remains a real diagnostic and therapeutic challenge for the clinicians, as there are still many important gaps in our knowledge in areas such as diagnostic criteria, most helpful biomarkers and management strategy. The pattern is even more complicated when an AOSD-MAS occurs. In that case, despite the significant improvement in survival with the HLH-94 protocol (which is currently the standard of care), mortality, as previously mentioned, remains high. Moreover, currently the diagnosis and treatment of AOSD-MAS are mainly based on the studies and clinical practice in s-JIA associated MAS, and transferring the pediatric classification criteria and treatment regimens to adult patients may not always be appropriate [46]. Thus, clinicians are strongly encouraged, if feasible, to enroll patients also in clinical trials testing HLH/MAS therapies or other clinical or research questions.
Ultimately, the research presented here stresses that a multidisciplinary team (including internists, rheumatologists, immunologists, pathologists, hematologists, radiologists, and infectious disease and critical care medicine specialists) is essential to give patients a chance of survival and recovery when serious life-threatening complications occur.
Author Contributions
Conceptualization, D.S. and C.S.; methodology, D.S.; software, F.B.; validation, C.B.; formal analysis, D.S. and C.S.; investigation, D.S., G.V., M.C.P., R.V., M.B. and L.D.P.; resources, D.S.; data curation, D.S.; writing—original draft preparation, C.B., F.B. and A.C.T.M.; writing—review and editing, D.S. and C.S.; visualization, D.S.; supervision, M.P.; project administration, M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This study was partially funded by the Italian Ministry of University and Research (MUR) program “Departments of Excellence 2023-2027”, AGING Project –Department of Translational Medicine, Università del Piemonte Orientale.
Institutional Review Board Statement
The study was conducted in strict accordance with the Declaration of Helsinki. Ethical approval was granted exemption by the local Ethics Committee (Comitato Etico Interaziendale Novara, https://comitatoetico.maggioreosp.novara.it/) both because it was considered as an observational study and all the procedures being performed, including genetic analyses, were part of the routine care and not a formal research protocol.
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Acknowledgments
C.S. would like to thank Gino Amisano and the Fondazione Valenza Anziani (Valenza, Italy) for partially funding his researcher position for studies in internal medicine/ geriatric medicine.
Conflicts of Interest
The authors declare no conflict of interest. The funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Efthimiou, P.; Kontzias, A.; Hur, P.; Rodha, K.; Ramakrishna, G.S.; Nakasato, P. Adult-Onset Still’s Disease in Focus: Clinical Manifestations, Diagnosis, Treatment, and Unmet Needs in the Era of Targeted Therapies. Semin Arthritis Rheum 2021, 51, 858–874. [Google Scholar] [CrossRef] [PubMed]
- Magadur-Joly, G.; Billaud, E.; Barrier, J.H.; Pennec, Y.L.; Masson, C.; Renou, P.; Prost, A. Epidemiology of Adult Still’s Disease: Estimate of the Incidence by a Retrospective Study in West France. Ann Rheum Dis 1995, 54, 587–590. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Ohta, A.; Tsunematsu, T.; Kasukawa, R.; Mizushima, Y.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Ota, T.; et al. Preliminary Criteria for Classification of Adult Still’s Disease. Journal of Rheumatology 1992, 19, 424–430. [Google Scholar] [PubMed]
- Arlet, J.B.; Huong, D.L.T.; Marinho, A.; Amoura, Z.; Wechsler, B.; Papo, T.; Piette, J.C. Reactive Haemophagocytic Syndrome in Adult-Onset Still’s Disease: A Report of Six Patients and a Review of the Literature. Ann Rheum Dis 2006, 65, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Bae, C.B.; Jung, J.Y.; Kim, H.A.; Suh, C.H. Reactive Hemophagocytic Syndrome in Adult-Onset Still Disease: Clinical Features, Predictive Factors, and Prognosis in 21 Patients. Medicine (United States) 2015, 94, e451. [Google Scholar] [CrossRef]
- Bergsten, E.; Horne, A.C.; Aricó, M.; Astigarraga, I.; Egeler, R.M.; Filipovich, A.H.; Ishii, E.; Janka, G.; Ladisch, S.; Lehmberg, K.; et al. Confirmed Efficacy of Etoposide and Dexamethasone in HLH Treatment: Long-Term Results of the Cooperative HLH-2004 Study. Blood 2017, 130, 2728–2738. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zerón, P.; López-Guillermo, A.; Khamashta, M.A.; Bosch, X. Adult Haemophagocytic Syndrome. In Proceedings of the The Lancet; Elsevier B.V., 2014, 383: 1503–1516. [CrossRef]
