Submitted:
01 January 2024
Posted:
03 January 2024
You are already at the latest version
Abstract
Pulmonary arterial hypertension (PAH) is a pathophysiological condition characterized by pulmonary vascular remodeling and elevated vascular resistance, which may ultimately result in cardiovascular failure and sudden cardiac death. In recent years, the occurrence of pregnancy complicated by pulmonary arterial hypertension has witnessed a notable increase, whereas the incidence of postpartum pulmonary arterial hypertension is comparatively rare. This study presents a case involving a 39-year-old female who exhibited severe pulmonary arterial hypertension, finally diagnosed as idiopathic pulmonary arterial hypertension, within 2 days postpartum. The diagnosis was made following a thorough clinical examination. This case report emphasizes the significance of promptly initiating targeted therapy as an initial step in effectively managing patients and the criticality of conducting a thorough assessment of multidisciplinary collaboration in postpartum management. Furthermore, the report has the potential to enhance awareness and education regarding the future management of similarly intricate conditions.
Keywords:
pulmonary arterial hypertension
; postpartum
; case report
Introduction
The diagnosis of pulmonary arterial hypertension (PAH) is established when the mean pulmonary artery pressure (PAP) exceeds 25 mmHg (1) There exist multiple classifications of pulmonary arterial hypertension (PAH), with the prevailing variant being pulmonary hypertension due to left heart disease (PH-LHD) (2). Pulmonary arterial hypertension (PAH) commonly manifests as difficulty breathing and has the potential to advance to the failure of the right ventricle, which poses considerable risks to both the mother and the fetus throughout pregnancy and the postpartum period. In recent years, the occurrence of pregnancy complicated by PAH has witnessed a notable increase, whereas the incidence of postpartum PAH is comparatively rare (1-5). In this report,we described the case of a postpartum patient with dyspnea who was eventually diagnosed with idiopathic PAH.
Case Presentation
A 39-year-old female patient, who was 2 days postpartum following an uneventful caesarean section for her second child, experienced sudden dyspnea following physical exertion, which was alleviated upon cessation of activity. The patient refuted the presence of symptoms such as fever, chest pain, abdominal pain, cough, and expectoration. The patient exhibited a state of previous good health and a lack of evident familial medical background. However, she had a brief history of drug use 3 years ago, which was manifested as six drug use episodes over two months. Furthermore, she currently presents with obesity, as indicated by her body mass index (BMI) of 31.2 kg/m2. The physical examination revealed the presence of generalized edema, with particular prominence observed in the bilateral lower extremities.
The patient initially disregarded the symptoms and subsequently sought medical evaluation and intervention. On September 6th, she presented to the nearby hospital due to progressively worsening dyspnea. The transthoracic echocardiography (TTE) demonstrated significant enlargement of the right atrium and right ventricle, as well as severe tricuspid valve regurgitation and PAH. Following diuresis, administration of antiasthmatic and anticoagulant therapies, the manifestation of dyspnea noticeably subsided. On September 8th, the patient was transferred to the municipal hospital, where TTE revealed an enlargement of the right heart, a reduction in systolic function, as well as severe tricuspid valve regurgitation and PAH. The findings from the computed tomography angiography (CTA) conducted on the same day provided additional evidence in favor of the presence of pulmonary artery dilatation (Figure 1A and Supplementary video 1). Consequently, the patient made the decision to be transferred to our cardiology department for further treatment and care.
Figure 1.
Manifestations of computed tomography pulmonary angiography, electrocardiogram, X-ray and pulmonary perfusion imaging on admission. Pulmonary computed tomography angiography conducted by external hospital indicated the presence of pulmonary artery dilatation. (B) Electrocardiogram showed q wave in lead V1 and right deviation of the electrical axis upon admission. (C) Chest X-ray demonstrated enlargement of the heart shadow. (D) Pulmonary perfusion imaging revealed the presence of multiple perfusion defects in both lung fields, with the left lung showing a more severe impairment than the right.
Figure 1.
Manifestations of computed tomography pulmonary angiography, electrocardiogram, X-ray and pulmonary perfusion imaging on admission. Pulmonary computed tomography angiography conducted by external hospital indicated the presence of pulmonary artery dilatation. (B) Electrocardiogram showed q wave in lead V1 and right deviation of the electrical axis upon admission. (C) Chest X-ray demonstrated enlargement of the heart shadow. (D) Pulmonary perfusion imaging revealed the presence of multiple perfusion defects in both lung fields, with the left lung showing a more severe impairment than the right.
