
Submitted:
14 November 2023
Posted:
23 November 2023
You are already at the latest version
Abstract
Cholesterol biosynthesis inhibitors (statins) protect hypercholesterolemic patients from developing active tuberculosis, suggesting that these drugs could help the host to control the pathogen at the initial stages of the disease. This work studies the effect of fluvastatin on the early response of healthy peripheral blood mononuclear cells (PBMC) to inactivated Mycobacterium tuberculosis (Mtb) H37Ra. We found that in fluvastatin-treated PBMC, most monocytes/macrophages became foamy cells that overproduce NLRP3 inflammasome components in the absence of immune stimulation, evidencing important cholesterol metabolism/immunity connections. When both fluvastatin-treated and untreated PBMC were exposed to Mtb, a subset of macrophages quickly captured large amounts of bacilli and died, concentrating the bacteria in necrotic areas. In fluvastatin-untreated cultures, most of the remaining macrophages became epithelioid cells that isolated these areas of cell death in granulomatous structures that barely produced IFN-gamma. By contrast, in fluvastatin-treated cultures, foamy macrophages surrounded the accumulated bacteria to degrade them, markedly activated caspase-1 and elicited a potent IFNgamma/cytotoxic response. In rabb-gammaits immunized with the same bacteria, fluvastatin therapy increased the tuberculin test response. We conclude that statins enhance macrophage effectiveness to control Mtb supported by adaptive immunity, offering a promising tool in the design of alternative therapies to fight tuberculosis.
Keywords:
tuberculosis
; statins
; host-directed therapy
; foam cells
; granulomas
; NLRP3 inflammasome
; mevalonate-kinase deficiencies
1. Introduction
The bacillus Mtb is the etiological agent of tuberculosis, one of the leading causes of death worldwide throughout history [1]. In response to Mtb infection, host immunity forms granulomas, an ancient response that restricts and kills the pathogen with variable efficacy [2]. Different authors have evidenced that virulent Mtb strains exploit early stages of granulomas formation for their proliferation and expansion, also restricting the development of a specific immune response, determining the course of the disease [3]. Studies in mice have revealed that in the incipient granuloma foamy macrophages (FM), recruited to clean the immune debris generated, become infected by Mtb. The pathogen interferes with lipid metabolism of these infected cells, converting them to lipid-laden FM (LL-FM) unable to contain the infection [4]. In these macrophages, the bacilli induce the accumulation of cholesterol, cholesteryl ester, and triacylglycerides, and use them as carbon and energy sources, and to upregulate genes associated with production of virulence factors, evasion of immunity and drug resistance. In this way, the pathogen persists latently in the infected macrophages [5,6]. In human granulomas, numerous LL-FM locate at the interface region nearest to the caseum where Mtb concentrates [7]. When they die, they deliver their lipidic cargo to these necrotic areas, creating a niche where the bacilli find a lipid-rich environment, surviving in them protected from host immunity [8]. Overtime, these LL-FM become key players for the persistence of Mtb in the granuloma, contributing to its destabilization and to bacilli dissemination, thus representing an important therapeutic target in fighting tuberculosis [9,10].
In the search of pre-existing drugs that could be efficient against Mtb, statins, the cholesterol biosynthesis inhibitors prescribed to hypercholesterolemic patients, have shown promising beneficial effects [11,12,13]. Different systematic reviews and meta-analyses, strongly associate the use of statins with a lower risk of developing active tuberculosis [14,15,16], suggesting that these drugs could help the host to control the pathogen at the first stages of the infection. At an experimental level, different studies have shown that statins reduce the severity of infectious diseases caused by Mtb and other pathogens not only by reducing cholesterol levels (both in the host and in pathogens), but also by exerting poorly known immunomodulatory actions [17,18,19]. Statins inhibit the activity of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme of cholesterol biosynthesis, also interfering in the synthesis of other isoprenoids, which are essential molecules for different cellular processes [20,21]. Two decades ago, we were pioneers in proposing that statins could be an adjuvant therapy for tuberculosis by promoting caspase-1 activation and the release of interleukin (IL)-1β and IL-18 [22], two essential ILs for Mtb control [23]. More recently, different studies have provided evidence that statins prevent the prenylation of some unknown proteins that negatively regulate the assembly of inflammasomes [24], the structures that activate caspase-1 [25]. Inflammasomes assembly requires two cell signals. Signal 1 (priming/licensing) is generated by membrane pattern recognition receptors (PRRs) after sensing extracellular pathogen associated molecular patterns (PAMPs), and/or danger associated molecular patterns (DAMPs). This signal induces up-regulation of the inflammasome components, pro-caspase-1 and pro-IL-1β. Signal 2 (assembly) is mediated by PAMPs, DAMPs, cellular stressors and even metabolic perturbations that induce intracellular events leading to a conformational rearrangement of a specific cytosolic PRR, initiating the activation of their corresponding inflammasome. Most of these structures oligomerize and assemble with ASC (apoptosis-associated speck-like protein containing a CARD), an adapter protein that recruits pro-caspase-1 to the assembling inflammasome. Then, the proximity of procaspases-1 induces their dimerization, initiating a complex autoproteolytic process that activates the enzyme [26,27,28]. Some authors have proposed that virulent Mtb strains impede the activation of the NLRP3 inflammasome, predisposing the host to acquire the disease [29,30,31]. Besides, human mutations linked to enhanced NLRP3 inflammasome activity limit Mtb growth in the infected macrophages [32].
The aim of this work was to study the effect of the drug fluvastatin on the early immune response elicited by peripheral blood mononuclear cells (PBMC) from healthy donors to inactivated Mtb H37Ra (as a source of Mtb PAMPs), in the context of NLRP3 inflammasome and caspase-1 activation. We found that, in fluvastatin untreated PBMC, macrophages respond to the bacteria by forming granulomatous structures that scarcely activate the NLRP3 inflammasome and caspase-1, resulting in low production of IFNγ. Unexpectedly, the drug induces a generalized conversion of monocytes/macrophages of PBMC into foamy cells overproducing NLRP3 and ASC in the absence of PAMPs, revealing new cholesterol metabolism/inflammation connections [33]. Based on it, we propose the existence of a cholesterol metabolism/inflammation integrated circuit regulating the activation of NLRP3 inflammasome, which could explain some autoinflammatory processes. In response to the bacteria these foamy macrophages did not form compact structures, but degraded the bacilli, markedly exacerbated caspase-1 activation and elicited a potent IFNγ/cytotoxic response. The effect of the drug on IFNγ production was confirmed in vivo by performing a tuberculin test to rabbits receiving or not fluvastatin therapy, and immunized with inactivated Mtb H37Ra. This work offers a useful experimental model to study the early events that initiate the formation of granulomas and the role that FM play in it, also opening new possibilities in the design of alternative therapies to fight not only tuberculosis, but other infectious diseases.
2. Materials and Methods
2.1. Mtb strain selection
We selected Mtb H37Ra as a source of Mtb PAMPs since this strain lacks virulence factors and shares most of their membrane proteins with their virulent counterpart Mtb H37Rv, what confer them the capacity to elicit a similar profile of cytokines [34,35]. Furthermore, differences in virulence between these strains have been attributed to their different abilities to interfere in the lipid metabolism of macrophages, regardless of their immunogenicity [36]. In addition as heat-killed or living Mtb H37Ra bacilli are equally effective inducers of a granulomatous response [37], thus we used an inactivated bacteria in order to differentiate immune effects mediated by the drug from those derived from a reduction in the availability of cholesterol by the bacteria.
2.2. Isolation of peripheral blood mononuclear cells (PBMC) and culture conditions
PBMC were isolated from blood buffy coats from de-identified samples (without any direct or indirect personal identifiers) of healthy blood donors, provided by the Hematology Service from Hospital La Paz and from Centro de Transfusiones de la Comunidad de Madrid (Madrid, Spain), in accordance to Spanish legislation (BOE-A-2007-12945, Ley 14/2007, de 3 de julio, de Investigación Biomédica). Protocols in the study were approved by the Hospital Universitario Ramón y Cajal Ethics Committee (approval code 188/09) in accordance with national and international guidelines. Buffy coats diluted 1:10 in sterile phosphate-buffer saline (PBS) were layered onto Lymphoprep (a ficoll medium at density 1.077 g/ml, from Nycomed, Oslom, Norway), to isolate PBMC following the method of Boyum et al. [38]. PBMC were then re-suspended at a final density of 2x106 cells/ml in RPMI 1640 (Gibco, CA, USA) supplemented with 10% heat inactivated fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 U/ml streptomycin and 10 μg gentamycin. Aliquots of 2ml were seeded on 12-well cell culture plates. Half of the wells were supplemented with 5 μM fluvastatin (Novartis Pharmaceutical, Basel, Switzerland) dissolved in DMSO to obtain a final concentration of 0.04% in the incubation medium. In the remaining wells we added the same amount of DMSO. Plates were incubated at 37°C in a humidified atmosphere containing 5% CO2 in air for 15h and, subsequently, half the wells of each condition were stimulated with 25 μg/ml of heat-inactivated Mtb H37Ra (Difco, DE, USA), re-suspended in PBS and the incubation was resumed for an additional 24h period. We analyze the evolution of the cells in the different cultures as follows: adherent cells from each well were scraped and harvested together with cells in suspension at 1, 4, 8, 12 and 24 hours after immune stimulation, which correspond to 16, 19, 21, 23, 25, 27 and 39 hours of incubation of both control and fluvastatin-treated cells. After that, aliquots of 100 μl from each one of the collected samples were cytocentrifuged onto microscope slides (Flex Immunohistochemistry microscope slides, DakoCytomation, Denmark) by using a Shandon Cytospin 4 cytocentrifuge (Thermo, UK). Cytospin preparations were stained with May-Grünwald-Giemsa solutions (Merck, NJ, USA) and analyzed under optical microscopy.
2.3. Transmission Electron Microscopy
PBMC were isolated and incubated under conditions described above. Thereafter, cells were processed for TEM following the method of Reynolds et al. [39]. Briefly, at the end of the incubation, cells from the different conditions were fixed in 2 % glutaraldehyde 4% paraformaldehyde in PBS overnight, post-fixed in 1% osmium tetroxide in water for 1h and dehydrated through a series of ethanol solutions (30%, 50%, 70%, 95%, 100%). After the last dehydration step, samples were incubated in a series of 2:1, 1:1, 1:2 ethanol and EPON resin mixture and finally embedded in EPON resin at 60ºC for 48h. Ultrathin sections (50-60 nm) were obtained using a diamond knife (Diatome, Hatfield, PA, USA) in an ultramicrotome (Leica Reichert Ultracut S, UK) and collected on 200-mesh copper grids. The sections were counterstained with 2% uranyl acetate in water for 20 min followed by a lead citrate solution for 10 min. Samples were analyzed using a transmission electron microscope (Jeol Jem1010 (100Kv) Tokyo, Japan), equipped with a digital camera (Gatan SC200 Pleasanton, CA, USA).
2.4. Immunolocalization of NLRP3 and ASC
Cultures were prepared as above and at the end of the incubation period, the cells were re-suspended. Aliquots of 100 μl of cells from each condition were cytocentrifuged onto microscopy slides and then immersed in 4% paraformaldehyde for 20 min at room temperature. After three washes with PBS, cells were permeabilized with 0,05% Triton X-100 in PBS for 5 min at 4°C, and washed three times in PBS. Thereafter, slides were incubated in a blocking solution (5% normal goat serum in PBS) for 30 min at room temperature, to prevent unspecific binding of specific antibodies. In the next step cells were incubated overnight, at 4ºC, with mouse monoclonal anti-NLRP3 antibody (Ag 20B-0014-C100, Adipogen Corp., San Diego, CA, USA) and rabbit polyclonal anti-ASC antibody (sc 22514R, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) diluted at 1/200 and 1/50 respectively, in 0,5% normal goat serum and 0,001% Triton X-100 in PBS. After primary antibodies incubation, cells were washed three times with PBS and then incubated for 45 min at room temperature with fluorescent-conjugated secondary antibodies diluted 1/400 in 0.5% normal goat serum in PBS. The following secondary antibodies were used: Alexa Fluor-568 goat anti-mouse IgG, and Alexa Fluor-488 goat anti-rabbit IgG (both from Molecular Probes, Eugene, OR, USA). Cell nuclei were stained with 3x10-5 M Hoechst 33342 (Sigma-Aldrich, St. Louis, MO, USA) incorporated to an aqueous mounting media containing 1 mg/ml p-phenylene diamine and 90% glycerol in PBS. Fluorescent immunostained images were acquired by using a Nikon Eclipse E 400 microscope (Nikon Corporation, Tokyo, Japan).
2.5. FAM-FLICA assay
We localize active caspase-1 in living cells, by performing a FAM-FLICA (Fluorescent Labeled Inhibitor of Caspases) assay (ImmunoChemistry Technologies Bloomington, MN, USA). The assay requires the use of DMSO to solve the fluorescent inhibitor probe FAM-YVAD, but high concentrations of this solvent cause cells damage and activate caspase-1 [40]. To optimize DMSO concentration in the cultures, we reduced 10 times the concentration of DMSO recommended by the supplier to solve the inhibitor. At the end of the incubation,10 μl of this solution was added to 300 μl of cell suspension (containing 6x105 cells) from each condition, and gently flicked the tubes. The cells were incubated at 37°C for 1h protected from light. To ensure an even distribution of the substrate, cells were gently re-suspended every 15 min. After that, cells were centrifuged at 200 x g for 5 min, the pellets were washed twice in 1,5 ml of PBS with 0.04% DMSO and finally were re-suspended in 300 μl of PBS. Thereafter, aliquots of 50μl and 100 μl of cells in suspension from each condition were cytocentrifuged onto microscopy slides as above mentioned. Cell nuclei were stained with 3x10-5 M Hoechst 33342 (Sigma-Aldrich, St. Louis, MO, USA) incorporated to an aqueous mounting media containing 1 mg/ml p-phenylene diamine and 90% glycerol in PBS. Cytospin preparations were then analyzed under fluorescence microscopy by using a Nikon Eclipse E 400 microscope (Nikon Corporation, Tokyo, Japan).
Mtb emits autofluorescence that can be used as a tool for detection of the bacilli in biological samples [41] but, in our study, green autofluorescence interferes with specific fluorescence emitted by FAM-YVAD. To analyze the results of the assay, we merged red and green autofluorescence. This way we can localize active caspase-1 (green fluorescence) and visualize simultaneously the bacteria (as merged yellow fluorescence). To prevent autofluorescence removal during washed procedures, we used a detergent-free washing solution, after evaluating that it did not change assay specificity. It is important to say that such colocalization could not be performed by using Zeihl-Neelsen acid-fast stain because solvents used in this technique suppress specific ligand fluorescence. On the other hand, immunolocalization of the bacteria requires cell permeabilization and washes that could affect FAM-FLICA specificity.
2.6. Dynamic of caspase-1 activation
A fluorimetric cleavage micro-assay, previously described by us [42], was used to evaluate caspase-1 activation over time. PBMC from the different conditions were collected at 1, 4, 6, 8, 10, 12, and 24 hours after Mtb stimulation. At each time, adherent cells were scraped, mixed with the cells in suspension and centrifuged. The collected pellets were lysed via three consecutive freeze-thaw cycles in lysis buffer (25 mM HEPES pH 7.5, 5 mM EDTA, 5 mM MgCl2, 0.1% Nonidet P-40, supplemented with 1 mM PMSF, 1 µg/ml aprotinin and 50 µg/ml antipain), using 50 µl for every 12x106 cells. Then, lysates were centrifuged at 10.000 x g for 15 min at 4°C, and the supernatants collected for protein concentration determination. For each sample, a total of 100 mg of protein in 50 ml of lysis buffer was added to 175 ml of reaction solution (25 mM HEPES pH 7.5, 5 mM EDTA, 5 mM MgCl2, 22.9% glycerol, 0.15% CHAPS, 11.5 mM DTT, 175.5 mM NaCl). The prepared samples were placed in microtiter plates Fluoronunc F16 black polysorp (Nalge Nunc International, Rochester, NY USA), in order to incubate them at 37°C for 2h in the presence of 100 µM Ac-WEHD-AMC (Calbiochem, CA, USA), a specific substrate for caspase-1 that emits fluorescence after its enzymatic cleavage by this protease. Thereafter caspase-1 activity was quantified in each condition by measuring the emitted fluorescence in a fluorometric plate reader (Spectrafluor, Tecan, Spain), using an excitation wavelength of 380 nm and an emission wavelength of 465 nm. The specificity of the reaction was assessed by adding equimolar amounts of AC-YVAD-CHO (Bachem, Bubendorf, Switzerland), a specific caspase-1 inhibitor, obtaining a 95% of inhibition of the reaction.
