Submitted:
06 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
Cerebellar ataxias are a wide heterogeneous group of movement disorders. Within this broad umbrella of diseases, there are both genetics and sporadic forms. The clinical presentation of these conditions can exhibit a diverse range of symptoms across different age groups, spanning from pure cerebellar manifestations to sensory ataxia and multisystemic diseases. Over the last few decades, advancements in our understanding of genetics and molecular pathophysiology related to both dominant and recessive ataxias have propelled the field forward, paving the way for in-novative therapeutic strategies aimed at preventing and arresting the progression of these diseas-es. Nevertheless, the rarity of certain forms of ataxia continues to pose challenges, leading to lim-ited insights into the etiology of the disease and the identification of target pathways. Addition-ally, the lack of suitable models hampers efforts to comprehensively understand the molecular foundations of disease pathophysiology and test novel therapeutic interventions. In the following review, we describe the epidemiology, symptomatology, and pathological progression of heredi-tary ataxia, including both the prevalent and less common forms of these diseases. Furthermore, we illustrate the diverse molecular pathways and therapeutic approaches currently undergoing investigation in both pre-clinical studies and clinical trials. Finally, we address the existing and anticipated challenges within this field, encompassing both basic research and clinical endeavors.
Keywords:
Ataxia
; cerebellum
; therapy
1. Introduction
The term ataxia derives from Greek, a- without and taxis- order, and it refers to poor coordination of movements. Hereditary ataxias (HA) are a group of heterogeneous neurodegenerative diseases (NDD) that involve the degeneration of cerebellar and extracerebellar related circuits, leading to the development of progressive abnormal gait, dysarthria and ataxia. Within this large family of movement disorders, other structures involved in balance and gait control might also be affected, for instance some forms of autosomal recessive cerebellar ataxias involve the degeneration of proprioceptive neurons located in the dorsal root ganglia (DRG) and loss of function of muscles spindles leading to loss of proprioception [1,2].
Given the broad spectrum of symptoms and phenotypic heterogeneity that characterize HA, their classification is challenging. A rather conservative classification can be on the mode of inheritance, autosomal dominant cerebellar ataxias (ADCA) mainly known as spinocerebellar ataxia (SCA), autosomal recessive cerebellar ataxias (ARCA) and X-linked [2]. For example, among the ADCA category, SCA3 is the most common, together with SCA1, 2, 6, 7 and 17, are caused by a CAG trinucleotide expansion leading to a polyQ expansion in the encoded protein [3]. Friedreich’s ataxia (FA) is one of the most common ARCA caused by an instable GAA expansion, followed by Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), ataxia telangiectasia (AT) and more recently cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS) [4,5,6]. Among the X-linked ataxia, Fragile X-associated tremor/ataxia syndrome (FXTAS), caused by a CGG repeat expansion in the premutation range in the 5’ noncoding region of the fragile X mental retardation 1 (FMR1) gene is the most common [7]. Finally, episodic ataxias (EA), mostly inherited in an autosomal dominant manner, where the most common are EA1 and EA2 are associated with mutation in the KCNA1 potassium channel and CACNA1A calcium channel, respectively [8,9]. The classification presented above is a traditional approach to describing ataxias. However, for the purpose of this review, we suggest a classification centered on gene function and the pathways impacted by various mutations, as these factors may hold greater significance in terms of treatment considerations (Figure 1). Indeed, different types of ataxias display impairments in neuronal excitability and calcium homeostasis [10], among these SCA1 [11,12], as well as EA1 and EA2 as these mutations are directly impinging onto channels that are involved in excitability and calcium homeostasis. Different mutations such as the STUB1 found in ARCA patients are involved in the ubiquitin proteasomes systems, therefore impairing proteostasis [13]. Moreover, PolyQ expanded ATXN3 has been found to be involved in the autophagy pathway [14]. Mitochondrial dysfunction also seems to be an appealing candidate for future therapeutical interventions. Indeed, oxidative stress, lipid oxidation, mitochondria dysfunction, and ATP production deficits are common denominators of different HA such as FA, ARSACS, ARCA2, SCA1, SCA3, SCA7 to list a few [15,16,17,18,19,20]. Finally, many HA including SCA1,2,3, 36 and many others are characterized by nucleotide repeat expansion, which might altered RNA metabolisms leading to RNA toxicity via sequestration of RNA binding proteins and RNA foci formation within vulnerable neurons [21,22,23,24].
Given the broad spectrum of symptoms, the different genetic pattern of inheritance, the various molecular pathways implicated, finding a resolutive treatment remains challenging. Nonetheless, during the past decade, encouraging advances in terms of basic as well clinical research have been achieved. In this review, we provide further insights in the field of HA, from autosomal dominant to X-linked, and discuss the major achievements in terms of promising preclinical models and therapeutical approaches. Finally, we will touch upon the main challenges and limiting factors that represent an obstacle to the achievement of an effective treatment for these conditions.
2. Autosomal Dominant Hereditary Ataxias
ADCA is an heterogenous group of genetically NDD, which are numbered chronologically in order of discovery of the genetic loci. All SCA share common features including cerebellar dysfunctions leading to progressive motor incoordination, dysarthria, unsteady gait and ataxia, commonly accompanied by pyramidal and extrapyramidal tracts impairments [25]. In some forms, non-motor symptoms such as cognitive and retinal degeneration are also present [25]. ADCAs are classified based on their clinical manifestation in type I, II and III. Typically, ADCA type I (SCA1,2,3,4,8,10,12,23,25,27,28,32,36), the clinical manifestations involve cerebellar symptoms accompanied by different neurological manifestations such as cognitive impairment and seizure; ADCAs type II (SCA7) also present retinal degeneration, while type III (SCA5,6,11,26,30,31) display pure cerebellar symptomatology [26,27].
The prevalence of ADCA is approximately between 1- 5:100,000, nevertheless it should be noted that there is great variability based of geographical and ethnical groups, with an incidence of 3:100,000 in the European population. Symptoms usually arise during the 4th decade of life, mainly with progressive loss of motor coordination, hyperreflexia, and spasticity mostly due to the degeneration of cerebellar Purkinje cells and brainstem. To date, more than 49 SCAs have been classified based on 37 gene mutations that include repeat expansions in both coding and non-coding gene regions, point mutations and deletions [2].
A CAG trinucleotide repeat expansion in different genes is the principal cause of the most common SCAs including SCA1,2,3,6,7,17, leading to an abnormal polyQ tract within the encoded protein. There is an inverse correlation between the size of the repeat and the age of onset [32]. Furthermore, due to the genetic instability of the CAG expansion, with a tendency to expand further when transmitted to the next generation, a phenomenon named anticipation, that is the tendency for disease to worsen from generation to generation, is present [32,33,34]. Moreover, the encoded proteins arising from gene mutations play roles in various physiological processes, such as transcription regulation, proteostasis, excitability, mitochondrial metabolism, and calcium homeostasis. These collectively contribute to the development of diseases and neuronal degeneration. In the following paragraphs, we describe the most common SCAs with their clinical and neuropathological features.
2.1. SCA1
SCA1 (OMIM # 164400) is characterized by ataxia, dysarthria and eventual deterioration of bulbar functions, with typically the onset in the 3rd-4th decade of life. One third of SCA1 patients also present cognitive impairments [35,36]. Respiratory failure is the main cause of death [37]. SCA1 patients usually present loss of Purkinje neurons (PN) and deep cerebellar nuclei (DCN) neurons located in the dentate nucleus, olivopontocerebellar atrophy and brainstem. Within the spinal cord, the anterior horns, Clarke’s column and the spinocerebellar tract are mainly affected [3,31,38,39].
SCA1 is caused by a CAG trinucleotide expansion in the coding region of the ATXN1 gene, encoding the ataxin-1 protein [40].The ataxin-1 protein is widely express throughout the central nervous system, and it shuttles between the cytoplasm and the nucleus via its nuclear localization signal (NLS) domain. Ataxin-1 is involved in RNA metabolism [41] and can act as transcriptional repressor together with the co-repressor Capicua (CIC). The pathological PolyQ expansion leads to the retention of the protein in the nuclear compartment leading to its aggregation and possible toxicity. Interestingly, a single amino acid substitution in the NLS sequence is able to abolish its pathogenicity [41,42]. In physiological conditions, ATXN1 protein forms a complex with the transcriptional repressor capicua (CIC), thereby directly modulating its activity. The gain of function of this complex can lead to PN degeneration [43,44]. Finally, phosphorylation defects of different residues of the Ataxin-1 protein such as Serine 776 (Ser776) in the C-terminal domain are implicated in disease pathophysiology as it allows its interaction with different binding partners including splicing factors [45,46,47,48].
2.2. SCA2
SCA2 (OMIM # 183090) patients manifest a broad spectrum of symptoms including cerebellar ataxia accompanied by oculomotor impairments and parkinsonism due to the degeneration of dopamine releasing neurons in the substantia nigra [49,50]. In addition, some degree of cognitive impairments has been observed in patients associated by sleep disturbances [51]. Finally, patients often manifest peripheral neuropathy [52]. Analogous to SCA1, SCA2 patients present the same pattern of degeneration within the olivopontocerebellar structure, however the deep cerebellar nuclei structures remain relatively spared by the pathology [53]. SCA2 is caused by a CAG trinucleotide expansion in the coding region of the ATXN2 gene, encoding the ataxin-2 protein [49]. Unaffected individuals have 13 to 31 CAG repeats, whereas affected individuals have over 37 repeats [54]. Interestingly, long CAG expansions within the normal range are a risk factor for amyotrophic later sclerosis (ALS) and fronto-temporal dementia (FTD) patients, both familial and sporadic cases [55,56,57]. Ataxin-2 protein is implicated in RNA metabolism, by interacting with polyA-binding protein (PABP) as well as transactive response (TAR) DNA-binding protein 43 (TDP43), which also bind to RNA, suggesting an interplay of these proteins within common pathways. This probably reflects a common risk factor for both SCA2 and ALS-FTD pathophysiology [58,59,60].
2.3. SCA3 (Machado–Joseph Disease)
SCA3 (OMIM # 109150) is considered one of the most common forms of SCA, nevertheless there is great variance in the prevalence between different countries, that can span from nearly 0% to more than 55% of the population in countries such as Portugal [61,62]. The onset of the pathology can vary from very young age to adulthood, which reflects different pattern of disease progression, therefore SCA3 can be classified based on clinical observation in 4 subtypes [39,63,64,65]. SCA3 type I involves patients with early onset between the age of 10 to 30 years and is characterized by nominal ataxia with the involvement of pyramidal and extrapyramidal signs. Type II, the most common, appears between the second and fifth decade of life with patients developing progressive ataxia accompanied by pyramidal signs, while type III appears later in life and mainly involves peripheral neuropathy that leads to muscles atrophy. Finally, type IV occurs at different ages and patients experience a strong parkinsonism component. Differently from SCA1 and SCA2, the cerebellar cortex is relatively spared, but degeneration occurs in the deep cerebellar nuclei, in particular the dentate nucleus [66,67]. Moreover, degeneration of the cerebral cortex as well as the substantia nigra, leading to parkinsonism, as well as other subthalamic nuclei is observed in some patients [39,68]. Furthermore, patients suffer of mild cognitive impairments [69]. The unstable CAG repeat that characterize SCA3 leads to a PolyQ expansion in the encoded Ataxin-3 protein. Normal range is up to 44 repeats while SCA3 patients have between 52-86 repeats. Under physiological condition, ataxin-3 shuttles between the nucleus and the cytoplasm, and is a deubiquitinating enzyme involved in protein quality control regulation [70,71,72]. In SCA3, mutant Ataxin-3 accumulates in the nucleus of neurons [73].
2.4. SCA6
SCA6 (OMIM # 183086) is considered a pure cerebellar ataxia where cerebellar PN of the cerebellar cortex are affected while other brain regions are relatively spared by the pathology compared to other SCAs [74]. On average, symptoms onset appear between the 4th and 5th decade of life, and include progressive imbalance and cerebellar ataxia accompanied by dysarthria and nystagmus [75,76]. Moreover, in some patient, basal ganglia dysfunction has been reported [75,76]. The highest prevalence is reported in Japan [77], whereas it is quite low in Europe and North America. The CAG pathological expansion, that ranges from 20 to 33 repeats, affects the CACNA1A gene [78,79,80] that encodes for the α1A subunit of the P/Q-type voltage-gated calcium channel Cav2.1, which is particularly abundant in the vulnerable PN [76]. In addition to full-length CACNA1A, a shorter form through the use of a ribosomal entry site generates the transcription factor α1ACT with the polyQ repeat at the C-terminal domain, involved in PN development [81].
2.5. SCA7
SCA7 (OMIM # 164500) patients experience cerebellar ataxia, dysarthria, and spasticity [82]. A peculiar characteristic of SCA7 is retinal degeneration, leading eventually to blindness [83]. A small percentage of patients manifest cognitive impairment and psychosis [84,85]. Adult onset SCA7 patients harbor between 37 to 50 repeats, whereas non-pathological repeats are found to be between 4 to 17 CAG [82]. As for other type of SCAs, juvenile forms of SCA7 are more aggressive in terms of symptomatology and prognosis. Adolescent SCA7 is characterized by extensive visual loss at first involving macular degeneration, without initial cerebellar ataxia symptoms [82]. Infantile SCA7, that manifests at birth or within the few first months, is caused by extremely long expansions reaching 460 repeats [87], characterized by congenital malformations resulting in multiorgan failure [82]. SCA7 is characterized by a CAG expansion in the Ataxin-7 gene [86]. ATXN7 is a core component of the Spt-Ada-Gcn5 acetyltransferase (SAGA) complex which function as a chromatin modifying transcriptional coactivator complex [88]. Ataxin-7 directly interacts with GCN5, the histone acetyltransferase component of STAGA/SPT3 complex, mediating the interaction with the cone-rod homeobox (CRX) transactivator of photoreceptor genes [89]. Finally, Ataxin-7 protein shuttles between nucleus and cytoplasm, and when it localize at the cytoplasm it has been shown to be involved in microtubule organization [90].
2.6. SCA17
To date, less than 100 families have been reported to be affected by SCA17 (OMIM # 607136) worldwide. Symptoms onset can vary from infancy to 75 years [91] and include cerebellar ataxia, pyramidal signs as well as psychiatric manifestation and dementia [92], caused by degeneration of the cerebral cortex, cingulate and hippocampal gyri as well as cerebellar PN and substantia nigra degeneration [39]. SCA 17 also called Huntington disease like-4 because of similarity in symptomatology with Huntington disease (HD), is caused by a CAG expansion in the coding region of the TATA box binding protein (TBP) gene [93]. The PolyQ stretches can vary in size causing different degrees of penetrance, with normal alleles containing 25 to 40 trinucleotide repeats expansion, whereas expansions containing more than 50 trinucleotides repeats are considered pathological with full penetrance [94]. TBP is an important factor for the initiation complex of the RNA polymerase II, which is essential for the initiation of transcription [34]. In SCA17, the PolyQ expansion affects multiple cellular processes, from Notch signaling pathway to ER stress response, by impairing different transcription factors via gain or loss of function mechanisms [95].
2.7. SCA27B (GAA-FGF14 Ataxia)
Recently, a heterozygous GAA trinucleotide expansion within intron 1 of the fibroblast growth factor 14 (FGF14) gene has rbeen identify in patient with late onset spinocerebellar ataxia (SCA27B - OMIM # 620174) [96]. The expansion was firstly identified in European and French-Canadian families, after long-range genome sequencing in families with unidentified hereditary ataxia. GAA expansion >250 within the FGF14 gene accounts for 10 to 61% of unsolved ataxia cases, in different cohorts analyzed in Australia, Europe and India [96]. Patient symptoms mainly manifest as cerebellar ataxia accompanied by downbeat nystagmus in 42 % of the patients [96] and appear during the sixth decade of life. Nevertheless, there might be a possibility of earlier onset of GAA-FGF14 ataxia [96]. Diagnosis of this form of ataxia remains challenging because of the long repeat expansion, that already in healthy individuals can go up to 300 repeats [96]. Nevertheless, Bonnet and colleagues have recently developed a new approach that combined long range PCR, bidirectional repeat-primed PCRs and Sanger sequencing for the detection of GAA-FGF14 expansion [97].
Post-mortem analysis of two French Canadian patients showed marked vermis atrophy and PN loss accompanied by astrogliosis. Finally, in both post-mortem tissue and iPSCs derived motoneurons, FGF14 transcript and protein level were found significantly reduced compared to controls, suggesting that the GAA expansion might lead to reduce FGF14 transcription, similarly to Friedreich Ataxia [96]. Moreover, Fgf14 knock-out mice display ataxia phenotype [98]. FGF14 is particularly abundant in the cerebellum and regulates the rhythmic firing property of the PN, by regulating voltage-gated sodium channels that are found in the initial axon segment [99,100]. Therefore, GAA-FGF14 might also be considered as a channelopathy.
3. Autosomal Recessive Cerebellar Ataxias
Autosomal recessive cerebellar ataxias (ARCAs) are a group of rare diseases with clinical and genetic heterogeneity. ARCAs clinical spectrum is heterogeneous and complex (often multisystemic), and the symptoms usually manifest during childhood or adolescence with a severe impact on daily life of patients [101]. ARCAs are characterized by abnormal development and progressive neurodegeneration of the cerebellum and/or its associated afferents tracts, often accompanied by damage of other neurological (e.g., corticospinal tract, basal ganglia, peripheral nerve) or non-neurological systems (e.g., heart, muscles, pancreas) [102,103]. To date, 92 genetic diseases with autosomal recessive inheritance show ataxia as the main clinical feature or combine ataxia with other predominant movement disorders [104]. However, due to genetic pleiotropy this number could be even higher. In fact, genes linked to autosomal dominant ataxia have also recently been found to cause autosomal recessive ataxia in case of biallelic inheritance (e.g., AFG3L2 / SPAX5 [105], SPTBN2 / SPARCA1 [106], ITPR1 / Gillespie Syndrome [107], OPA1 / Syndromic DOA or Behr phenotype [108,109]). Finally, other 89 non-ataxic autosomal recessive diseases have been reported in which occasionally cerebellar ataxia phenotype has been documented [104].
The worldwide prevalence of ARCAs is estimated around 3-6/100.000 individuals [29,110]. Due to the lack of molecular diagnosis, the accessibility to genetic tests, and regional founder effect and consanguinity, this is just an estimation. Among all the ARCAs, FA affects 1:29.000-50.000 people, with the highest prevalence in Caucasians and European countries [111,112,113], and appears to be the most common, even though the recently described CANVAS is predicted to have similar frequency [4,101,114].
In the upcoming sections, we delineate the clinical, genetic, and molecular traits of prevalent ARCAs, along with less common mutations that, despite their rarity, exhibit shared pathophysiological traits.
3.1. Friedreich Ataxia
Friedreich ataxia (FA - OMIM #229300) was first described in the nineteenth century by Nicholaus Friedreich. The onset of clinical symptoms appears before the age of 25 years (mean age of onset is 10-15 years) and the disease progression leads to wheelchair dependence within 10 years of disease onset. The classic presentation consists of progressive gait and limb ataxia, dysarthria, polyneuropathy and sensory loss [115]. Moreover, non-neurological symptoms such as scoliosis (~80% of the patients), diabetes (~30%), and hypertrophic cardiomyopathy (>66%) have been reported [116]. Heart failure is, indeed, a common cause of death for FA patients [103,117]. Differentially to other ARCAs, FA is considered an afferent ataxia because of the degeneration of the large sensory neurons in the dorsal root ganglia (DRG) and posterior column. In addition, the degeneration of the spinocerebellar and corticospinal tracts is well documented, and the lesions in the dentate nucleus cause the major cerebellar phenotype [5,118], while PN dysfunction and cerebral abnormalities are less involved in FA pathophysiology compared to other ARCAs [119,120].
