
Submitted:
27 December 2023
Posted:
27 December 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
A change in biological properties of the SARS-CoV-2 virus can enhance its transmissibility and virulence, complicate the disease course, and reduce the effectiveness of anti-viral therapy and vaccination. We have studied 16 genetic lineages of the Omicron variant. The viral reproduction course was analyzed based on plaque morphology in the agar layer and the maximum titer in Vero E6 cells. BALB/c mice and Syrian hamsters were used as animal models. Viral load and infectious SARS-CoV-2 titer were determined in the nasal tissue and lungs. The severity of infectious process was assessed using histological methods. An increased plug size was observed in CH.1.1 and BQ.1.2.1 strains after six passages. This was due to substitutions D614G, H655Y, and N764K in the coronavirus S protein. The same changes in gene variants Gamma and BA.1 were caused by the substitution R682W, which is not present in lineages CH.1.1 and BQ.1.2.1. A similar substitution, namely R682P, was observed in the BA.5.1 strain. We characterized BA.1 (as a reference), BA.5.2, and XBB.3 strains in vivo as representatives of the main circulating genetic lineages. For the BA.1 strain, ID50 was 1.3 and 14 TCD50 in Syrian hamsters and BALB/c mice, respectively. An insignificant increase in the BA.5.2 virulence compared to that of BA.1 was noted (ID50 was 07. and 10 TCD50, respectively). The XBB.3 pathogenicity in laboratory animals is similar to that of BA.1 (ID50 was 1.8 and 15 TCD50, respectively). Experiments showed a consistent decrease in the virulence of Omicron strains compared to previously circulating SARS-CoV-2 variants. A co-evolutionary change in amino acid sequences, affecting the conformation of the coronavirus S protein and its surface epitopes in representatives of different genetic lineages, was noted. The emergence of potential proteolysis sites and alterations of the furin cleavage site have been observed in numerous Omicron coronavirus variants.
Keywords:
COVID-19
; SARS-CoV-2 virus
; Omicron variant
; coronavirus evolution
; Vero E6 cells
; BALB/c mice
; Syrian hamster
1. Introduction
From the moment of the first encounter with the causative agent of Severe Acute Respiratory Syndrome (SARS) in December 2019, the World Health Organization (WHO) has been conducting molecular and biological monitoring of newly emerging coronavirus variants. It enables the identification of priority areas for studying the new coronavirus as well as the development and refinement of anti-epidemiological measures against COVID-19 (https://www.who.int/activities/tracking-SARS-CoV-2-variants, accessed on 10 September 2023).
Gene variants carrying key mutations that affect the biological properties of the coronavirus, such as increased ability to transmit infection, virulence, changes in disease course, decreased effectiveness of anti-viral therapy and vaccination, and sensitivity of diagnostic tests, have been designated by the WHO as variants of concern (VOCs). Greek alphabet letters were proposed as easy-to-pronounce and easy-to-memorize variant names (https://www.who.int/news/item/31-05-2021-who-announces-simple-easy-to-say-labels-for-sars-cov-2-variants-of-interest-and-concern, accessed on 10 September 2023).
Dynamic monitoring of mutational variability in coronaviruses identified in the territory of the Russian Federation has been conducted by institutions of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing (Rospotrebnadzor) since March 2020 [1]. During this period, five COVID-19 infection waves caused by the Alpha, Beta, Gamma, Delta, and Omicron VOCs were reported.
The first Omicron variant was discovered in the country in December 2021 and remained the dominant variant since January 2022 [2].
To date, the Omicron variant has completely displaced all other variants, spreading and continuously evolving into several sublineages, some of which have become dominant throughout the world. A change in biological properties is most often associated with amino acid substitutions in the S protein, which serves as a major selection target.
The first discovered genetic lineage of the Omicron variant, namely BA.1, rapidly displaced the Delta variant due to its high transmissibility and ability to evade the immune response [3].
BA.2 shares a similar structure of the receptor-binding domain (RBD) with BA.1; however, each of them also has unique substitutions. BA.2 has eight substitutions, while BA.1 has 13 substitutions [4]. The rapid spread of the BA.2 lineage is also due to an increase in its virulence, accompanied by a simultaneous decrease in pathogenicity [5].
The S proteins in BA.4 and BA.5, which were discovered almost simultaneously, are highly homologous to those in BA.2. In addition to the same mutations as in BA.2, lineages A.4 and BA.5 contain mutations 69-70del, L452R, and F486V, as well as a reverse substitution characteristic of the wild-type virus (Q493). BA.4 and BA.5 exhibit similar mutational patterns in the 5’-terminal genomic region (from ORF1ab to the E gene) but diverge in the 3’ region (from the M gene to the 3'-terminus). It has been suggested that BA.4 and BA.5 may have diverged because of recombination, and a possible location of the breakpoint between genes E and M has been proposed [6].
BA.5.1 differs from BA.2 in the presence of the amino acid substitution L452R, which stabilizes the S1 subunit and enhances its affinity for the ACE2 receptor. However, the R682P substitution in the furin cleavage site reduces virulence.
The BA.5 variant became dominant worldwide at the beginning of 2022 and is still present in one of its sublineages, namely B.1.5.2. The emergence of the latter is associated with restoration of the furin site through the P682R substitution [4].
The BQ.1.1 (Cerberus) lineage is one of the direct derivatives of BA.5. BQ.1.1 strains have substitutions in the S protein similar to those in BA.5, but they also carry additional substitutions: R346T, K444T, N460K, and Q493R. These substitutions contribute to an increased affinity for ACE2 and the virus ability to evade the immune response. Moreover, some mutations, such as L452R and F486V, provide a two-fold increase in infectivity [7].
The CL.1 lineage is one of the early BA.5.1 sublineages. It is believed to have originated in Russia. CL.1 is distinguished by mutations K150E, K444N, and N460K in the S protein. To date, this sublineage is practically absent (https://github.com/cov-lineages/pango-designation/issues/1187, accessed on 10 September 2023).
CK.1 is a descendant of the BA.5.2 lineage. It is distinguished by the unique substitution N460K. Its further evolution led to the emergence of sublineages with additional alterations in the S protein conformation (F157L, G181V, S255F, R346T, N460K, and E654K). These changes allowed for a greater antibody evasion, facilitated cleavage of the furin site, and increased the transmissibility of the virus. Despite this, the lineage has not received significant distribution (https://github.com/cov-lineages/pango-designation/issues/1467, accessed on 10 September 2023).
However, in addition to BA.5, BA.2 descendants are actively spreading and evolving. Among them, the most well-studied sublineage is BA.2.75 (Centaurus); it differs from the ancestral lineage in having four substitutions in the RBD: D339H, G446S, N460K, and R493Q. The affinity of RBDs in BA.2.75 coronaviruses for hACE2 was shown to be 4–6 times higher than that of other Omicron variants. It has the highest binding activity among the circulating SARS-CoV-2 strains detected to date. This could possibly explain why BA.2.75 and its descendants have a distribution advantage over BA.4/BA.5 [8].