2.7. Kinetic analysis of cytokines emission
We evaluated the production over time of IL-1β, IL-18, IL-10, and IL-12, by cells maintained at the different incubation conditions of this study. We followed the same experimental protocol than for caspase-1 activation dynamic but in this case, we collected cell supernatants. Cytokine quantification was performed by Enzyme-Linked Immunosorbent Assay (ELISA), using commercially available kits and following the manufacturer’s indications. For IL-1β and IFNγ determination, different kits were used and were supplied by R&D Systems (MN USA), and Genzyme (MA, USA), as well as ELI-PAIR kits from Diaclone Research (France). ELISA kits for IL-18 were supplied by MBL International Corporation (MA, USA). ELISA kits for IL-12 and IL-10 were supplied by R&D Systems (MN, USA) and by Diaclone Research (France).
2.8. Lactate dehydrogenase (LDH) determination
To measure cell death occurring under the different incubation conditions along the incubation period, we prepared cultures following the same experimental protocol than for the kinetic studies described above, but using phenol red-free RPMI 1640. After immune stimulation, supernatants were collected at the same intervals than above and LDH activity was measured by using the CytoTox 96 Kit (Promega, WI, USA), according to the manufacturer’s instructions. Cytotoxicity was expressed as a percentage of the maximum LDH release in control conditions.
2.9. Contribution of IL-12, IL-1β and IL-18 to IFNγ production
PBMC were placed on 12-well plates and treated or not with 5μM fluvastatin as above. To different wells from both conditions, we added specific antibodies against: IL-1β (1,25 μg/ml, C20 sc 1250), IL-18 (1 μg/ml, C18 sc-6177) or IL-12 p35 (0,5 μg/ml, C19 sc-1280), all of them from Santa Cruz Biotechnology, TX USA. To other wells we added the specific inhibitor of Caspase-1, Ac-YVAD-CHO (Bachem AG, Switzerland) at a concentration of 100 μM. Thereafter, plates were incubated for 15h and then, half of the aliquots from each condition were stimulated with 25 μg/ml of heat-inactivated Mtb H37Ra, and the incubation was resumed for a further 24h. At the end of the process, supernatants were collected and IFNγ quantified by ELISA (R&D Systems, MN USA).
In an independent study, PBMC were treated or not with 5 M fluvastatin as above and supplemented with recombinant human IL-12 (Pepro Tech, Ec LTD, UK) at the following concentrations: 0,01; 0,02; 0,10; 10 or 100 ng/ml. Thereafter, cells were incubated according to our basic experimental protocol. At the end of the process, supernatants were collected for IFNγ quantification by ELISA (R&D Systems, MN, USA).
2.10. Intracellular IFNγ determination by flow cytometry
For surface staining, PBMC from the different conditions were incubated at a density of 8x105 cells/100 μl for 30 min with mixtures of fluorochrome-conjugated mAbs (or isotype-matched controls). The following conjugated mAbs were used: anti-human CD3 APC, anti-human CD8 PerCP and anti-human CD56 PE, all obtained from Becton Dickinson (BD Biosciences, CA, USA). Then, cells were
fixed and permeabilized by using the Intra Stain kit from DAKO (Denmark). We avoided the use of protein transport inhibitors in our cytometry study, because its interference with fluvastatin [43].
Once permeabilized, the cells were washed and incubated for 30 min with anti-human IFNγ conjugated to FITC from Becton Dickinson (BD Biosciences, CA, USA). Four colour analyses were performed using a FACSCalibur flow cytometer from Becton Dickinson (BD Biosciences, CA, USA), with 10.000 events collected for each tube. The cells were gated using the forward scatter (FSC) and side scatter (SSC) to select the lymphocyte population. The intracellular production of IFNγ was studied on 3 different cells types: CD3+CD8+ cells, CD3+CD8- cells and CD3-CD56+ cells. The analysis was performed using the CellQuest Pro TM Software from Becton Dickinson (BD Biosciences, San Diego, USA).
2.11. Rabbit treatment and immunization
Five New Zealand white rabbits (2 months old and weighing 2 Kg) were fed with a standard chow diet and another five were fed with the same food but supplemented with 2mg/kg/day of fluvastatin (Novartis Pharmaceutical, Basel, Switzerland) as follows: the drug was dissolved in acetone and mixed with food, allowing the solvent to evaporate from the mixture before offering it to animals. After 15 days of treatment, all rabbits were immunized by intramuscular inoculation of 5 mg of heat-inactivated Mtb H37Ra emulsified in incomplete Freund adjuvant (DIFCO). Fifteen days later rabbits were boosted with the same doses. Forty-five days later we performed a tuberculin skin test (PPD Evans, AJ Vaccines, Copenhagen Denmark), by injecting an intradermal dose of 2 tuberculin units (TU- 0.1 ml). The reaction was read 72 h after injection by measuring with a caliper the skin fold thickness in the area of inoculation. Animal experimentation was performed in accordance with Spanish legislation on the protection of animals used for experimentation and other scientific purposes (Real Decreto 22311988), that was in force when the study was done. All the experimental protocols were reviewed and approved by a Scientific Committee from Hospital Ramón y Cajal (https://www.irycis.org/es/)
2.12. Statistical analysis
Results were expressed as mean ± SEM from several independent experiments indicated in each figure. For parametric data, a Student’s t-test, or one or two-way ANOVA followed by the Newman–Keuls multiple comparison test were performed. Differences were considered significant when p≤ 0.05. Statistical analysis was performed with the Graph Pad Prism software (La Jolla, CA, USA).
3. Results
3.1. Fluvastatin converts macrophages to foamy cells unable to form granulomatous structures in response to Mtb H37Ra
We started this study by analyzing under light microscopy the effects of fluvastatin on PBMC and on the response that these cells elicit against inactivated Mtb H37Ra. PBMC were treated or not with the statin for 15h. Next, we added the bacteria to half of the wells from each condition, and resumed the incubation for another 24h. At the end of this incubation, macrophages from drug-untreated cultures (control condition) showed numerous small vacuoles (Figure 1A), but in fluvastatin-treated cultures most macrophages became highly vacuolated rounded cells, with an eccentric and concave nucleus. Many of these macrophages tightly attached lymphocytes and/or monocytes (see Figure 1B), which here we will call “small cells”. In PBMC exposed only to Mtb H37Ra, macrophages formed compact aggregates enclosing necrotic cores (Figure 1C 1), similar to granulomas generated by viable Mtb H37Rv in PBMC from healthy donors [44]. Thus, we will refer to these aggregates as granuloma-like structures (GLS). In this condition, a few macrophages became highly vacuolated cells (Figure 1C(2)) alike those shown in Figure 1B. In fluvastatin-treated cultures the stimulation with the bacteria did not generate GLS, but instead, vacuolated macrophages formed cellular aggregates surrounding smaller necrotic areas (Figure 1D). All these vacuolated macrophages presented morphological characteristics of FM, which are a group of scavenger cells that perform homeostatic functions in human tissues and fluids [45,46]. Figure S1 shows the similarity between the vacuolated macrophages generated in this work and conventional FM that remove erythrocytes (erythrophages) in a hemorrhagic pleural effusion sample prepared for diagnostic at Hospital Ramón y Cajal. Here we will refer as FM to those vacuolated macrophages observed in cultures only exposed to the bacteria and as FFM to those macrophages transformed by fluvastatin.
A temporal evolution study revealed that GLS started to be formed just after adding the bacteria. During the first hour of immune stimulation, a few macrophages, including FM, surrounded small amorphous masses. Overtime more macrophages were recruited and some of them died forming necrotic areas (Figure 2A(1,2)). Thereafter, new macrophages reached these areas of cell death and formed incipient GLS (Figure 2A(3)). A few hours later, numerous macrophages enclosed them into compacted GLS (Figure 2A(4,5)). The number of FM in these cultures increased progressively and after 4h of adding the bacteria they represented 2-4% from total macrophages. These cells incorporated to GLS, impeding the final count of the transformed macrophages. At the end of the incubation period most of GLS were highly compacted, intensely stained and showed few FM (Figure 2A(5)). However, a reduced number of GLS enclosed smaller necrotic cores and had numerous FM (Figure 2A(6)). The formation of GLS was not a synchronized process and we were able to observe incipient granulomas at all times of the incubation period. By contrast, in cultures treated with fluvastatin and exposed to the bacilli, a few FFM initiated the formation of necrotic foci (Figure 2B(1,2)) that barely grew over time and were surrounded by more FFM. At the end of the incubation period, they formed loose cellular aggregates (Figure 2B(3–5)), but occasionally we saw some more compacted structures with less FFM (Figure 2B(6)). In the absence of bacteria both, untreated or fluvastatin-treated cultures did not form compacted cellular aggregates (Figure S2). These findings indicate that the drug not only deeply change the morphology of macrophages, but also prevents the activation of a granulomatous response against the bacteria.
3.2. Ultrastructural study of macrophages
To define more precisely the changes occurred in macrophages from the different conditions, we performed a transmission electron microscopy study. Compared to macrophages from control cultures (Figure 3A), most macrophages exposed to Mtb H37Ra increased in size, contained a large and elongated nucleus and showed ruffled membranes with projections ranging from small and extended pseudopodia to voluminous and irregular masses (Figure 3B(1)). These macrophages captured the bacteria in phagosomes that barely degraded them, and interlocked with each other through a labyrinthine network of membrane projections (Figure 3B(2.3)). All these cellular characteristics are coincident with those of epithelioid cells from tuberculous granulomas [47] and support that heat-killed or living Mtb H37Ra are equally effective inducers of these granulomas [37]. In the same culture, a few macrophages preserved their size and showed large cytoplasmic vacuoles containing not identifiable particles and debris (Figure 3B(4,6)). These cells emitted numerous short membrane protrusions that captured material from their surrounding microenvironment, forming a lattice of small vacuoles at the edge of the cell. Some of these macrophages enclosed non-degraded bacteria in phagosomes (Figure 3B(7)), but they also showed other bacilli captured together with debris and delivered into vacuoles, where they interacted with vesicular organelles with a size and morphology alike secondary lysosomes described by Armstrong and Hart [48]. Furthermore, signs of bacteria degradation at the contact points of this interaction were visible, indicating that these formations were in fact, degrading organelles. This type of phagocytosis, resembling macropinocytosis, is associated to the cleaning of necrotic debris at sites of injury and inflammation [49]. Thus, we consider that these vacuolated macrophages are FM performing scavenger task.
FFM resulting from fluvastatin treatment had numerous large vacuoles (Figure 3C(1)), and emitted short membrane protrusions that engulfed not identifiable material from their surrounding microenvironment (Figure 3C(2)). However, as in this condition immune debris was barely generated, and most macrophages were able to remove it, their vacuoles scarcely contained detritus and were apparently empty. A few of these FFM contained vesicular organelles within their vacuoles (Figure 3C(3)) like those shown in Figure 3B(8). We appreciated a tight association between the cytoplasmic membranes of FM and their attached cells (Figure 3C(4,5)). Another feature of these macrophages was that they frequently subdivided vacuoles by emitting filopodia-like projections (Figure 3C(6)). In fluvastatin-treated cultures and exposed to the bacteria, FFM were not converted to epithelioid cells and only emitted small membrane protrusions. These activated FFM showed few vacuoles and their cytoplasm was filled with debris and a high number of vesicular organelles, both intermingled with filopodia-like projections (Figure 3D(1)). Similar organelles were present at the periphery of these macrophages (Figure 3D(2)). Alike FM, these FFM captured some bacteria in phagosomes (Figure 3D(3)) and engulfed others along debris through a similar phagocytic process, but delivered them into a cytoplasm filled with organelles that could degrade them (Figure 3D(3,4)). Supporting this possibility, preserved bacterial cells were infrequently visualized in these cells, although their cytoplasm contained dark spots that due to their morphology and size could correspond to partially digested bacteria (Figure 3D(5,6)). Many of these cells presented numerous lipid bodies (Figure 3D(5)) alike those observed in macrophages infected whit the bacillus of Calmette-Guerin [50].
3.3. FFM overproduce NLRP3 and ASC and the bacteria promotes the co-localization of both proteins
To analyze if statins could interfere in the formation of NLRP3 inflammasomes, we studied by immunofluorescence the distribution and colocalization of ASC and NLRP3 in cells from the different conditions. In control cultures, a few macrophages showed some ASC inside the nucleus, and had a few NLRP3 distributed as small dots at perinuclear location (Figure 4). In cultures exposed to the bacteria, most macrophages in GLS had augmented ASC in their nuclei without evidence of an increase of NLRP3, although it appeared concentrated as a single dot near the nucleus (Figure 4, Mtb H37Ra upper row), an event considered as one of the initial step for the assembly of the inflammasome [51]. In this condition, some FM (cells with an eccentric and concave nucleus) formed ring-shaped aggregates of NLRP3, and partially moved ASC from the nucleus to co-localize with them (Figure 4, Mtb H37Ra bottom row). Unexpectedly, FFM showed a considerable increase of NLRP3 in their cytoplasm, forming several ring-shaped aggregates alike those seen in FM, despite the absence of exogenous PAMPs (Figure 4, fluvastatin row). The number of these formations was highly variable among FFM from the same culture and in cultures from different donors. ASC was also increased in the nuclei of all these cells, but co-localization with NLRP3 was only observed in some of them. In fluvastatin-treated cultures exposed to the bacteria, FFM massively released ASC from the nucleus to partially co-localize with NLRP3 in numerous ring-shaped aggregates (Figure 4, fluvastatin + Mtb H37Ra row). Besides, these macrophages released particles where ASC and NLRP3 partially co-localized (Figure 4, fluvastatin + Mtb H37Ra), indicating that they could assemble at the periphery of organelles that can be released out of the cell.
3.4. Fluvastatin exacerbates caspase-1 activation in response to Mtb PAMPs
Active caspase-1 was localized in cells by performing a FAM-FLICA assay. In control cultures, there was specific green fluorescence as dots in broken cells, inside dismantled nuclei or, as observed by others [52], forming large extracellular aggregates (Figure 5A(1,2)). In fluvastatin-treated cultures, some FFM showed specific fluorescence delineating either a ring at a perinuclear position, or several rings or arcs dispersed in the cytoplasm (Figure 5B(1,2)), alike NLRP3 aggregates seen in Figure 4. Unexpectedly, in PBMC exposed to the bacteria, a small subset of macrophages captured large amounts of bacilli emitting red and green autofluorescence (see the Methods section). Merging both fluorescences, the bacteria were delineated in yellow but we could not detect specific green fluorescence, indicating the absence of active caspase-1 in these macrophages (Figure 5C(1–3)). These cells associated to accumulate the captured bacilli in a central core (Figure 5(C4)), comparable to the amorphous masses shown in Figure 2A. Other macrophages, with very low bacterial load, surrounded these foci forming incipient GLS, without activating caspase-1 (Figure 5C(5)). In bigger GLS, numerous macrophages tightly compacted large amounts of yellow bacteria in a central core (Figure 5C(6)). In this condition, only a few macrophages, which could be FM due to their morphology, showed specific green fluorescence enclosed in a few round formations (Figure 5C(7,8)) with a similar size and cytoplasmic distribution as NLRP3/ASC aggregates (Figure 4, Mtb H37Ra lower row). These coincidences and the fact that in some organelles specific fluorescence accumulated in rings at their edges (Figure 5C(7)), indicate that the NLRP3 inflammasome activates caspase-1 at the periphery of these formations and that, once active, the enzyme is released inside them. In fluvastatin-treated cultures exposed to the bacteria, most FFM markedly increased the number of round formations enclosing active caspase-1 (Figure 5D(1,2)) at the same proportions as NLRP3/ASC ring-shaped aggregates (Figure 4). These data indicate that FFM assembled the inflammasome and activated caspase-1 through the same pathway as FM, but that this process was exacerbated in the former. Most of these activated FFM did not show intact bacteria, but had bacilli remnants in the same formations as active caspase-1, sometimes forming yellow arcs at their edges (Figure 5D(1)). Such colocalization and the fact that these formations had a similar number, size and cellular distribution as degrading organelles (Figure 3D), indicate that both correspond to the same structure. In this condition, a few macrophages preserved their ability to capture large amounts of bacteria (Figure 5D(2)), also accumulating them in necrotic areas. Some of these macrophages harbored the bacteria in their broken nuclei (Figure 5D(3)). None of these macrophages activated caspase-1. Close to them, other FFM showed numerous round formations with large amounts of specific green fluorescence unmasking a faint red autofluorescence emitted by small fragments of the bacilli (Figure 5D(4)), indicating that these FFM fully degraded the bacteria accumulated by the former, supporting TEM observations in present work.
