
Submitted:
21 November 2023
Posted:
22 November 2023
You are already at the latest version
Abstract
Induced pluripotent stem cells (iPSC) are derived from reprogrammed adult somatic cells. These adult cells are manipulated in vitro to express genes and factors essential for acquiring and maintaining embryonic stem cell (ESC) properties. This technology is widely applied in many fields, and there has been much attention to developing iPSC-based disease models to validate drug discovery platforms and study pathophysiological molecular processes underlying disease onset. Especially in neurological diseases, there is a great need for iPSC-based technological research, as these cells can be obtained from each patient and carry the individual’s bulk of genetic mutations and properties. Moreover, iPSC can differentiate into multiple cell types. These are essential characteristics since the study of neurological diseases is affected by limited access to injured sites, in vitro models composed of various cell types, the complexity of reproducing the brain’s anatomy, ethical issues, and the fact that post-mortem cell culture is challenging. Neurodegenerative diseases enormously impact global health due to their high incidence, symptoms severity, and usually lack of effective therapies. Recently, analyses using disease-specific iPSC-based models confirmed their efficacy for testing multiple drugs. This review summarizes the advances in iPSC technology used in disease modeling and drug testing with a primary focus on neurodegenerative diseases, including Parkinson’s and Alzheimer’s diseases.
Keywords:
induced pluripotent stem cells
; disease modeling
; neurodegenerative diseases
1. Introduction
1.1. Basic concepts
Neurodegenerative diseases are characterized by the progressive loss of neuron and glial populations. They are classified according to clinical features, such as anatomic distribution of tissue degeneration and/or primary molecular abnormality. Among neurodegenerative diseases, Parkinson’s disease (PD) and Alzheimer’s disease (AD); multiple sclerosis, amyotrophic lateral sclerosis; and Huntington’s disease represent a significant social and economic burden due to their high incidence, symptoms severity, and usually no effective therapies. Moreover, their incidence in developed countries is rising, at least partially because of the proportional increase in the elderly population. It has been published that the number of individuals with PD may double in 2030, compared with 2005 (1), and another study estimated that the number of amyotrophic lateral sclerosis cases worldwide would increase by 69% from 2015 to 2040 (2). The pathogenic mechanisms possibly involved in this increase are not fully understood, but aging, genetic propensity, increased protein misfolding, and apoptosis of neural cells have all been implicated (3).
Due to the neurodegenerative diseases’ high complexity, no in vivo or in vitro models reproduce their phenotype. In the case of animal models, more suitable for experimental studies, like mice or rats, there are significant physiological differences, making it challenging to represent the diseases’ etiology and symptoms. Moreover, conventional approaches to studying neuronal diseases based on the patient’s brain tissue collection can only be obtained post-mortem. Although no model for neuronal disease study is perfect, induced pluripotent stem cell (iPSC)-based technology complements and improves previous models. Thus, iPSC and gene editing technologies make it possible to use somatic cells from the patient, such as skin or blood, to produce a disease model containing the patient’s genetic context.
Our brain comprises neurons and glial cells, which are scaffold cells divided into three types: oligodendrocytes, microglia, and astrocytes. Each of these cells is important for optimizing the brain function. Oligodendrocytes tightly surround neurons' axons to form the myelin sheath. Microglia cells act on developing and maintaining neuronal networks and play a role in injury repair. They also have immune properties as they can detect damage-associated molecular pattern (PAMPs) components from damaged neurons and produce inflammatory mediators. Astrocytes are star-shaped glial cells that anatomically provide stromal support and hold neurons. They can also release gliotransmitters like glutamate to send signals to neighboring neurons. In addition, through end-feet connections, astrocytes expand or narrow blood vessels, controlling the flow of nutrients and oxygen to the brain (4).
Embryonic stem cells (ESC) have pluripotent plasticity, so they can differentiate into brain cells or any specialized cell in the human body, yielding the generation of drug test platforms. However, using these cells involves ethical issues (5). Thus, some milestones revolutionized the field, allowing the discovery of cellular reprogramming to generate induced pluripotent cells. Gurdon et al., followed by Wilmut et al., showed that the oocytes’ cytoplasm contains factors capable of reprogramming somatic cells to a pluripotent stage, generating individuals with normal postnatal development. Another paradigm break was demonstrated when Gehring et al. and Davis et al. presented the concept of master regulatory genes whose expression modulation can completely convert one cell type into another (6,7,8,9,10,11,12). Adding to this, ES's technical isolation and culture conditions were defined, and this set of remarkable breakthroughs allowed the iPSC to emerge. IPSC are generated by the induced expression of the reprogramming transcription factors OCT3/4, SOX2, KLF4, and c-MYC, constituting the first reprogramming cocktail (13). The first in vitro iPSC reprogramming method was developed by Takahashi and Yamanaka, and it has been improved by methods with higher efficiency and yield (14,15,16). iPSC can differentiate into any cell type in the human body.
Currently, coupled with improvements in reprogramming techniques, this technology has brought much knowledge about multiple disease pathologies and helped the development of more accurate therapeutic methods and advances in regenerative medicine (17). The advent of iPSC makes it possible to generate any cells of interest from the patient’s somatic cells and thus develop patient-specific drug testing models. Reprogramming somatic human cells from patients with neurodegenerative diseases and healthy people (as controls) into human iPSC and then differentiating these cells into various brain cell types has provided an unprecedented opportunity to study disease mechanisms. Before this advance, transgenic animal models made clinical correlation difficult because of the primary difference between species. A more detailed study of diseases that affect the human brain is hampered because of the invasive and challenging procedures to obtain living material suitable for cell culture. Moreover, the ability to generate neural cultures from post-mortem human brains depends significantly on the brain tissue quality (18).
