
Submitted:
30 November 2023
Posted:
01 December 2023
You are already at the latest version
Abstract
.Cellular senescence has emerged as an important driver of aging and age-related disease in the kidney. The activity of β-galactosidase at pH 6 (SA-β-Gal) is a classic maker of senescence in cellular biology. However the predictive role of kidney tissue SA-β-Gal on eGFR loss in chronic kidney disease (CKD) is still not understood. We retrospectively studied the expression of SA-β-Gal in kidney biopsies obtained in a cohort [n=29] of incident patients who were followed up for 3 years as for standard of care. SA-β-Gal staining was approximately 4-fold higher in the tubular compartment of patients with CKD vs. controls [26.0 ± 9 vs. 7.4 ± 6% positive tubuli in patients vs. controls; p < 0.025]. Tubular expressions of SA-β-Gal, but not proteinuria at the time of biopsy correlated with eGFR loss at follow-up; moreover, SA-β-Gal expression in more than 30% of kidney tubules was associated with fast progressive kidney disease. In summary, our study shows that SA-β-Gal is upregulated in the kidney tubular compartment of adult patients affected by CKD, and suggests that tubular SA-β-Gal is associated with accelerated loss of renal function.
Keywords:
senescence
; cell
; chronic kidney disease
; kidney tubules
1. Introduction
Cellular senescence has emerged as an important driver of aging and age-related disease in the kidney [1,2,3]. The term “cellular senescence” has been used to describe an irreversible growth arrest associated with functional and morphological changes, including chromatin organization, gene transcription, and protein secretion [1,2]. In vitro senescent cells have several peculiar characteristics, such as increased cell size and enzymatic activity of the lysosomal hydrolase senescence-associated β-galactosidase [SA-β-Gal] [4], upregulation of the cyclin-dependent kinase inhibitors p16INKA and/or p21cip1 and prosurvival pathways to resist apoptosis [1,2]. In addition, senescent cells may develop a senescence-associated secretory phenotype [SASP], a distinctive secretome consisting of various proinflammatory molecules, metalloproteases and growth factors [4,5]. Cell senescence is induced both as a physiological response during development or in response to multiple stressors including genotoxic injury, hypoxia, cellular stress, oncogene activation, mitochondrial dysfunction or nutrient deprivation (3). The consequences of accelerated cell senescence in kidney disease are twofold. First, senescence is thought to cause a loss of tissue-repair capacity because of cell cycle arrest; this is especially relevant to cells with low replication rates. Second, senescent cells produce proinflammatory and matrix-degrading molecules in the SASP to accelerate glomerulosclerosis and interstitial fibrosis [5,6]. As a matter of fact, different experimental models of CKD identified that accumulation of senescent cells arrested later the G2/M phase triggers the SASP phenotype [7]. This appears to be more pronounced in tubular epithelial cells and affects the tubulointerstitial space [8]. The definition of cell senescence in the kidney has practical implications in view of the new “senolytic” therapies which, by eliminating accumulated senescent cells, could potentially recover tissue function [3,9]. However, it is currently complicated to define unique markers which permit both the identification and quantification of senescent cells [9]. Available studies are based on the presence/absence of cell-cycle arrest markers [3,9]; however this may not be sufficient, as the increase in nuclei positively immunostaining for CDK4/6 inhibitors such as p16INKA does not always coincide with the upregulation of p16INKA expression in vivo [6,10,11]. Even if there are many potential markers of senescence, their specificity is context specific, and is likely influenced by variety of factors including cell type and organism developmental stage [11]; for this reason it has been proposed that the determination of tissue senescence needs the inclusion three approaches: staining of cell-cycle arrest markers (SA-β-Gal or lypofuscin), quantifying the effects of senescence-associated protein release, and excluding coexisting cellular proliferation [11]. Recently, in addition to the study of CDK4/6 inhibitors, other phenotypic markers, among which those of altered mitochondrial dynamics and its secretome, and telomere shortening, have been proposed to identify senescent cells [11,12].
SA-β-Gal is a lysosomal hydrolase, which cleaves terminal β-d-galactose residues [13]. The activity of SA-β-Gal is a classic maker of senescence in cellular biology [4]. The regulation of SA-β-Gal activity is independent of DNA synthesis and reflects changes in cell function that accompany senescence [4]. SA-β-Gal detection is easy and relatively fast to perform on freezed samples of kidney [9,14] and has been proposed as a screening test for subsequent use of more complex and complementary techniques [9]. However, SA-β-Gal is not a completely specific marker of cellular senescence [15]. In patients with diabetic nephropathy, we observed that SA-β-Gal staining in the tubular compartment was directly related to the nuclear + cytoplasmic expression of p16INK4A, but not to nuclear p16INK4A. When we examined the SA-β-Gal and p16INK4A coexpression, some tubule cells showed a complete coexpression of these two markers, while others showed an isolated expression of SA-β-Gal or p16INK4A (13). This suggests that the aging phenotypes are differently expressed at the same time in the same tissue. In addition, the predictive role of SA-β-Gal on eGFR loss is still not known, since in studies obtained in small cohorts of patients with glomerular diseases, telomere shortening activity [16] and p16INKA expression but not SA-β-Gal, were related to subsequent eGFR loss [17]. In this study, we tested the hypothesis that elevated SA-β-Gal activity in kidney biopsies is a predictor of subsequent eGFR loss in patients with CKD. With this in mind, we retrospectively studied the expression of SA-β-Gal in kidney biopsies obtained in a cohort of incident kidney patients who were followed up for 3 years as for the standard of care.