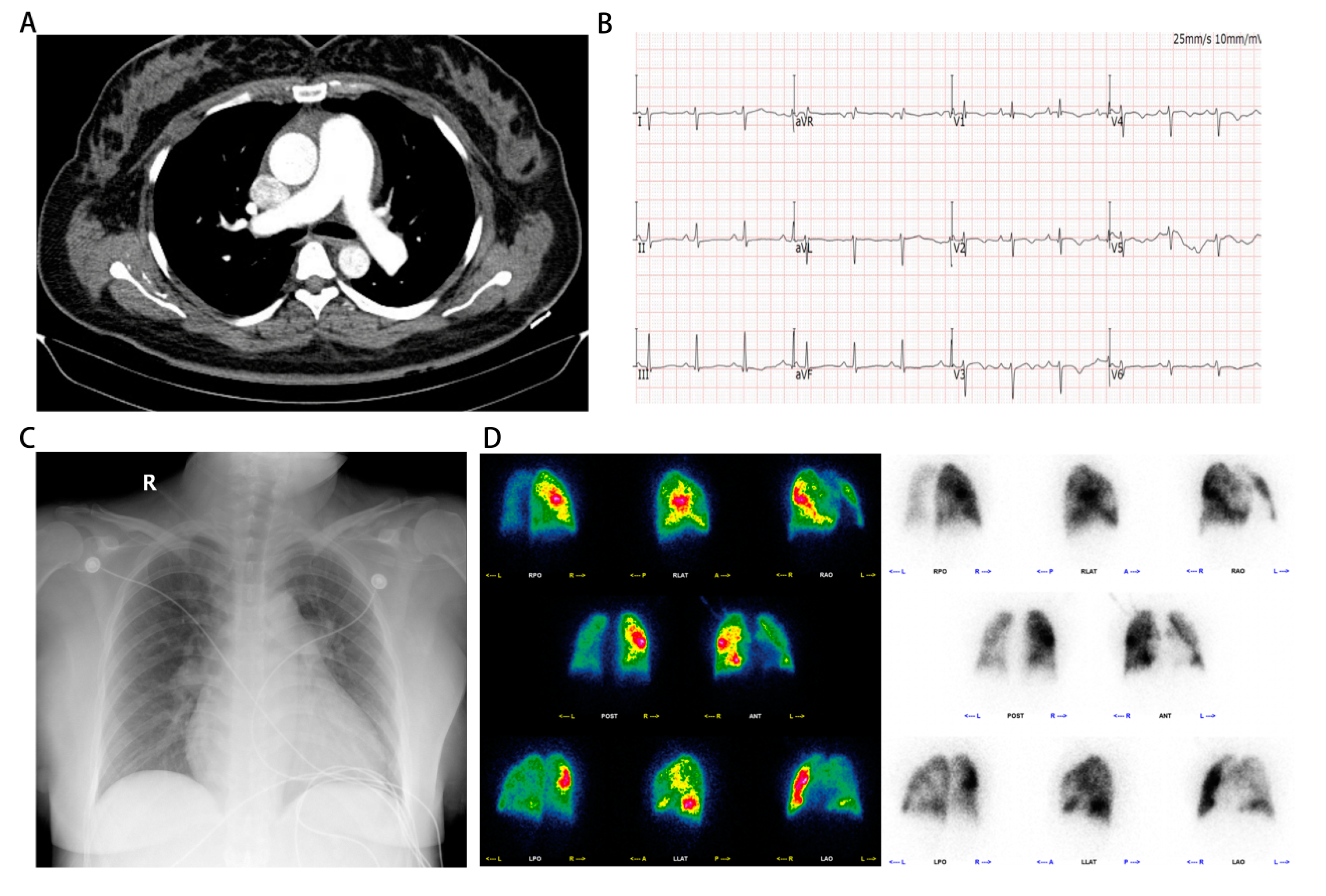
On admission, the patient's percutaneous oxygen saturation was measured at 96% while utilizing a nasal cannula at a flow rate of 3 L/min. Additionally, her blood pressure was recorded as 108/82 mmHg. Notably, her heart rate was observed to be 92 beats per minute, indicating a relatively elevated rate. As a consequence of experiencing breathlessness and impaired speech fluency, her cardiac function was classified as grade III according to the New York Heart Association (NYHA) classification system. During the physical examination, bilateral lung auscultation revealed coarse breath sounds without the presence of lung rales. Additionally, the patient exhibited normal heart sounds, along with a grade III/VI holosystolic murmur at the base of the heart that indicated tricuspid regurgitation. No abnormalities were detected in the abdomen, and there was no evidence of edema in the lower limbs.
Upon admission, a twelve-lead electrocardiogram (ECG) indicated the presence of a q wave in lead V1 and right deviation of the electrical axis (Figure 1B). Additionally, a pulmonary computerized tomography (CT) scan revealed poor ventilation in the upper lobe of the left lung. Besides, a chest X-ray demonstrated enlargement of the heart shadow (Figure 1C). On the first day of hospitalization, TTE revealed left atrial and right heart enlargement(La, 36 mm; Ra-L, 74.3 mm; Ra-M, 53.4 mm) with left ventricular ejection fraction of 62%, a dilated main pulmonary artery (26.8 mm), and severe regurgitation of the tricuspid valve, with an estimated pulmonary artery systolic blood pressure of 95 mmHg(Supplementary video 2-5). The abdominal ultrasound examination revealed an augmented inner diameter of the inferior vena cava, which indicated right heart failure. In the case of severe heart failure, a combination of bumetanide and spironolactone was used for treatment. Considering the presence of postpartum severe PAH, a decision was made to initiate sildenafil at a dosage of 12.5 mg thrice daily to mitigate pulmonary artery pressure.
