Submitted:
28 December 2023
Posted:
29 December 2023
You are already at the latest version
Abstract
Sporadic inclusion body myositis (sIBM) is the most common muscle disease of older people and is clinically characterized by slowly progressive asymmetrical muscle weakness, predominantly affecting quadriceps, deep finger flexors and foot extensors. At present there are no enduring treatments for this relentless disease that eventually leads to severe disability and wheelchair dependency. Although sIBM is considered a rare muscle disorder, its prevalence is certainly higher as the disease is often undiagnosed or misdiagnosed. The histopathogical phenotype of sIBM muscle biopsy includes muscle fiber degeneration and endomysial lymphocytic infiltrates that mainly consist of cytotoxic CD8+ T cells surrounding nonnecrotic muscle fibers expressing MHCI. Muscle fiber degeneration is characterized by vacuolization and accumulation of congophilic misfolded multi-protein aggregates, mainly in their non-vacuolated cytoplasm. Many players have been identified in sIBM pathogenesis including environmental factors, autoimmunity, abnormalities of protein transcription and processing, accumulation of several toxic proteins, impairment of autophagy and ubiquitin proteasome system, oxidative and nitrative stress, endoplasmic reticulum stress, myonuclear degeneration, and mitochondrial dysfunction. Aging has been also proposed as a contributor to the disease. However, the interplay between these processes and the primary event that leads to the coexistence of autoimmune and degenerative changes is still under debate. Here, we outline our current understanding of disease pathogenesis, focusing on degenerative mechanisms, and discuss the possible involvement of aging.
Keywords:
sporadic inclusion body myositis (sIBM)
; muscle fiber degeneration
; protein aggregation
; inflammation
; aging
; inflammaging.
1. Introduction
Sporadic inclusion body myositis (sIBM) is a chronic progressive muscle disease that primarily affects people 50 years and older [1,2]. It is the most common acquired myopathy in the elderly with a prevalence ranging between 5 to 180 per million, depending on the geographical area [3,4,5,6]. sIBM is more common in males than females (2:1) and is associate with higher morbidity and mortality [5,6]. The disease presents with muscle weakness affecting mainly quadriceps and finger flexor [6]. At present there are no disease-modifying therapies for this progressive disease that eventually leads to severe disability [7]. Despite common clinical characteristics, the phenotype can be variable, and the diagnosis relies on the combination of clinical evaluation, laboratory tests, and pathologic changes in muscle biopsy [6]. Histological hallmarks include both degenerative features such as protein aggregation in myofibers, and autoimmune aspects such as endomysial infiltration by T cells [8,9].
sIBM is classified among the idiopathic inflammatory myopathies (IIM) along with dermatomyositis (DM), polymyositis (PM), and immune-mediated necrotizing myopathy (IMNM), but the lack of response to immunosuppressive treatment by sIBM patients have raised questions about the relevance of immune processes in disease pathogenesis [8,10,11].
Even though several studies have uncovered the processes participating to the degenerative and immune responses occurring in the disease, the relationship between these two aspects remains still unknown. Improving our knowledge of the pathogenic mechanisms is necessary to better understand this disorder, identify therapeutic targets, and design effective therapies for patients. Here we provide an overview on the clinic, histopathology, and disease mechanisms of sIBM and discussed the contribution of skeletal muscle and immune system aging in the disease.
2. Diagnosis of sIBM
2.1. Clinical aspects
sIBM is clinically characterized by slowly progressive asymmetrical muscle weakness, predominantly affecting quadriceps, deep finger flexors and foot extensors [1,8]. Pharyngeal muscles become often impaired resulting in dysphagia [12]. Head drop and camptocormia may occur and facial muscles are occasionally affected [13,14]. Late stages of the disease are characterized by weakness and atrophy of distal and proximal muscles, and possible impairment of neck flexors and extensors [15]. Sensory function is normal and deep tendon reflexes are preserved, but when atrophy of major muscles occurs the latter can be abolished [16]. Disease progression is usually slow. Variability in the clinical presentation and lack of specific features/muscle involvement in some patients represent challenges for a prompt and correct diagnosis, making laboratory tests and histological analysis of muscle biopsy fundamental tools in the diagnostic workup [7].
2.2. Laboratory Studies
CK levels are frequently normal or only mildly elevated, usually less than 10 times the upper normal values [9,17].
Autoantibodies are considered a useful tool in the diagnosis of IIM. Based on their specificity, autoantibodies in IIM are traditionally classified in myositis-specific (MSA) or myositis-associated antibodies (MAA), with the latter occurring also in other systemic autoimmune rheumatic diseases [18]. In 2011, Salajegheh et al. showed that autoantibodies targeting a ∼44 kDa human muscle protein (referred as Mup44) occur in serum of 52% of patients with sIBM (n=25) with 100% specificity for the disease [19]. In 2013, two independently studies identified Mup44 as cytosolic 5′-nucleotidase 1A (cN1A), which is an enzyme highly expressed in skeletal muscle that catalyzes the dephosphorylation of adenosine monophosphate into adenosine and phosphate [20,21]. Even if absent or extremely rare in healthy controls, antibodies against cN1A have been detected also in primary Sjögren’s syndrome (pSS) and systemic lupus erythematosus (SLE) making them not specific for sIBM [22,23]. Nevertheless, anti-cN1A antibodies are considered a valuable diagnostic biomarker for sIBM because they occur in 33 to 76% of patients with sIBM and in less than 5% of patients with PM, DM or non-autoimmune neuromuscular diseases [23]. It has been reported that the specificity of anti-cN1A to distinguish sIBM from other IIM, neuromuscular diseases and autoimmune disorders ranges between 87 and 100% and between 74.6 and 92% to discriminate sIBM from other types of IIM [24].Contradictory observations have been made regarding the association of anti-cN1A antibodies with a more severe phenotype and increased mortality risk independent of age, gender, comorbidities, and the presence of dysphagia [20,23,24,25,26,27,28,29,30,31]. Even though there is no cure for sIBM, antibodies against cN1A are helpful in the diagnostic workup to reduce misdiagnosis with PM and consequent administration steroids that worsen sIBM progression after discontinuation [10,15].
2.3. Magnetic Resonance Imaging (MRI)
In recent years, muscle MRI has emerged as an important tool in the diagnosis of muscle disorders due to its ability to highlight affected muscle by revealing atrophy, fat infiltration and signs of inflammation [32]. In sIBM, MRI changes have been reported in both upper and lower limbs, are more severe in distal segments, and usually have an asymmetric pattern [32,33,34,35]. Fat infiltration is commonly seen in MRI scans [33,35,36,37]. It has been observed a distinctive MRI pattern that can discriminate between sIBM and other myopathies, namely the relative sparing of the rectus femoris, hamstrings and adductor muscles and the extent and asymmetry of fatty infiltration, mostly seen in the adductor magnus [38]. Furthermore, flexor digitorum profundus seem to be involved in sIBM patients on imaging even before its weakness become clinically detectable [38].
2.4. Pathological characteristics
sIBM muscle biopsy shows both degenerative and inflammatory features (Figure 1). Histopathological changes detectable by light microscopy include:
- 1)
- rimmed vacuoles, namely irregular vacuoles of variable size and shape, bordered by basophilic granular deposits that occur in nonnecrotic muscle fibers. Rimmed vacuoles can be a rather rare finding and are visible in 0.4-6.4% of the fibers [9]
- 2)
- eosinophilic cytoplasmic inclusions visible at hematoxylin and eosin (H&E) and modified Gomori trichrome staining [17]
- 3)
- cytoplasmic accumulation of aggregated/misfolded proteins referred as amyloid deposits or protein inclusions, that occur in 60 to 80% of the sIBM vacuolated muscle fibers, usually in non-vacuolated areas of the fiber [1,17]. These β-pleated-sheet amyloid inclusions are detectable by fluorescence-enhanced Congo red staining. Several proteins have been found within these aggregates including amyloid-β precursor protein (AβPP), amyloid-β (Aβ), phosphorylated-tau (p-tau), ubiquitin, to name a few [8]. Notably, TDP-43 immunopositivity has been reported in over 60% sIBM patients and is considered a hallmark of sIBM pathology [39,40]
- 4)
- endomysial lymphocytic infiltrates consisting predominantly of macrophages and CD8+T cells that invade nonnecrotic muscle fibers expressing MHC class I on the sarcolemma [17]
- 5)
- 6)
- angulated muscle fibers of small caliber suggesting a neurogenic process [43]
Ultrastructural analysis reveals characteristic degenerative changes such as:
3. Degenerative processes in sIBM pathogenesis
Skeletal muscle of patients with sIBM is characterized by several degenerative processes including cytoplasmic accumulation of misfolded/toxic proteins, vacuolization, impairment of the ubiquitin-proteasome system and autophagy, oxidative and nitrative stress, endoplasmic reticulum stress, mitochondrial dysfunction, aging, and abnormalities of protein transcription and processing [8] (Figure 2).
3.1. Protein aggregation
Protein aggregation represents the main degenerative feature of sIBM. Protein misfolding/unfolding was initially discovered with the detection of cytoplasmic congophilic deposits indicating the presence of amyloid inclusions [44]. This observation was followed by the identification of Aβ protein, tau, and ubiquitin in the aggregates [45,46,47,48,49]. Aβ peptides, mainly Aβ40 and Abβ42, are produced from AβPP through sequential proteolytic cleavage by the β-secretases β-site of the APP cleaving enzyme 1 and 2 (BACE1 and BACE2) and the γ-secretase complex [50]. It has been shown that among the different forms of Aβ proteins, the more cytotoxic Aβ42 is predominantly aggregated in sIBM muscle fibers and occurs in the form of oligomers [51]. Increased plasma Aβ42 was also reported in patients with sIBM [52]. Moreover, the proteases involved in AβPP processing were found to be significantly increased and/or accumulated in sIBM [49,51,53,54,55]. Phosphorylated tau has also been detected in cytoplasmic aggregates where it was found to colocalize with kinases involved in its phosphorylation such as extracellular signal-regulated kinase (ERK), cyclin-dependent kinase 5 (CDK5), glycogen synthase kinase 3β (GSK-3β), and casein kinase 1 [56,57,58,59,60,61].
The detection of Aβ and p-tau have initially received a lot of attention due to their already known involvement in Alzheimer’s disease. Nevertheless, over decades, several other proteins have been found as abnormally accumulated in the cytoplasm of sIBM muscle fibers in the form of aggregates/inclusions, and many of them are linked to different neurodegenerative disorders. These include proteins prone to unfold/misfold such as α-synuclein, presenilin 1, and prion, proteins involved in endoplasmic reticulum (ER) stress (GRP78 and GRP94) and oxidative/nitrative stress (nitrotyrosine, NOS and SOD1), protein-folding homeostasis (heath shock proteins, 26S proteasome components, ubiquitin, p62/ SQSTM1), and RNA metabolism (RNA polymerase II, FUS, TDP-43, VCP, c-Jun, NFkB hnRNPA1, hnRNPA2/B1, hnRNPC1/C2 and hnRNPH) [8,62,63,64].
Even though the list of proteins that are abnormally accumulated in sIBM aggregates has been growing over the years, the pathogenic events that drive the abnormal cytoplasmic localization and aggregation of these proteins as well as their relationship with the immune component of sIBM still remain unknown. A combination of processes likely contributes to the protein aggregation: these include increased protein translation and specific posttranslational modification, impaired protein disposal via ubiquitin proteasome system (UPS) and autophagy pathway, and altered cellular environment due, for example, to oxidative stress and aged milieu that promote damage and misfolding/unfolding of proteins and interfere with protein quality control systems [40].
3.2. Impairment of ubiquitin proteasome system (UPS) and autophagy
In the cell, several events can trigger protein misfolding such as protein mutation, harsh cellular environment and metabolic stress [65,66]. Misfolded proteins can have deleterious consequences for the cells due to the loss of specific function/s or the acquisition of toxic activities and the tendency to form insoluble aggregates [65,66]. Cells are equipped with protein quality control mechanisms that aim to maintain protein homeostasis (or proteostasis) by refolding, degrading, or segregating misfolded proteins [65,66]. Molecular chaperons constitute a group of proteins that bind to unstructured proteins including nascent polypeptides on ribosomes and abnormally exposed hydrophobic regions, to prevent protein misfolding and facilitated the acquisition of correct protein conformation [67]. A few studies are reported in the literature that suggest a possible alteration of molecular chaperones in sIBM [68,69]. HSP70 immunopositive inclusions have been observed in sIBM muscle [68]. Specifically, HSP70 immunoreactivity was found to colocalized with Aβ and p-tau signal mostly in vacuolated muscle fibers. It has also been found that HSP70 protein is upregulated in sIBM muscles and co-immunoprecipitates with Aβ [68]. This observation has been interpreted as a possible role of HSP70 in Aβ folding, although it cannot be excluded that this interaction is the result of unspecific binding due to protein unfolding/misfolding. αB-crystallin (αBC), a member of the small heat shock protein family, has also be found accumulated in sIBM muscle fibers and in other muscle diseases [69,70]. Evidence for the association between αBC with AβPP and Aβ oligomers have been shown both in sIBM muscle and in AβPP-overexpressing cultured human muscle fibers. It has been proposed that the binding of αBC to Aβ oligomers might hinder the segregation Aβ oligomers into non-toxic aggregates and, therefore, prolong their toxic effects [68,70].
Failure of molecular chaperones to assist protein folding results in the presence of unfolded/misfolded proteins in the cell that can have toxic and harmful effects. In addition, other types of protein damages resulting, for example, by oxidation, accumulation of proteins in insoluble aggregates can be cytotoxic. Therefore, unfolded/misfolded, damaged, and aggregated proteins need to be eliminated to avoid, or minimize, cellular toxicity. Two major systems are responsible for protein degradation, namely the ubiquitin proteasome system (UPS) and the autophagy pathway. Alterations in both processes have been described in sIBM and considered important players in pathogenesis of the disease [40].