2. Materials and Methods
2.1. Tissue Samples
We reviewed renal biopsies obtained at the Department of Internal Medicine, Nephrology Division, Genoa University, and IRCCS AOU San Martino, Genoa, Italy from May 2007 to June 2011, in which SA-β-Gal staining was available. The review included subjects aged 18 years or older, with or without type 2 diabetes, with a urinary protein excretion 150-10000 mg/day, and an eGFR of 25–120 mL/min per 1·73m2. All biopsies were not for research but for clinical reasons. Results obtained for FSGS have been recently published [17]. The study was part of a larger study on cell senescence and the kidney sponsored by the Ministero dell’Università e della Ricerca Scientifica [MIUR] [Progetti Finalizzati per la Ricerca di Base-FIRB 2002] and approved by the Ethical Committee of the Department of Internal Medicine [14,18]. The procedures were in accordance with the Declaration of Helsinki. All kidney biopsy specimens were analyzed by the same renal pathologist [FC]. Estimated GFR was assessed by CKD-EPI formula [18]. One hundred fifteen consecutive kidney biopsy cases in which SA-β-Gal activity was available were assessed for eligibility [Figure 1].
Sixty-six cases with eGFR<25 ml/min or insufficient tissue availability were excluded from further analysis. After initial evaluation 20 cases resulted to be lost at follow up [36 months]. At the end of follow up 22 biopsies were available, including IgA nephropathy [n=5], 7 FSGS [n=5], membranous glomerulonephritis [n=5], minimal change disease [n=1], chronic glomerulonephritis [n=1], hypertensive nephroangiosclerosis [n=2], stage II lupus nephritis [n=1], diabetic kidney disease [n=2]. Normal portions of kidney tissue from nephrectomies for renal carcinoma [n = 13, 8 males and 5 females, 60 ± 4 years] were examined as a control group. Baseline clinical and demographic characteristics of subjects are shown in Table 1.
The average urine protein excretion was 2.7 ±1.0 g/day; urine protein excretion was 150-200 mg/day for 4 cases, 9 subjects presented with nephrotic (> 3 g/day) proteinuria. The mean eGFR was 80 ± 5.7 mL/min 1.73 m2 and eGFR was less than 60 mL/min/1.73 m2 for 11 participants (38%). Routine clinical visits, including clinical labs, proteinuria, and eGFR estimate, were carried out basally, after 1-6 months from biopsy, and then at least annually for 3 years. This timeline represents standard of practice. Twenty-two subjects received single or double RAS blockade, and 16 subjects received statins. Thirteen subjects received steroids/immunosuppressive therapy. Four patients (28% of cases) doubled their serum creatinine and 2 subjects reached ESRD at the end of follow-up. Two patients deceased during follow up.
2.1.2. SA-β-Gal Activity on Cryopreserved Kidney Biopsies
Biopsies were embedded in OCT Compound (Tissue-Tek OCT compound by Sakura, Torrance, CA), flash-freezed in cold isopentane (Merck Group, Milan, Italy) and stored at -80° C. The frozen tissues were sectioned by a -20°C Cryostat (5 µm) and fixed in 0.5% glutaraldehyde (Merck Group) for 10 minutes at room temperature. SA-β-Gal staining was performed as described by Dimri et al. [4,14]: All reagents were purchased by Merck Group, Milan, Italy. Slides were observed at microscope and SA-β-Gal staining was detected at 20× magnification as blue precipitate in tubuli. SA-β-Gal quantification was performed by ImageJ software and expressed as the percentage of SA-β Gal positive tubuli as a function of total numbers of tubuli [13]. In brief, for analysis, the captured images were converted to gray scale and the threshold parameters were adjusted to ensure that only positive tubuli were evaluated [13].
2.1.3. Statistical Analysis
Data are presented as means ± SEM. Parameters not normally distributed were logarithmically transformed for statistical analysis. Comparisons between groups were made by ANOVA. Comparisons of proportions were made using the χ2-test or Fisher’s exact test whenever appropriate. Relationships between parameters were analyzed using simple regression analysis or the Spearman test, as required. Multivariate logistic regression analysis was performed to assess which parameters significantly related in the simple regression analysis were independent predictors of eGFR loss at follow-up. The Statview statistical package [Cary, NC, USA] was used for the analysis. A 2-tailed p value < 0.05 was considered statistically significant.
3. Results
3.1. SA-β-Gal Staining in the Tubular Compartment
The SA-β-Gal signal was predominantly confined to tubular cells, and it was also only weakly detected in glomerular parietal cells, podocytes, and vascular endothelial cells, in accordance with findings previously observed in diabetic kidney disease [13]. Tubular SA-β-Gal staining was approximately 2.6-fold higher in biopsies from patients with CKD patients vs. controls [26 ± 4 vs. 10 ± 6% positive tubuli in patients vs. controls; p < 0.025] [Figure 2A,B].
About 63% of biopsies had >10% of tubuli expressing SA-β-Gal. Mean eGFR was 80 ±5.7 mL/min/1.73 m2 at baseline, 75±5 mL/min/1.73 m2 at year 1, 70±6.1 mL/min/1.73 m2 at year 2, and 69±6.7 mL/min/1.73 m2 at year 3. Mean change in eGFR from baseline to year 3 was −3.54 [95% CI, −0.598 to −6.48] mL/min/1.73 m2/year.
3.2. Clinical Correlates of SA-β-Gal Expression in Kidney Tubules
Table 2 shows the associations between SA-β-Gal expression and clinical findings in subjects with CKD. By univariate linear regression analysis, SA-β-Gal was not related to age, BMI, serum creatinine, eGFR, PAS, PAD, total cholesterol, LDL cholesterol and Tryglicerides. Surprisingly, in this cohort of non-selected patients, SA-β-Gal expression was only as a trend [r=0.10, p= NS] related to logproteinuria at the time of the biopsy.