On the day of admission, the laboratory results indicated elevated levels of NT-proBNP (4020 pg/ml), C-reactive protein (12.3 mg/L), procalcitonin (0.128 ng/ml), and D-dimer (2.3 mg/L). Troponin I (TnI) and coagulation function tests yielded normal results. Arterial blood gas analysis revealed hypoxemia accompanied by respiratory alkalosis (pH, 7.48, pO2, 76 mmHg, pCO2, 29 mmHg, and Lac, 1.9 mmol/L), which is likely attributed to hyperventilation. Furthermore, the individual exhibited compromised liver and kidney function, as evidenced by an albumin level of 28g/L, an alanine transaminase (ALT) level of 147 IU, and a creatinine level of 110 umol/L. Although the neutrophil ratio was elevated at 64.6%, the overall blood count appeared to be within the normal range.
According to the findings from previous echocardiography, there was an absence of manifestations of structural heart disease in the patient under examination. Furthermore, the patient exhibited normal left heart diastolic and systolic functions. Consequently, it is improbable that the patient's pulmonary hypertension can be attributed to left heart disease. Additionally, pulmonary perfusion imaging was conducted, revealing the presence of multiple perfusion defects in both lung fields, suggesting a more severe impairment in the left lung (Figure 1D). In this particular case, it was necessary to establish a differential diagnosis for pulmonary embolism. To achieve this purpose, the patient underwent computed tomography pulmonary angiography (CTPA) and was promptly administered prophylactic anticoagulation therapy. Subsequently, the CTPA images revealed an enlargement of the pulmonary artery without any apparent filling defect, thereby contradicting the diagnosis of pulmonary embolism (Figure 2D,E and Supplementary video 10).
In addition, we have enhanced various examinations, such as the rheumatic immune blood screen, the human immunodeficiency virus (HIV) screen, and the multiple pathogens screen, to investigate potential underlying factors contributing to pulmonary hypertension. In the case of this particular patient, tests for legionella pneumophila A IgM antibody, mycoplasma and chlamydia IgM antibody, adenovirus IgM antibody, respiratory syncytial virus IgM antibody, influenza virus A and B IgM antibody, and parainfluenza virus antibody yielded negative results. Furthermore, DNA testing for Epstein-Barr virus and cytomegalovirus also returned negative results. The findings from these laboratory tests have effectively ruled out the presence of pulmonary arterial hypertension resulting from rheumatic immune diseases and infection.
Figure 2.
The images of pulmonary angiography and computed tomography pulmonary angiography. (A-C) The images of pulmonary angiography indicated the presence of pulmonary artery dilatation in left front tilt 30 degrees, right front tilt 30 degrees, forward and backward position 0 degree. (D, E) The images of computed tomography pulmonary angiography showed an enlargement of the pulmonary artery without any apparent filling defect.
Figure 2.
The images of pulmonary angiography and computed tomography pulmonary angiography. (A-C) The images of pulmonary angiography indicated the presence of pulmonary artery dilatation in left front tilt 30 degrees, right front tilt 30 degrees, forward and backward position 0 degree. (D, E) The images of computed tomography pulmonary angiography showed an enlargement of the pulmonary artery without any apparent filling defect.
