
Submitted:
01 December 2023
Posted:
04 December 2023
You are already at the latest version
Abstract
The Totiviridae family of viruses has a unique genome consisting of double-stranded RNA with two open reading frames that encode the capsid protein (Cap) and the RNA-dependent RNA polymerase (RdRpol). Most virions in this family are isometric in shape, approximately 40 nm in diameter, and lack envelope. There are five genera within this family, including Totivirus, Victorivirus, Giardiavirus, Leishmaniavirus, and Trichomonasvirus. While Totivirus and Victorivirus primarily infect fungi, Giardiavirus, Leishmaniavirus, and Trichomonasvirus infect diverse hosts, including protists, insects, and vertebrates. Recently, new totivirus-like species have been discovered in fish and plant hosts, and through metagenomic analysis, a novel totivirus-like virus (named Tianjin totivirus) has been isolated from bat guano. Interestingly, Tianjin totivirus causes cytopathic effects in insect cells but cannot grow in mammalian cells, suggesting that it infects insects consumed by insectivorous bats. In this study, we used next-generation sequencing and identified totivirus-like viruses in liver tissue from Molossus molossus bats in the Amazon region of Brazil. Comparative phylogenetic analysis based on the RNA-dependent RNA polymerase region revealed that the viruses identified in Molossus bats belong to two distinct phylogenetic clades, possibly comprising different genera within the Totiviridae family. Notably, the mean similarity between Tianjin totivirus and the totiviruses identified in Molossus bats is less than 18%. These findings suggest that the diversity of totiviruses in bats is more extensive than previously recognized and highlight the potential for bats to serve as reservoirs for novel toti-like viruses.
Keywords:
Metagenomics
; Totivirus
; Bats
; Molossus
; Amazon region.
1. Introduction
Bats are unique animals due to their extensive viral diversity, which distinguishes them from other species [1,2,3,4,5,6]. They have long been associated with many viral families and genera, such as Paramyxoviridae, Filoviridae and Rhabdoviridae [7,8,9,10,11,12,13,14,15,16,17]. Their ability to fly long distances and their diverse feeding habits make it easier for them to acquire and spread viruses across remote areas and to transmit them to other species. Additionally, their social structures and behaviors contribute to virus transmission and persistence within bat populations [4,18,19,20]. However, changes in the environment, such as urbanization, agricultural intensification, and deforestation, have altered the composition and dynamics of bat communities [21]. Studies have shown an overall decline in species richness and relative abundance associated with urbanization [22]. Nevertheless, insectivorous bats tend to thrive in large urban environments [4,18,19]. Furthermore, the diversity of bat habitats can influence both microbe transmission and persistence in bat communities [2,22,23,24].
Viral metagenomics research in bats has mainly focused on North American and Eurasian bat communities [21,25,26,27]. However, studies in the Amazon region have enabled the identification of numerous viruses using conventional techniques or high-throughput sequencing [7,8,9,12,14,16,28,29]. For instance, a metagenomic study conducted in French Guiana found various RNA viruses in fecal samples of Molussus bats (also known as velvety free-tailed bat or Pallas's mastiff bat), including some short sequences of viruses belonging to the Totiviridae family [30]. Another study performed in carcasses of deceased bats in Germany, also found short sequence of totiviruses [31]. Researchers in China were able isolate one toti-like virus in insect cells from guano samples of the insectivorous Myotis bats [32].
Recent studies have shed light on the diverse range of insect viruses found in bat droppings through Next Generation Sequencing (NGS) [21,33]. NGS has facilitated the isolation and characterization of several new totivirus-like viruses that have yet to be classified by the International Committee on Taxonomy of Viruses (ICTV). The Totiviridae family is a diverse group of RNA viruses that infect both protozoa and fungi, with five identified genera to date. The family comprises 28 species, each of which contains a single molecule of double-stranded RNA (dsRNA) ranging in size from 4.6 to 7.0 kbp. The virus genome consists of two frames, with the 5' ORF encoding the capsid protein (CP) and the 3' ORF encoding the RNA-dependent RNA polymerase (RdRpol) gene [34].
Viruses in the Totiviridae family are remarkably diverse in their ability to infect hosts ranging from fungi to protozoa [35,36,37,38,39,40,41,42,43]. The current recognized genera include Giardiavirus, Leishmaniavirus, Totivirus, Victorivirus, and Trichomonasvirus [35,44,45,46,47,48,49]. While those in the genera Totivirus and Victorivirus infect fungi, those in Giardiavirus, Leishmaniavirus, and Trichomonasvirus infect protozoa. Recently, new viruses have been identified in shrimp, fish, and mosquitoes that have similar genomic structures and morphology but low similarity to members of other genera in the Totiviridae family [47,50]. Researchers have also proposed three new genera within the Totiviridae family, namely Artivirus, Pistolvirus, and Tricladiviris [39,45,48,49,51,52]. For instance, Artivirus has been found to infect arthropods such as the Atlantic blue crab [53] and the mosquito Armigere subalbatus totivirus [54]. Pistolvirus has been found to infect fish species such as the Atlantic salmon and golden carp [39,52]. Tricladiviris has been found to infect planaria. Additionally, new toti-like viruses have been discovered in Eysarcoris guttigerus, with genomic characteristics similar to Sanya orius sauteri totivirus 2. Arboreal ants Camponotus yamaokai also carry a virus that is potentially a new totivirus, but it is not related to the Totiviruses identified in arthropods [55].
In our study, we utilized a metagenomic next-generation sequencing (NGS) approach to comprehensively examine viruses present in the insectivorous Molossus molossus bats that were captured in the urban areas of Santarém city, located in northern Brazil. Our analysis yielded forty-seven contigs, including three near-complete genomes, which showed an average amino acid identity of 40% with a closely related totivirus-like previously reported.
2. Materials and Methods
2.1. Sample collection
We captured a total of 47 bats in Caranazal (latitude 2°26'10"S and longitude 54°43'49"W), a municipality in the Santarém region of Pará state in the Lower Amazon Mesoregion. These bats were identified to Molossus molossus (Family Molossidae) based on external characters. To obtain the sequences, individual bats were euthanized for sample collection. We administered xylazine hydrochloride (1mg/kg) and ketamine hydrochloride (1-2mg/kg) via intramuscular injection to induce anaesthesia, followed by intracardiac phenobarbital (40mg/kg) once the animals had lost consciousness. The liver samples were then collected for further analysis. Details of samples and composition of pools are in Table S1.
To carry out this research, approval was obtained from the Animal Use Ethics Committee of the Federal University of Western Pará (CEUA/UFOPA) under number 0220220128 and from the Biodiversity Information and Authorization System (SISBIO - 18313-1) for capturing Chiroptera. The necropsy was performed at the Animal Morphology Laboratory of the Federal University of Western Pará, following the institutional biosafety norms.
