
Submitted:
23 February 2024
Posted:
26 February 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The novel coronavirus disease (COVID-19) pandemic has resulted in over 720 million confirmed cases and 7 million deaths worldwide, with insufficient treatment options. Innumerable efforts are being made around the world for faster identification of therapeutic agents to treat the deadly disease. Postacute sequelae of SARS-CoV-2 infection or COVID-19 (PASC), also called Long COVID, is still being understood and lacks treatment options as well. A growing list of drugs are being suggested by various in silico, in vitro and ex vivo models, however currently only two treatment options are widely used: the RNA-dependent RNA polymerase (RdRp) inhibitor remdesivir, and the main protease inhibitor nirmatrelvir in combination with ritonavir. Computational drug development tools and in silico studies involving molecular docking, molecular dynamics, entropy calculations and pharmacokinetics can be useful to identify new targets to treat COVID-19 and PASC, as shown in this work and our recent paper that identified alendronate as a promising candidate. We have now investigated all bisphosphonates which can bind competitively to nidovirus RdRp-associated nucleotidyl (NiRAN) transferase domain, and systematically down selected seven candidates (CHEMBL608526, CHEMBL196676, CHEMBL164344, CHEMBL4291724, CHEMBL4569308, CHEMBL387132, CHEMBL98211), two of which closely resemble the approved drugs minodronate and zoledronate. This work and our recent paper together provide an in silico mechanistic explanation for alendronate and zoledronate users having dramatically reduced odds of SARS-CoV-2 testing, COVID-19 diagnosis, and COVID-19-related hospitalizations, and indicate that similar observational studies with minodronate could be valuable.
Keywords:
Alendronate
; COVID-19
; Minodronate
; MM-GBSA
; Molecular docking
; Molecular dynamics
; RdRp
; SARS-CoV-2
; Virtual screening
; Zoledronate
1. Introduction
1.1. Objectives of this Work
Coronaviruses are responsible for causing seasonal respiratory tract infections in people and are associated with common cold symptoms [1]. The highly pathogenic human coronaviruses (HCoVs) such as severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV), Middle East respiratory syndrome–related coronavirus (MERS-CoV) and the novel SARS-CoV-2 cause infection to epithelial cells of the bronchi and pneumocytes, which could lead to life-threatening lung injuries [2]. Among these three, the SARS-CoV-2 virus which emerged in December 2019 exhibits faster human-to-human transmission, and resulted in over 780 million confirmed cases and 7 million reported deaths due to the novel coronavirus disease (COVID-19) [3]. Long COVID, also known as postacute sequelae of SARS-CoV-2 infection or COVID-19 (PASC) [4,5], may affect typically 3.1% of the population, especially those aged 35 to 69 years, females, people living in more deprived areas, those working in social care, those aged 16 years or over who were not working and not looking for work, and those with another activity-limiting health condition or disability [6].
With vaccines unable to completely stop the transmission of this disease, so far, the standard drugs of choice for treatment have been the emergency use authorized (EUA) drugs, nirmatrelvir and remdesivir. Nirmatrelvir is an orally available main protease (Mpro) inhibitor [7], while remdesivir is an RdRp inhibitor that is administered parenterally. The EUA status of remdesivir was revoked on April 2022 [8], and approval as a supplemental new drug application has been given in August 2023 [9]. However, a recent study published in The Lancet shows that while remdesivir could reduce the risk it had an insignificant effect in ventilated COVID-19 patients [10]. Nirmatrelvir, a main protease inhibitor, exhibited promising antiviral effects but was susceptible to rapid degradation. To mitigate this issue, ritonavir, a protease inhibitor, was incorporated in a combined formulation which received FDA approval as the first oral antiviral pill PAXLOVID [11]. Other drugs that have shown promise include molnupiravir [12], favipiravir [13] and fluvoxamine [14,15]. However, molnupiravir and favipiravir are no longer recommended as the former drug has poor clinical outcomes [16,17,18] and the latter showed ineffective in viral clearance [19,20,21]. Although fluvoxamine has been shown to have both immunomodulatory effects [22] as well as antiviral effects [14,23,24], it is clear that this drug can only be used in combination and not in its own [14,22].
Therefore, the quest to identify potential molecules to treat COVID-19 and PASC is still ongoing. We developed a systematic approach to catalogue all FDA/TGA approved drugs available in the Compounds Australia collection [25]; down-select 214 likely candidates [26]; evaluate in vitro/ex vivo efficacy of the top 12 leads [14]; and screen the 214 candidates using molecular modeling techniques to identify additional leads [27]. In our latter list is present a bisphosphonate molecule called alendronate, and we came across a parallel study which observed that humans who used alendronate (and another bisphosphonate called zoledronate) had lower odds ratios (typically 0.21-0.29) of testing for SARS-CoV-2 infection, COVID-19 diagnosis and COVID-19-related hospitalization [28]. We also noted a patent in which alendronate in combination with dexamethasone showed increased clearance of SARS-CoV-2 δ-strain by macrophages [29]. While the observational study compared 450,366 users of alendronate and zoledronate with non-users and presented compelling real-world evidence in humans (compared to in vitro/in vivo studies), it neither provided any molecular-level explanation for its observations nor systematically consider all bisphosphonates, and there could be other leads which are even more promising than alendronate or zoledronate warranting future observational studies in humans. This paper aims to address these three points by extending our molecular modeling techniques [27] to all bisphosphonates in a systematic manner.
1.2. Virological Context
SARS-CoV-2 employs a multi-subunit machinery for replication and transcription. Non-structural proteins (Nsp's) produced as cleavage products due to the open reading frame 1a and 1b (ORF1a and ORF1b) facilitate viral replication and transcription [30]. One of these, known as Nsp12 or RNA-dependent RNA polymerase (RdRp), catalyzes the synthesis of viral RNA and plays a central role in the replication and transcription cycle of SARS-CoV-2 with Nsp7 and Nsp8 as co-factors [31,32]. Therefore, Nsp12 is considered a primary target for antiviral agents, with the potential for treating COVID-19 [33], and possibly other coronaviral diseases because it is a highly conserved motif. For example, sequence alignment results from the literature shows 96% common identity between SARS-CoV and SARS-CoV-2 [34].
In this work, as we shall be using RdRp as a key target let us first describe its components: it has the Nsp12 catalytic subunit, two accessory subunits (Nsp8 and Nsp7), and more than two turns of RNA template-product duplex [35]. The RdRp domain is analogous to a cupped right hand, consisting of the finger ‘F’ (amino acid residues 398–581, 628–687), palm (amino acid residues 582–627, 688–815) and thumb (amino acid residues 816–919) subdomains found in all single-subunit polymerases [36]. An in-depth structural analysis depicts the Nsp12 subunit binding to the first turn of RNA between its F and thumb sub-domains. The core protein consists of a single chain of 942 amino acids. The active site comprises of five conserved Nsp12 elements that are found in the palm motif. The amino acids Asp760 and Asp761 (Figure 1) are necessary for synthesis, which binds to the 3′ end of the RNA. The RNA template is positioned by the supplementary Nsp12 finger motif. The second turn is positioned by two copies of Nsp8 that bind to the cleft on the opposite sides. As RNA exits, large helical extensions of Nsp8 protrude and create positively charged sliding poles that are necessary for coronaviruses to replicate their lengthy genomes [37]. Structural stability requires two Zinc (Zn) ions interacting with the residues present in the N-terminal domain (His295, Cys301, Cys306, Cys310) and finger domain (Cys487, His642, Cys645, Cys646). The presence of Zn in this site indicates its crucial role in stabilizing the overall 3D structure of the protein [38]. The binding of drugs to the amino acid residues in motif F of RdRp averts the entry of the substrate and divalent cations into the central active site cavity, thereby inhibiting the catalytic activity of the enzyme and preventing the RNA replication [39].
1.3. Medicinal Chemistry Context
In the literature, when databases of molecules including synthetic and natural origin were screened against RdRp [40,41,42,43,44,45,46,47] together with other non-structural proteins (Nsp's) inhibitors [48,49,50,51], these studies were looking for better alternatives to remdesivir. Few studies also reported the screening of analogues of different scaffolds such as quinolines [47], cytidines [52] and andrographolides [53]. It’s also common for repurposing efforts to propose drugs that are yet to be tested in vitro [27,33,45,46,47,54,55,56,57,58,59,60].
As RdRp is a key target, this paper looks at a class of small molecules called bisphosphonates (BPs) for the following reasons: 1. BP scaffolds exhibit competitive binding to the nidovirus RdRp-associated nucleotidyl (NiRAN) transferase domain [61]; 2. BPs benefit the age groups that also experience severe forms of COVID-19 [62,63,64]; 3. Our prior experience with an immunomodulatory drug, fluvoxamine, in different (in silico, in vitro and ex vivo) studies [14]; 4. Real-world observations have indicated that users of alendronate and another BP, zoledronate, exhibit a 3-5 fold reduction in the incidence of SARS-CoV-2 testing, COVID-19 diagnosis, and related hospitalization during the pandemic [28,65]; 5. In our recent screening of all FDA-approved drugs from the Compounds Australia collection, alendronate emerged as a lead compound, with our in silico studies providing a mechanistic explanation for its potential efficacy via RdRp target [27]. However, our modeling was confined to top-hit compounds with various scaffolds. Thus, we aim to rerun our model on the database of BPs to methodologically identify which other BP drugs might emerge as potential hit molecules.
BPs are a class of small-molecule drugs that have two phosphonate groups. They are categorized into nitrogen-containing (amino-BPs) and nitrogen-free BPs (non-amino-BPs). They are mainly used to treat osteoporosis, Paget's disease of the bone, and to lower high calcium levels in people with cancer [66]. In addition, amino-BPs control the activation, expansion, and function of a significant portion of human γδT cells (i.e., it reduces the amount of circulation γδT cells), as well as neutrophils, monocytes and macrophages. They can also modify the dendritic cell's ability to present antigens to the immune system. Results from animal experiments indicate that both amino-BPs and non-amino-BPs have strong adjuvant-like effects of increasing antibody and T-cell responses to viral antigens. [67]. Such a range of immunomodulatory effects and the drug’s binding with RdRp inspired us to investigate BPs as prospective COVID-19 and/or PASC drug candidates [28,61,67].