- Jordan, M.B.; Allen, C.E.; Weitzman, S.; Filipovich, A.H.; McClain, K.L. How I Treat Hemophagocytic Lymphohistiocytosis. Blood 2011, 118, 4041–4052. [Google Scholar] [CrossRef]
- Filipovich, A.H. Hemophagocytic Lymphohistiocytosis (HLH) and Related Disorders. Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program 2009, 1, 127–131. [Google Scholar] [CrossRef]
- Trottestam, H.; Horne, A.; Aricò, M.; Egeler, R.M.; Filipovich, A.H.; Gadner, H.; Imashuku, S.; Ladisch, S.; Webb, D.; Janka, G.; et al. Chemoimmunotherapy for Hemophagocytic Lymphohistiocytosis: Long-Term Results of the HLH-94 Treatment Protocol. Blood 2011, 118, 4577–4584. [Google Scholar] [CrossRef]
- Henter, J.-I.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatr Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef]
- Rosée, P. La; Horne, A.C.; Hines, M.; Greenwood, T.V.B.; Machowicz, R.; Berliner, N.; Birndt, S.; Gil-Herrera, J.; Girschikofsky, M.; Jordan, M.B.; et al. Recommendations for the Management of Hemophagocytic Lymphohistiocytosis in Adults. Blood 2019, 133, 2465–2477. [Google Scholar]
- Oen, K.; Duffy, C.N.M.; Tse, S.M.L.; Ramsey, S.; Ellsworth, J.; Chédeville, G.; Chetaille, A.L.; Saint-Cyr, C.; Cabral, D.A.; Spiegel, L.R.; et al. Early Outcomes and Improvement of Patients with Juvenile Idiopathic Arthritis Enrolled in a Canadian Multicenter Inception Cohort. Arthritis Care Res (Hoboken) 2010, 62, 527–536. [Google Scholar] [CrossRef]
- Gerfaud-Valentin, M.; Maucort-Boulch, D.; Hot, A.; Iwaz, J.; Ninet, J.; Durieu, I.; Broussolle, C.; Sève, P. Adult-Onset Still Disease: Manifestations, Treatment, Outcome, and Prognostic Factors in 57 Patients. Medicine (United States) 2014, 93, 91–99. [Google Scholar]
- Yamamoto, T. Cutaneous Manifestations Associated with Adult-Onset Still’s Disease: Important Diagnostic Values. Rheumatol Int 2012, 32, 2233–2237. [Google Scholar] [CrossRef] [PubMed]
- Ichida, H.; Kawaguchi, Y.; Sugiura, T.; Takagi, K.; Katsumata, Y.; Gono, T.; Ota, Y.; Kataoka, S.; Kawasumi, H.; Yamanaka, H. Clinical Manifestations of Adult-Onset Still’s Disease Presenting with Erosive Arthritis: Association with Low Levels of Ferritin and Interleukin-18. Arthritis Care Res (Hoboken) 2014, 66, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Pouchot, J.; Sampalis, J.S.; Beaudet, F.; Carette, S.; Décary, F.; Salusinsky-Sternbach, M.; Hill, R.O.; Gutkowski, A.; Harth, M.; Myhal, D.; et al. Adult Still’s Disease: Manifestations, Disease Course, and Outcome in 62 Patients. Medicine (United States) 1991, 70, 118–136. [Google Scholar]
- Nguyen, K.H.Y.; Weisman, M.H. Severe Sore Throat as a Presenting Symptom of Adult Onset Still’s Disease: A Case Series and Review of the Literature. Journal of Rheumatology 1997, 24, 592–597. [Google Scholar]
- Gerfaud-Valentin, M.; Jamilloux, Y.; Iwaz, J.; Sève, P. Adult-Onset Still’s Disease. Autoimmun Rev 2014, 13, 708–722. [Google Scholar] [CrossRef]
- Cheema, G.S.; Quismorio, F.P. Pulmonary Involvement in Adult-Onset Still’s Disease. Curr Opin Pulm Med 1999, 5, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Drouot, M.H.; Hachulla, E.; Houvenagel, E.; Hatron, P.Y.; Flipo, R.M.; Goullard, L.; Ducloux, G.; Devulder, B. Complications cardiaques de la maladie de Still de l’adulte: de la péricardite à la tamponade parfois révélatrice [Cardiac Complications in Adult Onset Still Disease: From Pericarditis to Tamponade as Manifestations]. Rev Med Interne 1994, 15, 740–743. [Google Scholar] [CrossRef]
- Chung, J.W.; Suh, Y.J.; Song, H.J.; Choi, J.H.; Park, H.S.; Cho, S.R.; Suh, C.H. Pure Red Cell Aplasia and Adult-Onset Still’s Disease. Clin Rheumatol 2004, 23, 368–370. [Google Scholar] [CrossRef]
- Fautrel, B. Adult-Onset Still Disease. Best Pract Res Clin Rheumatol 2008, 22, 773–792. [Google Scholar] [CrossRef]
- Kontzias, A.; Efthimiou, P. Adult-Onset Still’s Disease: Pathogenesis, Clinical Manifestations and Therapeutic Advances. Drugs 2008, 68, 319–337. [Google Scholar] [CrossRef]
- Khobragade, A.K.; Chogle, A.R.; Ram, R.P.; Mascarenhas, J.; Kothari, S.; Kawadkar, S.; Deshpande, S.S.; Nair, D.; Makhija, J. Reversible Posterior Leukoencephalopathy Syndrome in a Case of Adult Onset Still’s Disease with Concurrent Thrombotic Thrombocytopenic Purpura: Response to High Dose Immunoglobulin Infusions. Journal of Association of Physicians of India 2012, 60, 59–62. [Google Scholar] [PubMed]
- Saito, K.; Temmoku, J.; Sumichika, Y.; Yoshida, S.; Takano, E.; Watanabe, S.; Matsumoto, H.; Fujita, Y.; Matsuoka, N.; Asano, T.; et al. Adult-Onset Still’s Disease with Acute Kidney Injury Requiring Hemodialysis: A Case Report and Literature Review. Internal Medicine 2023, 62, 2901–2906. [Google Scholar] [CrossRef]