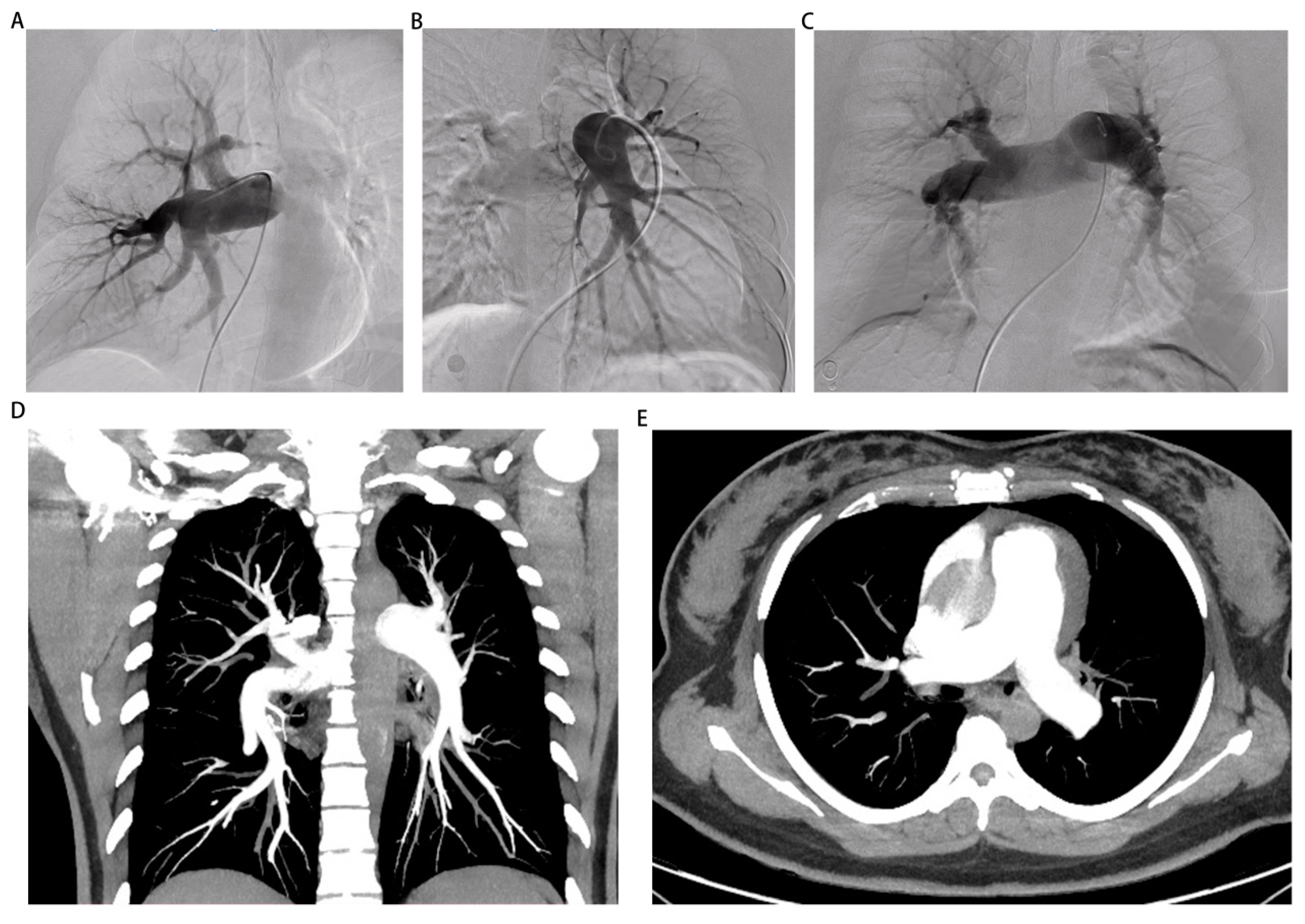
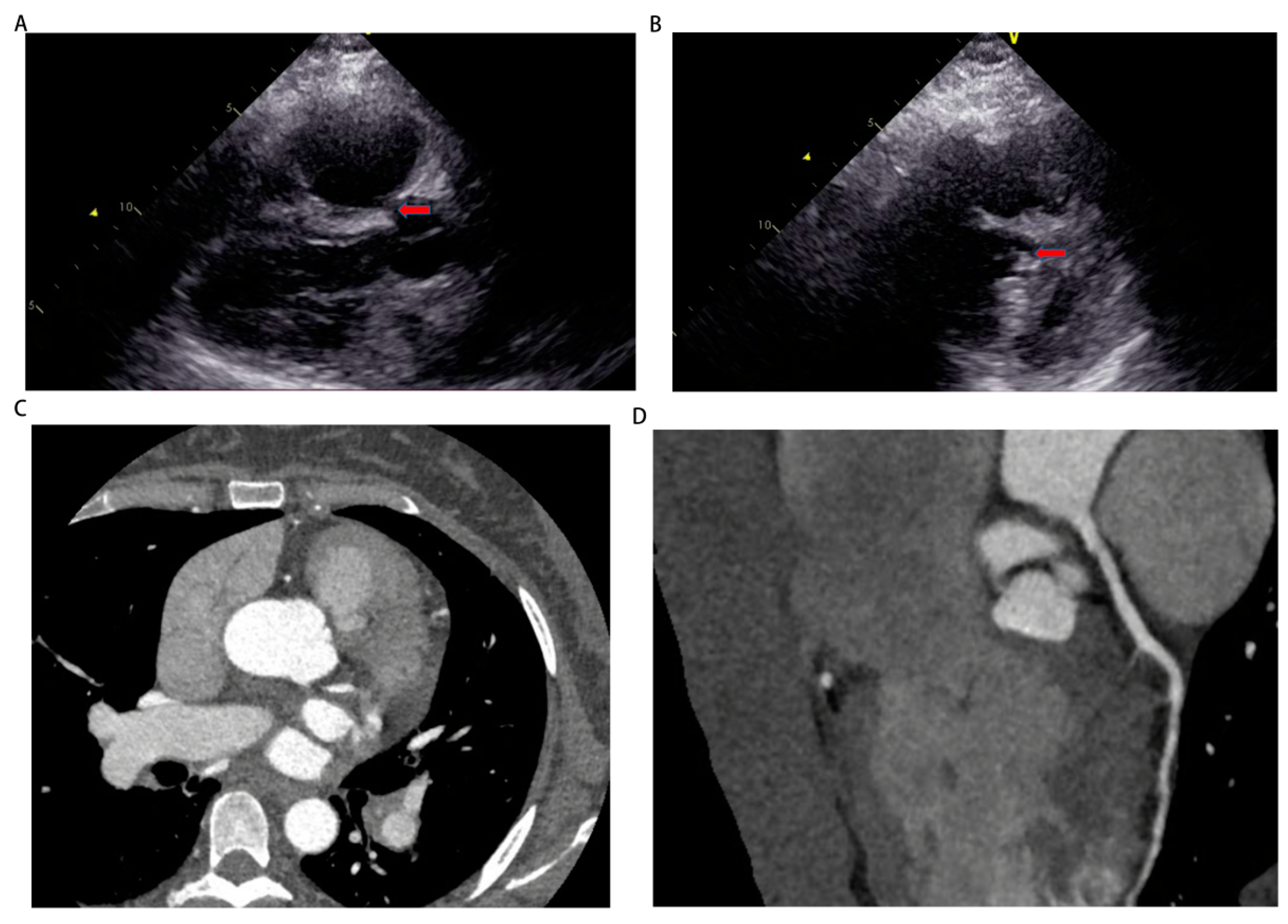
On September 13th, we performed right heart catheterization and pulmonary angiography, yielding the following results: a baseline mean pulmonary artery pressure of 50 mmHg, a mean pulmonary artery pressure of 47 mmHg after inhalation of nitric oxide, a mean pulmonary capillary wedge pressure (PCWP) of 7 mmHg, a pulmonary vascular resistance of 19.99 woods units, and a cardiac output of 2.7 L/min (Figure 2A-C, Table 1 and Supplementary video 6-8). These findings align with the diagnosis of pre-capillary pulmonary hypertension. However, the repeat echocardiography on September 16th revealed a discontinuity of approximately 5mm in the upper part of the interventricular septum and subarterial ventrieular septal defect to be excluded (Figure 3A,B). Thus, we further refined cardiac CTA (Figure 3C,D and Supplementary video 11) then the results showed no obvious atrioventricular shunt or coronary artery stenosis. Combined with the pulmonary artery time delay angiography (Left front tilt 90 degrees, Supplementary video 9), no aorta to pulmonary artery or atrioventricular shunt was observed, we can basically rule out the possibility of pulmonary hypertension caused by congenital heart disease. Finally, it is also possible for this patient to suffer from toxic pulmonary hypertension as a result of her drug use. Nevertheless, given the long interval between drug use and patient onset, as well as the time this patient with no special discomfort, we eliminated toxin-induced pulmonary arterial hypertension.
Figure 3.
The images of echocardiography and cardiac computed tomography angiography. (A, B) Echocardiography on September 16th showed a discontinuity of approximately 5 mm in the upper part of the interventricular septum and subarterial ventrieular septal defect to be excluded. (C, D) Cardiac computed tomography angiography showed no obvious atrioventricular shunt or coronary artery stenosis.
