
Submitted:
15 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
In response to various stressors, cardiac chambers undergo structural remodeling. Long-term exposure of the right ventricle (RV) to pressure or volume overload leads to its maladaptive remodeling, associated with RV failure and increased mortality. While left ventricular adverse remodeling is well understood and therapeutic options are available or emerging, RV remodeling remains underexplored and no specific therapies are currently available. Accumulating evidence implicates a role of mast cells in RV remodeling. Mast cells produce and release numerous inflammatory mediators, growth factors and proteases that can adversely affect cardiac cells, thus contributing to cardiac remodeling. Recent experimental findings suggest that mast cells might represent a potential therapeutic target. This review examines the role of mast cells in cardiac remodeling with specific focus on RV remodeling and explores the potential efficacy of therapeutic interventions targeting mast cells to mitigate adverse RV remodeling.
Keywords:
mast cells
; right ventricle
; cardiac remodeling
1. Introduction
Cardiac remodeling refers to changes in size, mass, geometry, and function of the heart, which develop in the course of various cardiovascular pathologies. It initially represents an adaptive process of the heart in response to mechanical, neurohumoral, or other stressors, with the primary aim of preserving cardiac function [1]. However, sustained exposure to pathological factors triggers development of maladaptive cardiac remodeling. Cardiac remodeling can affect either the left or right ventricle (RV) or both ventricles and is associated with unfavorable outcomes and treatment responses [1,2]. It is now recognized as an important aspect of cardiovascular disease progression and is emerging as a therapeutic target in heart failure therapy [1]. Currently approved therapies have been efficient in reducing mortality and morbidity in left heart failure patients [3,4,5]. Both pharmacological and non-pharmacological therapies beneficially impacted prognosis in heart failure patients by modulating the cardiac remodeling process [5,6,7]. However, despite advances in treatment, the burden of heart failure remains high, emphasizing the ongoing need for further research and development of novel management strategies [3].
Cardiac remodeling develops in response to stressors such as volume or pressure overload. Such unfavorable hemodynamic conditions occur in a variety of diseases including hypertension, valvular pathologies, chronic pulmonary diseases, obesity, and metabolic disorders. To counteract the sustained rise in wall stress caused by excessive pressure and/or volume load the myocardium undergoes phenotypic and functional transformations, which encompass a sequence of molecular, cellular and interstitial alterations. Cardiomyocytes become enlarged due to new contractile protein synthesis, contributing to increased size and mass of the affected cardiac chamber [8,9]. In addition to cardiomyocyte changes, cardiac remodeling involves alterations in other cell types and extracellular matrix organization. Fibroblast activation and proliferation results in amplified synthesis of extracellular matrix proteins [10]. Changes in coronary microvascular endothelial cells lead to alterations in coronary microvasculature and blood supply to the heart [11]. One of the important features of cardiac remodeling is recruitment and accumulation of diverse inflammatory and immune cells within the myocardium [12,13], which can mediate both protective and deleterious effects [12].
2. Right Ventricular Remodeling
Recent studies have provided strong evidence to recognize the pivotal role of the RV in cardiovascular pathologies [14]. Functional and structural changes of the RV define prognosis in patients with various cardiovascular diseases including congenital heart disease [15], pulmonary arterial hypertension [16], myocardial infarction [17], advanced left heart failure [18] and stable coronary artery disease [19]. The recognition of the RV as a critical player in the progression of cardiovascular and respiratory conditions has sparked increased research attention towards the RV in recent years [20,21,22].
Pressure overload is a key pathogenetic factor for RV remodeling and dysfunction, which is associated with the release and subsequent accumulation of a myriad of bioactive molecules in the circulation and within the cardiac tissue [23,24]. These bioactive molecules have the potential to directly impose deleterious effects on the RV and modulate its response to pressure or volume overload [13].
Similar to the remodeling of the left ventricle, RV remodeling in response to pressure or volume overload is intricately associated with alterations in the function of cardiomyocytes, fibroblasts, endothelial cells, and various immune and inflammatory cells [25,26,27]. These cells are critically involved in cardiac remodeling and their dysfunction can be caused by a multitude of factors and mediators synthesized and released by various cells within the RV myocardium [27]. Interactions between different cell types play a vital role in determining the overall outcome.
Another hallmark of RV remodeling is the increased production and deposition of extracellular matrix proteins. Ultimately, this alters the microenvironment of the myocardial cells and leads to a rise in tissue stiffness and expansion of the intercellular space within the heart. Additionally, it creates a space for the build-up and storage of growth factors and inflammatory mediators.
The pathogenesis of the deleterious events during the course of RV remodeling involves a complex interplay of various processess and signalling pathways, including inflammation, extracellular matrix synthesis, calcium homeostasis, endothelial cell permeability, nitric oxide (NO) synthesis, endothelial-mesenchymal transition, matricellular protein synthesis, growth factor signalling pathways such as the transforming growth factor-β (TGF-β) and apelin. These interactions collectively dictate the fate of the RV under pressure or volume overload, and a better understanding of these intricate processes is essential to develop effective therapeutic strategies for prevention or management of RV dysfunction and failure.
3. Mast Cell Biology
A rapidly growing body of evidence has implicated mast cells in a large variety of pathophysiologic processes other than allergic responses, such as tissue repair, organ remodeling, pathological fibrosis, and angiogenesis [28]. Mast cells are non-circulating immune cells that mature in the peripheral tissues in the presence of the c-kit ligand stem cell factor from bone marrow-derived precursors [29]. They represent multifunctional granular cells localized in various tissues throughout the body [30]. The cytoplasm of mast cells contains 50-200 large granules that store diverse mediators, cytokines, and neutral proteases [30].
Mast cells can be activated either by immunologic or nonimmunologic ways. The immunologic way of mast cell activation involves an interaction of the antigen-specific antibodies or the antigen with the corresponding mast cell receptors [31]. The non-immunologic activation of mast cells is mediated by substances such as neuropeptides, basic compounds, cytokines, and certain drugs [31]. Both mechanisms lead to mast cell degranulation and release of mediators (Figure 1).
Upon activation, mast cells release a large array of biologically active substances, which are broadly classified into three categories: namely pre-formed mediators (histamine, proteoglycans, serotonin, proteases), lipid-derived mediators (prostaglandins and leukotrienes), and newly synthesized mediators (interleukins IL-1, 3, 4, 5, 6, 8, 10, 13, 16, tumor necrosis factor α (TNF-α), and vascular endothelial growth factor (VEGF)) [32]. Mast cell proteases play a critical role in extracellular matrix remodeling and include tryptase, chymase, carboxypeptidase, and cathepsin G. These proteases are involved in the degradation of extracellular matrix components, such as collagen, elastin, and proteoglycans, and activation of matrix-degrading matrix metalloproteinases (MMPs) [33,34].
4. Mast Cells in Healthy Hearts
Besides cardiomyocytes, endothelial cells and fibroblasts, heart tissue contains a number of other cell types, including various immune and inflammatory cells [35,36]. The immune cells in the healthy myocardium comprise mast cells, macrophages, T lymphocytes (CD4+ and CD8+), natural killer cells, eosinophils, B cells, dendritic cells, neutrophils, monocytes, and plasma cells. These immune cells are involved in the maintenance of tissue homeostasis, regulation of inflammation, tissue repair, and heart protection against potential pathogens [36].
Cardiac mast cells are generally located in the interstitial space between cardiomyocytes and are closely associated with coronary vessels [37] and nerves [38]. They constitute only a small part of the immune cells present within the heathy myocardium. According to a recent single-cell RNA sequencing analysis mast cells constituted less than 1% of all CD45+ immune cells in the healthy mouse myocardium [39]. A relatively low abundance of mast cells in the myocardium was consistently demonstrated in histological studies in rodent and human heart tissues. In normal hearts, mast cells density ranges from 0.6 cells/mm2 in C57/BL/6 mice, to 1.4 cells/mm2 in Wistar Kyoto rats [40] and to 6.8 cells/mm2 in dogs [41].
Mast cell distribution exhibits significant variation within the wall of mouse hearts; they are most prevalent in the epicardium (50%) and myocardium (45%), with fewer numbers in the endocardium (5%) [42]. In contrast, in healthy rats, mast cell numbers appear to be similar in both the subepicardial and subendocardial layers of the left ventricle [43]. In mouse and rat hearts, mast cell density in the RV is approximately two-fold higher than in the left ventricle and interventricular septum [42,44]. There are also important differences between rats and mice in the mast cell density. In healthy rat hearts, mast cell density in the RV is two-fold lower than that in the left ventricle [44], whereas in healthy mouse hearts, the RV has approximately two-fold higher mast cell density compared to the left ventricle and interventricular septum [42]. Furthermore, aging in rats is associated with an increase in mast cell density within the myocardium of both ventricles [44].
Mast cells are known for their heterogeneity, so that the phenotypes of cardiac mast cells differ from those found in other tissues. In contrast to mast cells found in human skin and lungs, mast cells in human hearts demonstrate slightly different patterns of mediator release and synthesis [45]. Upon cross-linking of IgE receptors on human heart mast cells, histamine, tryptase, leukotriene C4, and prostaglandin D2 are released, although differ quantitatively and qualitatively compared to skin and lung mast cells [45]. Mast cells heterogeneity is also evident from the patterns of mediators release. Human heart mast cells respond to C5a and protamine by releasing histamine in a similar way as skin mast cells; however, they differ from lung mast cells in this aspect [45]. Further, human heart mast cells do not respond to substance P and morphine, which activate skin mast cells [45]. Overall, the unique phenotypes and response patterns of cardiac mast cells in comparison to mast cells in other tissues suggest that these cells might play specific roles in cardiac physiology and immune responses within the heart.
Increasing evidence suggests that cardiac mast cells might play a role in physiological conditions in the absence of injury [46]. Differences in cardiac structure and function between healthy mast cell-competent and mast cell deficient rats at different ages suggest that mast cells may be an important factor to maintain myocardial homeostasis in healthy rats [47].
5. Mast Cells in Right Ventricular Physiology and Remodeling
Various immune and inflammatory cells are crucially involved in cardiac remodeling [13]. Notably, the composition of immune cells in the RV differs from that in the left ventricle in physiological as well as in pathological conditions [48]. A substantial body of evidence has accumulated supporting the involvement of mast cells in left heart hypertrophy and failure. However, the role of mast cells in RV hypertrophy and failure remains insufficiently explored.