- Bagnari, V.; Colina, M.; Ciancio, G.; Govoni, M.; Trotta, F. Adult-Onset Still’s Disease. Rheumatol Int 2010, 30, 855–862. [Google Scholar] [CrossRef]
- Giacomelli, R.; Ruscitti, P.; Shoenfeld, Y. A Comprehensive Review on Adult Onset Still’s Disease. J Autoimmun 2018, 93, 24–36. [Google Scholar] [CrossRef]
- Zafran, M.; Wassef, N. Complex Presentation of Adult-Onset Still’s Disease. BMJ Case Rep 2019, 12, e228210. [Google Scholar] [CrossRef]
- Tsuboi, H.; Segawa, S.; Yagishita, M.; Toko, H.; Honda, F.; Kitada, A.; Miki, H.; Ohyama, A.; Hagiwara, S.; Kondo, Y.; et al. Activation Mechanisms of Monocytes/Macrophages in Adult-Onset Still Disease. Front Immunol 2022, 13, 953730. [Google Scholar] [CrossRef]
- Li, X.; Qu, B.; Nie, Y.; Zhu, G.; Li, W.; Mu, F. Clinical Features of Macrophage Activation Syndrome in the Adult Northern Chinese Population. Lupus 2014, 23, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Macovei, L.A.; Burlui, A.; Bratoiu, I.; Rezus, C.; Cardoneanu, A.; Richter, P.; Szalontay, A.; Rezus, E. Adult-Onset Still’s Disease—A Complex Disease, a Challenging Treatment. Int J Mol Sci 2022, 23, 12810. [Google Scholar] [CrossRef]
- Mehta, P.; Cron, R.Q.; Hartwell, J.; Manson, J.J.; Tattersall, R.S. Silencing the Cytokine Storm: The Use of Intravenous Anakinra in Haemophagocytic Lymphohistiocytosis or Macrophage Activation Syndrome. Lancet Rheumatol 2020, 2, e358–e367. [Google Scholar] [CrossRef] [PubMed]
- Fardet, L.; Galicier, L.; Lambotte, O.; Marzac, C.; Aumont, C.; Chahwan, D.; Coppo, P.; Hejblum, G. Development and Validation of the Hscore, a Score for the Diagnosis of Reactive Hemophagocytic Syndrome. Arthritis and Rheumatology 2014, 66, 2613–2620. [Google Scholar] [CrossRef] [PubMed]
- Henter, J.I.; Samuelsson-Horne, A.C.; Aricò, M.; Maarten Egeler, R.; Elinder, G.; Filipovich, A.H.; Gadner, H.; Imashuku, S.; Komp, D.; Ladisch, S.; et al. Treatment of Hemophagocytic Lymphohistiocytosis with HLH-94 Immunochemotherapy and Bone Marrow Transplantation. Blood 2002, 100, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, Z.; Hao, Z.; Li, L.; Lu, J.; Kang, H.; Lu, Y.; You, Y.; Li, L.; Chen, Q.; et al. Requirement for Etoposide in the Treatment of Pregnancy Related Hemophagocytic Lymphohistiocytosis: A Multicenter Retrospective Study. Orphanet J Rare Dis 2019, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, J.; Jing, X.; Zhang, Y.; Tang, H.; Wu, J. Etoposide Combined with Ruxolitinib for Refractory Hemophagocytic Lymphohistiocytosis during Pregnancy: A Case Report and Literature Review. Hematology (United Kingdom) 2019, 24, 751–756. [Google Scholar] [CrossRef]
- Masood, A.; Wahab, A.; Iqbal, Q.; Davis, J.; Ehsan, H.; Hashmi, H. Efficacy and Safety of Allogeneic Hematopoietic Stem Cell Transplant in Adults with Hemophagocytic Lymphohistiocytosis: A Systematic Review of Literature. Bone Marrow Transplant 2022, 57, 866–873. [Google Scholar] [CrossRef]
- Sarwar, M.R.; Iftikhar, S.; Saqib, A. Availability of Anticancer Medicines in Public and Private Sectors, and Their Affordability by Low, Middle and Highincome Class Patients in Pakistan. BMC Cancer 2018, 18, 14. [Google Scholar] [CrossRef]
- Montecucco, A.; Biamonti, G. Cellular Response to Etoposide Treatment. Cancer Lett 2007, 252, 9–18. [Google Scholar] [CrossRef]
- Cowell, I.G.; Sondka, Z.; Smith, K.; Lee, K.C.; Manville, C.M.; Sidorczuk-Lesthuruge, M.; Rance, H.A.; Padget, K.; Jackson, G.H.; Adachi, N.; et al. Model for MLL Translocations in Therapy-Related Leukemia Involving Topoisomerase IIβ-Mediated DNA Strand Breaks and Gene Proximity. Proc Natl Acad Sci U S A 2012, 109, 8989–8994. [Google Scholar] [CrossRef]
- Bailly, C. Etoposide: A Rider on the Cytokine Storm. Cytokine 2023, 168, 156234. [Google Scholar] [CrossRef]
- Cron, R.Q.; Goyal, G.; Chatham, W.W. Cytokine Storm Syndrome. Annu Rev Med 2023, 74, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Sung, L.; King, S.M.; Carcao, M.; Trebo, M.; Weitzman, S.S. Adverse Outcomes in Primary Hemophagocytic Lymphohistiocytosis. J Pediatr Hematol Oncol 2002, 24, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.A.; Tyrrell, P.; Valani, R.; Benseler, S.; Abdelhaleem, M.; Weitzman, S. Experience With Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome at a Single Institution. J Pediatr Hematol Oncol 2009, 31, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, T.; Ye, S.; Lv, L.; Chen, S.; Wang, X.; Bao, C.; Fu, Q. Short-Term, Low-Dose Etoposide in Refractory Adult-Onset Still’s Disease-Associated Macrophage Activation Syndrome. Clin Rheumatol 2022, 41, 2817–2823. [Google Scholar] [CrossRef]
Figure 1.