2. Results and Discussion
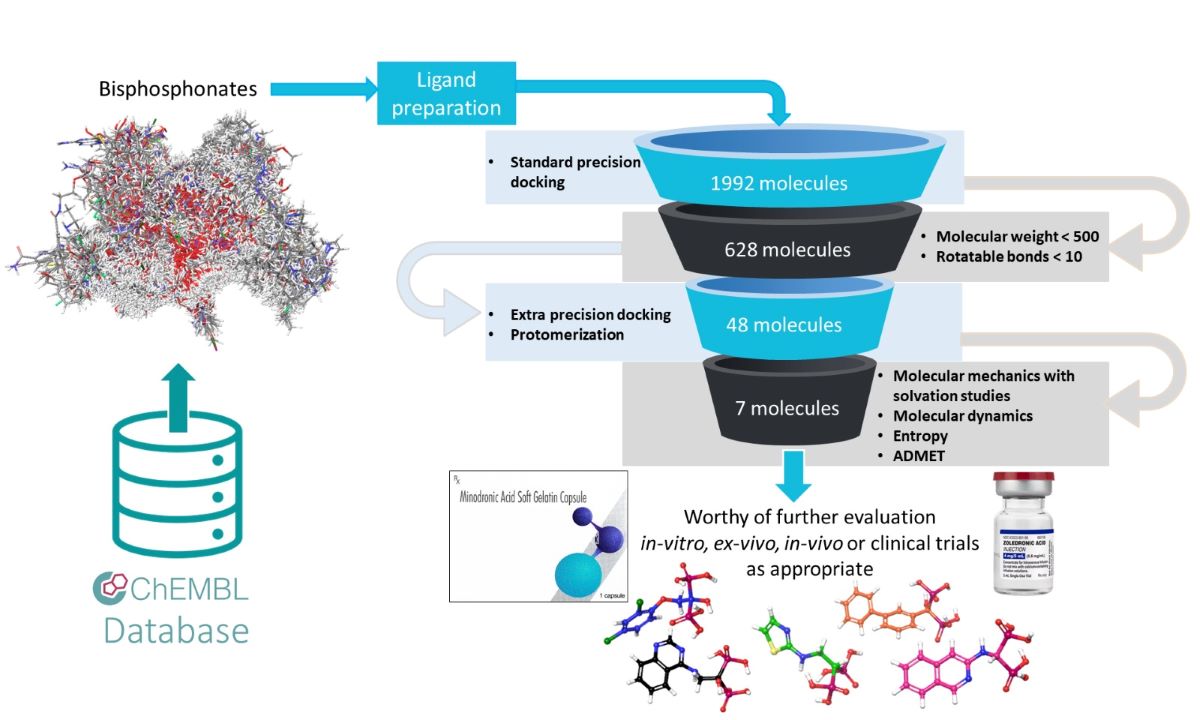
To methodologically identify which BPs are worthy of evaluation against COVID-19 in human observational studies, all the BP molecules (synthetic and approved) obtained from the ChEMBL database were docked into the binding site of the RdRp protein (PDB ID: 6M71) using the ligand docking wizard of the glide module of Schrödinger software. The test molecules were docked using the SP mode and we obtained 1,992 molecules, signifying that all test molecules have occupied the active site pocket. Larger molecules >500 Daltons find it harder to absorb as smaller molecules are more absorbable. Hence, we used molecular weight (<500 Daltons) to filter out large molecules. The number of drugs that passed the filter was 1,398. To further reduce this number, a filter corresponding to the number of rotatable bonds was applied, as the drugs having fewer rotatable bonds are acceptable [68]. With this filter, 628 molecules were obtained, which were further subjected to molecular docking using the extra precision (XP) mode.
All the molecules were binding to the active site with varying glide scores; hence, a cut-off docking score of -9.0 kcal/mol was chosen for down-selection as it is 2-3 times that of comparables (remdesivir -3.27 kcal/mol; favipiravir -3.44 kcal/mol; molnupiravir -4.93 kcal/mol). Concurrently we also enabled protomerization so that the uncharged BPs can be ionized at pH 7.0 ± 2.0. This is because BPs contain two phosphonate groups attached to a carbon or a nitrogen atom that can undergo ionization depending on the environment’s pH. For instance, alendronate, a bone resorption inhibitor used for treating osteoporosis exhibits multi-level ionization for the dissociation of 4-hydroxyl groups present on the phosphorous atom, resulting in a total of 4 dissociation constants at varying pH [69]. Similarly, at a pH ranging from 5 to 9, these BPs may exist as a mixture of protonated or ionized forms. Hence, considering the ionization behaviour at this stage is crucial to evaluate their binding mechanism with the target residues. Therefore, to improve the accuracy and reliability of our docking studies further, and to allow for a comprehensive exploration of ligand’s behavior in different biological environments, protomers at pH 7.0 ± 2.0 were generated using Epik module for the top 14 compounds that met the -9.0 kcal/mol docking score cut-off. This resulted in a total of 48 protomers, using which the XP mode of molecular docking was carried out. The list of molecules obtained after the cut-off and their corresponding best protomers are shown in Table 1.
2.1. MM-GBSA Studies
These top 14 molecules obtained using a cut-off value of <-9.0 kcal/mol from molecular docking studies, and their respective protomers, were subjected to MM-GBSA studies to evaluate the binding free energy of the ligand, and the molecules with the least score when compared to remdesivir were selected. We see that seven candidates which are either uncharged molecules or charged protomers (highlighed in bold font in Table 2 along with their ChEMBL ID’s) have lower binding energy than that of remdesivir (-40.32 kcal/mol). For these 7 molecules with lower energy compared to remdesivir, we further analyzed their interaction pattern as depicted in Figure 2 and Table 3.
2.2. Molecular Dynamics Results
The top ligands with the least binding energy from MM-GBSA studies were analyzed by molecular dynamics studies (Figure 3). The clinically used drug remdesivir was also subjected to dynamics simulations for comparative analysis, and showed a stable RMSD plot for the entire duration (5.0 Å-7.0 Å from the initial 5 ns until the end). Additionally, the fluctuations in the active site region of its corresponding protein were low, resulting in a stable RMSD and RMSF plots of the protein (Figure 3 and Figure 4). Significant interactions were observed by the residues of the palm domain that are necessary to bind with the RNA. The residual interactions include Asp618 (63%, water-mediated), Asp623 (70%, water-mediated), and Asp760 (90%, H-bond) (supplementary file, S1). In the case of compound CHEMBL164344, the RMSD plot has deviations till 10.0Å for the initial 18 ns, during which strong interactions were observed with the residues of the palm domain, Thr556, Asp623, and Arg624; later, these interactions gradually decreased, and the interactions with two residues of F domain, Asp452 (81%, H-bond) and Tyr455 (61%, π-π stacking) increased over the time. This has caused a decrease in the RMSD from 12.0 Å at 20th ns to 7.5 Å at 100th ns. In the case of its protomer, the ligand has stable deviations for the initial 25 ns (RMSD 4.0 Å – 6.0 Å), later increasing to 16 Å till 45 ns and then attaining equilibrium until the end with RMSD ranging between 11 Å and 13 Å. The residues that significantly participated in the interactions include Tyr455 (73% π-π stacking, 68% H-bond), Lys551 (69%, H-bond), Arg553 (92%, H-bond), Arg555 (35%, H-bond), and Lys621 (33%, π-cation). It is noted that the protomer’s RMSF is similar to that of the uncharged molecule (Figure 4). Change of phenoxymethyl (as in CHEMBL164344) with biphenyl group (as in CHEMBL196676) results in significantly more interactions with the residues Asp452 (81%, H-bond) of F domain and Asp623 (85%, H-bond) of palm domain, as well as inconsistent water-mediated interactions with Asp760 (27%). Initially, the interactions with Arg553, Thr556, and Asp623, as observed in the docked complex, remained for 18 ns, during which no significant deviations were observed (RMSD between 1.0 Å – 3.0 Å); later, the interactions with Asp452, Lys621 along with Asp623 had changed the ligand's conformation, resulting in the increase in the RMSD ranging between 8.0 Å and 7.0 Å which retained till 70 ns, and then a gradual decrease was observed until the end of the simulation (last frame RMSD is 5.4 Å) (Figure 3).
The quinazoline-4-amine compound (CHEMBL4291724) shows greater stability in terms of its RMSD plot as equilibrium is attained after 20 ns until the end of the simulation with RMSD ranging between 10.0 Å and 12.0 Å (Figure 3). Most of the interactions are between the phosphonic acid groups and the residues of the palm domain, which include Asp623, Thr680, and Asp760, with a contribution of 50%, 39%, and 86%, respectively (Supplementary File, S1). In the case of CHEMBL387132, a 2-amino isoquinoline compound, the RMSD plot is comparatively lower than other complexes, as the overall deviations for 100 ns were between 3.0 Å and 6.4 Å. Additionally, strong interactions were observed with Asp623 for the first 30 ns and then with Asp760 of the palm domain, contributing to an overall interaction of 30% and 64%, respectively. A residue from the F domain, Lys545, has a total of 33% H-bond interactions with the ligand. (Figure 3, Supplementary File, S1).
The 2-amino thiazole containing BP (CHEMBL4569308) showed a stable RMSD plot for initial 45 ns (between 1.0 Å and 2.0 Å), had a slight increment over the next few time frames and then attained equilibrium from 60 ns until the end with deviations between 2.5 Å and 4.0 Å. The stability is observed due to the significant involvement of thiazole ring in the interactions with Asp623 (99%, H-bond; 71% water-mediated H-bond) and Arg624 (89%, π-cation; 52% and 40% H-bond). Other residues such as Lys545 (68%, H-bond), Arg553 (76%, 82%, H-bond) and Arg555 (68%, 41%, H-bond) showed moderate interactions with the oxygen atoms of the compound. The imidazole compound (CHEMBL98211) attained stability from 20 ns until the end with RMSD between 5.5 Å and 7.0 Å. However, there were slight deviations observed between 45 ns-55 ns and 85 ns-90 ns with RMSD ranging between 4.0 Å and 7.0 Å. These deviations could be due to the intermittent water-mediated interactions with Asp623 (72%). The significantly contributing residues in the interactions include Lys545 (96%, H-bond), Arg555 (77%, 67%, H-bond) and Arg624 (73%, 49%, H-bond) (Supplementary File, S1). Other residues such as Thr556 and Lys621 contributed with 52% of water-mediated and 48% of H-bond interactions, respectively. The imidazo[1,2-α]pyridine-4-ium compound (CHEMBL608526) exhibited the most stable RMSD plot among all the studied complexes. For the initial 30 ns, its RMSD was between 3.0 Å and 4.0 Å, later there was a gradual decrease until 60 ns, and then equilibrium was attained until the end with RMSD ranging between 1.0 Å and 2.0 Å. There were several residues that significantly contributed to the interactions during the simulation which include Asp450 (91%, H-bond), Lys545 (99%, 68%, H-bond), Arg553 (100%, 99%, H-bond), Arg555 (100%, 93%, H-bond), Asp623 (102%, water-mediated H-bond) and Arg624 (101%, 84%, H-bond interactions). The other residues such as Lys551, Thr556, Ala558 and Ser682 contributed moderately with 63%, 43%, 33% and 60% interactions, respectively. The negative control cinnamaldehyde showed highly unstable RMSD plot with high deviations (up to 90.0 Å), and interactions with <10% contributions (Supplementary File, S1).
The protein RMSD plot shows a similar pattern of deviations with all the hit ligands, indicating their stability with the ligands during the MD simulation. Similarly, the RMSF plot shows that interacting residues in the active site have acceptable fluctuations (<2.0 Å) (Figure 4). From this, we infer that 5 out of our top 7 ligands behave in a similar fashion to that of remdesivir, with the active site residues forming strong bonds, and most of the interactions by our “hit compounds” attributable to bisphosphonic acid groups. Therefore, there is a good probability that these compounds may emerge as potential RdRp inhibitors if evaluated in vitro and/or ex vivo.
2.3. Estimation of Entropic Contribution by gmx_MMPBSA
The MMPBSA analysis helps to understand entropic contribution between protein and ligand during the molecular dynamic simulations [70]. It refers to the degree of randomness in a system and can guide to understand the entropic contributions of the ligand in the active site of the protein. Since MMPBSA analysis module is not available in Schrödinger software, the gmx_MMPBSA tool was used to determine the entropic contributions of the ligands, protein, and the protein-ligand complexes [71]. The change in free energy (ΔG) of the complex is then calculated using the following equation:
In the above equation (1), the Gcomplex represents the free energy of the protein-ligand complex in water, while Gprotein and Gligand represent the free energies of the protein and ligand respectively in water. The free energy of the total system (ΔGbinding) can be obtained by adding the interaction entropy (I.E. = - TΔS) to the change in total energy (ΔEMM) of the system [72]. ΔEMM is the summation of various change in energies such as van der Waals (ΔEvdW), electrostatic columbic (ΔEEL), electrostatic potential (ΔEPB) and non-polar (ΔENP). Applying equation (3), the free energy of the system (ΔGbinding) was calculated and tabulated in Table 4 and represented graphically in Figure 5.
