
Submitted:
18 December 2023
Posted:
19 December 2023
You are already at the latest version
Abstract
Epitranscriptomic RNA modifications play a crucial role in the posttranscriptional regulation of gene expression. N6-methyladenosine (m6A) is the most prevalent internal modification of eukaryotic RNA and plays a pivotal role in RNA fate. RNA m6A modification is regulated by a group of cellular proteins, methyltransferases (writers) and demethylases (erasers), which add and remove the methyl group from adenosine, respectively. m6A modification is recognized by a group of cellular RNA-binding proteins (readers) that specifically bind to m6A-modified RNA, mediating effects on RNA stability, splicing, transport, and translation. The functional significance of m6A modification of viral and cellular RNA is an active area of virology research. In this review, we summarize and analyze current literature on m6A modification of HIV-1 RNA, the multifaceted functions of m6A in regulating HIV-1 replication, and the role of viral RNA m6A modification in evading the innate immune responses to infection. Furthermore, we briefly discuss future directions and therapeutic implications of mechanistic studies of HIV-1 epitranscriptomic modifications.
Keywords:
Epitranscriptomic RNA modification
; N6-methyladenosine
; HIV-1
; virus-host interactions
; innate immunity
1. Introduction
Post-transcriptional RNA modification is an important mechanism of regulating gene expression. N6-methyladenosine (m6A) is the most prevalent RNA modification present internally within eukaryotic mRNAs and influences RNA stability, translation, splicing, and subcellular localization [1,2,3,4]. Viral RNA has been known to contain m6A for nearly 50 years [5]. However, advances in the technology for detecting and mapping sites of modified RNA have resulted in rapid progress in understanding how m6A influences virus replication, virus-host interactions, and antiviral immunity [6].
The literature describing m6A modification of viral RNA and how m6A pathway proteins regulate viral infections has grown at a remarkable pace over the last several years. This review will focus on the current knowledge of how m6A modifications influence diverse aspects of the life cycle of human immunodeficiency virus type-1 (HIV-1). We discuss the topology of m6A modification on HIV-1 RNA, the roles of m6A writers, readers, and erasers in regulating HIV-1 replication, and how m6A affects the induction of innate immunity in response to HIV-1 infection.
2. Cellular m6A regulating proteins and their functions
The addition of a methyl group to the N6 position of adenosine is catalyzed by a multi-subunit methyltransferase complex, also referred to as the m6A writer complex. The catalytic core is formed by a heterodimer of methyltransferase-like 3 (METTL3) and methyltransferase-like 14 (METTL14), with Wilms tumor 1-associating protein (WTAP) serving as a scaffold protein that stabilizes the methyltransferase complex [7,8]. Additional components of the methyltransferase complex that may direct the complex to sites of m6A modification include RNA binding motifs protein 15 (RBM15) and KIAA1429, also known as vir-like m6A methyltransferase associated (VIRMA) [9,10]. However, the function of each protein associated with the methyltransferase complex is not fully understood.
The writer complex exhibits preference for modification of adenosine in the consensus sequence 5’-DRACH-3' (D=A/G/U, R=A/G, H= U/A/C) in mammalian mRNAs [11]. Transcriptome-wide m6A mapping techniques have revealed that m6A is most common in 3’ UTRs and long exons of mRNA [11]. Although DRACH motifs occur frequently, only ~10% of them are m6A methylated in cells, suggesting that there are context-dependent signals for the methylation of specific DRACH motifs [11].
m6A modification is reversible through the action of demethylases, or erasers. These enzymes include alkB homolog 5 (ALKBH5) and fat mass and obesity-associated protein (FTO), and both enzymes can remove m6A modification [12,13]. However, since the discovery of these enzymes, it has been shown that FTO exhibits a strong preference for demethylating N6, 2’-O-methyladenosine (m6Am) residues next to the 5’ cap, rather than m6A at internal sites of mRNA [14]. Therefore, while expression of the individual eraser enzymes can be manipulated to affect overall changes in m6A levels, the role of each endogenous eraser in the context of virus infection in cells remain unclear.
The functional consequence of m6A modification is determined by the binding of m6A-specific RNA binding proteins, or readers. Readers that are most often studied in the context of m6A function are the YTH domain-containing family of proteins, YTHDC1 and YTHDF1-3. YTHDC1 regulates the nuclear export of mature mRNA [3]. The cytoplasmic YTHDF1-3 proteins were initially determined to be functionally distinct. YTHDF1 promotes translation, while YTHDF2 mediates degradation of m6A-modified RNAs [1]. YTHDF3 acts in synergy with YTHDF1 and YTHDF2 to enhance translation or mediate RNA decay [15]. However, more recent studies suggest that YTHDF1-3 are functionally redundant and mediate RNA degradation [16]. The discrepancies regarding YTHDF protein functions have been reviewed elsewhere [17,18,19]. While YTHD family readers are often the focus of studies designed to elucidate the role of m6A in viral replication, many other potential reader proteins have also been described (reviewed in [17]). These less studied m6A readers also deserve attention in investigating the mechanisms of m6A in regulating cellular gene expression and virus replication.
A major challenge in determining the function of m6A in regulating virus replication is the ability to specifically remove m6A on viral RNA, especially transcripts, while leaving the modification of cellular RNA unperturbed. Almost all studies to date have utilized overexpression or knockdown/knockout of writers, readers, and erasers to infer the function of viral RNA m6A during infection. However, these approaches will also lead to changes in the m6A modification of host cell RNA, which may have indirect effects on virus replication. Such indirect effects must always be carefully considered during the interpretation of these studies. Additional approaches, including mutations of m6A site of viral genomes, are often used in confirming specific effects [20,21,22,23,24].
3. Mapping m6A modification sites on HIV-1 RNA
Several studies have reported the location of m6A in HIV-1 RNA from infected cells or purified virions (Figure 1A,B). The techniques, virus strains, and cell types used among studies are summarized in Table 1. In the first report, methylated RNA immunoprecipitation sequencing (MeRIP-seq) was used to determine m6A modified regions of HIV-1 RNA from infected MT4 CD4+ T cells [25]. meRIP-seq utilizes fragmented RNA for m6A-specific immunoprecipitation followed by high-throughput sequencing and has a resolution of ~100-200 nt [11]. This analysis identified 14 regions predicted to contain m6A modifications [25] (Figure 1A). In contrast to subsequent reports, this data analysis did not include the 5’ and 3’ UTR sequences, which led to the absence of input or m6A signal mapping to these regions. Peaks mapped primarily to coding sequences and exhibited overlap with several splicing regulatory sequences. Unique to this study was the identification of a peak located in env and rev response element (RRE), which was the focus of subsequent functional analysis. Methylation of predicted sites in the RRE was confirmed by primer extension assay in infected cells [25]. However, none of the subsequent studies reported by other groups found m6A to be present in the RRE [26,27].
The next report identified sites of m6A modification in HIV-1 RNA from CEM-SS CD4+ T cells infected with single-cycle HIV-1 [26]. This study employed photo-crosslinking-assisted m6A sequencing (PA-m6A-seq), which modifies the meRIP protocol through the addition of 4-thiouridine (4SU) labeling of viral RNA followed by UV crosslinking of an m6A-specific antibody to the labeled RNA prior to sequencing. This approach improves the resolution of sites of m6A modification to ~20 nt due to the introduction of U to C mutations at the sites of crosslink between antibody and 4SU [32]. In this analysis, all predicted sites of methylation mapped to the 3’ UTR ~15% of the genome (Figure 1A). The PA-m6A-seq results were compared to photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) performed to identify the binding sites for YTHDF reader proteins on the HIV-1 RNA. Comparison of reader binding sites in HIV-1 RNA from HEK293T cells transfected with an HIV-1 proviral plasmid and m6A peaks from infected CEM-SS cells reveal four regions in common, located in env/rev, nef, and the 3’ UTR. The authors also sought to assess the topology of reader binding sites in different HIV-1 strains. PAR-CLIP data to assess reader binding sites in RNA of HIV-1BaL and HIV-1JR-CSF show that these primary isolates have the four binding sites in common with HIV-1NL4-3, while also possessing additional reader binding peaks. This illustrates the importance of considering cell-type and HIV-1 strain-specific differences in m6A modification of viral RNA. This group also reported the location of m6A modifications in HIV-1 genomic RNA (gRNA) purified from virions compared to cell-associated RNA from CEM-SS cells. PA-m6A-seq revealed major peaks in env/rev, nef, and 3’ UTR of gRNA, consistent with the results of m6A mapping to cell-associated viral RNA (Figure 1B) [28]. Interestingly, additional peaks were detected in several coding sequences of virion RNA including pol, env, and vpr.
Another study using meRIP-seq confirmed the presence of major m6A peaks in env/rev, nef, and the 3’ UTR in HIV-1 RNA from infected Jurkat and primary CD4+ T cells (Figure 1A) [27]. This analysis also identified additional smaller but prominent peaks in the 5’ UTR and gag. Cell-associated HIV-1 RNA collected from HEK293T cells transfected with an HIV-1 proviral plasmid showed similar sites of m6A modification as viral RNA from infected CD4+ T cells, with an additional major peak in tat. These results suggest that in addition to cell type differences, it is possible that patterns of HIV-1 m6A modification differ in virus producer cells and target cells. Validation of m6A modification at the predicted sites was performed by crosslinking-immunoprecipitation sequencing (CLIP-seq) to identify the binding sites of YTHDF1-3 proteins on HIV-1 RNA isolated from infected HeLa/CD4 cells. CLIP-seq analysis revealed reader binding sites in env/rev, nef, and the 5’ UTR, providing further support for the presence of m6A at these locations. Of note, reader binding was detected in several coding regions of the HIV-1 genome, sites that were not detected by meRIP-seq. This could be due to different cell types used for meRIP-seq and CLIP-seq or due to more sensitive and robust detection using a crosslinking approach.