3.3. Predictors of eGFR Loss at 36 Months
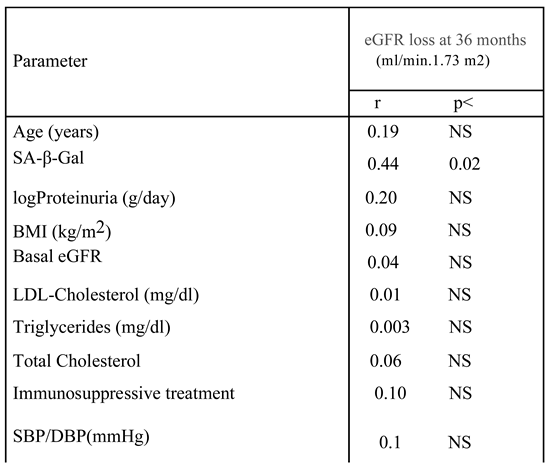
At univariate analysis, eGFR loss at 36 months was predicted by SA-β-Gal expression in kidney tubules, but not by baseline logproteinuria [Table 3, Figure 4 and Figure 5].
No association was observed between the use of immunosuppressive therapy, other clinical parameters as well, and eGFR loss. At visual inspection of the regression line [Figure 4], a tubulat SA-β-Gal percent expression greater than 30% was associated with fast (>3.5 ml/min/year) progression.
4. Discussion
Two issues are addressed in this study, which bear discussion. The first is the overexpression of SA-β-Gal in the tubular compartent of a large percentage of kidney biopsies at the time of diagnosis [ ˜63% of biopsies had >10% of tubuli expressing SA-β-Gal], a finding that suggests that an increase in SA-β-Gal activity in the tubular compartment is an early event in different kidney diseases. The second is the association between the overexpression of SA-β-Gal in the tubular compartment and eGFR loss at the follow-up, a finding which gives strength to the hypothesis that SA-β-Gal activity in kidney biopsies at the time of diagnosis is predictive of subsequent loss of function.
Besides its recognized role as marker of cell senescence [4,13] at high levels SA-β-Gal can damage basement membranes through hydrolyzation of proteoglycans glycosidic bonds [20]. Even as low as 10% of senescent cells in tissues appear to be sufficient to cause damage and dysfunction in studies in vitro and in vivo models [21]. However, no information is available in humans on the percentage of tubular SA-β-Gal expression which is associated to kidney damage. In our study, contrarywise to in vitro studies [21], a low (10-20%) expression of SA-β-Gal in kidney tubules was associated with limited or no eGFR loss at follow up. From the study of the regression line between SA-β-Gal activity and loss of eGFR, a “fast” progression rate. i.e. a eGFR loss > 3.5 ml/min year, was observed when the percentage of SA-β-Gal in tubuli was >30%. Our data therefore suggest that in human kidnay disease the predictive cut-off for tubular SA-β-Gal detection is greater than suggested by in vitro studies. Clearly, our data are preliminary, and ad hoc studies need to be performed in larger studies.
In addition, the association between SA-β-Gal activity in kidney tubules and accelerated disease progression, besides being statistically significant, was not really strong and the increase in SA-β-Gal activity explained only about 20% (r2=0.19) of changes in eGFR 3 years after the biopsy. We have to consider that some of the results reported here may have been the resultant not of “chronic” but of “acute” cell senescence. As a matter of fact, cell senescence may develop during an acute response after injury as a mechanism of tissue repair [21]; this kind of “acute senescence” is a tightly controlled process which participates in repair mechanisms and limits fibrosis. In contrast, in chronic diseases senescent cells accumulate in the kidney in response to a variety of stressors such as metabolic stress, telomere shortening [22] oncogenic mutations, inflammation and mitochondrial dysfunction [23]. These stressors promote cell-cycle arrest via pathways either dependent or independent of the DNA damage response. Differently from the “acute senescence” setting, in chronic diseases senescent cells are scarcely removed by apoptosis or immune clearance, and are increasingly considered to be mediators of disease progression. In a previous study in FSGS patients [17], while a tubular p16INK4A expression was associated with tubule atrophy, tubular SA-β-Gal staining was associated with interstitial fibrosis. These findings raise the possibility that p16INK4A and SA-β-Gal play different roles or, alternatively, are an expression of different mechanisms leading to chronic tubulointerstitial changes. However, considering the not impressive predictive role of SA-β-Gal on disease progression, one could argue that its role as a general marker of senescence [10] needs to be reconsidered.
A pressure-dependent association between increased cellular senescence and hypertension has been demonstrated in hypertensive rats [24]. Angiotensin II, a major player in hypertension and its complications, is increasingly recognized as a mediator of cell senescence [25]; in addition antihypertensive treatment blunts hypertensive-related cell senescence in deoxycorticosterone acetate-salt-treated rats [26]. However, it currently unknown if removal of senescent cells does indeed can treat hypertension or protects from hypertensive organ damage in humans. In our study we were not able to observe an association was between tubular SA-β-Gal expression and blood pressure levels; however almost all subjects were on treatment with RAAS inhibitors, which might have modified the expression or activity of SA-β-Gal. In addition, we could not find an association between age and SA-β-Gal activity. As a matter of fact, we studied an adult population, since younger subjects were not included in the study.
Functional kidney decline results in an accumulation of metabolites interfering with many cellular pathways including cellular senescence [27]. In our study, as also previously observed in a cohort of patients with diabetic nephropathy [14], eGFR was not related to tubular SA-β-Gal, suggesting that renal failure per se is not primary responsible for senescence activation at initial CKD stages. We observed an upregulation of SA-β-Gal activity also in biopsies from subjects with normal or subnormal GFR suggesting that the acceleration of senescence is an early phenomenon and that conditions different from a decreased eGFR may account for the observed changes.