Activation of mast cells in response to pressure overload occurs early after exposure to the stressor, as evidenced by increased mast cell degranulation in RV of mice 3 days following pulmonary artery banding (PAB) surgery [49]. The timing of mast cells accumulation in the course of RV remodeling might depend on the type of the overload and is summarized in Figure 2. In a rat aortocaval fistula model of volume overload, mast cell density in the RV significantly increased in the first days after surgery, returning to normal values already on the third day and did not change thereafter over 56 days [50], suggesting an important role of mast cells in the initial stages of volume overload-induced RV remodeling. In contrary, in mice subjected to PAB, RV mast cell density significantly increased two weeks after induction of the pressure overload and remained elevated at three weeks post-surgery [49]. Observations in PAB rats showed that mast cell density in the RV was significantly increased as late as 200 days following pressure overload induction [51].
The source of increased mast cell numbers in RV remodeling remains to be elucidated. In left ventricular remodeling induced by angiotensin II infusion, the increase in mast cell density was shown to result from the maturation of pre-existing immature mast cells [52]. Similarly, resident mast cells within the RV myocardium might undergo proliferation and maturation during RV remodeling. Alternatively, circulating mast cell progenitors might be recruited to the RV myocardium in response to volume or pressure overload.
Involvement of mast cells in RV remodeling is corroborated by findings of a progressive accumulation of mast cells in RV in spontaneously hypertensive rats in advanced stages of the disease, which is associated with development of RV hypertrophy and fibrosis [53]. The evidence for the pathological role of mast cells in adverse RV remodeling was further provided in experiments exploiting mast cell-deficient mice and pharmacological inhibition of mast cells activation [54]. Treatment of wild-type mice subjected to pressure overload by PAB with the mast cell stabilizer cromolyn significantly improved RV remodeling and function [54]. Similarly, mast cell-deficient KitW/KitW-v mice subjected to PAB showed minimal RV dilation and preserved RV function [54]. These beneficial changes in RV remodeling were associated with reduced gene expressions in the RV of proinflammatory cytokines, such as TNF-α and IL-6 [54]. Notably, both TNF-α and IL-6 were previously shown to be involved in pressure overload-induced left ventricular remodeling [55,56] and mast cells were found to be the main source of IL-6 in pressure-overloaded myocardium [39].
Accumulating evidence suggests that mast cells may also play a protective role in myocardial remodeling [57,58]. Indeed, mast cells release a wide range of mediators with multiple potential functions, so that may counteract the effects of each other with the resulting beneficial or detrimental outcome depending on the disease model. In addition, mast cells may be beneficial or harmful, depending on the stage of the disease [59]. The discrepancy may also be explained by the functional heterogeneity of mast cell populations involved in pathological processes [60]. Indeed, the secondary increase in mast cells in failing myocardium under mechanical ventricular support was associated with an increase in chymase-negative mast cells and a change in phenotypic expression in mast cells with decreased myocardial basic fibroblast growth factor (bFGF) levels [61]. Moreover, mast cells and their granules exert protective effects in the acute phase after myocardial infarction in rats via improving cardiomyocyte survival and vascularization [62].
6. Mechanisms of Mast Cell-Mediated Effects on the Heart
6.1. Effects of Mast Cells on Cardiomyocytes
Mediators released by activated mast cells may exert direct effects on cardiomyocytes and impact their survival, apoptosis, contraction, and arrhythmogenesis. Experiments with adoptive transfer of bone marrow-derived mast cells from wild-type mice or corresponding knockout mice to mast cell-deficient KitW-sh/W-sh mice with diabetic cardiomyopathy demonstrated a role of mast cell-derived IL6 and TNF-α in promoting cardiomyocyte death [63]. The mast cell-derived mediator histamine, which is present in the human heart at high concentrations, was shown to reduce cell viability and induce cardiomyocyte apoptosis [64]. In line with these observations, pharmacological inhibition or genetic disruption of histamine H2 receptor slowed heart failure progression in mice subjected to pressure overload through reduction of myocardial apoptosis [64]. In vitro experiments confirmed that activation of histamine H2 receptors increases apoptosis in isolated neonatal rat cardiomyocytes [64]. Mast cell proteasese can induce cardiomyocyte apoptosis through other mechanisms [65,66]. Thus, mouse mast cell protease-4 promoted cardiomyocyte apoptosis via degradation of insulin-like growth factor-1, which serves as a survival factor for cardiomyocytes [66].
In cardiac volume overload, chymase uptake by cardiomyocytes resulted in myosin degradation and cardomyocyte dysfunction [67]. In contrast, mast cell deficiency led to reduced contractility and myofilament Ca2+ sensitization after myocardial infarction in mice suggesting an important role of cardiac mast cells in preserving postischemic cardiac function [57].
Evidence suggests that cardiac mast cells can modulate cardiac electrical activity. Non-specific activation of resident mast cells by secretagoges affected the ability of pacemaker cardiomyocytes to generate spontaneous action potentials in the sinoatrial node and caused a shift in activation pattern [68].
Mast cells are the main source of renin in both human and rodent myocardium [38,69]. Renin derived from mast cells in ischemia/reperfusion activated cardiac renin-angiotensin-aldosterone system (RAAS) and caused excessive norepinephrine release leading to arrhythmias [69]. Concordantly, mast cell-deficient mouse hearts exhibited reduced renin release during reperfusion and were markedly protected from ischemia-reperfusion-induced arrhythmias [69].
6.2. Mast Cells and Extracellular Matrix Modulation
Myocardial fibrosis plays a pivotal role in the pathogenesis and progression of various cardiac diseases [70]. Elucidating the precise mechanisms underlying myocardial fibrosis development is crucial for understanding disease pathogenesis and identifying novel therapeutic targets [71]. Myocardial fibrosis involves an interplay of versatile processes, including synthesis and turnover of extracellular matrix proteins, activation and proliferation of cardiac fibroblasts, as well as the synthesis of diverse inflammatory mediators and growth factors with potent pro-fibrotic properties [72]. These intricate events are meticulously orchestrated by various cell types residing within the myocardium, including various immune cells [72,73,74].
Recent studies have implicated mast cells as significant contributors to myocardial fibrosis [75]. In patients with idiopathic dilated cardiomyopathy, a notable association was demonstrated between mast cell density and the severity of myocardial fibrosis [76]. Similarly, mast cell density positively correlated with the percentage of collagen fibers in the left ventricle in hypertensive heart disease patients [77]. Remarkably, mast cells were localized near areas of myocardial fibrosis in end-stage heart failure patients [61]. Co-culture of healthy fibroblasts with mast cells isolated from cardiac tissues of heart failure patients increased their collagen production [78] further supporting the potential role of mast cells in myocardial fibrogenesis. Accordingly, reduction in myocardial fibrosis after long-term left ventricular assist device support (>40 days) was associated with a decreased ratio of chymase-positive to total mast cell numbers [61].
Experimental studies provided further evidence to support the link between mast cell activation and myocardial fibrosis development. In Dahl salt-sensitive rats, degranulated mast cells were found to localize near fibrotic regions in the myocardium [79]. Notably, mast cells underwent degranulation during the progression from heart hypertrophy to failure in those rats by releasing pro-fibrotic mediators and inducing augmented myocardial fibrosis [79]. Activation of the ɛPKC signaling pathway in mast cells was responsible for mast cell degranulation and subsequent TGF-β release [79]. In line with these findings, in spontaneously hypertensive rats, expression of mast cell-derived TGF-β and bFGF significantly increased during the transition from cardiac hypertrophy to heart failure and was associated with an exaggerated myocardial fibrosis [80].
Chymase inhibition with chymostatin reduced myocardial active TGF-β1 levels in rats subjected to pressure overload by transverse aortic constriction (TAC), suggesting that activation of latent TGF-β1 is one of the pathways by which cardiac mast cell-derived chymase contributes to myocardial fibrosis [81]. Besides TGF-β1 activation, chymase induced TGF-β1 expression in a dose-dependent fashion in rat cardiac fibroblasts [82]. Further experiments confirmed that mast cell-derived chymase promote cardiac fibroblast proliferation and collagen synthesis via the TGF-β/Smad pathway [82]. Importantly, chymase inhibition siginificantly suppressed cardiac fibrosis in cardiomyopathic hamsters [83]. Chymase expression in left ventricular tissue correlated with increased mast cell density of terminal heart failure patients [84], suggesting that mast cell-derived chymase in heart failure might represent a potential therpeutic target.
Another pathway, by which mast cells can contribute to myocardial fibrosis is the activation of the RAAS in the heart. Mast cells are recognized as a significant source of renin in the myocardium, which consequently converts angiotensinogen to angiotensin I [38,85]. Next, cathepsin G [86] and chymase [87,88] derived from activated mast cells convert angiotensin I into angiotensin II. Angiotensin II subsequently exerts pro-fibrotic effects on the myocardium [89] through angiotensin II receptors, which are present on cardiac fibroblasts and mediate multiple pro-fibrotic effects [89,90].
Activated mast cells release a number of other factors, including histamin [64] and tryptase [40,91], that might account for their pro-fibrotic activities on cardiac fibroblasts. Treatment of isolated cardiac fibroblasts with tryptase induced increased collagen synthesis and fibroblast proliferation [40,91] . Collagen production was enhanced via activation of the protease-activated receptor-2 and ERK1/2 signaling [91]. The relevance of in vitro findings was further corroborated by reduction of myocardial fibrosis in spontaneously hypertensive rats treated with a tryptase inhibitor [91].
MMP/tissue inhibitors of metalloproteinases system plays a key role in the modulation of the extracellular matrix in pathological conditions. Several lines of evidence implicate mast cells in the regulation of this system in volume overload-induced cardiac remodeling (Figure 3). A strong correlation was reported between mast cell density and MMP activity in hearts of rats subjected to volume overload [50]. Application of the secretagogue compound 48/80 [92] or endothelin-1 [93] in isolated rat hearts caused cardiac mast cell degranulation and activation of MMPs, leading to collagen degradation and moderate ventricular dilation. Furthermore, in a model of volume overload induced by aortocaval fistula, which encodes a mast cell secretagogue substance P and neurokinin A, mice lacking TAC1 gene were protected from adverse left ventricular remodeling associated with attenuated myocardial MMP activity and collagen degradation due to reduced mast cell degranulation [94]. Mast cell activation along with enhanced MMP activity and increased collagen degradation was also reported in a model of dyslipidemic dilated cardiomyopathy induced by a high-fat diet in ApoE-/- mice [95]. Co-culture experiments further supported the role of mast cells in increased MMP-2 activity and expression in fibroblasts [96]. The importance of mast cells in mediating MMP activation and collagen degradation is emphasized by findings of attenuated MMP-2 activity and ameliorated left ventricular dilation in mast cell-deficient rats subjected to aortocaval fistula-induced volume overload [97].
6.3. Mast Cells and Myocardial Vascularization
Most cardiac pathologies are associated with changes in myocardial vascularization [98,99]. Mast cells are present in the wall of both microvessels and larger coronary arteries, suggesting their potential involvement in both micro- and macrovascular pathologies of the coronary arteries (Figure 4).