BN.1 (Hydra) is a descendant of BA.2.75; it has a high potential for evading the immune response after infection and vaccination due to a large number of substitutions in ORF1a and F490S in the S protein. However, the restoration of deletions H69del and V70del leads to reduced transmissibility. In addition, it is believed that it will continue to evolve and spread, dominating over other circulating lineages [9].
CH.1 (Orthrus) is a descendant of BA.2.75. It has a reverse substitution D1199N in the S protein trimerization site. This substitution results in a greater evasion of the immune response, although to a lesser extent than that of BQ.1.1 strains [10].
Recombination is the primary driver of RNA virus evolution. As previously mentioned, several recombinant SARS-CoV-2 variants have been documented during the COVID-19 pandemic. XBB.1 (Gryphon) is the most studied lineage; it has derived from two A.2 sublineages, namely BJ.1 (Argus) and BM.1.1.1 (Mimas) [11]. The genetic variability and ability to distribute in the human population of this recombinant lineage are slightly higher than those of the ancestral lineages. However, due to the presence of reverse substitutions G614D, Y655H, and K764N, which decrease the affinity of the RBD for the human ACE2 receptor compared to BA.2 lineage coronaviruses, XBB.1 strains show neither danger signs nor a high capacity for demographic expansion [12].
XBB.1.5 (Kraken) carries the same point mutations in the S protein as its predecessor XBB.1 (K417N, S477N, N501Y, and P681H). The only exception is a rather rare substitution, F486P, which increases the affinity of the coronavirus S protein for the human ACE2 receptor. This substitution provides an advantage in reproduction and, consequently, increases the transmissibility [13].
XBB.1.9 (Hyperion) is currently the only lineage that exhibits a significant growth advantage compared to XBB.1.5. However, the data available for XBB.1.9 do not indicate any differences in disease severity and/or clinical manifestations compared to the original Omicron variant [14]. It carries mutations R408S and F486P in the S protein as well as mutation G8* in ORF8. The viral protein encoded by the SARS-CoV-2 open reading frame 8 (ORF8) directly interacts with MHC-I (major histocompatibility complex class I) and suppresses its function. SARS-CoV-2-infected cells are much less susceptible to lysis by cytotoxic T cells. Mutations affecting the ORF8 protein contribute to a milder disease course, despite an increase in viral production [15].
XBB.3 was detected in the Russian Federation in October 2022. This lineage is distinguished by an absence of substitutions G142D, K417N, and L452R, which enhance reproduction and spread. However, unlike the previous XBB lineages, it carries substitutions D614G, H655Y, and N764K, which enhance the infectivity (https://github.com/cov-lineages/pango-designation/issues/1201, accessed on 10 September 2023).
We studied the biological properties of new coronavirus genetic lineages in order to assess the effect of resulting amino acid substitutions on their epidemic potential and confirm the mathematical modeling data on the course of the infectious process.
An in vitro study of the SARS-CoV-2 coronavirus was conducted using African green monkey kidney Vero E6 cells, which are characterized by a high level of ACE2 expression [16]. The reproductive course was assessed based on two parameters: the maximum infectious virus titer in the culture media and the plaque size, which is an indirect indicator of the rate of coronavirus spread between neighboring cells in a cell monolayer [17].
In vivo studies determined the virulence of the main genetic lineages for Syrian hamsters, which were showed to be the most susceptible animals [18]. In addition, BALB/c mice were used in the experiments as the model for selective susceptibility [19].
The aim of the study is to investigate the biological characteristics of the strains belonging to the Omicron variant lineages of the SARS-CoV-2 coronavirus that have been identified in the territory of the Russian Federation.
2. Materials and methods
2.1. Viruses and cell cultures.
Strains isolated from COVID-19 patients in the Russian Federation and belonging to genetic lineages of the SARS-CoV-2 Omicron variant were used in the study. Vero E6 cells were obtained from the collection of the State Research Center of Virology and Biochemistry “Vector” (Koltsovo, Novosibirsk region, Russia) and cultured in the DMEM medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, HyClone, Logan, UT, USA) and antibiotic-antimycotic (Gibco, Grand Island, NY, USA) in a 5% CO2 atmosphere at 37ºC. DMEM supplemented with 2% fetal bovine serum was used as the maintenance medium for virus propagation. SARS-CoV-2 virus stocks were stored at −70°C.
2.2. Animals
Inbred Mus musculus strain BALB/c mice (both sexes, aged 8 weeks old, weighing 18–22 g) and outbred Mesocricetus auratus Syrian hamsters (both sexes, aged 6 weeks old, weighing 80–110 g) were used in experiments. Animals were procured from the Laboratory Animal Nursery of the State Research Center of Virology and Biotechnology “Vector” (Koltsovo, Novosibirsk region, Russia). The authors confirm compliance with national and international guidelines for the humane care and ethical use of animals in accordance with the Consensus Author Guidelines for Animal Use (IAVES, 23 July, 2010). All in vivo experimental procedures were reviewed and approved by the bioethics committee of the State Research Center of Virology and Biotechnology “Vector” (application of the State Research Center for Virology and Biotechnology “Vector”/06-06.2021, approved by the protocol No. 3 of 06/15/2021).
2.3. Biosafety
All experiments involving infectious virus samples were conducted in a biosafety level 3 laboratory with all applicable national certifications and approvals required to study SARS-CoV-2.
2.4. Measurement of virus infectious titer
Cells were cultured in a 96-well culture plate. After removing the growth medium from the plate wells, sequential tenfold dilutions of the virus-containing solution in maintenance medium were added in triplicates. Plates were incubated for four days in a 5% CO2 atmosphere at 37°C and then stained with crystal violet. Specific damage to the cell culture monolayer in the well was considered a cytopathic effect (CPE). Virus titer was calculated using the Reed–Meunch method and expressed as lg TCD50/ml [20]. To determine the maximum infectious titer 96 hrs post-infection, cultural medium samples from each well were titrated in Vero E6 monolayer cells as described above.
2.5. Study of plaque morphology in the agar layer
The experiment was conducted on day 3 in 24-well culture plates with a multiplicity of infection (MOI) of 0.001 TCD50/cell for each coronavirus strain using the standard technique [21].
Vero E6 cells were visualized using an Epson Perfection V700 Photo flatbed scanner (Seiko Epson, Suwa, Nagano, Japan). The image was analyzed, and plaque size was calculated using an open-source and free image-editing software GIMP (GNU Image Manipulation Program, the GIMP team, GIMP 2.10, https://www.gimp.org).