Glucose reabsorption by the proximal tubule has been proposed as one of the mechanisms leading to accelerated cell senescence is in kidney tubules [17,26]. The process of continuous glucose reabsorption by proxymal tubules is now considered one of the mechanisms of tubulointerstitial damage and CKD progression both in diabetic and in non-diabetic disease [28,29]. In previous studies we observed also that human proximal tubule cells exposed to high glucose enter into a senescent, nonreplicative state in which they are metabolically active but do not respond to mitogenic stimuli [17]. Cells are characterized by enhanced expression of senescence markers, including SA-β-Gal, and different sets of genes, including negative regulators of the cell cycle [mainly p16INKA]. Interestingly, these changes can be prevented by pre-treatment with phloryzin, an inhibitor od SGLT action [26, unpublished].
In a previous study [13] in patients with diabetic nephropathy we observed that SA-β-Gal expression in kidney tubules was inversely related to BMI, suggesting a role of metabolic stress on inducing premature cell senescence. However, we could not observe this association in patients with nondiabetic CKD, which suggests an interaction between obesity and diabetic milieu to accelerate cell senescence.
Finally, we could not find an association between proteinuria and SA-β-Gal in tubular cells. A possible explanation is that, besides proteinuria, several other stressors [6,9] may cooperate to increase SA-β-Gal activity in kidney disease.
Our study has several strenghts, since it includes a real life renal biopsy sample cohort from incident patients with quite long follow-up. However, our study has also several limitations. The cohort studied is small, and possibly statistically underpowered to detect all the existing clinical associations of tubular cell senescence. In addition, virtually all patients received RAAS blockers and many were also on treatments with statins. Both drugs are known to posses a “senostatic” action, i.e. they can “downregulate” the senescent phenotype without removing senescent cells [30,31,32].
5. Conclusions
In summary, our study shows that SA-β-Gal is upregulated in the kidney tubular compartment adult patients CKD of different etiologies, and that SA-β-Gal expression >30% in kidney tubuli is associated with accelerated loss of renal function. Therefore, our results reinforce the hypothesis that SA-β-Gal overexpression contributes to accelerated loss of eGFR in CKD. This study provides a clinical basis for the use of SA-β-Gal as a marker of kidney senescence, in CKD, a common clinical condition that is still associated with an unfavorable prognosis. However our results cast some doubt on the use of SA-β-Gal as a unique screening marker of cell senescence in the human kidney.
Author Contributions
Conceptualization, Francesca Viazzi; Formal analysis, Daniela Verzola and Emanuele Parodi; Investigation, Francesca Costigliolo; Resources, Daniela Picciotto and Valentina Zanetti; Writing – original draft, Pasquale Esposito and Giacomo Garibotto; Writing – review & editing, Antonella Sofia, Gabriele Gaggero and Michela Saio.All authors approved the manuscript. G.G. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
Results obtained in this study were originally supported by a grant from the Ministero dell’Università e della Ricerca Scientifica e Tecnologica, Progetti Finalizzati per la Ricerca di Base [FIRB] 2001 entitled “Meccanismi molecolari di genesi e progressione della nefropatia diabetica e dell’invecchiamento del rene” and by grants from the University of Genoa (Finanziamento di Ateneo).
Institutional Review Board Statement
This study was approved by the Institutional Research Ethics Committee of Department of Internal Medicine, University of Genoa, Italy. This study was performed in compliance with the Helsinki Declaration.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The authors are willing to make available individual data on request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Melk A., Schmidt BM., Vongwiwatana A, et al. Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am J Transplant 2005; 6:1375-82.
- Schmitt R., Melk A. Molecular mechanisms of renal aging. Kidney Int 2017; 92:569-579.
- Docherty M.H.; Baird D.P.; Hughes J., et al. Cellular Senescence and Senotherapies in the Kidney: Current Evidence and Future Directions. Front. Pharmacol 2020;11: 755-69.
- Dimri G.,P.; Lee X., Basile G., et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92:9363-67.
- Coppé J.,P.; Patil C.,K.; Rodier F., et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One 2010;5:e9188.
- Childs B.,G.; Durik M.; Baker D.,J. et al. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 2015; 21:1424-35.
- Yang L.; Besschetnova T.,Y.; Brooks C.,R. et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 2010;16:535–543.
- Melk A.; Schmidt B.,M.; Takeuchi O.; Sawitzki B.; Rayner D.,C.; Halloran P.,F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int 2004;65:510–20.
- Docherty M.,H. ; O’ Sullivan E.,D. ; Bonventre J.,V. et al. Cellular Senescence in the Kidney. J Am Soc Nephrol 2019 ;30: 726–736.
- Gorgoulis V.; Adams P.,D.; Alimonti A.; Bennett D.,C.; Bischof O.; Bishop C.; Campisi J.; Collado M.; Evangelou K.; Ferbeyre G.; Gil J,; Hara E.; Krizhanovsky V.; Jurk D,; Maier A.,B. ; Narita M.; Niedernhofer L.; Passos J.,F. ; Robbins P.,D.; Schmitt C.,A. ; Sedivy J. ; Vougas K. ; von Zglinicki T. ; Zhou D.; Serrano M.; Demaria M. Cellular Senescence: Defining a Path Forward. Cell 2019 ;179:813-827. [CrossRef]
- Gurung R.,L.; Yiamunaa M.; Liu S. et al. Short Leukocyte Telomere Length Predicts Albuminuria Progression in Individuals With Type 2 Diabetes. Kidney Int Rep 2018;3:592-601.
- 12. Krishna DR, Sperker B, Fritz P, Klotz U. Does pH 6 β-galactosidase activity indicate cell senescence? Mech Ageing Dev 1999;109:113–123. [CrossRef]
- Verzola D., Gandolfo M,.T.; Gaetani G.; et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol Renal Physiol 2008;295:F1563-73.