The UPS consists of multiple components that act synergistically to remove aberrant proteins [71]. E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin-protein ligases act sequentially to tag proteins that need to be eliminated with multi-ubiquitin chains [71]. Shuttle factors are then responsible for the delivery of ubiquitinated proteins to the 26S proteasome, the molecular machine responsible for the degradation [71]. The 26S proteasome is multiprotein complex composed of a 20S catalytic subunit and one or two 19S regulatory subunits [71]. The regulatory complex unfolds the ubiquitin-conjugated proteins so that they can enter the 20S core where they are subjected to different proteolytic activities of various subunits of the 20S core [71]. Abnormalities in the UPS in sIBM is supported by the increased expression and accumulation of 26S proteasome subunits and decreased activity of proteasomal proteolytic enzymes [72,73]. The impairment of the UPS is also supported by the observation that multiprotein aggregates in sIBM have features of aggresome, an extreme form of protein aggregates [74]. Aggresome formation is a protective mechanism activated under chronic UPS inhibition that aims to sequester misfolded proteins to prevent their possible toxic effects and eliminate them through autophagy [67].
Autophagy is an intracellular catabolic pathway through which components that cannot be degraded by the UPS, such as part of the cytoplasm, protein aggregates, malfunctioning organelles, or invading pathogens, are eliminated and recycled by the cell [75]. These components are first segregated in double lipid bilayer structures known as autophagosomes and then delivered to lysosomes to be degraded by lysosomal hydrolases into macromolecules such as aminoacidic, nucleotides, and sugars that can be reused by the cell [75]. Autophagy allows the elimination of damaged components and the maintenance of the proper number of functional organelles. Furthermore, the cell can exploit autophagy to self-digest some of its structure to survive under conditions of low nutrient and stress [75]. Several studies have provided evidence that impairment of autophagy contributes to the accumulation of proteins in the form of aggregates in muscle fibers of patients with sIBM. A histological hallmark of sIBM is the presence of cytoplasmic vacuoles. The autophagic nature of these vacuoles is supported by their positivity for acid phosphatase and other lysosomal enzyme as wells as by their content consisting of membranous debris that could arise from remnants of impaired organelles [76,77]. The first indication of a possible involvement of autophagy in sIBM muscles came from the detection of increased mRNA or protein levels of some lysosome proteins including M6PR, clathrin and hApg5 and hApg12 in sIBM muscles [77]. Later, Atg8/LC3 positive autophagic vacuoles containing AβPP and Aβ were detected in muscle fibers from sIBM patients [78]. These AβPP/Aβ and Atg8/LC3 double-positive vacuoles were almost exclusively detected in degenerating muscle fibers and occurred in a subset of muscle fibers expressing major histocompatibility complex (MHC) class II and surrounded by CD4+and CD8+ T cells. This finding indicates a possible involvement of autophagy in generation of antigens for MHC presentation to invading T cells [78]. Other factors involved in autophagy have been found to be altered in sIBM muscles. Increased transcript and protein levels of p62/SQSTM1, a factor involved in targeting ubiquitinated proteins for proteasomal and lysosomal degradation, were detected in sIBM muscle and found to colocalize with p-tau positive inclusions. Based on the increase of p62 that occurs following pharmacological inhibition of proteasomal or lysosomal protein degradation, it has been proposed that defective protein degradation through these two processes might participate in sIBM pathology [79]. In support to this scenario, a decrease in the activity of the lysosomal enzymes cathepsin D and B was found in sIBM muscles [76].
Micro-tubule-associated protein 1 light chain 3B (LC3) is detected, under normal conditions, in the cytoplasm as LC3I form. After induction of autophagy, LC3I is conjugated to phosphatidylethanolamine to generated LC3II which is relocated to autophagosomes and autolysosomes and is therefore considered a marker of autophagy [80]. LC3II and a decreased ratio of phosophorylated p70S6 kinase to total p70S6 kinase were considered an indication of autophagy induction in sIBM muscles [76]. According to this interpretation, the accumulation of LC3II and its binding partner p62 in sIBM muscle fibers could be the result of increased autophagosome formation [81].
Like p62/SQSTM1, NBR1 is another carrier protein involved in the transport of ubiquitinated proteins for autophagic degradation. It has been found that NBR1 protein is increased and accumulated in sIBM muscle in aggregates immunopositive to p62, ubiquitin, and phosphorylated tau [82].
Increased expression levels of the autophagy markers Beclin 1, ATG5 and LC3 and of the endosomal marker clathrin were detected in sIBM muscle and attributed to increased autophagosomal and endosomal structure due to increased autophagosome formation and/or to impaired late steps of the autophagy pathway [83]. It has also been shown that Beclin 1 associates with p-tau in the sarcoplasm of s-IBM myofibers and that lymphocytes preferentially surround Beclin 1 fibers [83].
An upregulation of components of the chaperone-mediated autophagy (CMA), a form of lysosomal pathway responsible for the degradation of proteins carrying the KFERQ motif, have been reported in sIBM muscle [84]. Hsc70, involved in the recognition and binding of protein carrying the KFERQ motif, and LAMP2A, which binds Hsc70-chargo protein complex at the level of the lysosome, unfolds and transfers target proteins within the lysosome, are both increased in sIBM muscle [84]. Interestingly, α-synuclein, which contains a KFERQ motif and is a CMA substrate, was found to be physically associated with LAMP2A and Hsc70 [84]. These observations might indicate that CMA is induced in sIBM muscle to remove misfolded proteins and avoid the formation of insoluble protein aggregates. However, since these proteins are targeted to defective lysosomal structure, as suggested by the decreased lysosomal enzyme activity, they are not properly cleared but accumulated in aggregates [84].
Overall, these studies support that impaired proteostasis occurs in sIBM and contributes to protein accumulation by reducing protein disposal and recycling.
3.3. Endoplasmic reticulum (ER) stress and Unfolded Protein Response (UPR)
The endoplasmic reticulum (ER) plays a critical role in the processing, folding, posttranslational modification, and exporting of newly synthesized proteins into the secretory pathway [85,86]. Under certain conditions, ER protein folding capacity become saturated and unfolded/misfolded proteins accumulate in the ER lumen compromising the ER homeostasis, a state referred as ER stress [85,86]. In attempt to cope with unfolded/misfolded protein loading, cell triggers a set of signaling pathways, collectively known as the unfolded protein response (UPR), that ultimately regulate gene expression to increase ER abundance and protein folding machinery and to repress protein synthesis [85,86]. UPR involves three major signaling pathways, each of which is activated by specific ER stress sensor proteins that reside in the ER membrane such as ATF6 (activating transcription factor 6), IRE1 (inositol requiring enzyme 1) and PERK (double-stranded RNA-activated protein kinases (PKR)-like ER kinase) [85,86]. An additional pathway to relieve ER stress is the ER-associated degradation (ERAD) by which unfolded/misfolded proteins undergo retrotranslocation into the cytosol to be degraded by UPS [86].
ER stress, UPR activation, and ERAD induction have been documented in muscle fibers of sIBM patients highlighting the significant role of ER stress in the pathogenesis of sIBM. Increased expression of several ER chaperones including calnexin, calreticulin, BiP/GRP78, GRP94, and ERp72 have been found in muscle specimens of patients with sIBM [87]. Furthermore, both in sIBM muscle and in the AβPP-overexpressing cultured human muscle fibers, those chaperones physically interact with Aβ suggesting their possible role in Aβ folding process and/or in Aβ protein clearance [87]. The presence of the processed ATF6 N-terminal fragment, which mediates the upregulation of UPR target genes including BiP/GRP78, the production of the alternative spliced form of XBP1, which leads to the transcriptional activation of chaperones and folding proteins involved in ERAD, the increased expression of ATF4 protein, and the augmented levels of CHOP mRNA, which is regulated by the oligomerization of PERK and subsequent phosphorylation of the transcription factor eIF2α, were detected in muscle biopsies of sIBM patients, suggesting that all the three main UPR pathways are activated in sIBM [88].
Many other proteins implicated in ER stress have also been found to be altered in sIBM muscle. Upregulation at mRNA and protein level of Herp (homocysteine-induced ER protein), an ER stress-inducible protein normally localized to the ER membrane, and its focal accumulation in the cytoplasm of affected fibers together with Aβ, the ER chaperone BiP/GRP78, and the 20S β2 proteasome subunit was detected in muscle of patients with sIBM [89]. Furthermore, Herp was found to be overexpressed both at mRNA and protein level in ER stress-induced cultured human muscle fibers, where the protein presented a diffuse staining, and increased at protein level in proteasome-inhibited cultured human muscle fibers, where it forms cytoplasmic aggregates [89]. These observations led the authors to speculate that the increased expression of HERP is induced in sIBM muscle fibers by both ER stress and proteasome inhibition to stimulate the ER folding capacity and misfolded protein removal [89].
In cultured human muscle fiber ER stress has been demonstrated to regulate the expression of MstnPP (myostatin precursor protein) via the transcription factor NF-κB that become activated during UPR [90]. In skeletal muscle MstnPP is posttranslationally processed primarily into myostatin (Mstn), a member of the transforming growth factor beta (TGF-β) superfamily that negatively regulates muscle development and growth. Increased expression of MstnPP, at both the mRNA and protein levels, was observed in muscle of patients with sIBM [91,92]. Myostatin dimer were detected in sIBM muscle and found to colocalize and physically interacts with Aβ/AβPP [91,92]. These findings led to the hypothesis that an aberrant aggregation of MstnPP/Mstn contribute to muscle fiber atrophy and weakness in patients with sIBM.
3.4. Mitochondrial abnormalities
Mitochondrial alterations have been widely described in muscle of patients with sIBM. These alterations include mitochondrial proliferation, COX-negative fibers, and mitochondrial DNA (mtDNA) changes, and occur with a greater extent and frequency in sIBM patients compared to healthy aged-matched controls [93].
Mitochondria proliferation is detectable by the modified Gomori trichrome stain which highlights the accumulation of abnormal mitochondria (red stain) in the subsarcolemmal region of the muscle fibers that look overall irregular in shape and are, therefore, referred as “ragged red fibers” (RRFs). In patients with sIBM, RRFs represent 1% of the total fibers whereas in the elderly RRFs occur with a frequency of about 0.3% [93]. Accumulations of mitochondria might represent a compensatory response to the impaired functionalities of these organelles. Increased percentage of COX negative fibers in sIBM patients compared to healthy aged-matched controls has been reported in several studies [94,95]. Reported frequencies of COX negative fibers ranges between 0.5% to 5% of total fibers in sIBM muscles whereas they are only occasionally observed in healthy subjects with similar age [94]. Impaired oxidative phosphorylation (OXPHOS) has been further supported by biochemical studies that detected a 30% decreased in COX activity, confirming the COX deficit observed by histological analysis [95].
An impairment of mitochondrial function has been detected also in primary myoblasts isolated from patients with sIBM. sIBM primary myoblasts displayed reduced oxygen consumption rate (OCR) and intracellular ATP, but increased extracellular acidification rate (ECAR), compared to normal myoblasts, indicating the activation of glycolysis to compensate for the reduced oxidative phosphorylation [96].
Abnormalities in the expression of some of the mitochondrial respiratory complexes has been observed in sIBM muscles. A reduction in the protein levels of complex I, III, and, with more variability, complex IV, that are partly encoded by mtDNA, was documented in sIBM muscle supporting a functional alteration of the respiratory chain [97].
Mitochondrial function is tightly linked to mitochondrial dynamics and morphology. Abnormalities in mitochondria structure have also been observed by ultrastructural analysis on sIBM muscles [94,96]. Structural alterations include mitochondria enlargement, loss of cristae, and paracrystalline inclusions [94,96]. Some of these structural abnormalities might results from disturbances of the mitochondrial dynamics occurring in sIBM muscle. Alteration of the expression of mitofusins (Mfn), which control fusion of the outer mitochondrial membrane, and of optic atrophy 1 (OPA1), which regulates fusion of the inner mitochondrial membrane, have been found in sIBM and contribute to the abnormalities of the mitochondrial network [95]. A decrease in Opa1 and Drp1 mRNA levels were reported in sIBM patient myoblasts compared with normal control myoblasts, whereas the expression levels of mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) were found to be unchanged [96]. Time-lapse imaging of mitochondria revealed a decreased mitochondrial mobility in primary myoblasts obtained from sIBM patients compared to myoblasts from healthy subjects [96]. Alterations in mitochondrial biogenesis in sIBM is suggested by the altered expression of important regulators of this process [93,98]. The non-coding microRNAs (miRNAs) miR-133 is markedly reduced in sIBM whereas gene expression of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) is increased in sIBM compared to control muscles [93,98]. Interestingly, metabolomics studies performed on peripheral blood revealed that patients with sIBM share metabolic changes with subjects affected by primary mitochondrial myopathies [99]. Notably, a greater degree of lymphocytic and macrophage infiltration has been observed in sIBM muscle fibers with more respiratory chain dysfunction. A strong positive correlation between the degree of inflammation, mitochondrial changes and atrophy has been found, suggesting that the mitochondrial dysfunctions have a role in sIBM progression [100].
As part of mitochondrial pathology, a remarkable mtDNA depletion and the presence of mtDNA alterations have been found in sIBM [42,94,96,97,101,102]. mtDNA deletions have been detected in muscle from sIBM patients with different techniques [94,101]. PCR and Southern blot analysis detected the occurrence of multiple mtDNA deletions in muscle tissue of IBM patients [94,101]. In situ hybridization using different mtDNA probes revealed that deleted mtDNA accumulates in COX deficient muscle fibers in patients with sIBM [94]. PCR analysis performed on isolated single muscle fibers have detected mtDNA with only one type of deletion in each COX-deficient muscle fiber, suggesting that clonal expansion of deleted mtDNA might occurs in each COX negative fiber [101]. The 4977 bp “common deletion”, which deletes between nucleotides 8,470 and 13,447 of the human mtDNA, is frequently detected COX deficient fibers in sIBM muscles [42,101]. This mutation leads to the loss all or part of the genes encoding four subunits of complex I, one subunit of complex IV, two subunits of complex V and five tRNA genes and, as expected, has a major impact on mitochondrial functionality [103].