Regarding applicability, iPSCs are particularly important because they can be generated from somatic cells obtained by minimally invasive procedures and from any patient. Moreover, they potentially reproduce the cellular mechanisms of diseases in vitro, can be differentiated into transplantable cells and tissues compatible with the donor, and can be used to produce cellular platforms suitable for drug tests and screening. Another advantage is that iPSC carry all naturally occurring individuals’ genetic mutations and characteristics, which may affect the pathogenic outcome. In this case, unique pathogenic genetic mutations can be corrected using CRISPR/Cas9, for example, to produce healthy isogenic cells or organoids (19,20).
Constructing multiple levels of neural circuits composed of various cell types is essential, best reproducing functional, molecular, and cellular responses. For example, these complex structures modeling the individual’s genetic characteristics can be produced using 3D bioprinters. In this case, iPSC-derived terminally differentiated cell types could be anatomically organized in printed organoids (21). Organoids are miniaturized three-dimensional structures containing multiple cell types and more accurately represent analogs of human organs or tissues. Monolayer cell subtype analysis provides an excellent tool for studying lineage-specific disease mechanisms. However, in the human body, it does not work this way, and diseases that are influenced by the environment and genetics are best studied in multi-lineage or multi-system. Multicell analyses or integrated platforms offer many advantages over monolayers. They can be of the following types: microfluidic organs and body chips, organoids and assembloids, 2D co-culture, tissue engineering and bioprinting, chimeras, and humanized animals (22). Over the last decade, 3D organoid technology has become more prevalent in stem cell research, and human brain organoids derived from iPSC are increasingly being used in neurological disease modeling and therapeutic discovery and tests (23). These 3D brain organoids display critical features of brain-specific cytoarchitecture and network properties that can be used to study complex neural network phenomena in these neurodegenerative disease models. These organoids have been generated to model various brain parts, including the forebrain, midbrain, cerebellum, cortex, and hippocampus (24,25). Depending on the disease, different modeling methods, strategies, and cell types can be used, allowing selected compounds for drug development and testing. Furthermore, iPSC offer a nearly unlimited source of human cells available in public repositories and shared between laboratories and Health Units (26).
1.2. Neurodegenerative disease modeling by iPSC technology
For modeling neurological disorders from healthy individuals, somatic cells (blood, skin, etc.) are reprogrammed in vitro in colonies of iPSC. At this stage, genome-editing techniques can create additional isogenic cell lines containing specific pathological mutations or transgenes that reproduce a given disease etiology. Conversely, pathogenic genetic alterations can be corrected to generate control iPSC lines when the cells are obtained from patients with neurodegenerative disorders. Then, the iPSC lines of interest are induced to differentiate into neural cells, including neurons, glial cells, or neural progenitor cells. At the iPSC stage, self-organizing tissue cytosystems, or organoids, can also be created in a three-dimensional culture using or not 3D printers (Figure 1). In the case of a specific neurological disease, in AD, for example, neural cells differentiated from iPSC with familial AD background can show several AD-like phenotypes that can be tested in vitro. These phenotypes include, for example, amyloid β peptide production and, for three-dimensional culture, tau pathology, amyloid plaques, and synaptic dysfunction. Then, potentially therapeutic small molecules or alternative treatments can be tested directly in human neural cells.
Several attempts were made to direct pluripotent ESC toward a culture of neural cells carrying pathogenic mutations. It was then observed that the stem cells cultured under standard conditions, containing TGF-β-related negative inducers and without specific factors to maintain pluripotency, had no induced neural differentiation (27). Then, specific TGF-β antagonists were used to prevent the Suppressor of Mothers Against Decapentaplegic (SMAD) transcription factors family signaling, and there was a robust and significant improvement in the differentiation method. However, these methods did not recreate human neurogenesis and had important limitations in disease modeling. Then, improvements were made, and iPSC differentiation into the cerebral cortex was recapitulated in vivo, leading to the generation of all cortical projections of neurons in a pre-determined temporal order. This procedure enabled functional studies about the development of the human cerebral cortex and the generation of ex vivo individual-specific cortical networks for disease modeling (28). There have also been improvements in the methodology for generating electrophysiological active neurons without the need for co-culture with astrocytes or specialized media (29,30).
Generating isogenic pair cell lines is usually important in iPSC-based disease modeling. These paired control cell lines consist of introducing a given mutation of interest into normal iPSCs or correcting a disease-associated genetic modification to generate a cognate normal lineage. This is one of the best approaches for assessing the biological effects of one or several disease-associated mutations. CRISPR-Cas9-mediated gene editing is an excellent tool for modeling some monogenic diseases or studying the contribution of single or several gene variants associated with a given pathology (30) (Figure 2).
Obtaining subtypes of neurons, such as dopaminergic and motor neurons, is necessary to model neurodegenerative diseases. Motor neurons are important in modeling diseases such as amyotrophic lateral sclerosis (ALS), and several protocols have been established to generate these neurons from iPSC (31). Although ALS is a very complex disease, motor neurons originating from iPSCs may help resolve how genes selectively impact motor neuron biology and whether they rely on common pathways to cause neuronal degeneration (32). Dopaminergic neurons have already been generated from iPSCs and helped the study of PD (33). A preclinical study using a primate model of PD indicated that human iPSC-derived dopaminergic progenitors were clinically applicable for treating patients (34). Other neuron subtypes have also been developed and could be used in disease modeling and cell therapy (35).
iPSC can also be used to generate astrocytes via an intermediate neural progenitor (36). This type of cell is very important in neurological and psychiatric diseases. Jones et al. reported developing a human iPSC-derived astrocyte model created from healthy subjects and patients with early-onset familial AD or the late-onset sporadic form (37). Those astrocytes could reproduce several phenotypes found in vivo, features that could be employed for effective disease modeling (38). In this context, astrocytes derived from AD patients showed a typical pathological phenotype, with a less complex morphological appearance and abnormal localization of key functional astroglial markers (39).