- Debacq-Chainiaux F.; Erusalimsky J.,D.; Campisi J.; Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009;4:1798–1806. [CrossRef]
- Liu J.; Yang J.,R.; He Y.,N.; Cai G.,Y.; Zhang J.,G.; Lin L.,R.; et al. Accelerated senescence of renal tubular epithelial cells is associated with disease progression of patients with immunoglobulin A (IgA) nephropathy. Transl Res 2012;159: 454–63.
- Verzola D.; Saio M.; Picciotto D. et al. Cellular Senescence Is Associated with Faster Progression of Focal Segmental Glomerulosclerosis. Am J Nephrol 2021;51:950-58.
- Verzola D.; Gandolfo M.,T.; Ferrario F.; et al. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int 2007;72:1262-72.
- O’Callaghan C.,A.; Shine B.; Lasserson D.,S. Chronic kidney disease: a large-scale population-based study of the effects of introducing the CKD-EPI formula for eGFR reporting. BMJ Open 2011; 1:e000308. 10.1136/bmjopen-2011-000308.
- Li M.;Yang M.; Zhu W.,H. Advances in fluorescent sensors for β-galactosidase. Mater Chem Front 2021;5:763–774.
- 20. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nature Reviews Molecular Cell Biology 2014; 15: 482–496.
- D’Adda di Fagagna F.; Reaper P.,M.; Clay-Farrace L. et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003;426:194-198.
- Miwa S.; Kashyap S.; Chini E.; von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J Clin Invest 2022;132[13]:e158447. [CrossRef]
- Chiu C.,L. Does telomere shortening precede the onset of hypertension in spontaneously hypertensive mice?. Hypertension 2008; 52:123–129.
- Cicalese S.,M.; da Silva J.,F.; Priviero F.,R. et al. Vascular Stress Signaling in Hypertension. Circulation Research 2021;128:969–992.
- Westhoff J.,H.; Hilgers K.,F.; Steinbach M.,P. et al. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension 2008;52:123-9.
- Garibotto G.; Carta A.; Picciotto D.; Viazzi F.; Verzola D. Toll-like receptor-4 signaling mediates inflammation and tissue injury in diabetic nephropathy. J Nephrol 2017;30: 719-727. [CrossRef]
- White W.,E.; Yaqoob M.,M.; Harwood S.,M. Aging and uremia: is there cellular and molecular crossover? World J Nephrol 2015; 4:19–30.