Recently, deep sequencing of mtDNA by whole genome sequencing (WGS) has led to the identification of somatic mtDNA variants (deletions, duplications, and single nucleotide variants) [97] An increase of all these mtDNA alterations has been found in sIBM muscle compared to age-matched controls [97]. mtDNA copy numbers was also increased in muscle samples from sIBM patients, as reported in other studies [96,97]. Novel variants in nuclear genes involved in mtDNA maintenance have been found in sIBM. New variants of POLG and C10orf2 and a significantly higher frequency of an RRM2B variant were reported in IBM patients, suggesting that alterations in mechanisms involved in mtDNA maintenance might contribute to the disease [41].
3.5. Oxidative stress
The possible causative role of oxidative and nitrative stress in sIBM pathogenesis was first proposed by Askanas and Engel [104]. Nitric oxide (NO) is a short-lived free radical which plays a significant physiological role in different biological processes including vasodilation, antimicrobial activity of activated macrophages, transcriptional and post-transcriptional regulation of genes [105]. NO generation is catalyzed by three distinct isoforms of nitric oxide synthase (NOS): endothelial (eNOS), neuronal (nNOS) and inducible (iNOS) [106]. Nitric oxide (NO) can react with superoxide, a biologically relevant reactive oxygen species (ROS), to generate peroxynitrite, an highly reactive nitrogen species (RNS), which in turn leads to the conversion of tyrosine to 3-nitrotyrosine (3-NT), either free or part of a polypeptide chain [105]. The nitration of tyrosine can compromise the functional and/or structural integrity of target proteins [105]. The occurrence of nitrative stress in sIBM is supported by the abnormal accumulation of nitrotyrosine and of two NOS isoforms, nNOS and iNOS, in vacuolated fibers of sIBM muscle [107].
Since superoxide can be toxic, mammalian cells contain an enzymatic protective system against ROS-mediated damage which mainly include manganese superoxide dismutase (MnSOD) and copper-zinc superoxide dismutase (CuZnSOD). MnSOD, located in the mitochondria, and CuZnSOD, located in the cytoplasm, are scavenging enzymes that transform superoxide anions into hydrogen peroxide [108]. MnSOD and CuZnSOD were found to be increased and accumulated in vacuolated muscle fibers of patients with sBM but while MnSOD colocalized with nitrotyrosine and occasionally with p-tau, CuZnSOD was associated with ubiquitin suggesting that the two enzymes could have a different role in fiber protection [109,110]. It has been therefore hypothesized that while MnSOD exerts a protective effect in muscle fibers against the oxidative and nitrative stress, CuZnSOD participates in ubiquitination and clearance of inclusions in affected fibers [109,110]. Oxidative stress could also act as a key upstream trigger of AβPP overexpression in sIBM muscle through the upregulation of transcription factor NF-kB and redox factor-1 (Ref-1) [107,111].
Other factors that exert a protective function against the harmful effects of oxidative stress have been found in sIBM muscle. Increased mRNA and protein levels of insulin-like growth factor I (IGF-I), a pleiotropic growth factor with endocrine, paracrine and autocrine functions, and members of one of two main IGF-I signaling pathways, namely phosphoinositide-3-kinase (PI3K) and Akt, were detected in affected but not regenerating muscle fibers containing Aβ inclusions [112]. Based on the upregulation of IGF-I mRNA and protein that occurs in primary muscle cultures treated with Aβ(25-35) peptide, it has been postulated that IGF-I overexpression represent a protective response in vulnerable fibers to Aβ toxicity and that oxidative stress could play an important role in leading to increased expression of IGF-I [112]. DJ-1 is a small ubiquitously expressed protein that dimerizes under physiological conditions and plays several cellular functions including antioxidant response, chaperone activity, autophagy regulation, mitochondrial homeostasis, transcription regulation and neuroinflammation reduction [113]. DJ-1 was reported to be increased at both mRNA and protein level and highly oxidized in muscle of patients with sIBM [114]. It has also been shown that DJ protein forms cytoplasmic aggregates only in a small percentage of abnormal muscle fibers and is overexpressed as a 46 kDa dimer in addition to the 23 kDa monomer in the soluble fraction of sIBM muscle while the mitochondrial-enriched fraction contains only monomer. These observations have been interpreted as a possible role of DJ-1 in counteracting oxidative stress and mitochondria dysfunction in abnormal muscle fibers [114].
It is conceivable that multiple mechanisms can lead to generation of ROS/RNS and oxidative/nitrative stress in sIBM, one potential explanation is that mitochondrial dysfunction documented in muscle of sIBM patients causes increased ROS production with consequent oxidative stress and cellular damage.
3.6. Nuclear degeneration
The possible involvement of the nuclear compartment in sIBM has been reported in the literature [115,116]. Changes in the nuclear shape, release of filamentous structures from the nucleus to the cytoplasm, and alterations in the heterochromatin were observed in sIBM muscles and suggested that nuclear damage is involved in the disease [2,117,118,119]. The detection of the nuclear proteins emerin and lamin A/C within the rimmed vacuoles and the nuclear acid binding proteins, such as TDP-43 and other hnRNPs, in the sarcoplasm further support the idea that myonuclear damage contributes to protein mislocalization and accumulation in the cytoplasm [64,120,121]
Nevertheless, the exact nature of these nuclear alterations and their role in sIBM pathogenesis remain still unknown.
4. Aged skeletal muscle milieu
Aging is the progressive decline of cellular and physiological functions over time that lead to reduced survival [122]. Aging has been identified as a risk factor for a variety of human diseases including cancer, cardiovascular and neurodegenerative diseases [122]. sIBM mainly affects the elderly, with age at diagnosis over 50, strongly pointing to a key role of age-associated changes in the onset and progression of the disease.
Skeletal muscle, like all other tissues, undergoes age-dependent structural and biochemical changes that progressively impact its physiological functions. During aging, skeletal muscle experiences a reduction in protein synthesis [123], infiltration of adipose tissue and fibrosis [124], misregulation of proteostasis [125], dysfunctional autophagy and mitophagy [126], mitochondrial alterations [127], altered nuclear shape and spatial disorganization of nuclei [128], changes in nuclear-cytoplasmic signaling [129], decrease satellite cells number and function [130], increased ROS generation [131], DNA damage [132], and chronic inflammation [133]. Many of the processes contribute to the age-dependent decrease in myofiber size that causes loss of muscle mass and strength in older adults, a condition known as sarcopenia [134].
Several alterations affecting skeletal muscle during aging have been observed in skeletal muscle of patients with sIBM, usually to a greater extent than in skeletal muscle of aged-matched healthy subjects. Aging is associated with impaired protein homeostasis or proteostasis [122]. Proteostasis is maintained through mechanisms that ensure adequate proper synthesis, folding, and degradation [65]. These mechanisms work together to ensure a continuous supply of functional proteins and prevent the accumulation of damaged and malfunctioned proteins. Decrease in the production of protein chaperones and in the activity of the autophagy-lysosomal and ubiquitin proteasome systems are responsible for the loss of protein homeostasis during aging. Impaired proteostasis has been documented in sIBM and might contribute to protein aggregation [135].
Altered nuclear architecture and changes in nucleocytoplasmic signaling have been reported in skeletal muscle during aging [136]. Structural alterations have been observed in sIBM myonuclei, suggesting that impairment of nuclear architecture and function due to aging may contribute to sIBM pathology [118,119]
A decrease in the number and functionality of satellite cells (SCs) is characteristic of skeletal muscle aging [137]. An increased number of Pax7+ SCs has been reported in muscle specimens from sIBM patients indicating an ongoing repair process [138]. However, the expression of the myogenic regulatory factors MyoD and myogenin is altered in the disease and could indicate a delay and/or decrease in SC activation and myoblast proliferation, which could be due to replicative senescence of SCs after repeated cycles of activation [139]. These abnormalities in the myogenic program could contribute to the poor muscle repair in sIBM [139].
mtDNA is particularity susceptible to mutating during aging due to the highly oxidative mitochondrial environment, lack of protection from histone proteins, and low efficiency of mtDNA repair mechanisms compared to genomic DNA [140]. As a result, the somatic mutation rate of mtDNA is estimated to be 10 to 20 times higher than that of nuclear DNA [141]. Oxidative damage to mtDNA has been shown to occur during aging in skeletal muscle, leading to the accumulation of mtDNA mutations over time [142,143,144,145]. The 4977-bp “common deletion” is the most frequent age-associated mtDNA mutation and is also found in sIBM muscles [146]. This and other age-related mtDNA mutations lead to defective respiratory enzymes and, consequently, reduced oxidative phosphorylation and increased ROS production [145]. These alterations trigger oxidative stress and oxidative damage of the mitochondria, which contribute to further damaging mtDNA, perpetuating a vicious cycle.
Aging is characterized by changes in many molecular and cellular processes [122]. Although these age-associated alterations and the establishment of an aged milieu are clearly part of sIBM pathology, their contribution to sIBM pathogenesis remains poorly understood.
5. Inflammation in sIBM pathogenesis
The involvement of the immune system in sIBM is supported by the presence of endomysial immune cell infiltration and circulating autoantibodies. Although the role of inflammation in the pathogenesis of sIBM has been under discussion for a long time, many studies have provided data in to support the involvement of both innate and adaptive immunity in the disease.
5.1. T cells
Infiltrating immune cells are mostly represented by cytotoxic CD8+T cells that surround non-necrotic muscle fibers and are often localized in proximity of MHCI-expressing myofibers [17]. It has been estimated that CD8+T cells are about fivefold more abundant than CD4+T cells in sIBM muscle [147]. Several studies have shown that CD8+ T cells have restricted expression of T cell receptor (TCR) Vα/Vβ genes in blood and muscle of sIBM patients and are clonally expanded, suggesting that muscle-specific antigen drives the inflammatory response in the disease [148,149,150,151,152,153,154,155,156]. Interestingly, the expression of Vβ genes was found to be different in muscle-infiltrating lymphocytes and peripheral blood, indicating that T cells expand in situ in response to antigens presented by MHCI-expressing muscle cells or they are selectively recruited to the muscle [151,152]. Identical T cell clones have been found in different muscles and persist for years, indicating that they continuously react to muscle antigens perpetuating the inflammatory response over time [150,157].
T cell activation requires antigen presentation through MHC and the interaction between CD28 on T cells with costimulatory molecules on antigen presenting cells (APCs). Expression of the costimulatory molecule BB-1, ICOS, and B7-H3 by muscle fibers has been demonstrated in sIBM and suggests that myofibers may present the antigen and activate T cells expressing the appropriate TCR [158,159,160,161].
Regarding phenotypic and functional features, infiltrating CD8+T cells express markers of highly differentiated effector cells, have proinflammatory and cytotoxic features, and minimal or no proliferative ability [153,162,163,164,165]. Specifically, expression of the cytolytic molecules perforin, granulysin, and granzyme B, and natural killer (NK) markers CD16 or CD56, have been detected in CD8+T cells invading skeletal muscle, indicating they share functional and phenotypic features of NK cells [160,166,167,168]. The occurrence of Th1 immune response has been supported by increased expression levels of the cytokines and chemokines CXCL-9, CXCL-10, IL-12, CCL-2, and IL-1RA and by a higher number of IFN-γ expressing CD8+CD28−T cells in the blood of patients with sIBM [164]. These findings indicate that cytotoxic T cells respond to yet unknown muscle antigen/s and contribute to the myofiber damage that occurs in sIBM [9].
On the other hand, several observations show that, in sIBM, T cells are highly differentiated and have reduced effector functions. In this regard, it has been found that peripheral blood and muscle infiltrating T cells are effector memory T (TEM) cells and terminally differentiated effector memory T (TEMRA), as indicated by the lack of expression of CD28 and by the acquisition of CD224 and CD57 expression [9,164]. Loss of CD28 and gain of CD57 in T cells are known to be associated with decreased proliferative capacity, terminal differentiation, senescence, and clonal exhaustion, a state occurring with chronic antigen stimulation [169,170]. Exhausted T-cell have decreased ability to respond to cytokines, have lost their effector functions, and have upregulated inhibitory receptors (IRs) such as programmed cell death protein1 (PD-1), lymphocyte activation gene-3 (LAG-3), T-cell immunoglobulin domain and mucin domain-3 (TIM-3), and T-cell immune receptor with Ig and ITIM domains (TIGIT) [171,172]. The main function of IRs is to negatively regulate the activation and the effector functions of T cells and are responsible for fine-tuning T cell activity [172]. IRs have a key role in controlling immune responses and establishing T cell tolerance [172]. Expression of PD-1 has been reported on T cells infiltrating sIBM muscle [173]. PD1 ligands PD-L1 and PD-L2 have been detected on infiltrating macrophages and on myofibers, respectively, and possibly form immunologic synapses with PD1 expressed by lymphocytes [173]. In support of an exhausted phenotype of T cells in sIBM, the mRNA levels of the exhaustion markers PD1, EOMES, TBX21, LAG3, CD244, TIM3, and KLRG1 were found to be increased in sIBM muscle [173]. Taken together, these data indicate that, in sIBM, persistent exposure to antigens drives muscle infiltrating T cells toward a state of exhaustion characterized by reduced or absent effector functions and proliferation ability. These observations suggest that the T cell-mediated cytotoxic damage of muscle fibers likely play a role in the early stages of the disease, before T cell exhaustion occurs.
Regulatory T cells (Tregs) represent a subpopulation of CD4+T cells which have the main function to suppress immune responses [174]. Their immune suppressive activity is crucial for keeping immune reactions against invading pathogens under control and maintaining immune tolerance toward self-antigens [174]. Decreased number and function of Tregs has been associated with loss of tolerance and autoimmune diseases [175]. Studies have demonstrated that Tregs are able to control local immune responses during regeneration after skeletal muscle injury [176,177]. Although Tregs suppression activity in sIBM was found to be normal, a reduction in their number was observed in the blood and skeletal muscle of sIBM patients and proposed as a factor contributing to the autoimmune response to skeletal muscle in this disease [164].