Oligodendrocytes and their precursors are responsible not only for the generation of myelin in the central nervous system but also for the metabolic support of neurons and have a critical trophic function (40). Several groups developed protocols to obtain these cells from iPSCs, which were initially based on studies using ESCs (41,42). Then, an optimized and specific protocol was developed for obtaining oligodendrocytes from iPSC. This protocol consisted of seven steps with an average duration of 150 days; thus, myelinogenic oligodendrocytes could be obtained (43). In this study, oligodendrocyte precursor cells derived from iPSC were grafted to neonatal myelin-deficient shiverer mice and induced robust brain myelination and substantially increased survival (43). The authors improved the protocol for obtaining these cells and reduced the time required for cell differentiation (44). Following the same strategy, other authors showed the applicability of stem cells-derived oligodendrocytes, which led to remyelination and rescue of irradiated rats, modeling brain tumor radiation therapy with excellent therapeutic relevance (45).
Microglia cells reside in the central nervous system and play an essential role in the development and homeostasis of various neurological and psychiatric diseases. Human iPSCs were differentiated into microglia-like cells by exposure to multiple factors, such as IL-34, TGFβ-1, and CX3CL1, and co-cultured with astrocytes (46,47). It was also shown that human microglia-like cells derived from iPSCs migrated and secreted cytokines in response to inflammatory stimuli. Moreover, they robustly phagocytose central nervous system substrates, including amyloid-β (Aβ) fibrils, brain-derived tau oligomers, and human synaptosomes, similar to conventional microglia cells (48). These cells were also used to study the effects of Aβ fibrils and brain-derived tau oligomers over AD-related gene expression. Moreover, they can be used to study mechanisms involved in synaptic pruning (47). The microglial-like cells derived from iPSC were similar to conventional microglia at the transcriptome level and also responded to inflammatory stimuli (47). Figure 3 shows the most relevant neural cell types for modeling the main neurodegenerative diseases.
3D brain organoids derived from iPSCs can at least partially recapitulate the rearrangement of brain cytoarchitecture, an essential feature for studying the pathogenesis of brain diseases. Lancaster et al. pioneered the development of brain organoids from human pluripotent stem cells. The authors modeled microcephaly, a complex disease to be reproduced in mice (49). After this study, other groups used the same approach to generate brain organoids, which helped model different neurological disorders. These studies using patient-derived brain organoids revealed novel insights into molecular and genetic mechanisms in microcephaly, autism, and AD (50). A simplified and fast protocol was described for brain organoid induction from human iPSCs (51). Recently, the generation of arcuate organoids from human iPSC was shown to model the development of the human hypothalamic arcuate nucleus. Since the existing organoid models do not resolve fine brain subregions, such as different nuclei in the hypothalamus (52), the use of organ-specific progenitor cells highlights the potential of iPSCs in other fields besides regenerative medicine.
1.3. Drug testing in neurodegenerative diseases using iPSC
One of the prerequisites for drug screening using iPSC is targeting a relevant cellular phenotype in a given disease. In the first reports of drug screening using iPSC, neural crest precursors derived from iPSC were generated from individuals with familial dysautonomia. This disease is a rare and fatal genetic disorder affecting neural crest lineages. It is caused by mutations in the gene coding for the IkB kinase complex-associated protein (IKBKAP), resulting in a splicing defect and a dysfunctional truncated protein. In that work, 6,912 small compounds were tested, and one of them, known as SKF-86466, was found to improve disease-specific aberrant splicing (53). In another work on drug screening, iPSC derived from patients with sporadic amyotrophic lateral sclerosis and healthy individuals were used and differentiated into motor neurons. The authors de novo identified aggregation of TAR DNA-binding protein 43 (TDP-43) in patients' motor neurons. Using a high-content drug screen, they found a compound that reduced TDP-43 aggregation (54). Other authors used a patient-derived model of iPSC with spinal muscular atrophy (SMA) to validate specific drugs, and the hit compound was further evaluated in a mouse model. Administration of this compound to mice led to increased survival of motor neuron (SMN) protein levels, motor function improvement, and neuromuscular circuit protection (55). This inherited motor neuron disease is caused by SMN deficient expression and results in severe muscle weakness
Another application is drug repositioning using disease-specific iPSC. In this case, drugs already approved for specific diseases are tested to find new applications for other conditions. One of the examples of this type of approach showed that iPSC-derived motor neurons produced from amyotrophic lateral sclerosis patients, harboring SOD1 (superoxide dismutase 1) mutations, displayed a reproducible, disease-related phenotype and a reduced delayed-rectifier potassium channel activity (56,57). iPSC-based new evidence showed that correcting motor neuron physiology using the already approved antiepileptic ezogabine drug, a Kv7 potassium channel agonist, reduced neuronal excitability and improved cell survival (56,57). Drug discovery using iPSC from patients with multiple genetic forms of a neurodegenerative disease is of great value, as it allows testing the responsiveness of drugs in numerous patients.