FA is caused by a non-coding GAA trinucleotide expansion within the FXN gene encoding frataxin. Most patients (96%) harbour a homozygous GAA triplet expansion (ranging from 40 to 1000 triplets) within the first intron of FXN gene. While the remaining 4% of the cases are compound heterozygotes presenting a single allele GAA expansion and a more clssical mutation in the other allele [103,117,121]. In contrast with autosomal dominant cerebellar ataxia, FA is not subject to the anticipation phenomenon and, to date, together with CANVAS, is the only ARCA presenting nucleotide expansion as disease mechanism. The GAA expansion alters the epigenetic pattern (heterochromatin conformation) and/or promotes the formation of RNA/DNA hybrids (R-loops) resulting in a drastic decrease of FXN gene transcription and, therefore, reduced levels of frataxin (approximately estimated at 5-30% of normal levels) [117,122]. Frataxin is a ubiquitously expressed nuclear-encoded mitochondrial protein located in the mitochondrial matrix. Although, frataxin has been suggested to be involved in a variety of pathways link to iron metabolism, the only function widely accepted of frataxin is its essential role as an allosteric regulator of Fe-S clusters biogenesis. Since Fe-S clusters are crucial elements in oxidative phosphorylation, iron metabolism, protein translation and DNA repair/replication, frataxin deficiency leads to low levels of Fe-S cluster biogenesis and therefore triggers mitochondrial dysfunction, iron accumulation, oxidative stress and most likely DNA damage accumulation [123,124,125].
3.2. CANVAS
Cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS – OMIM #614575) is a common adult-onset and slowly progressive ataxia disorder characterized by imbalance (80% of the patients), sensory neuropathy (almost 100%), bilateral vestibular impairment (60%), chronic cough (>60%) and occasionally autonomic dysfunction [126]. The average age of onset has been reported to be 60 years old [127]. CANVAS has recently been linked to a biallelic AAGGG repeat expansion in the second intron of RFC1 (Replication factor C subunit 1) gene [128]. The allelic carrier frequency of the AAGGG repeat expansion in healthy controls has been documented around 0,7%, a percentage similar to FA healthy carriers suggesting that CANVAS may represent a frequent cause of late-onset ataxia with an estimated prevalence of 1:20.000 individuals [128]. The abnormal expansion has been identified in the poly(A) tail of an AluSx3 element and differs in both size (pathological size ranges from 400 to 2.000 repeats) and nucleotide sequence from the reference (AAAAG). Interestingly, the high polymorphism of this locus leads to other expanded non-pathogenic motifs: AAAAG extended pentanucleotide whose average size ranged from 15 to 200 repeats, AAAGG repetition ranged from 600 to 1.000 [128]. Lately, four novel pathogenic pentanucleotides configurations, both in homozygous and in compound heterozygous state with AAGGG, have been associated with CANVAS disease [129]. These expanded regions were sequenced and registered as AGGGC, AAGGC, AAGAG and AGAGG, with the extended region always found longer than 600 repeats [129]. Alu elements are repetitive (300 bps long) and highly conserved sequences within the primate genomes, whose 3’-end has an A-rich region critical for its amplification mechanism. While the pathogenic mechanism of the repeat is unclear, it has been hypothesized that the extended AAGGG expansion can induce the inactivation process of poly(A) tail of the retrotransposon AluSx3 [128]. Furthermore, the existence of truncating mutation in trans with the AAGGG repeat supports the existence of a loss-of-function mechanism [130]. Despite these reports, both transcript and proteins levels are not affected in patient samples [128,129]. More recently, the formation of G-quadruplexes has been reported to occur with the pathogenic motifs, suggesting toxic-DNA and toxic RNA modes of pathogenesis or possible R-loops formation [131]. RFC1 encodes the large subunit of replication factor C, a DNA polymerase accessory protein involved in DNA replication and repair. It is a DNA-dependent ATPase that binds in a structure-specific manner to the 3’ end of a primer hybridized to a template DNA. Therefore, in the presence of ATP, RFC1 activates the DNA polymerase δ and ε to promote the coordinated synthesis of both strands during replication or after DNA damage.
3.3. ARSACS
Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS - OMIM #270550) is an early-onset cerebellar ataxia initially described in 1979 by Bouchard and colleagues [132]. The disease name is given by the region of Charlevoix-Saguenay-Lac-Saint-Jean (CSLS), in Québec (Canada), where the highest frequency of healthy carriers (1:22 individuals) was registered [132,133]. Within this population, in 2000 Engert et al. described for the first time two mutations in the SACS gene. One of these two mutations, g.8844delT, belongs to a major haplotype shared by 96% of ARSACS families descending from an ancestral founder in the population that settled first in Québec [134]. Nowadays, ARSACS has been diagnosed worldwide, with the identification of more than 200 mutations spread all over the SACS gene, and until recently (before the description of CANVAS) it was considered the second most common recessive ataxia after FA [6]. Most of the patients are homozygous for SACS mutations (45,2%), followed by compound heterozygous (32,9%) and all types of mutation have been reported, with missense substitutions being the most represented, however macro-deletions, frameshifts changes and nonsense mutations have also been observed [135]. No genotype-phenotype correlation has been documented [136]. ARSACS is clinically characterized by early-onset (the mean for the age of onset is 3 years) and progressive cerebellar ataxia (observed in 78,9% of the patients), associated with spasticity (78,1%) and sensory-motor axonal peripheral neuropathy (73,7%) [137]. The main feature is the progressive degeneration of the cerebellar vermis (remarkable cerebellar atrophy and PN loss) documented by MRI and post-mortem analysis. Other typical symptoms include dysarthria, distal amyotrophy (likely a consequence of neuropathy), limb weakness, sensory loss, pyramidal signs, and cerebellar eye manifestations (e.g., nystagmus) supported by bilateral demyelination of both corticospinal and dorsal spinocerebellar tracts [132,133,135]. Moreover, ocular coherence tomography analysis detected a peculiar increase of the peripapillary retinal nerve fiber layer (RNFL) thickness in ARSACS patients, suggesting it as a putative disease diagnostic tool [138,139]. The SACS gene encodes for sacsin, a cytosolic protein highly expressed in the brain (with the highest expression in PN), skeletal muscle, heart and dermal fibroblasts [134]. While sacsin function is still largely unknown, molecular studies suggest a role of sacsin in protein quality control [140,141] affecting intermediate filament remodelling [142,143,144]. However, the involvement in the alteration of mitochondrial dynamics, ultrastructure and bioenergetics is not yet fully clarified [15,144,145].
3.4. Ataxia-Telangiectasia
Ataxia-telangiectasia (AT – OMIM #208900) was the first early-onset recessive for which a gene had been identified, and AT is considered a quite common ARCA, accounting 3-5% of all ARCA patients. The characteristic clinical symptoms appear between 1 and 4 years of age, including progressive neuronal degeneration (mainly cerebellum, but also cortex and peripheral system), accompanied by oculomotor apraxia (almost 100% of the patients), extrapyramidal movements (chorea and dystonia, >80%), immunodeficiency (60-80%) and cancer susceptibility (in particular leukaemia and lymphoma, 85%) [146,147]. From a genetic point of view, AT patients are mainly compound heterozygotes. The vast majority carry nonsense or frameshift mutations over the entire ATM gene, resulting in protein loss-of-function. While the few missense mutations identified affect splicing event [147]. The ATM is a serine/threonine kinase that safeguard the genome integrity from double strand break activating more than 20 proteins involved in DNA damage response (e.g., phosphatidylinositol 3-kinase-related protein kinase (PIKK) family which phosphorylates in turn several substrates, like H2AX) and cell cycle regulation (e.g., protein kinase Chk2 and p53) [148,149,150].
3.5. ARCA2
Autosomal recessive ataxia type 2 (ARCA2 – OMIM #612016), also identified as COQ8A-ataxia, is a rare form of multisystemic ataxia characterized by slowly progressive early-onset ataxia, combined with variable symptoms including exercise intolerance (25% of the patients), dystonia (28%), epilepsy (32%) and intellectual disability (49%) [151,152,153]. In most of the cases, disease onset occurs before the age of 6 [154]. ARCA2 is caused by biallelic loss-of-function mutations in COQ8A (also known as ADCK3), a mitochondrial atypical kinase/ATPase whose biochemical function remains so far not completely understood. To date, almost 50 pathogenic variants of different nature (missense, nonsense, frameshift and deletion) have been identified all along the gene with missense mutations harbouring mainly in the COQ8A active site impacting ATPase activity, protein stability and/or nucleotide binding capacity [154]. COQ8A is located in the matrix face of the inner mitochondrial membrane where it ensure the integrity of complex Q (enzymes involved in the CoQ biosynthesis) and has been suggested to regulate CoQ biosynthesis [155,156,157]. COQ8A deficiency affects CoQ10 levels in different tissues resulting in inefficient oxidative phosphorylation and alterations in mitochondrial homeostasis, and oxidative stress due to the lack of antioxidant power [17,158].
4. Episodic Ataxias
Episodic ataxia (EA) is a rare group of autosomal dominant inherited disorders associated to a heterogeneous clinical spectrum manifesting with recurrent cerebellar dysfunction (i.e., spells of truncal ataxia and incoordination) of variable duration and often accompanied by ictal and interictal symptoms [2]. The overall prevalence has been estimated less than 1:100.000 individuals, probably underrated because of unidentified causative genes and lack of genetic tests [9,159]. To date, eight different subtypes have been described with recent proposal for two additional ones, however the causative gene has been identified only for five (EA1 / KCNA1, EA2 / CACNA1A, EA5 / CACNB4, EA6 / SLC1A3 and EA8 / UBR4). Interestingly, four out of the five genes are ion channel protein involved in the excitatory neurotransmission [159]. Clinically, the patients show ataxia and vertigo symptoms starting from the early childhood [9,159,160]. EA1 (OMIM #160120) is associated with mutation in KCNA1 gene encoding the potassium voltage-gated channel α1 subunit Kv1.1, highly expressed in the cerebellum and crucial for the formation of GABAergic synapses on the PNs. Among the 63 KCNA1 mutations described, the most are missense mutations leading to a loss-of-function mechanism. The symptoms appear between 2 and 10 years of age and the attacks can last from seconds to minutes, with variable frequency [9,159]. CACNA1A encoding the alpha 1A subunit of the P/Q-type voltage-gated calcium channel (Cav2.1) has been identified to be mutated in EA2 (OMIM #108500). In most of the cases, nonsense and frameshift mutation leading to premature stop codon have been reported [161]. Cav2.1 is highly expressed in PNs and granule cells in the cerebellum conducting the synaptic transmission. The age of onset is very early (2-20 years of age), but, different from EA1, the intermittent spells of ataxia are longer, lasting several hours and in some cases even days [9,159].
5. X-Linked Degenerative Ataxias
Long CGG repeat expansion in the 5’ non-coding region of the FMR1 gene are associated to Fragile X syndrome (FXS), a common hereditary form of mental retardation and autism. Interestingly, carriers of relatively short CGG repeat expansion (55-200, premutation range) results in a distinct inherited degenerative disorder affecting ageing individuals, mainly men. This is the major X-linked ataxia disease identified as Fragile X-associated tremor/ataxia syndrome (FXTAS – OMIM #300623) [7,162,163]. Typically, symptoms of FXTAS appear in the fifth decade of life (however the premutation CGG repeat length was demonstrated to correlate with age of onset) [164] accompanied by progressive cerebellar ataxia (85% of all the patients), tremor (70%), and cognitive decline (approximately 50%), and associated with psychiatric manifestations (50%) and peripheral neuropathy (60%) [165]. Interestingly, FMR1 premutation female carriers are associated with an increased risk for primary ovarian insufficiency without developing ataxia and tremor [7,163,166]. From a molecular point of view, in FXTAS patients, FMRP (fragile X mental retardation protein) amount is normal, while FMR1 mRNA level is drastically elevated in brain and leukocytes [167], leading to toxicity (through the production of FRMpolyG by Repeat-associated non-AUG (RAN) translation) and causing cellular injury responsible for the symptoms (intranuclear inclusions and DNA damage through R-loops formation) [163].
6. Congenital Ataxias
Congenital cerebellar ataxias are a relatively small group of non-progressive autosomal recessive ataxias characterized by early-onset cerebellar malformations resulting in motor incoordination and developmental delay, with additional CNS structural defects leading to hypotonia, intellectual disabilities and apraxia [168,169]. Among the inherited congenital cerebellar ataxias, the most common is Joubert syndrome (OMIM - #213300). It is a clinical and genetic heterogeneous disorder with onset at the neonatal stage and characterized by cerebellum and brainstem malformations (the so-called molar tooth sign), and non-neurological malformation (mainly kidney, retina and liver) has been reported. The major symptoms are ataxia, global development delay, breathing impairments, and ocular motor apraxia. The overall prevalence is 1:80.000-100.000 and mutations identified reside in genes encoding for primary cilium proteins, where INPP5E, TMEM216, AHI1, NPHP1, CEP290, TMEM67 and CC2D2A are the most frequent mutated genes [170]. Since primary cilium is an immotile organelle sensing extracellular signals that are transduced within the cell during crucial steps of development and for cell function, Joubert syndrome is part of the large family of ciliopathies [170].
7. Different Approaches to Ameliorate Disease Outcome
To date, no efficient treatment to halt disease progression has been identified for HA. However, various research fields are actively exploring diverse therapeutic approaches to address these profoundly disabling diseases. In this view, considerable steps forward in understanding pathological mechanisms underlying these diseases have been achieved using preclinical disease models, from rodent to human induced pluripotent stem cells (iPSCs).
One of the major challenges remains the low prevalence of these diseases [4,30], which makes it difficult to recruit patients for clinical trials. Clinical heterogeneity as well as age of onset represents another constraint for the recruitment of patients. Indeed, despite the same genetic mutations or expansions, such as CAG repeats in SCAs, substantial differences are present within the different disease groups [171]. This variability justifies the different approaches that are now under investigation to counteract specific disease mechanisms.
The principal therapeutical strategies developed in cerebellar ataxias pre-clinical and clinical studies (summary in Table 1) are:
- Genome editing strategies to correct the pathological mutation;
- Antisense oligonucleotides (ASO) or small RNA structures to interfere with repeat expansion translation or R-loop formation;
- Gene therapy approaches to rescue the levels of disease-mutated genes or key pathway regulators;
- Disease protein homeostasis to restore physiological protein levels;
- Pharmacological treatments, either to target specific pathophysiological mechanisms, to reduce toxic metabolites, or to supplement crucial compounds.
7.1. Genome Editing Strategies to Correct Pathological Mutation
The incredible improvements in the genome editing technology provide interesting perspectives to treat genetic disorders. Indeed, the recent advances in the CRISPR-Cas system offer the possibility to correct disease mutations by introducing a frameshift mutation that disables mutant gene function, by the excision of large expansion repeats or by base or prime editing [172]. Genome editing has been efficient in reducing the main disease phenotypes and rescuing the molecular defects in several animal models of neurodegenerative disorders (e.g., ALS, HD, AD, PD and Fragile-X syndrome) [173,174,175,176,177]. To date, in the context of cerebellar ataxia, the potentiality of CRISPR-mediated genome editing has been successfully reported mainly in cellular models. The feasibility in deleting the polyQ expansion of ATXN3 was demonstrated in induced pluripotent stem cells (iPSCs) [178]. The corrected iPSC-clones can maintain normal functionality, such as pluripotency properties and the neuronal cell differentiation capacity, and ATXN3-modified protein preserved the ubiquitin-binding ability without the formation of toxic aggregates [178]. Similarly, two different groups successfully proved the efficacy of the targeted excision of different fragments containing the GAA repeats to restore FXN mRNA and frataxin protein levels in different iPSC clones [179,180]. However, they showed deleting exclusively the expanded CAG tract is not always sufficient in reverting the pathological FA hallmarks (e.g. axonal spreading and synaptic machinery organization). In fact, it is known that the intronic regions flanking the GAA repeats also contribute to the abnormal DNA methylation on FXN promoter resulting in a transcriptionally non-permissive heterochromatin state [179]. More studies, in particular with in vivo evidence, are necessary to fully understand the feasibility of translating these approaches to the clinic. This is the goal of biotechnology companies that are currently developing and optimizing these strategies. Earlier this year, Prime Medicine announced to have gotten proof-of-concept that the genome-editing removal of expanded GAA repeats [181] corrects FXN hypermethylation restoring normal protein expression (https://investors.primemedicine.com/news-releases/news-release-details/prime-medicine-announces-recent-progress-and-highlights-2023). Similarly, CRISPR Therapeutics and Capsida Biotherapeutics are developing CRISPR/Cas-based genome editing technique for in vivo treatment of FA and ALS (further information at: https://crisprtx.gcs-web.com/news-releases/news-release-details/crispr-therapeutics-and-capsida-biotherapeutics-announce). Moreover, AAV-CRISPR editing, and target long-read sequencing has been used in transgenic SCA2 mouse models [182]. Other nucleases have also been used in cerebellar ataxia field for genome editing. Zinc-finger nucleases (ZFN), a versatile tool for precise genetic modifications, efficiently corrected FA cell phenotypes, such as decreasing aconitase activity and intracellular ATP levels in iPSC-differentiated neurons and cardiomyopathy features in cardiomyocytes [183]. Overall, the genome editing technologies for the correction of mutation are the ideal treatment in polynucleotide expansion ataxia diseases, but several challenges, including delivery, off-target effects, indel formation by nonhomologous end joining, toxicity and safety concerns need to be addressed and further investigate to efficiently transfer it to in vivo experimentation.
7.2. Antisense Oligonucleotides (ASO) or Small RNA Structures to Interfere with Repeat Expansion Translation or R-Loop Formation
A second level to tackle the root cause of the disease is the mutant mRNA transcript. This can be addressed taking advantage of (1) antisense oligonucleotides (ASOs), single stranded oligonucleotides analogues binding pre-mRNA or mRNA affecting the protein expression, or (2) RNA interference (RNAi) strategies using double stranded-RNA sequences to decrease the mRNA expression. This latter class accounts small interfering RNAs (siRNAs), short harpin RNAs (shRNAs) and artificial microRNAs (miRNAs). All these molecules bind the specific mRNA target sequence resulting in its degradation or stabilization or preventing the formation of detrimental structures such as R-loops. Experimental results involving RNAi methods showed promising preclinical efficacy in several cerebellar ataxias. The stereotaxic injection of AAV expressing shRNAs or miRNAs targeting ATXN1 resulted in significant downregulation of ataxin-1 preventing motor defects and improving molecular and cellular neuropathological phenotypes in different SCA1 mouse models [184,185,186]. Based on the successful outcomes from the rodent models, AAV-ATXN1 miRNAs have been tested in non-human primates. Deep cerebellar nuclei (DCN) injection allowed for efficiently transduction of the cells of interest with significant reduction of the endogenous ATXN1 mRNA amount [187].