5.2. Plasma cells and antibody-mediated immune response
Several studies have provided evidence for a muscle antigen-driven B- and plasma cell-mediated immune response and humoral immunity in sIBM. Differentiated CD138+ plasma cells, but not CD19+ or CD20+ B cells, and increased expression of immunoglobulin gene transcripts were detected in skeletal muscle of patients with sIBM [178]. Analysis of the variable region of Ig H chain gene transcript obtained from sIBM muscles demonstrated that B cells and their descendent plasma cells undergo oligoclonal expansion, isotype switching, and accumulate somatic mutations [179]. These processes usually occur in secondary lymphoid organs where B cells migrate after exposure to antigen to maturate and differentiate into antibody-producing plasma cells [179]. Interestingly, it has been observed that in sIBM muscles inflammatory infiltrates of T cells, B cells, plasma cells, and myeloid dendritic cells, assemble in lymphoid structures that could support the in situ maturation of B cells into antibody-secreting plasma cells [180]. Furthermore, increased serum levels of B-cell activating factor (BAFF), a cytokine involved in the survival, maturation and differentiation of B cell, were detected in the serum of patients with sIBM [181]. These findings indicate that local maturation of B cells to antibody-producing plasma cells occur in muscle of sIBM patients in response to muscle antigen/s [179].
The presence of a humoral immune response in sIBM were further supported by the detection of antibodies against cN1A, as described previously in this manuscript. The pathogenic role of the anti- cN1A was supported by the finding that mice immunized with anti-cN1A IgG isolated from patients show increased sarcoplasmic aggregation of p62 and LC3, degenerative features of sIBM muscle [30].
Overall, these findings support that in sIBM B cells become activated and mature into plasma cells that produces antibodies directed against a muscle antigen.
5.3. Macrophages and dendritic cells
Macrophages and myeloid dendritic cells (DCs) have also been detected in sIBM muscle [147,182]. Proteomic studies have shown that CD74, CD163, and STAT1 are increased in sIBM muscle and immunohistochemical analysis revealed these molecules are enriched in macrophages [183]. Specifically, CD74 was found to be expressed in CD68+ macrophages and the sarcolemma. CD163 was detected in endomysial macrophages, and STAT1 was found on macrophages that displayed phagocytic activity [183]. The interferon-induced protein ISG15 and IRF8 were found to be strongly expressed on MHCII+ macrophages, indicating that these cells are in an activated state [183].
DCs are also infiltrating sIBM muscle. Study of the two population of DCs, namely myeloid DC (mDCs) and plasmacytoid DC (pDCs), revealed that mDCs surround and invade healthy myofibers [184,185]. The close colocalization of mDCs with T cells has been interpreted as a possible role of mDCs in antigen presentation to T cells [184,185].
These studies show that, despite the expression of MHC on muscle fibers of sIBM patients and their possible contribution to antigen presentation, professional antigen presenting cells (APCs) also play a role in the pathogenesis of sIBM [186,187,188]. However, further efforts are needed to better characterize the phenotype and function of macrophages and dendritic cells in sIBM.
6. Aging of the immune system and sIBM
Relevant to sIBM pathology, the immune system also undergoes notable changes with age, a process called inflammaging [189]. The development and function of the immune cells involved in the pathogenesis of sIBM have been shown to be affected by aging. The sIBM muscle is infiltrated by CD8+T cells and macrophages, immune cells that undergo important changes with age.
T cells development occurs in the thymus, where bone marrow derived progenitors differentiate and undergo positively and negatively selections to give rise to a functional and self-tolerant T cell repertoire [190]. During aging T cell production in the thymus declines from approximately 16% to <1% over a lifetime [191,192]. In adults and the elderly, the decrease in the production of naïve thymic T cells is partially compensated by the proliferation of peripheral T cells which, although sufficient to maintain a compartment of naïve CD4+ T cells, does not support an adequate pool of peripheral naïve CD8+ T cells that undergo greater severe decline during aging [193]. After antigen stimulation, naïve T cells become activated and differentiate into effector and memory T cells. Aging is accompanied by a shift in the composition of the T cell pool from naïve to memory T cells, accumulation of terminally differentiated T cells, restriction of the T cell receptor (TCR) repertoire, and decrease ability to generate a CD8+T cell response [194,195,196,197]. The expression of CD28, which is involved in T cell survival, activation, and proliferation, by T cells declines during aging and contribute to the reduced immune responses in the elderly [197]. These agie-related changes in T cells have also been described in sIBM and aging likely contribute to these alterations in the disease. Skeletal muscle Tregs are important for maintaining immune tolerance within skeletal muscle and controlling immune response and tissue regeneration after skeletal muscle injury [198]. Although an increase in Treg in secondary lymphoid organs occurs during aging, a reduced recruitment of Treg cells into injured muscle, a decrease in their proliferation and retention at the injury site has been reported in old mice [199]. Interestingly, sIBM is associated with peripheral Tregs deficiency which could promote autoimmune-like responses and halt skeletal muscle repair in this disease [164].
sIBM muscles are infiltrated by macrophages expressing type I/II interferon markers and are highly phagocytic [183]. A decline in the expression of surface molecules, including MHC-II and toll like receptors (TLRs), which impair their antigen presentation capacity, and a decrease in secretion of interleukin 6 (IL6) and tumor necrosis factor α (TNFα) after appropriate stimuli have been reported during aging [200]. Accordingly, a decline in proinflammatory macrophages, which have the main role in supporting the inflammatory response, and an increase in anti-inflammatory macrophages, which are involved in the resolution of inflammation and tissue healing, were observed in aged skeletal muscle [201]. Further characterization of the phenotype of skeletal muscle-infiltrating macrophage in sIBM could help to better understand their contribution to the disease.
Aging of the immune system likely influences the immune processes occurring in sIBM, and studies are needed to understand how aging alters the function of immune cells that infiltrate skeletal muscle and how these alterations contribute to the pathogenesis of sIBM.
7. Interplay between degeneration and inflammation
The occurrence of both degenerative and inflammatory processes in sIBM has given rise to the still debated question of which of these two aspects drives the pathogenesis of the disease.
Inflammation has been proposed as the main driver of disease and responsible for the degenerative alterations [9]. The role of inflammation in causing degenerative changes in sIBM is supported by the observation that inflammatory cytokines induce the formation of protein aggregates and the expression of nitrative stress markers in cultured human myoblasts [202,203,204,205]. Studies in a transgenic mouse model of IBM have shown that inflammatory stimuli increase CD8+ T cell infiltration, AβPP and Aβ42 levels, and tau phosphorylation in skeletal muscle supporting the view of inflammation as the primary cause of sIBM [206].
On the other hand, lack of response to immunosuppressive and immunomodulatory therapies in sIBM has been raised as a main argument against the primary role of inflammation in the disease [40]. In agreement with this interpretation, a recent study demonstrated that deletion of T cells in an sIBM xenograft model does not rescue the degenerative features [207]. However, based on this finding, we cannot exclude a role of T cells in establishing the degenerative phenotype and that T cell depletion in early stage of the disease may have positive impact. Also, the exhausted phenotype of T cells infiltrating sIBM muscle could provide an explanation for the lack of response to immunosuppressive treatment and suggests that the cytotoxic T cell mediated muscle fiber damage could play a role in early stages, when the disease is not yet diagnosed.
Despite the well-recognized crosstalk between inflammation and degeneration in sIBM, it is not yet known which of these two processes is the primum movens. The causative relationship between the two aspects is also yet to be understood. Investigation of these aspects is not only scientifically intriguing but also paramount to develop effective therapies for this relentlessly progressive and disabling disorder.
8. Conclusions
sIBM is a multifactorial disorder characterized by the coexistence of inflammation and degeneration. The pathological features of the disease have been extensively characterized, and alterations in specific cellular pathways have been proposed as contributors to disease pathogenesis. Notably, many of the cellular and molecular processes that are altered in sIBM are also affected by aging, which likely has an influence on disease onset and progression.
Despite some cellular and molecular mechanisms have been uncovered, the etiology of the disease is still unknown and the causal relationship between autoimmune and degenerative responses has not yet been established. Further studies are needed to illuminate these aspects with the long-term goal of designing effective therapies for this progressive and debilitating disease.
Author Contributions
Conceptualization, V.G. and G.V.; writing, original draft preparation, V.G., M.C., P.T. and G.V.; writing, review and editing, V.G., M.C., P.T. and G.V.; drawing figures, M.C.; supervision and funding acquisition, G.V. All authors have read and agreed to the published version of the manuscript.
Funding
Work supported by #NEXTGENERATIONEU (NGEU) and funded by the Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006) – A Multiscale integrated approach to the study of the nervous system in health and disease (DN. 1553 11.10.2022).
Institutional Review Board Statement
Not applicable.
Acknowledgments
The authors would like to thank Alessandro Dinoto for his contribution in drafting Figure 2. Figure 2 was created with BioRender.com and Alessandro Dinoto own a full licence to publish.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Engel, W.K.; Askanas, V. Inclusion-body myositis: clinical, diagnostic, and pathologic aspects. Neurology 2006, 66, S20–S29. [Google Scholar] [CrossRef]
- Carpenter, S.; Karpati, G.; Heller, I.; Eisen, A. Inclusion body myositis: a distinct variety of idiopathic inflammatory myopathy. Neurology 1978, 28, 8–17. [Google Scholar] [CrossRef]