1.4. Advances based on iPSCs for specific conditions
1.4.1. Parkinson disease
PD is the second most common degenerative disease, affecting 2 to 3% of the population over 65 years. Age is the most important risk factor for developing PD, and men are more susceptible than women, with a prevalence ratio of approximately 3:2. The pathological hallmark of PD is neuronal loss in the substantia nigra, which causes dopamine deficiency, and intracellular inclusions containing α-synuclein aggregates (58). This protein is encoded by the SNCA gene, whose duplications or triplications are associated with familial PD. Resting tremors, rigidity, akinesia, and postural reflex disturbance are all PD’s tetralogy (59). The history of PD treatment using cell-based therapies began in the late 1970s and early 1980s when several groups showed that dopaminergic neurons harvested from the developing fetal midbrain could survive in grafts transplanted to PD animal models (60). It was also revealed that the grafted cells could restore brain functionality in a PD rat model (60). Thus, several studies were done using neural cells from different origins, and later studies of dopaminergic neuron transplantation from iPSCs emerged as a therapeutic possibility for PD patients. In 2016, the International Stem Cell Corporation started the first approved clinical trial using iPSC to treat PD patients (61,62).
IPSC were used to generate dopaminergic neurons, and they were obtained from a patient with SNCA triplication and from an unaffected first-degree relative as a control. The patient’s neurons produced twice the amount of α-synuclein protein compared to the unaffected relative, recapitulating in vitro the PD pathology (63). Several groups have then generated dopaminergic neurons from PD patients and healthy controls using iPSC and compared molecular pathways that might differ. Among these pathways, some were susceptible to therapeutic modulation (38). For example, iPSC-derived dopamine neurons revealed differences between monozygotic twins discordant for PD. The affected twin’s neurons showed a lower dopamine level, increased monoamine oxidase B expression, and impaired intrinsic network activity. The targeted monoamine oxidase B inhibitors treatment normalized α-synuclein and dopamine levels, confirming this system’s suitability for drug testing (64). Recently, another case was reported in which autologous dopaminergic neurons were generated from iPSC obtained from a patient with idiopathic PD. These neurons were implanted back into the patient’s putamen (left hemisphere), followed by a right hemisphere implantation after six months. Exams of positron-emission tomography suggested graft survival and clinical control of PD symptoms after surgery, which improved at eighteen to twenty-four months after implantation (65). This form of cell therapy showed promising results and is currently one of the best options for slowing or halting PD progression (66) With the advances in cell reprogramming, iPSC have great potential for treating region-specific neurodegenerations such as PD. In addition, as the cells are patient-specific, the chances of an immune response after transplantation are significantly reduced.
1.4.2. Alzheimer disease
Clinically, AD is the most prevalent cause of dementia, characterized by memory loss and alterations in personality and rational thinking. AD significantly impacts society as it affects millions worldwide, accounting for 60–80% of all patients with dementia. This disease is part of the proteinopathies group and is characterized by amyloid-β (Aβ) peptide deposition as amyloid plaques and the protein tau as neurofibrillary tangles (67). AD is a very complex disease and is still not fully understood. In recent years, a third pathogenic component was involved in disease onset and progression: the neuroinflammatory response primarily mediated by microglia (68).
Initially, a model of familial AD was established using iPSC generated from autologous fibroblasts. It is known that mutations in the PS1 (Presenilin 1), PS2 (Presenilin 2), and the APP (amyloid precursor protein) genes account for most of the familial early onset cases of AD. The enhanced production of pathological Aβ leads to a greater tendency to form fibrillary amyloid deposits (69). In the experimental model established by the authors, patient-derived differentiated neurons increase Aβ42 secretion, recapitulating the pathological mechanism of familial AD associated with PS1 and PS2 mutations. This model was then exploited to test drugs that could repair the genetic mutation. In another work, the authors described the generation of iPSC lines from patients harboring familial AD based on the APP gene mutation (V717I). A significant increase in APP expression and Aβ level was observed during neural differentiation and maturation. Moreover, an increase in the total levels of phosphorylated tau was observed in those genetically manipulated neurons. These studies using human neurons revealed unpredicted effects of the most common familial AD APP gene mutation (70). Other authors used this same model to evaluate therapeutic candidates for AD and tested over 1,000 compounds for their ability to reduce Aβ load within the cells in culture. They obtained 27 promising candidates, and the list was narrowed to six leading compounds. Then, three candidates were combined to improve the anti-Aβ effect (bromocriptine, cromolyn, and topiramate) as an anti-Aβ cocktail. The results suggested that this iPSC approach could also be used for drug development (71). Xu et al. used the same technology based on iPSC-derived neurons to examine a chemical library containing hundreds of compounds. They found numerous small molecules that could be effective blockers against Aβ1-42 toxicity, including a cyclin-dependent kinase 2 inhibitor (72). This study screened the Aβ toxicity using iPSC-derived neurons for the first time, providing an excellent example of how iPSC can be used for disease modeling and high-throughput compound analysis (72).
Israel et al. tested neurons differentiated from iPSC obtained from patients with familial and sporadic AD, and the results suggested a direct correlation between Aβ precursor protein proteolytic processing, but not Aβ, in glycogen synthase kinase-3β activation and tau phosphorylation in human neurons-derived iPSC in culture. This approach allowed for the link between Aβ and tau and additional pathological highlights in AD (73). Tau pathology is present in AD and other diseases whose clinical phenotype includes dementia. Iovino et al. showed that neurons derived from iPSC carrying a mutation in the MAPT gene showed abnormal tau expression, hyperphosphorylation of tau aggregates, and multiple disease phenotypes (74), and this system may be used for drug screening purposes.