Mast cells have been shown to induce left ventricular diastolic dysfunction in various animal models of heart failure [100,101]. Abnormal mast cell activation was identified as a cause of cardiac microvessel disease in Leprdb/db female mice, another experimental model of diastolic dysfunction associated with heart failure with preserved ejection fraction [102]. The cardiac microvessel disease was characterized by enhanced vessel permeability due to -disruption of endothelial adherens junctions by mast cell-derived histamine [102]. Histamin released by mast cells may interact with both H1 and H2 receptors to exert its effects on the cardiac endothelial cells and contribute to heart failure development [103]. Notably, the use of H2 receptor antagonists in the aging human population was associated with reduced risk for incident heart failure and favorable effects on the heart [104].
Increased serum tryptase levels were reported in patients with stable coronary artery disease, suggesting that chronic low-grade inflammation in atherosclerotic plaques triggers mast cell activation [105]. In vitro experiments demonstrated that activated mast cells might contribute to plaque erosion by inducing endothelial cell apoptosis [106,107]. Indeed, recent evidence implicated mast cells in plaque destabilization and atherosclerotic coronary complications. Mast cell density increased from 0 in controls to 2.3-2.7 per mm2 in the media layer of the coronary artery, with the highest numbers observed in unstable plaques of myocardial infarction patients [108]. In patients with myocardial infarction, segments with plaque rupture demonstrated significantly higher numbers of adventitial mast cells compared to segments with non-ruptured plaques or normal intima of the infarct-related coronary artery [109].
In contrast to abovementioned reports, accumulating evidence suggests that mast cells exert beneficial effects on maintaining cardiac microvascular homeostasis. In left anterior descending artery (LAD) occlusion experiments, implantation of wild-type mast cells into mast cell-deficient c-Kit KitW/W-v mice enhanced angiogenesis, improved cardiac function and decreased infarct size at early time points after myocardial infarction [110]. Indeed, in several in vitro studies mast cells promoted angiogenesis by stimulating endothelial cells to release angiogenic factors [111,112].
6.4. Mast Cells and Myocardial Inflammation
Myocardial inflammation in various cardiac pathologies is recognized as a significant contributor to adverse myocardial remodeling in both ventricles [13]. The importance of mast cells in this process is emphasized by the fact that they possess a variety of inflammatory mediators in their granules and able to rapidly generate more inflammatory agents upon activation [113]. Activated mast cells release a number of inflammatory factors, including interleukins (IL-1, IL-3, IL-4, IL-5, IL-6, IL-8, IL-10, IL-13, and IL-17), TNF-α, and chemokines (CXCL8/IL-8, CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, and CCL5/RANTES) [30].
Inflammatory mediators released by mast cells can enhance myocardial inflammation by activating further inflammatory processess. In canine experimental cardiac ischemia-reperfusion, mast cells primarily released TNF-α, which then exacerbated myocardial inflammation and cardiac injury by upregulating IL-6 in infiltrating leukocytes and initiating the cytokine cascade [114]. In line with this report, attenuated myocardial injury in mast cell-deficient KitW/KitW-v mice following myocardial ischemia-reperfusion was associated with lower serum IL-6 compared to their wild-type counterparts [115]. Importantly, mast cell stabilization with ketotifen and cromolyn sodium prevented an increase in myocardial TNF-α levels following reperfusion [116].
Markedly elevated myocardial TNF-α levels in response to cardiac volume overload were observed in wild-type rats at 5 days post-fistula; conversely, TNF-α was almost undetectable in hearts of mast cell-deficient rats [97]. In a similar model, protection from adverse left ventricular remodeling in mice lacking the TAC1 gene was associated with reduced mast cell degranulation and attenuated TNF-α expression [94].
Growing evidence suggests that inflammatory mediators released from mast cells are critically involved in the development of pressure overload-induced cardiac remodeling. In spontaneously hypertensive rats, left ventricular nuclear factor kappa-B and IL-6 expression in mast cells were already increased during the prehypertensive stages [80]. Stimulation of cardiac mast cell degranulation with the compound 48/80 in an ex-vivo Langendorff heart preparations resulted in increased expression of nuclear factor kappa-B and IL-6 mRNA in left ventricles [80]. These data are supported by recent findings of single-cell sequencing of immune infiltrates in left ventricles of mice subjected to pressure overload, which revealed that mast cells had the highest expression of IL-6 among all immune cells [39].
Mast cell-derived inflammatory medators have recently been implicated in the pathogenesis of cardiometabolic diseases. Streptozotocin-induced diabetic cardiomyopathy in mice is characterized by pathologic myocardial inflammation and accumulation of mast cells in the heart [63]. Remarkably, mast cell-deficient KitW-sh/W-sh mice were protected from diabetic cardiomyopathy [63]. Preserved cardioprotection following adoptive transfer of bone marrow-derived mast cells from Tnf-deficient but not wild-type mice into KitW-sh/W-sh mice treated with streptozotocin identified TNF-α released by mast cells as the key mediator in this pathology [63].
7. Mast Cells as a Therapeutic Target
Preclinical studies have provided evidence that mast cells may represent an attractive target in the prevention and treatment of heart diseases. The potential strategies might include inhibition of mast cell activation, targeting individual mediators released by mast cells, blockade of receptors for mast cell-derived mediators, restriction of mast cell growth, or induction of mast cell apoptosis [117]. Some of these strategies have been explored in in rodent models of heart failure.
Prevention of mast cell activation by stabilization of their membranes has demonstrated efficacy in improving myocardial remodeling in various experimental models of heart failure. In spontaneously hypertensive rats, mast cell stabilization with nedocromil prevented left ventricular fibrosis, inflammatory cell recruitment, and cytokine overexpression in the myocardium, without affecting blood pressure or left ventricular hypertrophy [40]. Mast cell stabilization with cromolyn sodium attenuated left ventricular remodeling and left ventricular diastolic dysfunction in ovariectomized Fischer rats [100], ameliorated left ventricular diastolic dysfunction in Leprdb/db female mice [102] and attenuated RV dilatation and improved RV function in mice subjected to PAB [54]. The class effects of these drugs were further supported by prevention the transition from compensated hypertrophy to heart failure in animals subjected to abdominal aortic banding using another mast cell stabilizing agent tranilast [118].
Mast cell-derived chymase has been identified as one of the key mast cell-specific targets in various cardiac pathologies [119]. Chymase inhibition improved cardiac remodeling in various animal models including rapid ventricular pacing-induced heart failure in dogs [88], LAD-occlusion-induced myocardial infarction in rats [120] and hamsters [121], ischemia-reperfusion cardiac remodeling in pigs [122] and pressure overload-induced left ventricular remodeling in rats [81]. Further, mice lacking mMCP-4, the mouse counterpart of human mast cell chymase, were protected from adverse cardiac remodeling and dysfunction in the LAD-occlusion model of myocardial infarction [66,123,124]. Mast cell chymase can limit the cardiac efficacy of the angiotensin converting enzyme (ACE) inhibitor therapy in rodents [125]. Therefore, combined chymase and ACE inhibition, compared to ACE inhibition alone, achieved better results in left ventricular function improvement, amelioration of adverse cardiac remodeling, and improvement of survival after myocardial infarction in hamsters [125]. The role of chymase inhibition in RV remodeling and dysfunction, however, remains unexplored warranting future studies.
Blocking receptors for mast cell-derived mediators such as histamin H2 receptor improved cardiac function in including TAC mice [64] and in dogs with pacemaker-driven tachycardia [126]. Interestingly, a large prospective observational cohort study of participants without cardiovascular disease at baseline showed that baseline use of H2 receptor antagonists was associated with a 62% lower risk of incident heart failure [104]. Beneficial effects of H2 receptor antagonists on all-cause mortality in patients with different clinical forms pulmonary hypertension suggest that these drugs exert direct effects on the right ventricle [127]. In line with these observations, H2 receptor antagonist use in the general population was associated with lower RV mass and smaller RV end-diastolic volume [128]. A meta-analysis revealed that H2 receptor antagonists may improve cardiac function in heart failure patients by decreasing myocardial oxygen demand due to negative inotropic and chronotropic effects [129].
8. Conclusions and Future Directions
In this comprehensive review, we have conducted a thorough examination of the existing literature to elucidate the role played by mast cells in the initiation and progression of myocardial remodeling with a particular focus on dissecting mast cell involvement in RV remodeling and dysfunction.
Mast cells are the major source of a whole host of biologically active substances including growth factors, proteases, cytokines, chemokines, polypeptides, biogenic amines, proteoglycans, and phospholipid metabolites [130]. The biological complexities of mast cells in the setting of cardiac remodeling are highlighted by several key observations: 1) Mast cell density in healthy myocardium is markedly lower compared to that of other immune cell types; 2) activation of mast cells triggers release of a diverse array of mediators from their granules and induces de novo synthesis of further factors; 3) effects of these mediators extend to various myocardial cell types, including cardiomyocytes, cardiac fibroblasts, cardiac endothelial cells, and other immune cells within the myocardium; 4) proteases released by mast cells play a substantial role in the activation of extracellular proteins and enzymes.
Experimental evidence clearly showed that mast cell deficiency is associated with mitigation of the extent of cardiac injury and remodeling. Moreover, pharmacological inhibition of mast cell activation demonstrated their ability to alleviate adverse cardiac remodeling. It is important to emphasize that most investigations were devoted to the role of mast cells in left ventricular remodeling. Consequently, our understanding of the specific roles of mast cells in RV remodeling remains insufficiently explored (Figure 5).