Figure 3.
The images of echocardiography and cardiac computed tomography angiography. (A, B) Echocardiography on September 16th showed a discontinuity of approximately 5 mm in the upper part of the interventricular septum and subarterial ventrieular septal defect to be excluded. (C, D) Cardiac computed tomography angiography showed no obvious atrioventricular shunt or coronary artery stenosis.
In summary, the patient received a diagnosis of idiopathic pulmonary arterial hypertension (IPAH). According to the 2015 European pulmona hypertension guidelines, she can be stratified into high-risk groups with triple therapy based on endothelin receptor antagonists, phosphodiesterase-5 inhibitors and selexipag. To optimize therapeutic outcomes, targeted medications were administered during the initial phase of treatment. Specifically, on September 14th, a daily dosage of 2.5 mg of ambrisentan in combination with sildenafil was prescribed to decrease pulmonary artery pressure. Subsequently, on September 16th, the dosage of sildenafil was increased to 25mg. In the interim, ventricular rate was managed through the administration of low-dose diltiazem, while hepatic protectors were employed to enhance liver functionality. Following a two-week course of treatment, notable improvements were observed in the patient's symptoms, with a marked reduction in shortness of breath and an elevation in cardiac function grade to grade II (as per the New York Heart Association classification). Consequently, the patient was discharged from the hospital. Then we added selexipag to the combination treatment since the patient's blood pressure was normal and stable after discharge. Follow-up for 1 month after discharge showed the patient had no significant shortness of breath after activity and the cardiac function was maintained at grade II.