Mapping sites of m6A modification at a single timepoint does not provide information about how modification states may change over the course of infection. One group investigated whether temporal changes in m6A modification of viral RNA occur at different times during a single-cycle HIV-1 infection [29]. meRIP-seq was performed using cell-associated RNA from SupT1 cells infected with single-cycle HIV-1 for 12, 24, or 36 hrs post-infection (hpi). Each of these time points is earlier than the previously reported analyses, all performed at 96 hpi. m6A peak mapping at these earlier time points again showed enrichment of m6A in the 3’ end of the RNA, with peaks also detected in pol, pol/vif, and vpu (Figure 1A). One notable change in m6A mapping over time revealed a large peak overlapping the RNA packaging signal ψ at 36 hpi but not earlier. RNA isolated from virions released from the same cells at 36 hpi retains m6A modification in the env/nef and 3’ UTR regions, as well at the peak overlapping ψ, but is largely lacking prominent peaks in the remaining coding regions (Figure 1B). These results suggest that m6A modification patterns differ between packaged gRNA and translated viral transcripts. The peak width defined for these analyses is quite broad and ranges from ~200-500 nt, while meRIP-seq involves fragmentation of RNA to 100-200 nt in length. This low resolution prevents meaningful prediction of specific DRACH motifs that may be modified.
Another report comparing m6A modification of infected-cell HIV-1 RNA compared to RNA purified from viral particles also showed that the cell-associated RNA has a different pattern of methylation than that of packaged gRNA [30]. RNA was prepared from HEK293T cells transfected with HIV-1 proviral plasmid or from supernatant virus. Consistent with all other reports, both viral RNA preparations exhibited peaks overlapping env/rev, nef, and the 3’ UTR (Figure 1A, B). In contrast to the results reported by Cristinelli et al. [29], a peak was observed in the 5’ UTR of intracellular RNA that was absent from virion RNA. The authors conclude that m6A in the 5’ UTR regulates genome packaging and focus on this phenotype for subsequent functional studies.
Techniques such as meRIP-seq and PA-m6A-seq reveal the population-level modification state of fragmented RNA, and therefore cannot provide information on the presence of m6A in individual viral genomes or transcripts. Therefore, these data do not provide insight into whether there is differential m6A deposition among specific splice isoforms. The most recent analysis of m6A sites in HIV-1 RNA was performed using a modification of direct RNA sequencing (DRS) that resulted in a remarkable improvement of full-length genome reads from 0.01% to 34.9% [31]. This approach allowed for full-length, single-molecule analysis at single-nucleotide resolution for both viral gRNA and splice isoforms. Additional stringency was provided by using three independent bioinformatics tools for the identification of m6A signals. Seven common sites were identified across all three analyses. Interestingly, two of these sites, both in pol, were not located in canonical DRACH motif sequences (Figure 1A). Four sites in the 1.2 kb of the 3’ HIV-1 genome coincide with the common peaks identified in env/rev and the 3’ UTR in each of the previous studies [26,27,28,29]. These m6A are located at 8079, 8110, 8975, and 8989 of HIV-1NL4-3 [31].
Almost all available data describing the location of m6A in HIV-1 RNA is derived from cell lines infected in vitro. One study demonstrated that the pattern of viral RNA modification is similar in a CD4+ T cell line and primary CD4+ T cells infected ex vivo, suggesting that these results might be relevant to HIV-1 infected individuals [27]. The only information regarding in vivo HIV-1 infection comes from a transgenic rat model of HIV-1 infection [33]. These animals harbor a gag/pol deleted provirus that expresses viral genes in microglia and astrocytes [34]. meRIP-seq of RNA isolated from the hippocampus of these animals revealed m6A in the 5’ UTR and nef and 3’UTR, consistent with cells infected in vitro and ex vivo (Figure 1A) [27,33].
Differences in m6A site prediction among studies can arise for several reasons, including biological, technical, and computational differences in the experimental approach. Such variables include HIV-1 strain, cell type, virus purification method, sequencing approach, data processing, and bioinformatic tools. In addition to the issue of low resolution provided by meRIP-seq, concerns have been raised about reproducibility of the method, even when using the same experimental conditions [35]. Future studies would benefit from the use of sequencing approaches that allow for the identification of m6A at single-nucleotide resolution.
Despite some inconsistencies among studies, m6A located in env (2 sites) and the 3’ UTR (2 sites) have been reproducibly identified across several data sets. In addition, adenosine residues in viral DRACH motifs that are predicted to contain m6A are highly conserved in the HIV-1 genome that is otherwise highly susceptible to mutation [25,26,31,36]. In addition, only a very small fraction of the nearly 250 DRACH motifs in the HIV-1 full-length RNA [31] appear to contain m6A, suggesting that modification of these sites is very highly specific. These data suggest that these m6A modifications are especially important in facilitating efficient virus replication.
4. HIV-1 infection modulates the cellular RNA m6A profile
Epitranscriptomic modification of mRNA represents an important layer of gene expression regulation in mammalian cells. While much focus has been placed on determining the m6A profile of HIV-1 RNA, another area of interest is how viral infection influences changes in m6A modification of host cell transcripts. This can provide mechanistic insights into how HIV-1 induces changes in host cell gene expression, either directly through the action of viral proteins or because of the intrinsic antiviral response to infection.
Infection of various cell types including differentiated monocytic, microglial and CD4+ T cell lines as well as primary CD4+ T cells with HIV-1 induces an upregulation of m6A levels in total cellular RNA, through mechanisms that remain unclear [25,29,37,38,39]. Upregulation of m6A in CD4+ T cells and microglial cells is not due to changes in the expression of m6A writers or erasers [37]. It is possible that methyltransferase complex activity is being regulated at the level of complex formation, stability, or enzymatic activity.
In CD4+ T cells, m6A upregulation is also observed in cells treated with the reverse transcriptase inhibitor nevirapine, heat inactivated virus, and gp120, the surface unit of the HIV-1 envelope protein [37]. These data demonstrate that m6A upregulation is independent of virus replication. m6A upregulation occurs after treatment of cells with recombinant gp120 proteins from CXCR4- and CCR5-tropic HIV-1 and is blocked by occlusion of the primary receptor CD4 with anti-CD4 antibodies [37]. Whether HIV-1 co-receptor binding to gp120 is also required for m6A upregulation of cellular RNA remains unknown. Analysis of m6A levels in polyadenylated and non-coding RNA reveal upregulation of m6A in both, including ribosomal RNA (rRNA) [37]. The 18S and 28S rRNA subunits both contain a single m6A that is modified by the methyltransferases METTL5 and ZCCHC4, respectively [40,41]. Ribosomes will form normally in the absence of METTL5 or ZCCHC4, but the overall translational efficiency is reduced [40,42]. This raises the interesting possibility that HIV-1 may enhance its protein translation through m6A modification of the ribosome; however, this possible mechanism has not yet been explored.
High throughput sequencing approaches such as meRIP-seq allow for the identification of changes in the relative abundance of m6A-modified host cell mRNAs. The first transcriptome-wide meRIP-seq defining the topology of m6A in human cells showed an enrichment of m6A near stop codons and in coding sequences, particularly long exons [11]. Mapping of m6A in cellular transcripts from HIV-1-infected CD4+ T cell lines compared to uninfected controls reveals no significant changes in the distribution of m6A deposition to 5’ UTR, exons, introns, or 3’ UTR, with the majority of m6A sites found in exons and the 3’ UTR as expected [25,27,29].
Changes in relative abundance of m6A-modified transcripts alone does not indicate differential methylation of the RNA, because these changes could simply be a result of differential expression of m6A-modified transcripts. Therefore, true differential m6A methylation of HIV-1 infected cells can only be determined by measuring modification level and transcript abundance and comparing those data to the same transcript in uninfected control cells. Applying this strategy, over 3,615 transcripts were found to be hypermethylated upon HIV-1 infection compared to hypermethylation of only 777 transcripts [29]. These data support an overall upregulation of m6A modification of cellular transcripts in SupT1 cells upon HIV-1 infection. Comparing m6A methylation patterns at 12, 24, and 36 hrs post-infection, only 2% of differentially methylated transcripts were common among all three time points [29]. This shows that m6A modification patterns of cellular RNA are dynamic and temporally regulated during HIV-1 infection.
A new analysis combining both RNA-seq and meRIP-seq data showed that changes in transcript methylation occurred disproportionately in coding sequences compared to the 3’ UTR in a microglial cell line during a single-cycle HIV-1 infection [38]. Over 2,000 transcripts were reported to be hypo- or hyper-methylated in infected cells, suggesting that HIV-1 infection induces changes in gene expression at the level of m6A modification. Pathway analysis of these transcripts show significant enrichment of genes involved in signal transduction, including the Ras and MAPK signaling pathways [38]. Further studies will be required to determine the functional effect of differential methylation on these signaling pathways during HIV-1 infection of microglia.