2.6. Virulence (ID50) assessment for SARS-CoV-2 genetic variants
Animals were pre-anesthetized with intramuscular anesthesia using Zoletil 100 (Virbac, Carros, France) and infected intranasally by inoculation of 10-fold serial dilutions of coronavirus. Nasal cavity and lung tissues were collected from BALB/c mice and Syrian hamsters 72 and 120 hrs post-infection, respectively. Tissue homogenates (10%) were obtained using a FastPrep-24 mechanical homogenizer (MP Biomedicals, Santa Ana, USA) and clarified by centrifugation at 6,000 rpm (Eppendorf MiniSpin plus, Germany). Aliquots of clarified samples were used for PCR analysis and determination of infectious virus concentration (CPE50/ml) by titration in Vero E6 cells. Homogenates were then immediately frozen and stored at −70°C.
2.7. RNA isolation and quantitative PCR
SARS-CoV-2 viral load in tissue homogenates was estimated using quantitative PCR with reverse transcription (RT-PCR). Viral RNA was extracted from samples and clarified using the RIBO-prep Kit (AmpliSens, Moscow, Russia). The Reverta-L Kit (AmpliSens, Moscow, Russia) was used to generate cDNA from a purified RNA template. The Vector-PCRrv-2019-nCoV-RG Kit (State Research Center for Virology and Biotechnology “Vector”, Koltsovo, Novosibirsk Region, Russia) was used to quantify SARS-CoV-2. Amplification was carried out using a Rotor-Gene 6000 PCR system (BioRad, Hercules, CA, USA) with detection of the PCR product quantity in the FAM channel. Viral load was determined based on cycle threshold (Ct) values. Serial dilutions of the standardized positive control supplied with the PCR reagent kit were used as the standard.
2.8. Sequencing
Total RNA was isolated using the RIBO-prep kit for DNA/RNA extraction from biological samples (AmpliSens, Moscow, Russia). The extracted RNA was immediately utilized to obtain total cDNA using the LunaScript RT SuperMix Kit (New England Biolabs, Ipswich, Massachusetts, USA). Whole-genome sequencing was carried out using the ARTIC v3 protocol (COVID-19 ARTIC v3 Illumina library construction and sequencing protocol V.5) with two pools of oligonucleotide primers to cover the complete SARS-CoV-2 genome. The Q5® Hot Start High-Fidelity 2X Master Mix kit (New England Biolabs, Ipswich, Massachusetts, USA) was used for amplification. The resulting dsDNA fragments were purified from unused components and reaction products using AMPure beads (Beckman Coulter, Brea, California, USA). The concentration of nucleic acids was measured using Qubit 3.0 and the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA). The dsDNA fragments were then used to prepare NGS libraries. The Y-shaped adapter ligation method (Illumina, San Diego, California, USA) was utilized to prepare libraries. Sequencing was performed on the MiSeq platform (Illumina, San Diego, California, USA) using the MiSeq Reagent Kit v2 (500-cycles; Illumina, San Diego, California, USA).
2.9. Bioinformatics analysis
Quality control was performed using the FastQC software (v. 0.11.9); removal of low-quality reads and adapters, as well as trimming were carried out using the Cutadapt software (v.2.8). Reads were aligned to the reference using the BWA-MEM software (v. 0.7.17).SAM/BAM files were processed and analyzed using Samtools (v. 1.9). The iVar software (v. 1.1; https://github.com/andersen-lab/ivar/) was used to extract consensus sequences from BAM files. SARS-CoV-2 complete genome sequence NC_045512.2 (Wuhan-Hu-1) was used as a reference. The obtained sequences were deposited in the GISAID database (EPI_SET ID: EPI_SET_230804ug, https://doi.org/10.55876/gis8.230804ug accessed on August 4, 2023). Multiple alignment of amino acid sequences of S genes was carried out using MAFFT (v. 7.475). Phylogenetic analysis of genome sequences was carried out using the UPGMA method and the Phylo.io Web resource [22]. Pangolin software (v.3.17) was used to assign the genetic lineages. All software programs used for analysis are freely available and distributed under a free open-source license.
2.10. Microscopy studies
For histological analysis, fragments of hamster lungs fixed in a 4% buffered paraformaldehyde solution were dehydrated in a series of solutions of increasing concentrations of ethanol, isopropanol, and xylene on a tissue processor Sakura Tissue-Tek II (Sakura Finetek, Alphen aan den Rijn, the Netherlands) using a standard technique. Samples were then placed in Gistomix embedding medium (BioVitrum, Saint Petersburg, Russia) by forming blocks. Sections of a 3–4 μm thickness were obtained from the blocks using a Leica RM2255 microtome (Leica, Wetzlar, Germany), mounted on fat-free glass slides, and air-dried. For staining, sections were deparaffinized in xylene, immersed in ethanol solutions of decreasing concentrations, and washed with distilled water. Hematoxylin and eosin staining was performed as follows: sections were stained with Ehrlich hematoxylin for 30 sec, washed with running water, and stained with eosin (1%) for 2 min.
2.11. Statistical data analysis
Basic statistical analyses, including calculations of the mean, standard deviation, and coefficient of variation of the mean Ct value, were performed using Excel (Microsoft Corp., Redmond, WA, USA). Statistical data analysis was conducted using the statistical software STATISTICA 13 (StatSoft Inc., Tulsa, OK, USA).
3. Results
A total of 16 SARS-CoV-2 coronavirus strains, isolated during different pandemic periods and belonging to different genetic lineages of the Omicron variant, were used in the study.
The resulting plaque sizes in the agar layer (0.43±0.32 mm2) and infectious titers (5.25±0.38 lg TCD50/100 μl) for the BA.1 strain in Vero E6 cells were one of the lowest among the studied strains.
The studied BA.2 strain is characterized by high transmissibility. The plaque size is 0.48±0.27 mm2, while the virus is produced in average titers (6.25±0.25 lg TCD50/100 μl). Descendants of BA.2.75, as well as BN.1.3 and CH.1.1 strains, are characterized by one of the highest infectious titers (7.0 ± 0.35 and 6.25 ± 0.38 lg TCD50/100 μl, respectively). At the same time, the plaque size varied significantly (0.8±0.58 and 4.53±3.25, respectively). BA.5.1 studied in Vero E6 cells was characterized as a high-titer strain (7.25 ± 0.25 lg TCD50/100 μl). The plaque size was 1.02±0.73 mm2. BA.5.1 descendants and strains CL.1 and CL.1.2 were characterized by average values of the maximum infectious titer (6.5 and 6±0.35 lg TCD50/100 μl, respectively) and plaque size (1.42±1.36 and 1. 02±0.43 mm2).