Timeline with relevant data from the clinical history.
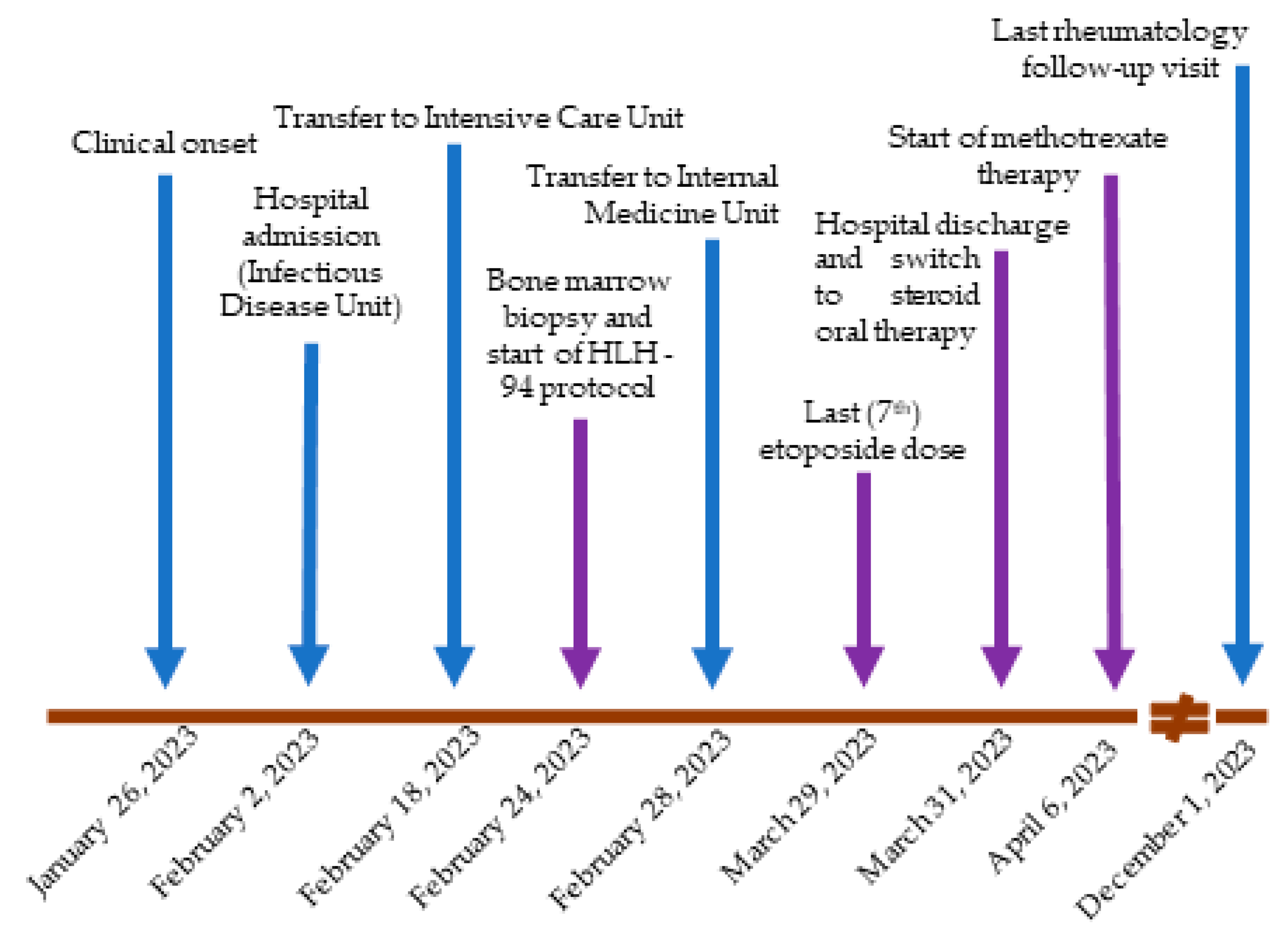
Figure 2.