The results reveal that the protomer of CHEMBL608526 has the least value (-75.62 kcal/mol), while CHEMBL196676 (-33.41 kcal/mol), CHEMBL164344 (-26.77 kcal/mol), CHEMBL4291724 (-26.38 kcal/mol) and CHEMBL4569308 (-22.97 kcal/mol) have values lower than remdesivir (-19.91 kcal/mol). The more negative values signify that the free energy of the complex is lower than the sum of the individual free energies of the protein and the ligand, as seen from equation (2). Therefore, these five candidates, highlighted in bold in Table 4, are worth evaluating in vitro and/or ex vivo. The two other candidates, CHEMBL387132 (-1.16 kcal/mol) and CHEMBL98211 (0.42 kcal/mol), have values close to zero and to the negative control cinnamaldehyde (-0.99 kcal/mol), but they may also be worth evaluating for the following reasons: 1. They perform as well as the 5 highlighted compounds in molecular dynamics studies, showing several interactions with the protein’s active site that are absent in the case of negative control (c.f. Figure 3, supplementary file S1); 2. ΔGbinding values can vary substantially, e.g., CHEMBL164344’s uncharged state value (-26.77 kcal/mol) is twice that of its protomer (-12.65 kcal/mol), therefore it may be premature to dismiss these two compounds; and 3. CHEMBL98211 resembles the approved drug zoledronate which has been observed to benefit COVID-19 human patients as described in Section 3.4 below. Although charged molecules are associated with electrostatic repulsions, higher degrees of freedom, and generally lower binding energies leading to more spontaneous binding, we have to exercise caution. This is because the relative contribution of the first term (enthalpy) versus the second term (entropy) in equation (3) is not straightforward in the case of proteins due to conformational effects in the latter and specific properties of a given system [73]. More studies are required with a range of molecules wherein the in silico predictions can be experimentally validated to determine whether free energy calculations for charged or uncharged molecules are better predictive and more useful for drug selection.
2.4. Results of in Silico Predicted ADMET Profiles
All 7 compounds have satisfactory pharmacokinetic and toxicity (ADMET) profiles compared to remdesivir (Table 5).
The intestinal absorption rate ranges from 34% to 72% for all the molecules except for CHEMBL164344 (16.77%). All compounds exhibited similar skin permeability; none acted as a P-glycoprotein (P-gp) substrate. A drug that acts as a substrate to P-gp implies low bioavailability, as evident from the studies on remdesivir [74]. The volume of distribution in steady state condition (Vdss) for all 7 compounds is less except for CHEMBL196676, as the latter possessed better tissue distribution value than remdesivir. The fraction of the drug that remains unbound with blood plasma protein is essential to pass through cell membranes; therefore, higher the fraction unbound (Fu) value, the higher is its distribution [75]. All 7 compounds had better Fu values than remdesivir (0.005 Fu), with CHEMBL608526 showing higher distribution pattern as its value was 0.811 Fu. Due to the presence of phosphonate groups, the compounds are likely to have poor permeability to the blood-brain barrier (BBB) or the central nervous system (CNS); however, CHEMBL387132 and CHEMBL196676 exhibited slightly higher permeability profiles than remdesivir. None of the molecules are likely to be metabolized by cytochrome P450, and all showed a good clearance rate. The maximum tolerated dose is higher than remdesivir and does not inhibit the human ether-a-go-go gene (hERG). The rat oral toxicity levels are higher than remdesivir, indicating high concentrations are required to cause toxicity.
3. Materials and Methods
As the objective of this study is to find a molecular-level explanation for [28] and identify any other leads by systematically analyzing all BPs, we downloaded from the ChEMBL database [76] a series of compounds having BPs, and carried out molecular docking studies to get all the “hit” molecules. These hits were further down-selected using molecular mechanics with generalized Born and surface area (MM-GBSA) studies and the top seven ligands were analyzed for their dynamic behavior via molecular dynamics studies followed by the entropic calculations using the software “gromacs” (v2022.4).
3.1. Protein Preparation
While the literature shows a high sequence identity (96%) between the RdRp proteins of SARS-CoV and SARS-CoV-2, we couldn’t find any report with MERS-CoV, so the latter’s RdRp domain (residues 4845–5412 from accession code A0A023YA54) was taken and multiple sequence alignment was carried out to reveal the conserved motifs (75%) for these three coronaviruses (Figure 1). To start screening BPs against SARS-CoV-2, RdRp protein (PDB: 6M71) [31] was selected and downloaded from the protein data bank (https://www.rcsb.org/). The protein was prepared using the protein preparation wizard of the glide module of the Schrödinger software (Schrödinger LLC., NY, v2022) [77]. Missing hydrogens and residues were added using the software’s prime module and pre-processed using the Epik module at pH 7.0±2.0. The protein was optimized by removing water molecules beyond 3 Å and subjected to minimization using the optimized potentials for liquid simulations 4 (OPLS4) force field [78].
3.2. Ligand Preparation
ChEMBL database has been used to download molecules containing BPs based on the Tanimoto similarity check (95%) [79]. These molecules were imported and prepared using Ligprep module of maestro (Schrödinger LLC., NY, v2022). The ionization state was set to neutral and chirality was determined from the 3D structure. The force field OPLS4 was employed to prepare the ligands.
3.3. Molecular Docking
For carrying out molecular docking studies, a grid box of 10 Å3 was generated using the maestro receptor grid generation wizard (Schrödinger) by specifying the binding (active) site residue Arg555, as no co-crystal ligand was available [80]. The ligands were docked using the ligand docking wizard of the glide module of Schrödinger with standard-precision (SP) mode initially. The molecules that were able to bind were subjected to filtration by applying the criterion of molecular weight (<500 Da) and number of rotatable bonds (<10) [68]. Molecules obtained from this filtration process were then subjected to docking with extra-precision (XP) mode [81].
3.4. MM-GBSA Calculations
Molecular mechanics with generalised Born and surface area solvation (MM-GBSA) studies help to calculate the ligand's binding-free energy (∆Gbind) value. The calculations were mainly based on the summation of differences in the minimization (∆EMM), solvation (∆GSolv), and surface area (∆GSA) energies of RdRp-ligand complex structure and free RdRp and ligand molecules [82]. The protein-ligand complex from the docking studies was used to calculate the binding free energy. The analysis was carried out in the prime module of Schrödinger software. OPLS4 force field with dielectric surface generalized Born (VSGB) continuum solvation model was used. Based on the following formula, the binding free energy of the ligand was calculated:
3.5. Molecular Dynamics Studies
The molecular dynamics (MD) simulation was accomplished to determine the ligand molecules' stability, confirmation and intermolecular interaction with the target RdRp protein (PDB ID: 6M71) [83]. The time-dependent modification of the complexes was estimated over 100 ns using the Desmond module. The MD simulation was executed at a constant temperature of 310 K using Nosé -Hoover chain thermostat, and constant pressure of 1.013 bar using Martyna-Tobias-Klein barostat [84,85]. The complete system was annealed and equilibrated using ensembles.
The down-selected ligand complexes were imported and prepared using the protein preparation wizard of the Desmond module (Schrödinger LLC., NY, v2020). The complex was solvated using the transferable intermolecular potential with 3 points (TIP3P) model and the grid boundary dimensions was set to 10 Å3 [86]. The complex model was electrically neutralized with Na+/Cl− ions, and built using a system builder wizard. The complete solvated model was minimized and molecular dynamics was carried out for 100 ns on all eight selected complexes (including the reference drug remdesivir, and the negative control cinnamaldehyde). The root mean square deviation (RMSD), root mean square fluctuation (RMSF) and interaction plots were used to interpret the stability of appropriate complexes.
3.6. Entropy Calculation for Molecular Dynamics Trajectories
The binding free energy of the protein-ligand complex was determined using the gmx_mmPBSA tool [71,87]. In this calculation, the molecular mechanism Poisson-Boltzmann surface area (MMPBSA) method with a dielectric model (“ipb” = 2) and a non-polar solvation model (“inp” = 1) were employed. The ionic strength of the surrounding medium was maintained at 0.15 M, and the temperature was set at 310 K. To calculate the entropy (-TΔS), the interaction entropy (I.E.) method was used [72]. This allowed us to evaluate the change in binding free energies with entropy contributions for the protein, ligand, and their complexes. The trajectories from the protein-ligand MD simulation in explicit water from the Desmond module were used to generate the gromacs trajectory file required for calculations using visual molecular dynamics (VMD) software. Additionally, topology files for protein and ligand were obtained separately by converting the *.cms files to *.gro and *.top files using the intermol software [88]. Since the initial frames during the dynamic simulation are involved in the equilibrium, the frames after 50 ns were considered for data analysis. To speed up the procedure and to get better averaging, we ran five independent MMPBSA calculations (10 ns each) for every complex from 50-100 ns and reported the average and standard deviation of these calculations [89].
3.7. ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) Studies
To determine the pharmacokinetic and pharmacodynamic profiles, the smiles format of the hit compounds was used and their parameters were predicted using the pkCSM web-tool [90]. All the absorption, distribution, metabolism, excretion and toxicity parameters obtained were compared with the standard reference drug remdesivir.
4. Conclusions
As mentioned in 1.1, we set out with three objectives for this work, all of which have been achieved. A recent, compelling study in humans reported that “prior bisphosphonate use was associated with dramatically reduced odds of SARS-CoV-2 testing, COVID-19 diagnosis, and COVID-19-related hospitalizations” without offering a molecular-level explanation for this phenomenon. These authors decided to investigate BPs (such as alendronate/alendronic acid and zoledronate) because pre-COVID-19 observational studies had reported a “decreased in-hospital mortality for patients in the ICU” [91] and a “reduced incidence of pneumoniae and pneumonia-related mortality in patients treated with amino-BPs versus controls” [92]. They found that 450,366 users of alendronate/alendronic acid and zoledronic acid, had “lower odds ratios (OR) of testing for SARS-CoV-2 infection (OR = 0.22; 95%CI:0.21–0.23; p<0.001), COVID-19 diagnosis (OR = 0.23; 95%CI:0.22–0.24; p<0.001), and COVID-19-related hospitalization (OR = 0.26; 95%CI:0.24–0.29; p<0.001)” [28,65]. An in vitro basis for these findings concerning alendronate/alendronic acid users was partially addressed by Huang et al. [29] wherein SARS-CoV-2 viral clearance was observed when used in combination with a glucocorticoid dexamethasone, while our recent work provided a partial, in silico mechanistic explanation pertaining to the RdRp target [27].
In this work, we have closed the gaps in knowledge by thoroughly investigating all BPs; and identified 7 promising molecules (c.f. Table 5), two of which closely resemble the approved drugs zoledronate and minodronate. CHEMBL602586 is very similar to minodronic acid, while CHEMBL98211 is close to zoledronic acid (supplementary file, S1). Minodronate and related ChEMBL ID’s (Table 4) are worthy of further investigation pertaining to COVID-19 and/or PASC. An in vitro/ex vivo analysis similar to McAuley et al. [14] may also be useful ahead of observational studies in humans with minodronate (similar to [28]), however this compound is only approved in Japan, therefore, such studies are best conducted there.