2.2. Processing of samples
To extract viral particles from liver tissue, we prepared pools of organ pieces from five different animals. These pools were named F1, F2 and so on. The tissue was first macerated in a tissue disruptor and then diluted in 500 µL of Hanks Buffered Saline Solution (HBSS). Next, the sample was added to a 2 mL tube containing lysis matrix C (MP Biomedicals, Santa Ana, CA, USA) and homogenized using a Vortex mixer. After removing the large debris, the supernatants were filtered through 0.45 µM filters (Merck Millipore, Billerica, MA, USA) to eliminate eukaryotic and bacterial cell particles. The resulting filtrates were placed in 1.5 mL screw cap tubes and centrifuged at 32,000 rpm for 5 minutes using a Beckman Coulter Optima LE-80 ultracentrifuge with a Heraeus Maximum rotor. This step allowed the viral particles to sediment, while the supernatant was carefully removed. The pellet (which may not be visible) was resuspended in 250 µL of PBS, and the samples were then ready for nuclease enzyme treatment. The filtrates were treated with DNase (concentration, 20 U/ml; Ambion) and RNase A (concentration, 0.1 mg/ml; Fermentas) at 37°C for 30 min to digest unprotected particle nucleic acids. Phi29 (Φ29) polymerase enzyme was also used for DNA circular amplification.
2.3. Nucleic acid extraction (DNA/RNA)
After sample preparation, viral nucleic acids were extracted using the QIAamp Viral RNA Mini Kit (QIAGEN GmbH, QIAGEN Strasse 1, 40724 Hilden, Germany), which purifies RNA and DNA, and the steps were followed according to the instructions of the manufacturer.
2.4. Preparation of libraries for the illumina platform
Library preparation was performed using the Nextera XT DNA Sample Preparation Kit (Illumina Inc.), following the manufacturer's guidelines. The Agilent 2100 Bioanalyzer system and the KAPA kit were used to perform library quantification. For sequencing, samples were pooled (5 organ samples per pool). After preparing the libraries, they were sequenced on the Illumina NovaSeq-6000 platform to provide 250 bp (base pairs) paired reads (Illumina).
2.5. Reads trimming and contig classifiction
The raw reads from Illumina sequencing underwent a meticulous pre-processing procedure. Initially, terminal matched sequence records were excised from both ends. Concurrently, low-quality sequences, stemming from reads shorter than 100 base pairs, were excluded [56]. The removal of adapter and primer sequences was performed with precision using VecScreen, a tool based on BLAST (Basic Local Alignment Search Tool, version BLAST 2.14.0), employing default parameters.
Subsequent to the pre-processing steps, bioinformatics analysis ensued following a well-established protocol [57]. Utilizing the bioinformatics pipeline, no reads associated with human, plant, fungal, or bacterial sequences were identified, emphasizing the specificity of the analysis. The resulting contigs underwent comparative analysis using BLASTx and BLASTn to identify similarities to viral proteins and nucleotides, respectively. The GenBank genetic sequence database (http://www.ncbi.nlm.nih.gov, accessed on January 9, 2023) served as the reference for this comparison.
In addition to the comparative analysis of the predicted gene sequences via the BLASTx online program, known for its protein alignment capabilities using DNA sequences, the most promising outcomes from the BLAST searches were meticulously selected. To minimize the potential for random matches, E values (e-value) were defined for each search. Based on the best result, the sequences were chosen for further alignment.
For confirmation and additional classification, reads and/or contigs underwent alignment against a viral protein database (obtained from https://ftp.ncbi.nlm.nih.gov/refseq/release/viral/) using DIAMOND software version 2.1.8 [58]. All sequences generated in this study were deposited in GenBank with the accession numbers OR069303-OR069349.
All sequences included in this study exhibited similarity to reference sequences from GenBank classified in the Totiviridae family. Complete or almost complete genomes were aligned (using MAFFT version 7.520 [59], with further adjustments and editions performed using the Ugene tool kit version 4.6 [60].
2.6. Genome annotation
The RdRpol domains and motifs were predicted using InterProScan (https://www.ebi.ac.uk/interpro/search/sequence, accessed 10 January 2023) and Motif Finder (https://www.genome.jp/tools/motif, accessed January 12, 2023), respectively.
2.7. Genetic distances
The genetic distance and its standard error were calculated using the composite model of maximum likelihood plus gamma correction and bootstrapping with 1000 replications. Distances were calculated using the MEGA X software [61]. To estimate the similarity of the sequences, a paired method was used, implemented in the SDT program [62]. The initial realignment of sequences involved penalizing gaps, a process carried out with the MUSCLE algorithm [63]. Following the calculation of identity scores for each pair of sequences (pairwise scores), the NEIGHBOR component of PHYLIP [64] was employed to construct a tree. This rooted neighbor-joining phylogenetic tree organizes all sequences based on their likely degrees of evolutionary relatedness. The outcomes are illustrated in a graphical interface through a frequency distribution of paired identities, forming an identity matrix.
2.8. Phylogenetic analysis
Phylogenetic trees were constructed using the maximum likelihood approach, and branching support was estimated using a bootstrap test with 1000 iterations using the IQ-Tree tool [65]. The maximum clade credibility tree was obtained using a Bayesian coalescent approach, implemented in the Beast v1.10.4 software [66] (https://github.com/beast-dev/beast-mcmc). We assumed a fixed clock and a constant rate of population size, and assumed that the evolutionary rate of a given site in a gene-sequence alignment was constant during evolution (homotachy). The WAG+I+G evolutionary model were used with the number of gamma categories set to 4. The runs were initiated using a random starting tree and a chain length of 100,000,000, with echo state to screen every 10,000 iterations, log parameters every 10,000 iterations, and a burn-in of 10%. To ensure convergence and an effective sample size (ESS) greater than 200, we examined the results using Tracer v1.7.1 [67] (http://tree.bio.ed.ac.uk/software/tracer).
3. Results
3.1. Contigs quality and BlastX similarities
We successfully retrieved forty-seven contigs from three different pools (F1, F3, and F6), which were identified as totiviruses based on blast searches. The contigs' quality, sequence size, and blast similarities are summarized in Table S2. The general quality of contigs was high due to the large number of reads in the assembly of each contig. BlastX comparative analysis showed that contigs identified in liver samples of Molossus bats had low amino acid identity in the NCBI database (mean 48%), with coverage ranging from 15% to 97% with theirs best-hit reference sequence. Three of the contigs (F1_001 F1_002 and F1_003) represent near full length genome of totiviruses.
3.2. Genome annotation of totiviruses identified in bats
We have discovered three nearly complete genomes of totiviruses in liver samples collected from Molossus bats, specifically F1_001, F1_002 and F1_003. Using a BlastX search, we found that these two sequences share a degree of amino acid similarity with totiviruses previously identified in the helminth Schistocephalus solidus. To further explore this similarity, we compared the genome annotations of all these sequences (Figure 1). It is worth noting that the reference sequences MN803435 and MN803437, identified in Schistocephalus solidus [68] are related, but they have distinct genome maps. MN803437 has an additional open reading frame (orf), and the proteins have different sizes. For instance, the polymerase in MN803437 has 840 amino acids, while in MN803435, it has 953 amino acids. Both F1_001 and F1_003 contain three open reading frames (ORFs), with F1_003 having a notably short hypothetical polymerase sequence (only 486 amino acids). An important characteristic of these sequences is the presence of long non-coding intergenic regions, such as the 538bp region found between the second complete genome F1_002 has two hypothetical proteins, the first has 1456 residues and the second (probably RdRpol) has 1042 residues.