To maximize the mutant gene silencing and most of all preserve wild-type expression and function, an allele-specific shRNA was tested on the SCA3 rat model. Tolerability and the beneficial effects on the neuropathological SCA3-associated symptoms were documented [188]. Moreover, SCA3 mice were also used in the assessment of siRNA and ASOs strategies. The siRNAs complementary to ATXN3 injected in the DCN of SCA3 transgenic mice reached the target and downregulated ATXN3 preventing its aggregation [189,190]. More recently, the intracerebroventricular (ICV) delivery of ASOs targeting the mutant ATXN3 successfully restored the potassium channel-mediated PN dysfunction [191] and improved the motor ability of SCA3 mice in addition to a direct correlation with the rescue of specific neurometabolites (choline, and partially taurine, glutamine and N-acetylaspartate) previously found dysregulated [192].
Furthermore, in SCA6, a miRNA targeting the CACNA1A IRES, a newly recognized internal ribosomal entry site within the CACNA1A C-terminal coding region, reduced ataxia and PN degeneration in a mouse model [193,194]. In SCA7 artificial miRNA complementary to ATXN7 were injected into the DCN and into the retina of transgenic SCA7 mice. The DCN miRNA-delivery was found well tolerated and efficient in improving the motor phenotype and PN survival, and similarly retinal administration did not show any adverse event, but achieved satisfactory ATXN7 downregulation [195,196]. To date, no gene therapy involving the siRNA or shRNA have been pursued in clinical trials for HA, however very encouraging results have been obtained in rodent and non-human primate models [184,187,197].
Another interesting category of gene therapy is represented by ASOs, which have shown remarkable progress in the last decade. These short, single-stranded strings of nucleic acids spamming between 8 to 50 nucleotides in length, are designed to interfere with RNA via different mechanisms [198]. The first preclinical data using ASOs were obtained in superoxide dismutase 1 (SOD1) ALS rodent model in 2006 [199]. Few years later, it was intrathecally delivered in a clinical trial to SOD1-ALS patients [200] where was well tolerated. The safety and efficacy of intrathecal administration of ASOs paved the approval from the FDA of Spinraza for the treatment of spinal muscular atrophy (SMA) after a successful phase 3 clinical trial [201]. Encouraging results also comes from PolyQ disease such as HD, where Roche in partnership with Ionis Pharmaceuticals reported that ASOs safety and tolerance are suitable for further studies [202,203], Nevertheless, those trials were halted prematurely because of failure to lower mutant huntingtin [203]. Despite the unfavorable outcome from some clinical trials, ASOs therapy remains an interesting approach suitable for the treatment of certain PolyQ SCAs.
In fact, this strategy was tested also in the ataxia field. Scoles and colleagues screened in vitro 152 ASOs targeting human ATXN2 and found ASO7 being a potent and well tolerated molecule resulting in reduced ATXN2 expression. They proved it in two SCA2 mouse models corroborating these results with significant improvement in motor performances, restoration of the most dysregulated genes and proteins, and rescue of the physiological properties (e.g. PC firing) [204]. Similarly, in FA, the efficacy of the ASOs (complementary to the expanded region) in activating the expression of FXN gene and restoring frataxin protein levels near to the physiological levels in different cell lines has been demonstrated. However, the low potency of these compounds could not encourage any pre-clinical study. For this reason, anti-AAG gapmer oligonucleotides, constructs consisting of a central DNA portion flanked by chemically modified RNA with increased binding affinity and efficacy, showed potent results in FXN expression activation in patient-cell lines driving its test in animal models [205,206].
The same genetic modulation can also be addressed to effectors influencing the levels of the disease downstream into the pathogenetic pathways. This is the case of the nuclear mitogen and stress-activated protein kinase 1 (MSK1) that is involved in the phosphorylation activity of a specific amino acid of the mutant ataxin 1 [207]. The downregulation of critical components of this pathway leads to reduced levels of mutant ataxin 1, attenuating the neurodegeneration and improving the disease phenotype of a SCA1 mouse model [207].
7.3. Gene Therapy Approaches to Rescue the Levels of Disease-Mutated Genes or Key Pathway Regulators
Gene therapy is a powerful strategy to replace a defective or missing gene often delivered via viral vector. The significant progresses in the design of new vectors able to cross the BBB, targeting more efficiently specific cell types and deliver larger genetic information, open new perspectives in loss-of-function diseases like ARCAs. To date, this approach has been applied only to FA. The first results were obtained injecting intravenously an AAVrh.10 vector expressing human frataxin (AAVrh.10-CAG-FXN) in mice lacking FXN in cardiac and skeletal muscles. The administration not only prevented, but also led to a complete and rapid recovery of cardiac functionality and related pathology [208,209,210]. More recently, the post-symptomatic dual intravenously delivery of AAV9-CAG-FXN simultaneously with intracerebral delivery of AAVrh.10-CAG-FXN resulted in a rapid and full rescue of FA sensory neuropathy and ganglionopathy [211]. Lexeo Therapeutics has completed in June 2023 the first dose cohort in a phase1/2 clinical trial for AAV-based gene therapy for FA cardiomyopathy (NCT05445323).
7.4. Disease Protein Homeostasis to Restore Physiological Protein Levels
In FA, where the protein expression is drastically reduced, a protein replacement strategy using the transactivator of transcription (TAT) peptides has been successfully proven in mice and is now in clinical trial. The frataxin fusion protein (TAT-FXN) was delivered to different tissues, including heart, DRG and cerebellum, and to the mitochondria, boosting the respiratory chain activity, and positively increasing the life span of the conditional heart model with partially recovery of the cardiomyopathy [212]. This strategy is now in a phase 2 clinical trial sponsored by Larimar Therapeutics (NCT05579691). Another approach is to activate the transcription of the FXN gene with synthetic transcription elongation factor 1 (Syn-TEF1). This was originally assessed in iPSC-derived cardiomyocytes and neurons, and mouse xenografts [213] and further taken into clinical trials sponsored by Design Therapeutics (NCT05285540).
Moreover, histone deacetylase (HDAC), which are involved in gene expression, are very promising candidates for the treatments of different diseases from neurodegeneration to cancer therapy [214], and it has been also used for increasing frataxin expression. Several HDAC inhibitors have been tested in preclinical studies, for instance, 2-aminobenzamide class I HDAC inhibitors was found efficient in promoting frataxin transcription in vitro as well as in FA mouse models [215]. However, reduced brain penetrability and gastrointestinal toxicity were observed, and therefore modified isoforms are currently being optimized for further use in clinical trial. Finally, a class III HDAC inhibitor, nicotinamide (vitamin B3), was demonstrated having satisfactory bioavailability and safety, and upregulating frataxin levels in preclinical models [216]. The encouraging results that have been collected in preclinical in vitro and in vivo model for HA paved the road for a randomized, double-blind, placebo-controlled clinical trial involving SCA3 patients, to test the efficacy of valproic acid (a well-known HDAC inhibitor). In this clinical trial, a significant improvement in the SARA score was reported in treated patients [217]. Another HDAC3 inhibitor, RG2833, has been tested in clinical trial phase 1 for FA [218], where frataxin deficiency is the main driver of disease pathology. RG2833 noteworthily increase FXN levels, however was not pursued as a metabolite was found to be toxic [218].
Another field of thought involves halting the formation of toxic protein aggregates such as toxic Poly. Despite encouraging results in preclinical ataxia models, to our knowledge, none has yet been considered for clinical trial testing. In the SCAs, the synthesis of abnormal polyQ-expanded proteins that often accumulate forming toxic aggregates requires an enhanced protein quality control system. To this aim, the expression of molecular chaperones or quality control ubiquitin ligase represent a valid disease-modifying therapy suppressing disease features of several animal models of SCA1, SCA3, SCA6 and SCA7 [219,220,221,222]. For instance, heat shock protein 90 inhibitors have been shown to be effective in reducing PolyQ accumulation in SCA1 [219]. Furthermore, it has been proven that the autophagic/lysosomal pathways is impaired in vulnerable neurons in SCA7 mouse model, but more interestingly ATG12 (a gene closely associated to the early phases of authopagy), correlates with disease severity in peripheral blood mononuclear cells (PBMCs) [222].
7.5. Pharmacological Treatments to Target Specific Pathomechanism, Reduce Toxic Metabolites and Supplement Crucial Compounds
Molecular studies aimed to disclose the mechanisms involved in cerebellar ataxias highlighted a converging mitochondrial dysfunction phenotype. Mitochondrial defects can be primarily caused by disease gene mutation or a downstream pathogenic effect. Frataxin deficiency causes decreased ATP production and impaired Fe-S cluster assembly resulting in ROS production and oxidative stress. To counteract oxidative stress, antioxidant molecules are now under investigation in FA pre-clinical and clinical trials, including Omaveloxolone (Omav) that has been recently approved by FDA as the first approved FA treatment. Other antioxidant targeting medications have also showed beneficial effects in FA and are currently being tested in clinical trials (i.e.Vatiquinone). Controversial results were collected by administrating coenzyme Q10 (CoQ10) and Idebenone, a CoQ10 analogue scavenging inner mitochondrial membrane ROS, already in clinical trial although with not fully established benefits in FA patients [223]. However, diet supplemented with coenzyme Q10 (CoQ10) significantly recovers motor coordination in SCA3 mice, reducing PN loss and skeletal muscle atrophy, where CoQ10 efficacy was explained by the improved autophagy-mediated clearance of mutant ataxin 3 [224]. Chronic CoQ10 oral supplementation induce a mild motor improvement in ARCA2 patients [225] and in vitro studies correlated CoQ10 administration with the restoration of mitochondrial dysfunction and calcium dysregulation [17]. It is important to note that in the context of ARCA2 the use of CoQ10 and its analogues for therapeutical purpose have different meaning. In fact, Coq8a depletion directly impacts on CoQ10 biosynthesis and so the exogenous supplementation can compensate the defective levels of this molecule crucial for several physiological processes (e.g., electron chain transport, antioxidant power, regulation of cytoplasm pH, membrane fluidity…).
MitoQ, an antioxidant ubiquinone which specifically accumulates within the mitochondria, has been proposed to be a promising therapeutic drug for several neurodegenerative diseases including ataxias. MitoQ was described to prevent cell death in FA fibroblasts [226]. Moreover, pre- and post-symptomatic in vivo MitoQ administration restores mitochondrial morphology and function, and positively improved the motor coordination and gait imbalance in SCA1 mouse model, and attenuating PN degeneration [19]. Recently, MitoQ chronic treatment in ARSACS mice displayed beneficial effects in PN survival and DCN innervation, preventing motor decline associated with sacsin depletion [227].
Nicotinamide riboside supplementation was found efficient in mitigating AT phenotype. This treatment boosts NAD+ levels and, promoting mitophagy, prevents senescence and neuroinflammation finally resulting in attenuated PN loss and other neuropathological AT features such as dsDNA damage and mitochondrial dysfunction, followed by improved motor ability of Atm-/- mice [228].
Several studies have reported changes in PN intrinsic excitability and impairment in synaptic signalling pathways, preceding neurodegeneration in numerous cerebellar ataxia mouse models, suggesting the restoration of electrophysiological properties, both via genetic and pharmacological approaches, as an attractive therapeutical strategy for cerebellar ataxias. Potassium channels, which are key regulators of PN excitability and dendritic plasticity, are attractive targets. The FDA-approved drug 4-aminopyridine (4-AP) ameliorates motor coordination and restore PN firing in SCA1 and SCA6 mice [229,230]. PN activity is also under the fine control of the small conductance calcium-activated potassium channel (SK), and oral administration of positive modulator of SK2/3 channels (NS13001 and chlorzoxazone) provided significant improvements of motor deficits and neuropathological manifestation in SCA2 mice [231,232].
Finally, compounds acting on ion channel and glutamate transporters effectively improved ataxic phenotypes. The Ceftriaxone administration in SCA28 and ARSACS murine models has shown significant improvements in motor defects, mitigating PN loss by restoring calcium homeostasis and reducing neuroinflammation [144,233].
Regardless of the intrinsic differences among HA, there are common pathological pathways that are shared not only between HA but with other neurodegenerative diseases, which makes it appealing to retest the same pharmacological approach among other diseases (drug repurposing). For instance, Riluzole, a glutamate release inhibitor and activator of calcium activated potassium channels, one of the few drugs approved by the FDA for ALS treatment, has been tested in clinical trial for SCA2. The ATRIL study was a randomized, double-blind, placebo controlled, multicenter clinical trial study that was conducted on 45 SCA2 patients [234], but unfortunately did not show any benefit in the improvement of motor deficits, probably because the recruited patients had marked PN atrophy, but also because these modulators have poor capacity for bypassing the BBB. However, previous phase 2 clinical trials in mixed cohorts of patients with different cerebellar ataxias (SCA1, 2, 17, 28 and FA) reported encouraging results after Riluzole administration [235,236], indicating that further investigation are needed to prove the efficacy of this compound for ataxia treatment. Currently, Biohaven Pharmaceutical, Inc. has a pro drug of Riluzole, named Troriluzole in phase 3 clinical trial (NCT03701399) for different spinocerebellar ataxia [237], acting on ameliorating PN functionality.
Another molecule that is commonly used to halt nicotine addiction is Varenicline, a partial agonist at α4β2 nicotinic acetylcholine receptors, that was tested in a double-blind, placebo control trial in SCA3 patients, showing marked improvement in Scale for the Assessment and Rating of Ataxia (SARA) score compared to placebo control group [238]. An additional molecule acting on receptors that control neuronal excitability that has been tested in randomized double-blind placebo-controlled trial of 57 patients with FA and multiple system atrophy - cerebellar subtype (MSA-C) is Amantadine, a noncompetitive N-methyl-D-aspartate (NMDA) agonist that has been shown to be effective for the treatment of Parkinson disease [239]. NMDA showed positive outcome for MSA-C patients but not for FA. Interestingly, positive results of Amantadine treatment also came from a non-randomized open-label study (NCT00950196) that involved ataxia telangiectasia [240].
Another interesting compound that has been tested in an open label trial in FA and that gave some promising results in preclinical models of SCA3 is resveratrol, acting on sirtuin 1 (SIRT1), despite the fact that the underlying mechanisms is not yet understood [241]. Regarding FA, the resveratrol open label trial did not show improvement in FXN level, nevertheless positive clinical effect might exist with high dosage. Further assessment in a randomized placebo-controlled trial is needed to assess the beneficial effect of this compound [242]. However, the results of the Phase 2 study presented at the International Congress for Ataxia Research in November 2022 were reported to be negative. Interesting technology has been developed by Erydel, which is able to deliver intra-erythrocyte dexamethasone for ataxia telangiectasia (NCT02770807). In fact, a short-term trial was first pioneer in this sense, testing the administration of oral betamethasone, which showed corticosteroid side effects, therefore, to overcome this issue autologous erythrocytes were loaded with dexamethasone phosphate (DPS) (Erydel). Likewise, betamethasone, dexamethasone possesses significant anti-inflammatory potency and does not exhibit mineralocorticoid activity, and erythrocyte phosphatases convert DSP to dexamethasone, which is then released into the bloodstream [243]. Moreover, a case report study reported the positive effect of oral administration of dexamethasone in a patient with ataxia-telangiectasia, were an improvement of 7 points in the SARA score was observed over 28 days period of treatment [244]. This represent a huge improvement which is northworth it further investigastions.
Autophagy impairments seem to underlie or at least contribute to disease pathogenesis of different NDD. In this context lithium, which is known to induce autophagy and help the clearance of mutant huntingtin as well as alpha synuclein [245], was used to treat SCA1 preclinical mouse model, where it improved motor coordination without an increase in lifespan [246]. However, the same effect was not seen in SCA3 mouse model nor in a double-blind, placebo-controlled phase 2 clinical trial for SCA3. The treatment involving 63 patients was tolerated, but no beneficial effect was observed in Neurological Examination Score for Spinocerebellar Ataxia (NESSCA) [247]. Similarly, when tested on a small cohort of SCA2 patients no significant improvement was reported following the SARA score [248]. Despite the unfavorable outcomes in clinical trials, it remains crucial to report these results, as they could provide valuable insights for future therapeutic approaches, and new clinical trials design.
8. Ataxia FDA Approved Drugs and Treatments
As of today, no resolutive treatment is available to treat HA. Most of the intervention rely on palliative care to treat symptoms. Multiple drugs have so far been inconclusive in the treatment of ataxia and further studies are needed to assess the safety and the efficacy of the previously described pharmacological treatments (riluzole, varenicline, lithium., etc…). Vitamins and anti-oxidant nutrient supplements, for instance Vitamin E, coenzyme Q, resveratrol are often recommended and have been shown to improve some aspect of the pathology, in particular in ataxias that involve main mitochondria dysfunctions like FA [249], although data collected are still insufficient to support the use of mitochondrial enhancement in patients with ataxia. Other approaches involve the application of small magnetic field to modify cerebellar activity using transcranial magnetic stimulation (TMS). Although major trials are needed to support the safety and outcomes of long-term treatment with these kind of stimulations [250]. These techniques have already been approved by FDA for the treatment of major depression and certain types of migraines [237]. Importantly, patients are encouraged to follow physiotherapy and physical exercising, that can improve quality of life and independence while often coping with a long-lasting disease [237,251].
Finally, in early 2023, FDA approved the first therapeutic agent for the treatment of FA patients older than 16 years, Omaveloxolone (SKYCLARYS™). Omaveloxolone is a nuclear factor erythroid 2-related factor 2 (Nrf2) activator, which helps in improving mitochondrial function, restoring redox balance, and reducing inflammation. FA patient that participated in the MOXIe (NCT02255435) double-blind, randomized, placebo-controlled, parallel-group, phase 2 trial that determined the safety and efficacy of Omaveloxolone over 48 weeks, reported significant improvement in the Modified Friedreich's Ataxia Rating Scale (mFARS) compared to the placebo-controlled group [252,253]. The approval of this drug after a certain number of failed clinical trials that did not reach their primary endpoints for FA [254], gives hope to many other forms of HA and represents a milestone in the treatment of ataxias.
9. Main Challenges and Limiting Factors
One of the main challenges in developing efficient clinical treatment is the prevalence of HA, as majority are in the range of 1-3/100,000 [4]. Another important constraint is the heterogeneity between the different forms of ataxia, even when considering same genetic conditions, such as CAG repeat in SCAs. The brain structure that are affected can be quite different which reflect the broad spectrum of symptoms that patients experience, for example SCA7 mainly involves retinal degeneration, SCA6 presents relatively pure cerebellar involvement, while other forms present cerebellar and brainstem dysfunctions [171,255].
Another important feature to take into account is the variability in genetic expansion that characterize some forms of HA, where different size in repeats expansion are often associated with different symptomatology severity and disease onset [25,256]. In addition, different forms of HA can vary among geographical and ethnical groups [30], information that needs to be considered for patient recruitments into clinical trials. Importantly, the diagnosis of patients might take some time if no clear genetic cause is known, and the lack of efficient pre-symptomatic biomarkers in familial forms such as fluidic biomarkers that can be measure in the cerebrospinal fluid (CSF) or in the blood of patients as indicator of disease progression, makes it difficult to intervene before disease onset or to monitor the efficacy of a treatment.