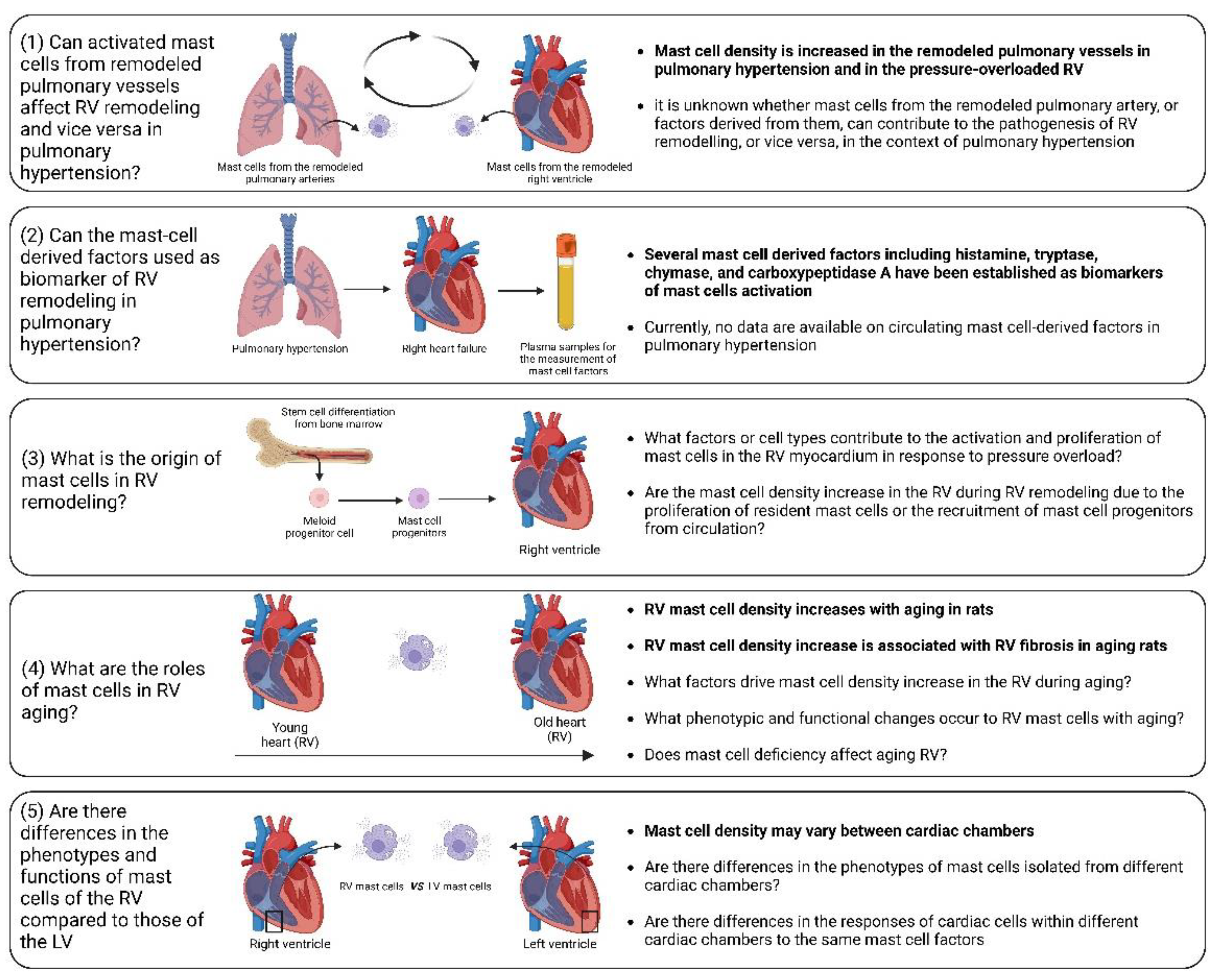
Despite recent significant advances, there are still unaddressed issues related to the precise role of mast cells in RV remodeling and dysfunction, including the following (Figure 5):
(1) Influence of activated mast cells from remodeled pulmonary vessels on RV remodeling and vice versa in pulmonary hypertension. It is conceivable that various factors released by activated mast cells in remodeled pulmonary arteries may be released into the circulation and transported to the RV myocardium. These factors can potentially alter the responses of the RV to pressure overload. Additionally, there is the evidence of increased mast cell activation in remodeled pulmonary arteries in pulmonary hypertension [131,132,133]. Similarly, it can be postulated that the release of various factors from activated mast cells within the RV myocardium may reach the pulmonary vasculature, where they could exacerbate the remodeling processes.
(2) Circulating mast-cell derived factors as biomarkers of RV remodeling. Mediators released by activated mast cells, including histamine, tryptase, chymase, and carboxypeptidase A, can be measured in the systemic circulation and have the potential to serve as markers of mast cell activation in a number of conditions [134,135].
(3) Origin of mast cells in the remodeled RV. It remains to be elucidated, whether mast cell density increase is caused by proliferation of resident mast cells or recruitment of mast cell progenitors from the circulation. To address this issue, reconstitution experiments with bone marrow-derived mast cells in mast cell-deficient mice subjected to PAB can be performed.
(4) Role of mast cells in RV ageing. A correlation between age-related myocardial fibrosis and the density of mast cells has been previously revealed. However, there are still unanswered questions regarding mast cells-derived factors that define RV myocardial fibrosis during aging. Furthermore, which specific factors drive the increase in mast cell density in the RV during aging and what are the associated phenotypic and functional changes in RV mast cells? What are the consequences of mast cell deficiency on healthy RV aging? To date, it remains uncertain, whether mast cell-deficient animals (mice or rats) maintain healthy aging of the RV.
(5) What factors govern mast cell activation and proliferation in the context of RV remodeling? Although it is clear that pressure overload is the main cause of mast cell activation and proliferation during cardiac remodeling, identifying the key factors that regulate this process could offer a means for scientists to prevent the onset of mast cell activation by inhibiting upstream triggers. Mast cell activation may occur due to direct mechanical strain, as previous research has indicated mast cell sense the mechanical properties of their microenvironment [136]. Another possible scenario is that cardiomyocytes may release specific mediators during the initial phases in response to pathological stimuli, promoting mast cell activation and growth. Ultimately, exploring this issue could bring us closer to understanding the mechanisms behind the disease and developing pharmacological treatments.
(6) Differential impact of mast cell-derived factors on the RV and left ventricle. It is unclear which of the factors released by mast cells might have RV-specific effects in comparison to the left ventricle. It is unclear whether the effects of mast cell-derived factors differ between cardiac chambers due to compartment-specific differences in mast cell phenotypes or due to the chamber-specific phenotypes of cardiac cells. These differences in the cells targeted by mast cell factors might be partially explained by variations in receptor density or in the activated signaling pathways.
In summary, mast cells represent pivotal cellular entities implicated in diverse cardiac pathologies. The multifaceted roles and functions of mast cells in the RV remain incompletely explored, emphasizing the necessity of future studies with a dedicated focus on dissecting the contribution of mast cells to RV remodeling.
Author Contributions
Conceptualization, A.M., and A.S.; writing—original draft preparation, A.M., and A.S.; writing—review and editing, A.M., Ab.M., Ak.S., R.T.S., and A.S.; visualization, A.M., and A.S.; supervision, A.S..; funding acquisition, R.T.S., and Ak.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), grant SCHE 691/6-1.
Institutional Review Board Statement
NA.
Informed Consent Statement
NA.
Data Availability Statement
NA.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cohn, J.N., R. Ferrari, and N. Sharpe, Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol 2000, 35, 569–582. [Google Scholar] [PubMed]
- Azevedo, P.S; et al. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq Bras Cardiol 2016, 106, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Pazos-López, P.; et al. The causes, consequences, and treatment of left or right heart failure. Vasc Health Risk Manag 2011, 7(237). [Google Scholar]
- Shah, A; et al. Heart Failure: A Class Review of Pharmacotherapy. P t 2017, 42, 464–472.
- Iacoviello, M., A. Palazzuoli, and E. Gronda, Recent advances in pharmacological treatment of heart failure. Eur J Clin Invest 2021, 51, e13624. [Google Scholar] [PubMed]
- Ishii, H; et al. Pharmacological intervention for prevention of left ventricular remodeling and improving prognosis in myocardial infarction. Circulation 2008, 118, 2710–2718.
- Kim, G.H., N. Uriel, and D. Burkhoff, Reverse remodelling and myocardial recovery in heart failure. Nat Rev Cardiol 2018, 15, 83–96. [CrossRef] [PubMed]
- Haque, Z.K. and D.Z. Wang, How cardiomyocytes sense pathophysiological stresses for cardiac remodeling. Cell Mol Life Sci 2017, 74, 983–1000. [CrossRef] [PubMed]
- Scarborough, E.A.; et al. Microtubules orchestrate local translation to enable cardiac growth. Nat Commun 2021, 12, 1547. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest 2017, 127, 1600–1612. [Google Scholar] [CrossRef]
- Segers, V.F.M., D.L. Brutsaert, and G.W. De Keulenaer, Cardiac Remodeling: Endothelial Cells Have More to Say Than Just NO. Front Physiol 2018, 9(382).
- Kologrivova, I.; et al. Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair After Myocardial Infarction. Front Immunol 2021, 12(664457). [Google Scholar] [CrossRef] [PubMed]
- Sydykov, A.; et al. Inflammatory Mediators Drive Adverse Right Ventricular Remodeling and Dysfunction and Serve as Potential Biomarkers. Front Physiol 2018, 9(609). [Google Scholar] [CrossRef] [PubMed]
- Tello, K.; et al. Pathophysiology of the right ventricle in health and disease: an update. Cardiovasc Res 2023, 119, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.K. and J.S. Ma, Right ventricular failure in congenital heart disease. Korean J Pediatr 2013, 56, 101–106. [CrossRef] [PubMed]
- Prisco, S.Z., T. Thenappan, and K.W. Prins, Treatment Targets for Right Ventricular Dysfunction in Pulmonary Arterial Hypertension. JACC Basic Transl Sci 2020, 5, 1244–1260. [CrossRef] [PubMed]
- Shahar, K.; et al. Time Dependence of the Effect of Right Ventricular Dysfunction on Clinical Outcomes After Myocardial Infarction: Role of Pulmonary Hypertension. J Am Heart Assoc 2016, 5.
- Dini, F.L.; et al. Right ventricular failure in left heart disease: from pathophysiology to clinical manifestations and prognosis. Heart Fail Rev 2023, 28, 757–766. [Google Scholar] [CrossRef]
- Sumin, A.N., E.V. Korok, and T.Y. Sergeeva, Impaired right ventricular filling in patients with a chronic coronary syndrome. Med Ultrason 2021, 23, 311–318.
- Edward, J.; et al. Right ventricular function across the spectrum of health and disease. Heart 2023, 109, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Mandoli, G.E.; et al. Right cardiac involvement in lung diseases: a multimodality approach from diagnosis to prognostication. J Intern Med 2021, 289, 440–449. [Google Scholar] [CrossRef]
- Mamazhakypov, A.; et al. Novel Therapeutic Targets for the Treatment of Right Ventricular Remodeling: Insights from the Pulmonary Artery Banding Model. Environ Res Public Health 2021, 18.
- Jabagi, H.; et al. Biomarkers in the Diagnosis, Management, and Prognostication of Perioperative Right Ventricular Failure in Cardiac Surgery-Are We There Yet? J Clin Med 2019, 8.