BA5.2 exhibited one of the lowest plaque sizes in the agar layer: 0.30 ± 0.22 mm2, while the virus was produced at the highest titers (7.5 lg TCD50/100 μl). Further BA.5.2 propagation resulted in decreased infectious titer and increased plaque size. For VA.5.2.6, the values were 6.75±0.38 lg TCD50/100µl and 0.78±0.38 mm2.
BA.5.3 descendant strains are characterized by average values of the maximum titer and plaque size in the agar layer. They are 6.75±0.25 lg TCD50/100 µl and 1.23±1.07 mm2 for BQ.1.1 and 6.75±0.38 lg TCD50/100 µl and 1.86±1.32 mm2 for BQ.1.2.1, respectively.
The coronavirus strain of one of the latest isolated lineages, namely XBB, has lower productivity (5–5.75 lg TCD50/100 µl) and growth rate compared to other genetic lineages. Plaque sizes are 0.27±0.14, 0.97±0.53, 1.26±0.86, and 1.58±0.41 mm2 for XBB.1, XBB.1.5, XBB.1.37, and XBB.1.9, respectively.
The coronavirus strain of one of the latest isolated lineages, namely XBB.3, was characterized by low rates of infectious virus accumulation in the culture medium in an in vitro experiment. The maximum concentration was noted after 96 hours, while the virus accumulated at low titers (5.0 lg TCD50/100 µl). The plaque size in the agar layer was 0.65±0.55 mm2.
BA.1, BA.2, and BA.5.1 (as a reference), as well as BA.5.2 and XBB.3, were characterized in vivo as representatives of the main genetic lineages.
No signs of lung consolidation were observed in animals infected with the Omicron variant BA.1. Alveolar wall infiltration with mild hemorrhage and edema were noted. An experiment in Syrian hamsters revealed a slight decrease in virulence (ID50 is 1.3 TCD50). In BALB/c mice, the ID50 was significantly lower than that obtained for other genetic variants, amounting to 14 TCD50.
A slight increase in virulence was noted in laboratory animals infected with the coronavirus genetic variant BA.2, (ID50 is 15 and 1.3 TCD50 for BALB/c mice and Syrian hamsters, respectively). Pathological changes in lung tissue similar to those under the effect of BA.1 were detected.
A study on the effects of BA.5.1 in animals revealed a slight increase in virulence (ID50 is 15 and 1.7 TCD50 for BALB/c mice and Syrian hamsters, respectively) and a pathogenic effect on lung tissue as the primary target organ compared to Omicron BA.2.
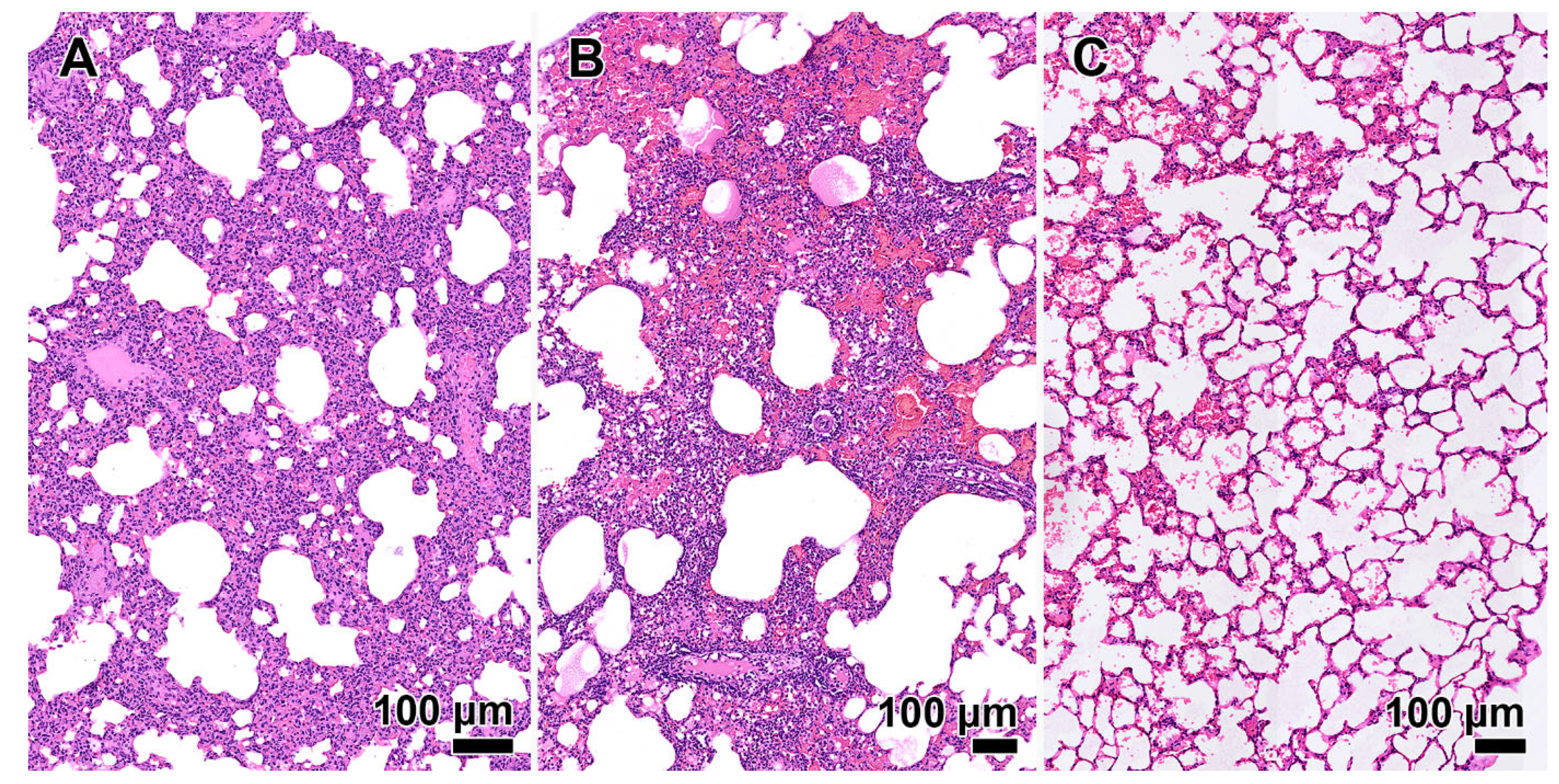
A study of the BA.5.2 strain in animals revealed a slight increase in virulence compared to BA.5.1 (ID50 is 10 and 0.7 TCD50 for BALB/c mice and Syrian hamsters, respectively). Analysis of histological preparations of lung tissue showed a reduction in the severity of pathomorphological changes caused by the infectious process. In the majority of animals, a moderate reduction in the lung aeration was observed in the form of dystelectasis (approximately 70% of the parenchymal area). Loci of moderate inflammatory cell infiltration were observed in the presence of pronounced hyperemia of the interalveolar wall capillaries. The infiltrates were predominantly composed of lymphoid elements, with a few neutrophilic granulocytes involved. In some animals, leukocytes were not only distributed diffusely throughout the parenchyma but also accumulated around individual blood vessels, indicating a tendency towards vasculitis. The bronchi remained unchanged.