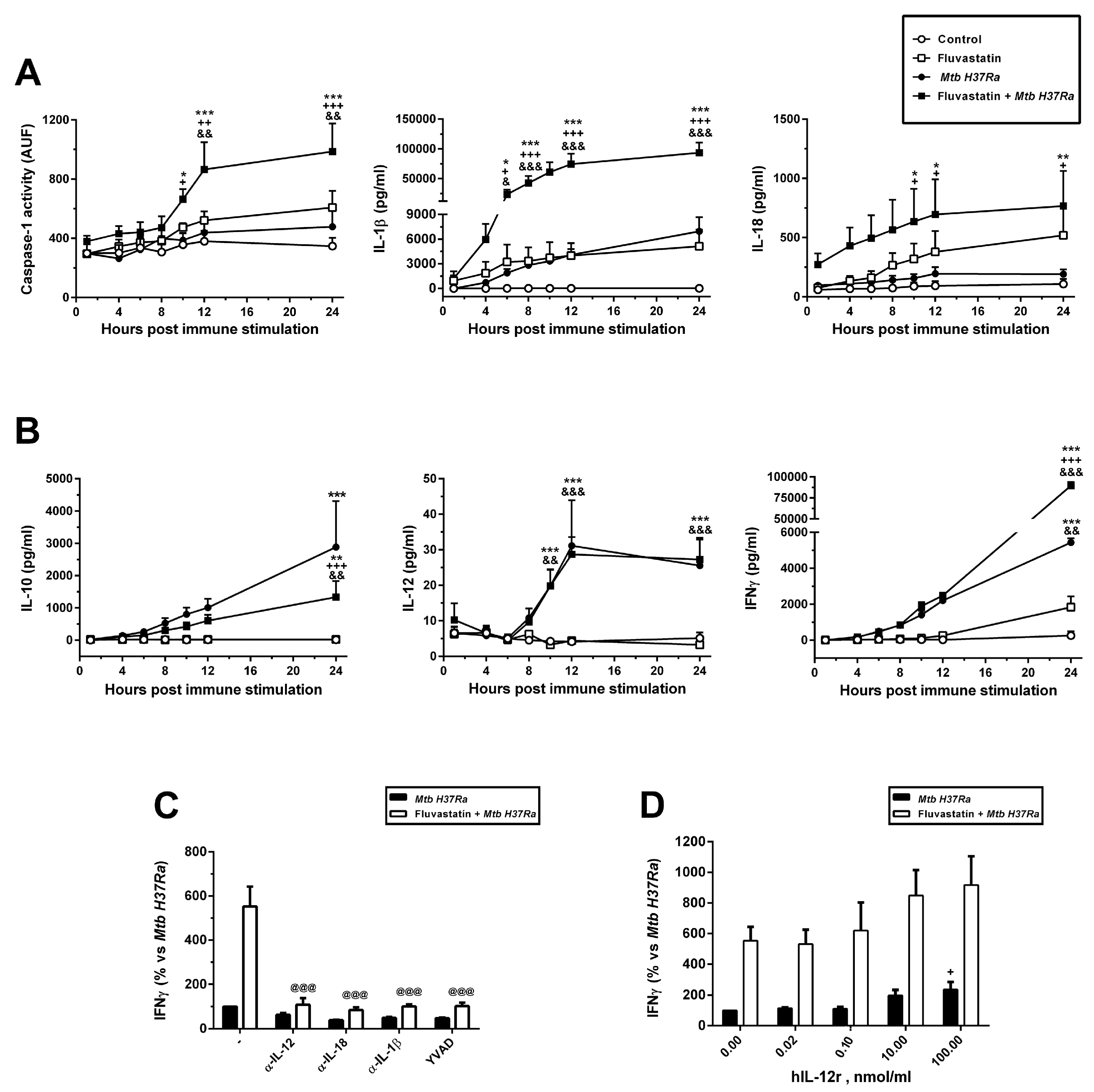
We also quantified caspase-1 activity in lysates from cells collected from each condition at different times of incubation. Results are presented in Figure 6A. In agreement with FAM-FLICA results, the control condition showed a basal caspase-1 activity sustained over time. In cultures exposed to the bacteria or treated with fluvastatin this basal activity did not augment, but when both were combined, the activity significantly increased from 10 hours of immune stimulation onwards.
3.5. Fluvastatin exacerbates the cellular response against Mtb H37Ra
After stimulation with the bacteria, we quantified the levels of IL-1β IL-18, IL-10, IL-12 and IFNγ in culture supernatants collected over time. As shown in Figure 6A,B, the levels of these cytokines were undetectable in supernatants from control cells. In cultures treated with fluvastatin, IL-1β and IL-18 production tended to augment but it was not statistically different from that of controls. In cultures stimulated with the bacteria, IL-10 and IFNγ increased progressively with time, and reached statistical significance vs. the control and fluvastatin conditions at the end of the incubation period, while IL-12 increased significantly after 10h of immune stimulation, and peaked two hours later (Figure 6B). By contrast, IL-1β and IL-18 production was not significantly stimulated (Figure 6A). When we combined fluvastatin and bacteria, the amount of IL-1β significantly increased at 6h of co-incubation, reaching later very high values. IL-18 levels augmented starting 10 hours after adding the bacteria (Figure 6A). On the other hand, IL-10 was significantly reduced without affecting IL-12 production. Nonetheless, IFNγ levels were markedly increased after IL-12 peaked (Figure 6B). Caspase-1 inhibition by its specific substrate YVAD, as well as the addition to the cultures of neutralizing antibodies targeting IL-1β or IL-18, prevented this peak of IFNγ (Figure 6C). Furthermore, it was necessary to supplement the cultures with high concentrations of recombinant human IL-12 to slightly increase the levels of IFNγ induced by the bacteria (Figure 6D). These results indicate that, in response to Mtb H37Ra, IL-12 is required for IFNγ production, but that the levels of ILs processed by caspase-1 determine the amount of cytokine produced.
Figure 6.
Dynamics of caspase-1 activation and production of cytokines. (A) Time course of caspase-1 activity in cell lysates expressed in arbitrary units of fluorescence, (AUF) and IL-1β and IL-18 concentrations in the media after immune stimulation. (B) Time course of the release of IL-10, IL-12 and IFNγ to the media. (C) Effect of neutralizing antibodies independently targeting IL-12, IL-18 or IL-1β and the caspase-1 inhibitor YVAD on IFNγ release induced by the bacteria in untreated (black bars) or fluvastatin-treated cultures (white bars). (D) Dose-response effect of human recombinant IL-12 on the production of IFNγ induced by the bacteria alone (black bars), or in combination with the bacteria (white bars). In A and B results represent the mean ± SEM of 6 independent experiments. In C and D values are normalized by IFNγ produced by fluvastatin-untreated PBMC in the presence of bacteria (100%) and represent the mean ± SEM of 4 independent experiments. *p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001 vs control, +p ≤ 0.05; ++p ≤ 0.01, +++p ≤ 0.001 vs Mtb H37Ra, &p ≤ 0.05; &&p ≤ 0.01, &&&p ≤ 0.001 vs fluvastatin, @@@p ≤ 0.001 vs fluvastatin + Mtb H37Ra (one-way (C and D) or two-way ANOVA (A and B) +Newman-Keuls).
Figure 6.
Dynamics of caspase-1 activation and production of cytokines. (A) Time course of caspase-1 activity in cell lysates expressed in arbitrary units of fluorescence, (AUF) and IL-1β and IL-18 concentrations in the media after immune stimulation. (B) Time course of the release of IL-10, IL-12 and IFNγ to the media. (C) Effect of neutralizing antibodies independently targeting IL-12, IL-18 or IL-1β and the caspase-1 inhibitor YVAD on IFNγ release induced by the bacteria in untreated (black bars) or fluvastatin-treated cultures (white bars). (D) Dose-response effect of human recombinant IL-12 on the production of IFNγ induced by the bacteria alone (black bars), or in combination with the bacteria (white bars). In A and B results represent the mean ± SEM of 6 independent experiments. In C and D values are normalized by IFNγ produced by fluvastatin-untreated PBMC in the presence of bacteria (100%) and represent the mean ± SEM of 4 independent experiments. *p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001 vs control, +p ≤ 0.05; ++p ≤ 0.01, +++p ≤ 0.001 vs Mtb H37Ra, &p ≤ 0.05; &&p ≤ 0.01, &&&p ≤ 0.001 vs fluvastatin, @@@p ≤ 0.001 vs fluvastatin + Mtb H37Ra (one-way (C and D) or two-way ANOVA (A and B) +Newman-Keuls).
Next, we analyzed the subsets of lymphocytes producing IFNγ by flow cytometry. In cultures exposed to the bacteria, treated or not with fluvastatin, the forward scatter versus side scatter dot plot showed only a gate of lymphocytes, indicating that most macrophages had been incorporated to GLS or to cellular aggregates. Despite the impossibility to use in this study protein transport inhibitors (see material and methods), we detected in this gate a low number of lymphocytes CD3+/CD8+, CD3+/CD8- and CD3-/CD56+ (NK) T cells that produced IFNɣ in response to the bacteria, and the number of positive cells increased in all these populations in PBMC from the same donors treated with fluvastatin and bacteria (Figure 7A). Since NK and CD3+/CD8+ cells are cytotoxic, we measured LDH activity in supernatants to evaluate cellular death. In control cultures, basal LDH activity was maintained through the incubation period, and it was not increased by the exposure to the bacteria (Figure 7B). Fluvastatin treatment slightly increased this activity, but when combined with the bacteria it augmented significantly (Figure 7B).
We also evaluated the effect of fluvastatin on IFNɣ production in response to bacteria in vivo by performing a tuberculin test in rabbits immunized with the inactivated Mtb H37Ra. As shown in Figure 7C, rabbits immunized with the bacteria were tuberculin-negative. However, the same protocol of immunization in combination with fluvastatin therapy, markedly increased the size of the induration area (Figure 7C).
4. Discussion
The regulation of the early immune events that determine the resolution or perpetuation of tuberculosis is presently seen as a therapeutic target to control the disease, but studying in vivo this early interaction has important technological limitations [53]. Here we see that, in response to inactivated Mtb H37Ra, monocytes/macrophages from healthy donors polarize their functions towards different phenotypes that cooperate with each other to form granulomatous structures. This is a stepwise process, which, as we will argue below, mimics early stages in the formation of tuberculous granulomas, providing valuable information on this regard.
It is generally agreed that, before tuberculous granulomas begin to organize, virulent Mtb strains accumulate and proliferate in necrotic areas, evading immunity [54]. Some authors have proposed that these foci of death are generated by minimally microbicidal macrophages that allow the pathogen to grow intracellularly [3]. Then, Mtb virulence factors block apoptosis in these infected macrophages, inducing different necrosis-like processes that results in the release of viable pathogens into an extracellular milieu where, paradoxically, they grow and spread further, contributing to disease progression [55,56,57]. In the present work, a small subset of macrophages quickly captures large amounts of inactivated Mtb to the point of death, also forming necrotic areas where the bacilli accumulate. Given that in our study Mtb lacks virulence factors and cannot infect macrophages or proliferate inside them, the formation of these necrotic areas cannot be ascribed to the virulent activity of the bacteria. We thought that, in healthy people, a subset of macrophages from the blood could be functionally predetermined (or perhaps trained) to detect free mycobacteria and, regardless of their virulence, concentrate them in an inhospitable space to impede their survival. It could represent a not described strategy of innate immunity to quickly prevent the uncontrolled spread of free mycobacteria to other parts of the body, however virulent Mtb strains would evade it because they have learned to survive in this space [8]. Herein we also observed that, in only 24h of immune stimulation, most of the remaining macrophages become epithelioid cells that enclose these foci of dead cells in compact GLS, suggesting that these macrophages undergoing necrosis could emit some unknown signal that initiate the formation of granulomas. In agreement with our results, near a century ago it was observed that, when blood drops from a range of species were exposed to tuberculous bacilli, monocytes became epithelioid cells, and some of them even evolved overtime to giant multinuclear cells [58]. Our own observations and those in the latter work, show that blood macrophages are important players in the initiation of a granulomatous response against Mtb, without requiring specific tissue factors. As occurs in tuberculoid lesions in vivo [3], central cores in GLS expand over time, suggesting that necrotizing macrophages continue replenishing necrotic areas with bacteria and debris once the structure is formed. In line with this, in animals superinfected with an isogenic Mtb virulent strain, the new infectious bacilli are rapidly transported within some macrophages to the necrotic centers of the preexisting granulomas, where they die and the bacilli proliferate [59]. It could be of interest to study whether a defective response of these necrotizing macrophages could underlie in some cases of miliary tuberculosis [60].
Another initial key event influencing on the progression of granuloma is the conversion of FM to LL-FM by the pathogen. The term FM was coined by pathologists to refer to a group of highly vacuolated macrophages, sharing morphological characteristics with foamy cells from atherosclerotic lesions, that perform scavenger functions in tissues and fluids to recover their homeostasis. These macrophages show heterogeneity between tissues and their number increases significantly in different pathologies [61]. The biogenesis and function of FM vary depending on the specific microenvironment of each disease [62,63], where these cells can accumulate not only lipids but also other products derived from the digestion of the waste engulfed from the damaged tissue, helping pathologists in the diagnosis [64]. It has been demonstrated that the formation of FM in tuberculous granulomas differs from that of FM in atherosclerotic lesions [65], although our knowledge on their immune function remains limited [66]. In the present work, simultaneously to the formation of necrotic foci, a subset of macrophages turns into FM and are quickly recruited to incipient necrotic areas. These FM enclose intact bacilli in phagosomes but, unlike epithelioid cells, they emit short protrusions to engulf other bacilli together with debris, which are degraded in large vacuoles with the help of some not identified vesicular organelle. Thus, as observed in vivo in the incipient granulomas [67], in our in vitro study some macrophages become FM in response to necrotic debris generated during the earliest immune response to Mtb. Overtime, these FM incorporate to GLS where they maintain their morphology, indicating that they do not respond to signals for their transformation into epithelioid cells. This particular response of FM would explain why these cells accumulate at the edge of the caseum from human granulomas [7], whereas most of the other macrophages recruited to the structure end up becoming epithelioid cells [68]. Because in our study the bacteria are dead, FM cannot convert macrophages to LL-FM, which allowed us to know how these cells would respond to Mtb PAMPs before being transformed by the pathogen. Here we found that these activated FM augment ASC in the nuclei and NLRP3 in the cytoplasm, the latter forming undescribed ring-shaped aggregates. In some of these cells, ASC partially leaves the nuclei to colocalize with NLRP3 aggregates and they show active caspase-1 inside undescribed round formations similar in number, size and cytoplasmic distribution to ASC/NLRP3 rings, indicating that FM assemble this inflammasome around organelles that retain the enzyme once activated. This pathway for NLRP3 inflammasome and caspase-1 activation has not been referred in other studies [27], but it shows analogy with a mechanism of inflammasome regulation which was not deeply studied [51]. Unlike FM, most of the cells forming GLS hardly augment levels of NLRP3 in response to Mtb PAMPs and, although ASC increases, it remains in the nuclei, an event that prevents caspase-1 activation [51]. Consequently, we detect poor levels of IL-1β and IL-18 in culture supernatants. Besides, these structures also act as active platforms for the release of very low amounts of IL-12 and high quantities of IL-10. This ILs pattern is similar to that produced by tuberculous granulomas in vivo, also resulting in the activation of a poor IFNγ production [69]
Altogether, this first part of results suggests that necrotizing macrophages would generate different warning signals to recruit macrophages from the blood, polarizing them toward different phenotypes which coordinately initiate the formation of granulomas. The simplicity of our experimental model offers many advantages to design complementary studies directed to decipher what signals transform macrophages into FM or into epithelioid cells and the role that they play in the formation of these structures.