- Pradhan, N.M., C. Mullin, and H.D. Poor, Biomarkers and Right Ventricular Dysfunction. Crit Care Clin 2020, 36, 141–153. [CrossRef] [PubMed]
- Kret, M. and R. Arora, Pathophysiological basis of right ventricular remodeling. J Cardiovasc Pharmacol Ther 2007, 12, 5–14. [CrossRef] [PubMed]
- Avazmohammadi, R.; et al. Interactions Between Structural Remodeling and Hypertrophy in the Right Ventricle in Response to Pulmonary Arterial Hypertension. J Biomech Eng 2019, 141, 0910161–09101613. [Google Scholar] [CrossRef] [PubMed]
- Lother, A. and P. Kohl, The heterocellular heart: identities, interactions, and implications for cardiology. Basic Res Cardiol 2023, 118, 30.
- Galli, S.J. and M. Tsai, Mast cells: versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. J Dermatol Sci 2008, 49, 7–19. [CrossRef] [PubMed]
- da Silva, E.Z., M.C. Jamur, and C. Oliver, Mast cell function: a new vision of an old cell. J Histochem Cytochem 2014, 62, 698–738. [CrossRef] [PubMed]
- Krystel-Whittemore, M., K.N. Dileepan, and J.G. Wood, Mast Cell: A Multi-Functional Master Cell. Front Immunol 2015, 6(620).
- Theoharides, T.C., I. Tsilioni, and H. Ren, Recent advances in our understanding of mast cell activation - or should it be mast cell mediator disorders? Expert Rev Clin Immunol 2019, 15, 639–656. [CrossRef] [PubMed]
- Moon, T.C., A.D. Befus, and M. Kulka, Mast cell mediators: their differential release and the secretory pathways involved. Front Immunol 2014, 5(569).
- Johnson, J.L.; et al. Activation of matrix-degrading metalloproteinases by mast cell proteases in atherosclerotic plaques. Arterioscler Thromb Vasc Biol 1998, 18, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.O.; et al. Mast cell chymase regulates extracellular matrix remodeling-related events in primary human small airway epithelial cells. J Allergy Clin Immunol 2022, 150, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Litviňuková, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.D.; et al. Myocardial Immune Cells: The Basis of Cardiac Immunology. J Immunol 2023, 210, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Marone, G.; et al. Immunological characterization and functional importance of human heart mast cells. Immunopharmacology 1995, 31, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.B.; et al. Mast cells: a unique source of renin. Proc Natl Acad Sci U S A 2004, 101, 13607–13612. [Google Scholar] [CrossRef] [PubMed]
- Martini, E.; et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation 2019, 140, 2089–2107. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; et al. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 2009, 53, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Gersch, C.; et al. Mast cells and macrophages in normal C57/BL/6 mice. Histochem Cell Biol 2002, 118, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Ingason, A.B. Distribution of mast cells within the mouse heart and its dependency on Mitf. Mol Immunol 2019, 105, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Engels, W.; et al. Transmural changes in mast cell density in rat heart after infarct induction in vivo. J Pathol 1995, 177, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Stamenov, N.; et al. Mast cells and basic fibroblast growth factor in physiological aging of rat heart and kidney. Biotech Histochem 2022, 97, 504–518. [Google Scholar] [CrossRef]
- Patella, V.; et al. Human heart mast cells: a definitive case of mast cell heterogeneity. Int Arch Allergy Immunol 1995, 106, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; et al. Cardiac Mast Cells: A Two-Head Regulator in Cardiac Homeostasis and Pathogenesis Following Injury. Front Immunol 2022, 13, 963444. [CrossRef] [PubMed]
- Kennedy, R.H., M. Hauer-Jensen, and J. Joseph, Cardiac function in hearts isolated from a rat model deficient in mast cells. Am J Physiol Heart Circ Physiol 2005, 288, H632–H637. [CrossRef] [PubMed]
- Gorr, M.W.; et al. Transcriptomic profiles reveal differences between the right and left ventricle in normoxia and hypoxia. Physiol Rep 2020, 8, e14344. [Google Scholar] [CrossRef] [PubMed]
- Luitel, H.; et al. Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle. Physiol Rep, 2017. 5.
- Brower, G.L.; et al. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am J Physiol Heart Circ Physiol 2002, 283, H518–H525. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; et al. Long-term pressure-induced cardiac hypertrophy: capillary and mast cell proliferation. Am J Physiol 1989, 257 (6 Pt 2), H1766-72.
- Widiapradja, A.; et al. Regulation of Cardiac Mast Cell Maturation and Function by the Neurokinin-1 Receptor in the Fibrotic Heart. Sci Rep 2019, 9, 11004. [Google Scholar] [CrossRef] [PubMed]
- Kotov, G.; et al. Changes in the number of mast cells, expression of fibroblast growth factor-2 and extent of interstitial fibrosis in established and advanced hypertensive heart disease. Ann Anat 2020, 232, 151564. [CrossRef] [PubMed]
- Sydykov, A.; et al. Genetic Deficiency and Pharmacological Stabilization of Mast Cells Ameliorate Pressure Overload-Induced Maladaptive Right Ventricular Remodeling in Mice. Int J Mol Sci 2020, 21.
- Sun, M.; et al. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation 2007, 115, 1398–1407. [CrossRef] [PubMed]
- Fang, G.; et al. Cadherin-11-Interleukin-6 Signaling between Cardiac Fibroblast and Cardiomyocyte Promotes Ventricular Remodeling in a Mouse Pressure Overload-Induced Heart Failure Model. Int J Mol Sci 2023, 24.
- Ngkelo, A.; et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J Exp Med 2016, 213, 1353–1374. [Google Scholar] [CrossRef]
- Joseph, J.; et al. Protective role of mast cells in homocysteine-induced cardiac remodeling. Am J Physiol Heart Circ Physiol 2005, 288, H2541–H2545. [Google Scholar] [CrossRef]
- Scandiuzzi, L.; et al. Mouse mast cell protease-4 deteriorates renal function by contributing to inflammation and fibrosis in immune complex-mediated glomerulonephritis. J Immunol 2010, 185, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Cildir, G.; et al. The transcriptional program, functional heterogeneity, and clinical targeting of mast cells. J Exp Med 2017, 214, 2491–2506. [Google Scholar] [CrossRef] [PubMed]
- Akgul, A.; et al. Role of mast cells and their mediators in failing myocardium under mechanical ventricular support. J Heart Lung Transplant 2004, 23, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.S.; et al. The novel role of mast cells in the microenvironment of acute myocardial infarction. J Mol Cell Cardiol 2011, 50, 814–825. [Google Scholar] [CrossRef] [PubMed]
- He, A.; et al. Mast cell-deficiency protects mice from streptozotocin-induced diabetic cardiomyopathy. Transl Res 2019, 208, 1–14. [CrossRef]
- Zeng, Z.; et al. Disruption of histamine H2 receptor slows heart failure progression through reducing myocardial apoptosis and fibrosis. Clin Sci (Lond) 2014, 127, 435–448. [Google Scholar] [CrossRef]
- Hara, M.; et al. Mast cells cause apoptosis of cardiomyocytes and proliferation of other intramyocardial cells in vitro. Circulation 1999, 100, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Tejada, T.; et al. IGF-1 degradation by mouse mast cell protease 4 promotes cell death and adverse cardiac remodeling days after a myocardial infarction. Proc Natl Acad Sci U S A 2016, 113, 6949–6954. [Google Scholar] [CrossRef]
- Powell, P.C.; et al. Chymase uptake by cardiomyocytes results in myosin degradation in cardiac volume overload. Heliyon 2019, 5, e01397. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, V.S.; et al. Inflammatory degranulation of the cardiac resident mast cells suppresses the pacemaking and affects activation pattern in the sinoatrial node. Translational Research in Anatomy 2022, 26, 100170. [CrossRef]
- Mackins, C.J.; et al. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest 2006, 116, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Egemnazarov, B.; et al. Right ventricular fibrosis and dysfunction: Actual concepts and common misconceptions. Matrix Biol 2018, 68-69, 507–521. [CrossRef] [PubMed]
- Jiang, W.; et al. Cardiac Fibrosis: Cellular Effectors, Molecular Pathways, and Exosomal Roles. Front Cardiovasc Med 2021, 8, 715258. [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc Res 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G., G. Marone, and P. T. Kovanen, Cardiac Mast Cells: Underappreciated Immune Cells in Cardiovascular Homeostasis and Disease. Trends Immunol 2020, 41, 734–746. [Google Scholar] [PubMed]
- Levick, S.P.; et al. Cardiac mast cells: the centrepiece in adverse myocardial remodelling. Cardiovasc Res 2011, 89, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Legere, S.A.; et al. Mast Cells in Cardiac Fibrosis: New Insights Suggest Opportunities for Intervention. Front Immunol 2019, 10, 580. [CrossRef] [PubMed]
- Batlle, M.; et al. Correlation between mast cell density and myocardial fibrosis in congestive heart failure patients. Transplant Proc 2007, 39, 2347–2349. [Google Scholar] [CrossRef] [PubMed]
- Juliano, G.R.; et al. Analysis of mast cells and myocardial fibrosis in autopsied patients with hypertensive heart disease. Rev Port Cardiol (Engl Ed) 2020, 39, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Skrabal, C.A.; et al. Interaction between isolated human myocardial mast cells and cultured fibroblasts. J Surg Res 2004, 118, 66–70. [Google Scholar] [CrossRef]
- Palaniyandi, S.S., K. Inagaki, and D. Mochly-Rosen, Mast cells and epsilonPKC: a role in cardiac remodeling in hypertension-induced heart failure. J Mol Cell Cardiol 2008, 45, 779–786. [CrossRef] [PubMed]
- Shiota, N.; et al. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J Hypertens 2003, 21, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Li, J., S. Jubair, and J.S. Janicki, Estrogen inhibits mast cell chymase release to prevent pressure overload-induced adverse cardiac remodeling. Hypertension 2015, 65, 328–334. [CrossRef] [PubMed]
- Zhao, X.Y. Chymase induces profibrotic response via transforming growth factor-beta 1/Smad activation in rat cardiac fibroblasts. Mol Cell Biochem 2008, 310, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Takai, S.; et al. A novel chymase inhibitor, 4-[1-([bis-(4-methyl-phenyl)-methyl]-carbamoyl)3-(2-ethoxy-benzyl)-4-oxo-azetidine-2-yloxy]-benzoic acid (BCEAB), suppressed cardiac fibrosis in cardiomyopathic hamsters. J Pharmacol Exp Ther 2003, 305, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Batlle, M.; et al. Increased expression of the renin-angiotensin system and mast cell density but not of angiotensin-converting enzyme II in late stages of human heart failure. J Heart Lung Transplant 2006, 25, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Schnee, J.M. and W.A. Hsueh, Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc Res 2000, 46, 264–268. [CrossRef]