5. HIV-1 m6A suppresses the induction of type I interferon (IFN-I) in macrophages
Internal 2’-O-methylation of HIV-1 RNA serves as an immune evasion strategy during HIV-1 infection of monocyte-derived macrophages and dendritic cells [43]. To test whether m6A also suppresses IFN-I in response to HIV-1 infection, viruses were produced in the presence of eraser (ALKBH5 or FTO) overexpression or knockout [44]. Differentiated monocytic U937 cells (macrophage-like cells) were then infected with HIV-1 containing low, normal, or high levels of m6A in the incoming RNA genome. The important advantage of manipulating m6A levels in producer cells rather than target cells is that it avoids the perturbation of cellular m6A levels so that observed phenotypes could be attributed specifically to m6A in the incoming HIV-1 gRNA. Using complementary approaches for modulating m6A, the authors demonstrate that the level of m6A in the HIV-1 genome is inversely correlated with the expression of IFN-α and IFN-β after transfection or infection of differentiated U937 cells [44]. The phenotype was observed in primary monocyte-derived macrophages transfected with HIV-1 RNA [44]. Taken together, the results suggest that m6A suppresses the expression of IFN-I by HIV-1 RNA. Delivery of RNA to cells by transfection does not recapitulate the pathway by which gRNA is delivered to cells during infection. Therefore, future mechanistic studies should be done using HIV-1 infected cells, particularly macrophages.
Viral RNA is detected in the host cell by protein sensors that recognize pathogen-associated molecular patterns in viral molecules to discriminate self from non-self RNA [45]. Binding of these sensors to their RNA ligands activates a signaling cascade to induce the expression of IFN-I. To determine which host cell cytosolic RNA sensor may be inhibited by m6A-modified HIV-1 RNA, U937 knockout (KO) or knockdown (KD) cell lines were generated lacking expression of retinoic acid-induced gene I (RIG-I) or melanoma differentiation-associated gene 5 (MDA5), respectively. These cells were transfected with a 42-mer HIV-1 derived RNA oligonucleotide with or without a single m6A. The sequence of the oligonucleotide corresponds to a portion of the HIV-1 5’ UTR of HIV-1, where potential m6A sites were predicted by meRIP-seq studies [25,27]. While the expression of IFN-α and IFN-β was lower in both RIG-I KO and MDA5 KD cells compared to control, RIG-I KO cells were no longer able to discriminate between oligonucleotides with or without m6A [44]. These data suggest that m6A may function in evading RIG-I sensing, and this would be consistent with a growing body of research in the field of innate immunity to RNA viruses [46,47,48,49]. Also, it was shown that the infection of differentiated U937 cells with m6A deficient HIV-1 induces more phosphorylation of IRF3 and IRF7 as compared to control viral particles. Based on this, it appears that the presence of m6A on HIV-1 RNA hinders IFN-1 production during the early stages of HIV-1 infection. However, a major limitation of this study is the use of RNA oligonucleotide transfection, especially given uncertainties regarding the exposure of HIV-1 genomes to the cytoplasm during transit to the nuclear pore [50,51,52,53]. It will be critical to determine whether m6A in the HIV-1 genome avoids detection by RIG-I in the context of infected cells, and if so, what is the underlying mechanisms. It is possible that other cellular RNA sensors might be involved in sensing m6A lower HIV-1 RNA.
6. m6A reader proteins negatively impact HIV-1 reverse transcription
The sequencing approaches described above demonstrate that m6A is present on viral genomes and transcripts. Therefore, m6A may impact both pre- and post-integration events in HIV-1 replication. Tirumuru et al. reported that YTHDF proteins have a negative impact on post-entry viral replication in several target cell types, including primary CD4+ T cells [27]. After virus entry, the measurable level of gag RNA drops relative to input, reflecting degradation of incoming genomes during reverse transcription (RT). The levels of gag RNA then increase after integration and transcription of new viral RNA from the provirus. However, gag RNA levels fail to increase in cells constitutively overexpressing YTHDF proteins, which is consistent with degradation of gRNA and a concomitant decrease in early and late RT products due to the lack of RT template [27,54]. Both cell-based and in vitro binding assays demonstrated binding of YTHDF proteins to HIV-1 RNA [27,54]. However, these binding assays were performed in vitro or post-lysis of infected cells. Therefore, it is not clear that the HIV-1 genome and transcripts interact with all three YTHDF proteins in cells during viral infection.
There are opposing results regarding whether YTHDF reader proteins are incorporated into HIV-1 particles. One study shows that both overexpressed and endogenous YTHDF3 are incorporated into virions in a nucleocapsid-dependent manner in both HEK293T cells transfected with HIV-1 proviral plasmid and infected A3R5-Rev-GFP cells (HIV Rev dependent reporter CD4+ T cells expressing CCR5) , but that the incorporated reader is then degraded by the viral protease [55]. In contrast, two other studies, both using highly purified virions, reported no detectable incorporation of FLAG-tagged YTHDF1-3 proteins into virus particles prepared from HEK293T cells transfected with HIV-1 proviral plasmid [54,56]. In one of these latter studies, YTHDF1-3 were not detected in virions even when HIV-1 protease was inhibited [56]. The reason for this discrepancy could be due to differences in HIV-1 purification methods. Future studies may address whether HIV-1 RNA is degraded in the presence of endogenous YTHDF proteins in CD4+ T cells or macrophages, and the underlying mechanisms.
7. m6A regulates HIV-1 RNA splicing and nuclear export
HIV-1 gene expression is driven by the viral 5’ long terminal repeat (LTR) of integrated provirus in a productively infected cell. The deposition of m6A on nascent transcripts occurs co-transcriptionally in the nucleus [57]. It is therefore possible for m6A to affect RNA splicing and nuclear export. The first study reporting the presence of m6A in HIV-1 RNA identified m6A enrichment in the RRE, with two potential methylation sites in hairpin loop IIB [25]. The RRE is a highly conserved and structured RNA element in HIV-1 RNA that is essential for the nuclear export of partially spliced or unspliced transcripts [58]. The authors hypothesized that m6A within the RRE may be required for efficient viral RNA export. Mutation of the predicted methylation site in the hairpin IIB bulge dramatically reduced the levels of env mRNA in HEK293T cells transfected with HIV-1 proviral DNA plasmid. Subcellular fractionation prior to measurement of env mRNA indicated a defect in nuclear export. However, Rev binding to these mutated RRE was not measured. A variety of in vitro assays to assess the structure and Rev binding properties of synthesized RRE IIB with or without m6A argued against a significant impact of m6A modification on IIB structure [59]. Further, a separate study made the same RRE mutations in a different HIV-1 laboratory strain and showed that when env and rev are provided in trans, there is no difference in viral gene expression in either producer or target cells [36]. The RRE was not identified as a site of m6A modification or reader binding in any subsequent studies [26,27,28]. Therefore, the function of m6A in the RRE remains to be confirmed.
The nuclear m6A reader YTHDC1 is known to be involved in RNA stability, splicing, and nuclear export of m6A-modified viral and cellular RNA [3,60,61]. Two independent studies assessed HIV-1 RNA splicing and nuclear export under conditions of overexpression or depletion of YTHDC1 during single-round infections [26,62]. Both studies found that YTHDC1 is required for not only maintaining overall viral RNA levels, but also for appropriate selection of splice sites. Splicing of HIV-1 transcripts is complex and involves the utilization of multiple splice donor and acceptor sites [63]. Specifically, YTHDC1 increased the use of more 3’ splice acceptor sites with a concomitant decrease in the use of splice acceptor sites further 5’ in the HIV-1 genome [56]. Manipulating the expression levels of YTHDC1 had no effect on the cellular NONO mRNA that is not m6A-modified [56]. This suggests that the observed changes in HIV-1 RNA upon YTHDC1 silencing or overexpression are due to the presence of m6A (Figure 2). Overexpressed and endogenous YTHDC1 bind to spliced and unspliced HIV-1 RNA in infected cells in a METTL3-dependent manner [62] (Figure 2). YTHDC1 binding sites were identified adjacent to HIV-1 splice acceptor (SA) sites 3 and 7 [56]. However, when the predicted m6A site near SA3 was mutated, there was no effect on the utilization of SA3 [56]. Therefore, whether YTHDC1 is acting directly on viral m6A is unclear. Both groups also reported that overexpression or depletion of YTHDC1 had no effect on the nuclear/cytoplasmic accumulation of unspliced, partially spliced, or completely spliced viral RNA [56,62]. These results argue against a role for YTHDC1 in HIV-1 viral RNA nuclear export, which is unexpected given the role of YTHDC1 in nuclear export of cellular mRNA [3].