In the current work, we have also made methodological improvements to molecular modelling studies of this nature. For example, in silico investigation of BP-containing molecules from the CHEMBL database identified 48 molecules with docking scores 2-3 times superior to remdesivir. Subsequent MM-GBSA analysis led to the selection of 7 molecules with notably better free energy scores than remdesivir. These 7 candidates underwent further assessment of their dynamics profiles and entropy calculations, resulting in the selection of 5 promising candidates for in vitro evaluation, comprising of 3 uncharged and 2 charged molecules. Our study confirms the importance of considering charged nature of molecules to assess protein binding and interactions in biological systems; while its uncharged state could still be relevant in the content of stability and metabolism. Notably, our investigation, exemplified by CHEMBL164344, revealed that despite charged and uncharged molecules having similar MM-GBSA scores, dynamic behavior, and total internal energies, their entropy contributions can differ significantly (for instance, CHEMBL164344’s values were -12.65 kcal/mol for charged and -26.77 kcal/mol for uncharged). Whether it is important to only consider the charged state for down-selection remains to be seen from future experimental and clinical trial observations. The general agreement between entropy and dynamics results, combined with in silico-predicted ADMET analysis, led us to predict that seven compounds (CHEMBL196676, CHEMBL164344, CHEMBL4291724, CHEMBL608526, CHEMBL4569308, CHEMBL98211, CHEMBL387132) are worthy of synthesis, in vitro and in vivo evaluation against RdRp.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, M.M.U-.R., M.S., and S.S.V.; methodology, M.M.U-.R., C.K.S., M.G., A.C. and P.V.K..; software, P.V.K., R.P.J., S.R.J.; validation, M.M.U-.R., M.G., M.S., and S.S.V.; formal analysis, M.M.U-.R., C.K.S., M.G., A.C., and P.V.K.; investigation, C.K.S., R.P.J., S.R.J.; resources, M.S., and S.S.V.; data curation, M.M.U-.R., and C.K.S.; writing—original draft preparation, M.M.U-.R., and C.K.S.; writing—review and editing, M.M.U-.R., M.G., M.S., and S.S.V.; visualization, M.M.U-.R., A.C. and M.S.; supervision, M.S., and S.S.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Any data not provided in this study will be shared upon request.
Acknowledgments
The authors are thankful to the Birla Institute of Technology and Science for providing the necessary facilities to conduct this research, and to the journal and the University of York for defraying the article processing charges.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mesel-Lemoine, M.; Millet, J.; Vidalain, P.-O.; Law, H.; Vabret, A.; Lorin, V.; Escriou, N.; Albert, M.L.; Nal, B.; Tangy, F. A Human Coronavirus Responsible for the Common Cold Massively Kills Dendritic Cells but Not Monocytes. J. Virol. 2012, 86, 7577–7587. [Google Scholar] [CrossRef]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A Brief Summary and Comparison of Severe Acute Respiratory Infections Caused by Three Highly Pathogenic Human Coronaviruses. Respir. Res. 2020, 21, 224. [Google Scholar] [CrossRef]
- WHO Coronavirus (COVID-19) Dashboard. WHO Coronavirus (COVID-19) Dashboard With Vaccination Data. Available online: https://covid19.who.int/ (accessed on 2 November 2023).
- Thaweethai, T.; Jolley, S.E.; Karlson, E.W.; Levitan, E.B.; Levy, B.; Mccomsey, G.A.; Mccorkell, L.; Nadkarni, G.N.; Parthasarathy, S.; Singh, U.; et al. Development of a Definition of Postacute Sequelae of SARS-CoV-2 Infection. JAMA 2023, 329, 1934–1946. [Google Scholar] [CrossRef] [PubMed]
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-Acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Office for National Statistics (ONS) Prevalence of Ongoing Symptoms Following Coronavirus (COVID-19) Infection in the UK: 2 February 2023. Available online: https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/bulletins/prevalenceofongoingsymptomsfollowingcoronaviruscovid19infectionintheuk/2february2023 (accessed on 6 November 2023).
- McDonald, E.G.; Lee, T.C. Nirmatrelvir-Ritonavir for COVID-19. CMAJ 2022, 194, E218. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA) Revocation of Emergency Use of a Drug During the COVID-19 Pandemic; Availability. Available online: https://www.federalregister.gov/documents/2022/07/26/2022-15956/revocation-of-emergency-use-of-a-drug-during-the-covid-19-pandemic-availability#citation-1-p44407 (accessed on 9 February 2024).
- Gilead. FDA Approves Veklury® (Remdesivir) to Treat COVID-19 in People With Mild to Severe Hepatic Impairment With No Dose Adjustment. Available online: https://www.gilead.com/news-and-press/press-room/press-releases/2023/8/fda-approves-veklury-remdesivir-to-treat-covid19-in-people-with-mild-to-severe-hepatic-impairment-with-no-dose-adjustment (accessed on 9 February 2024).
- Consortium, W.S.T. Remdesivir and Three Other Drugs for Hospitalised Patients with COVID-19: Final Results of the WHO Solidarity Randomised Trial and Updated Meta-Analyses. Lancet 2022, 399, 1941–1953. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA) FDA Approves First Oral Antiviral for Treatment of COVID-19 in Adults. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-antiviral-treatment-covid-19-adults (accessed on 6 November 2023).
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martín-Quirós, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef]
- Manabe, T.; Kambayashi, D.; Akatsu, H.; Kudo, K. Favipiravir for the Treatment of Patients with COVID-19: A Systematic Review and Meta-Analysis. BMC Infect. Dis. 2021, 21, 489. [Google Scholar] [CrossRef] [PubMed]
- McAuley, A.J.; Vuren, P.J. van; Mohammed, M.-U.-R.; Faheem, F.; Goldie, S.; Riddell, S.; Gödde, N.J.; Styles, I.K.; Bruce, M.P.; Chahal, S.; et al. Use of Human Lung Tissue Models for Screening of Drugs against SARS-CoV-2 Infection. Viruses 2022, 14, 2417. [Google Scholar] [CrossRef]
- Reis, G.; dos Santos Moreira-Silva, E.A.; Silva, D.C.M.; Thabane, L.; Milagres, A.C.; Ferreira, T.S.; dos Santos, C.V.Q.; de Souza Campos, V.H.; Nogueira, A.M.R.; de Almeida, A.P.F.G.; et al. Effect of Early Treatment with Fluvoxamine on Risk of Emergency Care and Hospitalisation among Patients with COVID-19: The TOGETHER Randomised, Platform Clinical Trial. Lancet Glob. Heal. 2022, 10, e42–e51. [Google Scholar] [CrossRef]
- Butler, C.C.; Hobbs, F.D.R.; Gbinigie, O.A.; Rahman, N.M.; Hayward, G.; Richards, D.B.; Dorward, J.; Lowe, D.M.; Standing, J.F.; Breuer, J.; et al. Molnupiravir plus Usual Care versus Usual Care Alone as Early Treatment for Adults with COVID-19 at Increased Risk of Adverse Outcomes (PANORAMIC): An Open-Label, Platform-Adaptive Randomised Controlled Trial. Lancet 2023, 401, 281–293. [Google Scholar] [CrossRef]
- Wise, J. Covid-19: Molnupiravir Does Not Cut Hospital Admissions or Deaths in Vaccinated People at High Risk, Trial Finds. BMJ 2022, o3055. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, M. Merck’s COVID Pill Loses Its Lustre: What That Means for the Pandemic. Nature 2021. [Google Scholar] [CrossRef] [PubMed]
- Batool, S.; Vuthaluru, K.; Hassan, A.; Bseiso, O.; Tehseen, Z.; Pizzorno, G.; Rodriguez Reyes, Y.; Saleem, F. Efficacy and Safety of Favipiravir in Treating COVID-19 Patients: A Meta-Analysis of Randomized Control Trials. Cureus 2023, 15, e33676. [Google Scholar] [CrossRef]
- Bosaeed, M.; Alharbi, A.; Mahmoud, E.; Alrehily, S.; Bahlaq, M.; Gaifer, Z.; Alturkistani, H.; Alhagan, K.; Alshahrani, S.; Tolbah, A.; et al. Efficacy of Favipiravir in Adults with Mild COVID-19: A Randomized, Double-Blind, Multicentre, Placebo-Controlled Clinical Trial. Clin. Microbiol. Infect. 2022, 28, 602–608. [Google Scholar] [CrossRef]
- Shah, P.L.; Orton, C.M.; Grinsztejn, B.; Donaldson, G.C.; Crabtree Ramírez, B.; Tonkin, J.; Santos, B.R.; Cardoso, S.W.; Ritchie, A.I.; Conway, F.; et al. Favipiravir in Patients Hospitalised with COVID-19 (PIONEER Trial): A Multicentre, Open-Label, Phase 3, Randomised Controlled Trial of Early Intervention versus Standard Care. Lancet Respir. Med. 2023, 11, 415–424. [Google Scholar] [CrossRef]