3.3. RNA-dependent RNA-polymerase of totiviruses
Due to the limited similarities between our contigs and the reference sequences, we opted to analyze the motifs of the RdRpol region of totiviruses. To achieve this, we utilized the cognate viruses identified through the Blast search, as well as additional contigs found in Molossus bats (Figure 2). To illustrate our findings, we have included the motifs of RdRpol from three contigs that belong to different groups of totiviruses (as determined by our phylogenetic analysis). In addition to these motifs, we have also shown the motifs of their respective best-hits. It is important to note that even though the RdRpol sequences identified in this study are highly divergent (more than 50%), all of the motifs of the polymerase are present. Typically, cognate sequences share the same pattern, with one exception being the sequence F1_001, which possesses an additional motif (indicated by an arrow in Figure 2a) when compared to its best-hit reference QJD26160, identified in Schistocephalus solidus.
3.4. Phylogenetic tree of RdRpol of totiviruses
Phylogenetic inference was performed using amino acid sequences of the RdRpol region to classify the sequences generated in this study. To this end, we utilized selected 53 reference sequences from the Totiviridae family. The resulting phylogenetic tree (Figure 3) was generated using a Coalescent Bayesian approach. The tree accurately depicts the major genera of totiviruses, including Giardiavirus, Trichomonavirus, Leishmaniairus, Victorivirus, and Totivirus. Furthermore, the recently proposed genera Artivirus, Pistolvirus and Tricladivirus are also identified in this tree. The sequences from this study were grouped into two distinct phylogenetic clades in the tree (indicated in blue color in the tree of Figure 3). Most sequences (i.e., F1_001, F1_006, F1_032, F1_066, F1_046, F1_052, F1_010, F1_012, F1_051, F1_042, F1_079, F1_045, F1_058, F1_043, F1_044, F1_036, F1_003, F1_062, F1_021, F1_007, F1_048, F1_081, F1_064, F1_008 and F1_002) clustered in a clade (here named Platyhelminthes) that includes the sequences QJD26156 and QJD26160 identified previously in the platyhelminthes Schistocephalus solidus in the United States in 2018. The sequences F1_071, F1_047, F1_067 and F1_080 form a cluster (named Insecta) with the reference UHR49805 that was identified in the insect Eysarcoris guttigerus in China in 2017.
3.5. RdRpol amino acid identity
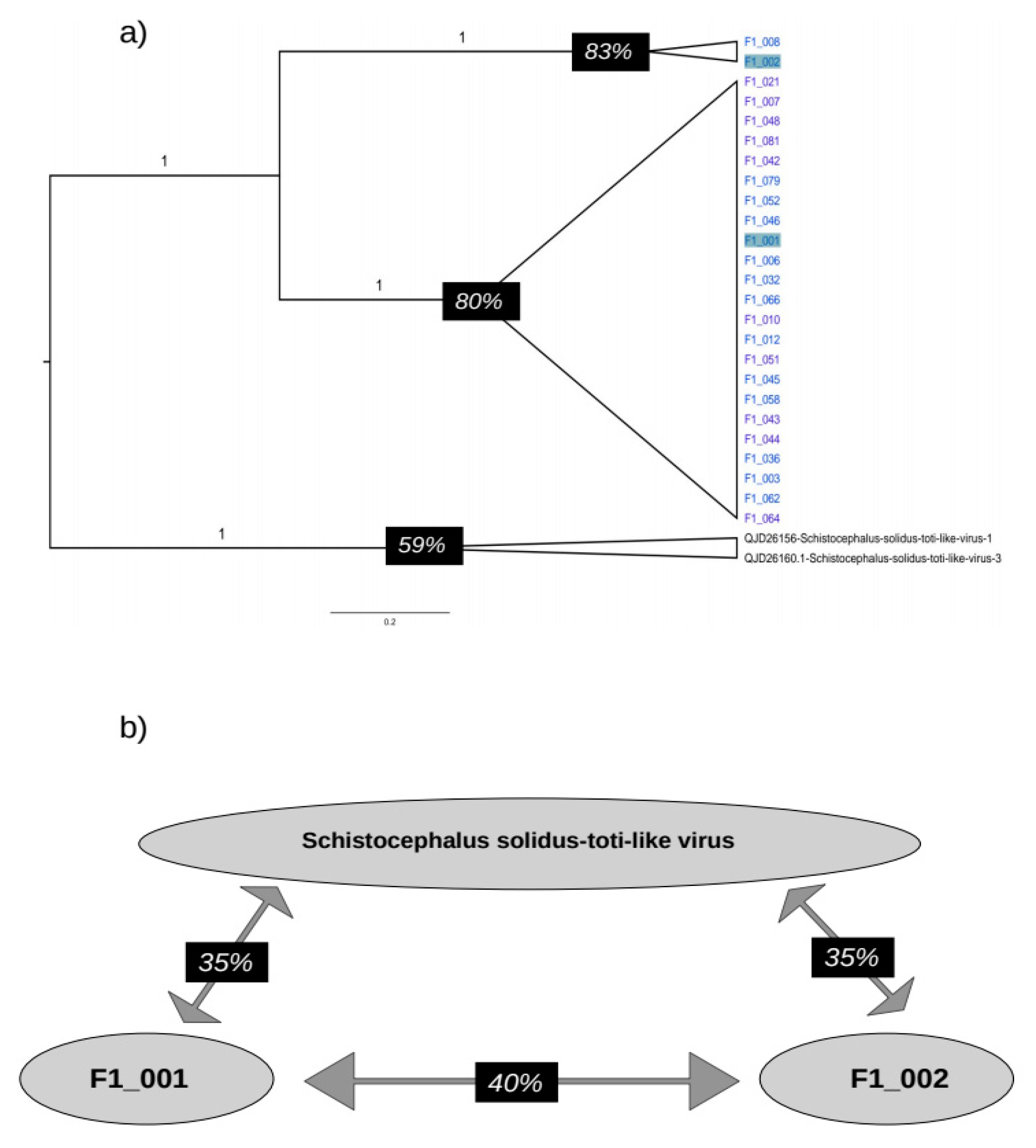
Our phylogenetic analysis revealed that the sequences in the Platyhelminthes clade generated in this study could be categorized into two subclades. To further examine the level of identity of the RdRpol of these sequences, we created a subtree containing the Platyhelminthes clade and used it to illustrate the level of identity (Figure 4). Amino acid identity between F1_008 and its sister sequence F1_002 was found to be 83%. The mean identity among sequences in the clade composed of F1_001, F1_006, F1_032, F1_066, F1_046, F1_052, F1_010, F1_012, F1_051, F1_042, F1_079, F1_045, F1_058, F1_043, F1_044, F1_036, F1_003, F1_062, F1_021, F1_007, F1_048, F1_081, and F1_064 is 80%. In contrast, the identity between the sequences identified in Schistocephalus solidus is only 59%. On the other hand, the mean identity of sequences identified in Schistocephalus solidus with F1_001 and F1_002 is 35%. The identity between F1_001 and F2_002 is 40%. Identity of all pairs of sequences generated in this study is shown in Figure S1 (supplementary material).
4. Discussion
Viral surveillance in bats has been conducted extensively in southern Brazil, but few studies have explored bat viruses in other regions. Previous investigations in Brazil have identified Flavivirus, Coronavirus, Arenavirus, Paramyxovirus, Adenovirus, Papillomavirus and Parvovirus in the bats Molossus molossus, Artibeus lituratus and Sturnira lilium [7,9,14,16,20,28,29,69,70].