- Jahanyar, J.; et al. Mast cell-derived cathepsin g: a possible role in the adverse remodeling of the failing human heart. J Surg Res 2007, 140, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; et al. An antiarrhythmic effect of a chymase inhibitor after myocardial infarction. J Pharmacol Exp Ther 2004, 309, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; et al. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation 2003, 107, 2555–2558. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; et al. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation 2000, 101, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Crabos, M.; et al. Characterization of angiotensin II receptors in cultured adult rat cardiac fibroblasts. Coupling to signaling systems and gene expression. J Clin Invest 1994, 93, 2372–2378. [Google Scholar] [PubMed]
- McLarty, J.L.; et al. Tryptase/Protease-activated receptor 2 interactions induce selective mitogen-activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension 2011, 58, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Chancey, A.L., G.L. Brower, and J.S. Janicki, Cardiac mast cell-mediated activation of gelatinase and alteration of ventricular diastolic function. Am J Physiol Heart Circ Physiol 2002, 282, H2152–H2158. [CrossRef] [PubMed]
- Murray, D.B.; et al. Endothelin-1 mediates cardiac mast cell degranulation, matrix metalloproteinase activation, and myocardial remodeling in rats. Am J Physiol Heart Circ Physiol 2004, 287, H2295–H2299. [Google Scholar] [CrossRef]
- Meléndez, G.C.; et al. Substance P induces adverse myocardial remodelling via a mechanism involving cardiac mast cells. Cardiovasc Res 2011, 92, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Hans, C.P.; et al. Opposing roles of PARP-1 in MMP-9 and TIMP-2 expression and mast cell degranulation in dyslipidemic dilated cardiomyopathy. Cardiovasc Pathol 2011, 20, e57–e68. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.; et al. Effects of mast cells on the behavior of isolated heart fibroblasts: modulation of collagen remodeling and gene expression. J Cell Physiol 2002, 191, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; et al. Protection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient rats. J Mol Cell Cardiol 2008, 45, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Frump, A.L.; et al. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2018, 314, L443–l460. [Google Scholar] [CrossRef] [PubMed]
- Luxán, G. and S. Dimmeler, The vasculature: a therapeutic target in heart failure? Cardiovasc Res 2022, 118, 53–64. [CrossRef] [PubMed]
- Wang, H.; et al. Mast Cell Inhibition Attenuates Cardiac Remodeling and Diastolic Dysfunction in Middle-aged, Ovariectomized Fischer 344 × Brown Norway Rats. J Cardiovasc Pharmacol 2016, 68, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Dona, M.S.I.; et al. Multi-omic analysis of the cardiac cellulome defines a vascular contribution to cardiac diastolic dysfunction in obese female mice. Basic Res Cardiol 2023, 118, 11. [Google Scholar] [CrossRef]
- Guimbal, S.; et al. Mast Cells Are the Trigger of Small Vessel Disease and Diastolic Dysfunction in Diabetic Obese Mice. Arterioscler Thromb Vasc Biol 2021, 41, e193–e207. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; et al. The Roles of Cardiovascular H(2)-Histamine Receptors Under Normal and Pathophysiological Conditions. Front Pharmacol 2021, 12, 732842. [CrossRef] [PubMed]
- Leary, P.J.; et al. Histamine H2 Receptor Antagonists, Left Ventricular Morphology, and Heart Failure Risk: The MESA Study. J Am Coll Cardiol 2016, 67, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Deliargyris, E.N.; et al. Mast cell tryptase: a new biomarker in patients with stable coronary artery disease. Atherosclerosis 2005, 178, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Heikkilä, H.M.; et al. Activated mast cells induce endothelial cell apoptosis by a combined action of chymase and tumor necrosis factor-alpha. Arterioscler Thromb Vasc Biol 2008, 28, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Lätti, S.; et al. Mast cell-mediated apoptosis of endothelial cells in vitro: a paracrine mechanism involving TNF-alpha-mediated down-regulation of bcl-2 expression. J Cell Physiol 2003, 195, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Kupreishvili, K.; et al. Mast cells are increased in the media of coronary lesions in patients with myocardial infarction and may favor atherosclerotic plaque instability. J Cardiol 2017, 69, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Laine, P.; et al. Association between myocardial infarction and the mast cells in the adventitia of the infarct-related coronary artery. Circulation 1999, 99, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; et al. The cardiac repair benefits of inflammation do not persist: evidence from mast cell implantation. J Cell Mol Med 2015, 19, 2751–2762. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, P.; et al. Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. J Pathol 2005, 205, 102–111. [Google Scholar] [CrossRef] [PubMed]
- de Souza Junior, D.A.; et al. Mast Cells Interact with Endothelial Cells to Accelerate In Vitro Angiogenesis. Int J Mol Sci 2017, 18.
- Theoharides, T.C.; et al. Mast cells and inflammation. Biochim Biophys Acta 2012, 1822, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; et al. Mast cell deficient W/Wv mice have lower serum IL-6 and less cardiac tissue necrosis than their normal littermates following myocardial ischemia-reperfusion. Int J Immunopathol Pharmacol 2007, 20, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Gilles, S.; et al. Release of TNF-alpha during myocardial reperfusion depends on oxidative stress and is prevented by mast cell stabilizers. Cardiovasc Res 2003, 60, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Paivandy, A. and G. Pejler, Novel Strategies to Target Mast Cells in Disease. J Innate Immun 2021, 13, 131–147. [CrossRef] [PubMed]
- Hara, M.; et al. Evidence for a role of mast cells in the evolution to congestive heart failure. J Exp Med 2002, 195, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S. and C.M. Ferrario, Chymase inhibitors for the treatment of cardiac diseases: a patent review (2010-2018). Expert Opin Ther Pat 2018, 28, 755–764. [CrossRef] [PubMed]
- Kanemitsu, H.; et al. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens Res 2006, 29, 57–64. [Google Scholar] [CrossRef]
- Jin, D.; et al. Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc Res 2003, 60, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, S.; et al. Chymase inhibition reduces infarction and matrix metalloproteinase-9 activation and attenuates inflammation and fibrosis after acute myocardial ischemia/reperfusion. J Pharmacol Exp Ther 2011, 339, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Houde, M.; et al. Mouse Mast Cell Protease 4 Deletion Protects Heart Function and Survival After Permanent Myocardial Infarction. Front Pharmacol 2018, 9, 868. [CrossRef] [PubMed]
- Wang, Y.; et al. Deficiency of mouse mast cell protease 4 mitigates cardiac dysfunctions in mice after myocardium infarction. Biochim Biophys Acta Mol Basis Dis 2019, 1865, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.C.; et al. Mast cell chymase limits the cardiac efficacy of Ang I-converting enzyme inhibitor therapy in rodents. J Clin Invest 2010, 120, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Takahama, H.; et al. A histamine H₂ receptor blocker ameliorates development of heart failure in dogs independently of β-adrenergic receptor blockade. Basic Res Cardiol 2010, 105, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Leary, P.J.; et al. H2 Receptor Antagonist Use and Mortality in Pulmonary Hypertension: Insight from the VA-CART Program. Am J Respir Crit Care Med 2018, 197, 1638–1641. [Google Scholar] [CrossRef] [PubMed]
- Leary, P.J.; et al. H2 receptor antagonists and right ventricular morphology: the MESA right ventricle study. Ann Am Thorac Soc 2014, 11, 1379–1386. [Google Scholar] [CrossRef]
- Zhang, J.; et al. Cardioprotective effect of histamine H2 antagonists in congestive heart failure: A systematic review and meta-analysis. Medicine (Baltimore) 2018, 97, e0409. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; et al. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev 2018, 282, 121–150. [Google Scholar] [CrossRef] [PubMed]
- Farha, S.; et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm Circ 2012, 2, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; et al. Mast cells promote lung vascular remodelling in pulmonary hypertension. Eur Respir J 2011, 37, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Bartelds, B.; et al. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest 2012, 141, 651–660. [Google Scholar] [CrossRef]
- Metcalfe, D.D.; et al. Biomarkers of the involvement of mast cells, basophils and eosinophils in asthma and allergic diseases. World Allergy Organ J 2016, 9, 7. [CrossRef]
- Kabashima, K.; et al. Biomarkers for evaluation of mast cell and basophil activation. Immunol Rev 2018, 282, 114–120. [Google Scholar] [CrossRef]
- Hu, K.K., M.A. Bruce, and M.J. Butte, Spatiotemporally and mechanically controlled triggering of mast cells using atomic force microscopy. Immunol Res 2014, 58, 211–217. [CrossRef]
Figure 1.
Mast cell activation factors and mast cell-derived mediators. A multitude of factors can induce mast cell activation and encompass tissue stretching due to pressure or volume overload, tissue inflammation, tissue injury, tissue fibrosis, tissue ischemia, tissue stiffness, allergies, and neurohormonal activation. Upon activation, mast cells release a diverse array of biologically active substances, including proteases (such as tryptase, chymase, cathepsin G, carboxypeptidase A3, dipeptidyl peptidase I (DPPI, cathepsin C), cathepsins L and S, and renin), proteoglycans (heparin), vasoactive amines (histamine and serotonin), cytokines/chemokines (such as TNFα, IL-2, IL-4, IL-5, IL-6, IL-8 [CXCL8], IL-13, CCL2 [MCP-1], CCL3 [MIP-1α], CCL4 [MIP-1β], and CCL5 [RANTES]), lipid mediators (LTB4, LTC4, PAF, PGE2, PGD2), and growth factors (VEGF, PDGF, TGFβ1, FGF-2).
Figure 1.
Mast cell activation factors and mast cell-derived mediators. A multitude of factors can induce mast cell activation and encompass tissue stretching due to pressure or volume overload, tissue inflammation, tissue injury, tissue fibrosis, tissue ischemia, tissue stiffness, allergies, and neurohormonal activation. Upon activation, mast cells release a diverse array of biologically active substances, including proteases (such as tryptase, chymase, cathepsin G, carboxypeptidase A3, dipeptidyl peptidase I (DPPI, cathepsin C), cathepsins L and S, and renin), proteoglycans (heparin), vasoactive amines (histamine and serotonin), cytokines/chemokines (such as TNFα, IL-2, IL-4, IL-5, IL-6, IL-8 [CXCL8], IL-13, CCL2 [MCP-1], CCL3 [MIP-1α], CCL4 [MIP-1β], and CCL5 [RANTES]), lipid mediators (LTB4, LTC4, PAF, PGE2, PGD2), and growth factors (VEGF, PDGF, TGFβ1, FGF-2).