Challenges are also found in developing the appropriate pre-clinical models. Indeed, as discussed, many promising preclinical results have not translated into clinic. Being able to develop humanized rodent models, or human-based models, such as iPSC, is the current challenge. While generation of induced cortical neuron and organoids have been achieved without major challenges [11,257,258,259,260,261], the generation of PN and cerebellar organoids needs further optimization as protocols available present low purity and maturity, and survival of PN in culture is still challenging [262,263,264,265]. These represent powerful tools for drug testing and the study of pathophysiological mechanisms underlying HA, allowing to investigate pathogenic variants while maintaining the genetic background of the patients. Moreover, the wide access to CRISPR-Cas genome editing allows to generate isogenic control from the same patient cell line [266], pointing toward the concept of personalized medicine.
Despite all the progress that has been made in a preclinical set up where a variety of molecules and gene therapy approaches have showed promising results, the step forward clinical trial and approved drug is challenging. Finally, innovation in clinical trial design will likely play a role in the future, for instance comparing multiple treated groups to a single placebo arm or cross-over arms will reduce the number of patients recruitment, and likely be more appealing to patients [267].
Author Contributions
writing—original draft preparation, F.P. and A.D.B.; writing—review and editing, H.P. All authors have read and agreed to the published version of the manuscript.”.
Funding
This research was funded by the ANR, as part of the TREAT-ARCA consortium under the frame of the European Joint Programme on Rare Diseases (EJP RD) 2020 and by the FRM MND202003011460 AtaxiaXplorer (to H.P.), and by the Christina Fondation Grant IF GR-078158 (to F.P.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pandolfo, M.; Manto, M. Cerebellar and Afferent Ataxias. Contin. Minneap. Minn. 2013, 19, 1312–1343. [CrossRef]
- Pilotto, F.; Saxena, S. Epidemiology of Inherited Cerebellar Ataxias and Challenges in Clinical Research. Clin. Transl. Neurosci. 2018, 2, 2514183X1878525. [CrossRef]
- Zoghbi, H.Y.; Orr, H.T. Glutamine Repeats and Neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [CrossRef]
- Palau, F.; Espinós, C. Autosomal Recessive Cerebellar Ataxias. Orphanet J. Rare Dis. 2006, 1, 47. [CrossRef]
- Koeppen, A.H. Friedreich’s Ataxia: Pathology, Pathogenesis, and Molecular Genetics. J. Neurol. Sci. 2011, 303, 1–12. [CrossRef]
- Coarelli, G.; Wirth, T.; Tranchant, C.; Koenig, M.; Durr, A.; Anheim, M. The Inherited Cerebellar Ataxias: An Update. J. Neurol. 2023, 270, 208–222. [CrossRef]
- Leehey, M.A. Fragile X-Associated Tremor/Ataxia Syndrome: Clinical Phenotype, Diagnosis, and Treatment. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2009, 57, 830–836. [CrossRef]
- Jen, J.C.; Graves, T.D.; Hess, E.J.; Hanna, M.G.; Griggs, R.C.; Baloh, R.W.; CINCH investigators Primary Episodic Ataxias: Diagnosis, Pathogenesis and Treatment. Brain J. Neurol. 2007, 130, 2484–2493. [CrossRef]
- Choi, K.-D.; Choi, J.-H. Episodic Ataxias: Clinical and Genetic Features. J. Mov. Disord. 2016, 9, 129–135. [CrossRef]
- Bushart, D.D.; Shakkottai, V.G. Ion Channel Dysfunction in Cerebellar Ataxia. Neurosci. Lett. 2019, 688, 41–48. [CrossRef]
- Pilotto, F.; Douthwaite, C.; Diab, R.; Ye, X.; Al Qassab, Z.; Tietje, C.; Mounassir, M.; Odriozola, A.; Thapa, A.; Buijsen, R.A.M.; et al. Early Molecular Layer Interneuron Hyperactivity Triggers Purkinje Neuron Degeneration in SCA1. Neuron 2023. [CrossRef]
- Ruegsegger, C.; Stucki, D.M.; Steiner, S.; Angliker, N.; Radecke, J.; Keller, E.; Zuber, B.; Rüegg, M.A.; Saxena, S. Impaired MTORC1-Dependent Expression of Homer-3 Influences SCA1 Pathophysiology. Neuron 2016, 89, 129–146. [CrossRef]
- Ronnebaum, S.M.; Patterson, C.; Schisler, J.C. Emerging Evidence of Coding Mutations in the Ubiquitin–Proteasome System Associated with Cerebellar Ataxias. Hum. Genome Var. 2014, 1, 14018. [CrossRef]
- Luo, H.; Todi, S.V.; Paulson, H.L.; Costa, M. do C. Regional and Age-Dependent Changes in Ubiquitination in Cellular and Mouse Models of Spinocerebellar Ataxia Type 3. Front. Mol. Neurosci. 2023, 16, 1154203. [CrossRef]
- Girard, M.; Larivière, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.M.; Duchen, M.R.; et al. Mitochondrial Dysfunction and Purkinje Cell Loss in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [CrossRef]
- Lynch, D.R.; Farmer, G. Mitochondrial and Metabolic Dysfunction in Friedreich Ataxia: Update on Pathophysiological Relevance and Clinical Interventions. Neuronal Signal. 2021, 5, NS20200093. [CrossRef]
- Manolaras, I.; Del Bondio, A.; Griso, O.; Reutenauer, L.; Eisenmann, A.; Habermann, B.H.; Puccio, H. Mitochondrial Dysfunction and Calcium Dysregulation in COQ8A-Ataxia Purkinje Neurons Are Rescued by CoQ10 Treatment. Brain J. Neurol. 2023, awad099. [CrossRef]
- Harmuth, T.; Weber, J.J.; Zimmer, A.J.; Sowa, A.S.; Schmidt, J.; Fitzgerald, J.C.; Schöls, L.; Riess, O.; Hübener-Schmid, J. Mitochondrial Dysfunction in Spinocerebellar Ataxia Type 3 Is Linked to VDAC1 Deubiquitination. Int. J. Mol. Sci. 2022, 23, 5933. [CrossRef]
- Stucki, D.M.; Ruegsegger, C.; Steiner, S.; Radecke, J.; Murphy, M.P.; Zuber, B.; Saxena, S. Mitochondrial Impairments Contribute to Spinocerebellar Ataxia Type 1 Progression and Can Be Ameliorated by the Mitochondria-Targeted Antioxidant MitoQ. Free Radic. Biol. Med. 2016, 97, 427–440. [CrossRef]
- Ward, J.M.; Stoyas, C.A.; Switonski, P.M.; Ichou, F.; Fan, W.; Collins, B.; Wall, C.E.; Adanyeguh, I.; Niu, C.; Sopher, B.L.; et al. Metabolic and Organelle Morphology Defects in Mice and Human Patients Define Spinocerebellar Ataxia Type 7 as a Mitochondrial Disease. Cell Rep. 2019, 26, 1189–1202.e6. [CrossRef]
- Li, P.P.; Moulick, R.; Feng, H.; Sun, X.; Arbez, N.; Jin, J.; Marque, L.O.; Hedglen, E.; Chan, H.Y.E.; Ross, C.A.; et al. RNA Toxicity and Perturbation of RRNA Processing in Spinocerebellar Ataxia Type 2. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 2519–2529. [CrossRef]
- Zhang, N.; Ashizawa, T. RNA Toxicity and Foci Formation in Microsatellite Expansion Diseases. Curr. Opin. Genet. Dev. 2017, 44, 17–29. [CrossRef]
- Fiszer, A.; Krzyzosiak, W.J. RNA Toxicity in Polyglutamine Disorders: Concepts, Models, and Progress of Research. J. Mol. Med. 2013, 91, 683–691. [CrossRef]
- Matsuzono, K.; Imamura, K.; Murakami, N.; Tsukita, K.; Yamamoto, T.; Izumi, Y.; Kaji, R.; Ohta, Y.; Yamashita, T.; Abe, K.; et al. Antisense Oligonucleotides Reduce RNA Foci in Spinocerebellar Ataxia 36 Patient IPSCs. Mol. Ther. Nucleic Acids 2017, 8, 211–219. [CrossRef]
- Durr, A. Autosomal Dominant Cerebellar Ataxias: Polyglutamine Expansions and Beyond. Lancet Neurol. 2010, 9, 885–894. [CrossRef]
- Fujioka, S.; Sundal, C.; Wszolek, Z.K. Autosomal Dominant Cerebellar Ataxia Type III: A Review of the Phenotypic and Genotypic Characteristics. Orphanet J. Rare Dis. 2013, 8, 14. [CrossRef]
- Duenas, A.M. Molecular Pathogenesis of Spinocerebellar Ataxias. Brain 2006, 129, 1357–1370. [CrossRef]
- Schöls, L.; Bauer, P.; Schmidt, T.; Schulte, T.; Riess, O. Autosomal Dominant Cerebellar Ataxias: Clinical Features, Genetics, and Pathogenesis. Lancet Neurol. 2004, 3, 291–304. [CrossRef]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology 2014, 42, 174–183. [CrossRef]
- Buijsen, R.A.M.; Toonen, L.J.A.; Gardiner, S.L.; van Roon-Mom, W.M.C. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 2019, 16, 263–286. [CrossRef]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H.T. Polyglutamine Spinocerebellar Ataxias — from Genes to Potential Treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [CrossRef]
- Honti, V.; Vecsei, L. Genetic and Molecular Aspects of Spinocerebellar Ataxias. Neuropsychiatr. Dis. Treat. 2005, 1, 125–133. [CrossRef]
- Maltecca, F.; Filla, A.; Castaldo, I.; Coppola, G.; Fragassi, N.A.; Carella, M.; Bruni, A.; Cocozza, S.; Casari, G.; Servadio, A.; et al. Intergenerational Instability and Marked Anticipation in SCA-17. Neurology 2003, 61, 1441–1443. [CrossRef]
- Nethisinghe, S.; Pigazzini, M.L.; Pemble, S.; Sweeney, M.G.; Labrum, R.; Manso, K.; Moore, D.; Warner, J.; Davis, M.B.; Giunti, P. PolyQ Tract Toxicity in SCA1 Is Length Dependent in the Absence of CAG Repeat Interruption. Front. Cell. Neurosci. 2018, 12, 200. [CrossRef]
- Filla, A.; Mariotti, C.; Caruso, G.; Coppola, G.; Cocozza, S.; Castaldo, I.; Calabrese, O.; Salvatore, E.; De Michele, G.; Riggio, M.C.; et al. Relative Frequencies of CAG Expansions in Spinocerebellar Ataxia and Dentatorubropallidoluysian Atrophy in 116 Italian Families. Eur. Neurol. 2000, 44, 31–36. [CrossRef]
- Donato, S.D.; Mariotti, C.; Taroni, F. Spinocerebellar Ataxia Type 1. In Handbook of Clinical Neurology; Elsevier, 2012; Volume 103, pp. 399–421. ISBN 978-0-444-51892-7.
- Orengo, J.P.; van der Heijden, M.E.; Hao, S.; Tang, J.; Orr, H.T.; Zoghbi, H.Y. Motor Neuron Degeneration Correlates with Respiratory Dysfunction in SCA1. Dis. Model. Mech. 2018, dmm.032623. [CrossRef]
- Robitaille, Y.; Schut, L.; Kish, S.J. Structural and Immunocytochemical Features of Olivopontocerebellar Atrophy Caused by the Spinocerebellar Ataxia Type 1 (SCA-1) Mutation Define a Unique Phenotype. Acta Neuropathol. 1995, 90, 572–581. [CrossRef]
- Seidel, K.; Siswanto, S.; Brunt, E.R.P.; den Dunnen, W.; Korf, H.-W.; Rüb, U. Brain Pathology of Spinocerebellar Ataxias. Acta Neuropathol. 2012, 124, 1–21. [CrossRef]
- Orr, H.T.; Chung, M.; Banfi, S.; Kwiatkowski, T.J.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.W.; Zoghbi, H.Y. Expansion of an Unstable Trinucleotide CAG Repeat in Spinocerebellar Ataxia Type 1. Nat. Genet. 1993, 4, 221–226. [CrossRef]
- Irwin, S.; Vandelft, M.; Pinchev, D.; Howell, J.L.; Graczyk, J.; Orr, H.T.; Truant, R. RNA Association and Nucleocytoplasmic Shuttling by Ataxin-1. J. Cell Sci. 2005, 118, 233–242. [CrossRef]
- Klement, I.A.; Skinner, P.J.; Kaytor, M.D.; Yi, H.; Hersch, S.M.; Clark, H.B.; Zoghbi, H.Y.; Orr, H.T. Ataxin-1 Nuclear Localization and Aggregation. Cell 1998, 95, 41–53. [CrossRef]
- Lam, Y.C.; Bowman, A.B.; Jafar-Nejad, P.; Lim, J.; Richman, R.; Fryer, J.D.; Hyun, E.D.; Duvick, L.A.; Orr, H.T.; Botas, J.; et al. ATAXIN-1 Interacts with the Repressor Capicua in Its Native Complex to Cause SCA1 Neuropathology. Cell 2006, 127, 1335–1347. [CrossRef]
- Rousseaux, M.W.C.; Tschumperlin, T.; Lu, H.-C.; Lackey, E.P.; Bondar, V.V.; Wan, Y.-W.; Tan, Q.; Adamski, C.J.; Friedrich, J.; Twaroski, K.; et al. ATXN1-CIC Complex Is the Primary Driver of Cerebellar Pathology in Spinocerebellar Ataxia Type 1 through a Gain-of-Function Mechanism. Neuron 2018, 97, 1235–1243.e5. [CrossRef]
- Emamian, E.S.; Kaytor, M.D.; Duvick, L.A.; Zu, T.; Tousey, S.K.; Zoghbi, H.Y.; Clark, H.B.; Orr, H.T. Serine 776 of Ataxin-1 Is Critical for Polyglutamine-Induced Disease in SCA1 Transgenic Mice. Neuron 2003, 38, 375–387. [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell 2010, 143, 1174–1189. [CrossRef]
- Serra, H.G.; Duvick, L.; Zu, T.; Carlson, K.; Stevens, S.; Jorgensen, N.; Lysholm, A.; Burright, E.; Zoghbi, H.Y.; Clark, H.B.; et al. RORα-Mediated Purkinje Cell Development Determines Disease Severity in Adult SCA1 Mice. Cell 2006, 127, 697–708. [CrossRef]
- de Chiara, C.; Menon, R.P.; Strom, M.; Gibson, T.J.; Pastore, A. Phosphorylation of S776 and 14-3-3 Binding Modulate Ataxin-1 Interaction with Splicing Factors. PloS ONE 2009, 4, e8372. [CrossRef]
- Antenora, A.; Rinaldi, C.; Roca, A.; Pane, C.; Lieto, M.; Saccà, F.; Peluso, S.; De Michele, G.; Filla, A. The Multiple Faces of Spinocerebellar Ataxia Type 2. Ann. Clin. Transl. Neurol. 2017, 4, 687–695. [CrossRef]
- Rüb, U.; Schöls, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.-W.; Deller, T. Clinical Features, Neurogenetics and Neuropathology of the Polyglutamine Spinocerebellar Ataxias Type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [CrossRef]
- Bürk, K. Cognition in Hereditary Ataxia. Cerebellum Lond. Engl. 2007, 6, 280–286. [CrossRef]
- Mutesa, L.; Pierquin, G.; Segers, K.; Vanbellinghen, J.F.; Gahimbare, L.; Bours, V. Spinocerebellar Ataxia Type 2 (SCA2): Clinical Features and Genetic Analysis. J. Trop. Pediatr. 2008, 54, 350–352. [CrossRef]
- Estrada, R.; Galarraga, J.; Orozco, G.; Nodarse, A.; Auburger, G. Spinocerebellar Ataxia 2 (SCA2): Morphometric Analyses in 11 Autopsies. Acta Neuropathol. 1999, 97, 306–310. [CrossRef]
- Vishwakarma, P.; Muthuswamy, S.; Agarwal, S. Current Molecular Insight to Reveal the Dynamics of CAG Repeating Units in Spinocerebellar Ataxia. Intractable Rare Dis. Res. 2018, 7, 79–86. [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 Intermediate-Length Polyglutamine Expansions Are Associated with Increased Risk for ALS. Nature 2010, 466, 1069–1075. [CrossRef]
- Van Damme, P.; Veldink, J.H.; van Blitterswijk, M.; Corveleyn, A.; van Vught, P.W.J.; Thijs, V.; Dubois, B.; Matthijs, G.; van den Berg, L.H.; Robberecht, W. Expanded ATXN2 CAG Repeat Size in ALS Identifies Genetic Overlap between ALS and SCA2. Neurology 2011, 76, 2066–2072. [CrossRef]
- Glass, J.D.; Dewan, R.; Ding, J.; Gibbs, J.R.; Dalgard, C.; Keagle, P.J.; Shankaracharya; García-Redondo, A.; Traynor, B.J.; Chia, R.; et al. ATXN2 Intermediate Expansions in Amyotrophic Lateral Sclerosis. Brain J. Neurol. 2022, 145, 2671–2676. [CrossRef]
- Kozlov, G.; Trempe, J.F.; Khaleghpour, K.; Kahvejian, A.; Ekiel, I.; Gehring, K. Structure and Function of the C-Terminal PABC Domain of Human Poly(A)-Binding Protein. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 4409–4413. [CrossRef]
- Shibata, H.; Huynh, D.P.; Pulst, S.M. A Novel Protein with RNA-Binding Motifs Interacts with Ataxin-2. Hum. Mol. Genet. 2000, 9, 1303–1313. [CrossRef]
- Watanabe, R.; Higashi, S.; Nonaka, T.; Kawakami, I.; Oshima, K.; Niizato, K.; Akiyama, H.; Yoshida, M.; Hasegawa, M.; Arai, T. Intracellular Dynamics of Ataxin-2 in the Human Brains with Normal and Frontotemporal Lobar Degeneration with TDP-43 Inclusions. Acta Neuropathol. Commun. 2020, 8, 176. [CrossRef]
- Juvonen, V.; Hietala, M.; Kairisto, V.; Savontaus, M.-L. The Occurrence of Dominant Spinocerebellar Ataxias among 251 Finnish Ataxia Patients and the Role of Predisposing Large Normal Alleles in a Genetically Isolated Population. Acta Neurol. Scand. 2005, 111, 154–162. [CrossRef]
- Vale, J.; Bugalho, P.; Silveira, I.; Sequeiros, J.; Guimarães, J.; Coutinho, P. Autosomal Dominant Cerebellar Ataxia: Frequency Analysis and Clinical Characterization of 45 Families from Portugal. Eur. J. Neurol. 2010, 17, 124–128. [CrossRef]
- Rosenberg, R.N. Machado-Joseph Disease: An Autosomal Dominant Motor System Degeneration. Mov. Disord. Off. J. Mov. Disord. Soc. 1992, 7, 193–203. [CrossRef]
- Bettencourt, C.; Santos, C.; Coutinho, P.; Rizzu, P.; Vasconcelos, J.; Kay, T.; Cymbron, T.; Raposo, M.; Heutink, P.; Lima, M. Parkinsonian Phenotype in Machado-Joseph Disease (MJD/SCA3): A Two-Case Report. BMC Neurol. 2011, 11, 131. [CrossRef]
- Riess, O.; Rüb, U.; Pastore, A.; Bauer, P.; Schöls, L. SCA3: Neurological Features, Pathogenesis and Animal Models. Cerebellum Lond. Engl. 2008, 7, 125–137. [CrossRef]
- Rüb, U.; Brunt, E.R.; Deller, T. New Insights into the Pathoanatomy of Spinocerebellar Ataxia Type 3 (Machado-Joseph Disease). Curr. Opin. Neurol. 2008, 21, 111–116. [CrossRef]
- Stefanescu, M.R.; Dohnalek, M.; Maderwald, S.; Thürling, M.; Minnerop, M.; Beck, A.; Schlamann, M.; Diedrichsen, J.; Ladd, M.E.; Timmann, D. Structural and Functional MRI Abnormalities of Cerebellar Cortex and Nuclei in SCA3, SCA6 and Friedreich’s Ataxia. Brain J. Neurol. 2015, 138, 1182–1197. [CrossRef]
- Wan, N.; Chen, Z.; Wan, L.; Tang, B.; Jiang, H. MR Imaging of SCA3/MJD. Front. Neurosci. 2020, 14, 749. [CrossRef]
- Ma, J.; Wu, C.; Lei, J.; Zhang, X. Cognitive Impairments in Patients with Spinocerebellar Ataxia Types 1, 2 and 3 Are Positively Correlated to the Clinical Severity of Ataxia Symptoms. Int. J. Clin. Exp. Med. 2014, 7, 5765–5771. [CrossRef]
- Doss-Pepe, E.W.; Stenroos, E.S.; Johnson, W.G.; Madura, K. Ataxin-3 Interactions with Rad23 and Valosin-Containing Protein and Its Associations with Ubiquitin Chains and the Proteasome Are Consistent with a Role in Ubiquitin-Mediated Proteolysis. Mol. Cell. Biol. 2003, 23, 6469–6483. [CrossRef]
- Liu, H.; Li, X.; Ning, G.; Zhu, S.; Ma, X.; Liu, X.; Liu, C.; Huang, M.; Schmitt, I.; Wüllner, U.; et al. The Machado-Joseph Disease Deubiquitinase Ataxin-3 Regulates the Stability and Apoptotic Function of P53. PLoS Biol. 2016, 14, e2000733. [CrossRef]
- Zeng, C.; Zhao, C.; Ge, F.; Li, Y.; Cao, J.; Ying, M.; Lu, J.; He, Q.; Yang, B.; Dai, X.; et al. Machado-Joseph Deubiquitinases: From Cellular Functions to Potential Therapy Targets. Front. Pharmacol. 2020, 11, 1311. [CrossRef]
- Bichelmeier, U.; Schmidt, T.; Hübener, J.; Boy, J.; Rüttiger, L.; Häbig, K.; Poths, S.; Bonin, M.; Knipper, M.; Schmidt, W.J.; et al. Nuclear Localization of Ataxin-3 Is Required for the Manifestation of Symptoms in SCA3: In Vivo Evidence. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 7418–7428. [CrossRef]
- Sasaki, H.; Kojima, H.; Yabe, I.; Tashiro, K.; Hamada, T.; Sawa, H.; Hiraga, H.; Nagashima, K. Neuropathological and Molecular Studies of Spinocerebellar Ataxia Type 6 (SCA6). Acta Neuropathol. 1998, 95, 199–204. [CrossRef]
- Casey, H.L.; Gomez, C.M. Spinocerebellar Ataxia Type 6. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993.