The emergence of next-generation sequencing technologies has allowed for the detection of many viruses in bats [6,25,26,27,71]. There are few reports of totiviruses in bats [27,32]. Recently, two contigs (2059bp and 627bp) of a totivirus-like virus were identified in the insectivorous bat Nyctalus noctula during a study on bat carcasses [31]. Similarly, short reads of totivirus were found in the feces of Molossus molossus bats in French Guiana in another study [30]. It is important to note that in metagenomics studies, it is not possible to determine the exact viral host due to the nature of next-generation sequencing approaches. This is also true for totiviruses identified in bats; their host can be ecto- or endoparasites, or even the food source consumed by these animals. The only totivirus-like virus that has been isolated in insect cells, named Tianjin totivirus, was detected in the guano of Myotis bats [32]. It is believed that this virus is likely to infect insects that are consumed by insectivorous bats.
We discovered 47 contigs in liver samples of Molossus bats that exhibit varying degrees of amino acid similarity with totivirus-like sequences previously detected in diverse hosts. Our phylogenetic analysis revealed twenty-five RdRpol sequences that are closely linked to the platyhelminthes Schistocephalus solidus found in the United States in 2018. Additionally, we identified four more RdRpol sequences associated with a totivirus reference discovered in the insect Eysarcoris guttigerus in China in 2017. It is crucial to highlight that the amino acid identity of RdRpol between our sequences and the totivirus-like reference is below 50%, indicating significant genetic divergence between these viruses. Notably, the Tianjin totivirus, isolated from guano bats, exhibits less than 18% amino acid identity in the RdRpol region compared to the totivirus-like sequences identified in Molossus bats. This observation strongly suggests that these viruses are ancient and possess host-specific characteristics.
In this study, we conducted next-generation sequencing to identify totivirus-like viruses in liver tissue obtained from Molossus molossus bats residing in the Amazon region of Brazil. Through comparative phylogenetic analysis of the RNA-dependent RNA polymerase region, we discovered that the viruses found in Molossus bats belong to two distinct phylogenetic clades, potentially representing different genera within the Totiviridae family. Viruses identified in Molossus molossus probably replicate in an organism that infects the liver of these bats.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Details of biological samples; Table S2: Quality of reads; Figure S1: Identity matrix of RdRpol of totiviruses.
Author Contributions
Conceptualization, W.U.A, L.R.R.R, L.F.M, E.L and A.C.d.C.; methodology, R.d.S.C, E.d.S.F.R, W.U.A, L.R.R.R, L.F.M, V.d.S.M, F.V, X.D, E.D, E.L and A.C.d.C.; investigation, R.d.S.C, E.d.S.F.R, W.U.A, L.R.R.R, L.F.M, V.d.S.M, F.V, R.P.P, X.D, E.D, E.L and A.C.d.C.; data curation, R.d.S.C, E.d.S.F.R, W.U.A, L.R.R.R, L.F.M, V.d.S.M, F.V, R.P.P, X.D, E.D, E.L and A.C.C.; writing—original draft preparation, E.d.S.F.R, E.L.; writing—review and editing, R.P.P.; supervision, E.L and A.C.d.C X.X.
Funding
E.d.S.F.R and R.d.S.C are supported by a scholarship provided by the Coordination for the Improvement of Higher Education Personnel—Brazil (CAPES).A.C.d.C is supported by a scholarship from HCFMUSP with funds donated by NUBANK under the #HCCOMVIDA scheme.
Acknowledgments
We thank Coordenação Geral de Laboratórios de Saúde Pública do Departamento de Articulação Estratégica da Secretaria de Vigilância em Saúde do Ministério da Saúde (CGLAB/DAEVS/SVS-MS), MP Biomedicals do Brasil, Zymo Research Inc. for the donation of reagents for this project. We thank Luciano Monteiro da Silva and Nilton Costa. We thank the Pró-reitoria de pesquisa e pós-graduação of UFPA for supporting the publication costs.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Brook, C.E.; Dobson, A.P. Bats as ‘Special’ Reservoirs for Emerging Zoonotic Pathogens. Trends in Microbiology 2015, 23, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Moratelli, R.; Calisher, C.H. Bats and Zoonotic Viruses: Can We Confidently Link Bats with Emerging Deadly Viruses? Mem. Inst. Oswaldo Cruz 2015, 110, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Olival, K.J.; Weekley, C.C.; Daszak, P. Are Bats Really “Special” as Viral Reservoirs? What We Know and Need to Know. In Bats and Viruses; Wang, L., Cowled, C., Eds.; Wiley, 2015; pp. 281–294 ISBN 9781118818732.
- Castelo-Branco, D.S.C.M.; Nobre, J.A.; Souza, P.R.H.; Diógenes, E.M.; Guedes, G.M.M.; Mesquita, F.P.; Souza, P.F.N.; Rocha, M.F.G.; Sidrim, J.J.C.; Cordeiro, R.A.; et al. Role of Brazilian Bats in the Epidemiological Cycle of Potentially Zoonotic Pathogens. Microbial Pathogenesis 2023, 177, 106032. [Google Scholar] [CrossRef] [PubMed]
- Poel, W.H.M.V.D.; Lina, P.H.C.; Kramps, J.A. Public Health Awareness of Emerging Zoonotic Viruses of Bats: A European Perspective. Vector-Borne and Zoonotic Diseases 2006, 6, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Van Brussel, K.; Holmes, E.C. Zoonotic Disease and Virome Diversity in Bats. Current Opinion in Virology 2022, 52, 192–202. [Google Scholar] [CrossRef]
- Asano, K.M.; Hora, A.S.; Scheffer, K.C.; Fahl, W.O.; Iamamoto, K.; Mori, E.; Brandão, P.E. Alphacoronavirus in Urban Molossidae and Phyllostomidae Bats, Brazil. Virol J 2016, 13, 110. [Google Scholar] [CrossRef]
- Bittar, C.; Machado, R.R.G.; Comelis, M.T.; Bueno, L.M.; Beguelini, M.R.; Morielle-Versute, E.; Nogueira, M.L.; Rahal, P. Alphacoronavirus Detection in Lungs, Liver, and Intestines of Bats from Brazil. Microb Ecol 2020, 79, 203–212. [Google Scholar] [CrossRef]
- Campos, A.C.A.; Góes, L.G.B.; Moreira-Soto, A.; De Carvalho, C.; Ambar, G.; Sander, A.-L.; Fischer, C.; Ruckert Da Rosa, A.; Cardoso De Oliveira, D.; Kataoka, A.P.G.; et al. Bat Influenza A(HL18NL11) Virus in Fruit Bats, Brazil. Emerg. Infect. Dis. 2019, 25, 333–337. [Google Scholar] [CrossRef]