- Price, M.A.; Barghout, V.; Benveniste, O.; Christopher-Stine, L.; Corbett, A.; de Visser, M.; Hilton-Jones, D.; Kissel, J.T.; Lloyd, T.E.; Lundberg, I.E.; et al. Mortality and Causes of Death in Patients with Sporadic Inclusion Body Myositis: Survey Study Based on the Clinical Experience of Specialists in Australia, Europe and the USA. J Neuromuscul Dis 2016, 3, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Needham, M.; Corbett, A.; Day, T.; Christiansen, F.; Fabian, V.; Mastaglia, F.L. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J Clin Neurosci 2008, 15, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Callan, A.; Capkun, G.; Vasanthaprasad, V.; Freitas, R.; Needham, M. A Systematic Review and Meta-Analysis of Prevalence Studies of Sporadic Inclusion Body Myositis. J Neuromuscul Dis 2017, 4, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Nagy, S.; Khan, A.; Machado, P.M.; Houlden, H. Inclusion body myositis: from genetics to clinical trials. J Neurol 2023, 270, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Skolka, M.P.; Naddaf, E. Exploring challenges in the management and treatment of inclusion body myositis. Curr Opin Rheumatol 2023, 35, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; Engel, W.K.; Nogalska, A. Sporadic inclusion-body myositis: A degenerative muscle disease associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. Biochim Biophys Acta 2015, 1852, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.A. Inclusion body myositis: clinical features and pathogenesis. Nat Rev Rheumatol 2019, 15, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, O.; Guiguet, M.; Freebody, J.; Dubourg, O.; Squier, W.; Maisonobe, T.; Stojkovic, T.; Leite, M.I.; Allenbach, Y.; Herson, S.; et al. Long-term observational study of sporadic inclusion body myositis. Brain 2011, 134, 3176–3184. [Google Scholar] [CrossRef]
- Danon, M.J.; Reyes, M.G.; Perurena, O.H.; Masdeu, J.C.; Manaligod, J.R. Inclusion body myositis. A corticosteroid-resistant idiopathic inflammatory myopathy. Arch Neurol 1982, 39, 760–764. [Google Scholar] [CrossRef]
- Oh, T.H.; Brumfield, K.A.; Hoskin, T.L.; Kasperbauer, J.L.; Basford, J.R. Dysphagia in inclusion body myositis: clinical features, management, and clinical outcome. Am J Phys Med Rehabil 2008, 87, 883–889. [Google Scholar] [CrossRef]
- Goodman, B.P.; Liewluck, T.; Crum, B.A.; Spinner, R.J. Camptocormia due to inclusion body myositis. J Clin Neuromuscul Dis 2012, 14, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Salam, S.; Morrow, J.M.; Howard, R.; Miller, J.A.L.; Quinlivan, R.M.; Machado, P.M. Two emerging phenotypes of atypical inclusion body myositis: illustrative cases. Clin Exp Rheumatol 2023, 41, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Naddaf, E. Inclusion body myositis: Update on the diagnostic and therapeutic landscape. Front Neurol 2022, 13, 1020113. [Google Scholar] [CrossRef]
- Beyenburg, S.; Zierz, S.; Jerusalem, F. Inclusion body myositis: clinical and histopathological features of 36 patients. Clin Investig 1993, 71, 351–361. [Google Scholar] [CrossRef]
- Dimachkie, M.M. Idiopathic inflammatory myopathies. J Neuroimmunol 2011, 231, 32–42. [Google Scholar] [CrossRef]
- Satoh, M.; Tanaka, S.; Ceribelli, A.; Calise, S.J.; Chan, E.K. A Comprehensive Overview on Myositis-Specific Antibodies: New and Old Biomarkers in Idiopathic Inflammatory Myopathy. Clin Rev Allergy Immunol 2017, 52, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Salajegheh, M.; Lam, T.; Greenberg, S.A. Autoantibodies against a 43 KDa muscle protein in inclusion body myositis. PLoS One 2011, 6, e20266. [Google Scholar] [CrossRef]
- Larman, H.B.; Salajegheh, M.; Nazareno, R.; Lam, T.; Sauld, J.; Steen, H.; Kong, S.W.; Pinkus, J.L.; Amato, A.A.; Elledge, S.J.; et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol 2013, 73, 408–418. [Google Scholar] [CrossRef]
- Pluk, H.; van Hoeve, B.J.; van Dooren, S.H.; Stammen-Vogelzangs, J.; van der Heijden, A.; Schelhaas, H.J.; Verbeek, M.M.; Badrising, U.A.; Arnardottir, S.; Gheorghe, K.; et al. Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann Neurol 2013, 73, 397–407. [Google Scholar] [CrossRef]
- Rietveld, A.; van den Hoogen, L.L.; Bizzaro, N.; Blokland, S.L.M.; Dähnrich, C.; Gottenberg, J.E.; Houen, G.; Johannsen, N.; Mandl, T.; Meyer, A.; et al. Autoantibodies to Cytosolic 5′-Nucleotidase 1A in Primary Sjögren’s Syndrome and Systemic Lupus Erythematosus. Front Immunol 2018, 9, 1200. [Google Scholar] [CrossRef]
- Herbert, M.K.; Stammen-Vogelzangs, J.; Verbeek, M.M.; Rietveld, A.; Lundberg, I.E.; Chinoy, H.; Lamb, J.A.; Cooper, R.G.; Roberts, M.; Badrising, U.A.; et al. Disease specificity of autoantibodies to cytosolic 5′-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann Rheum Dis 2016, 75, 696–701. [Google Scholar] [CrossRef]
- Diederichsen, L.P.; Iversen, L.V.; Nielsen, C.T.; Jacobsen, S.; Hermansen, M.L.; Witting, N.; Cortes, R.; Korsholm, S.S.; Krogager, M.E.; Friis, T. Myositis-related autoantibody profile and clinical characteristics stratified by anti-cytosolic 5′-nucleotidase 1A status in connective tissue diseases. Muscle Nerve 2023, 68, 73–80. [Google Scholar] [CrossRef]
- Lilleker, J.B.; Rietveld, A.; Pye, S.R.; Mariampillai, K.; Benveniste, O.; Peeters, M.T.; Miller, J.A.; Hanna, M.G.; Machado, P.M.; Parton, M.J.; et al. Cytosolic 5′-nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann Rheum Dis 2017, 76, 862–868. [Google Scholar] [CrossRef]
- Goyal, N.A.; Cash, T.M.; Alam, U.; Enam, S.; Tierney, P.; Araujo, N.; Mozaffar, F.H.; Pestronk, A.; Mozaffar, T. Seropositivity for NT5c1A antibody in sporadic inclusion body myositis predicts more severe motor, bulbar and respiratory involvement. J Neurol Neurosurg Psychiatry 2016, 87, 373–378. [Google Scholar] [CrossRef]
- Lucchini, M.; Maggi, L.; Pegoraro, E.; Filosto, M.; Rodolico, C.; Antonini, G.; Garibaldi, M.; Valentino, M.L.; Siciliano, G.; Tasca, G.; et al. Anti-cN1A Antibodies Are Associated with More Severe Dysphagia in Sporadic Inclusion Body Myositis. Cells 2021, 10. [Google Scholar] [CrossRef]
- Felice, K.J.; Whitaker, C.H.; Wu, Q.; Larose, D.T.; Shen, G.; Metzger, A.L.; Barton, R.W. Sensitivity and clinical utility of the anti-cytosolic 5′-nucleotidase 1A (cN1A) antibody test in sporadic inclusion body myositis: Report of 40 patients from a single neuromuscular center. Neuromuscul Disord 2018, 28, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Oyama, M.; Ohnuki, Y.; Inoue, M.; Uruha, A.; Yamashita, S.; Yutani, S.; Tanboon, J.; Nakahara, J.; Suzuki, S.; Shiina, T.; et al. HLA-DRB1 allele and autoantibody profiles in Japanese patients with inclusion body myositis. PLoS One 2020, 15, e0237890. [Google Scholar] [CrossRef] [PubMed]
- Tawara, N.; Yamashita, S.; Zhang, X.; Korogi, M.; Zhang, Z.; Doki, T.; Matsuo, Y.; Nakane, S.; Maeda, Y.; Sugie, K.; et al. Pathomechanisms of anti-cytosolic 5′-nucleotidase 1A autoantibodies in sporadic inclusion body myositis. Ann Neurol 2017, 81, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.A. Cytoplasmic 5′-nucleotidase autoantibodies in inclusion body myositis: Isotypes and diagnostic utility. Muscle Nerve 2014, 50, 488–492. [Google Scholar] [CrossRef]
- Zubair, A.S.; Salam, S.; Dimachkie, M.M.; Machado, P.M.; Roy, B. Imaging biomarkers in the idiopathic inflammatory myopathies. Front Neurol 2023, 14, 1146015. [Google Scholar] [CrossRef]
- Dion, E.; Cherin, P.; Payan, C.; Fournet, J.C.; Papo, T.; Maisonobe, T.; Auberton, E.; Chosidow, O.; Godeau, P.; Piette, J.C.; et al. Magnetic resonance imaging criteria for distinguishing between inclusion body myositis and polymyositis. J Rheumatol 2002, 29, 1897–1906. [Google Scholar] [PubMed]
- Phillips, B.A.; Cala, L.A.; Thickbroom, G.W.; Melsom, A.; Zilko, P.J.; Mastaglia, F.L. Patterns of muscle involvement in inclusion body myositis: clinical and magnetic resonance imaging study. Muscle Nerve 2001, 24, 1526–1534. [Google Scholar] [CrossRef]
- Guimaraes, J.B.; Zanoteli, E.; Link, T.M.; de Camargo, L.V.; Facchetti, L.; Nardo, L.; Fernandes, A. Sporadic Inclusion Body Myositis: MRI Findings and Correlation With Clinical and Functional Parameters. AJR Am J Roentgenol 2017, 209, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Ansari, B.; Salort-Campana, E.; Ogier, A.; Le Troter Ph, D.A.; De Sainte Marie, B.; Guye, M.; Delmont, E.; Grapperon, A.M.; Verschueren, A.; Bendahan, D.; et al. Quantitative muscle MRI study of patients with sporadic inclusion body myositis. Muscle Nerve 2020, 61, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Tasca, G.; Monforte, M.; De Fino, C.; Kley, R.A.; Ricci, E.; Mirabella, M. Magnetic resonance imaging pattern recognition in sporadic inclusion-body myositis. Muscle Nerve 2015, 52, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Cox, F.M.; Reijnierse, M.; van Rijswijk, C.S.; Wintzen, A.R.; Verschuuren, J.J.; Badrising, U.A. Magnetic resonance imaging of skeletal muscles in sporadic inclusion body myositis. Rheumatology (Oxford) 2011, 50, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Hiniker, A.; Daniels, B.H.; Lee, H.S.; Margeta, M. Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol Commun 2013, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; Engel, W.K.; Nogalska, A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol 2012, 71, 680–693. [Google Scholar] [CrossRef]
- Lindgren, U.; Roos, S.; Hedberg Oldfors, C.; Moslemi, A.R.; Lindberg, C.; Oldfors, A. Mitochondrial pathology in inclusion body myositis. Neuromuscul Disord 2015, 25, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Oldfors, A.; Moslemi, A.R.; Jonasson, L.; Ohlsson, M.; Kollberg, G.; Lindberg, C. Mitochondrial abnormalities in inclusion-body myositis. Neurology 2006, 66, S49–S55. [Google Scholar] [CrossRef]
- Vattemi, G.; Mirabella, M.; Guglielmi, V.; Lucchini, M.; Tomelleri, G.; Ghirardello, A.; Doria, A. Muscle biopsy features of idiopathic inflammatory myopathies and differential diagnosis. Auto Immun Highlights 2014, 5, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Gales, T.; Paul, L. Amyloid filaments in inclusion body myositis. Novel findings provide insight into nature of filaments. Arch Neurol 1991, 48, 1229–1234. [Google Scholar] [CrossRef]
- Askanas, V.; Serdaroglu, P.; Engel, W.K.; Alvarez, R.B. Immunolocalization of ubiquitin in muscle biopsies of patients with inclusion body myositis and oculopharyngeal muscular dystrophy. Neurosci Lett 1991, 130, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; Engel, W.K.; Alvarez, R.B. Light and electron microscopic localization of beta-amyloid protein in muscle biopsies of patients with inclusion-body myositis. Am J Pathol 1992, 141, 31–36. [Google Scholar]
- Askanas, V.; Engel, W.K.; Bilak, M.; Alvarez, R.B.; Selkoe, D.J. Twisted tubulofilaments of inclusion body myositis muscle resemble paired helical filaments of Alzheimer brain and contain hyperphosphorylated tau. Am J Pathol 1994, 144, 177–187. [Google Scholar] [PubMed]
- Askanas, V.; Alvarez, R.B.; Engel, W.K. beta-Amyloid precursor epitopes in muscle fibers of inclusion body myositis. Ann Neurol 1993, 34, 551–560. [Google Scholar] [CrossRef]
- Sarkozi, E.; Askanas, V.; Johnson, S.A.; Engel, W.K.; Alvarez, R.B. beta-Amyloid precursor protein mRNA is increased in inclusion-body myositis muscle. Neuroreport 1993, 4, 815–818. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromolecular Med 2010, 12, 1–12. [Google Scholar] [CrossRef]
- Nogalska, A.; D’Agostino, C.; Engel, W.K.; Klein, W.L.; Askanas, V. Novel demonstration of amyloid-β oligomers in sporadic inclusion-body myositis muscle fibers. Acta Neuropathol 2010, 120, 661–666. [Google Scholar] [CrossRef]
- Abdo, W.F.; van Mierlo, T.; Hengstman, G.J.; Schelhaas, H.J.; van Engelen, B.G.; Verbeek, M.M. Increased plasma amyloid-beta42 protein in sporadic inclusion body myositis. Acta Neuropathol 2009, 118, 429–431. [Google Scholar] [CrossRef]
- Vattemi, G.; Nogalska, A.; King Engel, W.; D’Agostino, C.; Checler, F.; Askanas, V. Amyloid-beta42 is preferentially accumulated in muscle fibers of patients with sporadic inclusion-body myositis. Acta Neuropathol 2009, 117, 569–574. [Google Scholar] [CrossRef]