Pomeshchik et al., 2020 developed a protocol to generate rapid hippocampal spheroids from human iPSC, which was used to model AD. The hippocampus is involved in forming new memories, emotions, and learning and is one of the first regions of the brain that atrophies in AD. In that work, the hippocampal spheroids generated from two AD patients carrying variations in the APP or PS1 genes exhibited cardinal cellular pathological features of AD, including loss of synaptic proteins and increased ratio of intracellular and extracellular Aβ42/Aβ40 peptides. The authors then developed a gene therapy approach to modulate gene expression in synaptic transmission. They showed that hippocampal spheroids from iPSC could be used to study the mechanisms underlying early pathogenic changes in the hippocampi of AD patients (75).
In animal models of AD, atrophic astrocytes were detected at the earliest stages of the disease, with the later appearance of hypertrophic reactive astrocytes in response to their proximity to extracellular accumulations of Aβ (76). The authors also analyzed the pathological potential of iPSC-derived astrocytes in AD. In this case, patient-derived induced astrocytes displayed a pathological phenotype, besides significantly less complex morphological appearance and abnormal localization of key functional astroglial markers (39). The authors reported the development of a human iPSC-derived astrocyte model created from healthy individuals and patients with AD. Astrocytes-derived iPSCs from AD patients showed a pronounced pathological phenotype, a general atrophic profile, and abnormal localization of key functional astroglial markers (39). This work provided a platform for further interrogation of early astroglial cell autonomic events in AD and the possibility of identifying new therapeutic targets for treating the disease. In another work, astrocytes differentiated from AD patients’ iPSC showed hallmarks of disease pathology, including increased Aβ production, altered cytokine release, and dysregulated Ca2+ homeostasis (77).
Microglia cells derived from iPSC have also been used to study their role in neurological diseases. Abud et al. showed that human microglial-like cells could be differentiated from iPSC. They secreted cytokines in response to inflammatory stimuli, migrated and underwent calcium transients, and avidly phagocytosed central nervous system substrates (47). Human microglia-like cells derived from familial AD patients were also analyzed, and the APOE4 genotype profoundly impacted several aspects of microglial function. This altered genotype impaired phagocytosis, migration, and metabolic activity but exacerbated their cytokine secretion (78).
3D-differentiated neuronal cells expressing familial AD mutations could also recapitulate the Aβ- and tau-dependent pathology. This model should also facilitate the development of more precise human neural cell models of other neurodegenerative disorders (79). Raja et al., using brain organoids derived from AD patients` iPSC recapitulated AD-like characteristics, such as amyloid aggregation, hyperphosphorylated tau protein, and endosome abnormalities. Thus, they found that treating patient-derived organoids with β- and γ-secretase inhibitors significantly reduced the cellular phenotype associated with AD (80). This 3D organoid system could also provide a platform to help develop new drug candidates for disease treatment.
1.4.3. Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the progressive degeneration of brain and spinal cord motor neurons. Its name reflects the degeneration of corticospinal motor neurons, as the descending axons in the lateral spinal cord seem scarred (lateral sclerosis), and there are diminished spinal motor neurons and muscle wasting (amyotrophy). Like most neurodegenerative diseases, it starts focally. Then it spreads, with symptoms beginning as subtle cramping or weakness in the limbs or bulbar muscles, progressing to the paralysis of almost all skeletal muscles. Death occurs typically three to five years after diagnosis (81).
Mutations in the TDP-43, C9ORF72, or SOD1 genes are most commonly related to familial ALS. Several groups have used iPSCs-derived motor neurons to test drugs for the familial forms of ALS, such as mutations in the Tar DNA binding protein-43 (TDP-43). It was published that iPSCs generated from an ALS patient differentiated into motor neurons carrying mutations in the TDP-43. In these samples, cytosolic aggregates formed similarly to those seen in post-mortem tissue from ALS patients. Four chemical compounds were then tested, and a histone acetyltransferase inhibitor, named anacardic acid, was found to have rescued the abnormal ALS motor neuron phenotype. Then, the authors suggested that anacardic acid may reverse ALS-associated phenotypes by downregulating TDP-43 mRNA expression. (82). In another work, fibroblast-derived iPSC generated from healthy donors or patients with sporadic ALS were induced to differentiate into neurons. Only the neurons obtained from the patients showed the disease phenotype. It was shown that motor neurons derived from three ALS patients’ iPSC had de novo TDP-43 aggregation and that the aggregates were similar to the ones observed in post-mortem tissue. Using this model, the authors identified that some FDA-approved small molecules, including Digoxin, could modulate TDP-43 aggregates (54). In another work, motor neurons carrying a mutation in the C9ORF72 gene, one of the genes responsible for the disease, were generated. Expansions of a hexanucleotide repeat (GGGGCC) in the noncoding region of the C9ORF72 gene are the most common cause of the familial form of ALS. In this case, the neurons showed altered expression of genes involved in membrane excitability, including DPP6, demonstrating a diminished capacity to fire continuous spikes upon depolarization compared to control motor neurons. Then, antisense oligonucleotides targeting the C9ORF72 transcript suppressed RNA foci formation and reversed gene expression alterations in motor neurons (83). RNA foci result from expanding RNA repeats, which are retained in the nucleus, assume unusual secondary folding, sequester some RNA-binding proteins, and can become toxic to the cell. SOD1 mutations induced a transcriptional signature with indications of increased oxidative stress, reduced mitochondrial function, altered subcellular transport, and activation of the endoplasmic reticulum stress (32). A system based on an optimized all-optical electrophysiology for high-throughput functional characterization was used for testing drugs on the disease phenotype with a mutation in the SOD1 (84).