- Canuti, M.; Eis-Huebinger, A.M.; Deijs, M.; De Vries, M.; Drexler, J.F.; Oppong, S.K.; Müller, M.A.; Klose, S.M.; Wellinghausen, N.; Cottontail, V.M.; et al. Two Novel Parvoviruses in Frugivorous New and Old World Bats. PLoS ONE 2011, 6, e29140. [Google Scholar] [CrossRef]
- Cibulski, S.P.; Teixeira, T.F.; De Sales Lima, F.E.; Do Santos, H.F.; Franco, A.C.; Roehe, P.M. A Novel Anelloviridae Species Detected in Tadarida Brasiliensis Bats: First Sequence of a Chiropteran Anellovirus. Genome Announc 2014, 2, e01028-14. [Google Scholar] [CrossRef] [PubMed]
- Cibulski, S.P.; De Sales Lima, F.E.; Teixeira, T.F.; Varela, A.P.M.; Scheffer, C.M.; Mayer, F.Q.; Witt, A.A.; Roehe, P.M. Detection of Multiple Viruses in Oropharyngeal Samples from Brazilian Free-Tailed Bats (Tadarida Brasiliensis) Using Viral Metagenomics. Arch Virol 2021, 166, 207–212. [Google Scholar] [CrossRef]
- Corman, V.M.; Rasche, A.; Diallo, T.D.; Cottontail, V.M.; Stöcker, A.; Souza, B.F.D.C.D.; Corrêa, J.I.; Carneiro, A.J.B.; Franke, C.R.; Nagy, M.; et al. Highly Diversified Coronaviruses in Neotropical Bats. Journal of General Virology 2013, 94, 1984–1994. [Google Scholar] [CrossRef]
- De Araujo, J.; Thomazelli, L.M.; Henriques, D.A.; Lautenschalager, D.; Ometto, T.; Dutra, L.M.; Aires, C.C.; Favorito, S.; Durigon, E.L. Detection of Hantavirus in Bats from Remaining Rain Forest in São Paulo, Brazil. BMC Res Notes 2012, 5, 690. [Google Scholar] [CrossRef]
- Razafindratsimandresy, R.; Jeanmaire, E.M.; Counor, D.; Vasconcelos, P.F.; Sall, A.A.; Reynes, J.-M. Partial Molecular Characterization of Alphaherpesviruses Isolated from Tropical Bats. Journal of General Virology 2009, 90, 44–47. [Google Scholar] [CrossRef]
- Lima, F.E.D.S.; Cibulski, S.P.; Dos Santos, H.F.; Teixeira, T.F.; Varela, A.P.M.; Roehe, P.M.; Delwart, E.; Franco, A.C. Genomic Characterization of Novel Circular ssDNA Viruses from Insectivorous Bats in Southern Brazil. PLoS ONE 2015, 10, e0118070. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats Host Major Mammalian Paramyxoviruses. Nat Commun 2012, 3, 796. [Google Scholar] [CrossRef]
- Jung, K.; Threlfall, C.G. Urbanisation and Its Effects on Bats—A Global Meta-Analysis.; Voigt, C.C., Kingston, T., Eds.; Springer International Publishing: Cham, 2016; pp. 13–33. [Google Scholar]
- Piló, L.B.; Calux, A.; Scherer, R.; Bernard, E. Bats as Ecosystem Engineers in Iron Ore Caves in the Carajás National Forest, Brazilian Amazonia. PLoS ONE 2023, 18, e0267870. [Google Scholar] [CrossRef] [PubMed]
- Wallau, G.L.; Barbier, E.; Tomazatos, A.; Schmidt-Chanasit, J.; Bernard, E. The Virome of Bats Inhabiting Brazilian Biomes: Knowledge Gaps and Biases towards Zoonotic Viruses. Microbiol Spectr 2023, 11, e04077-22. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the Bat Virome Catalog to Better Understand the Ecological Diversity of Bat Viruses and the Bat Origin of Emerging Infectious Diseases. ISME J 2016, 10, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Olivier, T.; Thébault, E.; Elias, M.; Fontaine, B.; Fontaine, C. Urbanization and Agricultural Intensification Destabilize Animal Communities Differently than Diversity Loss. Nat Commun 2020, 11, 2686. [Google Scholar] [CrossRef] [PubMed]
- Plowright, R.K.; Eby, P.; Hudson, P.J.; Smith, I.L.; Westcott, D.; Bryden, W.L.; Middleton, D.; Reid, P.A.; McFarlane, R.A.; Martin, G.; et al. Ecological Dynamics of Emerging Bat Virus Spillover. Proc. R. Soc. B. 2015, 282, 20142124. [Google Scholar] [CrossRef]
- Luis, A.D.; O’Shea, T.J.; Hayman, D.T.S.; Wood, J.L.N.; Cunningham, A.A.; Gilbert, A.T.; Mills, J.N.; Webb, C.T. Network Analysis of Host–Virus Communities in Bats and Rodents Reveals Determinants of Cross-species Transmission. Ecology Letters 2015, 18, 1153–1162. [Google Scholar] [CrossRef]
- Armero, A.; Li, R.; Bienes, K.M.; Chen, X.; Li, J.; Xu, S.; Chen, Y.; Hughes, A.C.; Berthet, N.; Wong, G. Myotis Fimbriatus Virome, a Window to Virus Diversity and Evolution in the Genus Myotis. Viruses 2022, 14, 1899. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Haskew, A.N.; Gates, J.E.; Huynh, J.; Moore, C.J.; Frieman, M.B. Metagenomic Analysis of the Viromes of Three North American Bat Species: Viral Diversity among Different Bat Species That Share a Common Habitat. J Virol 2010, 84, 13004–13018. [Google Scholar] [CrossRef]
- Hu, D.; Zhu, C.; Wang, Y.; Ai, L.; Yang, L.; Ye, F.; Ding, C.; Chen, J.; He, B.; Zhu, J.; et al. Virome Analysis for Identification of Novel Mammalian Viruses in Bats from Southeast China. Sci Rep 2017, 7, 10917. [Google Scholar] [CrossRef]
- Bueno, L.M.; Rizotto, L.S.; Viana, A.D.O.; Silva, L.M.N.; De Moraes, M.V.D.S.; Benassi, J.C.; Scagion, G.P.; Dorlass, E.G.; Lopes, B.L.T.; Cunha, I.N.; et al. High Genetic Diversity of Alphacoronaviruses in Bat Species (Mammalia: Chiroptera) from the Atlantic Forest in Brazil. Transbounding Emerging Dis 2022, 69. [Google Scholar] [CrossRef]
- Ramos, E.D.S.F.; Abreu, W.U.; Rodrigues, L.R.R.; Marinho, L.F.; Morais, V.D.S.; Villanova, F.; Pandey, R.P.; Araújo, E.L.L.; Deng, X.; Delwart, E.; et al. Novel Chaphamaparvovirus in Insectivorous Molossus Molossus Bats, from the Brazilian Amazon Region. Viruses 2023, 15, 606. [Google Scholar] [CrossRef] [PubMed]
- Salmier, A.; Tirera, S.; De Thoisy, B.; Franc, A.; Darcissac, E.; Donato, D.; Bouchier, C.; Lacoste, V.; Lavergne, A. Virome Analysis of Two Sympatric Bat Species (Desmodus Rotundus and Molossus Molossus) in French Guiana. PLoS ONE 2017, 12, e0186943. [Google Scholar] [CrossRef] [PubMed]
- Kohl, C.; Brinkmann, A.; Radonić, A.; Dabrowski, P.W.; Mühldorfer, K.; Nitsche, A.; Wibbelt, G.; Kurth, A. The Virome of German Bats: Comparing Virus Discovery Approaches. Sci Rep 2021, 11, 7430. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Y.; Ge, X.; Yuan, J.; Shi, Z. A Novel Totivirus-like Virus Isolated from Bat Guano. Arch Virol 2012, 157, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Call, L.; Nayfach, S.; Kyrpides, N.C. Illuminating the Virosphere Through Global Metagenomics. Annu. Rev. Biomed. Data Sci. 2021, 4, 369–391. [Google Scholar] [CrossRef]
- Totiviridae | ICTV. Available online: https://ictv.global/report_9th/dsRNA/Totiviridae (accessed on 23 November 2023).