Patient radiological aspect at total body computed tomography (CT) scan (a) Chest CT revealing a 22-mm pericardial effusion and bilateral pleural effusions; (b) Abdomen CT revealing severe hepatomegaly and moderate splenomegaly (before contrast); (c) Another image of the same abdominal aspect (portal venous phase).
Figure 2.
Patient radiological aspect at total body computed tomography (CT) scan (a) Chest CT revealing a 22-mm pericardial effusion and bilateral pleural effusions; (b) Abdomen CT revealing severe hepatomegaly and moderate splenomegaly (before contrast); (c) Another image of the same abdominal aspect (portal venous phase).
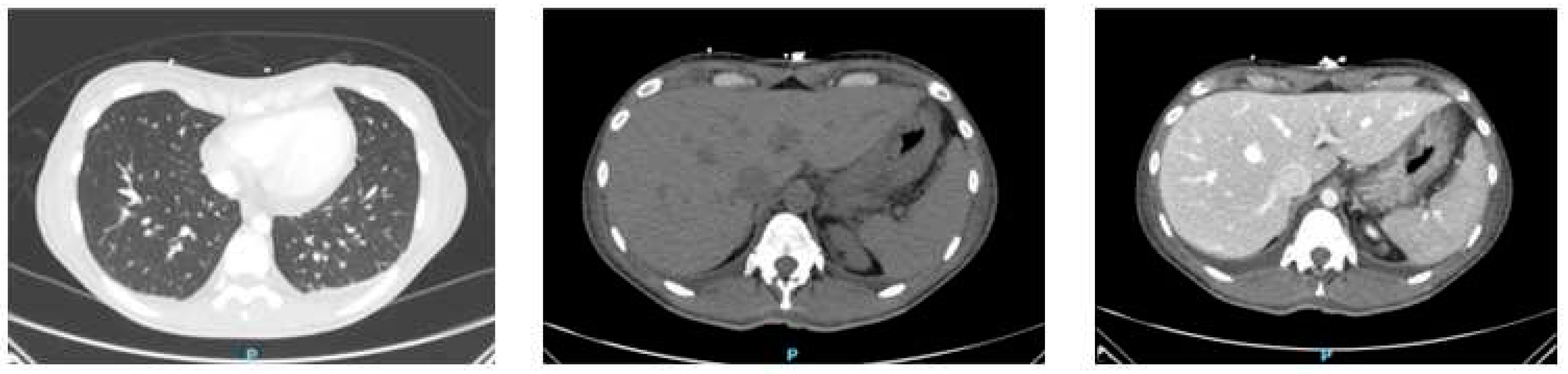
Figure 3.
Patient bone marrow biopsy and aspiration showing hemophagocytic syndrome. (a) Bone marrow is highly hypercellular and shows predominance of mature myeloid elements; megacariocytes are well represented; cells of eritroblastic lineage are heavily reduced (hematoxylin-eosin, 100x); (b) Macrophages are very numerous and collected in small aggregates or surrounding the adipocytes (CD68 immunostaining, 200x); (c) On smears of bone marrow, evidence of macrophages phagocytizing erythrocytes (May-Grṻnwald Giemsa, 300x).
Figure 3.
Patient bone marrow biopsy and aspiration showing hemophagocytic syndrome. (a) Bone marrow is highly hypercellular and shows predominance of mature myeloid elements; megacariocytes are well represented; cells of eritroblastic lineage are heavily reduced (hematoxylin-eosin, 100x); (b) Macrophages are very numerous and collected in small aggregates or surrounding the adipocytes (CD68 immunostaining, 200x); (c) On smears of bone marrow, evidence of macrophages phagocytizing erythrocytes (May-Grṻnwald Giemsa, 300x).
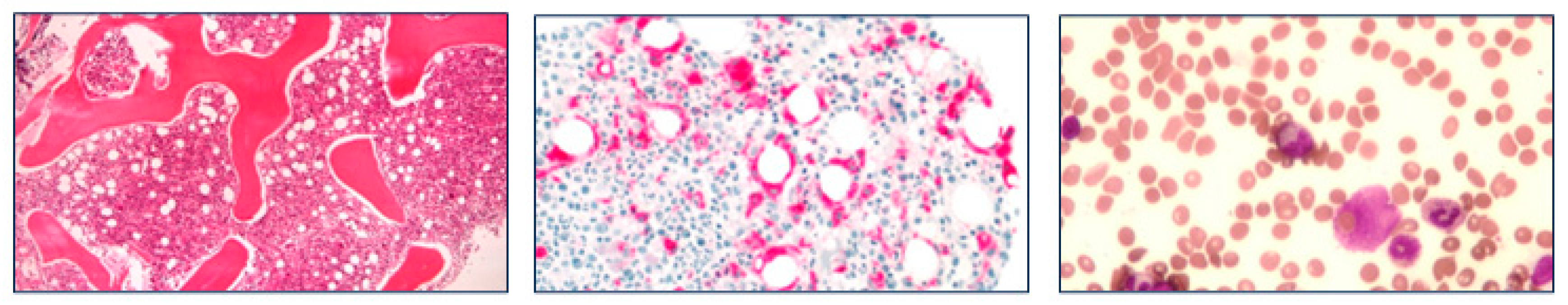

Table 1.