The XBB.3 virulence in laboratory animals is similar to that of BA.1 and BA.5.2 (ID50 is 15 and 1.8 TCD50 in BALB/c mice and Syrian hamsters, respectively). Histological preparations are characterized by either the absence or mild inflammatory response of the pulmonary parenchyma in the form of edema of the interalveolar septa and lymphocytic infiltration. Lung aeration is unchanged; a slight presence of alveolar hemorrhage is noted.
4. Discussion
The histological sections of Syrian hamster lung presented in this study demonstrate a change in the pathogenicity of coronaviruses of different Omicron variant lineages (Figure 1). The BA.1 lineage caused significant changes in the lung tissue of animals, similar to those observed in humans [23]. The severity of COVID-19 increased with the emergence of the BA.5.2 lineage, as confirmed by pathological changes in the lung tissues of the Syrian hamster. However, there have not been reports of a large number of severe COVID-19 cases in individuals without chronic diseases [6]. CPV lineages, which are currently predominant, are highly transmissible, although unable to cause severe pathomorphological changes in lung tissues of laboratory animals. This is confirmed by the absence of severe post-infectious complications in humans [11].
The BA.5.2 strain is the most pathogenic among the studied ones, which is attributed to a series of mutations in the S protein [24]. However, a low reproduction rate decreases the possibility of its transmission. At the same time, XBB lineages are distinguished by low values of the characterized parameters.
During cultivation, the strains of the three lineages acquired a number of amino acid substitutions that affected their biological properties. BQ.1.2.1 and CH.1 lineage strains were genetically different from the primary sample coronaviruses obtained from COVID-19 patients by the time of characterization. Changes in the BA.1 genome were discovered later, after long-term cultivation. An increase in the plaque area in the agar layer was associated with the occurrence of the R682W substitution in BA.1 [25]. Previously, this substitution was also discovered in the Gamma variant (EPI_ISL_6565014/EPI_ISL_18032149) after being cultured in Vero E6 cells for a long period. A similar substitution, namely R682P, was present in the BA.5.1 lineage strain throughout the entire cultivation period.
After six passages, CH.1.1 and BQ.1.2.1 lineage strains showed an increase in plaque size, which was attributed to the presence of adaptive substitutions D614G, H655Y, and N764K in the coronavirus S protein [26]. In contrast to the BQ.1.2.1 coronavirus, the S protein in the CH.1.1 lineage strain has an amino acid substitution R346T, which is characteristic of lineages BQ.1.1 and CBB [27]. At the same time, the K417N substitution, which was originally present in the coronavirus from the biological sample obtained from the patient, was not found. Cumulative mutations in the strain genome resulted in amino acid substitutions homologous to CHB.3 for CH.1.1 (Figure 3). This partially confirms the hypothesis regarding the emergence of multiple sublineages that have either identical or very similar RBDs. These sublineages share a common phenotype but do not belong to any specific lineage [28].
After isolating CK.1 from Vero E6 cells, the viral load in the culture medium was measured by RT-PCR. The Ct was 21.86 after the first passage and 24.39 after the second passage. The Ct did not decrease below 26 after subsequent passages and reached 13.91 only by the sixth passage.
After six passages, sequencing of the coronavirus revealed an original XBB.1.5 lineage, which differs from the previously isolated and sequenced ones. This eliminates the chance of potential sample contamination. The fluctuation of Ct values may indicate viral interference upon co-culturing of two genetic lineages of SARS-CoV-2 [29]. The presence of the XBB.1.5 lineage virus in the nasal wash sample used for culturing was not initially confirmed due to its low concentration. The sequencing detected its presence in the form of minor mutations, which have not been taken into consideration. This case may indicate an adaptive mode of the Omicron coronavirus evolution, according to which the genetic lineages with the optimal S protein conformation provided by amino acid substitutions R346T, D614G, H655Y, and N764K predominate.
Figure 4.
Phylogenetic tree of S-protein amino acid sequences of SARS-CoV-2 Omicron variant strains used in experiments. * – strains isolated from patients and altered upon cultivation in Vero E6 cells, ** – strains altered as a result of further cultivation in Vero E6 cells. *** – strain that completely displaced the CK.1 lineage upon co-culturing.
Figure 4.
Phylogenetic tree of S-protein amino acid sequences of SARS-CoV-2 Omicron variant strains used in experiments. * – strains isolated from patients and altered upon cultivation in Vero E6 cells, ** – strains altered as a result of further cultivation in Vero E6 cells. *** – strain that completely displaced the CK.1 lineage upon co-culturing.