- Fauver, J.R.; Grubaugh, N.D.; Krajacich, B.J.; Weger-Lucarelli, J.; Lakin, S.M.; Fakoli, L.S.; Bolay, F.K.; Diclaro, J.W.; Dabiré, K.R.; Foy, B.D.; et al. West African Anopheles Gambiae Mosquitoes Harbor a Taxonomically Diverse Virome Including New Insect-Specific Flaviviruses, Mononegaviruses, and Totiviruses. Virology 2016, 498, 288–299. [Google Scholar] [CrossRef]
- Guo, L.; Yang, X.; Wu, W.; Tan, G.; Fang, S.; Zhang, S.; Li, F. Identification and Molecular Characterization of Panax Notoginseng Virus A, Which May Represent an Undescribed Novel Species of the Genus Totivirus, Family Totiviridae. Arch Virol 2016, 161, 731–734. [Google Scholar] [CrossRef]
- Guo, L.; Yang, X.; Wu, W.; Tan, G.; Fang, S.; Zhang, S.; Li, F. Identification and Molecular Characterization of Panax Notoginseng Virus A, Which May Represent an Undescribed Novel Species of the Genus Totivirus, Family Totiviridae. Arch Virol 2016, 161, 731–734. [Google Scholar] [CrossRef]
- Khalifa, M.E.; MacDiarmid, R.M. A Novel Totivirus Naturally Occurring in Two Different Fungal Genera. Front. Microbiol. 2019, 10, 2318. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Du, J.; Wu, Z.; Zhang, W.; Fu, S.; Song, J.; Wang, Q.; He, Y.; Lei, W.; Xu, S.; et al. Identification and Genetic Analysis of a Totivirus Isolated from the Culex Tritaeniorhynchus in Northern China. Arch Microbiol 2020, 202, 807–813. [Google Scholar] [CrossRef]
- Løvoll, M.; Wiik-Nielsen, J.; Grove, S.; Wiik-Nielsen, C.R.; Kristoffersen, A.B.; Faller, R.; Poppe, T.; Jung, J.; Pedamallu, C.S.; Nederbragt, A.J.; et al. A Novel Totivirus and Piscine Reovirus (PRV) in Atlantic Salmon (Salmo Salar) with Cardiomyopathy Syndrome (CMS). Virol J 2010, 7, 309. [Google Scholar] [CrossRef]
- Okamoto, K.; Miyazaki, N.; Larsson, D.S.D.; Kobayashi, D.; Svenda, M.; Mühlig, K.; Maia, F.R.N.C.; Gunn, L.H.; Isawa, H.; Kobayashi, M.; et al. The Infectious Particle of Insect-Borne Totivirus-like Omono River Virus Has Raised Ridges and Lacks Fibre Complexes. Sci Rep 2016, 6, 33170. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; James, D.; Punja, Z.K. Co-Infection by Two Distinct Totivirus-like Double-Stranded RNA Elements in Chalara Elegans (Thielaviopsis Basicola). Virus Research 2005, 109, 71–85. [Google Scholar] [CrossRef]
- Qu, J.; Shi, N.; Yang, G.; Huang, B. Molecular Characterization of a Novel Totivirus Infecting the Basal Fungus Conidiobolus Heterosporus. Arch Virol 2021, 166, 1801–1804. [Google Scholar] [CrossRef] [PubMed]
- Mor, S.K.; Phelps, N.B.D. Molecular Detection of a Novel Totivirus from Golden Shiner (Notemigonus Crysoleucas) Baitfish in the USA. Arch Virol 2016, 161, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.E.W.; Takagi, Y.; Parent, K.N.; Cardone, G.; Nibert, M.L.; Baker, T.S. Three-Dimensional Structure of a Protozoal Double-Stranded RNA Virus That Infects the Enteric Pathogen Giardia Lamblia. J Virol 2015, 89, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- Procházková, M.; Füzik, T.; Grybchuk, D.; Falginella, F.L.; Podešvová, L.; Yurchenko, V.; Vácha, R.; Plevka, P. Capsid Structure of Leishmania RNA Virus 1. J Virol 2021, 95, e01957-20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.J.; Zhong, J.; Shang, H.H.; Pan, X.T.; Zhu, H.J.; Da Gao, B. The Complete Nucleotide Sequence and Genomic Organization of a Novel Victorivirus with Two Non-Overlapping ORFs, Identified in the Plant-Pathogenic Fungus Phomopsis Vexans. Arch Virol 2015, 160, 1805–1809. [Google Scholar] [CrossRef] [PubMed]
- Castón, J.R.; Luque, D.; Trus, B.L.; Rivas, G.; Alfonso, C.; González, J.M.; Carrascosa, J.L.; Annamalai, P.; Ghabrial, S.A. Three-Dimensional Structure and Stoichiometry of Helmintosporium victoriae190S Totivirus. Virology 2006, 347, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.P.; Ghabrial, S.A.; Fichorova, R.N.; Nibert, M.L. Trichomonasvirus: A New Genus of Protozoan Viruses in the Family Totiviridae. Arch Virol 2011, 156, 171–179. [Google Scholar] [CrossRef]
- Fraga, J.; Rojas, L.; Sariego, I.; Fernández-Calienes, A. Genetic Characterization of Three Cuban Trichomonas Vaginalis Virus. Phylogeny of Totiviridae Family. Infection, Genetics and Evolution 2012, 12, 113–120. [Google Scholar] [CrossRef]