- Vattemi, G.; Engel, W.K.; McFerrin, J.; Buxbaum, J.D.; Pastorino, L.; Askanas, V. Presence of BACE1 and BACE2 in muscle fibres of patients with sporadic inclusion-body myositis. Lancet 2001, 358, 1962–1964. [Google Scholar] [CrossRef]
- Vattemi, G.; Engel, W.K.; McFerrin, J.; Pastorino, L.; Buxbaum, J.D.; Askanas, V. BACE1 and BACE2 in pathologic and normal human muscle. Exp Neurol 2003, 179, 150–158. [Google Scholar] [CrossRef]
- Maurage, C.A.; Bussière, T.; Sergeant, N.; Ghesteem, A.; Figarella-Branger, D.; Ruchoux, M.M.; Pellissier, J.F.; Delacourte, A. Tau aggregates are abnormally phosphorylated in inclusion body myositis and have an immunoelectrophoretic profile distinct from other tauopathies. Neuropathol Appl Neurobiol 2004, 30, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Wilczynski, G.M.; Engel, W.K.; Askanas, V. Association of active extracellular signal-regulated protein kinase with paired helical filaments of inclusion-body myositis muscle suggests its role in inclusion-body myositis tau phosphorylation. Am J Pathol 2000, 156, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Wilczynski, G.M.; Engel, W.K.; Askanas, V. Cyclin-dependent kinase 5 colocalizes with phosphorylated tau in human inclusion-body myositis paired-helical filaments and may play a role in tau phosphorylation. Neurosci Lett 2000, 293, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Shinde, A.; Kawashima, S.; Nakamura, S.; Akiguchi, I.; Kimura, J. Inclusion body myositis: expression of extracellular signal-regulated kinase and its substrate. Neurology 2001, 56, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kannanayakal, T.J.; Mendell, J.R.; Kuret, J. Casein kinase 1 alpha associates with the tau-bearing lesions of inclusion body myositis. Neurosci Lett 2008, 431, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, C.; Nogalska, A.; Engel, W.K.; Askanas, V. In AbetaPP-overexpressing cultured human muscle fibers proteasome inhibition enhances phosphorylation of AbetaPP751 and GSK3beta activation: effects mitigated by lithium and apparently relevant to sporadic inclusion-body myositis. J Neurochem 2010, 112, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; Bilak, M.; Engel, W.K.; Alvarez, R.B.; Tomé, F.; Leclerc, A. Prion protein is abnormally accumulated in inclusion-body myositis. Neuroreport 1993, 5, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Plagnol, V.; Brady, S.; Simone, R.; Lashley, T.; Acevedo-Arozena, A.; de Silva, R.; Greensmith, L.; Holton, J.; Hanna, M.G.; et al. Widespread RNA metabolism impairment in sporadic inclusion body myositis TDP43-proteinopathy. Neurobiol Aging 2014, 35, 1491–1498. [Google Scholar] [CrossRef]
- Pinkus, J.L.; Amato, A.A.; Taylor, J.P.; Greenberg, S.A. Abnormal distribution of heterogeneous nuclear ribonucleoproteins in sporadic inclusion body myositis. Neuromuscul Disord 2014, 24, 611–616. [Google Scholar] [CrossRef]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and Functions of Spatial Protein Quality Control. Annu Rev Biochem 2017, 86, 97–122. [Google Scholar] [CrossRef]
- Chen, B.; Retzlaff, M.; Roos, T.; Frydman, J. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol 2011, 3, a004374. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Li, L.; Chin, L.S. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr Med Chem 2008, 15, 47–60. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K.; Nogalska, A. Inclusion body myositis: a degenerative muscle disease associated with intra-muscle fiber multi-protein aggregates, proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation. Brain Pathol 2009, 19, 493–506. [Google Scholar] [CrossRef]
- Banwell, B.L.; Engel, A.G. AlphaB-crystallin immunolocalization yields new insights into inclusion body myositis. Neurology 2000, 54, 1033–1041. [Google Scholar] [CrossRef]
- Wojcik, S.; Engel, W.K.; McFerrin, J.; Paciello, O.; Askanas, V. AbetaPP-overexpression and proteasome inhibition increase alphaB-crystallin in cultured human muscle: relevance to inclusion-body myositis. Neuromuscul Disord 2006, 16, 839–844. [Google Scholar] [CrossRef]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The ubiquitin-proteasome system in regulation of the skeletal muscle homeostasis and atrophy: from basic science to disorders. J Physiol Sci 2020, 70, 40. [Google Scholar] [CrossRef] [PubMed]
- Fratta, P.; Engel, W.K.; McFerrin, J.; Davies, K.J.; Lin, S.W.; Askanas, V. Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-beta precursor protein-overexpressing cultured human muscle fibers. Am J Pathol 2005, 167, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Martín, B.; Castaño, J.G.; Lucas, J.J.; Moreno, D.; Olivé, M. Proteasomal expression, induction of immunoproteasome subunits, and local MHC class I presentation in myofibrillar myopathy and inclusion body myositis. J Neuropathol Exp Neurol 2004, 63, 484–498. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D.J.; White, M.; Yan, W. HDAC inhibitor modulation of proteotoxicity as a therapeutic approach in cancer. Adv Cancer Res 2012, 116, 131–163. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Nogalska, A.; D’Agostino, C.; Terracciano, C.; Engel, W.K.; Askanas, V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol 2010, 177, 1377–1387. [Google Scholar] [CrossRef]
- Kumamoto, T.; Ueyama, H.; Tsumura, H.; Toyoshima, I.; Tsuda, T. Expression of lysosome-related proteins and genes in the skeletal muscles of inclusion body myositis. Acta Neuropathol 2004, 107, 59–65. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Schmidt, J.; Schmid, D.; Barthel, K.; Wrede, A.; Dalakas, M.C.; Münz, C. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol 2007, 61, 476–483. [Google Scholar] [CrossRef]
- Nogalska, A.; Terracciano, C.; D’Agostino, C.; King Engel, W.; Askanas, V. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol 2009, 118, 407–413. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol Biol 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett 2016, 21, 29. [Google Scholar] [CrossRef]
- D’Agostino, C.; Nogalska, A.; Cacciottolo, M.; Engel, W.K.; Askanas, V. Abnormalities of NBR1, a novel autophagy-associated protein, in muscle fibers of sporadic inclusion-body myositis. Acta Neuropathol 2011, 122, 627–636. [Google Scholar] [CrossRef]
- Girolamo, F.; Lia, A.; Amati, A.; Strippoli, M.; Coppola, C.; Virgintino, D.; Roncali, L.; Toscano, A.; Serlenga, L.; Trojano, M. Overexpression of autophagic proteins in the skeletal muscle of sporadic inclusion body myositis. Neuropathol Appl Neurobiol 2013, 39, 736–749. [Google Scholar] [CrossRef]
- Cacciottolo, M.; Nogalska, A.; D’Agostino, C.; Engel, W.K.; Askanas, V. Chaperone-mediated autophagy components are upregulated in sporadic inclusion-body myositis muscle fibres. Neuropathol Appl Neurobiol 2013, 39, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology (Basel) 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Vattemi, G.; Engel, W.K.; McFerrin, J.; Askanas, V. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol 2004, 164, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nogalska, A.; D’Agostino, C.; Engel, W.K.; Cacciottolo, M.; Asada, S.; Mori, K.; Askanas, V. Activation of the Unfolded Protein Response in Sporadic Inclusion-Body Myositis but Not in Hereditary GNE Inclusion-Body Myopathy. J Neuropathol Exp Neurol 2015, 74, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Nogalska, A.; Engel, W.K.; McFerrin, J.; Kokame, K.; Komano, H.; Askanas, V. Homocysteine-induced endoplasmic reticulum protein (Herp) is up-regulated in sporadic inclusion-body myositis and in endoplasmic reticulum stress-induced cultured human muscle fibers. J Neurochem 2006, 96, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Nogalska, A.; Wojcik, S.; Engel, W.K.; McFerrin, J.; Askanas, V. Endoplasmic reticulum stress induces myostatin precursor protein and NF-kappaB in cultured human muscle fibers: relevance to inclusion body myositis. Exp Neurol 2007, 204, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, S.; Engel, W.K.; McFerrin, J.; Askanas, V. Myostatin is increased and complexes with amyloid-beta within sporadic inclusion-body myositis muscle fibers. Acta Neuropathol 2005, 110, 173–177. [Google Scholar] [CrossRef]
- Sachdev, R.; Kappes-Horn, K.; Paulsen, L.; Duernberger, Y.; Pleschka, C.; Denner, P.; Kundu, B.; Reimann, J.; Vorberg, I. Endoplasmic Reticulum Stress Induces Myostatin High Molecular Weight Aggregates and Impairs Mature Myostatin Secretion. Mol Neurobiol 2018, 55, 8355–8373. [Google Scholar] [CrossRef]
- De Paepe, B. Sporadic Inclusion Body Myositis: An Acquired Mitochondrial Disease with Extras. Biomolecules 2019, 9. [Google Scholar] [CrossRef]
- Oldfors, A.; Larsson, N.G.; Lindberg, C.; Holme, E. Mitochondrial DNA deletions in inclusion body myositis. Brain 1993, 116 Pt 2, 325–336. [Google Scholar] [CrossRef]
- Catalán-García, M.; Garrabou, G.; Morén, C.; Guitart-Mampel, M.; Hernando, A.; Díaz-Ramos, À.; González-Casacuberta, I.; Juárez, D.L.; Bañó, M.; Enrich-Bengoa, J.; et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin Sci (Lond) 2016, 130, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, Y.; Izumi, R.; Koide, M.; Hagiwara, Y.; Kanzaki, M.; Suzuki, N.; Kikuchi, K.; Matsuhashi, T.; Akiyama, Y.; Ichijo, M.; et al. Mitochondrial dysfunction underlying sporadic inclusion body myositis is ameliorated by the mitochondrial homing drug MA-5. PLoS One 2020, 15, e0231064. [Google Scholar] [CrossRef] [PubMed]
- Hedberg-Oldfors, C.; Lindgren, U.; Basu, S.; Visuttijai, K.; Lindberg, C.; Falkenberg, M.; Larsson Lekholm, E.; Oldfors, A. Mitochondrial DNA variants in inclusion body myositis characterized by deep sequencing. Brain Pathol 2021, 31, e12931. [Google Scholar] [CrossRef] [PubMed]
- Georgantas, R.W.; Streicher, K.; Greenberg, S.A.; Greenlees, L.M.; Zhu, W.; Brohawn, P.Z.; Higgs, B.W.; Czapiga, M.; Morehouse, C.A.; Amato, A.; et al. Inhibition of myogenic microRNAs 1, 133, and 206 by inflammatory cytokines links inflammation and muscle degeneration in adult inflammatory myopathies. Arthritis Rheumatol 2014, 66, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Buzkova, J.; Nikkanen, J.; Ahola, S.; Hakonen, A.H.; Sevastianova, K.; Hovinen, T.; Yki-Järvinen, H.; Pietiläinen, K.H.; Lönnqvist, T.; Velagapudi, V.; et al. Metabolomes of mitochondrial diseases and inclusion body myositis patients: treatment targets and biomarkers. EMBO Mol Med 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, K.A.; Miller, J.; Grady, J.P.; Rocha, M.C.; Taylor, R.W.; Turnbull, D.M. Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol Appl Neurobiol 2015, 41, 288–303. [Google Scholar] [CrossRef]
- Oldfors, A.; Moslemi, A.R.; Fyhr, I.M.; Holme, E.; Larsson, N.G.; Lindberg, C. Mitochondrial DNA deletions in muscle fibers in inclusion body myositis. J Neuropathol Exp Neurol 1995, 54, 581–587. [Google Scholar] [CrossRef]
- Bhatt, P.S.; Tzoulis, C.; Balafkan, N.; Miletic, H.; Tran, G.T.T.; Sanaker, P.S.; Bindoff, L.A. Mitochondrial DNA depletion in sporadic inclusion body myositis. Neuromuscul Disord 2019, 29, 242–246. [Google Scholar] [CrossRef]
- Peng, T.I.; Yu, P.R.; Chen, J.Y.; Wang, H.L.; Wu, H.Y.; Wei, Y.H.; Jou, M.J. Visualizing common deletion of mitochondrial DNA-augmented mitochondrial reactive oxygen species generation and apoptosis upon oxidative stress. Biochim Biophys Acta 2006, 1762, 241–255. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K. Newest approaches to diagnosis and pathogenesis of sporadic inclusion-body myositis and hereditary inclusion-body myopathies, including molecular-pathologic similarities to Alzheimer disease. In Inclusion-Body Myositis and Myopathies. ; Cambridge, England: Cambridge University Press: 1998; Volume 3-78.