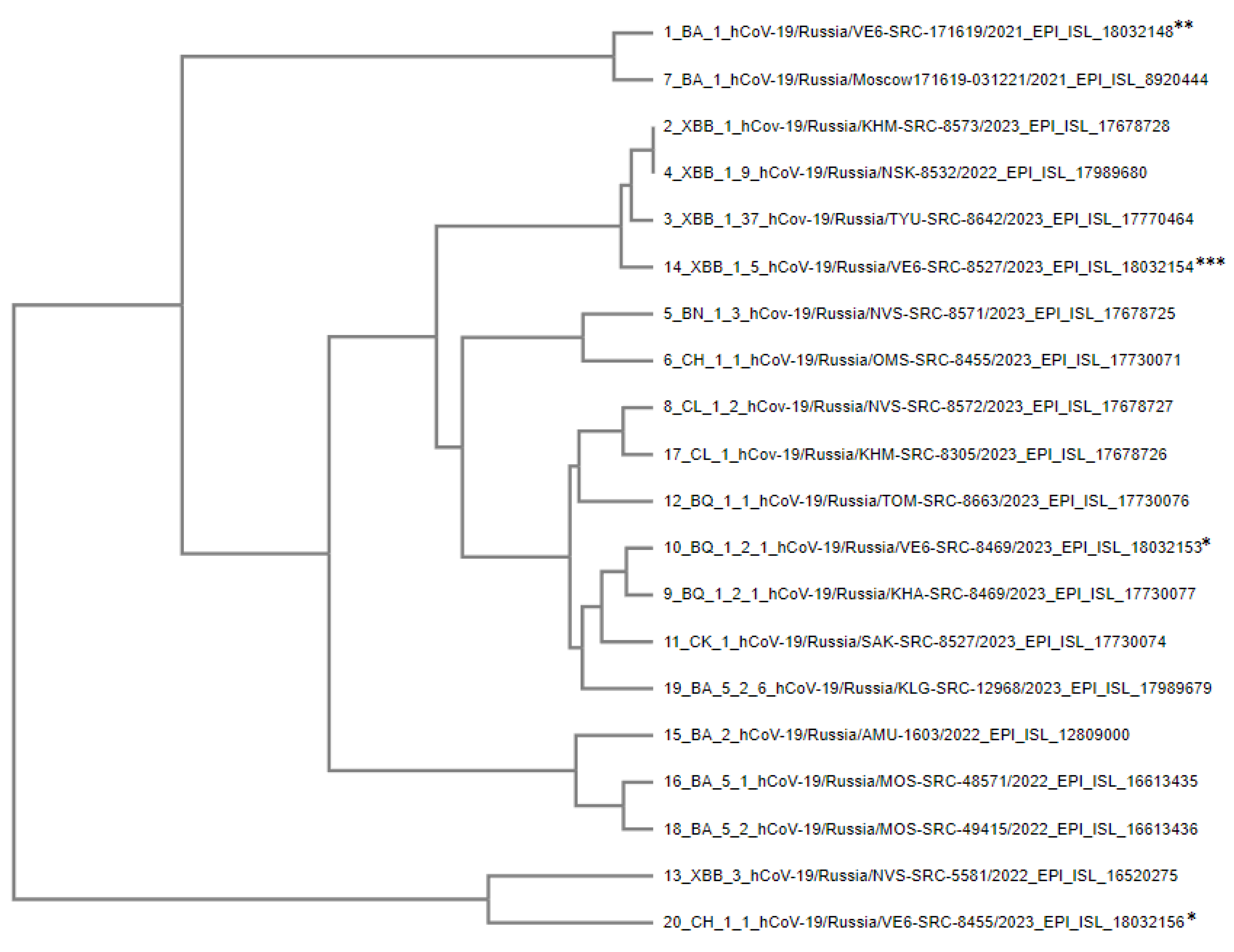
The phylogenetic relationship of amino acid sequences of the S protein of the studied coronavirus strains was analyzed using MAFFT (Multiple Alignment using Fast Fourier Transform) software, which utilizes more than 20 physicochemical and biochemical parameters of volume and polarity for each amino acid [30]. To assess the consistency of local topology in phylogenetic trees, the UPGMA algorithm was used, which makes it possible to rank lineages according to the degree of genetic variations from the hypothetical ancestor.
Branch topology demonstrates the affinity of amino acid sequences of the S protein of different genetic lineages. Representatives of some lineages are displayed closer to the ancestral strain, despite their later emergence. This fact may indicate a reverse evolution of coronavirus proteins and restoration of the surface epitope structure characteristic of earlier SARS-CoV-2 genetic lineages and variants.
5. Conclusions
Experiments revealed a consistent decrease in the virulence of Omicron lineage strains compared to the previously circulating SARS-CoV-2 variants. A co-evolutionary change in amino acid sequences affecting the conformation of the coronavirus S protein and its surface epitopes has been observed in representatives of various genetic lineages. An emergence of potential protease cleavage sites and changes in the furin cleavage site were detected in many Omicron variant coronaviruses. Such features of SARS-CoV-2 require additional studies on its biological properties in order to investigate potential changes in the course of coronavirus infection.
Author Contributions
A.V.S. and A.V.Z. designed the experiments; A.V.S., A.V.Z., G.A.K., I.A.D., T.V.B., T.V.T. and E.K.I. performed the experiments; M.P.L. conducted primary sample preparation; A.V.S., A.V.Z., G.A.K., I.A.D., M.E.A., T.V.B., T.V.T. and E.K.I. analyzed the data; S.A.B., O.S.T. and O.V.P. were responsible for supervision; A.V.S. and A.V.Z. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the state budget.
Institutional Review Board Statement
The animal study was conducted according to the guidelines of the Declaration of Helsinki, in compliance with the protocols and recommendations for the proper use and care of laboratory animals (ECC Directive 86/609/EEC, Ministry of Health of the Russian Federation Directive no. 708n/2010, and Ministry of Health of the Russian Federation Directive no. 267/2003). The protocol was approved by the Bioethics Committee of the Federal Budgetary Research Institution State Research Center of Virology and Biotechnology “Vector” (protocol code: SRC VB ”Vector”/06–06.2021; date of approval: 15 June 2021).
Conflicts of Interest
No authors report any conflict of interest. The founders had no role in the design of the study; in the collection, analyses, and interpretation of data; in the writing of the manuscript; and in the decision to publish the results.
References
- Komissarov:, A.B.; Safina, K.R.; Garushyants, S.K.; Fadeev, A.V.; Sergeeva, M.V.; Ivanova, A.A.; Danilenko, D.M.; Lioznov, D.; Shneider, O.V.; Shvyrev, N.; et al. Genomic epidemiology of the early stages of the SARS-CoV-2 outbreak in Russia. Nat Commun. 2021, 12, 649. [Google Scholar] [CrossRef]
- Akimkin, V.G.; Popova, A.Yu.; Khafizov, K.F.; Dubodelov, D.V.; Ugleva, S.V.; Semenenko, T.A.; Ploskireva, A.A.; Gorelov, A.V.; Pshenichnaya, N.Yu.; Yezhlova, E.B.; et al. COVID-19: evolution of the pandemic in Russia. Report II: dynamics of the circulation of SARS-CoV-2 genetic variants. Journal of microbiology, epidemiology and immunobiology = Zhurnal mikrobiologii, èpidemiologii i immunobiologii. 2022, 99, 269–286. [Google Scholar] [CrossRef]
- He, X.; He, C.; Hong, W.; Yang, J.; Wei, X. Research progress in spike mutations of SARS-CoV-2 variants and vaccine development. Med. Res. Rev. 2023, 43, 932–971. [Google Scholar] [CrossRef]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev Med Virol. 2022, 32, e2381. [Google Scholar] [CrossRef]
- Lyngse, F.P.; Kirkeby, C.T.; Denwood, M.; Christiansen, L.E.; Mølbak, K.; Møller, C.H.; Skov, R.L.; Krause, T.G.; Rasmussen, M.; Siebe, R.N.; et al. Transmission of SARS-CoV-2 Omicron VOC subvariants BA.1 and BA.2: Evidence from Danish Households. MedRxiv 2022, 2022.01.28.22270044. [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Ao, D.; He, X.; Hong, W.; Wei, X. The rapid rise of SARS-CoV-2 Omicron subvariants with immune evasion properties: XBB.1.5 and BQ.1.1 subvariants. MedComm. 2023, 4, e239. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Song, W.; Wang, L.; Liu, P.; Yue, C.; Jian, F.; Yu, Y.; Yisimayi, A.; Wang, P.; Wang, Y.; et al. Characterization of the enhanced infectivity and antibody evasion of Omicron BA.2.75. Cell Host Microbe 2022, 30, 1527–1539. e5. [Google Scholar] [CrossRef]
- Karyakarte, R.P.; Das, R.; Taji, N.; Yanamandra, S.; Shende, S.; Joshi, S.; Karekar, B.; Bawale, R.; Tiwari, R.; Jadhav, M.; et al. An Early and Preliminary Assessment of the Clinical Severity of the Emerging SARS-CoV-2 Omicron Variants in Maharashtra, India. Cureus 2022, 14, e31352. [Google Scholar] [CrossRef]
- Qu, P., Faraone, J.N.; Evans, J.P.; Zheng, Y.M.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Panchal, A.; et al. Extraordinary evasion of neutralizing antibody response by Omicron XBB.1.5, CH.1.1 and CA.3.1 variants. bioRxiv 2023, 2023.01.16.524244. [CrossRef]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat Commun. 2023, 14, 2800. [Google Scholar] [CrossRef]
- Scarpa F., Azzena I., Locci C., Casu M., Fiori P.L., Ciccozzi A. Angeletti, S.; Imperia, E.; Giovanetti, M.; Maruotti A.; et al. Molecular In-Depth on the Epidemiological Expansion of SARS-CoV-2 XBB.1.5. Microorganisms 2023, 11, 912. [CrossRef]
- Amin, R.; Pal, P.; Dhama, K.; Emran, T.B. Recent update on XBB.1.5 emerging novel mutation of COVID-19. Int J Surg. 2023, 109, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- UK Health Security Agency. Technical briefing #51. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1141754/variant-technical-briefing-51-10-march-2023.pdf (accessed on 10 September 2023).