An unexpected result of the present study is that, in the absence of immune stimulation, fluvastatin massively converts macrophages of PBMC to foamy cells (FFM) with the same morphology that FM induced by the bacteria in untreated PBMC, and that both perform comparable scavenger functions. Another important analogy between these macrophages is that both associate with small cells through close intercellular attachments, evidencing the existence of a communication among them that, at least in the case of FFM, cannot be linked to foreign antigens presentation. These similarities evidence an important participation of cholesterol metabolism in the conversion of resting macrophages to conventional FM. As schematized in Figure 8, fluvastatin inhibits HMG-CoA reductase, a key enzyme of the mevalonate pathway that converts HMG-CoA to mevalonate, which is subsequently phosphorylated by mevalonate kinases to generate isopentenyl-pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMAPP). These molecules are the precursors not only of sterols (mainly cholesterol) but also of isoprenoids such as geranyl-pyrophosphate, geranylgeranyl-pyrophosphate (GGPP) or farnesyl-pyrophosphate, that modulate numerous cell signaling processes [20] and cytoskeleton organization [21]. Thus, fluvastatin could simultaneously interfere with some of these processes to convert resting macrophages into FFM. Besides, cellular cholesterol depletion induced by statins can promote the synthesis of the sterol regulatory element-binding protein 2 (SREBP2), which binds to the SREBP cleavage-activating protein (SCAP) in the endoplasmic reticulum [70]. This dimer is transported to the Golgi apparatus, where SREBP2 undergoes two proteolytic cleavages to generate active SREBP2, a transcription factor that regulates the expression of different genes involved in cholesterol biosynthesis and uptake to recover cholesterol homeostasis in the cell [71].
A recent work has shown that statins interfere with the expression of several genes involved in both cholesterol homeostasis regulation and foamy cell formation [72]. Thus, to understand the complex metabolism/immunity interactions that transform macrophages to FM it would be necessary to carry out complementary studies.
Of note, despite the absence of immune stimulation, FFM overproduce ASC in the nucleus and NLRP3 in the cytoplasm, the latter forming numerous ring-shaped aggregates alike those observed in FM. This result indicates that the inhibition of cholesterol metabolism generates in macrophages an immune alert that mimics signal 1 mediated by Mtb PAMPs in FM. In a previous work performed in our lab, with the same experimental model, we demonstrate that GGPP regulates negatively caspase-1 autoprocessing [42]. Subsequently, several studies demonstrated that the inhibition of the mevalonate pathway by statins or bisphosphonates and mevalonate kinase deficiencies (MKD) (see Figure 8), leads to a shortage of GGPP that upregulates NALP3 expression [73,74,75,76]. Other works have evidenced that, in MKD patients, a compromised geranylgeranylation of as yet unidentified proteins allows a pathological activation of the NLRP3 inflammasome, which is responsible for autoinflammatory episodes mediated by caspase-1 [77,78]. Thus, in FFM from the present work, a fluvastatin-mediated reduction of GGPP would prevent the prenylation of this protein and promote NLRP3 expression. However, the high amounts of receptor observed in these cells suggest that the drug also prevents its down-regulation.
Figure 8.
Proposal for a cholesterol metabolism/immunity integrated circuit regulating NLRP3 inflammasome activation in macrophages. A) Schematic representation of the cholesterol biosynthesis pathway. The inhibition of this pathway by statins or bisphosphonates and mevalonate kinase deficiencies (MKD) reduces the synthesis of geranylgeranyl-PP (GGPP). B) In all these cases, a reduced geranylgeranylation of an as yet unidentified protein (? GG protein) induces overexpression of NLRP3 (blue arrow). According to Guo et al. [79] (red arrows), NLRP3 forms a ternary complex with SREBP2 and SCAP that is translocated from the endoplasmic reticulum (ER) to the Golgi apparatus. Two consecutive proteolytic cleavages of SREBP2 by S1P (site 1 protease) and S2P (site 2 protease) generate the active form of the transcription factor, which increases the expression of HMGCR, whereas the remaining SCAP-NLRP3 complex is released from the Golgi, optimizing the activation of the inflammasome. We propose that de novo synthesized HMG-CoA reductase allows the cell to restore GGPP levels to downregulate NLRP3 expression, thereby closing (discontinuous red lines) an undescribed cholesterol metabolism/immunity integrated circuit involved in the regulation of inflammation.
Figure 8.
Proposal for a cholesterol metabolism/immunity integrated circuit regulating NLRP3 inflammasome activation in macrophages. A) Schematic representation of the cholesterol biosynthesis pathway. The inhibition of this pathway by statins or bisphosphonates and mevalonate kinase deficiencies (MKD) reduces the synthesis of geranylgeranyl-PP (GGPP). B) In all these cases, a reduced geranylgeranylation of an as yet unidentified protein (? GG protein) induces overexpression of NLRP3 (blue arrow). According to Guo et al. [79] (red arrows), NLRP3 forms a ternary complex with SREBP2 and SCAP that is translocated from the endoplasmic reticulum (ER) to the Golgi apparatus. Two consecutive proteolytic cleavages of SREBP2 by S1P (site 1 protease) and S2P (site 2 protease) generate the active form of the transcription factor, which increases the expression of HMGCR, whereas the remaining SCAP-NLRP3 complex is released from the Golgi, optimizing the activation of the inflammasome. We propose that de novo synthesized HMG-CoA reductase allows the cell to restore GGPP levels to downregulate NLRP3 expression, thereby closing (discontinuous red lines) an undescribed cholesterol metabolism/immunity integrated circuit involved in the regulation of inflammation.
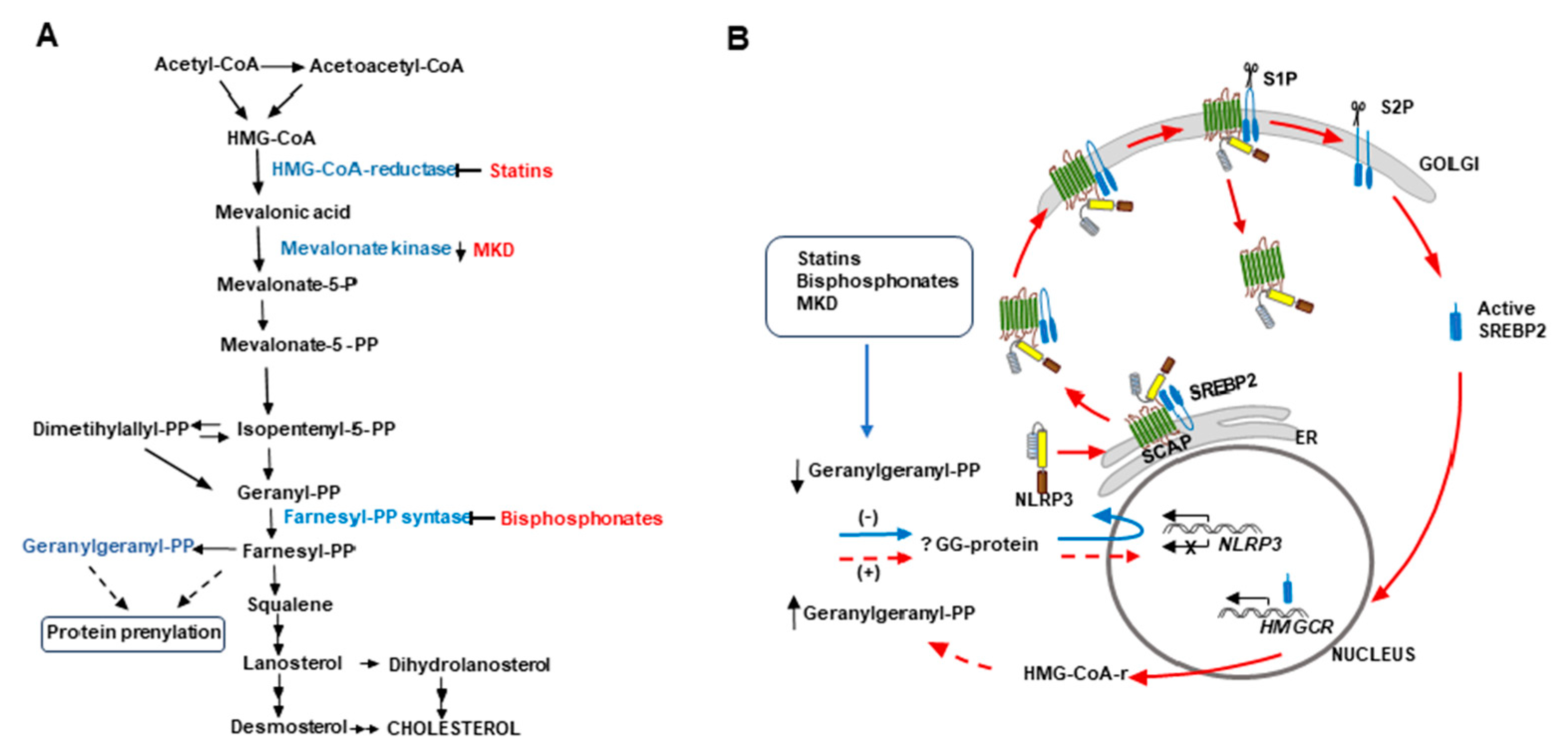
Guo et al. [79] have previously shown that, during immune activation of the NLRP3 inflammasome, SCAP-SREBP2 forms a ternary complex with NLRP3 in the endoplasmic reticulum that is translocated to the Golgi apparatus. The authors also demonstrated that this translocation and the subsequent first cleavage of SREBP2 are required for optimal NLRP3 inflammasome activation, which would take place after the release of the remaining SCAP-NLRP3 dimer from the Golgi apparatus (see Figure 8B). As mentioned above, a second cleavage of SREBP2 increases the expression of Hmgcr, among other of their target genes [79], although their role on NLRP3 inflammasome activation was not addressed. We propose that an upregulation of HMG-CoA reductase would augment the synthesis of mevalonic acid in order to reestablish GGPP levels in the cell, thereby downregulating NLRP3 expression. This would close an undescribed cholesterol metabolism/immunity integrated circuit that regulates inflammation (see Figure 8B). However, in FFM de novo synthesized HMG-CoA reductase is inhibited by fluvastatin so that these macrophages cannot recover GGPP levels, giving rise to a deregulated synthesis of NLRP3 that would explain its accumulation in these cells. Likewise, in macrophages from MKD patients, a shortage of GGPP would induce the synthesis of HMG-CoA reductase through this circuit, but, as in these patients mevalonic acid cannot be phosphorylated, the cell would not recover GGPP levels, thus not only accumulating NLRP3 but also increasing mevalonic acid levels and HMG-CoA reductase activity [78]. The existence of this circuit would support the proposal that exogenous supplementation of GGPP could help in the management of patients with MKD [80].
ASC is also overexpressed in FFM, most of it being retained in the nucleus, thus preventing inflammasome assemblage [51]. However, in some FFM, ASC moderately leaves the nucleus and partially colocalizes with NLRP3. In this condition, a few FFM show specific FAM-YVAD-FMK fluorescence delineating rings with a similar size and cytoplasmic distribution than NLRP3. During the process of caspase-1 activation, an intermediate form of the enzyme, partially active, is generated by intradimer proteolysis of its pro-form, which remains bound to the inflammasome [81]. Thus, this specific fluorescence observed in FFM could correspond to intermediate forms of caspase-1 that could be generated from constitutive pro-caspases-1 autoprocessing. These observations indicate that fluvastatin not only increases ASC and NLRP3, but also contributes to their assemblage to initiate caspase-1 activation, and that additional immune signals are required to fully activate the enzyme. Consequently, we scarcely detected increased caspase-1 activity in cellular lysates from this condition respect to those of control cells. The mechanism for fluvastatin-induced synthesis of ASC is unknown, but it evidences the existence of additional cholesterol metabolism/inflammation links. It could be interesting to study if MKD patients also overexpress this inflammasome component. New studies directed to decipher how statins regulate ASC and NLRP3 expression could help to understand other inflammatory pathologies linked to a deregulated cholesterol metabolism [24,82].
All the effects exerted by fluvastatin on healthy macrophages deeply affect their response to Mtb H37Ra. Although the drug does not impede that some macrophages accumulate the bacilli in necrotic areas, FFM do not respond to necrotic signals for epithelioid transformation, and thereby they do not form GLS, but instead they capture and destroy the accumulated bacteria. In response to Mtb PAMPs, FFM markedly increase the number of NLRP3 ring-shaped aggregates induced by the drug, providing additional evidences that NLRP3 expression is unregulated in these macrophages. It has been described that different cellular organelles, variable among different cells, can act as supports for the trafficking and assembly of the inflammasome components, thus mediating different responses [83]. In our study, FFM respond to Mtb by assembling the NLRP3 inflammasome and activating caspase-1 around numerous organelles that degrade the bacteria and that retain the enzyme once it has been activated. The presence of bacilli remnants forming arcs at the edges of these organelles suggest that bacilli degradation will provide inflammasomes with PAMPs to generate a signal 2 that fully activates caspase-1. This possibility would change the concept that NLRP3 does not directly interact with their ligands for its activation [84]. Comparing the response of FFM and FM to Mtb H37Ra, we conclude that both activate the same inflammatory pathway but that fluvastatin highly exacerbates it. We consider that this inflammatory response could be a characteristic of FM that remains unexplored because these macrophages have not yet been studied appropriately.
A recent study in primates evidences that only those granulomas able to activate T-cells and NK cells for IFNγ/cytotoxic response, can be associated to bacterial clearance, whereas in the others, the bacteria persist and grow [85]. The high levels of IL-1β and IL-18 produced by FFM against the bacteria activates a potent IFNγ /cytotoxic response, despite IL-12 levels remain very low. This agrees with the fact that low concentrations of IL-12 rend human T-cells highly responsive to IL-1β and IL-18 for IFNγ production [86]. The fact that YVAD and neutralizing antibodies targeting IL-1β or IL-18, independently prevent this peak of IFNγ supports the importance of caspase-1 in the activation of a protective response against the bacteria [23]. Furthermore, in our previous study [42], this potent inflammatory response promoted by fluvastatin against Mtb was prevented by geranylgeraniol, supporting the importance of this isoprenoid in controlling the inflammation mediated by the NLRP3 inflammasome. Our cytometry results involve T-cells and NK cells in this augmented IFNγ response, but we do not exclude the possibility that some “small cell” bound to FM can contribute to it. The fact that in fluvastatin-untreated PBMC exposed to the bacteria the same T-cells and NK-cells scarcely activate IFNγ production indicates that healthy people have enough specific immune cells to activate a protective IFNγ/cytotoxic response in a first encounter with Mtb, but that they require to be properly instructed by the infected macrophages. Our study evidences that in fluvastatin-untreated PBMC exposed to the bacteria, even the most incipient granulomas are unable to activate them, supporting the proposal that granuloma formation limits the effectiveness of adaptive immunity in controlling the pathogen [2]. Thus, we consider that control of Mtb infection requires not only generating specific T-cells, but also assisting macrophages for their activation. This consideration reinforces the idea that adaptive immunity against Mtb is difficult to improve with current vaccines [87] and ratifies the view of some authors that have questioned whether vaccination and immunity enhancing strategies, that only mimic the natural immune response to Mtb, can be able to control pulmonary tuberculosis [88]. In agreement with our in vitro results, rabbits immunized with heat-inactivated Mtb H37Ra, tuberculin test was negative, but when rabbits received fluvastatin treatment, the same immunization protocol significantly increased the induration area in this test, validating in vivo the potential of statins to increase the production of IFNγ in response to Mtb PAMPs. Together, our in vitro and in vivo results indicate that in hypercholesterolemic patients receiving statin therapy, these drugs would help FM to activate an efficient IFNγ/cytotoxic response that control the pathogen at the earliest stages of infection, thus preventing their conversion to LL-FM and the formation of granulomas. Furthermore, as FFM do not respond to signals for epithelioid transformation, these macrophages could reach the caseum of granulomas. It would explain why statins regress tuberculoid lesions in a mouse model with human-like necrotic pulmonary granulomas [89], and also accelerate Mycobacterium tuberculosis clearance in pulmonary TB in humans [90]. However, our findings also alert on the potential of statins to exacerbate pre-existing inflammatory diseases linked to a sterile activation of the NLRP3 inflammasome [91], as observed in diabetes mellitus type I patients [92].