Several independent studies reported m6A modification in the 3’ region of HIV-1 RNA overlap between env and the second exon of rev [26,27,28,29,30,33]. DRS identified the modified adenosine in a DRACH motif in the env/rev overlap at position 8079 of HIV-1NL4-3, downstream of SA7 [31]. Two other high confidence m6A are located in the 3’ UTR. These m6A represent three of the four most prominent sites of m6A modification, in agreement with other studies, and were mutated for functional studies. Interestingly, mutation of these sites individually had no significant effect on viral protein expression, the proportion of unspliced RNA, p24 release, or virion infectivity [31]. Only when all three adenosines were mutated was there a significant reduction in virus infectivity. The reduction in HIV-1 replication is likely due to a decrease in the abundance of unspliced RNA, which is necessary to produce infectious virions. The triple mutation also results in modest but significant increases in the proportion of partially and completely spliced viral RNA relative to total RNA [31]. This suggests functional redundancy of the individual m6A and that the modifications are involved in the regulation of splicing. This idea is further supported by the observation that completely spliced RNAs are more m6A-modified at these three predominant sites than unspliced RNAs [31]. It will be interesting to uncover the mechanisms by which these three m6A site modulate HIV-1 RNA splicing, whether other m6A are also functionally redundant, and how a single nucleotide modification can substitute for the function of another.
8. m6A enhances post-integration HIV-1 RNA abundance, stability, and translation
One strategy to indirectly test whether m6A inhibits or enhances HIV-1 replication is to manipulate the expression of writer complex components or m6A erasers in target cells. Silencing or overexpression of writers or erasers would presumably only effect viral RNA that is made post-integration; however, this has not been assessed in detail. Several studies that have used this approach agree that in general, m6A is required for efficient HIV-1 replication as measured by viral RNA, viral protein, and progeny particle release [25,27,30,56]. One exception to this general phenotype is a study using single-cycle HIV-1 transduction of HeLa cells [62]. The exact reason for the discrepancy is unknown but could be due to the kinetics of viral gene expression in this experimental system compared to replication competent HIV-1 entry by plasma membrane fusion.
Another area of controversy in the field of HIV-1 and m6A is the effect of readers on viral replication. One study measured HIV-1 RNA and protein levels in HEK293T producer cells overexpressing each YTHDF1-3 reader individually and found that in all cases, both HIV-1 RNA and protein levels were increased [26] (Figure 2). In addition, silencing of YTHDF2 decreases the half-life of HIV-1 RNA and cellular RNAs containing m6A, yet had no significant effect on cellular RNAs that are not though to be modified [56]. These results all suggest that m6A readers enhance HIV-1 replication by stabilizing viral RNA. The overexpressed readers are assumed to be acting on viral RNA, however, enhanced replication can also be due to changes in cellular gene expression that indirectly affect viral gene expression. Consistent results were reported after YTHDF2 overexpression or knockout in CEM T-cells infected with single-cycle HIV-1 [26,56]. In these cells there was a reduction in Gag protein levels and p24 levels of released virions, again suggesting that YTHDF2 is required for efficient viral gene expression (Figure 2). These results are in direct opposition to other reports that show a decrease in viral RNA and protein in infected HeLa, Jurkat, and primary CD4+ T cells [27,54]. As discussed above, this was concluded to be a result of inhibition of RT through binding of readers to incoming viral RNA leading ultimately to its degradation [27,54,55]. One possibility for these seemingly opposing results is that m6A has a negative effect on HIV-1 gRNA but a potential benefit at post-integration phases of infection. The use of different cell types, viruses, and length of infection make direct comparison of data from different groups imprecise.
Two potential functions for viral RNA m6A are destabilization of RNA or enhanced translation efficiency mediated by m6A readers [1,2]. To assess the functional significance of m6A modifications in the 3’-UTR of HIV-1 RNA, Kennedy et al. placed the wild-type or m6A-deficient (A to G mutated) forms of the HIV-1 3’-UTR downstream of a reporter gene [26]. Their results revealed that substituting A with G significantly reduced both RNA and protein levels of the reporter. These data argue against RNA destabilization or enhanced translation and indicate that m6A in the 3’ UTR of HIV-1 RNA enhance the abundance of an unrelated RNA (Figure 2). Recruiting any of the YTHDF readers to the 3’UTR of a reporter, independent of m6A, also resulted in enhanced expression of the reporter [26]. Taken together, these results suggest that m6A modification in the 3’UTR of HIV-1 mRNA enhances viral gene expression at the mRNA and protein levels.
9. m6A inhibits HIV-1 RNA packaging and reduces virion infectivity
Several groups reported the presence of m6A in the 5’ UTR of HIV-1 RNA [27,29,30]. The 5’ UTR is highly structured and contains several regulatory elements that are indispensable for efficient HIV-1 replication [64]. One important aspect of HIV-1 replication that is mediated by RNA sequences in the 5’ UTR is the selection of full-length RNA genomes for packaging into progeny virions [65]. There are two DRACH motifs of particular interest in the 5’UTR. One is in the primer binding site (PBS) required for the initiation of RT and another is near the dimer initiation sequence (DIS) that is involved in a monomer-dimer switch that regulates whether a given full-length HIV-1 RNA is translated (monomer) or selected for packaging (dimer) [54].
Mutation of either of these two adenosine residues led to a slight increase in p24 levels in HEK293T producer cells, yet reduced infectivity of the release virions when used to infect target cells [54]. RNA secondary structure prediction in silico showed an altered structure of the PBS but not the DIS because of the mutations [54]. However, due to the possibility of the disruption of primer binding for RT or Gag binding during particle assembly as a result of these mutations, the interpretation of these data with respect to m6A modification can be multifaceted.
Several lines of evidence suggest that m6A modification impairs packaging of gRNA. meRIP-seq data derived from intracellular viral RNA and virion RNA revealed a prominent peak representing m6A in the 5’ UTR of intracellular RNA, but not RNA from purified virus particles [30]. In addition, the m6A/A level of intracellular viral RNA was significantly higher than that in packaged RNA [30]. This raises the possibility that m6A is playing a role in the preferential selection of gRNA molecules for packaging. Overexpression of METTL3/14, which presumably increases m6A modification of viral RNA, resulted in higher levels of Gag protein and lower levels of RNA packaging, whereas silencing of the methyltransferase complex resulted in the opposite phenotype [30]. These results are consistent with the observed decrease in Gag protein in other studies where METTL3 is silenced [25,27]. Interestingly, the interaction between Gag and full-length viral RNA in the cytoplasm was decreased under conditions of METTL3/14 overexpression, further supporting the idea that increased methylation of viral RNA inhibits is ability to be packaged [30]. Unexpectedly, deletion of the two adenosine residues that are potential sites of methylation resulted in a significant increase in overall m6A content of viral RNA and a significant decrease in packaging efficiency of gRNA [30]. Due to deletion rather than mutation of these adenosines, it is unknown whether the proper secondary structure of the 5’ UTR was affected. Another study also demonstrated that virion RNA contains fewer m6A modifications that viral transcripts, however this study only reported modification of three adenosines in the 3’ ~1.4 kb of the viral genome. In addition, YTHDF proteins bind to m6A modified gRNA and form a complex with Gag in an RNA-dependent manner [27,54]. It is therefore possible that YTHDF binding prevents efficient binding of Gag and therefore inhibits packaging. Further experiments are needed to investigate this hypothesis.
HIV-1 Gag colocalizes in the nucleus with unspliced viral RNA [66]. Interestingly, Gag also colocalizes with FTO exclusively in the nucleus and there is an increase in viral RNA m6A levels in the absence of gag expression that can be rescued when Gag is provided in trans [30]. These data suggest that FTO mediates demethylation of viral RNA in a Gag-dependent manner (Figure 2). However, while ALKBH5 and FTO can both demethylate HIV-1 RNA when each demethylase is overexpressed, it is not known which of these proteins acts on viral RNA under conditions of endogenous expression levels. Emerging evidence supports a predominant role for FTO in the removal of methyl groups from m6Am and not m6A in human cells [67,68]. Further studies are needed to determine the molecular mechanisms of the removal of m6A from HIV-1 RNA and how this process is regulated to control translation or packaging of full-length gRNA.
10. m6A modulation may have therapeutic potential for HIV-1-infected individuals
Overall, it is clear that m6A modification of HIV-1 RNA is required for proper regulation of viral gene expression, although the molecular mechanisms remain to be elucidated. This raises the question of whether m6A deposition is a candidate target for anti-viral therapy or for new strategies towards HIV-1 functional cure. Indeed, the methylation inhibitor 3-deaza-adenosine was found to reduce HIV-1 replication nearly three decades ago [69]. Recently, compounds that are highly specific for activation or inhibition of components of the m6A methyltransferase complex have been developed [70,71]. Compounds that activate the METTL3/14/WTAP complex increase HIV-1 production in latently infected cells subject to reactivation [39]. Naturally, the use of compounds that enhance or inhibit m6A modification in a clinical setting would raise concerns about pleiotropic effects on the m6A modification of cellular RNA. Nevertheless, METTL3 inhibitors have shown promise in the treatment of cancers in animal models and are currently being tested in human clinical trials [71,72,73].
It will first be important to determine whether and how m6A regulates HIV-1 replication in infected individuals. A study of gene expression in peripheral blood mononuclear cells (PBMC) isolated from HIV-1-infected individuals showed significant correlation between the size of the latent HIV-1 reservoir and the expression of METTL3 [74]. Conversely, there was an inverse correlation between the size the latent reservoir and the expression of ALKBH5. These data suggest that higher levels of HIV-1 replication may correlate with higher levels of m6A in RNA from PBMC of HIV-1-infected patients. However, m6A levels were not measured in this study. An elevation of cellular m6A levels in HIV-1 infected individuals would be consistent with the results of in vitro and ex vivo infections. Future studies using blood or tissue CD4+ T cells collected from HIV-1 patients with viremia and on suppressive antiretroviral therapy will be useful in determining how m6A levels are affected by HIV-1 infection in vivo.