- Rhee J.,J.; Jardine M.,J.; Chertow G.,M.; Mahaffey K.,W. Dedicated kidney disease-focused outcome trials with sodium-glucose cotransporter-2 inhibitors: Lessons from CREDENCE and expectations from DAPA-HF, DAPA-CKD, and EMPA-KIDNEY. Diabetes Obes Metab 2020; (Suppl 1):46-54. [CrossRef]
- Rossing P.; Caramori M.,L.; Chan J.,C.,N.; Heerspink H.,J.,L.; Hurst C.; Khunti K. et al. Executive summary of the KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease: an update based on rapidly emerging new evidence. Kidney Int 2022; 102:990-999. [CrossRef]
- Xu M.; Pirtskhalava T.; Farr J.,N.; Weigand B.,M.; Palmer A.,K.; Weivoda M.,M. et al. Senolytics improve physical function and increase lifespan in old age. Nat Med 2018;24: 1246–56.
- Chan J.; Eide I.,A.;Tannæs T.,M. et al. Marine n-3 Polyunsaturated Fatty Acids and Cellular Senescence Markers in Incident Kidney Transplant Recipients: The Omega-3 Fatty Acids in Renal Transplantation (ORENTRA) Randomized Clinical Trial. Kidney Med 2021;3:1041-49.
- Koppelstaetter C.; Leierer J.; Rudnicki M.; et al. Computational Drug Screening Identifies Compounds Targeting Renal Age-associated Molecular Profiles. Comput Struct Biotechnol J 2019;17:843-853.
Figure 1.
Diagram of the study flow.
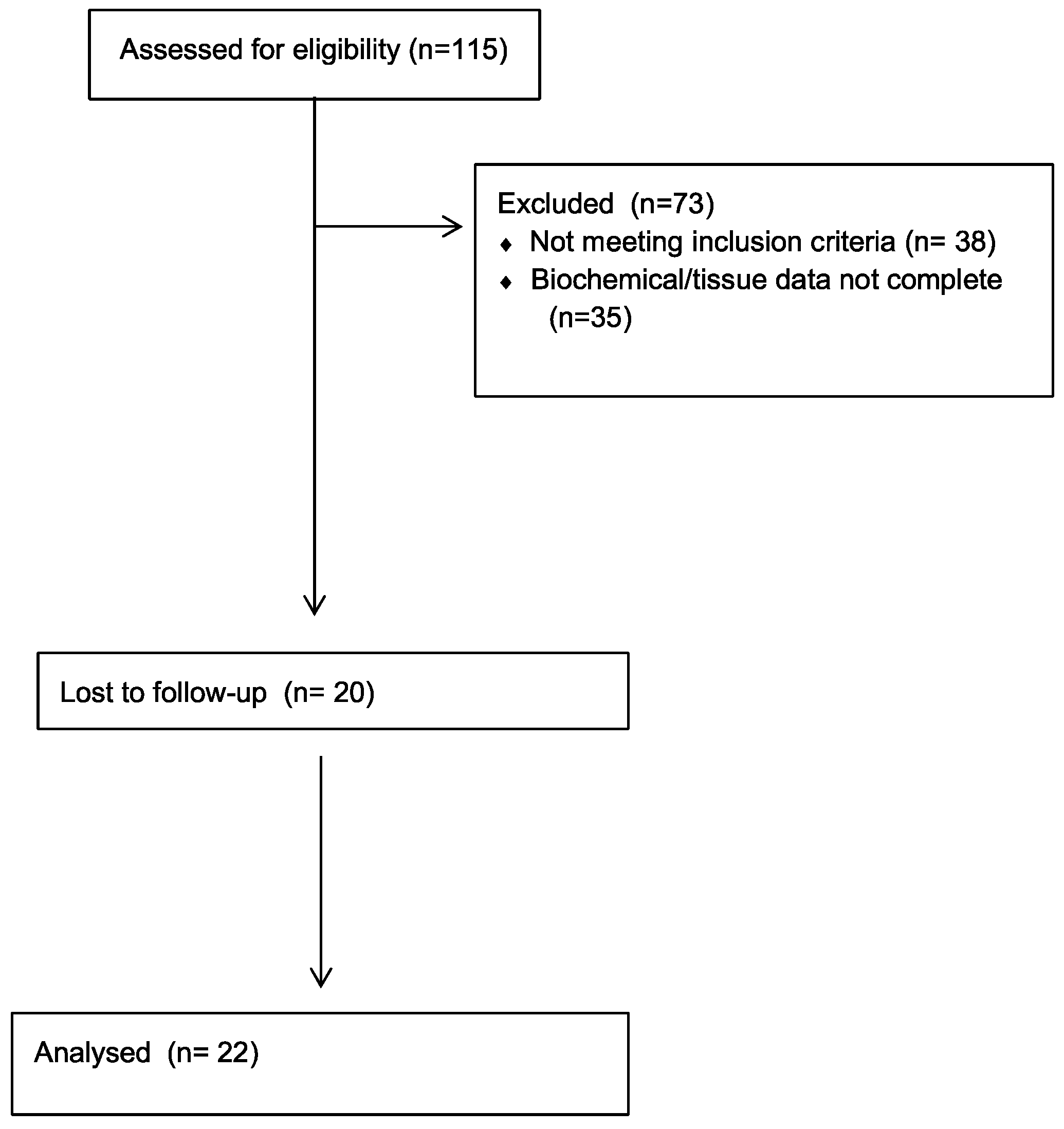
Figure 2.
(A, B). A) representative images of SA-β-Gal tubular expression in CKD patients. SA-β-Gal assay was approximately 2.6-fold higher [p < 0.025] in the tubular compartment of biopsies of CKD patients than in controls. Magnification × 200. SA-β-Gal, senescence-associated β-galactosidase; Scale bar= 150-75 µm. SA-β-Gal, senescence-associated β-galactosidase.
Figure 2.
(A, B). A) representative images of SA-β-Gal tubular expression in CKD patients. SA-β-Gal assay was approximately 2.6-fold higher [p < 0.025] in the tubular compartment of biopsies of CKD patients than in controls. Magnification × 200. SA-β-Gal, senescence-associated β-galactosidase; Scale bar= 150-75 µm. SA-β-Gal, senescence-associated β-galactosidase.
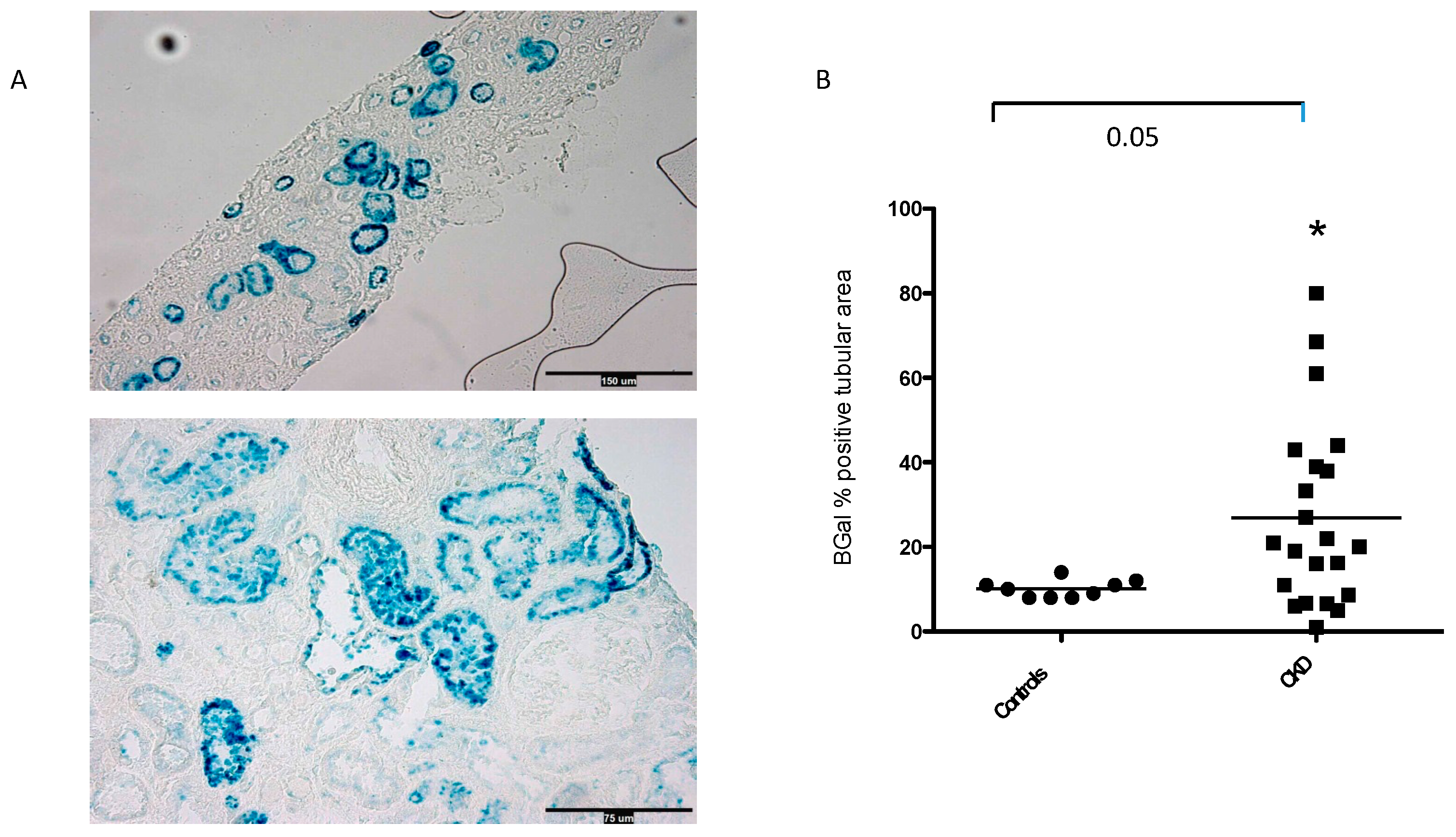
Figure 3.