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal Dominant Cerebellar Ataxia (SCA6) Associated with Small Polyglutamine Expansions in the Alpha 1A-Voltage-Dependent Calcium Channel. Nat. Genet. 1997, 15, 62–69. [CrossRef]
- Ikeuchi, T.; Takano, H.; Koide, R.; Horikawa, Y.; Honma, Y.; Onishi, Y.; Igarashi, S.; Tanaka, H.; Nakao, N.; Sahashi, K.; et al. Spinocerebellar Ataxia Type 6: CAG Repeat Expansion in Alpha1A Voltage-Dependent Calcium Channel Gene and Clinical Variations in Japanese Population. Ann. Neurol. 1997, 42, 879–884. [CrossRef]
- Riess, O.; Schöls, L.; Bottger, H.; Nolte, D.; Vieira-Saecker, A.M.; Schimming, C.; Kreuz, F.; Macek, M.; Krebsová, A.; Macek, M. Sen; et al. SCA6 Is Caused by Moderate CAG Expansion in the Alpha1A-Voltage-Dependent Calcium Channel Gene. Hum. Mol. Genet. 1997, 6, 1289–1293. [CrossRef]
- Shizuka, M.; Watanabe, M.; Ikeda, Y.; Mizushima, K.; Okamoto, K.; Shoji, M. Molecular Analysis of a de Novo Mutation for Spinocerebellar Ataxia Type 6 and (CAG)n Repeat Units in Normal Elder Controls. J. Neurol. Sci. 1998, 161, 85–87. [CrossRef]
- Ishikawa, K.; Tanaka, H.; Saito, M.; Ohkoshi, N.; Fujita, T.; Yoshizawa, K.; Ikeuchi, T.; Watanabe, M.; Hayashi, A.; Takiyama, Y.; et al. Japanese Families with Autosomal Dominant Pure Cerebellar Ataxia Map to Chromosome 19p13.1-P13.2 and Are Strongly Associated with Mild CAG Expansions in the Spinocerebellar Ataxia Type 6 Gene in Chromosome 19p13.1. Am. J. Hum. Genet. 1997, 61, 336–346. [CrossRef]
- Du, X.; Wang, J.; Zhu, H.; Rinaldo, L.; Lamar, K.-M.; Palmenberg, A.C.; Hansel, C.; Gomez, C.M. Second Cistron in CACNA1A Gene Encodes a Transcription Factor Mediating Cerebellar Development and SCA6. Cell 2013, 154, 118–133. [CrossRef]
- La Spada, A.R. Spinocerebellar Ataxia Type 7. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle (WA), 1993.
- Aleman, T.S.; Cideciyan, A.V.; Volpe, N.J.; Stevanin, G.; Brice, A.; Jacobson, S.G. Spinocerebellar Ataxia Type 7 (SCA7) Shows a Cone-Rod Dystrophy Phenotype. Exp. Eye Res. 2002, 74, 737–745. [CrossRef]
- Benton, C.S.; de Silva, R.; Rutledge, S.L.; Bohlega, S.; Ashizawa, T.; Zoghbi, H.Y. Molecular and Clinical Studies in SCA-7 Define a Broad Clinical Spectrum and the Infantile Phenotype. Neurology 1998, 51, 1081–1086. [CrossRef]
- Turk, K.W.; Flanagan, M.E.; Josephson, S.; Keene, C.D.; Jayadev, S.; Bird, T.D. Psychosis in Spinocerebellar Ataxias: A Case Series and Study of Tyrosine Hydroxylase in Substantia Nigra. Cerebellum Lond. Engl. 2018, 17, 143–151. [CrossRef]
- David, G.; Abbas, N.; Stevanin, G.; Dürr, A.; Yvert, G.; Cancel, G.; Weber, C.; Imbert, G.; Saudou, F.; Antoniou, E.; et al. Cloning of the SCA7 Gene Reveals a Highly Unstable CAG Repeat Expansion. Nat. Genet. 1997, 17, 65–70. [CrossRef]
- Ansorge, O.; Giunti, P.; Michalik, A.; Van Broeckhoven, C.; Harding, B.; Wood, N.; Scaravilli, F. Ataxin-7 Aggregation and Ubiquitination in Infantile SCA7 with 180 CAG Repeats. Ann. Neurol. 2004, 56, 448–452. [CrossRef]
- Cheon, Y.; Kim, H.; Park, K.; Kim, M.; Lee, D. Dynamic Modules of the Coactivator SAGA in Eukaryotic Transcription. Exp. Mol. Med. 2020, 52, 991–1003. [CrossRef]
- Palhan, V.B.; Chen, S.; Peng, G.-H.; Tjernberg, A.; Gamper, A.M.; Fan, Y.; Chait, B.T.; La Spada, A.R.; Roeder, R.G. Polyglutamine-Expanded Ataxin-7 Inhibits STAGA Histone Acetyltransferase Activity to Produce Retinal Degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 8472–8477. [CrossRef]
- Nakamura, Y.; Tagawa, K.; Oka, T.; Sasabe, T.; Ito, H.; Shiwaku, H.; La Spada, A.R.; Okazawa, H. Ataxin-7 Associates with Microtubules and Stabilizes the Cytoskeletal Network. Hum. Mol. Genet. 2012, 21, 1099–1110. [CrossRef]
- Monin, M.-L.; Tezenas du Montcel, S.; Marelli, C.; Cazeneuve, C.; Charles, P.; Tallaksen, C.; Forlani, S.; Stevanin, G.; Brice, A.; Durr, A. Survival and Severity in Dominant Cerebellar Ataxias. Ann. Clin. Transl. Neurol. 2015, 2, 202–207. [CrossRef]
- Toyoshima, Y.; Yamada, M.; Onodera, O.; Shimohata, M.; Inenaga, C.; Fujita, N.; Morita, M.; Tsuji, S.; Takahashi, H. SCA17 Homozygote Showing Huntington’s Disease-like Phenotype. Ann. Neurol. 2004, 55, 281–286. [CrossRef]
- Stevanin, G.; Brice, A. Spinocerebellar Ataxia 17 (SCA17) and Huntington’s Disease-like 4 (HDL4). Cerebellum Lond. Engl. 2008, 7, 170–178. [CrossRef]
- Nolte, D.; Sobanski, E.; Wissen, A.; Regula, J.U.; Lichy, C.; Müller, U. Spinocerebellar Ataxia Type 17 Associated with an Expansion of 42 Glutamine Residues in TATA-Box Binding Protein Gene. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1396–1399. [CrossRef]
- Yang, S.; Li, X.-J.; Li, S. Molecular Mechanisms Underlying Spinocerebellar Ataxia 17 (SCA17) Pathogenesis. Rare Dis. Austin Tex 2016, 4, e1223580. [CrossRef]
- Rafehi, H.; Read, J.; Szmulewicz, D.J.; Davies, K.C.; Snell, P.; Fearnley, L.G.; Scott, L.; Thomsen, M.; Gillies, G.; Pope, K.; et al. An Intronic GAA Repeat Expansion in FGF14 Causes the Autosomal-Dominant Adult-Onset Ataxia SCA27B/ATX-FGF14. Am. J. Hum. Genet. 2023, 110, 1018. [CrossRef]
- Bonnet, C.; Pellerin, D.; Roth, V.; Clément, G.; Wandzel, M.; Lambert, L.; Frismand, S.; Douarinou, M.; Grosset, A.; Bekkour, I.; et al. Optimized Testing Strategy for the Diagnosis of GAA-FGF14 Ataxia/Spinocerebellar Ataxia 27B. Sci. Rep. 2023, 13, 9737. [CrossRef]
- Wang, Q.; Bardgett, M.E.; Wong, M.; Wozniak, D.F.; Lou, J.; McNeil, B.D.; Chen, C.; Nardi, A.; Reid, D.C.; Yamada, K.; et al. Ataxia and Paroxysmal Dyskinesia in Mice Lacking Axonally Transported FGF14. Neuron 2002, 35, 25–38. [CrossRef]
- Bosch, M.K.; Carrasquillo, Y.; Ransdell, J.L.; Kanakamedala, A.; Ornitz, D.M.; Nerbonne, J.M. Intracellular FGF14 (IFGF14) Is Required for Spontaneous and Evoked Firing in Cerebellar Purkinje Neurons and for Motor Coordination and Balance. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 6752–6769. [CrossRef]
- Xiao, M.; Bosch, M.K.; Nerbonne, J.M.; Ornitz, D.M. FGF14 Localization and Organization of the Axon Initial Segment. Mol. Cell. Neurosci. 2013, 56, 393–403. [CrossRef]
- Traschütz, A.; Reich, S.; Adarmes, A.D.; Anheim, M.; Ashrafi, M.R.; Baets, J.; Basak, A.N.; Bertini, E.; Brais, B.; Gagnon, C.; et al. The ARCA Registry: A Collaborative Global Platform for Advancing Trial Readiness in Autosomal Recessive Cerebellar Ataxias. Front. Neurol. 2021, 12, 677551. [CrossRef]
- Anheim, M.; Tranchant, C.; Koenig, M. The Autosomal Recessive Cerebellar Ataxias. N. Engl. J. Med. 2012, 366, 636–646. [CrossRef]
- Synofzik, M.; Németh, A.H. Recessive Ataxias. Handb. Clin. Neurol. 2018, 155, 73–89. [CrossRef]
- Rossi, M.; Anheim, M.; Durr, A.; Klein, C.; Koenig, M.; Synofzik, M.; Marras, C.; van de Warrenburg, B.P.; International Parkinson and Movement Disorder Society Task Force on Classification and Nomenclature of Genetic Movement Disorders the Genetic Nomenclature of Recessive Cerebellar Ataxias. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 1056–1076. [CrossRef]
- Pierson, T.M.; Adams, D.; Bonn, F.; Martinelli, P.; Cherukuri, P.F.; Teer, J.K.; Hansen, N.F.; Cruz, P.; Mullikin for the Nisc Comparative Sequencing Program, J.C.; Blakesley, R.W.; et al. Whole-Exome Sequencing Identifies Homozygous AFG3L2 Mutations in a Spastic Ataxia-Neuropathy Syndrome Linked to Mitochondrial m-AAA Proteases. PLoS Genet. 2011, 7, e1002325. [CrossRef]
- Lise, S.; Clarkson, Y.; Perkins, E.; Kwasniewska, A.; Sadighi Akha, E.; Schnekenberg, R.P.; Suminaite, D.; Hope, J.; Baker, I.; Gregory, L.; et al. Recessive Mutations in SPTBN2 Implicate β-III Spectrin in Both Cognitive and Motor Development. PLoS Genet. 2012, 8, e1003074. [CrossRef]
- Gerber, S.; Alzayady, K.J.; Burglen, L.; Brémond-Gignac, D.; Marchesin, V.; Roche, O.; Rio, M.; Funalot, B.; Calmon, R.; Durr, A.; et al. Recessive and Dominant De Novo ITPR1 Mutations Cause Gillespie Syndrome. Am. J. Hum. Genet. 2016, 98, 971–980. [CrossRef]
- Nasca, A.; Rizza, T.; Doimo, M.; Legati, A.; Ciolfi, A.; Diodato, D.; Calderan, C.; Carrara, G.; Lamantea, E.; Aiello, C.; et al. Not Only Dominant, Not Only Optic Atrophy: Expanding the Clinical Spectrum Associated with OPA1 Mutations. Orphanet J. Rare Dis. 2017, 12, 89. [CrossRef]
- Othman, B.A.; Ong, J.E.; Dumitrescu, A.V. Biallelic Optic Atrophy 1 (OPA1) Related Disorder-Case Report and Literature Review. Genes 2022, 13, 1005. [CrossRef]
- Harding, A.E. Classification of the Hereditary Ataxias and Paraplegias. Lancet Lond. Engl. 1983, 1, 1151–1155. [CrossRef]
- Schulz, J.B.; Boesch, S.; Bürk, K.; Dürr, A.; Giunti, P.; Mariotti, C.; Pousset, F.; Schöls, L.; Vankan, P.; Pandolfo, M. Diagnosis and Treatment of Friedreich Ataxia: A European Perspective. Nat. Rev. Neurol. 2009, 5, 222–234. [CrossRef]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology 2014, 42, 174–183. [CrossRef]
- Vankan, P. Prevalence Gradients of Friedreich’s Ataxia and R1b Haplotype in Europe Co-Localize, Suggesting a Common Palaeolithic Origin in the Franco-Cantabrian Ice Age Refuge. J. Neurochem. 2013, 126 (Suppl 1), 11–20. [CrossRef]
- Fogel, B.L. Autosomal-Recessive Cerebellar Ataxias. Handb. Clin. Neurol. 2018, 147, 187–209. [CrossRef]
- Harding, A.E. Friedreich’s Ataxia: A Clinical and Genetic Study of 90 Families with an Analysis of Early Diagnostic Criteria and Intrafamilial Clustering of Clinical Features. Brain J. Neurol. 1981, 104, 589–620. [CrossRef]
- Pandolfo, M. Friedreich Ataxia: The Clinical Picture. J. Neurol. 2009, 256 (Suppl 1), 3–8. [CrossRef]
- Collins, A. Clinical Neurogenetics: Friedreich Ataxia. Neurol. Clin. 2013, 31, 1095–1120. [CrossRef]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Mazurkiewicz, J.E. Dorsal Root Ganglia in Friedreich Ataxia: Satellite Cell Proliferation and Inflammation. Acta Neuropathol. Commun. 2016, 4, 46. [CrossRef]
- Kemp, K.C.; Cook, A.J.; Redondo, J.; Kurian, K.M.; Scolding, N.J.; Wilkins, A. Purkinje Cell Injury, Structural Plasticity and Fusion in Patients with Friedreich’s Ataxia. Acta Neuropathol. Commun. 2016, 4, 53. [CrossRef]
- Selvadurai, L.P.; Harding, I.H.; Corben, L.A.; Georgiou-Karistianis, N. Cerebral Abnormalities in Friedreich Ataxia: A Review. Neurosci. Biobehav. Rev. 2018, 84, 394–406. [CrossRef]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 1996, 271, 1423–1427. [CrossRef]
- Groh, M.; Lufino, M.M.P.; Wade-Martins, R.; Gromak, N. R-Loops Associated with Triplet Repeat Expansions Promote Gene Silencing in Friedreich Ataxia and Fragile X Syndrome. PLoS Genet. 2014, 10, e1004318. [CrossRef]
- Colin, F.; Martelli, A.; Clémancey, M.; Latour, J.-M.; Gambarelli, S.; Zeppieri, L.; Birck, C.; Page, A.; Puccio, H.; Ollagnier de Choudens, S. Mammalian Frataxin Controls Sulfur Production and Iron Entry during de Novo Fe4S4 Cluster Assembly. J. Am. Chem. Soc. 2013, 135, 733–740. [CrossRef]
- Martelli, A.; Puccio, H. Dysregulation of Cellular Iron Metabolism in Friedreich Ataxia: From Primary Iron-Sulfur Cluster Deficit to Mitochondrial Iron Accumulation. Front. Pharmacol. 2014, 5, 130. [CrossRef]
- Synofzik, M.; Puccio, H.; Mochel, F.; Schöls, L. Autosomal Recessive Cerebellar Ataxias: Paving the Way toward Targeted Molecular Therapies. Neuron 2019, 101, 560–583. [CrossRef]
- Cortese, A.; Tozza, S.; Yau, W.Y.; Rossi, S.; Beecroft, S.J.; Jaunmuktane, Z.; Dyer, Z.; Ravenscroft, G.; Lamont, P.J.; Mossman, S.; et al. Cerebellar Ataxia, Neuropathy, Vestibular Areflexia Syndrome Due to RFC1 Repeat Expansion. Brain J. Neurol. 2020, 143, 480–490. [CrossRef]
- Szmulewicz, D.J.; Waterston, J.A.; MacDougall, H.G.; Mossman, S.; Chancellor, A.M.; McLean, C.A.; Merchant, S.; Patrikios, P.; Halmagyi, G.M.; Storey, E. Cerebellar Ataxia, Neuropathy, Vestibular Areflexia Syndrome (CANVAS): A Review of the Clinical Features and Video-Oculographic Diagnosis. Ann. N. Y. Acad. Sci. 2011, 1233, 139–147. [CrossRef]
- Cortese, A.; Simone, R.; Sullivan, R.; Vandrovcova, J.; Tariq, H.; Yau, W.Y.; Humphrey, J.; Jaunmuktane, Z.; Sivakumar, P.; Polke, J.; et al. Biallelic Expansion of an Intronic Repeat in RFC1 Is a Common Cause of Late-Onset Ataxia. Nat. Genet. 2019, 51, 649–658. [CrossRef]
- Dominik, N.; Magri, S.; Currò, R.; Abati, E.; Facchini, S.; Corbetta, M.; MacPherson, H.; Di Bella, D.; Sarto, E.; Stevanovski, I.; et al. Normal and Pathogenic Variation of RFC1 Repeat Expansions: Implications for Clinical Diagnosis. Brain J. Neurol. 2023, awad240. [CrossRef]
- Ronco, R.; Perini, C.; Currò, R.; Dominik, N.; Facchini, S.; Gennari, A.; Simone, R.; Stuart, S.; Nagy, S.; Vegezzi, E.; et al. Truncating Variants in RFC1 in Cerebellar Ataxia, Neuropathy, and Vestibular Areflexia Syndrome. Neurology 2023, 100, e543–e554. [CrossRef]
- Abdi, M.H.; Zamiri, B.; Pazuki, G.; Sardari, S.; Pearson, C.E. Pathogenic CANVAS-Causing but Not Nonpathogenic RFC1 DNA/RNA Repeat Motifs Form Quadruplex or Triplex Structures. J. Biol. Chem. 2023, 299, 105202. [CrossRef]
- Bouchard, J.P.; Barbeau, A.; Bouchard, R.; Bouchard, R.W. Electromyography and Nerve Conduction Studies in Friedreich’s Ataxia and Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS). Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1979, 6, 185–189. [CrossRef]
- Bouhlal, Y.; Amouri, R.; El Euch-Fayeche, G.; Hentati, F. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay: An Overview. Parkinsonism Relat. Disord. 2011, 17, 418–422. [CrossRef]
- Engert, J.C.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a Spastic Ataxia Common in Northeastern Québec, Is Caused by Mutations in a New Gene Encoding an 11.5-Kb ORF. Nat. Genet. 2000, 24, 120–125. [CrossRef]