6. Future Perspectives
Several predicted m6A sites in the HIV-1 RNA are in regions overlapping with structural features that are important for virus replication or near functional domains such as SA or NF-kB binding sites [25,26,31,54]. Mutation of these residues can therefore lead to perturbation of RNA secondary or tertiary structure, although structure is often not assessed in these studies. An alternative approach to investigating the role of m6A during viral replication is by manipulating the levels of expression of m6A writers or erasers in cells. However, this strategy is not sequence-specific and can also lead to indirect effects that are not straightforward to interpret. This can be partially addressed when investigating the role of m6A during the pre-integration stages of infection by using viruses produced in cells overexpressing erasers to specifically reduce m6A on viral RNA only without perturbing modification of host cells RNA [44,75]. However, this approach also does not allow for the specificity required for investigating individual m6A function. Nevertheless, in the absence of robust techniques allowing for better specificity, many groups have investigated how components of the m6A methyltransferase complex and m6A demethylases influence HIV-1 replication.
With rapid advancements in the technology for studying the role of epitranscriptomic modifications, mechanistic studies can be enhanced. Many m6A sequencing approaches have been developed that now allow for identification of m6A at single-nucleotide resolution, which is a vast improvement upon original meRIP-seq and even PA-m6A-seq approaches [76,77,78,79]. In addition, CRISPR/Cas9 technology has been harnessed for the sequence specific addition or removal of methyl groups to adenosines of interest [20,21,22,23,24,80]. Whether these techniques are robust enough to be of use for the delineation of m6A-dependent phenotypes in the context of viral infections remains to be confirmed. Regardless, these more powerful tools can be harnessed to explore the molecular mechanisms by which m6A modification of HIV-1 RNAs regulates various phases of the viral life cycle. These mechanistic studies will benefit our understanding of HIV-1 replication and viral persistence.
Author Contributions
Conceptualization, T.M., S.P., L.W.; writing —Original draft preparation, T.M., S.P., S.H.; writing —review and editing, T.M., S.P., L.W.; literature search, T.M., S.P., S.H., L.W.; supervision, L.W. T.M. and S.P. contributed equally. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health (NIH) of the USA, grant numbers R61 AI169659, R21 AI170070, and R21 AI159546.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Wang, X.; Zhao, B. S.; Roundtree, I. A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C., N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, (6), 1388-99. [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G. C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; Ren, B.; Pan, T.; He, C., N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, (7481), 117-20. [CrossRef]
- Roundtree, I. A.; Luo, G. Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; Xie, P.; He, E.; Shen, B.; He, C., YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife 2017, 6. [CrossRef]
- Zhao, X.; Yang, Y.; Sun, B. F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y. J.; Ping, X. L.; Chen, Y. S.; Wang, W. J.; Jin, K. X.; Wang, X.; Huang, C. M.; Fu, Y.; Ge, X. M.; Song, S. H.; Jeong, H. S.; Yanagisawa, H.; Niu, Y.; Jia, G. F.; Wu, W.; Tong, W. M.; Okamoto, A.; He, C.; Rendtlew Danielsen, J. M.; Wang, X. J.; Yang, Y. G., FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res 2014, 24, (12), 1403-19. [CrossRef]
- Lavi, S.; Shatkin, A. J., Methylated simian virus 40-specific RNA from nuclei and cytoplasm of infected BSC-1 cells. Proc Natl Acad Sci U S A 1975, 72, (6), 2012-6. [CrossRef]
- Williams, G. D.; Gokhale, N. S.; Horner, S. M., Regulation of Viral Infection by the RNA Modification N6-Methyladenosine. Annu Rev Virol 2019, 6, (1), 235-253. [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; Dai, Q.; Chen, W.; He, C., A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 2014, 10, (2), 93-5. [CrossRef]
- Ping, X. L.; Sun, B. F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W. J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y. S.; Zhao, X.; Li, A.; Yang, Y.; Dahal, U.; Lou, X. M.; Liu, X.; Huang, J.; Yuan, W. P.; Zhu, X. F.; Cheng, T.; Zhao, Y. L.; Wang, X.; Rendtlew Danielsen, J. M.; Liu, F.; Yang, Y. G., Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 2014, 24, (2), 177-89. [CrossRef]
- Patil, D. P.; Chen, C. K.; Pickering, B. F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S. R., m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, (7620), 369-373. [CrossRef]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; Wang, F.; Wang, X.; Shen, B.; Wang, Y.; Feng, X.; He, C.; Liu, J., VIRMA mediates preferential m(6)A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov 2018, 4, 10. [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; Sorek, R.; Rechavi, G., Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, (7397), 201-6. [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y. G.; He, C., N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 2011, 7, (12), 885-7.
- Zheng, G.; Dahl, J. A.; Niu, Y.; Fedorcsak, P.; Huang, C. M.; Li, C. J.; Vagbo, C. B.; Shi, Y.; Wang, W. L.; Song, S. H.; Lu, Z.; Bosmans, R. P.; Dai, Q.; Hao, Y. J.; Yang, X.; Zhao, W. M.; Tong, W. M.; Wang, X. J.; Bogdan, F.; Furu, K.; Fu, Y.; Jia, G.; Zhao, X.; Liu, J.; Krokan, H. E.; Klungland, A.; Yang, Y. G.; He, C., ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 2013, 49, (1), 18-29. [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A. V.; Patil, D. P.; Linder, B.; Pickering, B. F.; Vasseur, J. J.; Chen, Q.; Gross, S. S.; Elemento, O.; Debart, F.; Kiledjian, M.; Jaffrey, S. R., Reversible methylation of m(6)A(m) in the 5' cap controls mRNA stability. Nature 2017, 541, (7637), 371-375. [CrossRef]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B. S.; Ma, H.; Hsu, P. J.; Liu, C.; He, C., YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res 2017, 27, (3), 315-328. [CrossRef]
- Zaccara, S.; Jaffrey, S. R., A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell 2020, 181, (7), 1582-1595 e18. [CrossRef]
- Zaccara, S.; Ries, R. J.; Jaffrey, S. R., Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 2019, 20, (10), 608-624.
- Meyer, K. D.; Jaffrey, S. R., Rethinking m(6)A Readers, Writers, and Erasers. Annu Rev Cell Dev Biol 2017, 33, 319-342.
- Zou, Z.; Sepich-Poore, C.; Zhou, X.; Wei, J.; He, C., The mechanism underlying redundant functions of the YTHDF proteins. Genome Biol 2023, 24, (1), 17. [CrossRef]
- Liu, X. M.; Zhou, J.; Mao, Y.; Ji, Q.; Qian, S. B., Programmable RNA N(6)-methyladenosine editing by CRISPR-Cas9 conjugates. Nat Chem Biol 2019, 15, (9), 865-871. [CrossRef]
- Li, J.; Chen, Z.; Chen, F.; Xie, G.; Ling, Y.; Peng, Y.; Lin, Y.; Luo, N.; Chiang, C. M.; Wang, H., Targeted mRNA demethylation using an engineered dCas13b-ALKBH5 fusion protein. Nucleic Acids Res 2020, 48, (10), 5684-5694. [CrossRef]
- Wilson, C.; Chen, P. J.; Miao, Z.; Liu, D. R., Programmable m(6)A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat Biotechnol 2020, 38, (12), 1431-1440. [CrossRef]
- Chang, C.; Ma, G.; Cheung, E.; Hutchins, A. P., A programmable system to methylate and demethylate N(6)-methyladenosine (m(6)A) on specific RNA transcripts in mammalian cells. J Biol Chem 2022, 298, (11), 102525. [CrossRef]
- Shi, H.; Xu, Y.; Tian, N.; Yang, M.; Liang, F. S., Inducible and reversible RNA N(6)-methyladenosine editing. Nat Commun 2022, 13, (1), 1958. [CrossRef]
- Lichinchi, G.; Gao, S.; Saletore, Y.; Gonzalez, G. M.; Bansal, V.; Wang, Y.; Mason, C. E.; Rana, T. M., Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol 2016, 1, 16011. [CrossRef]
- Kennedy, E. M.; Bogerd, H. P.; Kornepati, A. V.; Kang, D.; Ghoshal, D.; Marshall, J. B.; Poling, B. C.; Tsai, K.; Gokhale, N. S.; Horner, S. M.; Cullen, B. R., Posttranscriptional m(6)A Editing of HIV-1 mRNAs Enhances Viral Gene Expression. Cell Host Microbe 2016, 19, (5), 675-85.
- Tirumuru, N.; Zhao, B. S.; Lu, W.; Lu, Z.; He, C.; Wu, L., N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. eLife 2016, 5.