Estimated GFR (eGFR) during the study (means, 5-95th percentiles). Mean eGFR was 80 ±5.7 mL/min/1.73 m2 at baseline, 75±5 mL/min/1.73 m2 at year 1, 70±6.1 mL/min/1.73 m2 at year 2, and 69±6.7 mL/min/1.73 m2 at year 3. Mean change in eGFR from baseline to year 3 was −3.54 [95% CI, −0.598 to −6.48] mL/min/1.73 m2/year.
Figure 3.
Estimated GFR (eGFR) during the study (means, 5-95th percentiles). Mean eGFR was 80 ±5.7 mL/min/1.73 m2 at baseline, 75±5 mL/min/1.73 m2 at year 1, 70±6.1 mL/min/1.73 m2 at year 2, and 69±6.7 mL/min/1.73 m2 at year 3. Mean change in eGFR from baseline to year 3 was −3.54 [95% CI, −0.598 to −6.48] mL/min/1.73 m2/year.
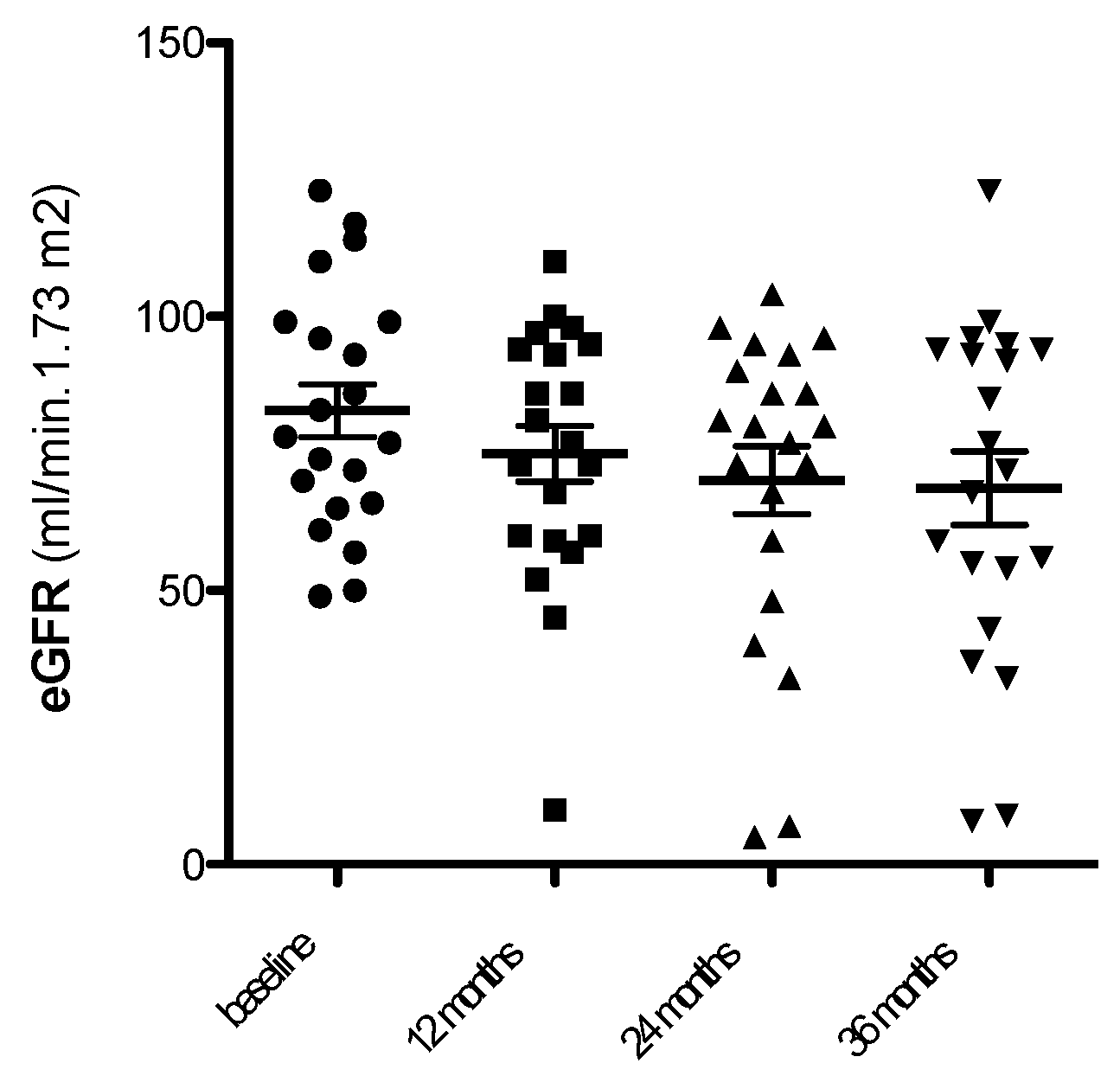
Figure 4.
Relationship between SA-β-Gal expression in kidney tubules and eGFR loss at follow up (3 years).
Figure 4.