Yamaguchi criteria for diagnosing adult-onset Still’s disease (AOSD) [3].
Table 1.
Yamaguchi criteria for diagnosing adult-onset Still’s disease (AOSD) [3].
| Major criteria |
| Fever ≥ 39°C, lasting ≥ 1 week Arthralgias or arthritis lasting ≥2 weeks Typical salmon-pink nonpruritic skin rash Peripheral blood leukocytosis ≥ 10,000/µL, with granulocytes ≥ 80% |
| Minor criteria |
| Pharyngodynia |
| Lymphadenopathy |
| Hepatomegaly and/or splenomegaly |
| Abnormal liver function tests |
| Negative tests for rheumatoid factor (RF) and antinuclear antibodies (ANA) |
| Suggested exclusion criteria: |
| Infections (especially sepsis and infectious mononucleosis) |
| Malignancies (especially malignant lymphoma) |
| Other rheumatic diseases (especially polyarteritis nodosa and rheumatoid vasculitis with extraarticular features) |
Table 2.
Main diagnostic (clinical, laboratory and histopathologic) criteria for hemophagocytic lymphohistocytosis (HLH) [6,8]. Diagnosis is generally made when 5 of the following 9 criteria used in the HLH-2004 trial are met, but some authors consider the following modified criteria sufficient: 3 of 4 clinical findings (HLH-2004 criteria 1 to 3 and hepatitis) plus abnormality of 1 of 4 immune markers (HLH-2004 criteria 5 to 8) 1 [8,9].
Table 2.
Main diagnostic (clinical, laboratory and histopathologic) criteria for hemophagocytic lymphohistocytosis (HLH) [6,8]. Diagnosis is generally made when 5 of the following 9 criteria used in the HLH-2004 trial are met, but some authors consider the following modified criteria sufficient: 3 of 4 clinical findings (HLH-2004 criteria 1 to 3 and hepatitis) plus abnormality of 1 of 4 immune markers (HLH-2004 criteria 5 to 8) 1 [8,9].
|
|
|
|
|
|
|
|
|
1 further laboratory and radiographic abnormalities in other organ systems not used in the HLH-2004 trial may include: liver function and coagulation abnormalities (including bleeding manifestations); neurologic abnormalities (such as seizures, mental status changes and ataxia); respiratory abnormalities (including acute respiratory distress syndrome); severe hypotension requiring the administration of one or more vasopressors; renal dysfunction with or without hyponatremia (including renal failure requiring dialysis); skin manifestations (including rashes, erythroderma, edema, petechiae, and purpura); clinical features of Kawasaki disease (including conjunctivitis, red lips and cervical lymphadenopathy) [7]. 2 hemoglobin <9 g/dL; platelets <100 x109/L; absolute neutrophil count <1.0 x109/L. 3 fasting triglycerides > 265 mg/dL. 4 < 150 mg/dL. 5 > 500 ng/mL (but a ferritin > 3000 ng/mL has a better specificity and positive predictive value). 6 two standard deviations above age-adjusted laboratory-specific normal values
Table 3.
Main patient laboratory findings. Bold values are outside local laboratory normal ranges.
| Admission | VP-16 1st dose |
VP-16 2nd dose |
VP-16 3rd dose |
VP-16 4th dose |
VP-16 5th dose |
VP-16 6th dose |
VP-16 7th dose |
Discharge | f/up 2 mos |
f/up 8 mos |
Local laboratory NR |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WBC | 10.84 | 12.97 | 3.05 | 2.99 | 2.36 | 6.65 | 3.91 | 9.16 | 4.55 | 6.64 | 7.76 | 4.50-11.00 x109/L |
| Neutrophils | 9.42 | 10.99 | 2.59 | 2.42 | 1.54 | 3.18 | 1.61 | 5.31 | 2.24 | 4.03 | 4.85 | 1.80-7.70 x109/L |
| Lymphocytes | 1.00 | 0.67 | 0.39 | 0.51 | 0.81 | 2.53 | 2.02 | 2.96 | 1.83 | 1.93 | 2.20 | 1.00-4.50 x109/L |
| Hb | 9.2 | 7.9 | 8.9 | 8.7 | 9 | 8.5 | 8.2 | 8.1 | 8.5 | 12.2 | 11.0 | 11.5-15.5 g/dL |
| PLTs | 296 | 30 | 36 | 46 | 56 | 76 | 238 | 176 | 152 | 197 | 191 | 130-400 x109/L |
| INR | 1.38 | 1.75 | 1.64 | 1.50 | 1.47 | 1.19 | 1.09 | 1.05 | 1.02 | 1.01 | 1.01 | 0.8-1.2 Units |
| Fibrinogen | 112 | 96 | 98 | 85 | 83 | 107 | 66 | 137 | 209 | / | / | 200-393 mg/dL |
| Glycemia | 158 | 183 | 162 | 88 | 76 | 70 | 106 | 73 | 79 | 80 | 78 | 74-100 mg/dL |
| Creatinine | 0.72 | 0.55 | 0.37 | 0.22 | 0.40 | 0.49 | 0.47 | 0.42 | 0.44 | 0.65 | 0.65 | 0.5-121 mg/dL |
| Na | 136 | 137 | 137 | 135 | 136 | 137 | 136 | 139 | 141 | 143 | 144 | 134-146 mmol/L |
| K | 3.5 | 4.4 | 4.0 | 4.2 | 4.1 | 4.1 | 4.0 | 4.0 | 4.0 | 3.8 | 4.0 | 3.4-4.5 mmol/L |