- Sabadashka, M.; Nagalievska, M.; Sybirna, N. Tyrosine nitration as a key event of signal transduction that regulates functional state of the cell. Cell Biol Int 2021, 45, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Mattila, J.T.; Thomas, A.C. Nitric oxide synthase: non-canonical expression patterns. Front Immunol 2014, 5, 478. [Google Scholar] [CrossRef]
- Yang, C.C.; Alvarez, R.B.; Engel, W.K.; Askanas, V. Increase of nitric oxide synthases and nitrotyrosine in inclusion-body myositis. Neuroreport 1996, 8, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med 2002, 33, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, Y.; Furuta, A.; Taniguchi, N.; Yamada, T.; Kira, J.; Iwaki, T. Increased expression of manganese superoxide dismutase is associated with that of nitrotyrosine in myopathies with rimmed vacuoles. Acta Neuropathol 2002, 103, 59–65. [Google Scholar] [CrossRef]
- Askanas, V.; Sarkozi, E.; Alvares, R.B.; McFerrin, J.; Engel, W.K.; Siddique, T. Superoxide dismutase-1 gene and protein in vacuolated muscle fibers of sporadic inclusion-body myositis, hereditary inclusion-body myopathy, and cultured human muscle after B-amyloid precursor protein gene transfer. Neurology 1996, 46, 73003. [Google Scholar]
- Broccolini, A.; Engel, W.K.; Alvarez, R.B.; Askanas, V. Redox factor-1 in muscle biopsies of patients with inclusion-body myositis. Neurosci Lett 2000, 287, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Broccolini, A.; Ricci, E.; Pescatori, M.; Papacci, M.; Gliubizzi, C.; D’Amico, A.; Servidei, S.; Tonali, P.; Mirabella, M. Insulin-like growth factor I in inclusion-body myositis and human muscle cultures. J Neuropathol Exp Neurol 2004, 63, 650–659. [Google Scholar] [CrossRef]
- Huang, M.; Chen, S. DJ-1 in neurodegenerative diseases: Pathogenesis and clinical application. Prog Neurobiol 2021, 204, 102114. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, C.; Nogalska, A.; Engel, W.K.; Wojcik, S.; Askanas, V. In inclusion-body myositis muscle fibers Parkinson-associated DJ-1 is increased and oxidized. Free Radic Biol Med 2008, 45, 773–779. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K. Molecular pathology and pathogenesis of inclusion-body myositis. Microsc Res Tech 2005, 67, 114–120. [Google Scholar] [CrossRef]
- Engel AG, H.R. Inclusion body myositis. In Myology, 3rd ed.; Engel AG, F.-A.C., Ed.; McGraw-Hill: New York, 2004; pp. 1367–1388. [Google Scholar]
- Nalbantoglu, J.; Karpati, G.; Carpenter, S. Conspicuous accumulation of a single-stranded DNA binding protein in skeletal muscle fibers in inclusion body myositis. Am J Pathol 1994, 144, 874–882. [Google Scholar] [PubMed]
- Greenberg, S.A.; Pinkus, J.L.; Amato, A.A. Nuclear membrane proteins are present within rimmed vacuoles in inclusion-body myositis. Muscle Nerve 2006, 34, 406–416. [Google Scholar] [CrossRef]
- Nakano, S.; Shinde, A.; Fujita, K.; Ito, H.; Kusaka, H. Histone H1 is released from myonuclei and present in rimmed vacuoles with DNA in inclusion body myositis. Neuromuscul Disord 2008, 18, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.A. Inflammatory myopathies: disease mechanisms. Curr Opin Neurol 2009, 22, 516–523. [Google Scholar] [CrossRef]
- Hernandez Lain, A.; Millecamps, S.; Dubourg, O.; Salachas, F.; Bruneteau, G.; Lacomblez, L.; LeGuern, E.; Seilhean, D.; Duyckaerts, C.; Meininger, V.; et al. Abnormal TDP-43 and FUS proteins in muscles of sporadic IBM: similarities in a TARDBP-linked ALS patient. J Neurol Neurosurg Psychiatry 2011, 82, 1414–1416. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Toth, M.J.; Matthews, D.E.; Tracy, R.P.; Previs, M.J. Age-related differences in skeletal muscle protein synthesis: relation to markers of immune activation. Am J Physiol Endocrinol Metab 2005, 288, E883–E891. [Google Scholar] [CrossRef]
- Brack, A.S.; Conboy, M.J.; Roy, S.; Lee, M.; Kuo, C.J.; Keller, C.; Rando, T.A. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007, 317, 807–810. [Google Scholar] [CrossRef]
- Fernando, R.; Drescher, C.; Nowotny, K.; Grune, T.; Castro, J.P. Impaired proteostasis during skeletal muscle aging. Free Radic Biol Med 2019, 132, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-Khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J Physiol 2018, 596, 3567–3584. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A 2005, 102, 5618–5623. [Google Scholar] [CrossRef]
- Malatesta, M.; Perdoni, F.; Muller, S.; Zancanaro, C.; Pellicciari, C. Nuclei of aged myofibres undergo structural and functional changes suggesting impairment in RNA processing. Eur J Histochem 2009, 53, e12. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.R.; Hsia, R.C.; Folker, E.S.; Lovering, R.M. Age-dependent changes in nuclear-cytoplasmic signaling in skeletal muscle. Exp Gerontol 2021, 150, 111338. [Google Scholar] [CrossRef] [PubMed]
- Day, K.; Shefer, G.; Shearer, A.; Yablonka-Reuveni, Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev Biol 2010, 340, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; McArdle, A. Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J Physiol 2011, 589, 2139–2145. [Google Scholar] [CrossRef]
- Szczesny, B.; Tann, A.W.; Mitra, S. Age- and tissue-specific changes in mitochondrial and nuclear DNA base excision repair activity in mice: Susceptibility of skeletal muscles to oxidative injury. Mech Ageing Dev 2010, 131, 330–337. [Google Scholar] [CrossRef]
- Perandini, L.A.; Chimin, P.; Lutkemeyer, D.D.S.; Câmara, N.O.S. Chronic inflammation in skeletal muscle impairs satellite cells function during regeneration: can physical exercise restore the satellite cell niche? FEBS J 2018, 285, 1973–1984. [Google Scholar] [CrossRef]
- Walston, J.D. Sarcopenia in older adults. Curr Opin Rheumatol 2012, 24, 623–627. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu Rev Biochem 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Pathak, R.U.; Soujanya, M.; Mishra, R.K. Deterioration of nuclear morphology and architecture: A hallmark of senescence and aging. Ageing Res Rev 2021, 67, 101264. [Google Scholar] [CrossRef] [PubMed]
- Conboy, I.M.; Rando, T.A. Aging, stem cells and tissue regeneration: lessons from muscle. Cell Cycle 2005, 4, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Hollemann, D.; Budka, H.; Löscher, W.N.; Yanagida, G.; Fischer, M.B.; Wanschitz, J.V. Endothelial and myogenic differentiation of hematopoietic progenitor cells in inflammatory myopathies. J Neuropathol Exp Neurol 2008, 67, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Wanschitz, J.V.; Dubourg, O.; Lacene, E.; Fischer, M.B.; Höftberger, R.; Budka, H.; Romero, N.B.; Eymard, B.; Herson, S.; Butler-Browne, G.S.; et al. Expression of myogenic regulatory factors and myo-endothelial remodeling in sporadic inclusion body myositis. Neuromuscul Disord 2013, 23, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev 2008, 129, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Lax, N.Z.; Turnbull, D.M.; Reeve, A.K. Mitochondrial mutations: newly discovered players in neuronal degeneration. Neuroscientist 2011, 17, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Katayama, M.; Tanaka, M.; Yamamoto, H.; Ohbayashi, T.; Nimura, Y.; Ozawa, T. Deleted mitochondrial DNA in the skeletal muscle of aged individuals. Biochem Int 1991, 25, 47–56. [Google Scholar]
- Chung, S.S.; Weindruch, R.; Schwarze, S.R.; McKenzie, D.I.; Aiken, J.M. Multiple age-associated mitochondrial DNA deletions in skeletal muscle of mice. Aging (Milano) 1994, 6, 193–200. [Google Scholar] [CrossRef]
- Kadenbach, B.; Münscher, C.; Frank, V.; Müller-Höcker, J.; Napiwotzki, J. Human aging is associated with stochastic somatic mutations of mitochondrial DNA. Mutat Res 1995, 338, 161–172. [Google Scholar] [CrossRef]
- Peterson, C.M.; Johannsen, D.L.; Ravussin, E. Skeletal muscle mitochondria and aging: a review. J Aging Res 2012, 2012, 194821. [Google Scholar] [CrossRef]
- Wei, Y.H.; Lu, C.Y.; Lee, H.C.; Pang, C.Y.; Ma, Y.S. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann N Y Acad Sci 1998, 854, 155–170. [Google Scholar] [CrossRef]
- Engel, A.G.; Arahata, K. Monoclonal antibody analysis of mononuclear cells in myopathies. II: Phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann Neurol 1984, 16, 209–215. [Google Scholar] [CrossRef]
- Salajegheh, M.; Rakocevic, G.; Raju, R.; Shatunov, A.; Goldfarb, L.G.; Dalakas, M.C. T cell receptor profiling in muscle and blood lymphocytes in sporadic inclusion body myositis. Neurology 2007, 69, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, M.; Wiesener, S.; Babbe, H.; Roers, A.; Wekerle, H.; Dornmair, K.; Hohlfeld, R.; Goebels, N. Clonal tracking of autoaggressive T cells in polymyositis by combining laser microdissection, single-cell PCR, and CDR3-spectratype analysis. Proc Natl Acad Sci U S A 2003, 100, 4090–4095. [Google Scholar] [CrossRef] [PubMed]
- Müntzing, K.; Lindberg, C.; Moslemi, A.R.; Oldfors, A. Inclusion body myositis: clonal expansions of muscle-infiltrating T cells persist over time. Scand J Immunol 2003, 58, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Fyhr, I.M.; Moslemi, A.R.; Tarkowski, A.; Lindberg, C.; Oldfors, A. Limited T-cell receptor V gene usage in inclusion body myositis. Scand J Immunol 1996, 43, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Fyhr, I.M.; Moslemi, A.R.; Lindberg, C.; Oldfors, A. T cell receptor beta-chain repertoire in inclusion body myositis. J Neuroimmunol 1998, 91, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, C.; Oldfors, A.; Tarkowski, A. Restricted use of T cell receptor V genes in endomysial infiltrates of patients with inflammatory myopathies. Eur J Immunol 1994, 24, 2659–2663. [Google Scholar] [CrossRef]
- O’Hanlon, T.P.; Dalakas, M.C.; Plotz, P.H.; Miller, F.W. The alpha beta T-cell receptor repertoire in inclusion body myositis: diverse patterns of gene expression by muscle-infiltrating lymphocytes. J Autoimmun 1994, 7, 321–333. [Google Scholar] [CrossRef]
- Fyhr, I.M.; Moslemi, A.R.; Mosavi, A.A.; Lindberg, C.; Tarkowski, A.; Oldfors, A. Oligoclonal expansion of muscle infiltrating T cells in inclusion body myositis. J Neuroimmunol 1997, 79, 185–189. [Google Scholar] [CrossRef]
- Bender, A.; Behrens, L.; Engel, A.G.; Hohlfeld, R. T-cell heterogeneity in muscle lesions of inclusion body myositis. J Neuroimmunol 1998, 84, 86–91. [Google Scholar] [CrossRef]
- Amemiya, K.; Granger, R.P.; Dalakas, M.C. Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time. Studies in repeated muscle biopsies. Brain 2000, 123 Pt 10, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Dalakas, M.C. Expression of the costimulatory molecule BB-1, the ligands CTLA-4 and CD28, and their mRNA in inflammatory myopathies. Am J Pathol 1999, 155, 453–460. [Google Scholar] [CrossRef]