Relationship between SA-β-Gal expression in kidney tubules and eGFR loss at follow up (3 years).
Figure 5.
Relationship between logproteinuria and eGFR loss at follow up (3 years).
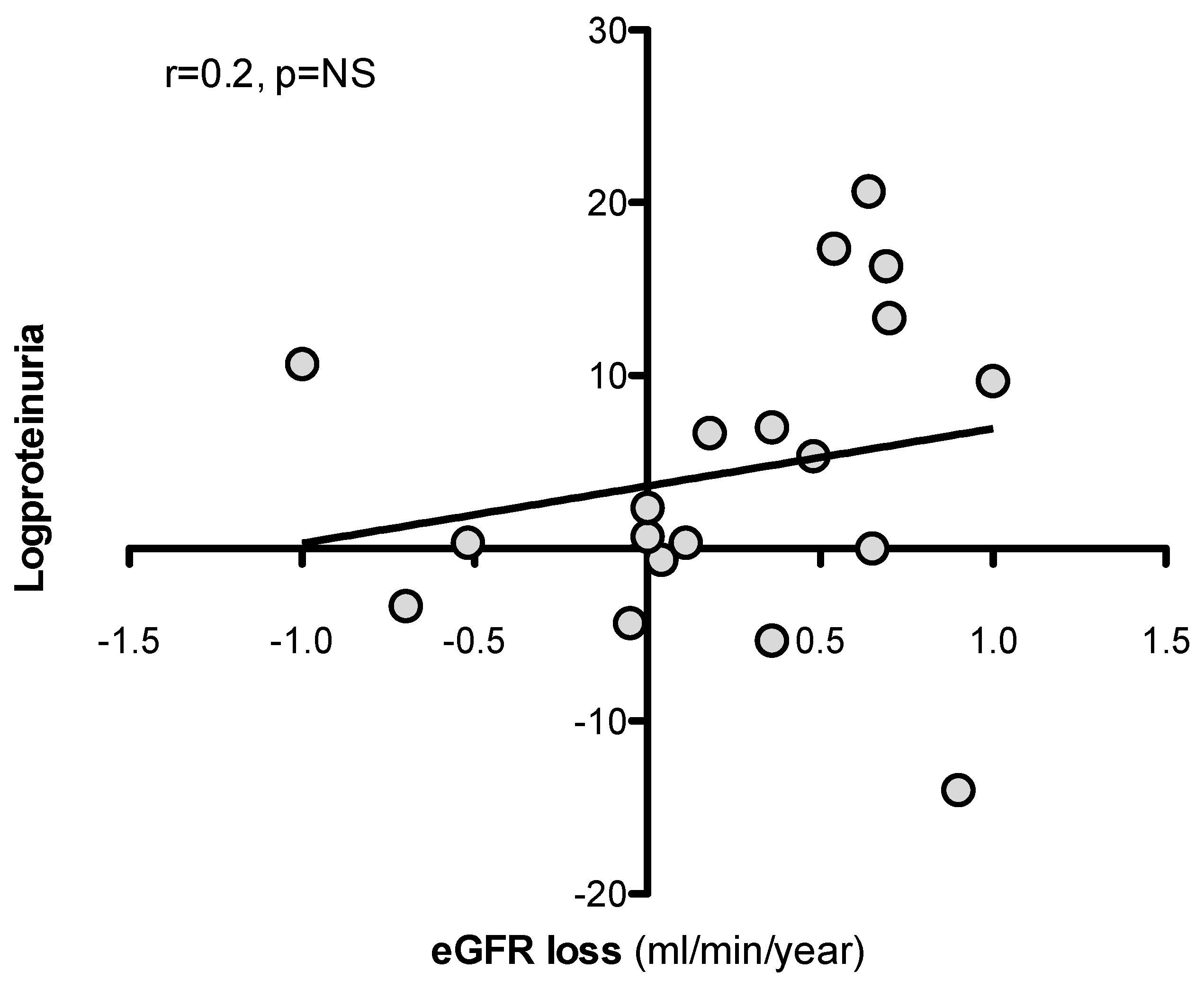

Table 2.
Univariate analysis of the correlation between clinical parameters and Senescence-associated β-galactosidase (SA-β-Gal) expression in kidney tubules (n=22).
Table 2.
Univariate analysis of the correlation between clinical parameters and Senescence-associated β-galactosidase (SA-β-Gal) expression in kidney tubules (n=22).
| Clinical characteristics | SA-β-Gal (% tubular area) | |
|---|---|---|
| r | p | |
| Age (years) | 0.08 | NS |
| Serum creatinine (mg/dl) | 0.05 | NS |
| eGFR (ml/min.1.73 m2) | −0.28 | NS |
| BMI (kg/m2) | 0.1 | NS |
| logProteinuria (g/day) | 0.27 | NS |
| LDL-Cholesterol (mg/dl) | 0.06 | NS |
| Triglycerides (mg/dl) | 0.11 | NS |
| Total Cholesterol | −0.19 | NS |
| SBP (mmHg) | 0.15 | NS |
| DBP(mmHg) | −0.09 | NS |
Abbreviations: eGFR=estimated Glomerular Filtration Rate,BMI= Body Mass Index,SBP= systolic blood pressure; DBP= diastolic blood pressure.
Table 3.
Univariate analysis of the correlation between Senescence-associated β-galactosidase (SA-β-Gal), clinical parameters and loss of eGFR at follow up (3 years) in patients with CKD (n=22).
Table 3.
Univariate analysis of the correlation between Senescence-associated β-galactosidase (SA-β-Gal), clinical parameters and loss of eGFR at follow up (3 years) in patients with CKD (n=22).
 |
Abbreviations: eGFR=estimated Glomerular Filtration Rate,BMI= Body Mass Index,SBP= systolic blood pressure; DBP= diastolic blood pressure.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