- Wang, H.; De Matos Filipe, D.; Okamoto, K. A Full-Length Infectious cDNA Clone of a dsRNA Totivirus-like Virus. Virology 2022, 576, 127–133. [Google Scholar] [CrossRef]
- Burrows, J.T.A.; Depierreux, D.; Nibert, M.L.; Pearson, B.J. A Novel Taxon of Monosegmented Double-Stranded RNA Viruses Endemic to Triclad Flatworms. J Virol 2020, 94, e00623-20. [Google Scholar] [CrossRef]
- Sandlund, L.; Mor, S.K.; Singh, V.K.; Padhi, S.K.; Phelps, N.B.D.; Nylund, S.; Mikalsen, A.B. Comparative Molecular Characterization of Novel and Known Piscine Toti-Like Viruses. Viruses 2021, 13, 1063. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Xu, L.; Bowers, H.; Schott, E.J. Characterization of Two Novel Toti-Like Viruses Co-Infecting the Atlantic Blue Crab, Callinectes Sapidus, in Its Northern Range of the United States. Front. Microbiol. 2022, 13, 855750. [Google Scholar] [CrossRef]
- Zhai, Y.; Attoui, H.; Mohd Jaafar, F.; Wang, H. -q.; Cao, Y. -x.; Fan, S. -p.; Sun, Y. -x.; Liu, L. -d.; Mertens, P.P.C.; Meng, W. -s.; et al. Isolation and Full-Length Sequence Analysis of Armigeres Subalbatus Totivirus, the First Totivirus Isolate from Mosquitoes Representing a Proposed Novel Genus (Artivirus) of the Family Totiviridae. Journal of General Virology 2010, 91, 2836–2845. [CrossRef]
- Sassa, Y.; Ono, S.; Takata, M.; Koyama, S.; Furuya, T.; Satoh, T.; Ohmatsu, T.; Nagai, M.; Sakai, C.; Mizutani, T.; et al. Identification, Characterization and Full-Length Sequence Analysis of a Novel dsRNA Virus Isolated from the Arboreal Ant Camponotus Yamaokai. Journal of General Virology 2015, 96, 1930–1937. [Google Scholar] [CrossRef]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An Ensemble Strategy That Significantly Improves de Novo Assembly of Microbial Genomes from Metagenomic Next-Generation Sequencing Data. Nucleic Acids Research 2015, 43, e46–e46. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. the UGENE team Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Molecular Biology and Evolution 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Research 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary Trees from DNA Sequences: A Maximum Likelihood Approach. J Mol Evol 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Molecular Biology and Evolution 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Systematic Biology 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Rosario, K.; Lucas, P.; Dheilly, N.M. Characterization of Viruses in a Tapeworm: Phylogenetic Position, Vertical Transmission, and Transmission to the Parasitized Host. ISME J 2020, 14, 1755–1767. [Google Scholar] [CrossRef]
- Lima, F.E.D.S.; Cibulski, S.P.; Elesbao, F.; Carnieli Junior, P.; Batista, H.B.D.C.R.; Roehe, P.M.; Franco, A.C. First Detection of Adenovirus in the Vampire Bat (Desmodus Rotundus) in Brazil. Virus Genes 2013, 47, 378–381. [Google Scholar] [CrossRef]
- Góes, L.G.B.; Campos, A.C.D.A.; Carvalho, C.D.; Ambar, G.; Queiroz, L.H.; Cruz-Neto, A.P.; Munir, M.; Durigon, E.L. Genetic Diversity of Bats Coronaviruses in the Atlantic Forest Hotspot Biome, Brazil. Infection, Genetics and Evolution 2016, 44, 510–513. [Google Scholar] [CrossRef]
- Aguilar Pierlé, S.; Zamora, G.; Ossa, G.; Gaggero, A.; Barriga, G.P. The Myotis Chiloensis Guano Virome: Viral Nucleic Acid Enrichments for High-Resolution Virome Elucidation and Full Alphacoronavirus Genome Assembly. Viruses 2022, 14, 202. [Google Scholar] [CrossRef]
Figure 1.
Genome map of totiviruses. Horizontal diagrams represent the location of hypothetical open reading frames (ORF) in the genome of totiviruses. Upper panels indicate the ORFs (cyan bars) detected in the genomes of sequences MN803435 and MN803437, identified in Schistocephalus solidus. Lower panels show the ORFs (orange bars) detected in the genomes of F1_001, F1_002 and F1_003 identified in Molossus bats.
Figure 1.
Genome map of totiviruses. Horizontal diagrams represent the location of hypothetical open reading frames (ORF) in the genome of totiviruses. Upper panels indicate the ORFs (cyan bars) detected in the genomes of sequences MN803435 and MN803437, identified in Schistocephalus solidus. Lower panels show the ORFs (orange bars) detected in the genomes of F1_001, F1_002 and F1_003 identified in Molossus bats.
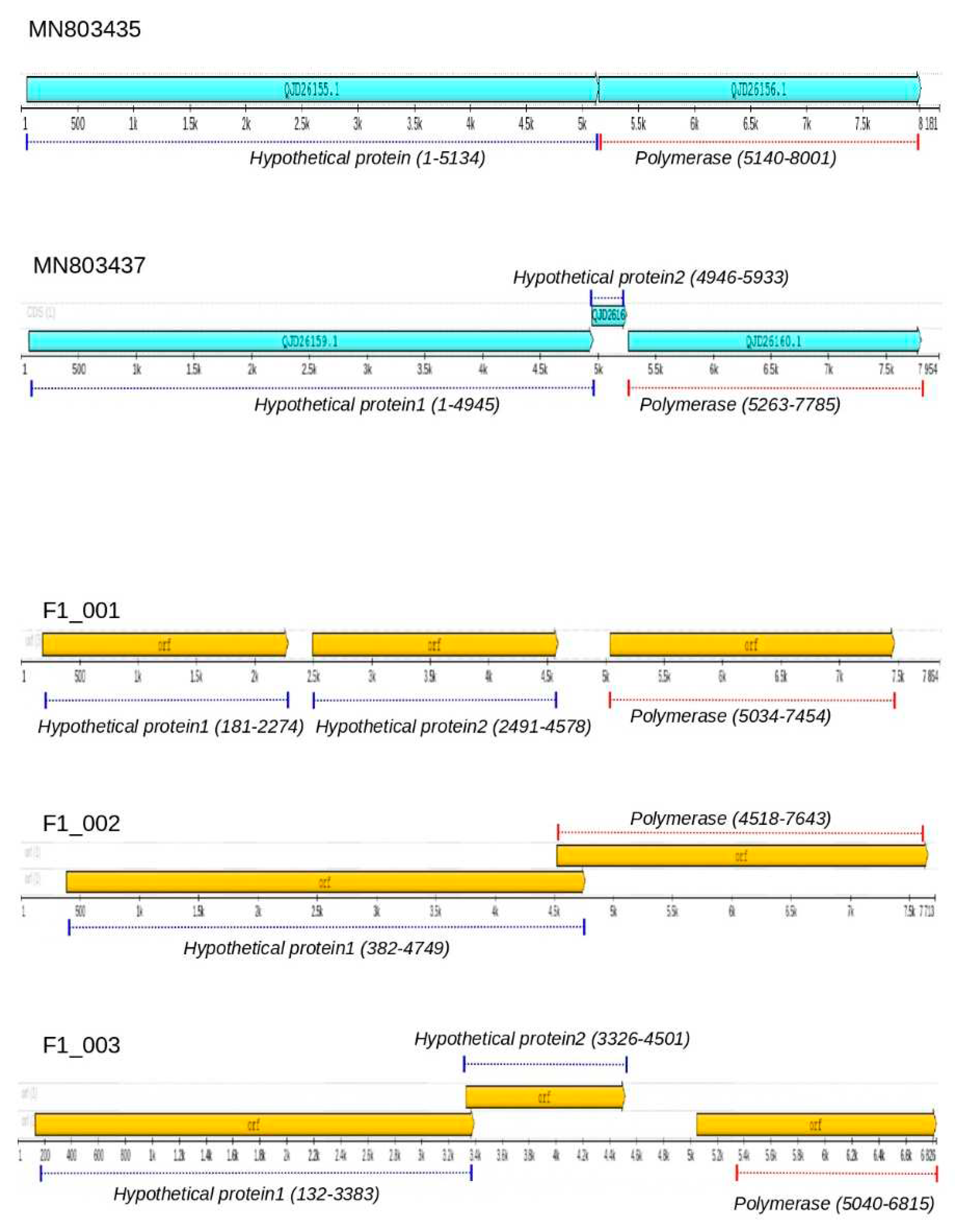
Figure 2.