- Behrens, L.; Kerschensteiner, M.; Misgeld, T.; Goebels, N.; Wekerle, H.; Hohlfeld, R. Human muscle cells express a functional costimulatory molecule distinct from B7.1 (CD80) and B7.2 (CD86) in vitro and in inflammatory lesions. J Immunol 1998, 161, 5943–5951. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Rakocevic, G.; Raju, R.; Dalakas, M.C. Upregulated inducible co-stimulator (ICOS) and ICOS-ligand in inclusion body myositis muscle: significance for CD8+ T cell cytotoxicity. Brain 2004, 127, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Waschbisch, A.; Wintterle, S.; Lochmüller, H.; Walter, M.C.; Wischhusen, J.; Kieseier, B.C.; Wiendl, H. Human muscle cells express the costimulatory molecule B7-H3, which modulates muscle-immune interactions. Arthritis Rheum 2008, 58, 3600–3608. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.M.; Fasth, A.E.; Zong, M.; Arnardottir, S.; Dani, L.; Lindroos, E.; Malmström, V.; Lundberg, I.E. Expanded T cell receptor Vβ-restricted T cells from patients with sporadic inclusion body myositis are proinflammatory and cytotoxic CD28null T cells. Arthritis Rheum 2010, 62, 3457–3466. [Google Scholar] [CrossRef]
- Lindberg, C.; Oldfors, A.; Tarkowski, A. Local T-cell proliferation and differentiation in inflammatory myopathies. Scand J Immunol 1995, 41, 421–426. [Google Scholar] [CrossRef]
- Allenbach, Y.; Chaara, W.; Rosenzwajg, M.; Six, A.; Prevel, N.; Mingozzi, F.; Wanschitz, J.; Musset, L.; Charuel, J.L.; Eymard, B.; et al. Th1 response and systemic treg deficiency in inclusion body myositis. PLoS One 2014, 9, e88788. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Pinkus, J.L.; Kong, S.W.; Baecher-Allan, C.; Amato, A.A.; Dorfman, D.M. Highly differentiated cytotoxic T cells in inclusion body myositis. Brain 2019, 142, 2590–2604. [Google Scholar] [CrossRef]
- Ikezoe, K.; Ohshima, S.; Osoegawa, M.; Tanaka, M.; Ogawa, K.; Nagata, K.; Kira, J.I. Expression of granulysin in polymyositis and inclusion-body myositis. J Neurol Neurosurg Psychiatry 2006, 77, 1187–1190. [Google Scholar] [CrossRef]
- Orimo, S.; Koga, R.; Goto, K.; Nakamura, K.; Arai, M.; Tamaki, M.; Sugita, H.; Nonaka, I.; Arahata, K. Immunohistochemical analysis of perforin and granzyme A in inflammatory myopathies. Neuromuscul Disord 1994, 4, 219–226. [Google Scholar] [CrossRef]
- Goebels, N.; Michaelis, D.; Engelhardt, M.; Huber, S.; Bender, A.; Pongratz, D.; Johnson, M.A.; Wekerle, H.; Tschopp, J.; Jenne, D.; et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest 1996, 97, 2905–2910. [Google Scholar] [CrossRef] [PubMed]
- Strioga, M.; Pasukoniene, V.; Characiejus, D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011, 134, 17–32. [Google Scholar] [CrossRef]
- Dzangué-Tchoupou, G.; Mariampillai, K.; Bolko, L.; Amelin, D.; Mauhin, W.; Corneau, A.; Blanc, C.; Allenbach, Y.; Benveniste, O. CD8+(T-bet+) cells as a predominant biomarker for inclusion body myositis. Autoimmun Rev 2019, 18, 325–333. [Google Scholar] [CrossRef]
- Gao, Z.; Feng, Y.; Xu, J.; Liang, J. T-cell exhaustion in immune-mediated inflammatory diseases: New implications for immunotherapy. Front Immunol 2022, 13, 977394. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Knauss, S.; Preusse, C.; Allenbach, Y.; Leonard-Louis, S.; Touat, M.; Fischer, N.; Radbruch, H.; Mothes, R.; Matyash, V.; Böhmerle, W.; et al. PD1 pathway in immune-mediated myopathies: Pathogenesis of dysfunctional T cells revisited. Neurol Neuroimmunol Neuroinflamm 2019, 6, e558. [Google Scholar] [CrossRef] [PubMed]
- Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Valentini, N.; Requejo Cier, C.J.; Lamarche, C. Regulatory T-cell dysfunction and its implication for cell therapy. Clin Exp Immunol 2023, 213, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef]
- Schiaffino, S.; Pereira, M.G.; Ciciliot, S.; Rovere-Querini, P. Regulatory T cells and skeletal muscle regeneration. FEBS J 2017, 284, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.A.; Bradshaw, E.M.; Pinkus, J.L.; Pinkus, G.S.; Burleson, T.; Due, B.; Bregoli, L.; O’Connor, K.C.; Amato, A.A. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology 2005, 65, 1782–1787. [Google Scholar] [CrossRef]
- Bradshaw, E.M.; Orihuela, A.; McArdel, S.L.; Salajegheh, M.; Amato, A.A.; Hafler, D.A.; Greenberg, S.A.; O’Connor, K.C. A local antigen-driven humoral response is present in the inflammatory myopathies. J Immunol 2007, 178, 547–556. [Google Scholar] [CrossRef]
- Salajegheh, M.; Pinkus, J.L.; Amato, A.A.; Morehouse, C.; Jallal, B.; Yao, Y.; Greenberg, S.A. Permissive environment for B-cell maturation in myositis muscle in the absence of B-cell follicles. Muscle Nerve 2010, 42, 576–583. [Google Scholar] [CrossRef]
- Krystufková, O.; Vallerskog, T.; Helmers, S.B.; Mann, H.; Putová, I.; Belácek, J.; Malmström, V.; Trollmo, C.; Vencovsky, J.; Lundberg, I.E. Increased serum levels of B cell activating factor (BAFF) in subsets of patients with idiopathic inflammatory myopathies. Ann Rheum Dis 2009, 68, 836–843. [Google Scholar] [CrossRef]
- Benveniste, O.; Stenzel, W.; Hilton-Jones, D.; Sandri, M.; Boyer, O.; van Engelen, B.G. Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the degenerative chicken. Acta Neuropathol 2015, 129, 611–624. [Google Scholar] [CrossRef]
- Roos, A.; Preusse, C.; Hathazi, D.; Goebel, H.H.; Stenzel, W. Proteomic Profiling Unravels a Key Role of Specific Macrophage Subtypes in Sporadic Inclusion Body Myositis. Front Immunol 2019, 10, 1040. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Pinkus, G.S.; Amato, A.A.; Pinkus, J.L. Myeloid dendritic cells in inclusion-body myositis and polymyositis. Muscle Nerve 2007, 35, 17–23. [Google Scholar] [CrossRef] [PubMed]
- de Padilla, C.M.; Reed, A.M. Dendritic cells and the immunopathogenesis of idiopathic inflammatory myopathies. Curr Opin Rheumatol 2008, 20, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Blumbergs, P.C.; Manavis, J.; Limaye, V.S. Major histocompatibility complex class I and II expression in idiopathic inflammatory myopathy. Appl Immunohistochem Mol Morphol 2013, 21, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Englund, P.; Lindroos, E.; Nennesmo, I.; Klareskog, L.; Lundberg, I.E. Skeletal muscle fibers express major histocompatibility complex class II antigens independently of inflammatory infiltrates in inflammatory myopathies. Am J Pathol 2001, 159, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Bartoccioni, E.; Gallucci, S.; Scuderi, F.; Ricci, E.; Servidei, S.; Broccolini, A.; Tonali, P. MHC class I, MHC class II and intercellular adhesion molecule-1 (ICAM-1) expression in inflammatory myopathies. Clin Exp Immunol 1994, 95, 166–172. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Spits, H. Development of alphabeta T cells in the human thymus. Nat Rev Immunol 2002, 2, 760–772. [Google Scholar] [CrossRef]
- Fagnoni, F.F.; Vescovini, R.; Passeri, G.; Bologna, G.; Pedrazzoni, M.; Lavagetto, G.; Casti, A.; Franceschi, C.; Passeri, M.; Sansoni, P. Shortage of circulating naive CD8(+) T cells provides new insights on immunodeficiency in aging. Blood 2000, 95, 2860–2868. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Mechanisms underlying T cell ageing. Nat Rev Immunol 2019, 19, 573–583. [Google Scholar] [CrossRef]
- Decman, V.; Laidlaw, B.J.; Dimenna, L.J.; Abdulla, S.; Mozdzanowska, K.; Erikson, J.; Ertl, H.C.; Wherry, E.J. Cell-intrinsic defects in the proliferative response of antiviral memory CD8 T cells in aged mice upon secondary infection. J Immunol 2010, 184, 5151–5159. [Google Scholar] [CrossRef]
- Saule, P.; Trauet, J.; Dutriez, V.; Lekeux, V.; Dessaint, J.P.; Labalette, M. Accumulation of memory T cells from childhood to old age: central and effector memory cells in CD4(+) versus effector memory and terminally differentiated memory cells in CD8(+) compartment. Mech Ageing Dev 2006, 127, 274–281. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Lee, W.W.; Weyand, C.M. Aging and T-cell diversity. Exp Gerontol 2007, 42, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Briceño, O.; Lissina, A.; Wanke, K.; Afonso, G.; von Braun, A.; Ragon, K.; Miquel, T.; Gostick, E.; Papagno, L.; Stiasny, K.; et al. Reduced naïve CD8(+) T-cell priming efficacy in elderly adults. Aging Cell 2016, 15, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heredero, G.; Gómez de Las Heras, M.M.; Escrig-Larena, J.I.; Mittelbrunn, M. Extremely Differentiated T Cell Subsets Contribute to Tissue Deterioration During Aging. Annu Rev Immunol 2023, 41, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ren, B.; Wang, D.; Lin, H. Regulatory T cells in skeletal muscle repair and regeneration: recent insights. Cell Death Dis 2022, 13, 680. [Google Scholar] [CrossRef] [PubMed]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Salam, N.; Rane, S.; Das, R.; Faulkner, M.; Gund, R.; Kandpal, U.; Lewis, V.; Mattoo, H.; Prabhu, S.; Ranganathan, V.; et al. T cell ageing: effects of age on development, survival & function. Indian J Med Res 2013, 138, 595–608. [Google Scholar] [PubMed]
- Cui, C.Y.; Driscoll, R.K.; Piao, Y.; Chia, C.W.; Gorospe, M.; Ferrucci, L. Skewed macrophage polarization in aging skeletal muscle. Aging Cell 2019, 18, e13032. [Google Scholar] [CrossRef] [PubMed]
- McCord, B.; Day, R.M. Cytotoxic immune cells do not affect TDP-43 and p62 sarcoplasmic aggregation but influence TDP-43 localisation. Sci Rep 2023, 13, 15935. [Google Scholar] [CrossRef]
- Schmidt, J.; Barthel, K.; Wrede, A.; Salajegheh, M.; Bähr, M.; Dalakas, M.C. Interrelation of inflammation and APP in sIBM: IL-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain 2008, 131, 1228–1240. [Google Scholar] [CrossRef] [PubMed]
- Adams, V.; Nehrhoff, B.; Späte, U.; Linke, A.; Schulze, P.C.; Baur, A.; Gielen, S.; Hambrecht, R.; Schuler, G. Induction of iNOS expression in skeletal muscle by IL-1beta and NFkappaB activation: an in vitro and in vivo study. Cardiovasc Res 2002, 54, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; Brown, T.; Becker, L.; Prager, M.; Giroir, B.P. Cytokine-induced expression of nitric oxide synthase in C2C12 skeletal muscle myocytes. Am J Physiol 1994, 267, R1020–R1025. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Trinh, D.N.; LaFerla, F.M. Inflammation induces tau pathology in inclusion body myositis model via glycogen synthase kinase-3beta. Ann Neurol 2008, 64, 15–24. [Google Scholar] [CrossRef]
- Britson, K.A.; Ling, J.P.; Braunstein, K.E.; Montagne, J.M.; Kastenschmidt, J.M.; Wilson, A.; Ikenaga, C.; Tsao, W.; Pinal-Fernandez, I.; Russell, K.A.; et al. Loss of TDP-43 function and rimmed vacuoles persist after T cell depletion in a xenograft model of sporadic inclusion body myositis. Sci Transl Med 2022, 14, eabi9196. [Google Scholar] [CrossRef]
Figure 1.
Histopathological features of sporadic inclusion body myositis (sIBM) muscle biopsy (A, B and C, hematoxylin and eosin; D, cytochrome c oxidase (COX)-succinate dehydrogenase): endomysial inflammatory infiltrate that invade nonnecrotic muscle fibers (B), rimmed vacuoles within muscle fibers (asterisks, A and C), eosinophilic cytoplasmic inclusion (arrow, A), ragged red fiber and COX-negative fibers (D), and angulated muscle fibers (A and C).
Figure 1.
Histopathological features of sporadic inclusion body myositis (sIBM) muscle biopsy (A, B and C, hematoxylin and eosin; D, cytochrome c oxidase (COX)-succinate dehydrogenase): endomysial inflammatory infiltrate that invade nonnecrotic muscle fibers (B), rimmed vacuoles within muscle fibers (asterisks, A and C), eosinophilic cytoplasmic inclusion (arrow, A), ragged red fiber and COX-negative fibers (D), and angulated muscle fibers (A and C).
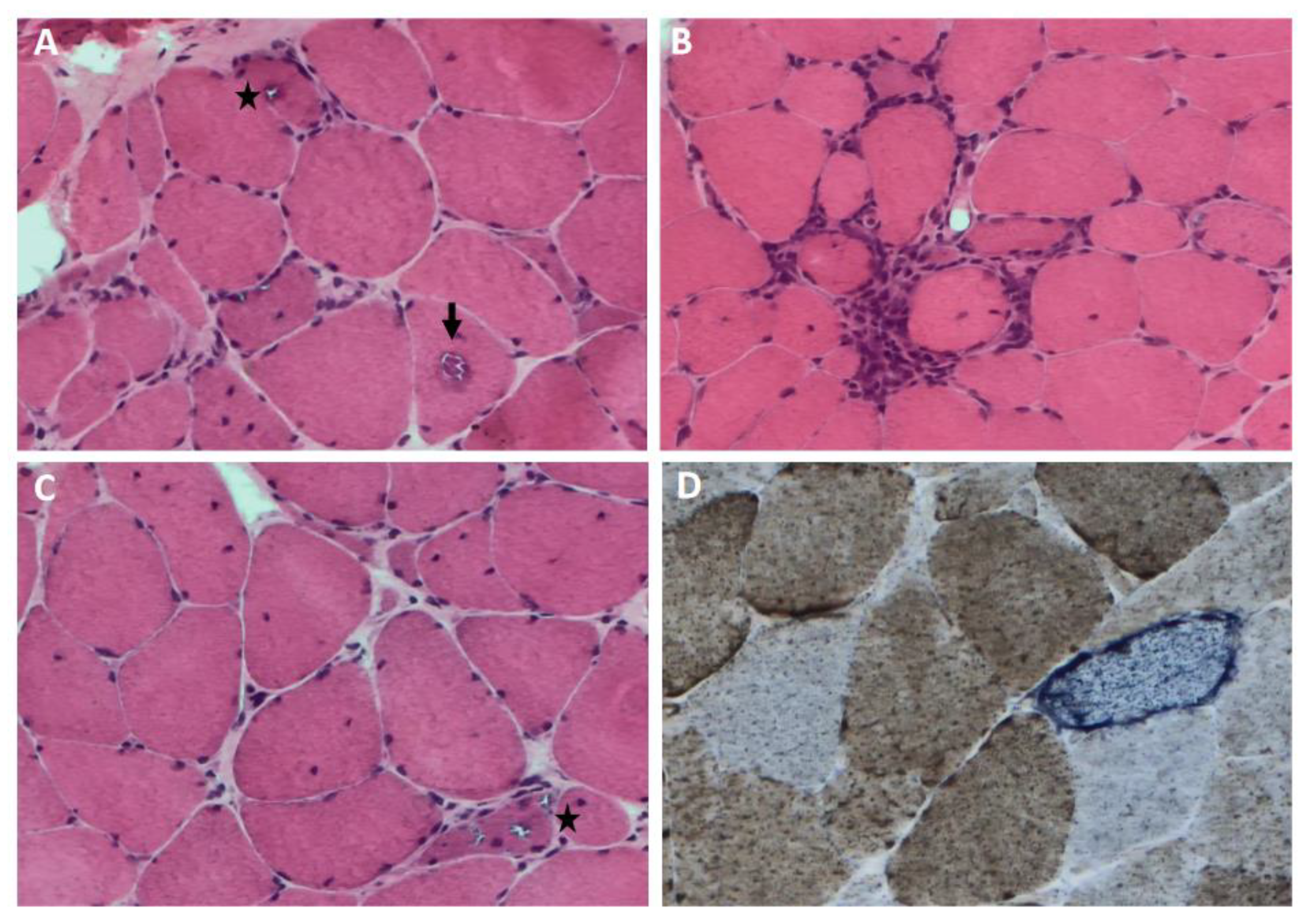
Figure 2.
Schematic representation of the main molecular mechanisms contributing to the pathogenesis of sporadic inclusion body myositis (sIBM).
Figure 2.
Schematic representation of the main molecular mechanisms contributing to the pathogenesis of sporadic inclusion body myositis (sIBM).
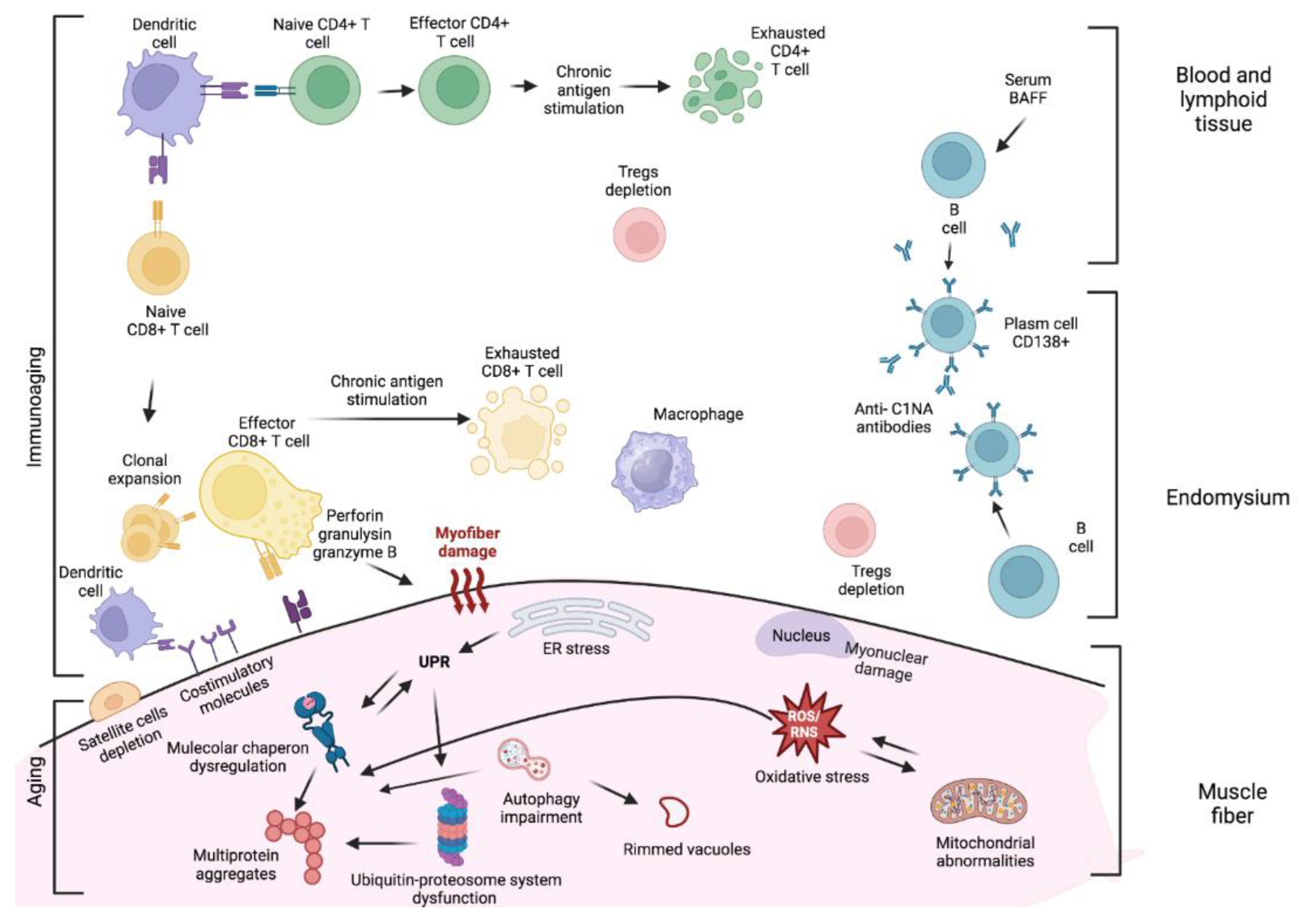
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