- Synofzik, M.; Soehn, A.S.; Gburek-Augustat, J.; Schicks, J.; Karle, K.N.; Schüle, R.; Haack, T.B.; Schöning, M.; Biskup, S.; Rudnik-Schöneborn, S.; et al. Autosomal Recessive Spastic Ataxia of Charlevoix Saguenay (ARSACS): Expanding the Genetic, Clinical and Imaging Spectrum. Orphanet J. Rare Dis. 2013, 8, 41. [CrossRef]
- Longo, F.; De Ritis, D.; Miluzio, A.; Fraticelli, D.; Baets, J.; Scarlato, M.; Santorelli, F.M.; Biffo, S.; Maltecca, F. Assessment of Sacsin Turnover in Patients With ARSACS: Implications for Molecular Diagnosis and Pathogenesis. Neurology 2021, 97, e2315–e2327. [CrossRef]
- Xiromerisiou, G.; Dadouli, K.; Marogianni, C.; Provatas, A.; Ntellas, P.; Rikos, D.; Stathis, P.; Georgouli, D.; Loules, G.; Zamanakou, M.; et al. A Novel Homozygous SACS Mutation Identified by Whole Exome Sequencing-Genotype Phenotype Correlations of All Published Cases. J. Mol. Neurosci. MN 2020, 70, 131–141. [CrossRef]
- Parkinson, M.H.; Bartmann, A.P.; Clayton, L.M.S.; Nethisinghe, S.; Pfundt, R.; Chapple, J.P.; Reilly, M.M.; Manji, H.; Wood, N.J.; Bremner, F.; et al. Optical Coherence Tomography in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Brain J. Neurol. 2018, 141, 989–999. [CrossRef]
- Ricca, I.; Morani, F.; Bacci, G.M.; Nesti, C.; Caputo, R.; Tessa, A.; Santorelli, F.M. Clinical and Molecular Studies in Two New Cases of ARSACS. Neurogenetics 2019, 20, 45–49. [CrossRef]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.M.; Prodromou, N.V.; Webb, T.R.; Gallo, J.-M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The Ataxia Protein Sacsin Is a Functional Co-Chaperone That Protects against Polyglutamine-Expanded Ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [CrossRef]
- Anderson, J.F.; Siller, E.; Barral, J.M. The Sacsin Repeating Region (SRR): A Novel Hsp90-Related Supra-Domain Associated with Neurodegeneration. J. Mol. Biol. 2010, 400, 665–674. [CrossRef]
- Duncan, E.J.; Larivière, R.; Bradshaw, T.Y.; Longo, F.; Sgarioto, N.; Hayes, M.J.; Romano, L.E.L.; Nethisinghe, S.; Giunti, P.; Bruntraeger, M.B.; et al. Altered Organization of the Intermediate Filament Cytoskeleton and Relocalization of Proteostasis Modulators in Cells Lacking the Ataxia Protein Sacsin. Hum. Mol. Genet. 2017, 26, 3130–3143. [CrossRef]
- Gentil, B.J.; Lai, G.-T.; Menade, M.; Larivière, R.; Minotti, S.; Gehring, K.; Chapple, J.-P.; Brais, B.; Durham, H.D. Sacsin, Mutated in the Ataxia ARSACS, Regulates Intermediate Filament Assembly and Dynamics. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 2982–2994. [CrossRef]
- Del Bondio, A.; Longo, F.; De Ritis, D.; Spirito, E.; Podini, P.; Brais, B.; Bachi, A.; Quattrini, A.; Maltecca, F. Restoring Calcium Homeostasis in Purkinje Cells Arrests Neurodegeneration and Neuroinflammation in the ARSACS Mouse Model. JCI Insight 2023, 8, e163576. [CrossRef]
- Bradshaw, T.Y.; Romano, L.E.L.; Duncan, E.J.; Nethisinghe, S.; Abeti, R.; Michael, G.J.; Giunti, P.; Vermeer, S.; Chapple, J.P. A Reduction in Drp1-Mediated Fission Compromises Mitochondrial Health in Autosomal Recessive Spastic Ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 2016, 25, 3232–3244. [CrossRef]
- Chun, H.H.; Gatti, R.A. Ataxia-Telangiectasia, an Evolving Phenotype. DNA Repair 2004, 3, 1187–1196. [CrossRef]
- Perlman, S.L.; Boder Deceased, E.; Sedgewick, R.P.; Gatti, R.A. Ataxia-Telangiectasia. Handb. Clin. Neurol. 2012, 103, 307–332. [CrossRef]
- Shiloh, Y. ATM and Related Protein Kinases: Safeguarding Genome Integrity. Nat. Rev. Cancer 2003, 3, 155–168. [CrossRef]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The Histone Guardian of the Genome. DNA Repair 2004, 3, 959–967. [CrossRef]
- Chang, J.R.; Ghafouri, M.; Mukerjee, R.; Bagashev, A.; Chabrashvili, T.; Sawaya, B.E. Role of P53 in Neurodegenerative Diseases. Neurodegener. Dis. 2012, 9, 68–80. [CrossRef]
- Lagier-Tourenne, C.; Tazir, M.; López, L.C.; Quinzii, C.M.; Assoum, M.; Drouot, N.; Busso, C.; Makri, S.; Ali-Pacha, L.; Benhassine, T.; et al. ADCK3, an Ancestral Kinase, Is Mutated in a Form of Recessive Ataxia Associated with Coenzyme Q10 Deficiency. Am. J. Hum. Genet. 2008, 82, 661–672. [CrossRef]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Boddaert, N.; Desguerre, I.; de Lonlay, P.; de Baulny, H.O.; et al. CABC1 Gene Mutations Cause Ubiquinone Deficiency with Cerebellar Ataxia and Seizures. Am. J. Hum. Genet. 2008, 82, 623–630. [CrossRef]
- Eto, M.; Watanabe, K.; Ishii, K. Apolipoprotein E Alleles and Hyperlipoproteinemia in Japan. Clin. Genet. 1988, 34, 246–251. [CrossRef]
- Traschütz, A.; Schirinzi, T.; Laugwitz, L.; Murray, N.H.; Bingman, C.A.; Reich, S.; Kern, J.; Heinzmann, A.; Vasco, G.; Bertini, E.; et al. Clinico-Genetic, Imaging and Molecular Delineation of COQ8A-Ataxia: A Multicenter Study of 59 Patients. Ann. Neurol. 2020, 88, 251–263. [CrossRef]
- Floyd, B.J.; Wilkerson, E.M.; Veling, M.T.; Minogue, C.E.; Xia, C.; Beebe, E.T.; Wrobel, R.L.; Cho, H.; Kremer, L.S.; Alston, C.L.; et al. Mitochondrial Protein Interaction Mapping Identifies Regulators of Respiratory Chain Function. Mol. Cell 2016, 63, 621–632. [CrossRef]
- Stefely, J.A.; Reidenbach, A.G.; Ulbrich, A.; Oruganty, K.; Floyd, B.J.; Jochem, A.; Saunders, J.M.; Johnson, I.E.; Minogue, C.E.; Wrobel, R.L.; et al. Mitochondrial ADCK3 Employs an Atypical Protein Kinase-like Fold to Enable Coenzyme Q Biosynthesis. Mol. Cell 2015, 57, 83–94. [CrossRef]
- Stefely, J.A.; Licitra, F.; Laredj, L.; Reidenbach, A.G.; Kemmerer, Z.A.; Grangeray, A.; Jaeg-Ehret, T.; Minogue, C.E.; Ulbrich, A.; Hutchins, P.D.; et al. Cerebellar Ataxia and Coenzyme Q Deficiency through Loss of Unorthodox Kinase Activity. Mol. Cell 2016, 63, 608–620. [CrossRef]
- Cullen, J.K.; Abdul Murad, N.; Yeo, A.; McKenzie, M.; Ward, M.; Chong, K.L.; Schieber, N.L.; Parton, R.G.; Lim, Y.C.; Wolvetang, E.; et al. AarF Domain Containing Kinase 3 (ADCK3) Mutant Cells Display Signs of Oxidative Stress, Defects in Mitochondrial Homeostasis and Lysosomal Accumulation. PloS ONE 2016, 11, e0148213. [CrossRef]
- Hassan, A. Episodic Ataxias: Primary and Secondary Etiologies, Treatment, and Classification Approaches. Tremor Hyperkinetic Mov. N. Y. N 2023, 13, 9. [CrossRef]
- Jen, J.C.; Wan, J. Episodic Ataxias. Handb. Clin. Neurol. 2018, 148, 521–529. [CrossRef]
- Martínez-Monseny, A.F.; Edo, A.; Casas-Alba, D.; Izquierdo-Serra, M.; Bolasell, M.; Conejo, D.; Martorell, L.; Muchart, J.; Carrera, L.; Ortez, C.I.; et al. CACNA1A Mutations Causing Early Onset Ataxia: Profiling Clinical, Dysmorphic and Structural-Functional Findings. Int. J. Mol. Sci. 2021, 22, 5180. [CrossRef]
- Brussino, A.; Gellera, C.; Saluto, A.; Mariotti, C.; Arduino, C.; Castellotti, B.; Camerlingo, M.; de Angelis, V.; Orsi, L.; Tosca, P.; et al. FMR1 Gene Premutation Is a Frequent Genetic Cause of Late-Onset Sporadic Cerebellar Ataxia. Neurology 2005, 64, 145–147. [CrossRef]
- Cabal-Herrera, A.M.; Tassanakijpanich, N.; Salcedo-Arellano, M.J.; Hagerman, R.J. Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS): Pathophysiology and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 4391. [CrossRef]
- Tassone, F.; Adams, J.; Berry-Kravis, E.M.; Cohen, S.S.; Brusco, A.; Leehey, M.A.; Li, L.; Hagerman, R.J.; Hagerman, P.J. CGG Repeat Length Correlates with Age of Onset of Motor Signs of the Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS). Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2007, 144B, 566–569. [CrossRef]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.; Grigsby, J.; Zhang, L.; Brunberg, J.A.; Greco, C.; Des Portes, V.; Jardini, T.; Levine, R.; et al. Fragile X Premutation Tremor/Ataxia Syndrome: Molecular, Clinical, and Neuroimaging Correlates. Am. J. Hum. Genet. 2003, 72, 869–878. [CrossRef]
- Cronister, A.; Schreiner, R.; Wittenberger, M.; Amiri, K.; Harris, K.; Hagerman, R.J. Heterozygous Fragile X Female: Historical, Physical, Cognitive, and Cytogenetic Features. Am. J. Med. Genet. 1991, 38, 269–274. [CrossRef]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated Levels of FMR1 MRNA in Carrier Males: A New Mechanism of Involvement in the Fragile-X Syndrome. Am. J. Hum. Genet. 2000, 66, 6–15. [CrossRef]
- Bertini, E.; Zanni, G.; Boltshauser, E. Nonprogressive Congenital Ataxias. Handb. Clin. Neurol. 2018, 155, 91–103. [CrossRef]
- Steinlin, M. Non-Progressive Congenital Ataxias. Brain Dev. 1998, 20, 199–208. [CrossRef]
- Romani, M.; Micalizzi, A.; Valente, E.M. Joubert Syndrome: Congenital Cerebellar Ataxia with the Molar Tooth. Lancet Neurol. 2013, 12, 894–905. [CrossRef]
- Brooker, S.M.; Edamakanti, C.R.; Akasha, S.M.; Kuo, S.; Opal, P. Spinocerebellar Ataxia Clinical Trials: Opportunities and Challenges. Ann. Clin. Transl. Neurol. 2021, 8, 1543–1556. [CrossRef]
- Duan, W.; Urani, E.; Mattson, M.P. The Potential of Gene Editing for Huntington’s Disease. Trends Neurosci. 2023, 46, 365–376. [CrossRef]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In Vivo Genome Editing Improves Motor Function and Extends Survival in a Mouse Model of ALS. Sci. Adv. 2017, 3, eaar3952. [CrossRef]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-Mediated Gene Editing Ameliorates Neurotoxicity in Mouse Model of Huntington’s Disease. J. Clin. Invest. 2017, 127, 2719–2724. [CrossRef]
- Duan, Y.; Ye, T.; Qu, Z.; Chen, Y.; Miranda, A.; Zhou, X.; Lok, K.-C.; Chen, Y.; Fu, A.K.Y.; Gradinaru, V.; et al. Brain-Wide Cas9-Mediated Cleavage of a Gene Causing Familial Alzheimer’s Disease Alleviates Amyloid-Related Pathologies in Mice. Nat. Biomed. Eng. 2022, 6, 168–180. [CrossRef]
- Yoon, H.H.; Ye, S.; Lim, S.; Jo, A.; Lee, H.; Hong, F.; Lee, S.E.; Oh, S.-J.; Kim, N.-R.; Kim, K.; et al. CRISPR-Cas9 Gene Editing Protects from the A53T-SNCA Overexpression-Induced Pathology of Parkinson’s Disease In Vivo. CRISPR J. 2022, 5, 95–108. [CrossRef]
- Lee, B.; Lee, K.; Panda, S.; Gonzales-Rojas, R.; Chong, A.; Bugay, V.; Park, H.M.; Brenner, R.; Murthy, N.; Lee, H.Y. Nanoparticle Delivery of CRISPR into the Brain Rescues a Mouse Model of Fragile X Syndrome from Exaggerated Repetitive Behaviours. Nat. Biomed. Eng. 2018, 2, 497–507. [CrossRef]
- Ouyang, S.; Xie, Y.; Xiong, Z.; Yang, Y.; Xian, Y.; Ou, Z.; Song, B.; Chen, Y.; Xie, Y.; Li, H.; et al. CRISPR/Cas9-Targeted Deletion of Polyglutamine in Spinocerebellar Ataxia Type 3-Derived Induced Pluripotent Stem Cells. Stem Cells Dev. 2018, 27, 756–770. [CrossRef]
- Mazzara, P.G.; Muggeo, S.; Luoni, M.; Massimino, L.; Zaghi, M.; Valverde, P.T.-T.; Brusco, S.; Marzi, M.J.; Palma, C.; Colasante, G.; et al. Frataxin Gene Editing Rescues Friedreich’s Ataxia Pathology in Dorsal Root Ganglia Organoid-Derived Sensory Neurons. Nat. Commun. 2020, 11, 4178. [CrossRef]
- Li, Y.; Li, J.; Wang, J.; Zhang, S.; Giles, K.; Prakash, T.P.; Rigo, F.; Napierala, J.S.; Napierala, M. Premature Transcription Termination at the Expanded GAA Repeats and Aberrant Alternative Polyadenylation Contributes to the Frataxin Transcriptional Deficit in Friedreich’s Ataxia. Hum. Mol. Genet. 2022, 31, 3539–3557. [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [CrossRef]
- Simpson, B.P.; Yrigollen, C.M.; Izda, A.; Davidson, B.L. Targeted Long-Read Sequencing Captures CRISPR Editing and AAV Integration Outcomes in Brain. Mol. Ther. J. Am. Soc. Gene Ther. 2023, 31, 760–773. [CrossRef]
- Li, Y.; Polak, U.; Bhalla, A.D.; Rozwadowska, N.; Butler, J.S.; Lynch, D.R.; Dent, S.Y.R.; Napierala, M. Excision of Expanded GAA Repeats Alleviates the Molecular Phenotype of Friedreich’s Ataxia. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1055–1065. [CrossRef]
- Xia, H.; Mao, Q.; Eliason, S.L.; Harper, S.Q.; Martins, I.H.; Orr, H.T.; Paulson, H.L.; Yang, L.; Kotin, R.M.; Davidson, B.L. RNAi Suppresses Polyglutamine-Induced Neurodegeneration in a Model of Spinocerebellar Ataxia. Nat. Med. 2004, 10, 816–820. [CrossRef]
- Keiser, M.S.; Geoghegan, J.C.; Boudreau, R.L.; Lennox, K.A.; Davidson, B.L. RNAi or Overexpression: Alternative Therapies for Spinocerebellar Ataxia Type 1. Neurobiol. Dis. 2013, 56, 6–13. [CrossRef]
- Keiser, M.S.; Boudreau, R.L.; Davidson, B.L. Broad Therapeutic Benefit after RNAi Expression Vector Delivery to Deep Cerebellar Nuclei: Implications for Spinocerebellar Ataxia Type 1 Therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 588–595. [CrossRef]
- Keiser, M.S.; Kordower, J.H.; Gonzalez-Alegre, P.; Davidson, B.L. Broad Distribution of Ataxin 1 Silencing in Rhesus Cerebella for Spinocerebellar Ataxia Type 1 Therapy. Brain J. Neurol. 2015, 138, 3555–3566. [CrossRef]
- Alves, S.; Nascimento-Ferreira, I.; Auregan, G.; Hassig, R.; Dufour, N.; Brouillet, E.; Pedroso de Lima, M.C.; Hantraye, P.; Pereira de Almeida, L.; Déglon, N. Allele-Specific RNA Silencing of Mutant Ataxin-3 Mediates Neuroprotection in a Rat Model of Machado-Joseph Disease. PloS ONE 2008, 3, e3341. [CrossRef]
- Rodríguez-Lebrón, E.; Costa, M. do C.; Luna-Cancalon, K.; Peron, T.M.; Fischer, S.; Boudreau, R.L.; Davidson, B.L.; Paulson, H.L. Silencing Mutant ATXN3 Expression Resolves Molecular Phenotypes in SCA3 Transgenic Mice. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1909–1918. [CrossRef]
- Costa, M. do C.; Luna-Cancalon, K.; Fischer, S.; Ashraf, N.S.; Ouyang, M.; Dharia, R.M.; Martin-Fishman, L.; Yang, Y.; Shakkottai, V.G.; Davidson, B.L.; et al. Toward RNAi Therapy for the Polyglutamine Disease Machado-Joseph Disease. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1898–1908. [CrossRef]
- Bushart, D.D.; Zalon, A.J.; Zhang, H.; Morrison, L.M.; Guan, Y.; Paulson, H.L.; Shakkottai, V.G.; McLoughlin, H.S. Antisense Oligonucleotide Therapy Targeted Against ATXN3 Improves Potassium Channel-Mediated Purkinje Neuron Dysfunction in Spinocerebellar Ataxia Type 3. Cerebellum Lond. Engl. 2021, 20, 41–53. [CrossRef]