The possibility to pharmacologically regulate host cholesterol metabolism to generate pro-inflammatory FM able to destroy the bacteria with the support of adaptive immunity, reinforce the proposal that stains could be an adjuvant therapy in treating tuberculosis. However, as the course of tuberculosis shows different clinical manifestations and outcomes according to patient’s immune status [93], the efficacy of statins could be variable among patients. In fact, these therapies could result less effective in patients with some deficiencies in IFNγ production, whereas, on the contrary, in patients developing post-primary tuberculosis statins could exacerbate the necrotizing hypersensitivity reaction (Koch’s phenomenon) [94], thereby complicating the pathology.
Thus, the possibility of modulating cholesterol metabolism to help macrophages in fighting the bacteria provides a valuable tool to design innovative therapies to treat tuberculosis, although it may require specific adaptations in some patients. It even opens an unexplored possibility to generate in vitro efficient pro-inflammatory FM that could be adoptively transferred to the hosts to help them in the control of different infectious processes.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization: M.T. M-V; Methodology: M.T. M-V; J.M.; Validation: M.T. M-V, J.M. E.B and D.G-C.; Formal Analysis, J.M. and E.B.; Investigation, M.T. M-V, J.M., E.B., D.R., A.A., C.C., F.U., R. G-G. D.G-C.; Resources, M.T. M-V; E.B. and D.G-C; Visualization: M.T. M-V., J.M., and E.B.; Writing – Original Draft Preparation, M.T. M-V; Writing – Review & Editing, M.T. M-V, J.M., E.B., D.R., A.A., C.C., R. G-G. D.G-C; Project Administration, M.T. M-V, D.G-C.; Funding Acquisition, M.T. M-V, D.G-C.
Funding
This work was funded by Fondo de Investigación Sanitaria, Instituto de Salud Carlos III to D.G-C. and M.T.M-V (Grant 00/0229) and by Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS) to M.T.M-V (188/09). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
The present study was conducted according to the principles expressed in the Helsinki Declaration of 1983. Studies with buffy coats from anonymous de-identified samples of healthy blood donors were approved by the Research Committee of the Blood Transfusion Centre of the Comunidad de Madrid (Madrid, Spain) and the Research Ethics Committee of the Hospital Universitario Ramón y Cajal (approval code 188/09) in accordance with national and international guidelines. All procedures associated with animal experimentation were performed following the Council Directive 86/609/EEC of 24 November 1986 and in accordance with Spanish Real Decreto 22311988, that was in force at the time in which the study was performed. The study was reviewed and approved by the Scientific Committee of the Hospital Universitario Ramón y Cajal.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets generated and/or analyzed in the current study are available from the corresponding author on reasonable request.
Acknowledgments
Authors thank to Purification Dominguez for her help in comparing FM with histiocytes present in pleural hemorrhagic effusion, to Covadonga Aguado for sample preparations for TEM, to Marina Fernandez for providing us the tuberculin test, and to Mercedes Salaices for her support in the ultrastructural analysis of the samples. We are very grateful to the Hematology Service from Hospital La Paz as well as Centro de Transfusiones de la Comunidad de Madrid (Madrid, Spain) for providing us buffy coats. Special thanks to Novartis for kindly giving us fluvastatin as a gift. We also thank to Professor Fernando Baquero and Drs. Garbiñe Roy and Luisa María Villar for their valuable comments and advices during the execution of the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Organization, W.H. .Global Tuberculosis reports Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (accessed on 4).
- Cohen, S.B.; Gern, B.H.; Urdahl, K.B. The Tuberculous Granuloma and Preexisting Immunity. Annu Rev Immunol 2022, 40, 589-614. [CrossRef]
- Pagan, A.J.; Ramakrishnan, L. Immunity and Immunopathology in the Tuberculous Granuloma. Cold Spring Harbor perspectives in medicine 2014, 5. [CrossRef]
- Soldevilla, P.; Vilaplana, C.; Cardona, P.J. Mouse Models for Mycobacterium tuberculosis Pathogenesis: Show and Do Not Tell. Pathogens 2022, 12. [CrossRef]
- Wilburn, K.M.; Fieweger, R.A.; VanderVen, B.C. Cholesterol and fatty acids grease the wheels of Mycobacterium tuberculosis pathogenesis. Pathogens and disease 2018, 76. [CrossRef]
- Pawelczyk, J.; Brzostek, A.; Minias, A.; Plocinski, P.; Rumijowska-Galewicz, A.; Strapagiel, D.; Zakrzewska-Czerwinska, J.; Dziadek, J. Cholesterol-dependent transcriptome remodeling reveals new insight into the contribution of cholesterol to Mycobacterium tuberculosis pathogenesis. Scientific reports 2021, 11, 12396. [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffe, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog 2008, 4, e1000204. [CrossRef]
- Sarathy, J.P.; Dartois, V. Caseum: a Niche for Mycobacterium tuberculosis Drug-Tolerant Persisters. Clinical microbiology reviews 2020, 33. [CrossRef]
- Shim, D.; Kim, H.; Shin, S.J. Mycobacterium tuberculosis Infection-Driven Foamy Macrophages and Their Implications in Tuberculosis Control as Targets for Host-Directed Therapy. Front Immunol 2020, 11, 910. [CrossRef]
- Brandenburg, J.; Marwitz, S.; Tazoll, S.C.; Waldow, F.; Kalsdorf, B.; Vierbuchen, T.; Scholzen, T.; Gross, A.; Goldenbaum, S.; Holscher, A.; et al. WNT6/ACC2-induced storage of triacylglycerols in macrophages is exploited by Mycobacterium tuberculosis. J Clin Invest 2021, 131. [CrossRef]
- Fatima, S.; Bhaskar, A.; Dwivedi, V.P. Repurposing Immunomodulatory Drugs to Combat Tuberculosis. Front Immunol 2021, 12, 645485. [CrossRef]
- Battah, B.; Chemi, G.; Butini, S.; Campiani, G.; Brogi, S.; Delogu, G.; Gemma, S. A Repurposing Approach for Uncovering the Anti-Tubercular Activity of FDA-Approved Drugs with Potential Multi-Targeting Profiles. Molecules 2019, 24. [CrossRef]
- Kim, H.; Shin, S.J. Revolutionizing control strategies against Mycobacterium tuberculosis infection through selected targeting of lipid metabolism. Cell Mol Life Sci 2023, 80, 291. [CrossRef]
- Li, X.; Sheng, L.; Lou, L. Statin Use May Be Associated With Reduced Active Tuberculosis Infection: A Meta-Analysis of Observational Studies. Frontiers in medicine 2020, 7, 121. [CrossRef]
- Su, V.Y.; Pan, S.W.; Yen, Y.F.; Feng, J.Y.; Su, W.J.; Chen, Y.M. Statin use and impact on tuberculosis risk. Expert review of anti-infective therapy 2021, 19, 1093-1098. [CrossRef]
- Duan, H.; Liu, T.; Zhang, X.; Yu, A.; Cao, Y. Statin use and risk of tuberculosis: a systemic review of observational studies. International journal of infectious diseases: IJID: official publication of the International Society for Infectious Diseases 2020, 93, 168-174. [CrossRef]
- Parihar, S.P.; Guler, R.; Brombacher, F. Statins: a viable candidate for host-directed therapy against infectious diseases. Nat Rev Immunol 2019, 19, 104-117. [CrossRef]
- Tahir, F.; Bin Arif, T.; Ahmed, J.; Shah, S.R.; Khalid, M. Anti-tuberculous Effects of Statin Therapy: A Review of Literature. Cureus 2020, 12, e7404. [CrossRef]
- Guerra-De-Blas, P.D.C.; Torres-Gonzalez, P.; Bobadilla-Del-Valle, M.; Sada-Ovalle, I.; Ponce-De-Leon-Garduno, A.; Sifuentes-Osornio, J. Potential Effect of Statins on Mycobacterium tuberculosis Infection. Journal of immunology research 2018, 2018, 7617023. [CrossRef]
- Wang, M.; Casey, P.J. Protein prenylation: unique fats make their mark on biology. Nature reviews. Molecular cell biology 2016, 17, 110-122. [CrossRef]
- Bifulco, M. Role of the isoprenoid pathway in ras transforming activity, cytoskeleton organization, cell proliferation and apoptosis. Life sciences 2005, 77, 1740-1749. [CrossRef]
- Montero, M.T.; Hernandez, O.; Suarez, Y.; Matilla, J.; Ferruelo, A.J.; Martinez-Botas, J.; Gomez-Coronado, D.; Lasuncion, M.A. Hydroxymethylglutaryl-coenzyme A reductase inhibition stimulates caspase-1 activity and Th1-cytokine release in peripheral blood mononuclear cells. Atherosclerosis 2000, 153, 303-313. [CrossRef]
- Ma, J.; Zhao, S.; Gao, X.; Wang, R.; Liu, J.; Zhou, X.; Zhou, Y. The Roles of Inflammasomes in Host Defense against Mycobacterium tuberculosis. Pathogens 2021, 10. [CrossRef]
- Dang, E.V.; Cyster, J.G. Loss of sterol metabolic homeostasis triggers inflammasomes - how and why. Curr Opin Immunol 2019, 56, 1-9. [CrossRef]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: guardians of the body. Annu Rev Immunol 2009, 27, 229-265. [CrossRef]
- Kesavardhana, S.; Kanneganti, T.D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol 2017, 29, 201-210. [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cellular & molecular immunology 2021, 18, 1141-1160. [CrossRef]
- Ross, C.; Chan, A.H.; von Pein, J.B.; Maddugoda, M.P.; Boucher, D.; Schroder, K. Inflammatory Caspases: Toward a Unified Model for Caspase Activation by Inflammasomes. Annu Rev Immunol 2022, 40, 249-269. [CrossRef]
- Master, S.S.; Rampini, S.K.; Davis, A.S.; Keller, C.; Ehlers, S.; Springer, B.; Timmins, G.S.; Sander, P.; Deretic, V. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 2008, 3, 224-232. [CrossRef]
- Wawrocki, S.; Druszczynska, M. Inflammasomes in Mycobacterium tuberculosis-Driven Immunity. The Canadian journal of infectious diseases & medical microbiology = Journal canadien des maladies infectieuses et de la microbiologie medicale 2017, 2017, 2309478. [CrossRef]
- Rastogi, S.B., V. Interaction of Mycobacteria With Host Cell Inflammasomes. frontiers in immunology 2022, 13. [CrossRef]
- Eklund, D.; Welin, A.; Andersson, H.; Verma, D.; Soderkvist, P.; Stendahl, O.; Sarndahl, E.; Lerm, M. Human Gene Variants Linked to Limit Intramacrophage Growth of Mycobacterium tuberculosis. J Infect Dis 2014, 209, 749-753.
- Cardoso, D.; Perucha, E. Cholesterol metabolism: a new molecular switch to control inflammation. Clin Sci (Lond) 2021, 135, 1389-1408. [CrossRef]
- Malen, H.; De Souza, G.A.; Pathak, S.; Softeland, T.; Wiker, H.G. Comparison of membrane proteins of Mycobacterium tuberculosis H37Rv and H37Ra strains. BMC Microbiol 2011, 11, 18. [CrossRef]
- Zhang, M.; Gong, J.; Lin, Y.; Barnes, P.F. Growth of virulent and avirulent Mycobacterium tuberculosis strains in human macrophages. Infect Immun 1998, 66, 794-799. [CrossRef]
- Singh, V.; Jamwal, S.; Jain, R.; Verma, P.; Gokhale, R.; Rao, K.V. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 2012, 12, 669-681. [CrossRef]
- Youmans, G.P.; Youmans, A.S. An Acute Pulmonary Granulomatous Response in Mice Produced by Mycobacterial Cells and Its Relation to Increased Resistance and Increased Susceptibility to Experimental Tuberculous Infection. J Infect Dis 1964, 114, 135-151. [CrossRef]
- Boyum, A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl 1968, 97, 77-89.
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. The Journal of cell biology 1963, 17, 208-212. [CrossRef]
- Xiang, Y.; Zhao, M.M.; Sun, S.; Guo, X.L.; Wang, Q.; Li, S.A.; Lee, W.H.; Zhang, Y. A high concentration of DMSO activates caspase-1 by increasing the cell membrane permeability of potassium. Cytotechnology 2018, 70, 313-320. [CrossRef]
- Patino, S.; Alamo, L.; Cimino, M.; Casart, Y.; Bartoli, F.; Garcia, M.J.; Salazar, L. Autofluorescence of mycobacteria as a tool for detection of Mycobacterium tuberculosis. Journal of clinical microbiology 2008, 46, 3296-3302. [CrossRef]
- Montero, M.T.; Matilla, J.; Gomez-Mampaso, E.; Lasuncion, M.A. Geranylgeraniol regulates negatively caspase-1 autoprocessing: implication in the Th1 response against Mycobacterium tuberculosis. J Immunol 2004, 173, 4936-4944. [CrossRef]
- Ivessa, N.E.; Gravotta, D.; De Lemos-Chiarandini, C.; Kreibich, G. Functional protein prenylation is required for the brefeldin A-dependent retrograde transport from the Golgi apparatus to the endoplasmic reticulum. J Biol Chem 1997, 272, 20828-20834. [CrossRef]
- Huang, Z.; Luo, Q.; Guo, Y.; Chen, J.; Xiong, G.; Peng, Y.; Ye, J.; Li, J. Mycobacterium tuberculosis-Induced Polarization of Human Macrophage Orchestrates the Formation and Development of Tuberculous Granulomas In Vitro. PLoS One 2015, 10, e0129744. [CrossRef]
- De Faramarz Naeim, P.N.R., Sophie Song, Ryan Phan. . Atlas of Hematopathology: Morphology, Immunophenotype, Cytogenetics, and Molecular Approaches. Histiocytic Disorders 2018, , 829-849. [CrossRef]
- MyPathologyReport / Foamy histiocytes Available online: https://www.mypathologyreport.ca/es/pathology-dictionary/foamy-histiocytes/ (accessed on May 2, 2023).
- Adams, D.O. The structure of mononuclear phagocytes differentiating in vivo. I. Sequential fine and histologic studies of the effect of Bacillus Calmette-Guerin (BCG). The American journal of pathology 1974, 76, 17-48.
- Armstrong, J.A.; Hart, P.D. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med 1971, 134, 713-740.
- Westman, J.; Grinstein, S.; Marques, P.E. Phagocytosis of Necrotic Debris at Sites of Injury and Inflammation. Front Immunol 2019, 10, 3030. [CrossRef]
- Melo, R.C.; Dvorak, A.M. Lipid body-phagosome interaction in macrophages during infectious diseases: host defense or pathogen survival strategy? PLoS Pathog 2012, 8, e1002729.