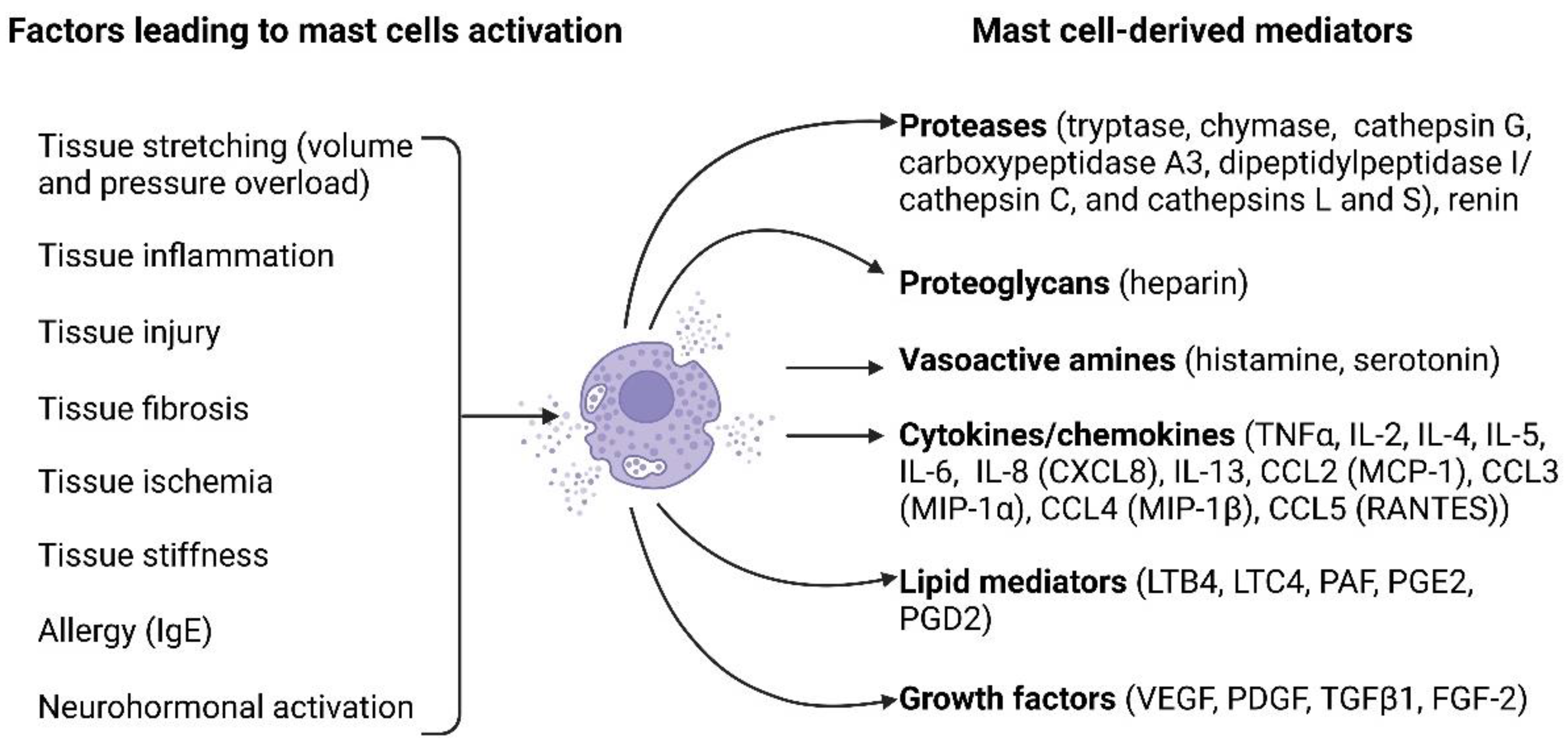
Figure 2.
Mast cell density and activation in experimental models of right ventricular (RV) remodeling. Mast cell density and activation were studied in mice and rats subjected to pulmonary artery banding (PAB) for various times up to 3 weeks and 28 weeks, respectively, and in rats subjected to aortocaval shunt for 8 weeks. Mast cell density was assessed in the RV tissue in all three models, while mast cell activation (degranulation) was evaluated only in the PAB mouse model. Mast cell density increased significantly in the RV of PAB mice and rats, starting at week 2 and remained elevated up to the end of the observation. In contrast, mast cell density in the RV of rats subjected to aortocaval shunt increased early during the first few days after surgery, returned to baseline values within the first week, and remained at these low values over the course of 8 weeks. Activation of mast cells (degranulation) in the RV of PAB mice occurred as early as three days after PAB surgery and persisted over the course of 3 weeks. Arrows indicate the time points of the measurements.
Figure 2.
Mast cell density and activation in experimental models of right ventricular (RV) remodeling. Mast cell density and activation were studied in mice and rats subjected to pulmonary artery banding (PAB) for various times up to 3 weeks and 28 weeks, respectively, and in rats subjected to aortocaval shunt for 8 weeks. Mast cell density was assessed in the RV tissue in all three models, while mast cell activation (degranulation) was evaluated only in the PAB mouse model. Mast cell density increased significantly in the RV of PAB mice and rats, starting at week 2 and remained elevated up to the end of the observation. In contrast, mast cell density in the RV of rats subjected to aortocaval shunt increased early during the first few days after surgery, returned to baseline values within the first week, and remained at these low values over the course of 8 weeks. Activation of mast cells (degranulation) in the RV of PAB mice occurred as early as three days after PAB surgery and persisted over the course of 3 weeks. Arrows indicate the time points of the measurements.
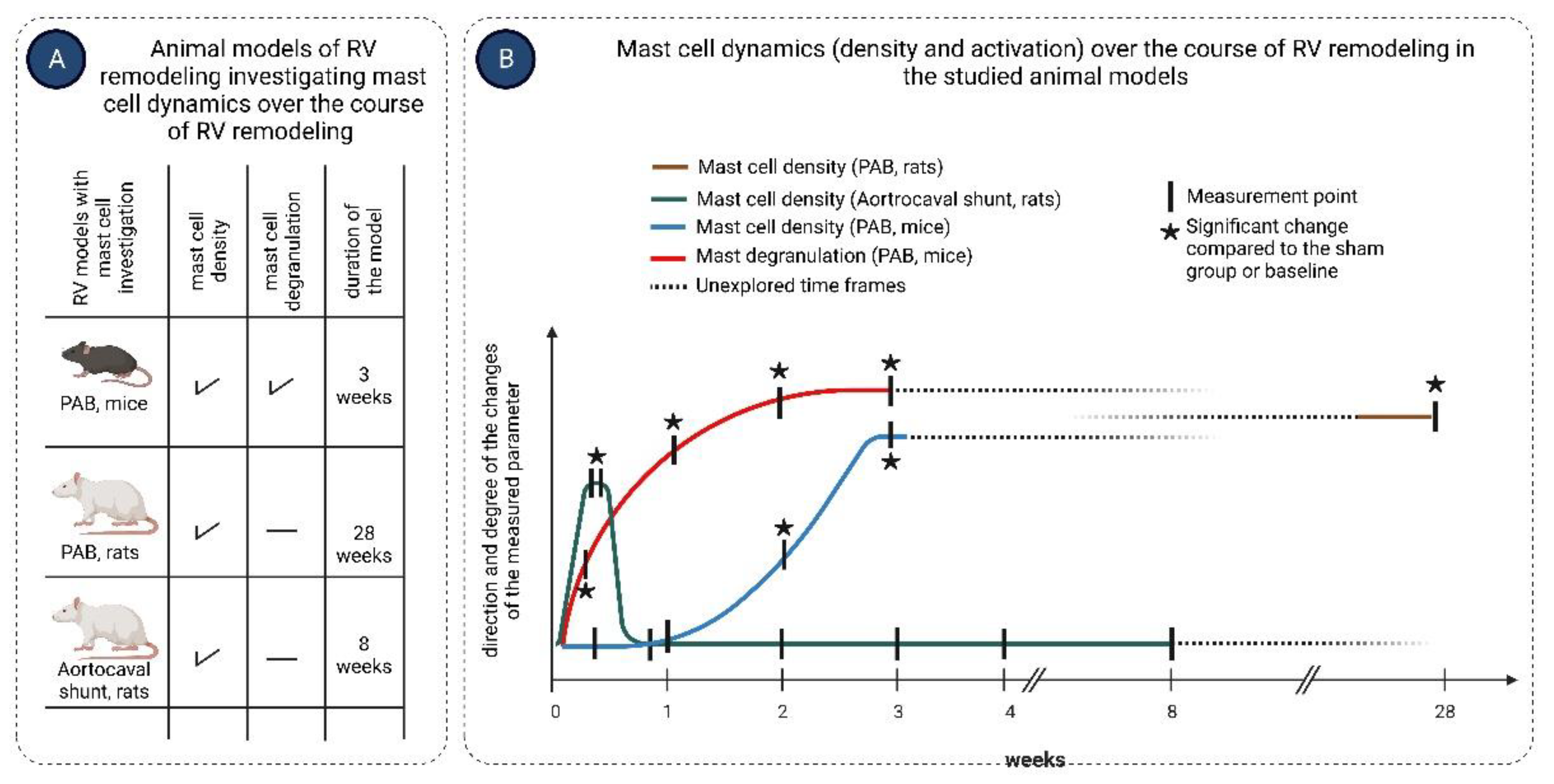
Figure 3.
Mast cell in cardiac fibrosis. (A) Mast cell density positively correlates with the degree of myocardial fibrosis in the left ventricular tissues isolated from patients with various cardiac pathologies, including idiopathic dilated cardiomyopathy, hypertensive heart disease, end-stage heart failure, and long-term LVAD support; (B) Mast cells isolated from cardiac tissues of heart failure patients stimulate activation of healthy cardiac fibroblasts, causing them to synthesize and release extracellular matrix molecules in co-culture experiments; (C) Mast cells activate the local RAAS in the myocardium. Renin produced by mast cells can convert angiotensinogen to angiotensin I. Cathepsin G and chymase derived from mast cells transform angiotensin I to angiotensin II. Angiotensin II stimulates cardiac fibroblasts to synthesize extracellular matrix molecules; (D) Several factors derived from mast cells, such as tryptase, chymase, histamine, TGF-β, and bFGF, activate cardiac fibroblasts to myofibroblasts and stimulate production of extracellular matrix molecules.
Figure 3.
Mast cell in cardiac fibrosis. (A) Mast cell density positively correlates with the degree of myocardial fibrosis in the left ventricular tissues isolated from patients with various cardiac pathologies, including idiopathic dilated cardiomyopathy, hypertensive heart disease, end-stage heart failure, and long-term LVAD support; (B) Mast cells isolated from cardiac tissues of heart failure patients stimulate activation of healthy cardiac fibroblasts, causing them to synthesize and release extracellular matrix molecules in co-culture experiments; (C) Mast cells activate the local RAAS in the myocardium. Renin produced by mast cells can convert angiotensinogen to angiotensin I. Cathepsin G and chymase derived from mast cells transform angiotensin I to angiotensin II. Angiotensin II stimulates cardiac fibroblasts to synthesize extracellular matrix molecules; (D) Several factors derived from mast cells, such as tryptase, chymase, histamine, TGF-β, and bFGF, activate cardiac fibroblasts to myofibroblasts and stimulate production of extracellular matrix molecules.