- McLoughlin, H.S.; Gundry, K.; Rainwater, O.; Schuster, K.H.; Wellik, I.G.; Zalon, A.J.; Benneyworth, M.A.; Eberly, L.E.; Öz, G. Antisense Oligonucleotide Silencing Reverses Abnormal Neurochemistry in Spinocerebellar Ataxia 3 Mice. Ann. Neurol. 2023. [CrossRef]
- Miyazaki, Y.; Du, X.; Muramatsu, S.-I.; Gomez, C.M. An MiRNA-Mediated Therapy for SCA6 Blocks IRES-Driven Translation of the CACNA1A Second Cistron. Sci. Transl. Med. 2016, 8, 347ra94. [CrossRef]
- Pastor, P.D.H.; Du, X.; Fazal, S.; Davies, A.N.; Gomez, C.M. Targeting the CACNA1A IRES as a Treatment for Spinocerebellar Ataxia Type 6. Cerebellum Lond. Engl. 2018, 17, 72–77. [CrossRef]
- Ramachandran, P.S.; Bhattarai, S.; Singh, P.; Boudreau, R.L.; Thompson, S.; Laspada, A.R.; Drack, A.V.; Davidson, B.L. RNA Interference-Based Therapy for Spinocerebellar Ataxia Type 7 Retinal Degeneration. PloS One 2014, 9, e95362. [CrossRef]
- Ramachandran, P.S.; Boudreau, R.L.; Schaefer, K.A.; La Spada, A.R.; Davidson, B.L. Nonallele Specific Silencing of Ataxin-7 Improves Disease Phenotypes in a Mouse Model of SCA7. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1635–1642. [CrossRef]
- Keiser, M.S.; Monteys, A.M.; Corbau, R.; Gonzalez-Alegre, P.; Davidson, B.L. RNAi Prevents and Reverses Phenotypes Induced by Mutant Human Ataxin-1. Ann. Neurol. 2016, 80, 754–765. [CrossRef]
- Rinaldi, C.; Wood, M.J.A. Antisense Oligonucleotides: The next Frontier for Treatment of Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [CrossRef]
- Smith, R.A.; Miller, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense Oligonucleotide Therapy for Neurodegenerative Disease. J. Clin. Invest. 2006, 116, 2290–2296. [CrossRef]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An Antisense Oligonucleotide against SOD1 Delivered Intrathecally for Patients with SOD1 Familial Amyotrophic Lateral Sclerosis: A Phase 1, Randomised, First-in-Man Study. Lancet Neurol. 2013, 12, 435–442. [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [CrossRef]
- van Roon-Mom, W.M.C.; Roos, R.A.C.; de Bot, S.T. Dose-Dependent Lowering of Mutant Huntingtin Using Antisense Oligonucleotides in Huntington Disease Patients. Nucleic Acid Ther. 2018, 28, 59–62. [CrossRef]
- Rook, M.E.; Southwell, A.L. Antisense Oligonucleotide Therapy: From Design to the Huntington Disease Clinic. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2022, 36, 105–119. [CrossRef]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense Oligonucleotide Therapy for Spinocerebellar Ataxia Type 2. Nature 2017, 544, 362–366. [CrossRef]
- Shen, X.; Wong, J.; Prakash, T.P.; Rigo, F.; Li, Y.; Napierala, M.; Corey, D.R. Progress towards Drug Discovery for Friedreich’s Ataxia: Identifying Synthetic Oligonucleotides That More Potently Activate Expression of Human Frataxin Protein. Bioorg. Med. Chem. 2020, 28, 115472. [CrossRef]
- Li, L.; Shen, X.; Liu, Z.; Norrbom, M.; Prakash, T.P.; O’Reilly, D.; Sharma, V.K.; Damha, M.J.; Watts, J.K.; Rigo, F.; et al. Activation of Frataxin Protein Expression by Antisense Oligonucleotides Targeting the Mutant Expanded Repeat. Nucleic Acid Ther. 2018, 28, 23–33. [CrossRef]
- Park, J.; Al-Ramahi, I.; Tan, Q.; Mollema, N.; Diaz-Garcia, J.R.; Gallego-Flores, T.; Lu, H.-C.; Lagalwar, S.; Duvick, L.; Kang, H.; et al. RAS-MAPK-MSK1 Pathway Modulates Ataxin 1 Protein Levels and Toxicity in SCA1. Nature 2013, 498, 325–331. [CrossRef]
- Perdomini, M.; Belbellaa, B.; Monassier, L.; Reutenauer, L.; Messaddeq, N.; Cartier, N.; Crystal, R.G.; Aubourg, P.; Puccio, H. Prevention and Reversal of Severe Mitochondrial Cardiomyopathy by Gene Therapy in a Mouse Model of Friedreich’s Ataxia. Nat. Med. 2014, 20, 542–547. [CrossRef]
- Belbellaa, B.; Reutenauer, L.; Monassier, L.; Puccio, H. Correction of Half the Cardiomyocytes Fully Rescue Friedreich Ataxia Mitochondrial Cardiomyopathy through Cell-Autonomous Mechanisms. Hum. Mol. Genet. 2019, 28, 1274–1285. [CrossRef]
- Munoz-Zuluaga, C.; Gertz, M.; Yost-Bido, M.; Greco, A.; Gorman, N.; Chen, A.; Kooner, V.; Rosenberg, J.B.; De, B.P.; Kaminsky, S.M.; et al. Identification of Safe and Effective Intravenous Dose of AAVrh.10hFXN to Treat the Cardiac Manifestations of Friedreich’s Ataxia. Hum. Gene Ther. 2023, 34, 605–615. [CrossRef]
- Piguet, F.; de Montigny, C.; Vaucamps, N.; Reutenauer, L.; Eisenmann, A.; Puccio, H. Rapid and Complete Reversal of Sensory Ataxia by Gene Therapy in a Novel Model of Friedreich Ataxia. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1940–1952. [CrossRef]
- Britti, E.; Delaspre, F.; Feldman, A.; Osborne, M.; Greif, H.; Tamarit, J.; Ros, J. Frataxin-Deficient Neurons and Mice Models of Friedreich Ataxia Are Improved by TAT-MTScs-FXN Treatment. J. Cell. Mol. Med. 2018, 22, 834–848. [CrossRef]
- Erwin, G.S.; Grieshop, M.P.; Ali, A.; Qi, J.; Lawlor, M.; Kumar, D.; Ahmad, I.; McNally, A.; Teider, N.; Worringer, K.; et al. Synthetic Transcription Elongation Factors License Transcription across Repressive Chromatin. Science 2017, 358, 1617–1622. [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent Developments of HDAC Inhibitors: Emerging Indications and Novel Molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [CrossRef]
- Soragni, E.; Gottesfeld, J.M. Translating HDAC Inhibitors in Friedreich’s Ataxia. Expert Opin. Orphan Drugs 2016, 4, 961–970. [CrossRef]
- Chan, P.K.; Torres, R.; Yandim, C.; Law, P.P.; Khadayate, S.; Mauri, M.; Grosan, C.; Chapman-Rothe, N.; Giunti, P.; Pook, M.; et al. Heterochromatinization Induced by GAA-Repeat Hyperexpansion in Friedreich’s Ataxia Can Be Reduced upon HDAC Inhibition by Vitamin B3. Hum. Mol. Genet. 2013, 22, 2662–2675. [CrossRef]
- Lei, L.-F.; Yang, G.-P.; Wang, J.-L.; Chuang, D.-M.; Song, W.-H.; Tang, B.-S.; Jiang, H. Safety and Efficacy of Valproic Acid Treatment in SCA3/MJD Patients. Parkinsonism Relat. Disord. 2016, 26, 55–61. [CrossRef]
- Soragni, E.; Miao, W.; Iudicello, M.; Jacoby, D.; De Mercanti, S.; Clerico, M.; Longo, F.; Piga, A.; Ku, S.; Campau, E.; et al. Epigenetic Therapy for Friedreich Ataxia. Ann. Neurol. 2014, 76, 489–508. [CrossRef]
- Ding, Y.; Adachi, H.; Katsuno, M.; Sahashi, K.; Kondo, N.; Iida, M.; Tohnai, G.; Nakatsuji, H.; Sobue, G. BIIB021, a Synthetic Hsp90 Inhibitor, Induces Mutant Ataxin-1 Degradation through the Activation of Heat Shock Factor 1. Neuroscience 2016, 327, 20–31. [CrossRef]
- Williams, A.J.; Knutson, T.M.; Colomer Gould, V.F.; Paulson, H.L. In Vivo Suppression of Polyglutamine Neurotoxicity by C-Terminus of Hsp70-Interacting Protein (CHIP) Supports an Aggregation Model of Pathogenesis. Neurobiol. Dis. 2009, 33, 342–353. [CrossRef]
- Tsou, W.-L.; Hosking, R.R.; Burr, A.A.; Sutton, J.R.; Ouyang, M.; Du, X.; Gomez, C.M.; Todi, S.V. DnaJ-1 and Karyopherin A3 Suppress Degeneration in a New Drosophila Model of Spinocerebellar Ataxia Type 6. Hum. Mol. Genet. 2015, 24, 4385–4396. [CrossRef]
- Alves, S.; Cormier-Dequaire, F.; Marinello, M.; Marais, T.; Muriel, M.-P.; Beaumatin, F.; Charbonnier-Beaupel, F.; Tahiri, K.; Seilhean, D.; El Hachimi, K.; et al. The Autophagy/Lysosome Pathway Is Impaired in SCA7 Patients and SCA7 Knock-in Mice. Acta Neuropathol. 2014, 128, 705–722. [CrossRef]
- Parkinson, M.H.; Schulz, J.B.; Giunti, P. Co-Enzyme Q10 and Idebenone Use in Friedreich’s Ataxia. J. Neurochem. 2013, 126 (Suppl 1), 125–141. [CrossRef]
- Wu, Y.-L.; Chang, J.-C.; Sun, H.-L.; Cheng, W.-L.; Yen, Y.-P.; Lin, Y.-S.; Chao, Y.-C.; Liu, K.-H.; Huang, C.-S.; Liu, K.-L.; et al. Coenzyme Q10 Supplementation Increases Removal of the ATXN3 Polyglutamine Repeat, Reducing Cerebellar Degeneration and Improving Motor Dysfunction in Murine Spinocerebellar Ataxia Type 3. Nutrients 2022, 14, 3593. [CrossRef]
- Schirinzi, T.; Favetta, M.; Romano, A.; Sancesario, A.; Summa, S.; Minosse, S.; Zanni, G.; Castelli, E.; Bertini, E.; Petrarca, M.; et al. One-Year Outcome of Coenzyme Q10 Supplementation in ADCK3 Ataxia (ARCA2). Cerebellum Ataxias 2019, 6, 15. [CrossRef]
- Jauslin, M.L.; Meier, T.; Smith, R.A.J.; Murphy, M.P. Mitochondria-Targeted Antioxidants Protect Friedreich Ataxia Fibroblasts from Endogenous Oxidative Stress More Effectively than Untargeted Antioxidants. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1972–1974. [CrossRef]
- Márquez, B.T.; Leung, T.C.S.; Hui, J.; Charron, F.; McKinney, R.A.; Watt, A.J. A Mitochondrial-Targeted Antioxidant (MitoQ) Improves Motor Coordination and Reduces Purkinje Cell Death in a Mouse Model of ARSACS. Neurobiol. Dis. 2023, 183, 106157. [CrossRef]
- Yang, B.; Dan, X.; Hou, Y.; Lee, J.-H.; Wechter, N.; Krishnamurthy, S.; Kimura, R.; Babbar, M.; Demarest, T.; McDevitt, R.; et al. NAD+ Supplementation Prevents STING-Induced Senescence in Ataxia Telangiectasia by Improving Mitophagy. Aging Cell 2021, 20, e13329. [CrossRef]
- Hourez, R.; Servais, L.; Orduz, D.; Gall, D.; Millard, I.; de Kerchove d’Exaerde, A.; Cheron, G.; Orr, H.T.; Pandolfo, M.; Schiffmann, S.N. Aminopyridines Correct Early Dysfunction and Delay Neurodegeneration in a Mouse Model of Spinocerebellar Ataxia Type 1. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 11795–11807. [CrossRef]
- Jayabal, S.; Chang, H.H.V.; Cullen, K.E.; Watt, A.J. 4-Aminopyridine Reverses Ataxia and Cerebellar Firing Deficiency in a Mouse Model of Spinocerebellar Ataxia Type 6. Sci. Rep. 2016, 6, 29489. [CrossRef]
- Egorova, P.A.; Zakharova, O.A.; Vlasova, O.L.; Bezprozvanny, I.B. In Vivo Analysis of Cerebellar Purkinje Cell Activity in SCA2 Transgenic Mouse Model. J. Neurophysiol. 2016, 115, 2840–2851. [CrossRef]
- Kasumu, A.W.; Hougaard, C.; Rode, F.; Jacobsen, T.A.; Sabatier, J.M.; Eriksen, B.L.; Strøbæk, D.; Liang, X.; Egorova, P.; Vorontsova, D.; et al. Selective Positive Modulator of Calcium-Activated Potassium Channels Exerts Beneficial Effects in a Mouse Model of Spinocerebellar Ataxia Type 2. Chem. Biol. 2012, 19, 1340–1353. [CrossRef]
- Maltecca, F.; Baseggio, E.; Consolato, F.; Mazza, D.; Podini, P.; Young, S.M.; Drago, I.; Bahr, B.A.; Puliti, A.; Codazzi, F.; et al. Purkinje Neuron Ca2+ Influx Reduction Rescues Ataxia in SCA28 Model. J. Clin. Invest. 2015, 125, 263–274. [CrossRef]
- Coarelli, G.; Heinzmann, A.; Ewenczyk, C.; Fischer, C.; Chupin, M.; Monin, M.-L.; Hurmic, H.; Calvas, F.; Calvas, P.; Goizet, C.; et al. Safety and Efficacy of Riluzole in Spinocerebellar Ataxia Type 2 in France (ATRIL): A Multicentre, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2022, 21, 225–233. [CrossRef]
- Ghanekar, S.D.; Kuo, S.-H.; Staffetti, J.S.; Zesiewicz, T.A. Current and Emerging Treatment Modalities for Spinocerebellar Ataxias. Expert Rev. Neurother. 2022, 22, 101–114. [CrossRef]
- Ristori, G.; Romano, S.; Visconti, A.; Cannoni, S.; Spadaro, M.; Frontali, M.; Pontieri, F.E.; Vanacore, N.; Salvetti, M. Riluzole in Cerebellar Ataxia: A Randomized, Double-Blind, Placebo-Controlled Pilot Trial. Neurology 2010, 74, 839–845. [CrossRef]
- Perlman, S.L. Update on the Treatment of Ataxia: Medication and Emerging Therapies. Neurother. J. Am. Soc. Exp. Neurother. 2020, 17, 1660–1664. [CrossRef]
- Zesiewicz, T.A.; Greenstein, P.E.; Sullivan, K.L.; Wecker, L.; Miller, A.; Jahan, I.; Chen, R.; Perlman, S.L. A Randomized Trial of Varenicline (Chantix) for the Treatment of Spinocerebellar Ataxia Type 3. Neurology 2012, 78, 545–550. [CrossRef]
- Botez, M.I.; Botez-Marquard, T.; Elie, R.; Pedraza, O.L.; Goyette, K.; Lalonde, R. Amantadine Hydrochloride Treatment in Heredodegenerative Ataxias: A Double Blind Study. J. Neurol. Neurosurg. Psychiatry 1996, 61, 259–264. [CrossRef]
- Nissenkorn, A.; Hassin-Baer, S.; Lerman, S.F.; Levi, Y.B.; Tzadok, M.; Ben-Zeev, B. Movement Disorder in Ataxia-Telangiectasia: Treatment with Amantadine Sulfate. J. Child Neurol. 2013, 28, 155–160. [CrossRef]
- Cunha-Santos, J.; Duarte-Neves, J.; Carmona, V.; Guarente, L.; Pereira de Almeida, L.; Cavadas, C. Caloric Restriction Blocks Neuropathology and Motor Deficits in Machado-Joseph Disease Mouse Models through SIRT1 Pathway. Nat. Commun. 2016, 7, 11445. [CrossRef]
- Yiu, E.M.; Tai, G.; Peverill, R.E.; Lee, K.J.; Croft, K.D.; Mori, T.A.; Scheiber-Mojdehkar, B.; Sturm, B.; Praschberger, M.; Vogel, A.P.; et al. An Open-Label Trial in Friedreich Ataxia Suggests Clinical Benefit with High-Dose Resveratrol, without Effect on Frataxin Levels. J. Neurol. 2015, 262, 1344–1353. [CrossRef]
- Leuzzi, V.; Micheli, R.; D’Agnano, D.; Molinaro, A.; Venturi, T.; Plebani, A.; Soresina, A.; Marini, M.; Ferremi Leali, P.; Quinti, I.; et al. Positive Effect of Erythrocyte-Delivered Dexamethasone in Ataxia-Telangiectasia. Neurol. Neuroimmunol. Neuroinflammation 2015, 2, e98. [CrossRef]
- Saberi-Karimian, M.; Beyraghi-Tousi, M.; Jamialahmadi, T.; Sahebkar, A. The Positive Short-Term Effect of Dexamethasone on Ataxia Symptoms in a Patient with Ataxia-Telangiectasia: A Case Report. Clin. Case Rep. 2022, 10, e05895. [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium Induces Autophagy by Inhibiting Inositol Monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [CrossRef]
- Watase, K.; Gatchel, J.R.; Sun, Y.; Emamian, E.; Atkinson, R.; Richman, R.; Mizusawa, H.; Orr, H.T.; Shaw, C.; Zoghbi, H.Y. Lithium Therapy Improves Neurological Function and Hippocampal Dendritic Arborization in a Spinocerebellar Ataxia Type 1 Mouse Model. PLoS Med. 2007, 4, e182. [CrossRef]
- Kieling, C.; Rieder, C.R.M.; Silva, A.C.F.; Saute, J. a. M.; Cecchin, C.R.; Monte, T.L.; Jardim, L.B. A Neurological Examination Score for the Assessment of Spinocerebellar Ataxia 3 (SCA3). Eur. J. Neurol. 2008, 15, 371–376. [CrossRef]