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yuet F.; al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Ι. Proc. Nat. Acad. Sci. USA 2021, 118, e2024202118. [CrossRef] [PubMed]
- Leist, S.R., Schäfer, A.; Martinez, D.R. Cell and animal models of SARS-CoV-2 pathogenesis and immunity. Dis Model Mech. 2020, 13, dmm046581. [CrossRef]
- Jeong, G.U.; Yoon, G.Y.; Moon, H.W.; Lee,W.; Hwang, I.; Kim, H.; Kim, K.-D.; Kim, C.; Ahn, D.-G.; Kim, B.-T.; et al. Comparison of Plaque Size, Thermal Stability, and Replication Rate among SARS-CoV-2 Variants of Concern. Viruses 2022, 14, 55. [CrossRef]
- Rosenke, K.; Meade-White, K.; Letko, M.; Clancy, C.; Hansen, F.; Liu, Y.; Okumuraa, A.; Tang-Huaua, T.-L.; Lic, R.; Saturday, G.; et al. Defining the Syrian hamster as a highly susceptible preclinical model for SARS-CoV-2 infection. Emerg. Microbes Infect. 2020, 9, 2673–2684. [Google Scholar] [CrossRef]
- Wang, J., Shuai, L., Wang, C.; Liu, R.; He, X.; Zhang, X.; Sun, Z.; Shan, D.; Ge, J.; Wang, X.; et al. Mouse-adapted SARS-CoV-2 replicates efficiently in the upper and lower respiratory tract of BALB/c and C57BL/6J mice. Protein Cell 2020, 11, 776–782. [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am J Hyg. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Case, J.B.; Bailey, A.L.; Kim, A.S.; Chen, R.E.; Diamond, M.S. Growth, detection, quantification, and inactivation of SARS-CoV-2. Virology 2020, 548, 39–48. [Google Scholar] [CrossRef]
- Robinson, O.; Dylus, D.; Dessimoz, C. Phylo.io: Interactive Viewing and Comparison of Large Phylogenetic Trees on the Web. Mol. Biol. Evol. 2016, 33, 2163–2166. [Google Scholar] [CrossRef]
- Märkl, B.; Dintner, S.; Schaller, T.; Sipos, E.; Kling, E.; Miller, S.; López, F.F.; Grochowski, P.; Reitsam, N.; Waidhauser, J.; et al. Fatal cases after Omicron BA.1 and BA.2 infection: Results of an autopsy study. Int J Infect Dis. 2023, 128, 51–57. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 spike mutations, L452R, T478K, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- Escalera, A.; Gonzalez-Reiche, A.S.; Aslam, S.; Mena, I.; Laporte, M.; Pearl, R.L.; Fossati, A.; Rathnasinghe, R.; Alshammary, H.; van de Guchte, A.; et al. Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell Host Microbe 2022, 30, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Maaroufi, H. The N764K and N856K mutations in SARS-CoV-2 Omicron BA. 1 S protein generate potential cleavage sites for SKI-1/S1P protease. BioRxiv 2022, 2022.01.21.477298. [CrossRef]
- Weng, S.; Shang, J.; Cheng, Y.; Zhou, H.; Ji, C.; Yang, R.; Wu, A. Genetic differentiation and diversity of SARS-CoV-2 Omicron variant in its early outbreak. Biosaf Health. 2022, 4, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Groenheit, R.; Galanis; I.; Sondén, K.; Sperk, M.; Movert, E.; Bacchus, P.; Efimova, T.; Petersson, L.; Rapp, M.; Sahlén, V.; et al. Rapid emergence of omicron sublineages expressing spike protein R346T. Lancet Reg Health Eur. 2023, 24, 100564. [CrossRef]
- Piret, J.; Boivin, G. Viral interference between respiratory virus. Emerg. Infect. Dis. 2022, 28, 273–281. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
Figure 1.
Data obtained for genetic lineage strains in Vero E6 cells: Blue columns – Maximum infectious titer (lg TCID50/100µl). Red columns – Plaque area sizes (mm2).
Figure 1.
Data obtained for genetic lineage strains in Vero E6 cells: Blue columns – Maximum infectious titer (lg TCID50/100µl). Red columns – Plaque area sizes (mm2).