Discussion
When pulmonary hypertension occurs in pregnant women, it typically has an unfavorable prognosis (3). Pulmonary hypertension can arise as a secondary condition to various diseases, including left-sided heart disease such as congenital heart disease and valvular heart disease, lung diseases, chronic thromboembolic disease, systemic conditions such as rheumatic immune diseases, infectious diseases, and drug usage (4). The diagnosis of IPAH can be established after ruling out other secondary causes. IPAH is a rare disease with an unknown cause, and it is more prevalent in women than in men (5). The features of IPAH encompass pulmonary artery vasoconstriction and vasomotor remodeling, which may ultimately leading to an augmented burden on the right ventricle.
This report elucidated the clinical manifestation of a patient who experienced profound pulmonary hypertension subsequent to childbirth. We hypothesize a potential causal association between elevated PAH and the act of delivery. The pertinent literature indicated that individuals within a 30-day postpartum period were particularly susceptible to the development of PAH (6). Despite its uncertain pathogenesis, the pathophysiology of postpartum PAH likely involves a multifactorial interplay. One of the most significant factors to consider was the alterations in cardiac output, which encompass autotransfusion from the uteroplacental circulation to the systemic circulation, as well as vasoconstriction resulting from involution postpartum (7, 8). Additionally, other factors such as pulmonary embolism should also be taken into account.
Previous studies have established a significant correlation between pulmonary embolism and cesarean section, identifying the latter as an independent risk factor (9).
The patient's manifestation of dyspnea and hypoxemia following a cesarean section, resembling the clinical symptoms of pulmonary embolism, which possibly suggests the occurrence of microthrombosis and subsequent obstruction of micro branches within the pulmonary vessel during the early stages of the disease (10). Therefore, the possibility of pulmonary embolism was not disregarded and a thorough examination was conducted utilizing a range of diagnostic methodologies, namely pulmonary perfusion imaging, CTPA, and pulmonary angiography. The results obtained from the pulmonary perfusion imaging revealed the presence of multiple areas with impaired blood perfusion in both lungs, with a particular emphasis on the left lung. Conversely, both CTPA and pulmonary angiography did not provide any conclusive evidence of thrombus formation. Consequently, the diagnosis of pulmonary embolism was excluded and it was postulated that this phenomenon was linked to pulmonary vasoconstriction (11).
Due to the unclear etiology, the patient was ultimately diagnosed with IPAH. Nevertheless, IPAH exhibits intricate pathogenesis and individuals afflicted with IPAH commonly experience an unfavorable prognosis (12, 13). The clinical management of IPAH primarily encompasses general treatment, symptomatic treatment, and targeted therapy. Additionally, in cases where patients do not respond to medical interventions, lung transplantation will become a necessary course of action. In recent years, significant progress has been made in the treatment of IPAH due to the widespread utilization of prostacyclin receptor agonists (such as epoprostenol, beraprost, and treprostinil), prostacyclin analogues, calcium channel blockers, endothelin receptor antagonists, and phosphodiesterase-5 inhibitors (such as sildenafil, tadalafil, and vardenafil) (14). Monotherapy or combination therapy involving the utilization of various drugs is a common approach in the treatment of IPAH. Epoprostenol, as a first-line drug, holds significant importance in this regard. However, treprostinil, despite its relatively longer duration of action, does not exhibit inferiority. A study has proposed that the prompt titration of treprostinil, in conjunction with oral sildenafil, post-administration, leads to acute enhancements in pulmonary hemodynamics and a notable increase in the overall survival rate of patients (15).
In relation to alternative therapeutic agents, such as calcium channel blockers, several scholars have posited that the timely administration of calcium channel blockers at the maximum tolerated dosage can effectively mitigate pulmonary hypertension and enhance prognosis, provided that the acute vascular response test yields positive results. This test entails a reduction in mean pulmonary artery pressure exceeding 10mmHg, with an absolute value below 40mmHg, while maintaining normal cardiac output during right heart catheterization in response to pulmonary vasodilators like nitric oxide (16, 17). Based on statistical data, it has been observed that around 10% of patients diagnosed with idiopathic or hereditary pulmonary hypertension exhibit positive results in the vascular response test (14). Furthermore, ambrisentan, as an exemplar of endothelin receptor antagonists, has demonstrated therapeutic potential in vasodilation, inhibition of vascular remodeling, and other mechanisms, with the most notable efficacy observed in patients with IPAH (18). Nevertheless, it is imperative to exercise caution when administering endothelin receptor antagonists, particularly bosentan, due to the potential for increased serum aminotransferase levels, particularly in cases of impaired liver function (19).
In our particular circumstance, as a result of the transient scarcity of treprostinil and the apprehension regarding the potential intolerance of the patient's low blood pressure to the therapeutic dosage of sildenafil, we initiated treatment with a 12.5mg dose of sildenafil to mitigate pulmonary artery pressure. Subsequently, we conducted a more comprehensive assessment of the vascular response, which regrettably yielded a negative outcome, indicating that the administration of calcium channel blockers was not anticipated to be effective in treating this patient. Consequently, a low dose of diltiazem was solely employed to regulate the ventricular rate. Following a series of treatments, the patient's blood pressure exhibited an initial increase and subsequently stabilized, prompting the administration of sildenafil at a dosage of 25mg.