- Courtney, D. G.; Tsai, K.; Bogerd, H. P.; Kennedy, E. M.; Law, B. A.; Emery, A.; Swanstrom, R.; Holley, C. L.; Cullen, B. R., Epitranscriptomic Addition of m(5)C to HIV-1 Transcripts Regulates Viral Gene Expression. Cell Host Microbe 2019, 26, (2), 217-227 e6. [CrossRef]
- Cristinelli, S.; Angelino, P.; Janowczyk, A.; Delorenzi, M.; Ciuffi, A., HIV Modifies the m6A and m5C Epitranscriptomic Landscape of the Host Cell. Frontiers in Virology 2021, 1. [CrossRef]
- Pereira-Montecinos, C.; Toro-Ascuy, D.; Ananias-Saez, C.; Gaete-Argel, A.; Rojas-Fuentes, C.; Riquelme-Barrios, S.; Rojas-Araya, B.; Garcia-de-Gracia, F.; Aguilera-Cortes, P.; Chnaiderman, J.; Acevedo, M. L.; Valiente-Echeverria, F.; Soto-Rifo, R., Epitranscriptomic regulation of HIV-1 full-length RNA packaging. Nucleic Acids Res 2022, 50, (4), 2302-2318. [CrossRef]
- Baek, A.; Lee, G.; Golconda, S.; Rayhan, A.; Manganaris, A.; Chen, S.; Tirumuru, R.; Yu, H.; Shihyoung, K.; Kimmel, C.; Zablocki, O.; Sullivan, M.; Addepalli, B.; Wu, L.; Kim, S., Single-RNA-level analysis of full-length HIV-1 RNAs reveals functional redundancy of m6As. Research Square 2023.
- Chen, K.; Lu, Z.; Wang, X.; Fu, Y.; Luo, G. Z.; Liu, N.; Han, D.; Dominissini, D.; Dai, Q.; Pan, T.; He, C., High-resolution N(6) -methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew Chem Int Ed Engl 2015, 54, (5), 1587-90.
- Fu, Y.; Zorman, B.; Sumazin, P.; Sanna, P. P.; Repunte-Canonigo, V., Epitranscriptomics: Correlation of N6-methyladenosine RNA methylation and pathway dysregulation in the hippocampus of HIV transgenic rats. PLoS One 2019, 14, (1), e0203566. [CrossRef]
- Reid, W.; Sadowska, M.; Denaro, F.; Rao, S.; Foulke, J., Jr.; Hayes, N.; Jones, O.; Doodnauth, D.; Davis, H.; Sill, A.; O'Driscoll, P.; Huso, D.; Fouts, T.; Lewis, G.; Hill, M.; Kamin-Lewis, R.; Wei, C.; Ray, P.; Gallo, R. C.; Reitz, M.; Bryant, J., An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc Natl Acad Sci U S A 2001, 98, (16), 9271-6. [CrossRef]
- McIntyre, A. B. R.; Gokhale, N. S.; Cerchietti, L.; Jaffrey, S. R.; Horner, S. M.; Mason, C. E., Limits in the detection of m(6)A changes using MeRIP/m(6)A-seq. Sci Rep 2020, 10, (1), 6590. [CrossRef]
- Lu, W.; Chen, S.; Yu, J.; Behrens, R.; Wiggins, J.; Sherer, N.; Liu, S. L.; Xiong, Y.; Xiang, S. H.; Wu, L., The Polar Region of the HIV-1 Envelope Protein Determines Viral Fusion and Infectivity by Stabilizing the gp120-gp41 Association. J Virol 2019, 93, (7). [CrossRef]
- Tirumuru, N.; Wu, L., HIV-1 envelope proteins up-regulate N (6)-methyladenosine levels of cellular RNA independently of viral replication. J Biol Chem 2019, 294, (9), 3249-3260. [CrossRef]
- Peng, Q.; Qiao, J.; Li, W.; You, Q.; Hu, S.; Liu, Y.; Liu, W.; Hu, K.; Sun, B., Global m6A methylation and gene expression patterns in human microglial HMC3 cells infected with HIV-1. Heliyon 2023, 9, (11), e21307. [CrossRef]
- Selberg, S.; Zusinaite, E.; Herodes, K.; Seli, N.; Kankuri, E.; Merits, A.; Karelson, M., HIV Replication Is Increased by RNA Methylation METTL3/METTL14/WTAP Complex Activators. ACS Omega 2021, 6, (24), 15957-15963.
- Ma, H.; Wang, X.; Cai, J.; Dai, Q.; Natchiar, S. K.; Lv, R.; Chen, K.; Lu, Z.; Chen, H.; Shi, Y. G.; Lan, F.; Fan, J.; Klaholz, B. P.; Pan, T.; Shi, Y.; He, C., N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol 2019, 15, (1), 88-94. [CrossRef]
- van Tran, N.; Ernst, F. G. M.; Hawley, B. R.; Zorbas, C.; Ulryck, N.; Hackert, P.; Bohnsack, K. E.; Bohnsack, M. T.; Jaffrey, S. R.; Graille, M.; Lafontaine, D. L. J., The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res 2019, 47, (15), 7719-7733. [CrossRef]
- Rong, B.; Zhang, Q.; Wan, J.; Xing, S.; Dai, R.; Li, Y.; Cai, J.; Xie, J.; Song, Y.; Chen, J.; Zhang, L.; Yan, G.; Zhang, W.; Gao, H.; Han, J. J.; Qu, Q.; Ma, H.; Tian, Y.; Lan, F., Ribosome 18S m(6)A Methyltransferase METTL5 Promotes Translation Initiation and Breast Cancer Cell Growth. Cell Rep 2020, 33, (12), 108544. [CrossRef]
- Ringeard, M.; Marchand, V.; Decroly, E.; Motorin, Y.; Bennasser, Y., FTSJ3 is an RNA 2'-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature 2019, 565, (7740), 500-504. [CrossRef]
- Chen, S.; Kumar, S.; Espada, C. E.; Tirumuru, N.; Cahill, M. P.; Hu, L.; He, C.; Wu, L., N6-methyladenosine modification of HIV-1 RNA suppresses type-I interferon induction in differentiated monocytic cells and primary macrophages. PLoS Pathog 2021, 17, (3), e1009421. [CrossRef]
- Rehwinkel, J.; Gack, M. U., RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol 2020, 20, (9), 537-551. [CrossRef]
- Lu, M.; Zhang, Z.; Xue, M.; Zhao, B. S.; Harder, O.; Li, A.; Liang, X.; Gao, T. Z.; Xu, Y.; Zhou, J.; Feng, Z.; Niewiesk, S.; Peeples, M. E.; He, C.; Li, J., N(6)-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat Microbiol 2020, 5, (4), 584-598.
- Lu, M.; Xue, M.; Wang, H. T.; Kairis, E. L.; Ahmad, S.; Wei, J.; Zhang, Z.; Liu, Q.; Zhang, Y.; Gao, Y.; Garcin, D.; Peeples, M. E.; Sharma, A.; Hur, S.; He, C.; Li, J., Nonsegmented Negative-Sense RNA Viruses Utilize N(6)-Methyladenosine (m(6)A) as a Common Strategy To Evade Host Innate Immunity. J Virol 2021, 95, (9).
- Qiu, W.; Zhang, Q.; Zhang, R.; Lu, Y.; Wang, X.; Tian, H.; Yang, Y.; Gu, Z.; Gao, Y.; Yang, X.; Cui, G.; Sun, B.; Peng, Y.; Deng, H.; Peng, H.; Yang, A.; Yang, Y. G.; Yang, P., N(6)-methyladenosine RNA modification suppresses antiviral innate sensing pathways via reshaping double-stranded RNA. Nat Commun 2021, 12, (1), 1582. [CrossRef]
- Li, N.; Hui, H.; Bray, B.; Gonzalez, G. M.; Zeller, M.; Anderson, K. G.; Knight, R.; Smith, D.; Wang, Y.; Carlin, A. F.; Rana, T. M., METTL3 regulates viral m6A RNA modification and host cell innate immune responses during SARS-CoV-2 infection. Cell Rep 2021, 35, (6), 109091.
- Burdick, R. C.; Li, C.; Munshi, M.; Rawson, J. M. O.; Nagashima, K.; Hu, W. S.; Pathak, V. K., HIV-1 uncoats in the nucleus near sites of integration. Proc Natl Acad Sci U S A 2020, 117, (10), 5486-5493. [CrossRef]
- Selyutina, A.; Persaud, M.; Lee, K.; KewalRamani, V.; Diaz-Griffero, F., Nuclear Import of the HIV-1 Core Precedes Reverse Transcription and Uncoating. Cell Rep 2020, 32, (13), 108201. [CrossRef]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E. M., Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat Microbiol 2020, 5, (9), 1088-1095. [CrossRef]
- Li, C.; Burdick, R. C.; Nagashima, K.; Hu, W. S.; Pathak, V. K., HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc Natl Acad Sci U S A 2021, 118, (10). [CrossRef]
- Lu, W.; Tirumuru, N.; St Gelais, C.; Koneru, P. C.; Liu, C.; Kvaratskhelia, M.; He, C.; Wu, L., N(6)-Methyladenosine-binding proteins suppress HIV-1 infectivity and viral production. J Biol Chem 2018, 293, (34), 12992-13005. [CrossRef]
- Jurczyszak, D.; Zhang, W.; Terry, S. N.; Kehrer, T.; Bermudez Gonzalez, M. C.; McGregor, E.; Mulder, L. C. F.; Eckwahl, M. J.; Pan, T.; Simon, V., HIV protease cleaves the antiviral m6A reader protein YTHDF3 in the viral particle. PLoS Pathog 2020, 16, (2), e1008305. [CrossRef]
- Tsai, K.; Bogerd, H. P.; Kennedy, E. M.; Emery, A.; Swanstrom, R.; Cullen, B. R., Epitranscriptomic addition of m(6)A regulates HIV-1 RNA stability and alternative splicing. Genes Dev 2021, 35, (13-14), 992-1004. [CrossRef]
- Knuckles, P.; Carl, S. H.; Musheev, M.; Niehrs, C.; Wenger, A.; Buhler, M., RNA fate determination through cotranscriptional adenosine methylation and microprocessor binding. Nat Struct Mol Biol 2017, 24, (7), 561-569. [CrossRef]
- Malim, M. H.; Hauber, J.; Le, S. Y.; Maizel, J. V.; Cullen, B. R., The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature 1989, 338, (6212), 254-7. [CrossRef]
- Chu, C. C.; Liu, B.; Plangger, R.; Kreutz, C.; Al-Hashimi, H. M., m6A minimally impacts the structure, dynamics, and Rev ARM binding properties of HIV-1 RRE stem IIB. PLoS One 2019, 14, (12), e0224850. [CrossRef]
- Shima, H.; Matsumoto, M.; Ishigami, Y.; Ebina, M.; Muto, A.; Sato, Y.; Kumagai, S.; Ochiai, K.; Suzuki, T.; Igarashi, K., S-Adenosylmethionine Synthesis Is Regulated by Selective N(6)-Adenosine Methylation and mRNA Degradation Involving METTL16 and YTHDC1. Cell Rep 2017, 21, (12), 3354-3363. [CrossRef]
- Kasowitz, S. D.; Ma, J.; Anderson, S. J.; Leu, N. A.; Xu, Y.; Gregory, B. D.; Schultz, R. M.; Wang, P. J., Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet 2018, 14, (5), e1007412. [CrossRef]
- N'Da Konan, S.; Segeral, E.; Bejjani, F.; Bendoumou, M.; Ait Said, M.; Gallois-Montbrun, S.; Emiliani, S., YTHDC1 regulates distinct post-integration steps of HIV-1 replication and is important for viral infectivity. Retrovirology 2022, 19, (1), 4.