- Siripongboonsitti, T.; Ungtrakul, T.; Tawinprai, K.; Nimmol, T.; Buttakosa, M.; Sornsamdang, G.; Jarrusrojwuttikul, T.; Silapant, P.; Mahanonda, N. Efficacy of Combination Therapy of Fluvoxamine and Favipiravir vs Favipiravir Monotherapy to Prevent Severe COVID-19 among Mild to Moderate COVID-19 Patients: Open-Label Randomized Controlled Trial (EFFaCo Study). Int. J. Infect. Dis. 2023, 134, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Lenze, E.J.; Mattar, C.; Zorumski, C.F.; Stevens, A.; Schweiger, J.; Nicol, G.E.; Miller, J.P.; Yang, L.; Yingling, M.; Avidan, M.S.; et al. Fluvoxamine vs Placebo and Clinical Deterioration in Outpatients With Symptomatic COVID-19. JAMA 2020, 324, 2292. [Google Scholar] [CrossRef]
- Calusic, M.; Marcec, R.; Luksa, L.; Jurkovic, I.; Kovac, N.; Mihaljevic, S.; Likic, R. Safety and Efficacy of Fluvoxamine in COVID-19 ICU Patients: An Open Label, Prospective Cohort Trial with Matched Controls. Br. J. Clin. Pharmacol. 2022, 88, 2065–2073. [Google Scholar] [CrossRef]
- Jain, H.A.; Agarwal, V.; Bansal, C.; Kumar, A.; Faheem, F.; Mohammed, M.-U.-R.; Murugesan, S.; Simpson, M.M.; Karpe, A.V.; Chandra, R.; et al. CoviRx: A User-Friendly Interface for Systematic Down-Selection of Repurposed Drug Candidates for COVID-19. Data 2022, 7, 164. [Google Scholar] [CrossRef]
- MacRaild, C.A.; Mohammed, M.U.R.; Faheem, *!!! REPLACE !!!*; Murugesan, S.; Styles, I.K.; Peterson, A.L.; Kirkpatrick, C.M.J.; Cooper, M.A.; Palombo, E.A.; Simpson, M.M.; et al. Systematic Down-Selection of Repurposed Drug Candidates for COVID-19. Int. J. Mol. Sci. 2022, 23, 11851. [Google Scholar] [CrossRef]
- Muzaffar-Ur-Rehman, M.; Suryakant, C.K.; Chandu, A.; Kumar, B.K.; Joshi, R.P.; Jadav, S.R.; Sankaranarayanan, M.; Vasan, S.S. Molecular Docking and Dynamics Identify Potential Drugs to Be Repurposed as SARS-CoV-2 Inhibitors. J. Comput. Biophys. Chem. 2023, 1–23. [Google Scholar] [CrossRef]
- Thompson, J.; Wang, Y.; Dreischulte, T.; Barreiro, O.; Gonzalez, R.J.; Hanč, P.; Matysiak, C.; Neely, H.R.; Rottenkolber, M.; Haskell, T.; et al. Association between Bisphosphonate Use and COVID-19 Related Outcomes. Elife 2023, 12, e79548. [Google Scholar] [CrossRef]
- Huang, B. Use Of A Nitrogen-Containing Bisphosphonate In Combination With A Glucocorticoid In Preventing Or Treating Viral Pneumonia. 2023, 1–26, US20230149429A1. [Google Scholar]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 Genome, Structure, Evolution, Pathogenesis and Therapies: Structural Genomics Approach. Biochim. Biophys. acta. Mol. basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-Dependent RNA Polymerase from COVID-19 Virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and Functions of Coronavirus Replication–Transcription Complexes and Their Relevance for SARS-CoV-2 Drug Design. Nat. Rev. Mol. Cell Biol. 2021, 23, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Gangadharan, S.; Ambrose, J.M.; Rajajagadeesan, A.; Kullappan, M.; Patil, S.; Gandhamaneni, S.H.; Veeraraghavan, V.P.; Nakkella, A.K.; Agarwal, A.; Jayaraman, S.; et al. Repurposing of Potential Antiviral Drugs against RNA-Dependent RNA Polymerase of SARS-CoV-2 by Computational Approach. J. Infect. Public Health 2022, 15, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, H.; Er, S.Y.; Choong, Y.K.; Gavor, E.; Sivaraman, J. Structural Basis of SARS-CoV-2- and SARS-CoV-Receptor Binding and Small-Molecule Blockers as Potential Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 465–493. [Google Scholar] [CrossRef]
- Bertolin, A.P.; Weissmann, F.; Zeng, J.; Posse, V.; Milligan, J.C.; Canal, B.; Ulferts, R.; Wu, M.; Drury, L.S.; Howell, M.; et al. Identifying SARS-CoV-2 Antiviral Compounds by Screening for Small Molecule Inhibitors of Nsp12/7/8 RNA-Dependent RNA Polymerase. Biochem. J. 2021, 478, 2425–2443. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural Basis for Inhibition of the RNA-Dependent RNA Polymerase from SARS-CoV-2 by Remdesivir. Science (80-.) 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV Nsp12 Polymerase Bound to Nsp7 and Nsp8 Co-Factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef]
- Ahmad, J.; Ikram, S.; Ahmad, F.; Rehman, I.U.; Mushtaq, M. SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp)–A Drug Repurposing Study. Heliyon 2020, 6, e04502. [Google Scholar] [CrossRef]
- El Sohaimy, S.; Abdo, N.; Shehata, M.; Moheyeldin, O. Inhibition of COVID-19 RNA-Dependent RNA Polymerase by Natural Bioactive Compounds: Molecular Docking Analysis. Egypt. J. Chem. 2021, 64, 1989–2001. [Google Scholar] [CrossRef]
- Zamzami, M.A. Molecular Docking, Molecular Dynamics Simulation and MM-GBSA Studies of the Activity of Glycyrrhizin Relevant Substructures on SARS-CoV-2 RNA-Dependent-RNA Polymerase. J. Biomol. Struct. Dyn. 2023, 41, 1846–1858. [Google Scholar] [CrossRef] [PubMed]
- Brunt, D.; Lakernick, P.M.; Wu, C. Discovering New Potential Inhibitors to SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) Using High Throughput Virtual Screening and Molecular Dynamics Simulations. Sci. Rep. 2022, 12, 19986. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Lu, W.; Jiang, H.; Yang, C.; Dong, X. Molecular Docking and Dynamics of Phytochemicals From Chinese Herbs With SARS-CoV-2 RdRp. Nat. Prod. Commun. 2022, 17. [Google Scholar] [CrossRef]
- Askari, F.S.; Ebrahimi, M.; Parhiz, J.; Hassanpour, M.; Mohebbi, A.; Mirshafiey, A. Digging for the Discovery of SARS-CoV-2 Nsp12 Inhibitors: A Pharmacophore-Based and Molecular Dynamics Simulation Study. Future Virol. 2022, 17, 743–759. [Google Scholar] [CrossRef] [PubMed]
- Uengwetwanit, T.; Chutiwitoonchai, N.; Wichapong, K.; Karoonuthaisiri, N. Identification of Novel SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) Inhibitors: From in Silico Screening to Experimentally Validated Inhibitory Activity. Comput. Struct. Biotechnol. J. 2022, 20, 882–890. [Google Scholar] [CrossRef]
- Chinnamadhu, A.; Ramakrishnan, J.; Suresh, S.; Ramadurai, P.; Poomani, K. Dynamics and Binding Affinity of Nucleoside and Non-Nucleoside Inhibitors with RdRp of SARS-CoV-2: A Molecular Screening, Docking, and Molecular Dynamics Simulation Study. J. Biomol. Struct. Dyn. 2022, 41, 10396–10410. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, F.A.; Alkarim, S.A.; Hawsawi, Y.M.; Abdulaal, W.H.; Albiheyri, R.; Kurdi, B.; Alguridi, H.; El-Magd, M.A. 25 (S)-Hydroxycholesterol Acts as a Possible Dual Enzymatic Inhibitor of SARS-CoV-2 M pro and RdRp–: An Insight from Molecular Docking and Dynamics Simulation Approaches. J. Biomol. Struct. Dyn. 2023, 41, 4744–4755. [Google Scholar] [CrossRef] [PubMed]
- Alexpandi, R.; De Mesquita, J.F.; Pandian, S.K.; Ravi, A.V. Quinolines-Based SARS-CoV-2 3CLpro and RdRp Inhibitors and Spike-RBD-ACE2 Inhibitor for Drug-Repurposing Against COVID-19: An in Silico Analysis. Front. Microbiol. 2020, 11, 1796. [Google Scholar] [CrossRef]
- Kushwaha, P.P.; Singh, A.K.; Bansal, T.; Yadav, A.; Prajapati, K.S.; Shuaib, M.; Kumar, S. Identification of Natural Inhibitors Against SARS-CoV-2 Drugable Targets Using Molecular Docking, Molecular Dynamics Simulation, and MM-PBSA Approach. Front. Cell. Infect. Microbiol. 2021, 11, 730288. [Google Scholar] [CrossRef]
- Shady, N.H.; Hayallah, A.M.; Mohamed, M.F.A.; Ghoneim, M.M.; Chilingaryan, G.; Al-Sanea, M.M.; Fouad, M.A.; Kamel, M.S.; Abdelmohsen, U.R. Targeting 3CLpro and SARS-CoV-2 RdRp by Amphimedon Sp. Metabolites: A Computational Study. Molecules 2021, 26, 3775. [Google Scholar] [CrossRef]
- Gajjar, N.D.; Dhameliya, T.M.; Shah, G.B. In Search of RdRp and Mpro Inhibitors against SARS CoV-2: Molecular Docking, Molecular Dynamic Simulations and ADMET Analysis. J. Mol. Struct. 2021, 1239, 130488. [Google Scholar] [CrossRef]
- Parihar, A.; Sonia, Z.F.; Akter, F.; Ali, M.A.; Hakim, F.T.; Hossain, M.S. Phytochemicals-Based Targeting RdRp and Main Protease of SARS-CoV-2 Using Docking and Steered Molecular Dynamic Simulation: A Promising Therapeutic Approach for Tackling COVID-19. Comput. Biol. Med. 2022, 145, 105468. [Google Scholar] [CrossRef]
- M A Kawsar, S.; Hosen, M.A.; Ahmad, S.; El Bakri, Y.; Laaroussi, H.; Ben Hadda, T.; Almalki, F.A.; Ozeki, Y.; Goumri-Said, S. Potential SARS-CoV-2 RdRp Inhibitors of Cytidine Derivatives: Molecular Docking, Molecular Dynamic Simulations, ADMET, and POM Analyses for the Identification of Pharmacophore Sites. PLoS ONE 2022, 17, e0273256. [Google Scholar] [CrossRef]