Different neural cell types, such as astrocytes, oligodendrocytes, and motor neurons, contribute to ALS pathology, so they should also be considered in developing reliable drug testing platforms. Some studies indicated that astrocytes may mediate motor neuron death in this context. It has also been shown that mutations in genes that encode essential autophagy factors impair autophagy and may lead to neurodegenerative conditions such as ALS (85). However, Madill et al. demonstrated that iPSC generated from an ALS patient differentiated into astrocytes that modulated the autophagy pathway in a non-cell autonomous manner. Data from this work suggested that the patient’s astrocytes may modulate motor neuron cell death by impairing autophagic mechanisms (86). Ferraiuolo et al. also showed the death of motor neurons induced by oligodendrocytes. In this case, the oligodendrocytes were mutated for SOD1 and caused the death of control motor neurons and hyperexcitability when co-cultured (87). Therefore, the study of ALS with cells derived from iPSC presented new strategies in the search for effective drugs for the disease.
1.4.4. Multiple sclerosis
Multiple sclerosis (MS) is a chronic central nervous system inflammatory disease of autoimmune etiology characterized by neuronal damage and axonal loss due to demyelination and subsequent degeneration. Activated T lymphocytes mediate this disease with the contribution of B lymphocytes and cells of the innate immune system (88). Clinical symptoms of MS are variable and typically result from the involvement of sensory, motor, visual, and brainstem pathways and include fatigue, spasticity, and gait instability (88). Researchers then developed a myelinating platform for drug screening using human pluripotent stem cells. For this, it was observed that the transcription factor SOX10 overexpression led to the generation of surface antigen O4-positive (O4+) and myelin basic protein-positive oligodendrocytes from pluripotent stem cells. Using this platform, the myelination of neurons by oligodendrocytes was demonstrated, and it could be applied for high-throughput screening to test the response of pro-myelinating drugs (89). Researchers also report that to discover compounds that increase the myelination of oligodendrocyte progenitor cells, they screened a library of small bioactive molecules on mouse pluripotent epiblast stem-cell-derived oligodendrocyte progenitor cells. With this test they found two drugs, miconazole and clobetasol, that were effective in promoting precocious myelination in organotypic cerebellar slice cultures, and in vivo in early postnatal mouse pups. Furthermore, both drugs enhanced the generation of human oligodendrocytes from human oligodendrocyte progenitor cells in vitro (90). These studies indicate that developing myelinating platforms for drug screening can bring discoveries that can be translated into clinical practice.
iPSC brain organoids have also been made for the study of MS. The authors reported cerebral organoids from iPSCs of healthy control subjects as well as from primary progressive MS, secondary progressive MS and relapsing-remitting MS patients to understand the pathological basis of the varied clinical phenotypic of MS. In fact, it was observed most notably in primary progressive MS, a decrease of proliferation marker Ki67 and a reduction of the SOX2+ stem cell pool associated with an increased expression of neuronal markers CTIP2 and TBR1 as well as a strong decrease of oligodendrocyte differentiation. The brain organoids developed in this study from iPSCs from MS patients provide important information about the effect of the patient's genetic background on neural cells and interactions. This approach may bring novel insights into the development of neural interactions that occur in MS patients (91).
1.4.5. Huntington’s disease
Huntington’s disease (HD) is an inherited neurodegenerative disorder characterized by neuropsychiatric symptoms, movement disability, and progressive cognitive impairment. Unfortunately, there is no effective therapy available for HD. The diagnosis is usually made by identifying an increased CAG repeat length in the huntingtin gene associated with the clinical condition. This genetic alteration results in the loss of GABAergic neurons in the striatum. At the cellular level, the mutant gene results in neuronal dysfunction and death through several mechanisms, including disruption of proteostasis, transcriptional and mitochondrial dysfunction, and direct toxicity of the mutant protein (92).
The authors reported the generation of iPSC from HD patients and healthy individuals. The microarray profile distinguished the lines of cells from healthy controls and patients as the gene expression profile showed CAG repeat expansion. The iPSC lines differentiated into neural cells showed disease-associated differences in electrophysiology, metabolism, cell death, and longer CAG repeat expansions. This is because the severity of these disease-associated phenotypes is directly influenced by the extension of CAG repeats. The strategy presented in that work provided a human stem cell platform for screening new therapeutic candidates for HD (93). Using iPSCs from HD patients, Xu et al. reported a genetic correction using CRISPR-Cas9 that reversed the phenotypic abnormality (94). The interaction between genome editing and iPSCs can expand HD cellular models and therapeutic target discovery. Different disease phenotypes, such as aggregation of the mutated huntingtin protein, cell death, and neuronal toxicity, can be effective for drug testing (95).
In a disease modeling work from iPSC, GABAergic medium spiny neurons were generated that outlined the HD pathology in vitro as evidenced by mutant huntingtin protein aggregation, increased number of lysosomes/autophagosomes, nuclear indentations, and enhanced neuronal death during cell aging. Furthermore, the EVP4593 drug, a quinazoline derivative, reduced the number of lysosomes/autophagosomes and was neuroprotective during cell aging. This approach provides a valuable tool for identifying candidate anti-HD drugs. (96). The severity of these disease-associated phenotypes was directly influenced by the extension of CAG repeats (93). In this line, researchers also reported a model to investigate changes in blood-brain barrier phenotype with expansion of CAG repeats using an isogenic pair of iPSC. These cells were differentiated into brain microvascular endothelial-like cells, which, due to CAG expansion, showed subtle changes in phenotype, including differences in cell turnover and immune cell adhesion. The results then pointed out that the expansion of CAGs contributes to changes in the blood-brain barrier in HD (97). Therefore, iPSC-derived cells provide a reliable model that allows drug testing and targeting of drug action to counteract HD pathology.