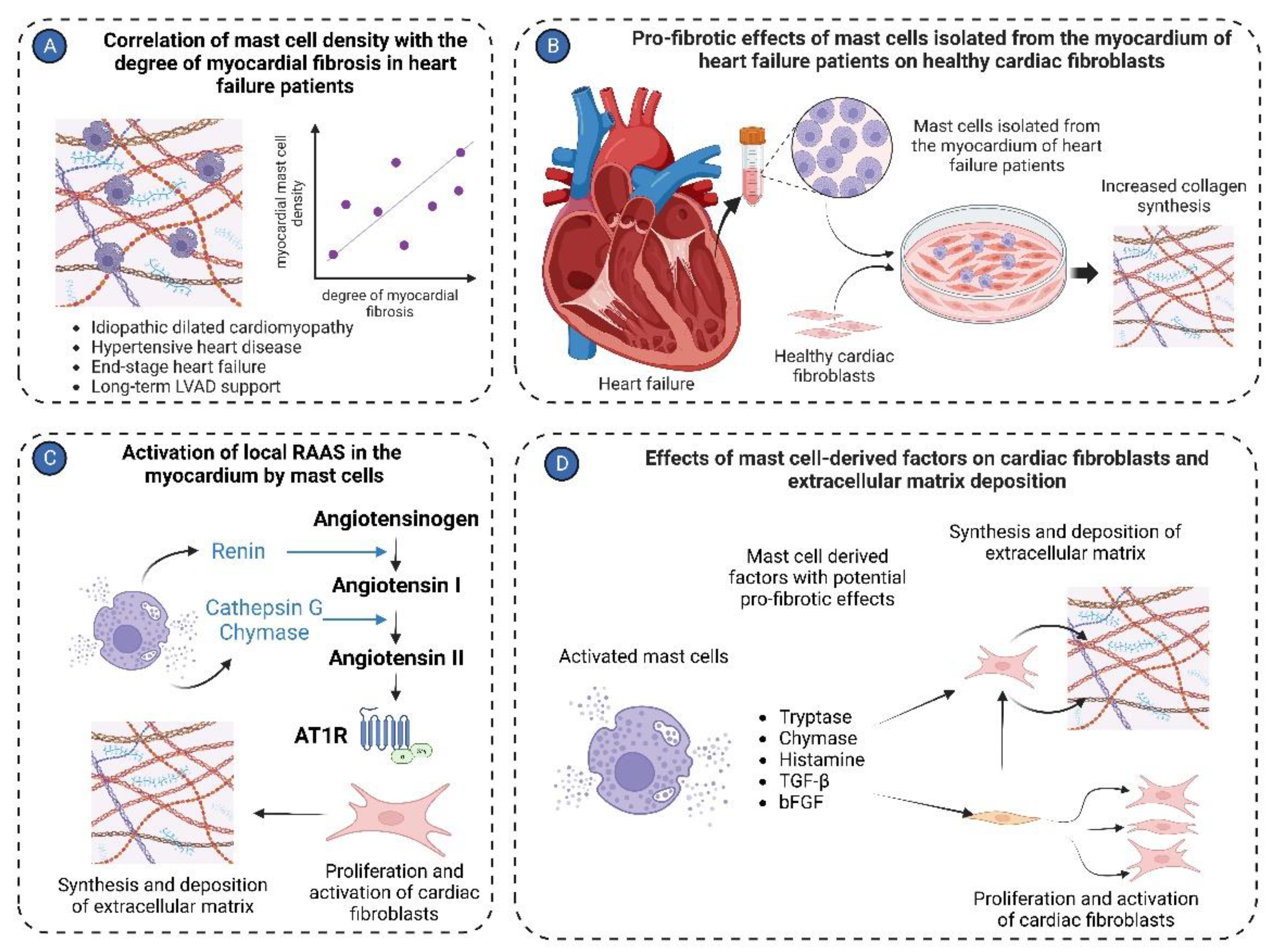
Figure 4.
Mast cells in cardiac vasculature. Mast cells in the cardiac vasculature have been implicated in the pathologies of both large coronary arteries and coronary microvasculature. (A) In large coronary arteries, mast cells are mainly involved in the pathogenesis of coronary atherosclerotic plaque formation and contribute to its instability, ultimately leading to the development of myocardial infarction; (B) In coronary microvessels, mast cells primarily interact with endothelial cells, modulating their functions. Histamine derived from mast cells increases the permeability of endothelial cells. Mast cell-derived TNF-α, IFN-γ, and IL-6 induce expression of endothelial cell adhesion molecules such as VCAM-1, ICAM-1, P-selectin, and E-selectin. Chymase and TNF-α derived from mast cells promote endothelial cell apoptosis.
Figure 4.
Mast cells in cardiac vasculature. Mast cells in the cardiac vasculature have been implicated in the pathologies of both large coronary arteries and coronary microvasculature. (A) In large coronary arteries, mast cells are mainly involved in the pathogenesis of coronary atherosclerotic plaque formation and contribute to its instability, ultimately leading to the development of myocardial infarction; (B) In coronary microvessels, mast cells primarily interact with endothelial cells, modulating their functions. Histamine derived from mast cells increases the permeability of endothelial cells. Mast cell-derived TNF-α, IFN-γ, and IL-6 induce expression of endothelial cell adhesion molecules such as VCAM-1, ICAM-1, P-selectin, and E-selectin. Chymase and TNF-α derived from mast cells promote endothelial cell apoptosis.
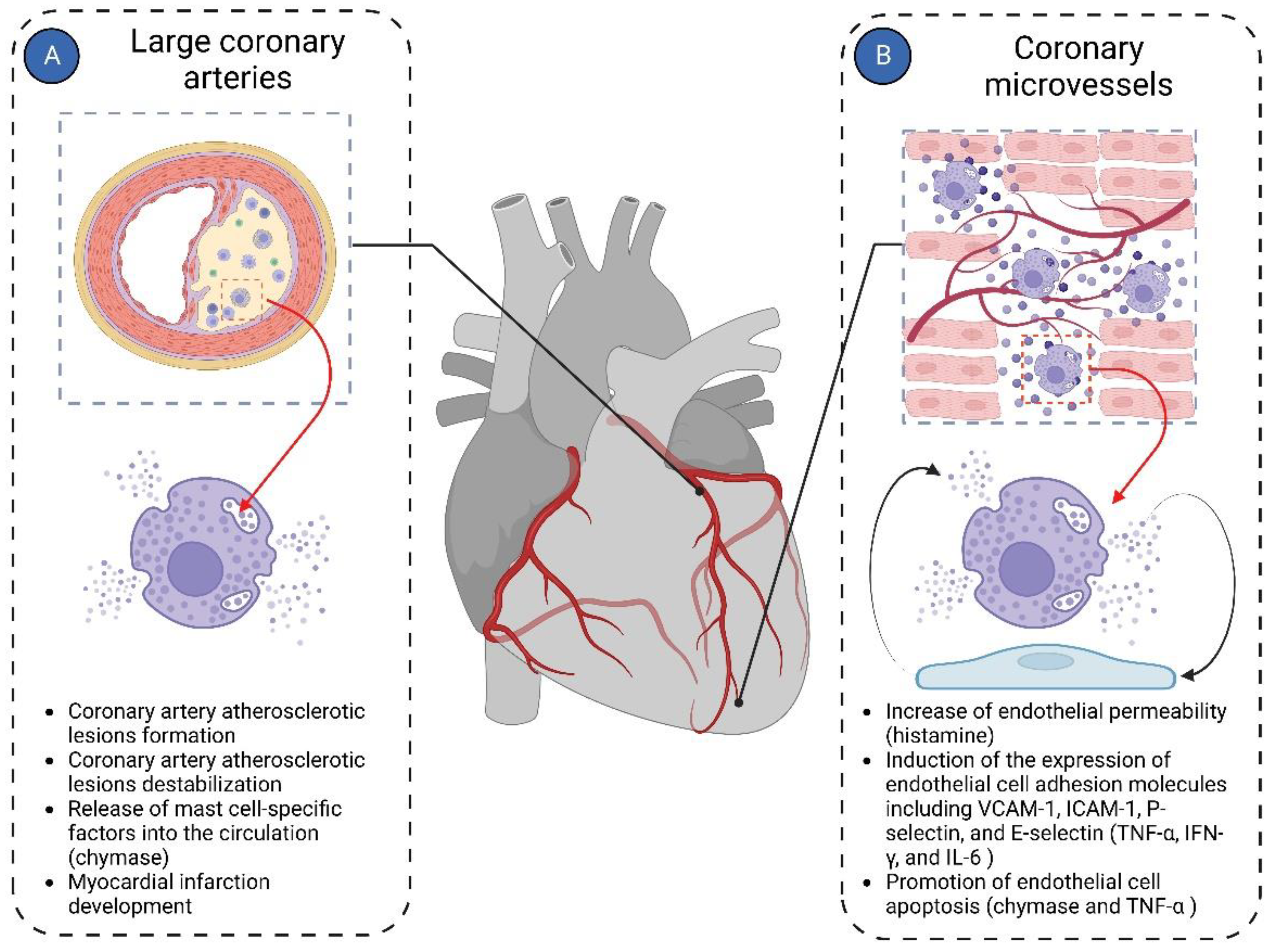
Figure 5.
Unaddressed issues related to the role of mast cells in right ventricular remodeling. (1) Influence of activated mast cells from remodeled pulmonary vessels on RV remodeling and vice versa in pulmonary hypertension; (2) Circulating mast cell-derived factors as a biomarker of RV remodeling in pulmonary hypertension; (3) Origin of mast cells in remodeled RV; (4) Role of mast cells in RV aging;. (5) Phenotypic and functional differences between mast cells in the RV and those in the left ventricle in health and disease. The known facts are highlighted in bold.
Figure 5.
Unaddressed issues related to the role of mast cells in right ventricular remodeling. (1) Influence of activated mast cells from remodeled pulmonary vessels on RV remodeling and vice versa in pulmonary hypertension; (2) Circulating mast cell-derived factors as a biomarker of RV remodeling in pulmonary hypertension; (3) Origin of mast cells in remodeled RV; (4) Role of mast cells in RV aging;. (5) Phenotypic and functional differences between mast cells in the RV and those in the left ventricle in health and disease. The known facts are highlighted in bold.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