| Triglycerides | 309 | / | / | 226 | 64 | / | / | / | / | / | / | < 150 mg/dL |
| CRP | 11.38 | 2.10 | 1.04 | 0.32 | 0.22 | / | / | 0.12 | / | 0.03 | 0.02 | 0-0.50 mg/dL |
| PCT | 0.3 | 0.2 | 0.3 | < 0.05 | / | / | / | / | / | / | < 0.05 | < 0.5 µg/L |
| Ferritin | 1059 | 1608 | 1338 | 553 | 574 | 628 | 531 | 580 | 555 | 19 | 38 | 13-150 µg/L |
| LDH | 970 | 1727 | 735 | 562 | 530 | 461 | 475 | 568 | / | 443 | 27 | 208-450 U/L |
| Total bilirubin | 0.55 | 1.63 | 1.58 | 1.24 | 1.36 | 1.43 | 1.00 | 0.66 | 1.05 | 0.70 | 0.81 | 0.30-1.20 mg/dL |
| AST | 49 | 590 | 100 | 39 | 27 | 21 | 27 | 30 | 28 | 24 | 19 | 0-40 U/L |
| ALT | 42 | 620 | 349 | 188 | 127 | 55 | 56 | 62 | 61 | 15 | 17 | 0-40 U/L |
| GGT | 74 | 199 | 288 | 239 | 177 | 93 | 79 | 85 | 85 | 26 | 34 | 0-50 U/L |
| ALP | 187 | 139 | 104 | 87 | 79 | 66 | 61 | 58 | 66 | 31 | 70 | 46-116 U/L |
ALP: alkaline phosphatase; ALT: alanine transaminase; AST: aspartate transaminase; CRP: C-reactive protein; GGT: gamma-glutamyl transferase; f/up: follow-up; Hb: hemoglobin; INR: international normalized ratio; K: potassium; LDH: lactate dehydrogenase; mos: months; Na: sodium; NR: normal range; PCT: procalcitonin; PLTs: platelets; VP-16: etoposide; WBC: white blood cells; /: not tested.
Table 4.
Results of other laboratory analyses performed during hospitalization. Bold values are outside local laboratory normal ranges.
Table 4.
Results of other laboratory analyses performed during hospitalization. Bold values are outside local laboratory normal ranges.
| Laboratory test | Result | Local laboratory NR |
|---|---|---|
| C3 / C4 | 1.45 / 0.19 | 0.90-1.80 / 0.10-0.40 g/L |
| Rheumatoid factor (RF) | negative | - |
| ANA | negative | - |
| dsDNA Abs | 9.5 | 0-27 IU/ml |
| Anti-ENA antibody screen 1 | < 3.6 | 0-20 CU |
| Autoimmune liver disease panel 2 | 0 | 0-6 CU |
| ANCA | negative | - |
| LA testing, SCT screening ratio | 0.94 | 0.77-1.20 |
| LA testing, dRVTT screening ratio | 1.06 | 0.70-1.20 |
| Anti-cardiolipin IgG | 0.7 | 0-20 CU |
| Anti.cardiolipin IgM | 7.5 | 0-20 CU |
| Anti-beta2glycoprotein IgG | 3.9 | 0-20 CU |
| Anti-beta2glycoprotein IgM | 1.8 | 0-20 CU |
| Beta-2 microglobulin | 2.40 | 1.16-2.52 mg/L |
| Serum IgG | 806 | 751-1560 mg/dL |
| Serum IgM | 222 | 48-220 mg/dL |
| Serum IgA | 182 | 80-400 mg/dL |
| Quantiferon-TB Gold Plus test 3 | negative | < 0.35 UI/mL |
| HIV Ab | negative | - |
| HBsAg | negative | - |
| HCV Ab | negative | - |
| ASLO | 45 | < 200 IU/mL |
| Anti-Parvovirus B19 IgG, index | 2.0 | 0.90-1.20 |
| Anti-Parvovirus B19 IgM, index | <0.10 | 0.90-1.10 |
| Widal-Wright 4 | < 1/80 | < 1/80 |
| Fecal calprotectin | 6.77 | < 50 µg/g |
| SARS-CoV-2 antigen rapid test 5 | negative | - |
| Serum Aspergillus antigen, ratio | 0.10 | 0.00-0.16 |
| CMV DNA | negative | - |
| EBV DNA | negative | - |
| Interleukin-6 | 2.7 | 0.0-4.4 pg/mL |
| Anti tTG IgA | 1.7 | 0.0-20.0 CU |
| TSH | 1.08 | 0.36-3.74 mIU/L |
| FT4 / FT3 | 8.0 / 1.2 | 9.0-17.0 / 2.7-4.4 ng/L |
| TG Ab / TPO Ab | 30 / 12 | 10-115 / 0-35 IU/mL |
Ab: antibodies; ANA: antinuclear antibodies; ANCA: antineutrophil cytoplasmic antibodies; ASLO: antibodies anti-streptolysin O; CMV: Cytomegalovirus; C3: complement component 3; C4: complement component 4; CU: chemiluminescent units; dRVVT: dilute Russell’s viper venom time; dsDNA Abs: anti-double-stranded deoxyribonucleic acid antibodies; EBV: Epstein–Barr virus; ENA: extractable nuclear antigen; FT4: thyroxine; FT3: triiodothyronine; IU: international units; HBsAg: hepatitis B surface antigen; HCV: hepatitis C virus; HIV: human immunodeficiency virus; Ig: immunoglobulin; LA: lupus anticoagulant; NR: normal range; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; SCT: silica clotting time; TB: tuberculosis; TG: thyroglobulin; TPO: tireoperoxidase; TSH: thyroid stimulating hormone; tTG: tissue transglutaminase. 1 including the following autoantibodies: anti-Sm (anti-Smith), anti-RNP (anti-ribonucleoprotein), anti-SSA (anti-Sjögren’s syndrome type A)/Ro60, anti-SSA/Ro52, anti-SSB (anti-Sjögren’s syndrome type B), anti-Jo-1 (anti-histidyl tRNA synthetase), anti-Scl 70 (anti-topoisomerase I). 2 including the following autoantibodies: anti-LKM1 (antibodies to liver/kidney microsome type 1); ASMA (anti-smooth muscle antibodies), AMA (anti-mitochondrial antibodies), anti-LC1 (anti-liver cytosolic antigen type 1), anti-SLA (anti-soluble liver antigen), anti-gp210 (anti-glycoprotein210), anti-SP100 (anti SP100 nuclear antigen). 3 interferon gamma dosage. 4 including the following antibodies: anti Typhus; anti Paratyphoid; anti Brucella. 5 nasal swab.