- Veerasamy, R.; Karunakaran, R. Molecular Docking Unveils the Potential of Andrographolide Derivatives against COVID-19: An in Silico Approach. J. Genet. Eng. Biotechnol. 2022, 20, 58. [Google Scholar] [CrossRef]
- Mohammed, A.O.; Abo-Idrees, M.I.; Makki, A.A.; Ibraheem, W.; Alzain, A.A. Drug Repurposing against Main Protease and RNA-Dependent RNA Polymerase of SARS-CoV-2 Using Molecular Docking, MM-GBSA Calculations and Molecular Dynamics. Struct. Chem. 2022, 33, 1553–1567. [Google Scholar] [CrossRef] [PubMed]
- Ribaudo, G.; Ongaro, A.; Oselladore, E.; Zagotto, G.; Memo, M.; Gianoncelli, A. A Computational Approach to Drug Repurposing against SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp). J. Biomol. Struct. Dyn. 2022, 40, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Baby, K.; Maity, S.; Mehta, C.H.; Suresh, A.; Nayak, U.Y.; Nayak, Y. Targeting SARS-CoV-2 RNA-Dependent RNA Polymerase: An in Silico Drug Repurposing for COVID-19. F1000Research 2020, 9, 1166. [Google Scholar] [CrossRef]
- Hosseini, M.; Chen, W.; Xiao, D.; Wang, C. Computational Molecular Docking and Virtual Screening Revealed Promising SARS-CoV-2 Drugs. Precis. Clin. Med. 2021, 4, 1–16. [Google Scholar] [CrossRef]
- El Hassab, M.A.; Hemeda, L.R.; Elsayed, Z.M.; Al-Rashood, S.T.; Abdel-Hamid Amin, M.K.; Abdel-Aziz, H.A.; Eldehna, W.M. Computational Prediction of the Potential Target of SARS-CoV-2 Inhibitor Plitidepsin via Molecular Docking, Dynamic Simulations and MM-PBSA Calculations. Chem. Biodivers. 2022, 19, e202100719. [Google Scholar] [CrossRef]
- Vesga, L.C.; Ruiz-Hernández, C.A.; Alvarez-Jacome, J.J.; Duque, J.E.; Rincon-Orozco, B.; Mendez-Sanchez, S.C. Repurposing of Four Drugs as Anti-SARS-CoV-2 Agents and Their Interactions with Protein Targets. Sci. Pharm. 2022, 90, 24. [Google Scholar] [CrossRef]
- Elfiky, A.A. Dual Targeting of RdRps of SARS-CoV-2 and the Mucormycosis-Causing Fungus: An in Silico Perspective. Future Microbiol. 2022, 17, 755–762. [Google Scholar] [CrossRef]
- Wang, B.; Svetlov, D.; Artsimovitch, I. NMPylation and De-NMPylation of SARS-CoV-2 Nsp9 by the NiRAN Domain. Nucleic Acids Res. 2021, 49, 8822–8835. [Google Scholar] [CrossRef] [PubMed]
- McClung, M.; Harris, S.T.; Miller, P.D.; Bauer, D.C.; Davison, K.S.; Dian, L.; Hanley, D.A.; Kendler, D.L.; Yuen, C.K.; Lewiecki, E.M. Bisphosphonate Therapy for Osteoporosis: Benefits, Risks, and Drug Holiday. Am. J. Med. 2013, 126, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Shen, Q.; Guo, W.; He, W.; Li, J.; Zhang, Y.; Wang, Y.; Zhou, Z.; Deng, D.; Ouyang, X.; et al. Clinical Characteristics of Elderly Patients with COVID-19 in Hunan Province, China: A Multicenter, Retrospective Study. Gerontology 2020, 66, 467–475. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, M.; Wang, Y.; Zheng, F.; Huang, Y.; Huang, K.; Yu, Q.; Cai, C.; Chen, D.; Tian, Y.; et al. Clinical Characteristics of Older and Younger Patients Infected with SARS-CoV-2. Aging (Albany. NY). 2020, 12, 11296–11305. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, V.; Iannacone, M. The Interplay of Drug Therapeutics and Immune Responses to SARS-CoV-2. Cell. Mol. Immunol. 2023, 21, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.T.; Clarke, B.L.; Khosla, S. Bisphosphonates: Mechanism of Action and Role in Clinical Practice. Mayo Clin. Proc. 2008, 83, 1032–1045. [Google Scholar] [CrossRef]
- Kunzmann, V.; Bauer, E.; Feurle, J.; Tony Florian Weißinger, H.-P.; Wilhelm, M.; Florian Weißinger, H.-P.; Wilhelm, M. Stimulation of Γδ T Cells by Aminobisphosphonates and Induction of Antiplasma Cell Activity in Multiple Myeloma. Blood 2000, 96, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Dou, H.; Zhang, X.; Uhagaze, D.S.; Ding, X.; Dong, Y. Determination of PKa Values of Alendronate Sodium in Aqueous Solution by Piecewise Linear Regression Based on Acid-Base Potentiometric Titration. J. Pharm. Anal. 2016, 6, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson−Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Liu, X.; Zhang, J.Z.H. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein-Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef]
- MacRaild, C.A.; Daranas, A.H.; Bronowska, A.; Homans, S.W. Global Changes in Local Protein Dynamics Reduce the Entropic Cost of Carbohydrate Binding in the Arabinose-Binding Protein. J. Mol. Biol. 2007, 368, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, S.C.J.; Kebriaei, R.; Dresser, L.D. Remdesivir: Review of Pharmacology, Pre-clinical Data, and Emerging Clinical Experience for COVID-19. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 659–671. [Google Scholar] [CrossRef]
- Cervelli, M.J.; Russ, G.R. Principles of Drug Therapy, Dosing, and Prescribing in Chronic Kidney Disease and Renal Replacement Therapy. In Comprehensive Clinical Nephrology; Elsevier, 2010; pp. 871–893. ISBN 9780323077668. [Google Scholar]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL Web Services: Streamlining Access to Drug Discovery Data and Utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for PK(a) Prediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Tanimoto, T.T. An Elementary Mathematical Theory of Classification and Prediction. In Proceedings of the Proc. IBM Internal Report; International Business Machines Corp; 1958; pp. 1–11. [Google Scholar]
- Koulgi, S.; Jani, V.; Uppuladinne, M.V.N.; Sonavane, U.; Joshi, R. Remdesivir-Bound and Ligand-Free Simulations Reveal the Probable Mechanism of Inhibiting the RNA Dependent RNA Polymerase of Severe Acute Respiratory Syndrome Coronavirus 2. RSC Adv. 2020, 10, 26792–26803. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, J.A. Molecular Dynamics Simulations of Biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Nosé, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tuckerman, M.E.; Tobias, D.J.; Klein, M.L. Explicit Reversible Integrators for Extended Systems Dynamics. Mol. Phys. 1996, 87, 1117–1157. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Klein, C.; Swails, J.M.; Yin, J.; Gilson, M.K.; Mobley, D.L.; Case, D.A.; Zhong, E.D. Lessons Learned from Comparing Molecular Dynamics Engines on the SAMPL5 Dataset. J. Comput. Aided. Mol. Des. 2017, 31, 147–161. [Google Scholar] [CrossRef]
- Ekberg, V.; Ryde, U. On the Use of Interaction Entropy and Related Methods to Estimate Binding Entropies. J. Chem. Theory Comput. 2021, 17, 5379–5391. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Lee, P.; Ng, C.; Slattery, A.; Nair, P.; Eisman, J.A.; Center, J.R. Preadmission Bisphosphonate and Mortality in Critically Ill Patients. J. Clin. Endocrinol. Metab. 2016, 101, 1945–1953. [Google Scholar] [CrossRef] [PubMed]
- Sing, C.; Kiel, D.P.; Hubbard, R.B.; Lau, W.C.; Li, G.H.; Kung, A.W.; Wong, I.C.; Cheung, C. Nitrogen-Containing Bisphosphonates Are Associated With Reduced Risk of Pneumonia in Patients With Hip Fracture. J. Bone Miner. Res. 2020, 35, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure of RdRp representing (a) Nsp 12 in complex with Nsp 7 and Nsp 8; (b) ribbon form representation of the Nsp 12 with different motifs; (c) the RdRp sequence alignment of MERS, SARS and SARS-CoV-2 representing the motifs highlighted in green, blue, and pink colors. The alignment shows highly conserved regions (*) among the three viruses (75%); the residues Asp760-761 are also conserved as they are essential for RNA synthesis.
Figure 1.
Structure of RdRp representing (a) Nsp 12 in complex with Nsp 7 and Nsp 8; (b) ribbon form representation of the Nsp 12 with different motifs; (c) the RdRp sequence alignment of MERS, SARS and SARS-CoV-2 representing the motifs highlighted in green, blue, and pink colors. The alignment shows highly conserved regions (*) among the three viruses (75%); the residues Asp760-761 are also conserved as they are essential for RNA synthesis.

Figure 2.
Representation of top hit molecules bound with the target protein (PDB ID: 6M71).
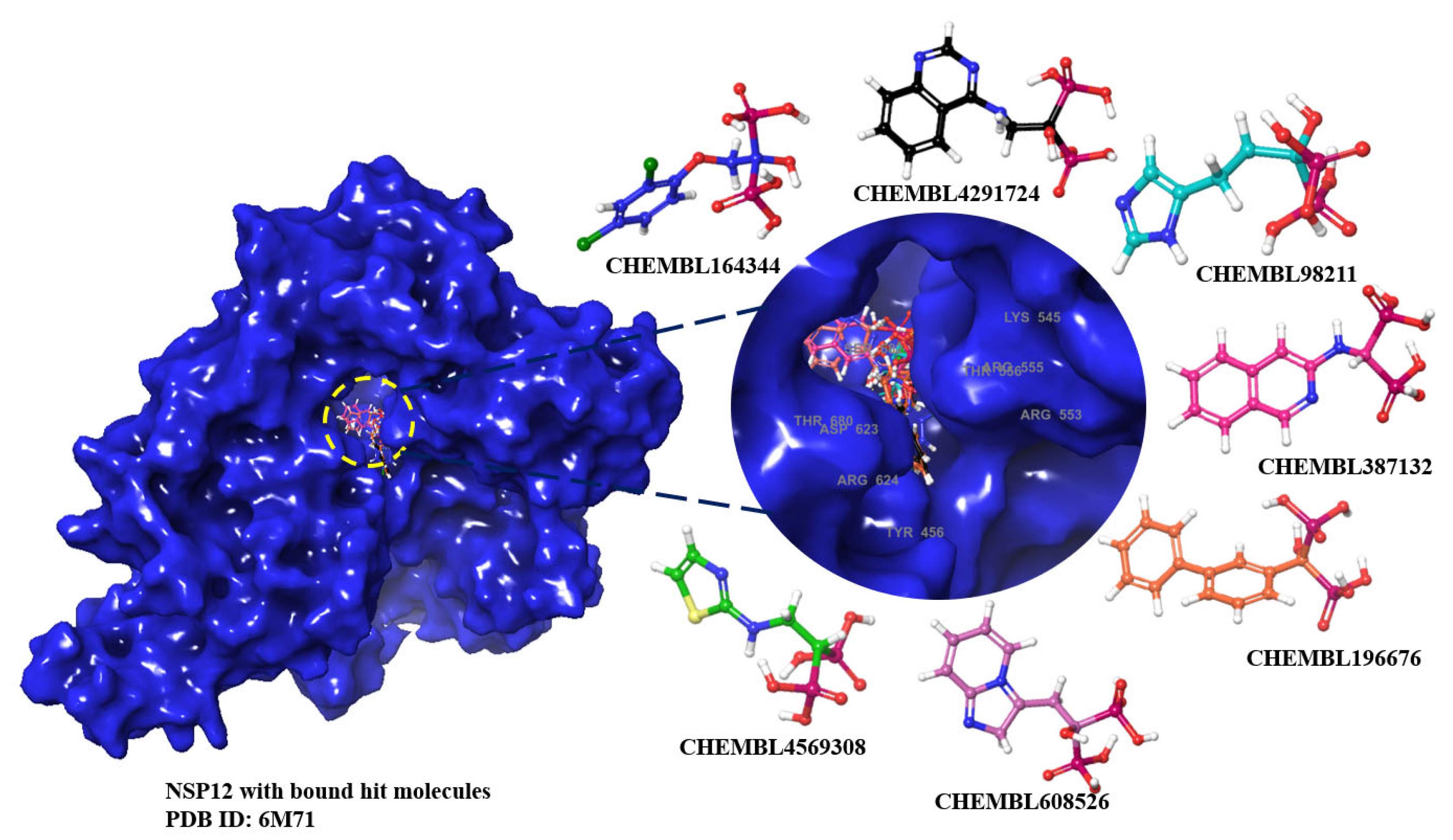
Figure 3.
RMSD plot of the hit ligands (above) and the protein (PDB ID: 6M71).