- Saccà, F.; Puorro, G.; Brunetti, A.; Capasso, G.; Cervo, A.; Cocozza, S.; de Leva, M.; Marsili, A.; Pane, C.; Quarantelli, M.; et al. A Randomized Controlled Pilot Trial of Lithium in Spinocerebellar Ataxia Type 2. J. Neurol. 2015, 262, 149–153. [CrossRef]
- Liu, J.; Wang, L. Mitochondrial Enhancement for Neurodegenerative Movement Disorders: A Systematic Review of Trials Involving Creatine, Coenzyme Q10, Idebenone and Mitoquinone. CNS Drugs 2014, 28, 63–68. [CrossRef]
- Grimaldi, G.; Argyropoulos, G.P.; Boehringer, A.; Celnik, P.; Edwards, M.J.; Ferrucci, R.; Galea, J.M.; Groiss, S.J.; Hiraoka, K.; Kassavetis, P.; et al. Non-Invasive Cerebellar Stimulation--a Consensus Paper. Cerebellum Lond. Engl. 2014, 13, 121–138. [CrossRef]
- Hartley, H.; Cassidy, E.; Bunn, L.; Kumar, R.; Pizer, B.; Lane, S.; Carter, B. Exercise and Physical Therapy Interventions for Children with Ataxia: A Systematic Review. Cerebellum Lond. Engl. 2019, 18, 951–968. [CrossRef]
- Lee, A. Omaveloxolone: First Approval. Drugs 2023, 83, 725–729. [CrossRef]
- Lynch, D.R.; Chin, M.P.; Delatycki, M.B.; Subramony, S.H.; Corti, M.; Hoyle, J.C.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Mathews, K.D.; et al. Safety and Efficacy of Omaveloxolone in Friedreich Ataxia ( MOXIE Study). Ann. Neurol. 2021, 89, 212–225. [CrossRef]
- Strawser, C.; Schadt, K.; Hauser, L.; McCormick, A.; Wells, M.; Larkindale, J.; Lin, H.; Lynch, D.R. Pharmacological Therapeutics in Friedreich Ataxia: The Present State. Expert Rev. Neurother. 2017, 17, 895–907. [CrossRef]
- Ashizawa, T.; Öz, G.; Paulson, H.L. Spinocerebellar Ataxias: Prospects and Challenges for Therapy Development. Nat. Rev. Neurol. 2018, 14, 590–605. [CrossRef]
- Shakkottai, V.; Paulson, H. Expanding the Genetic Basis of Ataxia. Nat. Genet. 2019, 51, 580–581. [CrossRef]
- Wong, M.M.K.; Watson, L.M.; Becker, E.B.E. Recent Advances in Modelling of Cerebellar Ataxia Using Induced Pluripotent Stem Cells. J. Neurol. Neuromedicine 2017, 2, 11–15. [CrossRef]
- Georges, P.; Boza-Moran, M.-G.; Gide, J.; Pêche, G.A.; Forêt, B.; Bayot, A.; Rustin, P.; Peschanski, M.; Martinat, C.; Aubry, L. Induced Pluripotent Stem Cells-Derived Neurons from Patients with Friedreich Ataxia Exhibit Differential Sensitivity to Resveratrol and Nicotinamide. Sci. Rep. 2019, 9, 14568. [CrossRef]
- Hansen, S.K.; Stummann, T.C.; Borland, H.; Hasholt, L.F.; Tümer, Z.; Nielsen, J.E.; Rasmussen, M.A.; Nielsen, T.T.; Daechsel, J.C.A.; Fog, K.; et al. Induced Pluripotent Stem Cell - Derived Neurons for the Study of Spinocerebellar Ataxia Type 3. Stem Cell Res. 2016, 17, 306–317. [CrossRef]
- Buijsen, R.A.M.; Hu, M.; Sáez-González, M.; Notopoulou, S.; Mina, E.; Koning, W.; Gardiner, S.L.; van der Graaf, L.M.; Daoutsali, E.; Pepers, B.A.; et al. Spinocerebellar Ataxia Type 1 Characteristics in Patient-Derived Fibroblast and IPSC-Derived Neuronal Cultures. Mov. Disord. Off. J. Mov. Disord. Soc. 2023. [CrossRef]
- He, L.; Wang, S.; Peng, L.; Zhao, H.; Li, S.; Han, X.; Habimana, J. de D.; Chen, Z.; Wang, C.; Peng, Y.; et al. CRISPR/Cas9 Mediated Gene Correction Ameliorates Abnormal Phenotypes in Spinocerebellar Ataxia Type 3 Patient-Derived Induced Pluripotent Stem Cells. Transl. Psychiatry 2021, 11, 479. [CrossRef]
- Chen, Y.; Bury, L.; Chen, F.; Aldinger, K.A.; Miranda, H.C.; Wynshaw-Boris, A. Generation of Advanced Cerebellar Organoids for Neurogenesis and Neuronal Network Development. Hum. Mol. Genet. 2023, ddad110. [CrossRef]
- Silva, T.P.; Fernandes, T.G.; Nogueira, D.E.S.; Rodrigues, C.A.V.; Bekman, E.P.; Hashimura, Y.; Jung, S.; Lee, B.; Carmo-Fonseca, M.; Cabral, J.M.S. Scalable Generation of Mature Cerebellar Organoids from Human Pluripotent Stem Cells and Characterization by Immunostaining. J. Vis. Exp. JoVE 2020. [CrossRef]
- Watson, L.M.; Wong, M.M.K.; Vowles, J.; Cowley, S.A.; Becker, E.B.E. A Simplified Method for Generating Purkinje Cells from Human-Induced Pluripotent Stem Cells. Cerebellum Lond. Engl. 2018, 17, 419–427. [CrossRef]
- Buchholz, D.E.; Carroll, T.S.; Kocabas, A.; Zhu, X.; Behesti, H.; Faust, P.L.; Stalbow, L.; Fang, Y.; Hatten, M.E. Novel Genetic Features of Human and Mouse Purkinje Cell Differentiation Defined by Comparative Transcriptomics. Proc. Natl. Acad. Sci. USA 2020, 117, 15085–15095. [CrossRef]
- Hommersom, M.P.; Buijsen, R.A.M.; van Roon-Mom, W.M.C.; van de Warrenburg, B.P.C.; van Bokhoven, H. Human Induced Pluripotent Stem Cell-Based Modelling of Spinocerebellar Ataxias. Stem Cell Rev. Rep. 2022, 18, 441–456. [CrossRef]
- Chow, S.-C.; Huang, Z. Innovative Design and Analysis for Rare Disease Drug Development. J. Biopharm. Stat. 2020, 30, 537–549. [CrossRef]
Figure 1.
Pathophysiological changes in Hereditary Ataxia (HA) and the corresponding treatments. The diagram depicts various pathophysiological alterations in ataxia and suggests treatments that address specific pathways. Proposed pharmacological strategies aim to regulate excitability and calcium homeostasis, (i.e. Riluzole, Amantadine and Verenicline), as well as target proteostasis, mitochondrial dysfunction and inflammation, with the recently FDA-approved drug Omaveloxolone. Additionally, alternative therapeutic methods like Antisense oligonucleotides and gene therapy can address distinct pathways within the proposed framework.
Figure 1.
Pathophysiological changes in Hereditary Ataxia (HA) and the corresponding treatments. The diagram depicts various pathophysiological alterations in ataxia and suggests treatments that address specific pathways. Proposed pharmacological strategies aim to regulate excitability and calcium homeostasis, (i.e. Riluzole, Amantadine and Verenicline), as well as target proteostasis, mitochondrial dysfunction and inflammation, with the recently FDA-approved drug Omaveloxolone. Additionally, alternative therapeutic methods like Antisense oligonucleotides and gene therapy can address distinct pathways within the proposed framework.
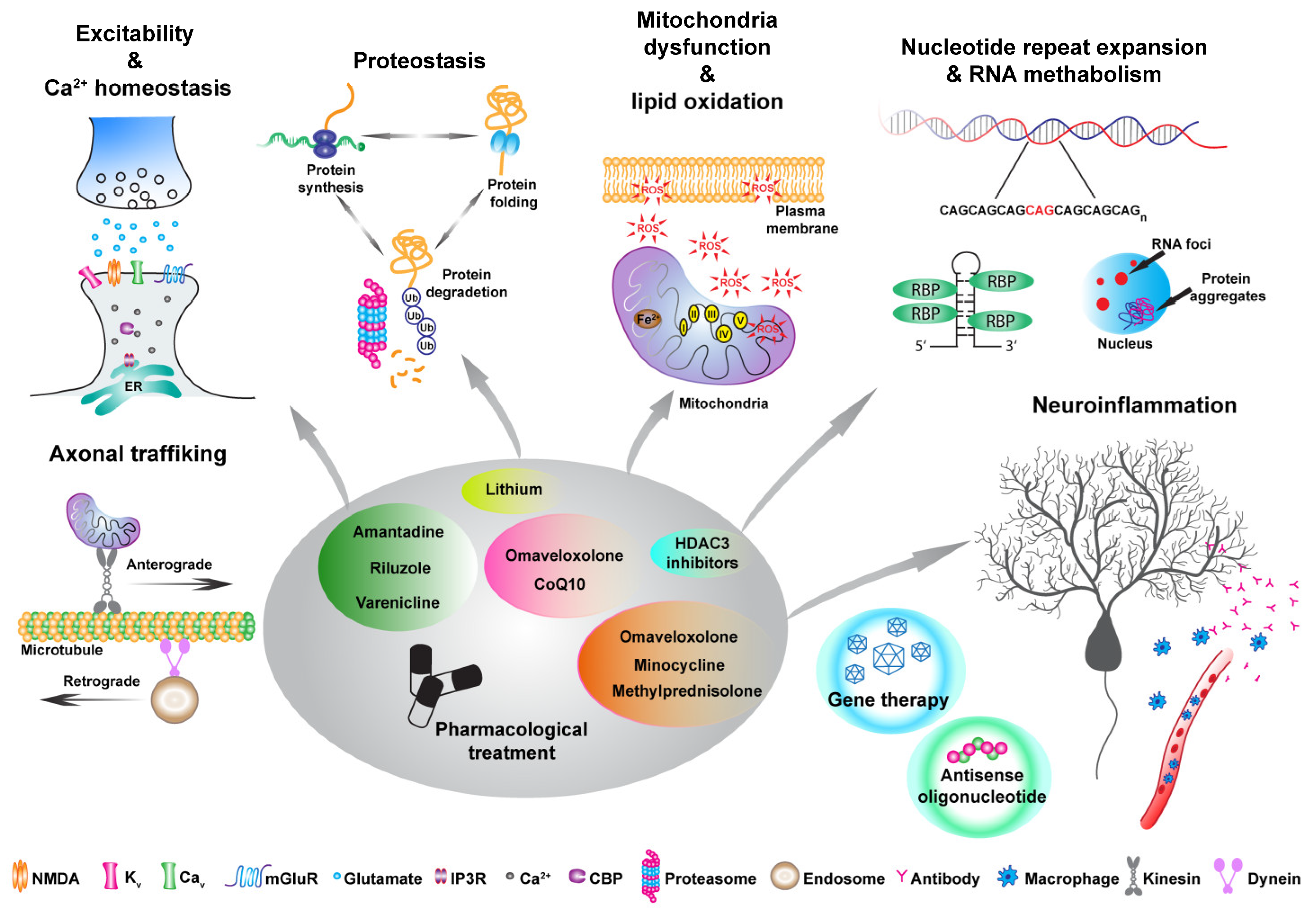

Table 1.
Summary of different treatment strategy and outcomes for HA.
| Strategy | Stage | Model | Outcomes | Reference | |
|---|---|---|---|---|---|
| Spinocerebellar ataxia type-1 (SCA1) | |||||
| Small RNA structures | shRNA and miRNA | Pre-clinical | SCA1 mouse models | Improvement of motor coordination, restoration of cerebellar morphology and absence of ataxin-1 inclusions | [184,185,186] |
| Small RNA structures | miRNA | Pre-clinical | SCA1 non-human primate | Reduction of endogenous ATXN1 mRNA | |
| Proteostasis | HSP inhibitors | Pre-clinical | Human SCA1 cell lines | Reduction of ataxin-1 aggregates | [219] |
| Pharmacological treatment | MitoQ | Pre-clinical | SCA1 mouse models | Restoration of mitochondrial function, attenuation of PN degeneration and improvement of motor coordination | [19] |
| Pharmacological treatment | 4-animopyridine | Pre-clinical | SCA1 mouse model | Restoration of PN firing, improvement of motor coordination, and partial protection against cell atrophy | [229] |
| Pharmacological treatment | Lithium | Pre-clinical | SCA1 mouse model | Improvement in motor coordination | [246] |
| Spinocerebellar ataxia type-2 (SCA2) | |||||
| Genome editing | CRISPR | Pre-clinical | SCA2 mouse models | / | [182] |
| Small RNA structure | ASO | Pre-clinical | SCA2 mouse models | Improvement of motor performance and restoration of physiological properties and deregulated genes and proteins | [204] |
| Pharmacological treatment | Chlorzoxazone | Pre-clinical | SCA2 mouse model | Normalization of PN firing and alleviation of ataxic phenotype | [231] |
| Pharmacological treatment | NS13001 | Pre-clinical | SCA2 mouse model | Improvement of motor ability and reduced PN degeneration | [232] |
| Spinocerebellar ataxia type-3 (SCA3) | |||||
| Genome editing | CRISPR | Pre-clinical | Human iPSCs | Restoration of ataxin-3 functionality without the formation of toxic aggregates | [178] |
| Small RNA structure | shRNA | Pre-clinical | SCA3 rat model | Reduced of ataxin-3 inclusions and prevention of neurodegeneration | [188] |
| Small RNA structure | siRNA | Pre-clinical | SCA3 mouse model | ATXN3 downregulation and prevention of its aggregation | [189,190] |
| Small RNA structure | ASO | Pre-clinical | SCA3 mouse model | Improvement of motor ability, restoration of PN dysfunction and rescue of altered neurometabolites | [191] |
| Gene activation | HDAC inhibitor, valproic acid | Clinical phase 1/2 | SCA3 patients | Patients treated with valproic acid improved locomotor function (SARA scale) | [217] |
| Pharmacological treatment | Coenzyme Q10 | Pre-clinical | SCA3 mouse model | Recovery of motor coordination, reduced PN degeneration and muscle atrophy | [224] |
| Pharmacological treatment | Varenicline | Clinical | SCA3 patients | Improvement in SARA scale score | [238] |
| Pharmacological treatment | Resveratrol | Clinical | SCA3 mouse model | Reduced motor incoordination | [241] |
| Spinocerebellar ataxia type-6 (SCA6) | |||||
| Small RNA structure | miRNA | Pre-clinical | SCA6 mouse model | Reduced ataxic phenotype and PN degeneration | [193,194] |
| Pharmacological treatment | 4-aminopyridine | Pre-clinical | SCA6 mouse model | Improvement of motor ability and restoration of PN firing | [230] |
| Spinocerebellar ataxia type-7 (SCA7) | |||||
| Small RNA structure | miRNA | Pre-clinical | SCA7 mouse model | Improvement of motor deficit, increased PN survival and ATXN7 downregulation | [195,196] |
| Spinocerebellar ataxia type-28 (SCA28) | |||||
| Pharmacological treatment | Ceftriaxone | Pre-clinical | SCA28 mouse model | Reduced PN degeneration and improvement of motor performance | [144] |
| Friedreich Ataxia (FA) | |||||
| Genome editing | CRISPR | Pre-clinical | Human iPSCs | Deletion of the expanded CAG tract is not always sufficient to revert the phenotype | [179,180] |
| Genome editing | ZFN | Pre-clinical | Human iPSC-derived neurons and cardiomyocytes | Decreased aconitase activity and ATP levels in iPSC-derived neurons and corrected the cardiomyopathy in cardiomyocytes | [183] |
| Small RNA structure | ASO | Pre-clinical | Human FA cell lines | Activation of FXN expression and consequent restoration of frataxin levels | [206] |
| Small RNA structure | Gapmer | Pre-clinical | Human FA cell lines | Activation of FXN expression | [205] |
| Gene therapy | Pre-clinical | FA mouse model | Complete and rapid recovery of cardiac functionality | [208,209,210] | |
| Gene therapy | Pre-clinical | FA mouse model | Complete and rapid rescue of sensory neuropathy and ganglionopathy | [211] | |
| Gene therapy | Clinical phase 1/2 | FA patients | Ongoing sponsored by Lexeo Therapeutics (NCT05445323) | ||
| Protein replacement | TAT peptides | Pre-clinical | FA mouse model | Decreased neurite degeneration and apoptotic markers resulting in increased cell survival. And restoration of mitochondrial features | [212] |
| Protein replacement | TAT peptides | Clinical phase 2 | FA patients | Ongoing sponsored by Larimar Therapeutics (NCT05579691) | |
| Gene activation | Syn-TEF1 | Pre-clinical | Human iPSC-derived neurons and cardiomyocytes | Activation of FXN expression | [213] |
| Gene activation | Syn-TEF1 | Clinical phase 1a | FA patients | Ongoing sponsored by Design Therapeutics (NCT05285540) | |
| Gene activation | HDAC inhibitors, 2-aminobenzamide | Pre-clinical | Human cell lines and FA mouse models | Activation of FXN expression | [215] |
| Gene activation | HDAC inhibitors, nicotinamide | Pre-clinical | FA mouse model | FXN upregulation | [216] |
| Gene activation | HDAC inhibitors, RG2833 | Clinical phase 1 | FA patients | Increased FXN levels, but a toxic metabolites was detected | [218] |
| Pharmacological treatment | Omaveloxolone | Clinical phase 2 | FA patients | Approved: sponsored by Biogen (NCT02255435) | [253] |
| Pharmacological treatment | Vatiquinone | Clinical phase 2/3 | FA patients | Ongoing sponsored by PTC Therapeutics (NCT04577352) | |
| Pharmacological treatment | MitoQ | Pre-clinical | FA cell lines | Reduced cell death | [226] |
| Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) | |||||
| Pharmacological treatment | MitoQ | Pre-clinical | ARSACS mouse model | Decreased PN degeneration, increased DCN innervation and prevention of motor decline | [227] |
| Pharmacological treatment | Ceftriaxone | Pre-clinical | ARSACS mouse model | Restoration of calcium homeostasis, reduced neuroinflammation and improvement of motor ability | [233] |
| Ataxia-telangiectasia (AT) | |||||
| Pharmacological treatment | Nicotinamide riboside | Pre-clinical | AT mouse model | Prevention of neuroinflammation, reduced mitochondrial dysfunction and PN death, and improvement in motor ability | [228] |
| Pharmacological treatment | Amantadine | Clinical phase 4 | AT patients | Improvement in ataxic phenotype, involuntary movements and parkinsonism symptoms | [240] |
| Pharmacological treatment | Dexamethasone | Clinical phase 3 | AT patients | Sponsored by Erydel (NCT02770807) | |
| Autosomal Recessive Ataxia tye-2 (ARCA2) | |||||
| Pharmacological treatment | Coenzyme Q10 | Clinical | ARCA2 patients | Mild improvement of motor features | [225] |
| Multi disease trials | |||||
| Pharmacological treatment | Riluzole | Clinical phase 2 | SCA1, SCA2, SCA17, SCA28 and FA patients | Improvement of ICARS scale score | [236] |
| Pharmacological treatment | Riluzole | Clinical phase 3 | SCA1, SCA2, SCA3, SCA6, SCA7, SCA8 and SCA10 patients | Ongoing sponsored by Biohaven Pharmaceutical, Inc. (NCT03701399) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