Table 5.
Characteristics of the clinical manifestations of adult-onset Still’s disease (AOSD).
| Fever [14] | Fever is almost always daily, with no spontaneous intercritical intervals. Very characteristically it is biphasic (2 peaks within the same day) with a sudden temperature increase (4C° in 4 hours). |
| Rash [15] | The rash is asymptomatic for what concerns itching or pain, occurs together with fever and disappears when the temperature returns to normal (fleeting). It presents as spots or maculopapules, usually on the trunk and extremities, rarely on the palmoplantar areas and on the face. The Koebner phenomenon (more intense in the areas of stress from the clothes) is frequently observed. |
| Arthritis [16] | Arthritis is typically transient and mild (however cases have been described in which severe synovitis led to joint destruction, so acute arthritis does not rule out the diagnosis, although it is not considered typical). It is a migrating oligoarticular manifestation, with preference, in order of probability, of: knees, wrists, metacarpophalangeal/proximal interphalangeal joints, ankles, elbows, shoulders. |
| Myalgia [17] | Myalgia is closely related to fever; the disease does not specifically attack the muscles, in fact electromyography and muscle biopsies in these cases are always normal. However, the dosage of muscle enzymes in the acute phases of the disease can show increases, albeit slight. |
| Pharyngitis [18] | Sore throat is a characteristic of the disease, especially in its onset before the occurrence of fever, and should always be sought in the patient’s anamnesis when there is a suspected diagnosis. It is a non-suppurative cricothyroid perichondritis or aseptic nonexudative pharyngitis of a purely inflammatory nature. |
| Lymphadenopathy [19] | Lymphadenopathy affects more than 2 out of 3 people with AOSD. it is typically a symmetrical lymphadenopathy involving the lymph nodes in the neck, which feel soft or stretchy to the touch. This phenomenon is due to benign B-cell hyperplasia of the pericortical zone. |
| Splenomegaly [19] | Acute splenomegaly is present in at least 33% of patients at diagnosis. It typically occurs in the absence of clinical hyperplenism and is not painful. |
| Liver disease [19] | Elevated liver necrosis rates are more frequent than hepatomegaly. In general, these are transient and non-dangerous conditions, even if cases in which fulminant hepatitis develops (all in patients treated with high doses of NSAIDs) have been reported. |
| Cardiac and pulmonary disease [20,21] | Cardiac (non-ischemic) involvements including arrhythmias and pericarditis have been described, as well as pulmonary infiltrates and pleural effusions, which together do not represent a significant proportion of patients, but which should be considered possible and should not confuse the physician in the differential diagnosis. |
| Hematologic manifestations [22] | Microangiopathic haemolytic anemia, haemolytic uremic syndrome, and thrombotic thrombocytopenic purpura have all occasionally been described, in addition to the fearsome MAS. |
|
Gastrointestinal symptoms [19,23] |
The presence of abdominal pain is highly variable in AOSD, generally in relation to the onset of fever; however, there are reports of pancreatitis and aseptic peritonitis. |
| Others [19,24,25,26] | Rare manifestations include: conjunctivitis, uveitis, aseptic meningitis, interstitial nephritis, glomerulonephritis, and secondary amyloidosis. |
AOSD: adult-onset Still’s disease; MAS: macrophage activation syndrome; NSAIDs: non-steroidal anti-inflammatory drugs.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