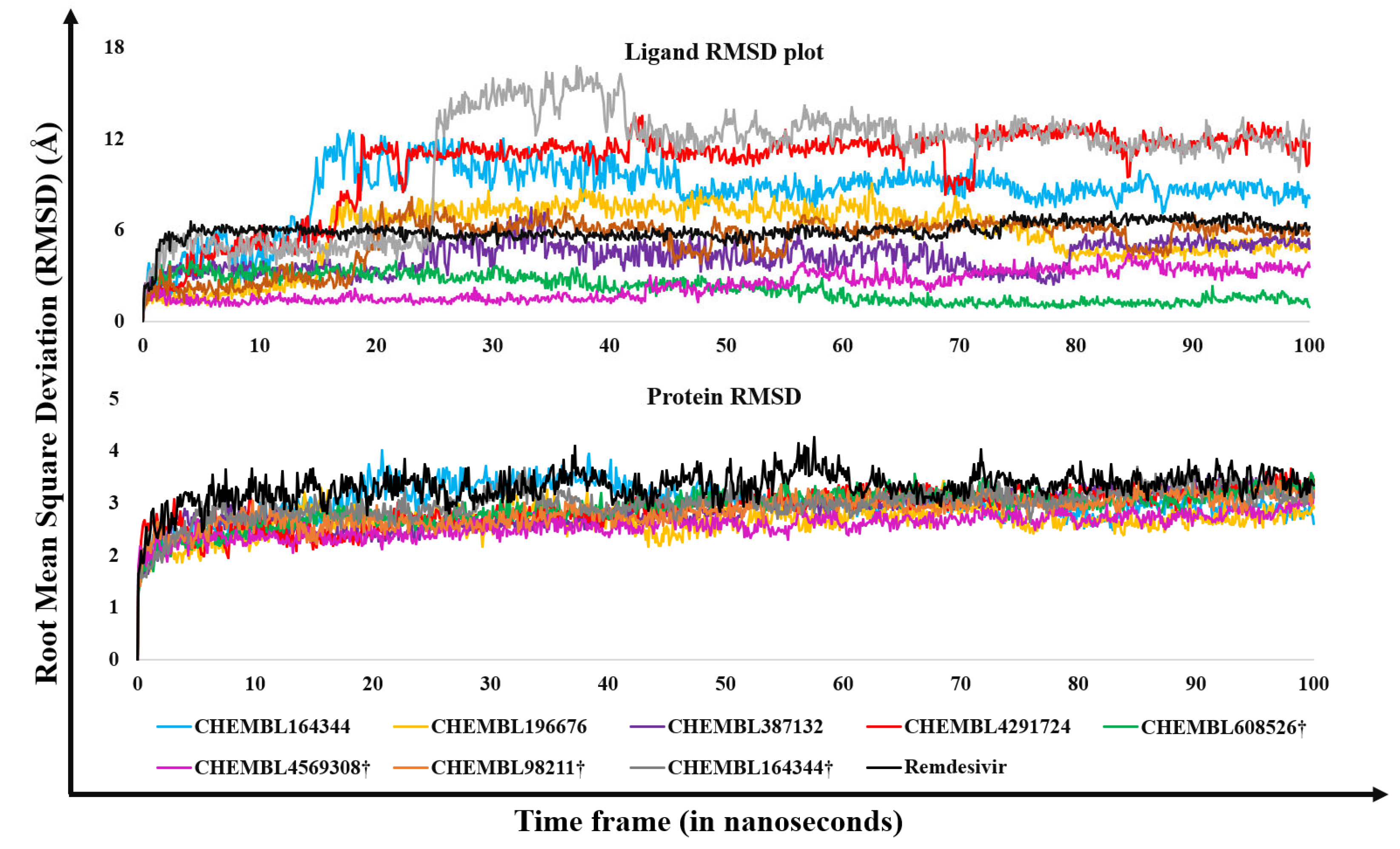
Figure 4.
RMSF plot of the protein (PDB ID 6M71) corresponding to top BP ligands, the reference drug (remdesivir) and the negative control (cinnamaldehyde). The fluctuations are shown in colored area plot, while the H-bond interactions are shown as histogram plot within the area. † Indicates protomers.
Figure 4.
RMSF plot of the protein (PDB ID 6M71) corresponding to top BP ligands, the reference drug (remdesivir) and the negative control (cinnamaldehyde). The fluctuations are shown in colored area plot, while the H-bond interactions are shown as histogram plot within the area. † Indicates protomers.
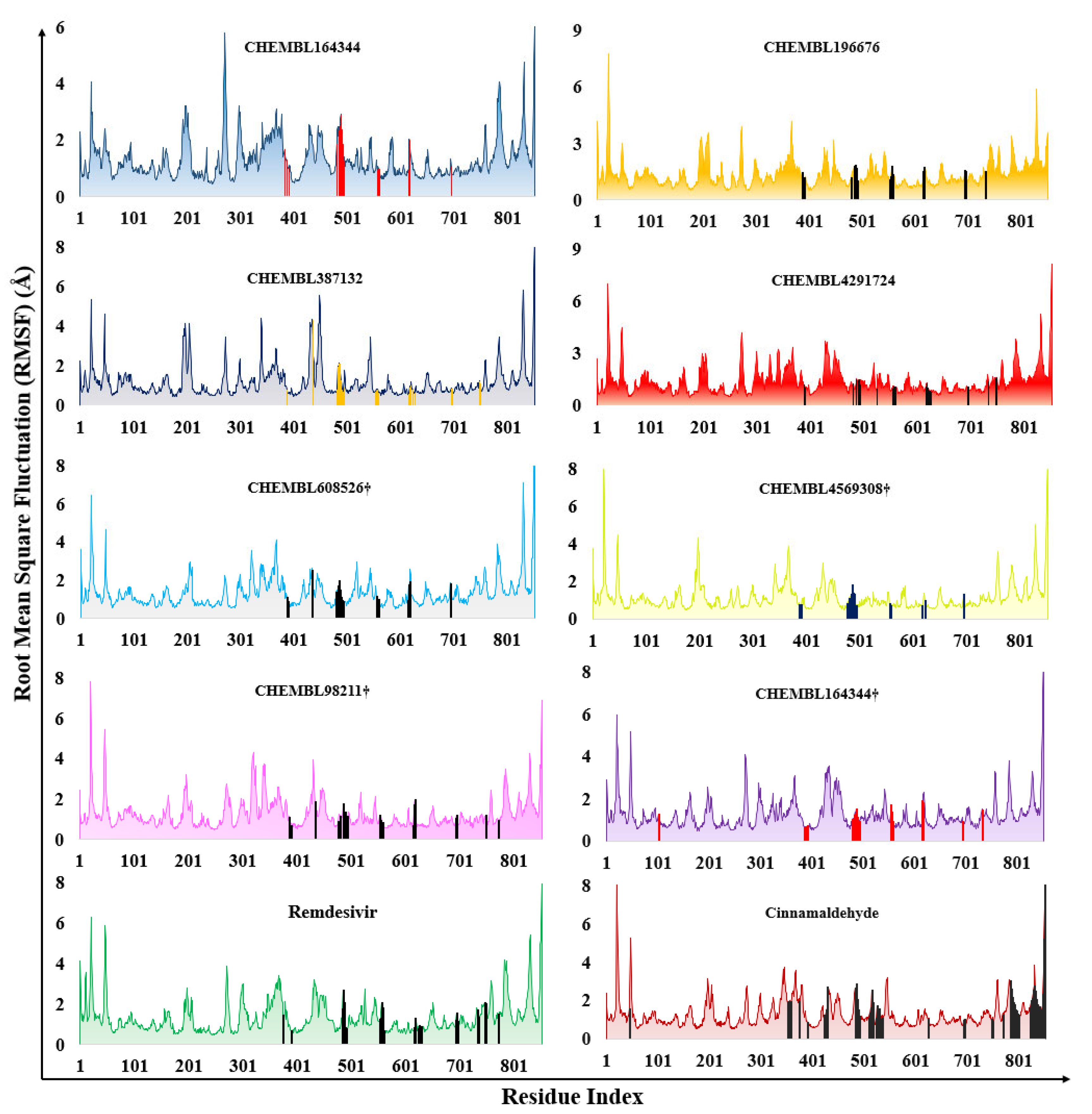
Figure 5.
Free energy plot showing the total energies of the system (histogram) and the binding free energies of the hit compounds (line graph) during the dynamics simulations; † = protomer form.
Figure 5.
Free energy plot showing the total energies of the system (histogram) and the binding free energies of the hit compounds (line graph) during the dynamics simulations; † = protomer form.
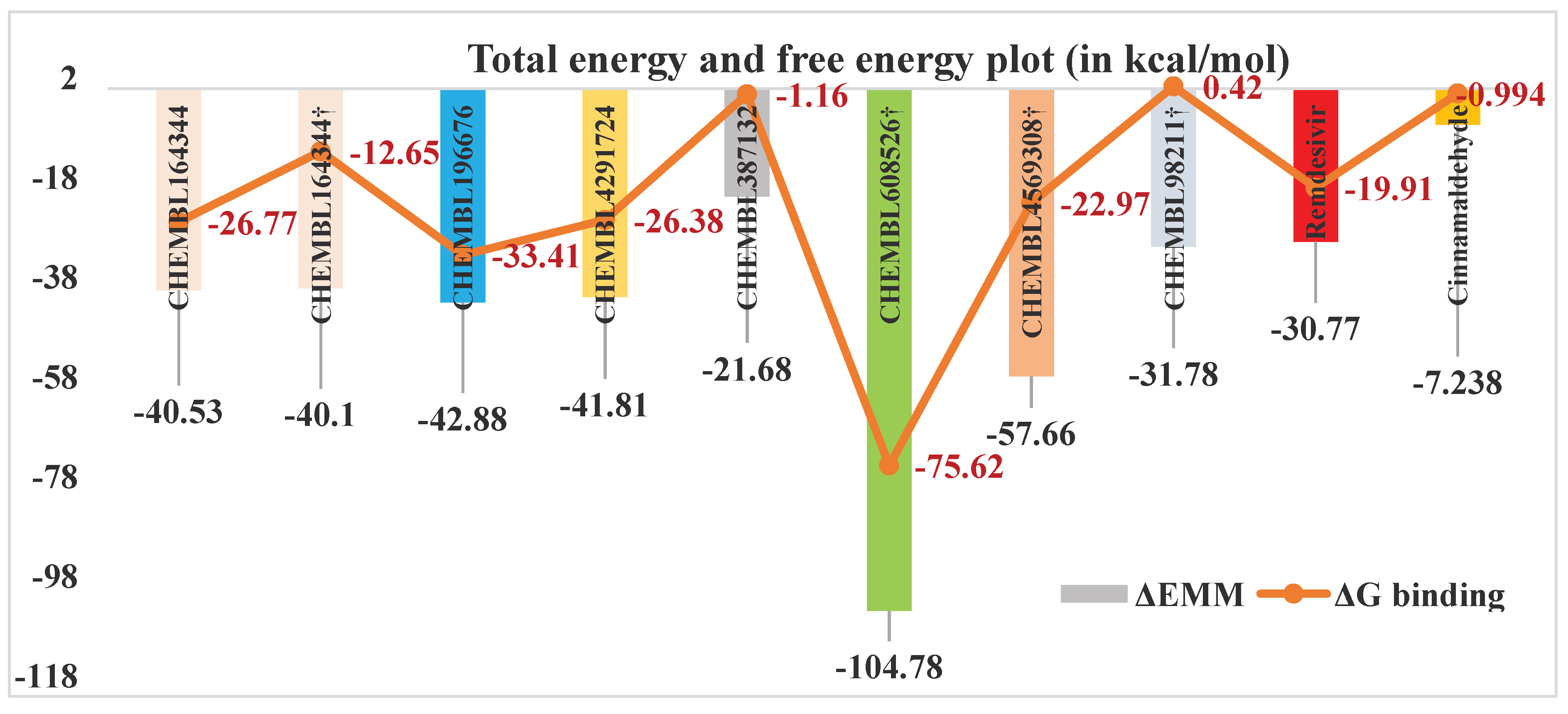

Table 1.