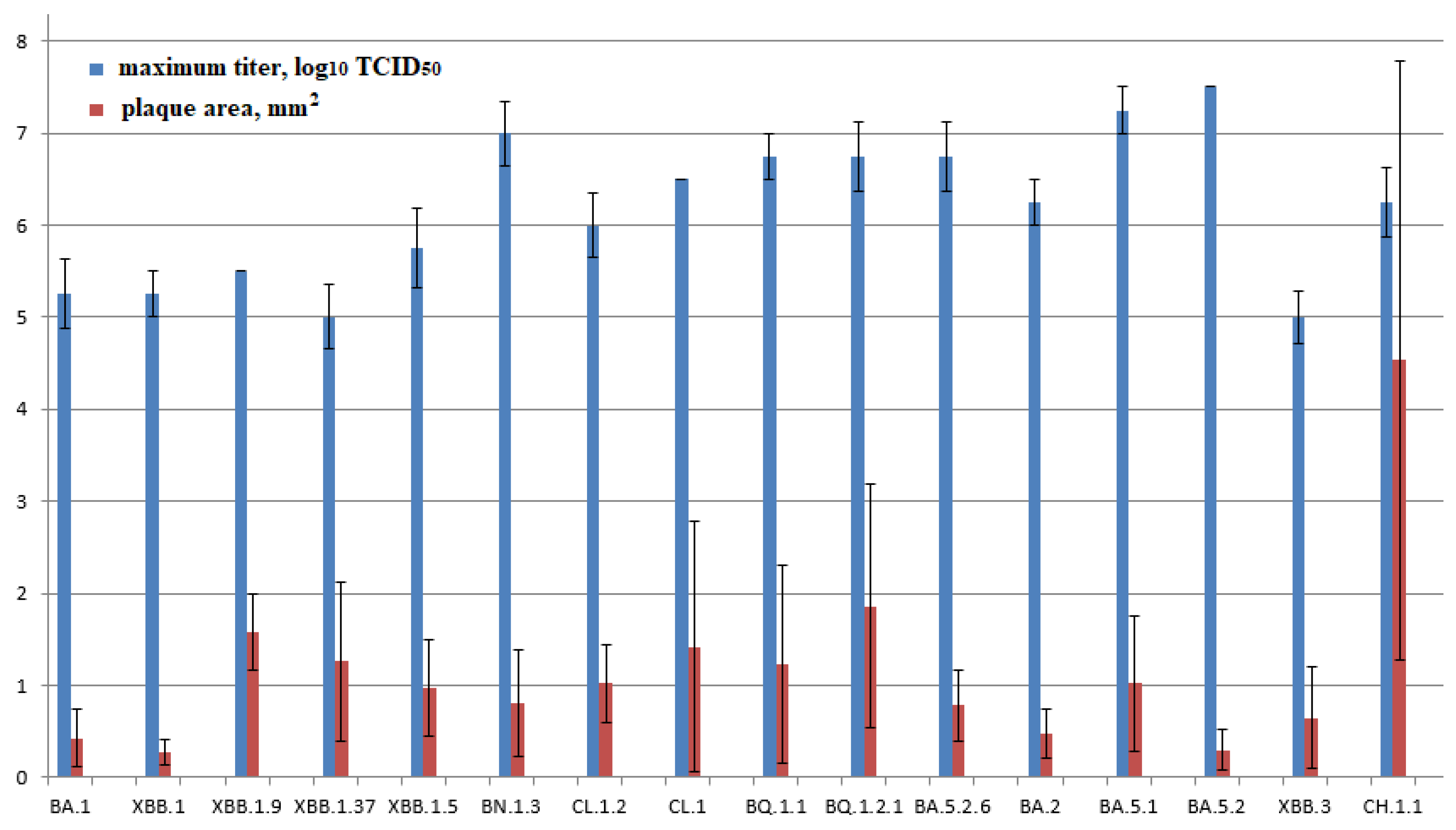
Figure 2.
Crystal violet-stained plaque assay plates. Morphology of plaques in Vero E6 monolayer cells 72 hours after infection with Omicron variant lineages.
Figure 2.
Crystal violet-stained plaque assay plates. Morphology of plaques in Vero E6 monolayer cells 72 hours after infection with Omicron variant lineages.
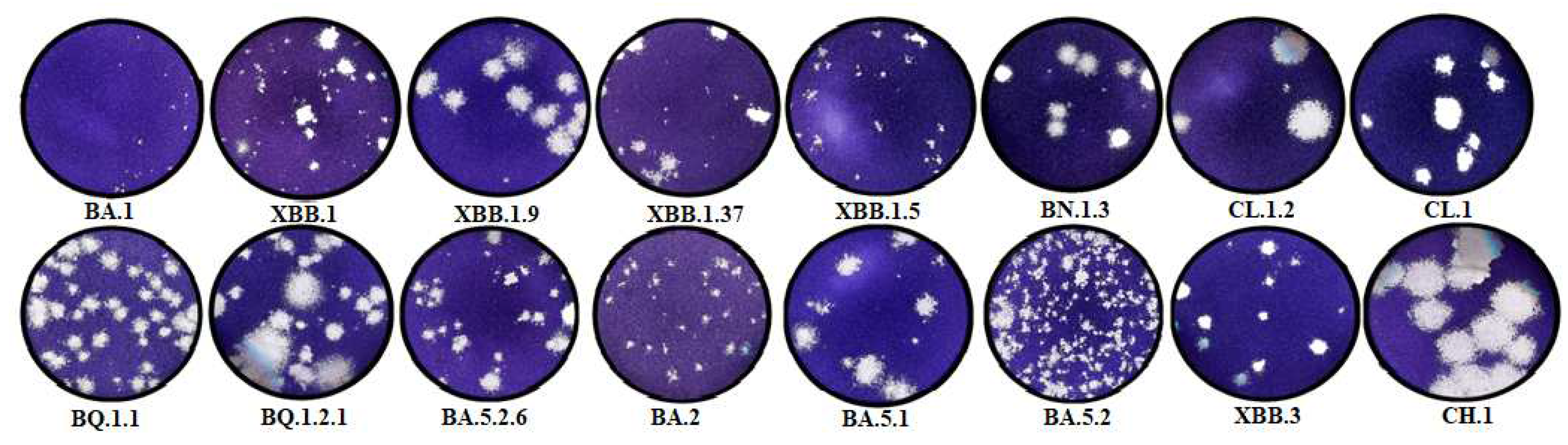
Figure 3.
Lung histological section of a Syrian hamster infected with genetic lineages of SARS-CoV-2 Omicron variant at a dose 30 ID50 (A–C). A – Omicron BA.1. Inflammatory infiltration of the interalveolar septa as manifestation of interstitial pneumonia. B – Omicron BA.5.2. Inflammatory infiltration of the interalveolar septa as manifestation of interstitial pneumonia, alveolar hemorrhage, and edematous fluid in the alveolar cavity. C – Omicron XBB.3. Lung aeration is unchanged, mild alveolar hemorrhage is observed. H&E staining. Scale bar is presented in the photomicrograph.
Figure 3.
Lung histological section of a Syrian hamster infected with genetic lineages of SARS-CoV-2 Omicron variant at a dose 30 ID50 (A–C). A – Omicron BA.1. Inflammatory infiltration of the interalveolar septa as manifestation of interstitial pneumonia. B – Omicron BA.5.2. Inflammatory infiltration of the interalveolar septa as manifestation of interstitial pneumonia, alveolar hemorrhage, and edematous fluid in the alveolar cavity. C – Omicron XBB.3. Lung aeration is unchanged, mild alveolar hemorrhage is observed. H&E staining. Scale bar is presented in the photomicrograph.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