- Emery, A.; Swanstrom, R., HIV-1: To Splice or Not to Splice, That Is the Question. Viruses 2021, 13, (2). [CrossRef]
- Berkhout, B., Multiple biological roles associated with the repeat (R) region of the HIV-1 RNA genome. Adv Pharmacol 2000, 48, 29-73.
- D'Souza, V.; Summers, M. F., How retroviruses select their genomes. Nat Rev Microbiol 2005, 3, (8), 643-55. [CrossRef]
- Tuffy, K. M.; Maldonado, R. J. K.; Chang, J.; Rosenfeld, P.; Cochrane, A.; Parent, L. J., HIV-1 Gag Forms Ribonucleoprotein Complexes with Unspliced Viral RNA at Transcription Sites. Viruses 2020, 12, (11). [CrossRef]
- Sun, H.; Li, K.; Zhang, X.; Liu, J.; Zhang, M.; Meng, H.; Yi, C., m(6)Am-seq reveals the dynamic m(6)Am methylation in the human transcriptome. Nat Commun 2021, 12, (1), 4778. [CrossRef]
- Garcia-Campos, M. A.; Edelheit, S.; Toth, U.; Safra, M.; Shachar, R.; Viukov, S.; Winkler, R.; Nir, R.; Lasman, L.; Brandis, A.; Hanna, J. H.; Rossmanith, W.; Schwartz, S., Deciphering the "m(6)A Code" via Antibody-Independent Quantitative Profiling. Cell 2019, 178, (3), 731-747 e16.
- Flexner, C. W.; Hildreth, J. E.; Kuncl, R. W.; Drachman, D. B., 3-Deaza-adenosine and inhibition of HIV. Lancet 1992, 339, (8790), 438. [CrossRef]
- Selberg, S.; Blokhina, D.; Aatonen, M.; Koivisto, P.; Siltanen, A.; Mervaala, E.; Kankuri, E.; Karelson, M., Discovery of Small Molecules that Activate RNA Methylation through Cooperative Binding to the METTL3-14-WTAP Complex Active Site. Cell Rep 2019, 26, (13), 3762-3771 e5. [CrossRef]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E. S.; Aspris, D.; Leggate, D.; Hendrick, A. G.; Webster, N. A.; Andrews, B.; Fosbeary, R.; Guest, P.; Irigoyen, N.; Eleftheriou, M.; Gozdecka, M.; Dias, J. M. L.; Bannister, A. J.; Vick, B.; Jeremias, I.; Vassiliou, G. S.; Rausch, O.; Tzelepis, K.; Kouzarides, T., Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, (7860), 597-601. [CrossRef]
- Guirguis, A. A.; Ofir-Rosenfeld, Y.; Knezevic, K.; Blackaby, W.; Hardick, D.; Chan, Y. C.; Motazedian, A.; Gillespie, A.; Vassiliadis, D.; Lam, E. Y. N.; Tran, K.; Andrews, B.; Harbour, M. E.; Vasiliauskaite, L.; Saunders, C. J.; Tsagkogeorga, G.; Azevedo, A.; Obacz, J.; Pilka, E. S.; Carkill, M.; MacPherson, L.; Wainwright, E. N.; Liddicoat, B.; Blyth, B. J.; Albertella, M. R.; Rausch, O.; Dawson, M. A., Inhibition of METTL3 Results in a Cell-Intrinsic Interferon Response That Enhances Antitumor Immunity. Cancer Discov 2023, 13, (10), 2228-2247. [CrossRef]
- Cully, M., Chemical inhibitors make their RNA epigenetic mark. Nat Rev Drug Discov 2019, 18, (12), 892-894. [CrossRef]
- Chen, Y.; Lin, S.; Lai, J.; Wang, Q.; Lin, J.; Ao, W.; Ye, H.; Han, X., Expression of RNA-m6A-related genes correlates with the HIV latent reservoir level and the CD4+ and CD8+T cell profiles of patients with AIDS. Cell Mol Biol (Noisy-le-grand) 2023, 69, (4), 125-132. [CrossRef]
- Phillips, S.; Baek, A.; Kim, S.; Chen, S.; Wu, L., Protocol for the generation of HIV-1 genomic RNA with altered levels of N (6)-methyladenosine. STAR Protoc 2022, 3, (3), 101616. [CrossRef]
- Linder, B.; Grozhik, A. V.; Olarerin-George, A. O.; Meydan, C.; Mason, C. E.; Jaffrey, S. R., Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 2015, 12, (8), 767-72. [CrossRef]
- Zhang, Z.; Chen, L. Q.; Zhao, Y. L.; Yang, C. G.; Roundtree, I. A.; Zhang, Z.; Ren, J.; Xie, W.; He, C.; Luo, G. Z., Single-base mapping of m(6)A by an antibody-independent method. Sci Adv 2019, 5, (7), eaax0250.
- Roberts, J. T.; Porman, A. M.; Johnson, A. M., Identification of m(6)A residues at single-nucleotide resolution using eCLIP and an accessible custom analysis pipeline. RNA 2021, 27, (4), 527-541. [CrossRef]
- Hu, L.; Liu, S.; Peng, Y.; Ge, R.; Su, R.; Senevirathne, C.; Harada, B. T.; Dai, Q.; Wei, J.; Zhang, L.; Hao, Z.; Luo, L.; Wang, H.; Wang, Y.; Luo, M.; Chen, M.; Chen, J.; He, C., m(6)A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat Biotechnol 2022, 40, (8), 1210-1219.
- Liu, X. M.; Qian, S. B., Targeted RNA m(6)A Editing Using Engineered CRISPR-Cas9 Conjugates. Methods Mol Biol 2021, 2298, 399-414.
Figure 1.
Summary of m6A mapping in HIV-1 RNA. The position of the m6A peaks on the HIV-1 genome is illustrated by a schematic diagram that is drawn to scale. The HIV-1NL4-3 strain genome was used as a reference to compare data reported by each publication. Black boxes represent predicted peak locations based on various sequencing approaches using RNA purified from (A) HIV-1-infected cells or (B) HIV-1 virions. Where possible, boxes represent exact reported peak width [25,26,27,28,31,33]. Other boxes are drawn based on peak calling maps from individual publications [29,30].
Figure 1.
Summary of m6A mapping in HIV-1 RNA. The position of the m6A peaks on the HIV-1 genome is illustrated by a schematic diagram that is drawn to scale. The HIV-1NL4-3 strain genome was used as a reference to compare data reported by each publication. Black boxes represent predicted peak locations based on various sequencing approaches using RNA purified from (A) HIV-1-infected cells or (B) HIV-1 virions. Where possible, boxes represent exact reported peak width [25,26,27,28,31,33]. Other boxes are drawn based on peak calling maps from individual publications [29,30].