- Bryan, N.B.; Dorfleutner, A.; Rojanasakul, Y.; Stehlik, C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 2009, 182, 3173-3182. [CrossRef]
- Baroja-Mazo, A.; Martin-Sanchez, F.; Gomez, A.I.; Martinez, C.M.; Amores-Iniesta, J.; Compan, V.; Barbera-Cremades, M.; Yague, J.; Ruiz-Ortiz, E.; Anton, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 2014, 15, 738-748. [CrossRef]
- Corleis, B.; Dorhoi, A. Early dynamics of innate immunity during pulmonary tuberculosis. Immunology letters 2020, 221, 56-60. [CrossRef]
- Cadena, A.M.; Flynn, J.L.; Fortune, S.M. The Importance of First Impressions: Early Events in Mycobacterium tuberculosis Infection Influence Outcome. mBio 2016, 7, e00342-00316. [CrossRef]
- Lerner, T.R.; Borel, S.; Greenwood, D.J.; Repnik, U.; Russell, M.R.; Herbst, S.; Jones, M.L.; Collinson, L.M.; Griffiths, G.; Gutierrez, M.G. Mycobacterium tuberculosis replicates within necrotic human macrophages. The Journal of cell biology 2017, 216, 583-594. [CrossRef]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Tak, U.; Sun, J.; Orihuela, C.J.; Niederweis, M. NAD(+) Depletion Triggers Macrophage Necroptosis, a Cell Death Pathway Exploited by Mycobacterium tuberculosis. Cell reports 2018, 24, 429-440. [CrossRef]
- Behar, S.M.; Divangahi, M.; Remold, H.G. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol 2010, 8, 668-674. [CrossRef]
- Lewis, M.R. The Formation of Macrophages, Epithelioid Cells and Giant Cells from Leucocytes in Incubated Blood. The American journal of pathology 1925, 1, 91-100 101.
- Cosma, C.L.; Humbert, O.; Sherman, D.R.; Ramakrishnan, L. Trafficking of superinfecting Mycobacterium organisms into established granulomas occurs in mammals and is independent of the Erp and ESX-1 mycobacterial virulence loci. J Infect Dis 2008, 198, 1851-1855. [CrossRef]
- Moule, M.G.; Cirillo, J.D. Mycobacterium tuberculosis Dissemination Plays a Critical Role in Pathogenesis. Front Cell Infect Microbiol 2020, 10, 65. [CrossRef]
- Available online: https://www.sciencedirect.com/topics/medicine-and-dentistry/foam-cell (accessed on 07/25/2023).
- Guerrini, V.; Gennaro, M.L. Foam Cells: One Size Doesn’t Fit All. Trends in immunology 2019, 40, 1163-1179. [CrossRef]
- Guerrini, V.; Prideaux, B.; Khan, R.; Subbian, S.; Wang, Y.; Sadimin, E.; Pawar, S.; Ukey, R.; Singer, E.A.; Xue, C.; et al. Heterogeneity of foam cell biogenesis across diseases. bioRxiv: the preprint server for biology 2023. [CrossRef]
- Rossi, G.; Cavazza, A.; Spagnolo, P.; Bellafiore, S.; Kuhn, E.; Carassai, P.; Caramanico, L.; Montanari, G.; Cappiello, G.; Andreani, A.; et al. The role of macrophages in interstitial lung diseases: Number 3 in the Series “Pathology for the clinician” Edited by Peter Dorfmuller and Alberto Cavazza. European respiratory review: an official journal of the European Respiratory Society 2017, 26. [CrossRef]
- Guerrini, V.; Prideaux, B.; Blanc, L.; Bruiners, N.; Arrigucci, R.; Singh, S.; Ho-Liang, H.P.; Salamon, H.; Chen, P.Y.; Lakehal, K.; et al. Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease-specific. PLoS Pathog 2018, 14, e1007223. [CrossRef]
- Agarwal, P.; Gordon, S.; Martinez, F.O. Foam Cell Macrophages in Tuberculosis. Front Immunol 2021, 12, 775326. [CrossRef]
- Cardona, P.J.; Llatjos, R.; Gordillo, S.; Diaz, J.; Ojanguren, I.; Ariza, A.; Ausina, V. Evolution of granulomas in lungs of mice infected aerogenically with Mycobacterium tuberculosis. Scand J Immunol 2000, 52, 156-163. [CrossRef]
- Pagan, A.J.; Ramakrishnan, L. The Formation and Function of Granulomas. Annu Rev Immunol 2018, 36, 639-665. [CrossRef]
- O’Garra, A.; Redford, P.S.; McNab, F.W.; Bloom, C.I.; Wilkinson, R.J.; Berry, M.P. The immune response in tuberculosis. Annu Rev Immunol 2013, 31, 475-527.
- Matsuda, M.; Korn, B.S.; Hammer, R.E.; Moon, Y.A.; Komuro, R.; Horton, J.D.; Goldstein, J.L.; Brown, M.S.; Shimomura, I. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes & development 2001, 15, 1206-1216. [CrossRef]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu Rev Biochem 2018, 87, 783-807. [CrossRef]
- Mahmoudi, A.; Atkin, S.L.; Jamialahmadi, T.; Sahebkar, A. Identification of key upregulated genes involved in foam cell formation and the modulatory role of statin therapy. Int Immunopharmacol 2023, 119, 110209. [CrossRef]
- Pontillo, A.; Paoluzzi, E.; Crovella, S. The inhibition of mevalonate pathway induces upregulation of NALP3 expression: new insight in the pathogenesis of mevalonate kinase deficiency. Eur J Hum Genet 2010, 18, 844-847. [CrossRef]
- Kaneko, J.; Okinaga, T.; Hikiji, H.; Ariyoshi, W.; Yoshiga, D.; Habu, M.; Tominaga, K.; Nishihara, T. Zoledronic acid exacerbates inflammation through M1 macrophage polarization. Inflammation and regeneration 2018, 38, 16. [CrossRef]
- Marcuzzi, A.; Piscianz, E.; Zweyer, M.; Bortul, R.; Loganes, C.; Girardelli, M.; Baj, G.; Monasta, L.; Celeghini, C. Geranylgeraniol and Neurological Impairment: Involvement of Apoptosis and Mitochondrial Morphology. International journal of molecular sciences 2016, 17, 365. [CrossRef]
- Zhang, Q.; Yu, W.; Lee, S.; Xu, Q.; Naji, A.; Le, A.D. Bisphosphonate Induces Osteonecrosis of the Jaw in Diabetic Mice via NLRP3/Caspase-1-Dependent IL-1beta Mechanism. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research 2015, 30, 2300-2312. [CrossRef]
- Mandey, S.H.; Kuijk, L.M.; Frenkel, J.; Waterham, H.R. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum 2006, 54, 3690-3695. [CrossRef]
- Politiek, F.A.; Waterham, H.R. Compromised Protein Prenylation as Pathogenic Mechanism in Mevalonate Kinase Deficiency. Front Immunol 2021, 12, 724991. [CrossRef]
- Guo, C.; Chi, Z.; Jiang, D.; Xu, T.; Yu, W.; Wang, Z.; Chen, S.; Zhang, L.; Liu, Q.; Guo, X.; et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity 2018, 49, 842-856 e847. [CrossRef]
- Jeyaratnam, J.; Frenkel, J. Management of Mevalonate Kinase Deficiency: A Pediatric Perspective. Front Immunol 2020, 11, 1150. [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med 2018, 215, 827-840. [CrossRef]
- Pizzuto, M.; Pelegrin, P.; Ruysschaert, J.M. Lipid-protein interactions regulating the canonical and the non-canonical NLRP3 inflammasome. Progress in lipid research 2022, 87, 101182. [CrossRef]
- Pandey, A.; Shen, C.; Feng, S.; Man, S.M. Cell biology of inflammasome activation. Trends Cell Biol 2021, 31, 924-939. [CrossRef]
- Ta, A.; Vanaja, S.K. Inflammasome activation and evasion by bacterial pathogens. Curr Opin Immunol 2021, 68, 125-133. [CrossRef]
- Gideon, H.P.; Hughes, T.K.; Tzouanas, C.N.; Wadsworth, M.H., 2nd; Tu, A.A.; Gierahn, T.M.; Peters, J.M.; Hopkins, F.F.; Wei, J.R.; Kummerlowe, C.; et al. Multimodal profiling of lung granulomas in macaques reveals cellular correlates of tuberculosis control. Immunity 2022, 55, 827-846 e810. [CrossRef]
- Tominaga, K.; Yoshimoto, T.; Torigoe, K.; Kurimoto, M.; Matsui, K.; Hada, T.; Okamura, H.; Nakanishi, K. IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int Immunol 2000, 12, 151-160. [CrossRef]
- Cardona, P.J. Revisiting the natural history of tuberculosis. The inclusion of constant reinfection, host tolerance, and damage-response frameworks leads to a better understanding of latent infection and its evolution towards active disease. Archivum immunologiae et therapiae experimentalis 2010, 58, 7-14. [CrossRef]
- Ehlers, S.; Schaible, U.E. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol 2012, 3, 411. [CrossRef]
- Dutta, N.K.; Bruiners, N.; Zimmerman, M.D.; Tan, S.; Dartois, V.; Gennaro, M.L.; Karakousis, P.C. Adjunctive Host-Directed Therapy With Statins Improves Tuberculosis-Related Outcomes in Mice. J Infect Dis 2020, 221, 1079-1087. [CrossRef]
- Adewole, O.O.; Omotoso, B.A.; Ogunsina, M.; Aminu, A.; Odeyemi, A.O.; Awopeju, O.F.; Ayoola, O.; Adedeji, T.; Sogaolu, O.M.; Adewole, T.O.; et al. Atorvastatin accelerates Mycobacterium tuberculosis clearance in pulmonary TB: a randomised phase IIA trial. Int J Tuberc Lung Dis 2023, 27, 226-228. [CrossRef]
- Molla, M.D.; Akalu, Y.; Geto, Z.; Dagnew, B.; Ayelign, B.; Shibabaw, T. Role of Caspase-1 in the Pathogenesis of Inflammatory-Associated Chronic Noncommunicable Diseases. Journal of inflammation research 2020, 13, 749-764. [CrossRef]
- Mansi, I.A.; Chansard, M.; Lingvay, I.; Zhang, S.; Halm, E.A.; Alvarez, C.A. Association of Statin Therapy Initiation With Diabetes Progression: A Retrospective Matched-Cohort Study. JAMA internal medicine 2021, 181, 1562-1574. [CrossRef]
- Lin, P.L.; Flynn, J.L. The End of the Binary Era: Revisiting the Spectrum of Tuberculosis. J Immunol 2018, 201, 2541-2548. [CrossRef]
- Hunter, R.L. The Pathogenesis of Tuberculosis-The Koch Phenomenon Reinstated. Pathogens 2020, 9. [CrossRef]
Figure 1.
Changes undergone by macrophages of PBMC under the different incubation conditions. (A) Macrophages in untreated PBMC show small vacuoles (control condition). (B) Fluvastatin treatment of PBMC induces a generalized conversion of monocytes/macrophages to highly vacuolated cells, similar to conventional foamy macrophages, here referred as FFM. Note how these macrophages tightly attach several monocytes and/or lymphocytes to them. (C) In the absence of fluvastatin, inactivated Mtb H37Ra induces in PBMC the formation of granuloma-like structures (GLS) and transforms a few macrophages to foamy cells alike FFM (D) In fluvastatin-treated cultures the exposition to Mtb H37Ra induces cellular aggregates of FFM rather than GLS. Cells were stained with May-Grünwald-Giemsa solutions and analyzed by optical microscopy.
Figure 1.
Changes undergone by macrophages of PBMC under the different incubation conditions. (A) Macrophages in untreated PBMC show small vacuoles (control condition). (B) Fluvastatin treatment of PBMC induces a generalized conversion of monocytes/macrophages to highly vacuolated cells, similar to conventional foamy macrophages, here referred as FFM. Note how these macrophages tightly attach several monocytes and/or lymphocytes to them. (C) In the absence of fluvastatin, inactivated Mtb H37Ra induces in PBMC the formation of granuloma-like structures (GLS) and transforms a few macrophages to foamy cells alike FFM (D) In fluvastatin-treated cultures the exposition to Mtb H37Ra induces cellular aggregates of FFM rather than GLS. Cells were stained with May-Grünwald-Giemsa solutions and analyzed by optical microscopy.
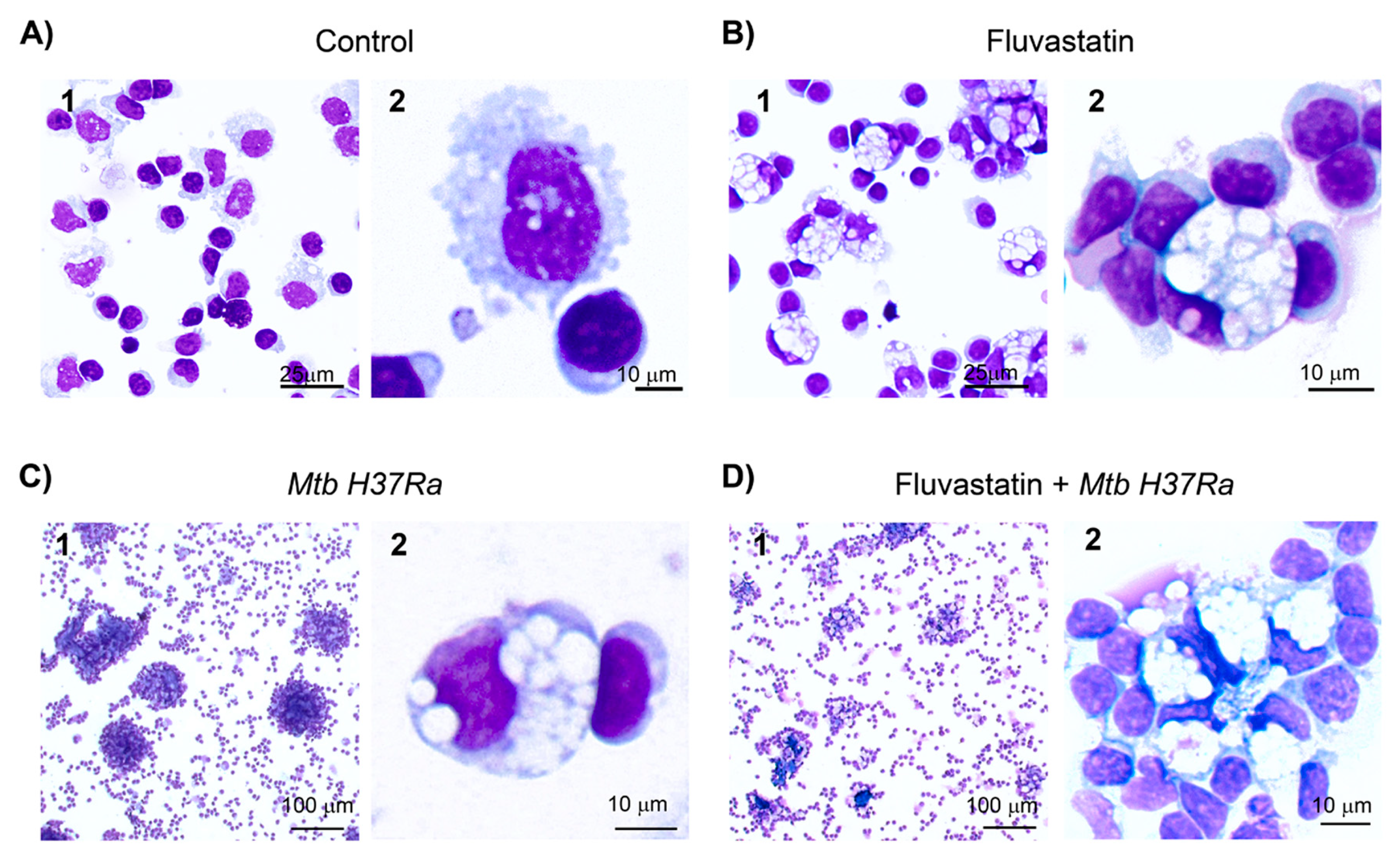
Figure 2.