2. Conclusions
The development of iPSC technology represents a breakthrough in the medical field. This discovery of neurodegenerative diseases has already resulted in huge advances in modeling this complex group of pathologies, enlightening the cellular and molecular mechanisms related to them as described here. The three major degenerative diseases PD, ALS, and AD are characterized by abnormal specific proteins inside and outside neurons: TDP-43 in ALS, α-synuclein in PD, and tau and β-amyloid in AD. In addition to the abnormal aggregation of proteins, as shown in Figure 4, other common endophenotypes, such as reduced mitochondrial activity, accumulation of reactive oxygen species (ROS), and enhanced inflammation, are found in these diseases and could be investigated using iPSC. The production of brain organoids formed by specialized neural cells derived from patient-specific iPSC has accelerated the development of drug discovery platforms to treat neurodegenerative diseases allowing the screening of new drugs, as well as the resignificance of already commercialized drugs intended to treat other target diseases, and also the development of neurodegenerative cell models. Even the familial and sporadic phenotypes related to neurodegenerative diseases were demonstrated using iPSC derived from patients and their corresponding genetically edited isogenic cell lineage (19).
Despite these significant advances, some open pathways are still under investigation to develop a universal and robust neuronal platform derived from iPSC for drug screening and studying these diseases' pathogenesis in vitro with human cells. Continuous developments are being made to challenge technical obstacles, such as improving culture differentiation protocols, aiming at increasing differentiation efficiency and cell maturation, combined with optimizations of the brain organoids technology (98,99). Multi-omics single-cell analysis may represent a new tool kit in a search for high-purity specific cell lineages that could recapitulate the intended phenotype and underlying mechanism of these diseases in a reproducible, robust, and consistent way (20,100,101). With the focus on the mimics of the patient disease, researchers are already using genetic induction of cellular aging, genetic edition, and small molecules to reproduce the patient phenotype, including late-onset disease manifestation, familial or sporadic form, and even the environmental factors that could be important in this scenario (102). Combining all these initiatives with bioinformatics and computational and statistical analysis, a pattern of clinical and biological features is under construction, highlighting the promising role of iPSC in drug discovery and neurodegenerative disease modeling.
Author Contributions
Conceptualization, writing, original draft preparation, D.G.B.; writing-review, T.H.K.-B.; writing—review and editing, A.H.-P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Instituto Oswaldo Cruz, Fundação Oswaldo Cruz.
Acknowledgments
The authors thank Samuel Iwao Horita for his excellent artwork. Figures Created with BioRender.com
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Kim, J.; Park, H.; Choi, H. Modelling neurodegenerative diseases with 3D brain organoids. Biol Rev Camb Philos Soc 2020, 95, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Hall, W.C.; LaMantia, A.-S.; White, L.E. Neuroscience, 5a edition ed.; 2012.
- Kamm, F.M. Ethical issues in using and not using embryonic stem cells. Stem Cell Rev 2005, 1, 325–330. [Google Scholar] [CrossRef] [PubMed]
- GURDON, J.B. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J Embryol Exp Morphol 1962, 10, 622–640. [Google Scholar] [CrossRef] [PubMed]
- Wilmut, I.; Schnieke, A.E.; McWhir, J.; Kind, A.J.; Campbell, K.H. Viable offspring derived from fetal and adult mammalian cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Gehring, W.J. Homeo boxes in the study of development. Science 1987, 236, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Smith, A.G.; Heath, J.K.; Donaldson, D.D.; Wong, G.G.; Moreau, J.; Stahl, M.; Rogers, D. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 1988, 336, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef]
- Aboul-Soud, M.A.M.; Alzahrani, A.J.; Mahmoud, A. Induced Pluripotent Stem Cells (iPSCs)-Roles in Regenerative Therapies, Disease Modelling and Drug Screening. Cells 2021, 10. [Google Scholar] [CrossRef]
- Verwer, R.W.; Dubelaar, E.J.; Hermens, W.T.; Swaab, D.F. Tissue cultures from adult human postmortem subcortical brain areas. J Cell Mol Med 2002, 6, 429–432. [Google Scholar] [CrossRef]
- Meijboom, K.E.; Abdallah, A.; Fordham, N.P.; Nagase, H.; Rodriguez, T.; Kraus, C.; Gendron, T.F.; Krishnan, G.; Esanov, R.; Andrade, N.S.; et al. CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro. Nat Commun 2022, 13, 6286. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fleck, J.S.; Martins-Costa, C.; Burkard, T.R.; Themann, J.; Stuempflen, M.; Peer, A.M.; Vertesy, Á.; Littleboy, J.B.; Esk, C.; et al. Single-cell brain organoid screening identifies developmental defects in autism. Nature 2023, 621, 373–380. [Google Scholar] [CrossRef]
- Nakazawa, T. Modeling schizophrenia with iPS cell technology and disease mouse models. Neurosci Res 2022, 175, 46–52. [Google Scholar] [CrossRef]
- Sharma, A.; Sances, S.; Workman, M.J.; Svendsen, C.N. Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell 2020, 26, 309–329. [Google Scholar] [CrossRef] [PubMed]