In relation to the suggestions pertaining to combination therapy, prior scholarly works have advocated for the utilization of sildenafil in conjunction with bosentan for advanced pulmonary arterial hypertension (PAH) (20). However, recent scholarly findings have indicated that the co-administration of bosentan may diminish the plasma levels of sildenafil and subsequently impact its therapeutic efficacy (21, 22). Conversely, the concomitant administration of ambrisentan with sildenafil exhibits minimal pharmacokinetic interaction, thereby rendering this combination therapy approach more effective (23). Due to the relatively low likelihood of drug-drug interactions associated with ambrisentan and the observed improvement in the patient's liver function, the administration of ambrisentan in conjunction with sildenafil was incorporated as a therapeutic approach for the management of pulmonary arterial hypertension (24). This patient was stratified as high-risk group based on the guidelines (14). A long-term prognostic study in Japan has shown that using triple targeted therapy early has more advantages in improving the prognosis of patients with medium-high risks (25). In this regard, D'Alto M has proposed that the addition of parenteral prostaglandin analogues based on dual oral endothelin receptor antagonists and phosphodiesterase-5 inhibitors is safer and more efficient than dual therapy alone (26). Moreover, Selexipag, as a representative of prostaglandin analogues, the subsequent study has further confirmed the safety and efficacy of sequential triple therapy with Selexipag in intermediate-high risk patinets (27). We started treatment with a combination of sildenafil and amrisentan due to low blood pressure during hospitalization. Encouragingly, the patient experienced notable amelioration of symptoms subsequent to the treatment, leading to an uneventful discharge. The patient's blood pressure returned to a stable level after discharge, so selexipag was added to the outpatient clinic in the later stage to strengthen targeted therapy.
This case report emphasizes the significance of conducting a thorough assessment of interdisciplinary cooperation in the postpartum care, as it plays a crucial role in enhancing the outcomes for patients diagnosed with IPAH.
Conclusions
The occurrence or exacerbation of pulmonary hypertension can be attributed to pregnancy. In the case of most maternal pulmonary hypertension diagnoses, early termination or avoidance of pregnancy is recommended. Idiopathic pulmonary arterial hypertension, a rare and malignant pulmonary vascular disease, continues to exhibit high mortality rates. Despite advancements in targeted therapy, the diagnosis and treatment of idiopathic pulmonary arterial hypertension remains complex. Initiating targeted therapy at an early stage is of utmost importance in effectively managing these patients.
This case report emphasizes the significance of meticulous evaluation of multidisciplinary collaboration in postpartum management through the introduction of patient assessment, investigation, treatment, and follow-up. In the context of a maternal hospitalized patient experiencing dyspnea following physical exertion, it is crucial to consider the potential occurrence of IPAH. Prompt identification of IPAH warrants early implementation of targeted therapy to enhance prognosis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
(I) Conception and design: Lianglong Chen and Shumei Li; (II) Administrative support: All authors; (III) Provision of study materials or patients: Feng Hu; (IV) Collection and assembly of data: Qianyao Lai; (V) Data analysis and interpretation: Yuansheng Wu; (VI) Manuscript writing: Feng Hu and Qianyao Lai; (VII); Final approval of manuscript: All authors.
Competing interests: The authors have no conflicts of interest to declare.
Funding
This study was supported by grants from the talent start-up capital program of Fujian Medical University Union Hospital (2023XH027), Cardiovascular Medicine Center of Fujian Province (080070102), and Funding for Top Hospital and Specialty Excellence of Fujian Province (212790530102).