Effect of fluvastatin in the time course response of PBMC to inactivated Mtb H37Ra exposition. (A). In cultures only exposed to the bacteria, two or a few macrophages initiate the formation of granuloma-like structures (GLS) by surrounding small amorphous masses (1-2). These central masses grow overtime and appear encircled by some broken nuclei and by new recruited macrophages (3). Thereafter, much more macrophages enclose these necrotic cores in compact GLS (4-5). Note that some FM incorporate to these structures (arrows in 3 and 4). At the end of the incubation most GLS are highly compacted and scarcely contain FM (5), however a few GLS are less compacted, enclose a smaller necrotic core, and incorporate more FM (6). (B). Similarly, in fluvastatin treated cultures exposed to inactivated Mtb H37Ra, two or more FFM surround an amorphous mass, but that evolve to small necrotic cores surrounded by numerous vacuolated macrophages (1-3). Later, more FFM surround them (4-5), forming small non compacted cells aggregates than enclose reduced necrotic areas (compare B5 and A5). Occasionally, some aggregates have large necrotic cores and less FFM (6). Cells were stained with May-Grünwald-Giemsa solutions and analyzed by optical microscopy.
Figure 2.
Effect of fluvastatin in the time course response of PBMC to inactivated Mtb H37Ra exposition. (A). In cultures only exposed to the bacteria, two or a few macrophages initiate the formation of granuloma-like structures (GLS) by surrounding small amorphous masses (1-2). These central masses grow overtime and appear encircled by some broken nuclei and by new recruited macrophages (3). Thereafter, much more macrophages enclose these necrotic cores in compact GLS (4-5). Note that some FM incorporate to these structures (arrows in 3 and 4). At the end of the incubation most GLS are highly compacted and scarcely contain FM (5), however a few GLS are less compacted, enclose a smaller necrotic core, and incorporate more FM (6). (B). Similarly, in fluvastatin treated cultures exposed to inactivated Mtb H37Ra, two or more FFM surround an amorphous mass, but that evolve to small necrotic cores surrounded by numerous vacuolated macrophages (1-3). Later, more FFM surround them (4-5), forming small non compacted cells aggregates than enclose reduced necrotic areas (compare B5 and A5). Occasionally, some aggregates have large necrotic cores and less FFM (6). Cells were stained with May-Grünwald-Giemsa solutions and analyzed by optical microscopy.
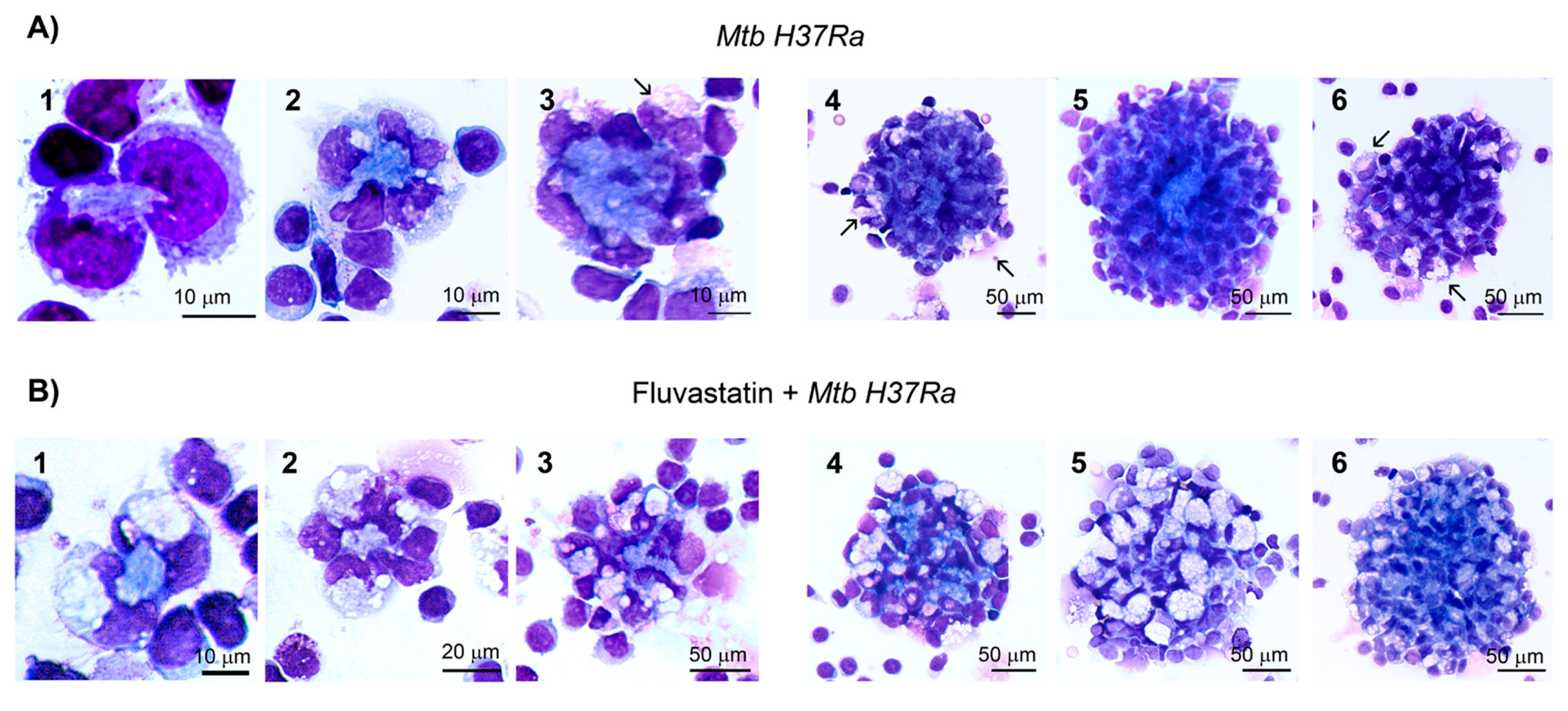
Figure 3.
Ultrastructural characteristics of macrophages. (A) Resting macrophages in control PBMC. (B) In inactivated Mtb H37Ra stimulated cultures, macrophages become epithelioid cells (1) that associate through a net of filopodia (2), and contain phagosomes that scarcely degrade bacteria (3). These cultures also show a few FM with large cytoplasmic vacuoles and some smaller ones at the margin of the cell (4 and 5, black arrows). Some of them FM enclose a few undegraded bacilli within phagosomes (white square in 6, enlarged in 7), and also phagocytose bacteria, along with debris, delivering them into the same vacuole (black square in 6, enlarged in 8). Note that in this vacuole three bacteria show disorganized membranes at the contact points with a vesicular organelle (8, black arrow). (C) FFM contain numerous vacuoles that in some cases are closely associated to the nuclei (1, white arrows). These vacuolated macrophages emit phagocytic filopodia (2, black arrows), and enclose vesicular organelles in their vacuoles (3). These cells tightly attach to a small cell through an intercellular membrane association (4, enlarged in 5), and emit filopodia-like projections to subdivide vacuoles (6). (D) FFM exposed to inactivated Mtb H37Ra do not show well defined vacuoles, and their cytoplasm is filled with numerous vesicular organelles and debris, apparently trapped by a poorly defined network of filopodia (1). These organelles can be released out of the cells to be recognized by filopodia emitted by other cells (2). FM also contain undegraded bacteria inside phagosomes (3, black arrow, enlarged in the inserted image), or in vacuoles where they show signs of degradation (3, white arrow, enlarged in 4). Note the presence in these macrophages of large lipid bodies in the cytoplasm (5) and nearby, partially degraded bacteria inside vacuoles (5, square and 6, black arrows).
Figure 3.
Ultrastructural characteristics of macrophages. (A) Resting macrophages in control PBMC. (B) In inactivated Mtb H37Ra stimulated cultures, macrophages become epithelioid cells (1) that associate through a net of filopodia (2), and contain phagosomes that scarcely degrade bacteria (3). These cultures also show a few FM with large cytoplasmic vacuoles and some smaller ones at the margin of the cell (4 and 5, black arrows). Some of them FM enclose a few undegraded bacilli within phagosomes (white square in 6, enlarged in 7), and also phagocytose bacteria, along with debris, delivering them into the same vacuole (black square in 6, enlarged in 8). Note that in this vacuole three bacteria show disorganized membranes at the contact points with a vesicular organelle (8, black arrow). (C) FFM contain numerous vacuoles that in some cases are closely associated to the nuclei (1, white arrows). These vacuolated macrophages emit phagocytic filopodia (2, black arrows), and enclose vesicular organelles in their vacuoles (3). These cells tightly attach to a small cell through an intercellular membrane association (4, enlarged in 5), and emit filopodia-like projections to subdivide vacuoles (6). (D) FFM exposed to inactivated Mtb H37Ra do not show well defined vacuoles, and their cytoplasm is filled with numerous vesicular organelles and debris, apparently trapped by a poorly defined network of filopodia (1). These organelles can be released out of the cells to be recognized by filopodia emitted by other cells (2). FM also contain undegraded bacteria inside phagosomes (3, black arrow, enlarged in the inserted image), or in vacuoles where they show signs of degradation (3, white arrow, enlarged in 4). Note the presence in these macrophages of large lipid bodies in the cytoplasm (5) and nearby, partially degraded bacteria inside vacuoles (5, square and 6, black arrows).
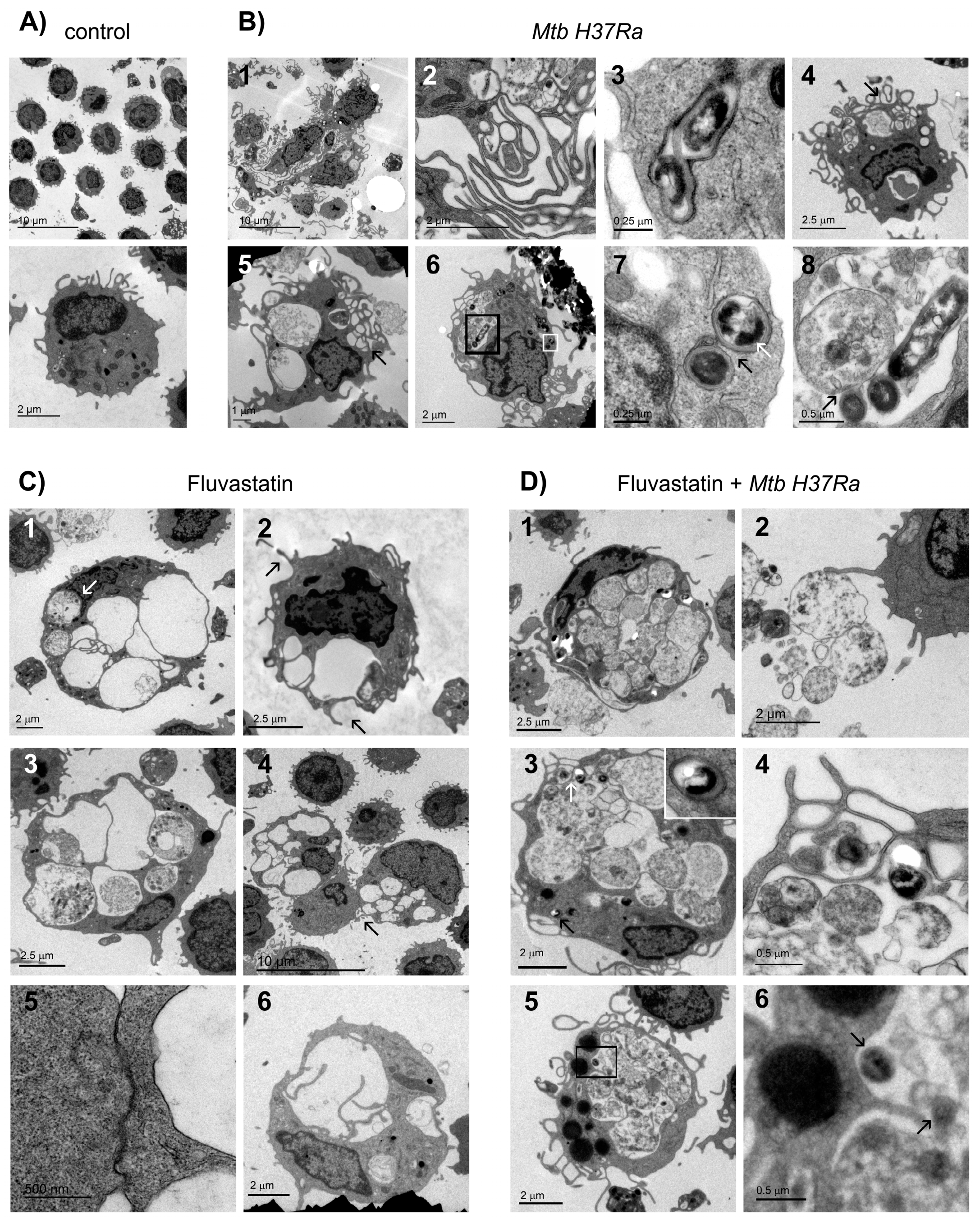
Figure 4.
Cellular localization of ASC and NLRP3 in macrophages from different experimental conditions. Microphotographs show immunodetection of ASC (green), NLRP3 (red) and Hoechst-stained nuclei (blue). Control row shows resting macrophages with some ASC inside the nucleus and NLRP3 forming small perinuclear dots. In Mtb H37Ra most cells in GLS show increased ASC in their nuclei and concentrate NLRP3 in single perinuclear dots (merged + nuclei column). In FM induced in response to the bacteria, NLRP3 forms a few ring-shaped aggregates in the cytoplasm. ASC increases in the nucleus, and partially moves to the cytoplasm to co-localize with NLRP3 (yellow in merged column). Under fluvastatin condition, FFM markedly increase ASC in the nucleus and NLRP3 in the cytoplasm where it forms numerous ring-shaped structures (merged + nuclei column). In response to inactivated Mtb H37Ra, FFM show high amounts of ASC that has moved from the nucleus to the cytoplasm to co-localize with NLRP3 (yellow in merged + nuclei column). Note the presence of free extracellular particles where ASC partially co-localize with NLRP3 (white arrow in merged column).
Figure 4.
Cellular localization of ASC and NLRP3 in macrophages from different experimental conditions. Microphotographs show immunodetection of ASC (green), NLRP3 (red) and Hoechst-stained nuclei (blue). Control row shows resting macrophages with some ASC inside the nucleus and NLRP3 forming small perinuclear dots. In Mtb H37Ra most cells in GLS show increased ASC in their nuclei and concentrate NLRP3 in single perinuclear dots (merged + nuclei column). In FM induced in response to the bacteria, NLRP3 forms a few ring-shaped aggregates in the cytoplasm. ASC increases in the nucleus, and partially moves to the cytoplasm to co-localize with NLRP3 (yellow in merged column). Under fluvastatin condition, FFM markedly increase ASC in the nucleus and NLRP3 in the cytoplasm where it forms numerous ring-shaped structures (merged + nuclei column). In response to inactivated Mtb H37Ra, FFM show high amounts of ASC that has moved from the nucleus to the cytoplasm to co-localize with NLRP3 (yellow in merged + nuclei column). Note the presence of free extracellular particles where ASC partially co-localize with NLRP3 (white arrow in merged column).