Molecular docking results of top 14 bisphosphonate ligands.
| S. No | ChEMBL ID | Score* | S. No | ChEMBL ID | Score* | S. No | ChEMBL ID | Score* | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CHEMBL1213265
|
-10.235 -7.111 † |
2 | CHEMBL608526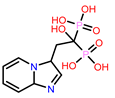
|
-9.706 -8.235 † |
3 | CHEMBL319144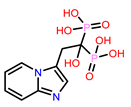
|
-9.657 -8.010 † |
|
| 4 | CHEMBL4802971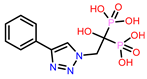
|
-9.355 -8.809 † |
5 | CHEMBL98211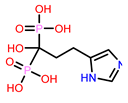
|
-9.347 -7.956 † |
6 | CHEMBL4291724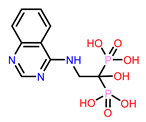
|
-9.308 -6.912 † |
|
| 7 | CHEMBL301247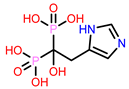
|
-9.219 -8.150 † |
8 | CHEMBL164344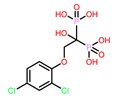
|
-9.213 -8.041 † |
9 | CHEMBL300361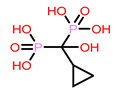
|
-9.151 -8.254 † |
|
| 10 | CHEMBL4289996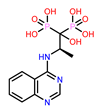
|
-9.119 -8.118 † |
11 | CHEMBL387132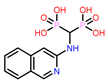
|
-9.11 -8.476 † |
12 | CHEMBL196676
|
-9.059 -7.558 † |
|
| 13 | CHEMBL4569308
|
-9.02 -8.208 † |
14 | CHEMBL338622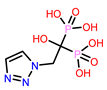
|
-9.014 -7.938 † |
15 | Cinnamaldehyde (Negative control) 
|
-1.707 | |
| 16 | Remdesivir (Reference drug) 
|
-3.270 |
17 | Favipiravir (Additional reference drug) 
|
-3.443 | 18 | Molnupiravir (Additional reference drug) 
|
-4.927 | |
* Represents docking score (in kcal/mol); † = protomer form.
Table 2.
MM-GBSA scores of the top-hit bisphosphonate ligands.
| S. no | Drugs | MM-GBSA dG Bind (kcal/mol) | |
|---|---|---|---|
| Uncharged state | at pH 7.0 ± 2.0 | ||
| 1. | CHEMBL1213265 | -7.74 | -33.25 |
| 2. | CHEMBL338622 | -24.14 | -26.52 |
| 3. | CHEMBL301247 | -24.22 | -27.73 |
| 4. | CHEMBL4289996 | -24.81 | -26.97 |
| 5. | CHEMBL98211 | -26.04 | -40.50† |
| 6. | CHEMBL300361 | -26.68 | -23.47 |
| 7. | CHEMBL608526 | -33.42 | -40.88† |
| 8. | CHEMBL319144 | -35.2 | -26.39 |
| 9. | CHEMBL4802971 | -36.77 | -39.13 |
| 10. | CHEMBL4569308 | -40.94† | -43.06† |
| 11. | CHEMBL4291724 | -41.51† | -34.06 |
| 12. | CHEMBL387132 | -43.28† | -38.34 |
| 13. | CHEMBL196676 | -44.14† | -37.10 |
| 14. | CHEMBL164344 | -46.65† | -46.73 |
| 15. | Favipiravir | -19.13 | -- |
| 16. | Molnupiravir | -34.13 | -- |
| 17. | Remdesivir | -40.32 | -- |
| 18. | Cinnamaldehyde | -30.05 | -- |
† MM-GBSA scores lower than that of remdesivir are highlighted in bold.
Table 3.
Molecular docking results of the hit ligands along with reference drug and a negative control.
Table 3.
Molecular docking results of the hit ligands along with reference drug and a negative control.
| S. No | ChEMBL ID | S. No | ChEMBL ID | S. No | ChEMBL ID | |
|---|---|---|---|---|---|---|
| 1 | CHEMBL4291724 Score: -9.308 kcal/mol 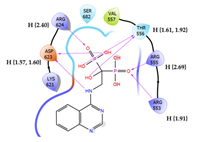
|
5 | CHEMBL608526 † Score: -8.235 kcal/mol 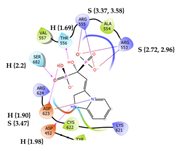
|
9 | Favipiravir Score: -3.443 kcal/mol 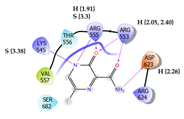
|
|
| 2 | CHEMBL164344 Score: -9.213 kcal/mol 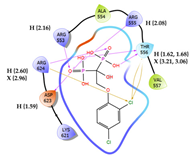
|
6. | CHEMBL98211† Score: -7.956 kcal/mol 
|
10 | Molnupiravir Score = -4.927 kcal/mol 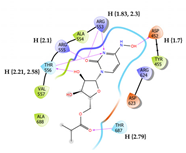
|
|
| 3 | CHEMBL387132 Score: -9.110 kcal/mol 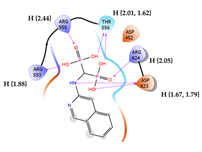
|
7 | CHEMBL4569308 † Score: -8.208 kcal/mol 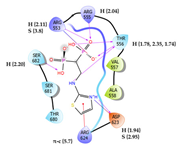
|
11 |
Cinnamaldehyde Score: -1.707 kcal/mol 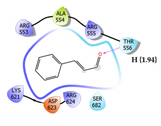
|
|
| 4 | CHEMBL196676 Score: -9.059 kcal/mol 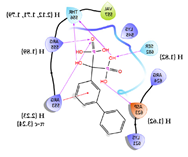
|
8 | Remdesivir Score: -3.270 kcal/mol 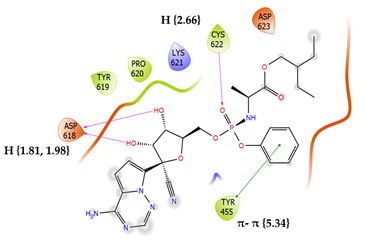
|
|||
H = Hydrogen bond; S = Salt bridge; π-c = pi-cation; π-π = pi-pi stacking; X = halogen bond; {} = bond length in Å.
Table 4.
Entropy results of the top hit compounds showing the entropy contributions, total energies of the system, and the total binding free energies of the ligand at different time frames.
Table 4.
Entropy results of the top hit compounds showing the entropy contributions, total energies of the system, and the total binding free energies of the ligand at different time frames.
| Compounds | I.E. = -TΔS | Total energy contributions (ΔEMM) | ΔGbinding | ||||
|---|---|---|---|---|---|---|---|
| ΔEvdW | ΔEEL | ΔEPB | ΔENP | ΔEMM = ∑ ΔE | |||
| CHEMBL196676 | 9.48 | -13.06 | -90.55 | 63.70 | -2.98 | -42.88 | -33.41 |
| CHEMBL164344 | 13.76 | -22.77 | -93.71 | 79.14 | -3.19 | -40.53 | -26.77 |
| CHEMBL4291724 | 15.43 | -13.47 | -123.47 | 97.86 | -2.73 | -41.81 | -26.38 |
| CHEMBL387132 | 20.52 | -20.15 | -87.58 | 88.93 | -2.88 | -21.68 | -1.16 |
| CHEMBL608526† | 29.16 | 6.03 | -620.27 | 512.53 | -3.13 | -104.78 | -75.62 |
| CHEMBL4569308† | 34.69 | -3.58 | -373.84 | 322.48 | -2.71 | -57.66 | -22.97 |
| CHEMBL164344† | 27.45 | -11.38 | -133.86 | 107.72 | -2.57 | -40.10 | -12.65 |
| CHEMBL98211† | 32.20 | 1.81 | -253.57 | 222.44 | -2.46 | -31.78 | 0.42 |
| Remdesivir | 10.86 | -52.27 | -64.85 | 92.31 | -5.95 | -30.77 | -19.91 |
| Cinnamaldehyde | 6.24 | -11.16 | -3.24 | 8.20 | -1.03 | -7.24 | -0.99 |
I.E. = Interaction entropy; ΔEMM = Total energy contributions of the system; ΔGbinding = binding free energy; † protomer form.
Table 5.
Results of in silico predicted ADMET profiles of hit bisphosphonate molecules.
| ADMET parameters | Hit molecules | |||||||||||||
| CHEMBL → | 4291724 | 164344 | 387132 | 196676 | 98211 | 608526 | 4569308 | Remdesivir | ||||||
| Absorption | WS (log mol/L) | -2.47 | -2.152 | -2.046 | -3.677 | -2.896 | -1.444 | -1.641 | -3.07 | |||||
| CP (log Papp in 10-6 cm/s) | -0.438 | -0.461 | 0.093 | 1.245 | -0.295 | 0.334 | -0.527 | 0.635 | ||||||
| IA (% Absorbed) | 42.008 | 16.774 | 71.627 | 38.34 | 24.799 | 34.854 | 43.351 | 71.109 | ||||||
| S.P. (log Kp) | -2.735 | -2.743 | -2.759 | -2.735 | -2.735 | -2.736 | -2.882 | -2.735 | ||||||
| P-glyco protein | Substrate | No | No | No | Yes | Yes | No | No | Yes | |||||
| Inhibitor | I | No | No | No | Yes | No | No | No | Yes | |||||
| II | No | No | No | No | No | No | No | No | ||||||
| Distribution | V.D. ss (log L/kg) | -0.768 | -0.558 | -0.814 | 0.578 | -0.84 | -0.439 | -0.641 | 0.307 | |||||
| F.U. (Fu) | 0.331 | 0.469 | 0.516 | 0.028 | 0.655 | 0.811 | 0.6 | 0.005 | ||||||
| BBB (log BB) | -2.302 | -2.284 | -1.86 | -1.908 | -2.489 | -1.522 | -2.541 | -2.056 | ||||||
| CNS (log P.S.) | -4.756 | -3.881 | -4.034 | -3.879 | -6.045 | -4.881 | -4.528 | -4.675 | ||||||
| Metabolism | CYP action | Substrate | 2D6 | No | No | No | No | No | No | No | No | |||
| 3A4 | No | No | No | No | No | No | No | Yes | ||||||
| Inhibition against 1A2, 2C19, 2C9, 2D6, 3A4 | No | No | No | No | No | No | No | No | ||||||
| Excretion | T.C. (log ml/min/kg) | 0.146 | 0.032 | -0.025 | -0.119 | 0.664 | 0.374 | 0.143 | 0.198 | |||||
| ROC | No | No | No | No | No | No | No | No | ||||||
| Toxicity | Ames assay | No | No | No | Yes | No | No | No | No | |||||
| MTD (log mg/kg/day) | 0.841 | 0.445 | 0.689 | 0.574 | -0.312 | 0.444 | 1.142 | 0.15 | ||||||
| hERG I inhibitor | No | No | No | No | No | No | No | No | ||||||
| hERG II inhibitor | No | No | No | Yes | No | No | No | Yes | ||||||
| Rat oral toxicity | Acute (LD50) (mol/kg) | 2.67 | 2.613 | 1.886 | 3.117 | 2.564 | 2.374 | 2.738 | 2.043 | |||||
| Chronic (LOAEL) (Log mg/kg_bw/day) |
3.195 | 3.597 | 3.052 | 3.404 | 4.773 | 3.253 | 3.826 | 1.639 | ||||||
| HT | Yes | No | Yes | No | No | Yes | No | Yes | ||||||
| SS | No | No | No | No | No | No | No | No | ||||||
| TT (log ug/L) | 0.285 | 0.285 | 0.288 | 0.285 | 0.285 | 0.285 | 0.285 | 0.285 | ||||||
| MT (log mM) | 2.992 | 1.686 | 2.938 | 0.427 | 2.709 | 2.355 | 2.875 | 0.291 | ||||||
WS: Water Solubility, CP: Caco2 permeability, IA: Intestinal absorption (human), S.P.: Skin Permeability, V.D. ss: Volume of distribution in steady state, F.U.: Fraction unbound (human), T.C.: Total Clearance, ROC: Renal OCT2 substrate, MTD: Max. Tolerated dose (human), LD50: Lethal dose at 50% concentration, LOAEL: Lowest observed Adverse effect level, HT: Hepatotoxicity, SS: Skin Sensitization, TT: T. Pyriformis toxicity, MT: Minnow toxicity.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