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- Simonneau G, Montani D, Celermajer D. S.; et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Charalampopoulos A, Lewis R, Hickey P; et al. Pathophysiology and Diagnosis of Pulmonary Hypertension Due to Left Heart Disease. Front Med (Lausanne). 2018, 5, 174. [Google Scholar] [CrossRef] [PubMed]
- Leopold JA, Maron BA. Molecular Mechanisms of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2016, 17, 761. [Google Scholar] [CrossRef] [PubMed]
- Taichman DB, Ornelas J, Chung L; et al. Pharmacologic therapy for pulmonary arterial hypertension in adults: CHEST guideline and expert panel report. Chest. 2014, 146, 449–475. [Google Scholar] [CrossRef]
- Droste AS, Rohde D, Voelkers M; et al. Endothelin receptor antagonist and airway dysfunction in pulmonary arterial hypertension. Respir. Res. 2009, 10, 129. [Google Scholar] [CrossRef] [PubMed]
- Escalante J. P., Diez A., Figueroa Casas M.; et al. Postpartum pulmonary hypertension. Medicina. 2015, 75, 44–47. [Google Scholar]
- Olsson K. M., Jais X. Birth control and pregnancy management in pulmonary hypertension. Semin. Respir. Crit. Care Med. 2013, 34, 681–688. [Google Scholar] [PubMed]
- Yang JZ, Fernandes TM, Kim NH; et al. Pregnancy and pulmonary arterial hypertension: a case series and literature review. Am. J. Obstet. Gynecol. MFM. 2021, 3, 100358. [Google Scholar] [CrossRef]
- Zhang L, Chen Y, Liu W; et al. Predictive value of D-dimer and analysis of risk factors in pregnant women with suspected pulmonary embolism after cesarean section. BMC Pulm. Med. 2021, 21, 391. [Google Scholar] [CrossRef]
- Villagra J, Shiva S, Hunter LA; et al. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007, 110, 2166–72. [Google Scholar] [CrossRef]
- O'Riordan D, Kiely DG, O'Driscoll BR. Reversible pulmonary artery perfusion abnormalities in the postpartum period as a precursor to the development of pulmonary arterial hypertension. Pulm. Circ. 2018, 8, 2045894018775190. [Google Scholar] [CrossRef] [PubMed]
- Xu J, Yang Y, Yang Y; et al. Identification of Potential Risk Genes and the Immune Landscape of Idiopathic Pulmonary Arterial Hypertension via Microarray Gene Expression Dataset Reanalysis. Genes (Basel). 2021, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Wang W, Jiang Z, Zhang D; et al. Comparative Transcriptional Analysis of Pulmonary Arterial Hypertension Associated With Three Different Diseases. Front. Cell Dev. Biol. 2021, 9, 672159. [Google Scholar] [CrossRef] [PubMed]
- Galiè N, Humbert M, Vachiery JL; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016, 37, 67–119. [Google Scholar] [PubMed]
- Wang T, Lu J, Li Q; et al. Rapid Titration of Intravenous Treprostinil to Treat Severe Pulmonary Arterial Hypertension Postpartum: A Retrospective Observational Case Series Study. Anesth. Analg. 2019, 129, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Demerouti E, Manginas A, Rammos S; et al. Postpartum pulmonary arterial hypertension: two cases covering a wide spectrum of presentations. Hellenic J. Cardiol. 2012, 53, 472–475. [Google Scholar]
- Ezedunukwe IR, Enuh H, Nfonoyim J; et al. Anticoagulation therapy versus placebo for pulmonary hypertension. Cochrane Database Syst. Rev. 2014, 2014, CD010695. [Google Scholar] [CrossRef] [PubMed]
- Liu C, Chen J, Gao Y; et al. Endothelin receptor antagonists for pulmonary arterial hypertension. Cochrane Database Syst. Rev. 2013, 2013, CD004434. [Google Scholar] [CrossRef]
- Wu S, Hoang HB, Yang JZ; et al. Drug-Drug Interactions in the Management of Patients with Pulmonary Arterial Hypertension. Chest. 2022, 162, 1360–1372. [Google Scholar] [CrossRef]
- Galiè N, Corris PA, Frost A; et al. Updated treatment algorithm of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2013, 62 (Suppl. 25), D60–D72. [Google Scholar] [CrossRef]
- Paul GA, Gibbs JS, Boobis AR; et al. Bosentan decreases the plasma concentration of sildenafil when coprescribed in pulmonary hypertension. Br. J. Clin. Pharmacol. 2005, 60, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Burgess G, Hoogkamer H, Collings L; et al. Mutual pharmacokinetic interactions between steady-state bosentan and sildenafil. Eur. J. Clin. Pharmacol. 2008, 64, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Spence R, Mandagere A, Dufton C; et al. Pharmacokinetics and safety of ambrisentan in combination with sildenafil in healthy volunteers. J. Clin. Pharmacol. 2008, 48, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Venitz J, Zack J, Gillies H; et al. Clinical pharmacokinetics and drug-drug interactions of endothelin receptor antagonists in pulmonary arterial hypertension. J. Clin. Pharmacol. 2012, 52, 1784–1805. [Google Scholar] [CrossRef] [PubMed]
- Kozu K, Sugimura K, Ito M; et al. Current status of long-term prognosis among all subtypes of pulmonary hypertension in Japan. Int. J. Cardiol. 2020, 300, 228–235. [Google Scholar] [CrossRef]
- D'Alto M, Constantine A, Balint OH; et al. The effects of parenteral prostacyclin therapy as add-on treatment to oral compounds in Eisenmenger syndrome. Eur. Respir. J. 2019, 54, 1901401. [Google Scholar] [CrossRef]
- Galiè N, Gaine S, Channick R; et al. Long-Term Survival, Safety and Tolerability with Selexipag in Patients with Pulmonary Arterial Hypertension: Results from GRIPHON and its Open-Label Extension. Adv. Ther. 2022, 39, 796–810. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