RdRpol profiles of totiviruses. The translated polymerase sequence of totiviruses is represented by horizontal bars that show the location of RdRpol motifs. The polymerase of F1_001 and F1_002 were compared to their best-hit reference QJD26160 (identified in Schistocephalus solidus), and the identified motifs are shown. In addition, F1_001 has an extra motif that is highlighted in blue and indicated by an arrow in this figure. In sequence F1_047, the motifs are compared to its cognate best-hit reference UHR49805, which was identified in the herbivorous insect Eysarcoris guttigerus that feeds on plants.
Figure 2.
RdRpol profiles of totiviruses. The translated polymerase sequence of totiviruses is represented by horizontal bars that show the location of RdRpol motifs. The polymerase of F1_001 and F1_002 were compared to their best-hit reference QJD26160 (identified in Schistocephalus solidus), and the identified motifs are shown. In addition, F1_001 has an extra motif that is highlighted in blue and indicated by an arrow in this figure. In sequence F1_047, the motifs are compared to its cognate best-hit reference UHR49805, which was identified in the herbivorous insect Eysarcoris guttigerus that feeds on plants.

Figure 3.
Phylogenetic tree of RdRpol of totiviruses. The translated RdRpol sequences were used to infer the tree, and branch support is based on bootstrap test, which is indicated by colors according to the scale in the upper part of the figure. Clades with high branch support are represented by triangles in the tree. The horizontal bar shows the scale of the tree in substitutions per site. Sequences identified in this study are indicated in blue color in the tree. The names above the branches indicate the main totivirus genera as per the International Committee on Taxonomy of Viruses (ICTV). Names inside parenthesis, such as Platyhelminthes and Insecta are not genera; rather, they represent clades identified in this study. Colored triangles represent different genera. Numbers indicate the amino acid identity among sequences.
Figure 3.
Phylogenetic tree of RdRpol of totiviruses. The translated RdRpol sequences were used to infer the tree, and branch support is based on bootstrap test, which is indicated by colors according to the scale in the upper part of the figure. Clades with high branch support are represented by triangles in the tree. The horizontal bar shows the scale of the tree in substitutions per site. Sequences identified in this study are indicated in blue color in the tree. The names above the branches indicate the main totivirus genera as per the International Committee on Taxonomy of Viruses (ICTV). Names inside parenthesis, such as Platyhelminthes and Insecta are not genera; rather, they represent clades identified in this study. Colored triangles represent different genera. Numbers indicate the amino acid identity among sequences.
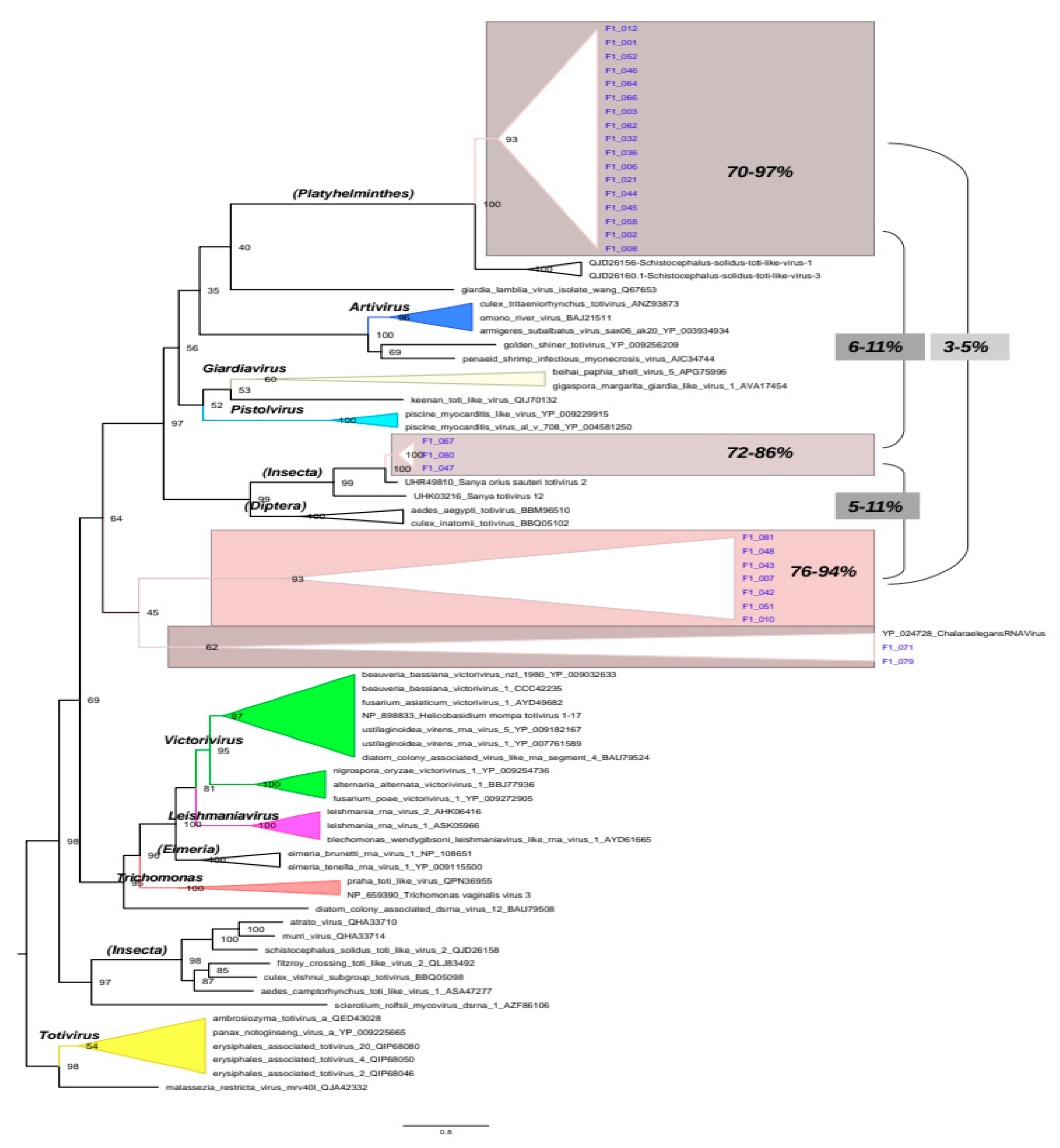
Figure 4.
Amino acid identity in the RdRpol of totiviruses. a) Subtree depicting the phylogenetic clade Platyhelminthes. Values above branches indicate the posterior probability. The identity within each clade is indicated by black rectangles in the tree. b) Identity of sequences identified in Schistocephalus solidus with F1_001 and F1_002.
Figure 4.
Amino acid identity in the RdRpol of totiviruses. a) Subtree depicting the phylogenetic clade Platyhelminthes. Values above branches indicate the posterior probability. The identity within each clade is indicated by black rectangles in the tree. b) Identity of sequences identified in Schistocephalus solidus with F1_001 and F1_002.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