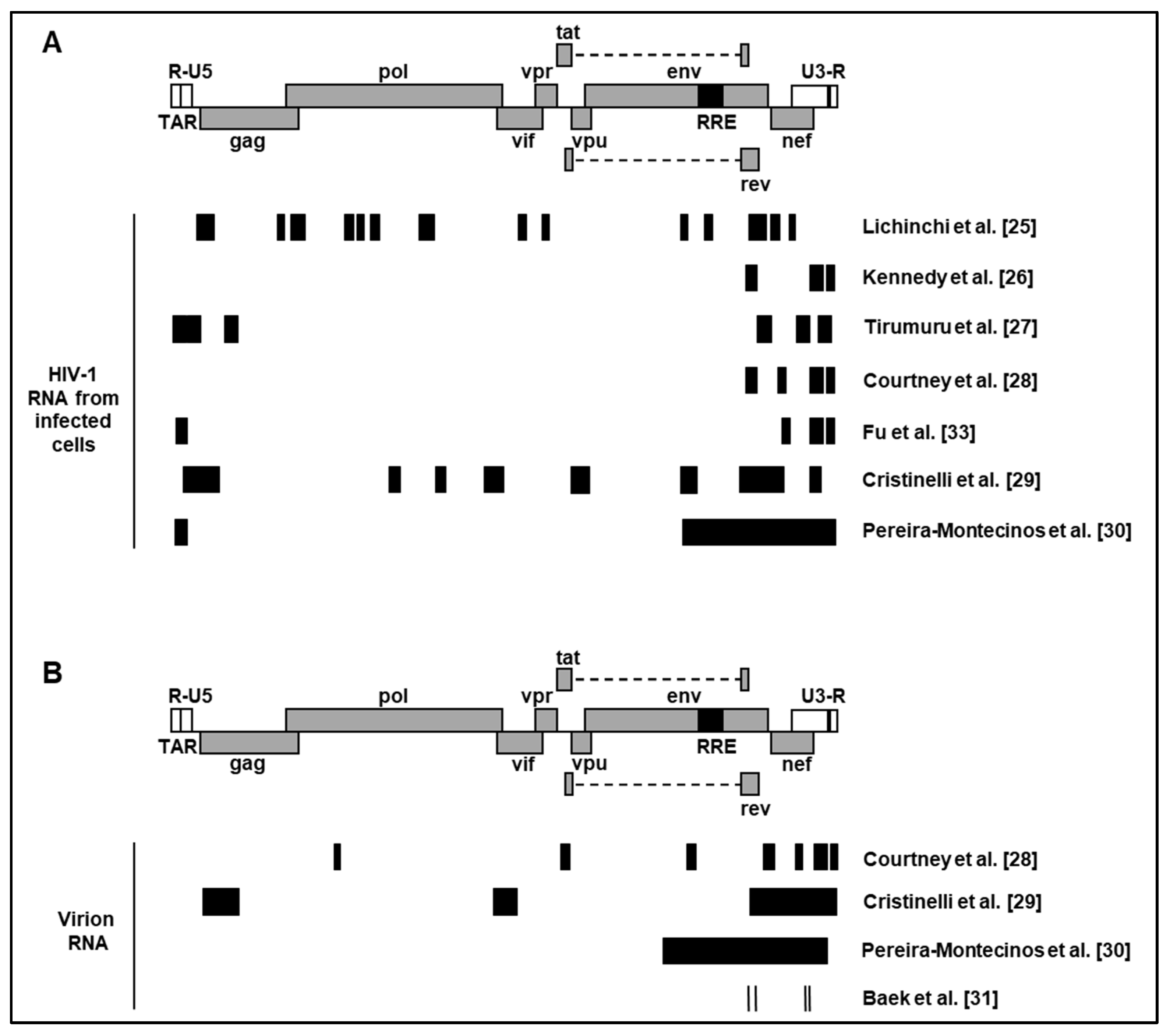
Figure 2.
Post-integration regulation of HIV-1 RNA by m6A regulators. (1) Viral RNA is methylated by the methyltransferase complex in the nucleus (for simplicity, only one m6A is shown on viral transcripts). (2) YTHDC1 regulates the splicing of full-length and incompletely spliced HIV-1 RNA but does not affect multiply spliced transcripts. (3) YTHDF1, 2 and 3 proteins positively regulate HIV-1 RNA abundance and viral protein synthesis. (4) Gag is imported in the nucleus and interacts with FTO to demethylate full-length HIV-1 RNA. (5 and 6) Gag preferentially interacts with full-length HIV-1 RNA and is packaged into progeny virions.
Figure 2.
Post-integration regulation of HIV-1 RNA by m6A regulators. (1) Viral RNA is methylated by the methyltransferase complex in the nucleus (for simplicity, only one m6A is shown on viral transcripts). (2) YTHDC1 regulates the splicing of full-length and incompletely spliced HIV-1 RNA but does not affect multiply spliced transcripts. (3) YTHDF1, 2 and 3 proteins positively regulate HIV-1 RNA abundance and viral protein synthesis. (4) Gag is imported in the nucleus and interacts with FTO to demethylate full-length HIV-1 RNA. (5 and 6) Gag preferentially interacts with full-length HIV-1 RNA and is packaged into progeny virions.
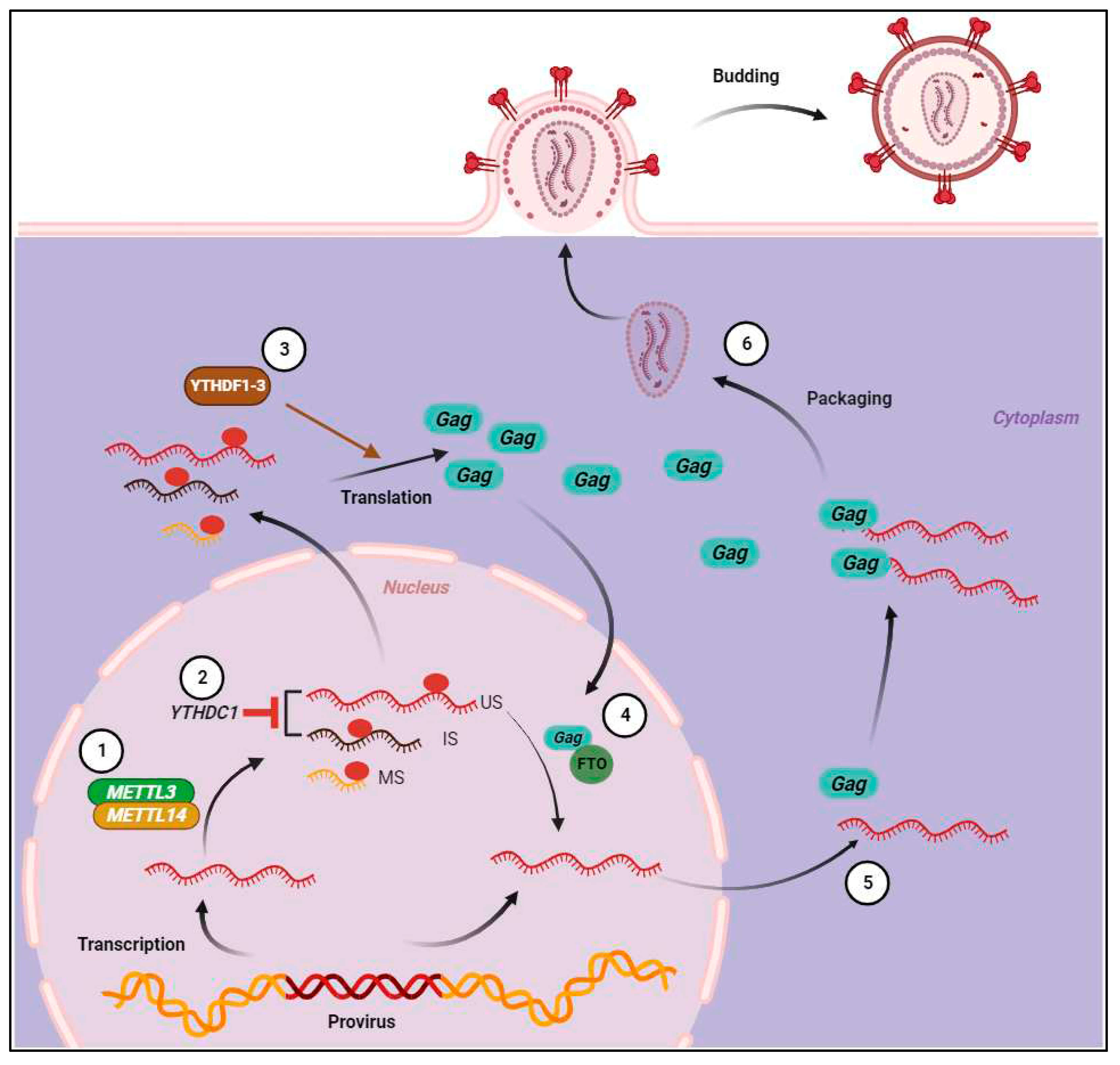

Table 1.
Methods for mapping m6A modification sites of HIV-1 RNA.
| HIV-1 Strain | Cell type | Sequencing method | References |
|---|---|---|---|
| LAI | MT4 | meRIP-seq3 | [25] |
| NL4-3 ΔEnv VSV-G1 | CEM | PA-m6A-seq | [26] |
| NL4-3 | Jurkat$Primary CD4+ T cells | meRIP-seq | [27] |
| NL4-3 | CEM | PA- m6A-seq4 | [28] |
| NL4-3 GFP ΔEnv VSV-G2 | SupT1 | meRIP-seq | [29] |
| NL4-3 ΔEnv VSV-G | HEK293T | meRIP-seq | [30] |
| NL4-3 | HEK293T | Direct RNA sequencing | [31] |
1 Single cycle VSV-G pseudotyped HIV-1NL4-3. 2 Single cycle VSV-G pseudotyped HIV-1NL4-3 with a GFP reporter. 3 Methylated RNA immunoprecipitation sequencing. 4 photo-crosslinking-assisted m6A sequencing.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