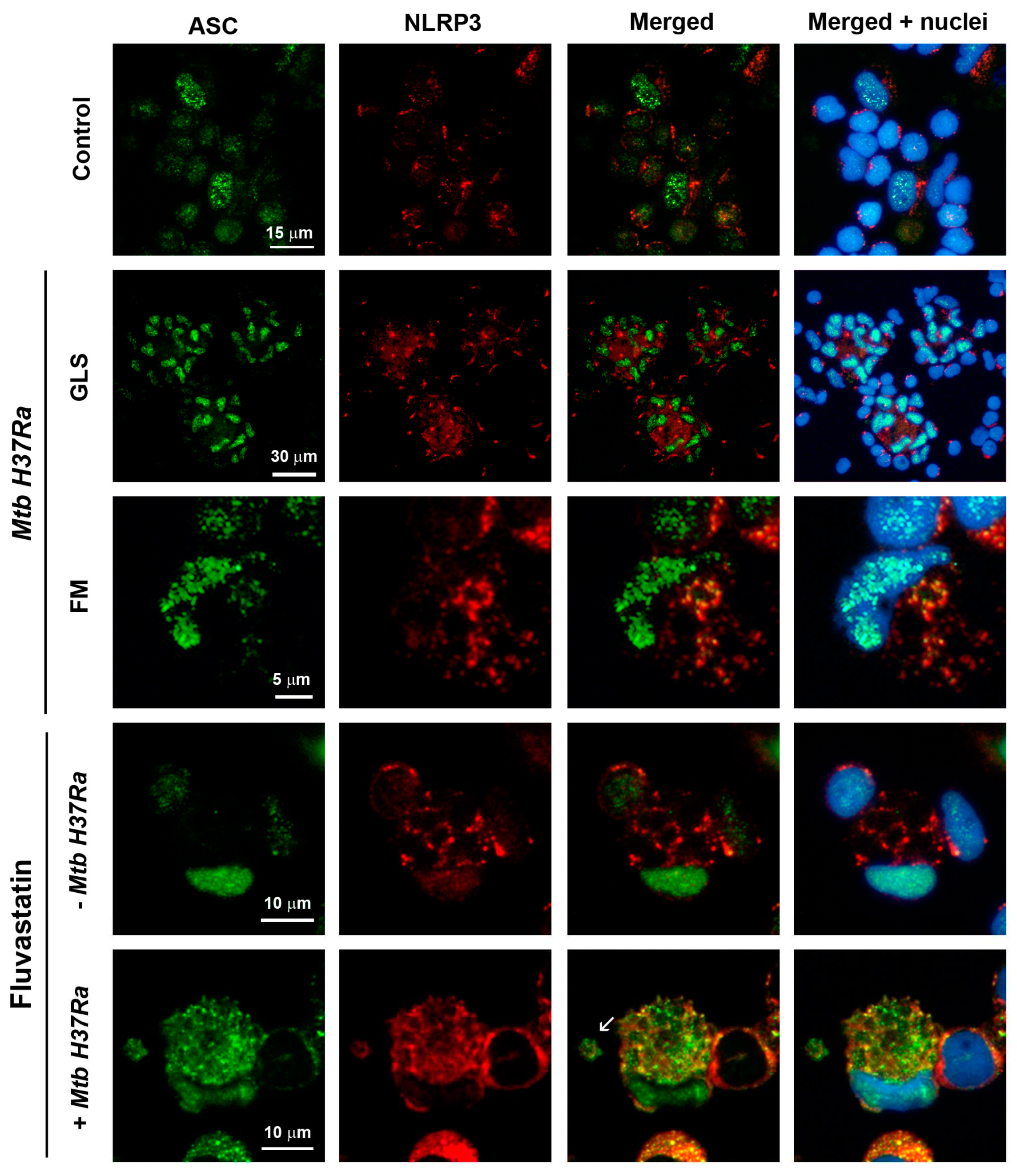
Figure 5.
Detection of active caspase-1 in PBMC by the FAM-FLICA assay. Green fluorescence is emitted by FAM-YVAD-FMK (specific ligand of caspase-1) and cells nuclei are stained with Hoeschst. (A) Control PBMC show specific green fluorescence as dots in the broken nuclei of some macrophages (1,2) or forming large extracellular aggregates (2, white arrows). (B) In fluvastatin-treated PBMC, a variable number of FFM show specific green fluorescence forming a ring near the nucleus (1) or several rings and arcs in the cytoplasm (2). (C) In PBMC exposed to inactivated Mtb H37Ra, a few macrophages show large amounts of bacteria emitting green (1) and red (2) autofluorescence. Merged 1 and 2 images delineate the bacteria in yellow and evidence the absence of specific green fluorescence in these cells (3). Several of these macrophages associate to accumulate the bacilli at a same point (4). In the incipient GLS, the accumulated bacteria form a central core that is surrounded by other macrophages with a very low bacterial load, and that do not show active caspase-1 either (5, 6). In this condition, only some FM show specific green fluorescence inside a few cytoplasmic round formations. Note that these cells do not contain intact yellow bacilli (7, 8). (D) In fluvastatin treated cultures exposed to inactivated Mtb H37Ra, most FFM show an important increase in the number of round formations containing specific green fluorescence (1). These macrophages barely show intact bacteria, but they contain some bacilli remnants inside these round formations enclosing the enzyme active (1) In some of these formations these remnants accumulate at their edges (fig1, white arrow). In the same cultures, a few macrophages accumulate large amounts of bacteria without activate caspase-1 (2). These macrophages also associate to accumulate non-degraded bacilli in a central area (3, yellow) in which some bacteria locate into their broken nuclei. Near them, other FFM contain numerous round formations filled with active caspase-1 (3, green) unmasking numerous red auto-fluorescent small particles indicating bacteria degradation (4, white arrow).
Figure 5.
Detection of active caspase-1 in PBMC by the FAM-FLICA assay. Green fluorescence is emitted by FAM-YVAD-FMK (specific ligand of caspase-1) and cells nuclei are stained with Hoeschst. (A) Control PBMC show specific green fluorescence as dots in the broken nuclei of some macrophages (1,2) or forming large extracellular aggregates (2, white arrows). (B) In fluvastatin-treated PBMC, a variable number of FFM show specific green fluorescence forming a ring near the nucleus (1) or several rings and arcs in the cytoplasm (2). (C) In PBMC exposed to inactivated Mtb H37Ra, a few macrophages show large amounts of bacteria emitting green (1) and red (2) autofluorescence. Merged 1 and 2 images delineate the bacteria in yellow and evidence the absence of specific green fluorescence in these cells (3). Several of these macrophages associate to accumulate the bacilli at a same point (4). In the incipient GLS, the accumulated bacteria form a central core that is surrounded by other macrophages with a very low bacterial load, and that do not show active caspase-1 either (5, 6). In this condition, only some FM show specific green fluorescence inside a few cytoplasmic round formations. Note that these cells do not contain intact yellow bacilli (7, 8). (D) In fluvastatin treated cultures exposed to inactivated Mtb H37Ra, most FFM show an important increase in the number of round formations containing specific green fluorescence (1). These macrophages barely show intact bacteria, but they contain some bacilli remnants inside these round formations enclosing the enzyme active (1) In some of these formations these remnants accumulate at their edges (fig1, white arrow). In the same cultures, a few macrophages accumulate large amounts of bacteria without activate caspase-1 (2). These macrophages also associate to accumulate non-degraded bacilli in a central area (3, yellow) in which some bacteria locate into their broken nuclei. Near them, other FFM contain numerous round formations filled with active caspase-1 (3, green) unmasking numerous red auto-fluorescent small particles indicating bacteria degradation (4, white arrow).
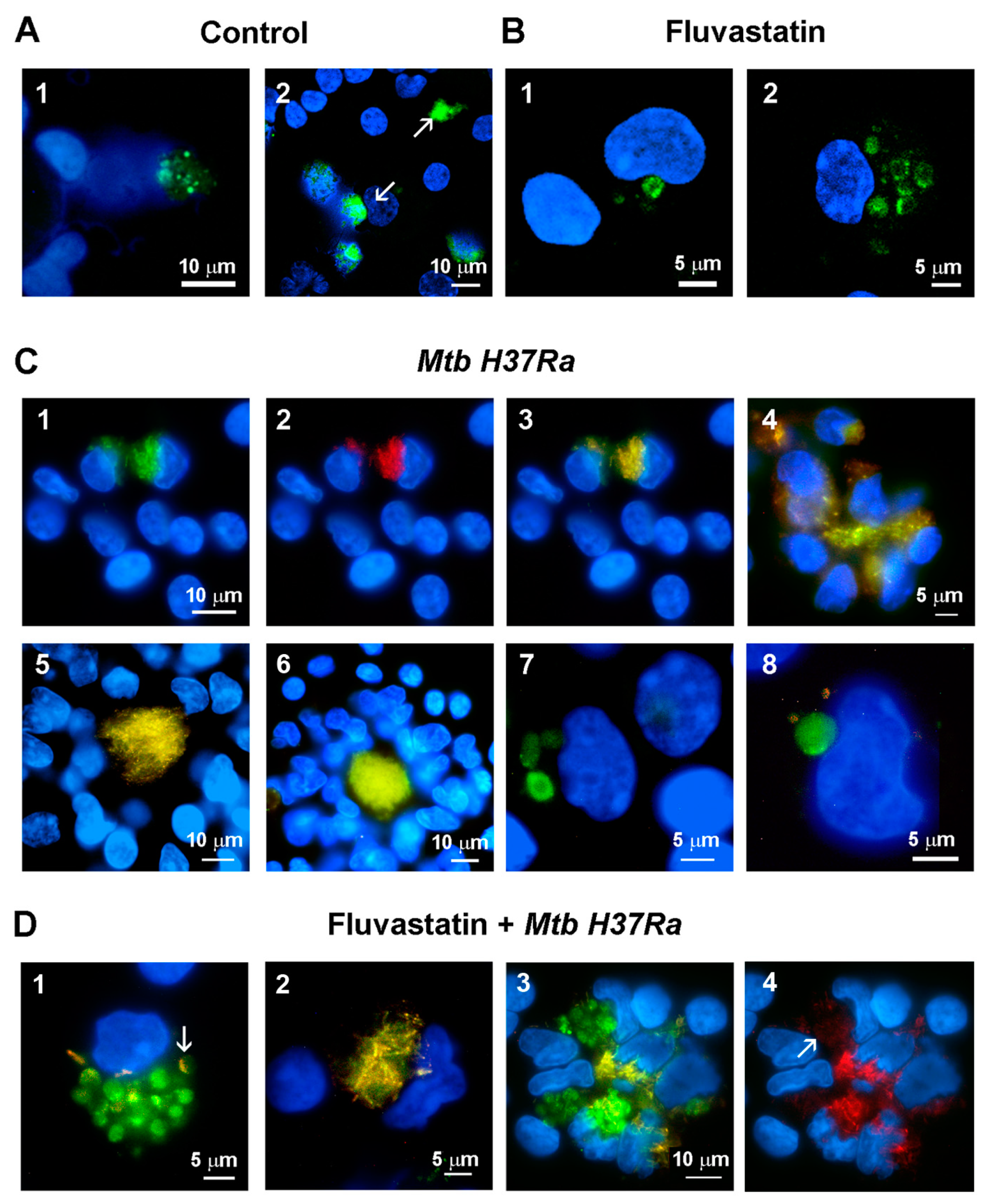
Figure 7.
Fluvastatin exacerbates the cellular response against Mtb H37Ra. (A) Percentage of CD3+CD8-, CD3+CD8+ and CD3-CD56+ cells that produce IFNγ in untreated (black circles) or fluvastatin-treated PBMC exposed to Mtb H37Ra (open circles). (B) Time-course of LDH activity in culture media. Note the significant increase in cytotoxicity promoted by fluvastatin in cells exposed to Mtb H37Ra. (C) Response to tuberculin test in rabbits immunized with Mtb H37Ra that previously received fluvastatin (open circles) or vehicle (black circles). Results in A and B represent the mean ± SE of 9 (A), or 5 (B) independent experiments. *p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001 vs control, +p ≤ 0.05; ++p ≤ 0.01, +++p ≤ 0.001 vs Mtb H37Ra, &p ≤ 0.05; &&p ≤ 0.01, &&&p ≤ 0.001 vs. fluvastatin (Two-way ANOVA+Neuman-Keuls). In C results represent the mean ± SEM of 5 independent rabbits for each experimental condition. +p ≤ 0.05; ++p ≤ 0.01 vs Mtb H37Ra (Unpaired t-test). .
Figure 7.
Fluvastatin exacerbates the cellular response against Mtb H37Ra. (A) Percentage of CD3+CD8-, CD3+CD8+ and CD3-CD56+ cells that produce IFNγ in untreated (black circles) or fluvastatin-treated PBMC exposed to Mtb H37Ra (open circles). (B) Time-course of LDH activity in culture media. Note the significant increase in cytotoxicity promoted by fluvastatin in cells exposed to Mtb H37Ra. (C) Response to tuberculin test in rabbits immunized with Mtb H37Ra that previously received fluvastatin (open circles) or vehicle (black circles). Results in A and B represent the mean ± SE of 9 (A), or 5 (B) independent experiments. *p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001 vs control, +p ≤ 0.05; ++p ≤ 0.01, +++p ≤ 0.001 vs Mtb H37Ra, &p ≤ 0.05; &&p ≤ 0.01, &&&p ≤ 0.001 vs. fluvastatin (Two-way ANOVA+Neuman-Keuls). In C results represent the mean ± SEM of 5 independent rabbits for each experimental condition. +p ≤ 0.05; ++p ≤ 0.01 vs Mtb H37Ra (Unpaired t-test). .
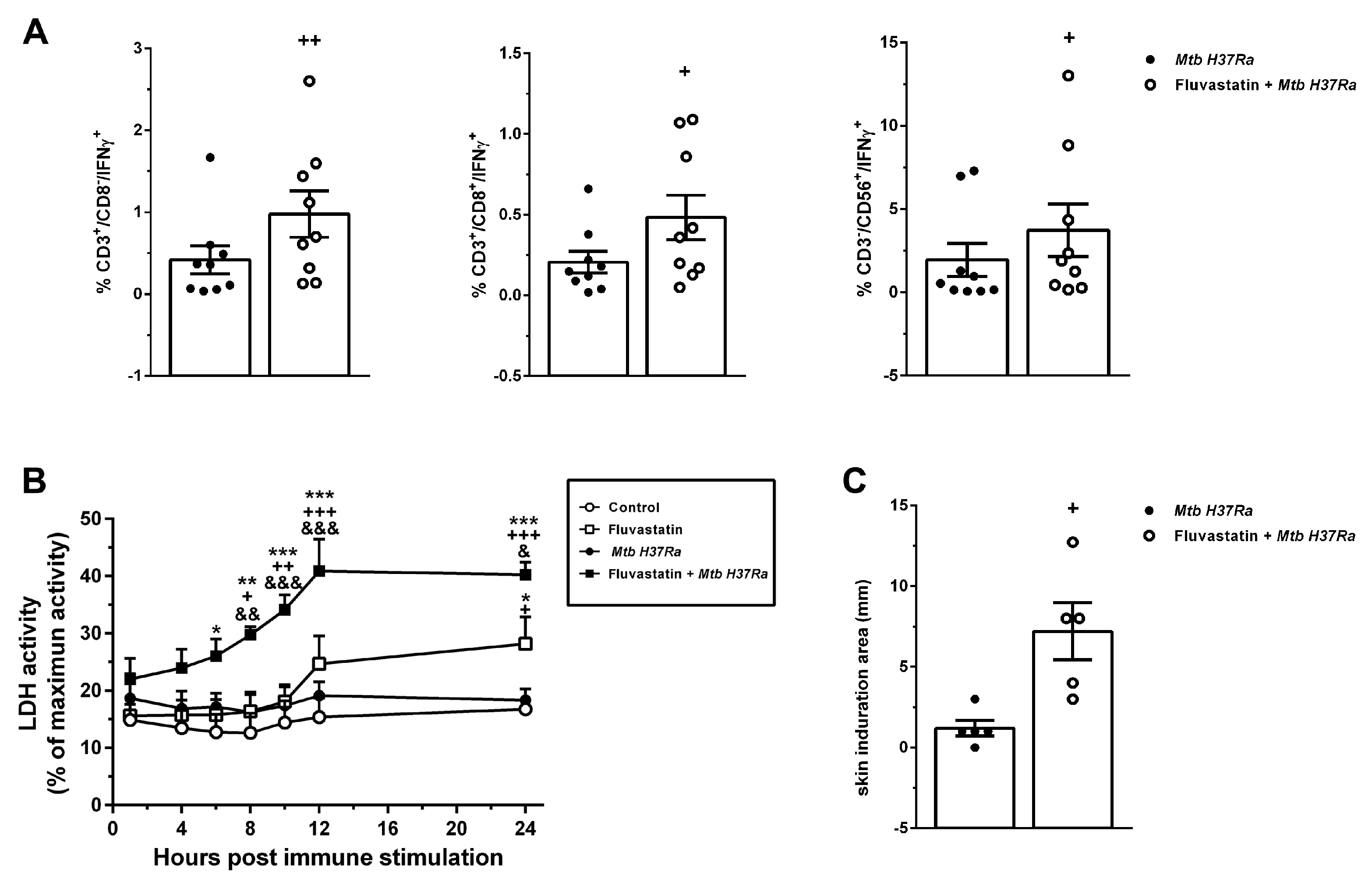
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