- Wang, H. Modeling Neurological Diseases With Human Brain Organoids. Front Synaptic Neurosci 2018, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, K.; Nishiyama, A.; Kawakami, H.; Hashimoto, K.; Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep 2015, 10, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, C.B.; Yang, A.; Lara, E.; McDonough, J.A.; Blauwendraat, C.; Peng, L.; Oguro, H.; Kanaujiya, J.; Zou, J.; Sebesta, D.; et al. A reference human induced pluripotent stem cell line for large-scale collaborative studies. Cell Stem Cell 2022, 29, 1685–1702. [Google Scholar] [CrossRef] [PubMed]
- Tropepe, V.; Hitoshi, S.; Sirard, C.; Mak, T.W.; Rossant, J.; van der Kooy, D. Direct neural fate specification from embryonic stem cells: a primitive mammalian neural stem cell stage acquired through a default mechanism. Neuron 2001, 30, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kirwan, P.; Smith, J.; Robinson, H.P.; Livesey, F.J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci 2012, 15, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Gunhanlar, N.; Shpak, G.; van der Kroeg, M.; Gouty-Colomer, L.A.; Munshi, S.T.; Lendemeijer, B.; Ghazvini, M.; Dupont, C.; Hoogendijk, W.J.G.; Gribnau, J.; et al. A simplified protocol for differentiation of electrophysiologically mature neuronal networks from human induced pluripotent stem cells. Mol Psychiatry 2018, 23, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Bardy, C.; van den Hurk, M.; Eames, T.; Marchand, C.; Hernandez, R.V.; Kellogg, M.; Gorris, M.; Galet, B.; Palomares, V.; Brown, J.; et al. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc Natl Acad Sci U S A 2015, 112, E2725–E2734. [Google Scholar] [CrossRef]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schüle, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson's disease model. Nature 2017, 548, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhang, S.C. Neural Subtype Specification from Human Pluripotent Stem Cells. Cell Stem Cell 2016, 19, 573–586. [Google Scholar] [CrossRef]
- Tcw, J.; Wang, M.; Pimenova, A.A.; Bowles, K.R.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.M.; Phatnani, H.; et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Reports 2017, 9, 600–614. [Google Scholar] [CrossRef]
- Jones, V.; Atkinson-Dell, R.; Verkhratsks, A. Aberrant iPSC-derived human astrocytes in Alzheimer's disease. Nature: cell death & disease, 2017; p e2696.
- Garcia-Leon, J.A.; Vitorica, J.; Gutierrez, A. Use of human pluripotent stem cell-derived cells for neurodegenerative disease modeling and drug screening platform. Future Med Chem 2019, 11, 1305–1322. [Google Scholar] [CrossRef] [PubMed]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant iPSC-derived human astrocytes in Alzheimer's disease. Cell Death Dis 2017, 8, e2696. [Google Scholar] [CrossRef]
- García-León, J.A.; Verfaillie, C.M. Stem Cell-Derived Oligodendroglial Cells for Therapy in Neurological Diseases. Curr Stem Cell Res Ther 2016, 11, 569–577. [Google Scholar] [CrossRef]
- Ogawa, S.; Tokumoto, Y.; Miyake, J.; Nagamune, T. Induction of oligodendrocyte differentiation from adult human fibroblast-derived induced pluripotent stem cells. In Vitro Cell Dev Biol Anim 2011, 47, 464–469. [Google Scholar] [CrossRef]
- Pouya, A.; Satarian, L.; Kiani, S.; Javan, M.; Baharvand, H. Human induced pluripotent stem cells differentiation into oligodendrocyte progenitors and transplantation in a rat model of optic chiasm demyelination. PLoS One 2011, 6, e27925. [Google Scholar] [CrossRef]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Douvaras, P.; Wang, J.; Zimmer, M.; Hanchuk, S.; O'Bara, M.A.; Sadiq, S.; Sim, F.J.; Goldman, J.; Fossati, V. Efficient generation of myelinating oligodendrocytes from primary progressive multiple sclerosis patients by induced pluripotent stem cells. Stem Cell Reports 2014, 3, 250–259. [Google Scholar] [CrossRef]
- Piao, J.; Major, T.; Auyeung, G.; Policarpio, E.; Menon, J.; Droms, L.; Gutin, P.; Uryu, K.; Tchieu, J.; Soulet, D.; et al. Human embryonic stem cell-derived oligodendrocyte progenitors remyelinate the brain and rescue behavioral deficits following radiation. Cell Stem Cell 2015, 16, 198–210. [Google Scholar] [CrossRef]
- Pandya, H.; Shen, M.J.; Ichikawa, D.M.; Sedlock, A.B.; Choi, Y.; Johnson, K.R.; Kim, G.; Brown, M.A.; Elkahloun, A.G.; Maric, D.; et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat Neurosci 2017, 20, 753–759. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer's disease. J Cell Biol 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Bendriem, R.M.; Wu, W.W.; Shen, R.F. 3D brain Organoids derived from pluripotent stem cells: promising experimental models for brain development and neurodegenerative disorders. J Biomed Sci 2017, 24, 59. [Google Scholar] [CrossRef]
- Tomaskovic-Crook, E.; Crook, J.M. Clinically Amendable, Defined, and Rapid Induction of Human Brain Organoids from Induced Pluripotent Stem Cells. Methods Mol Biol 2019, 1576, 13–22. [Google Scholar] [CrossRef]
- Huang, W.K.; Wong, S.Z.H.; Pather, S.R.; Nguyen, P.T.T.; Zhang, F.; Zhang, D.Y.; Zhang, Z.; Lu, L.; Fang, W.; Chen, L.; et al. Generation of hypothalamic arcuate organoids from human induced pluripotent stem cells. Cell Stem Cell 2021, 28, 1657–1670. [Google Scholar] [CrossRef]
- Lee, G.; Ramirez, C.N.; Kim, H.; Zeltner, N.; Liu, B.; Radu, C.; Bhinder, B.; Kim, Y.J.; Choi, I.Y.; Mukherjee-Clavin, B.; et al. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol 2012, 30, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, M.F.; Martinez, F.J.; Wright, S.; Ramos, C.; Volfson, D.; Mason, M.; Garnes, J.; Dang, V.; Lievers, J.; Shoukat-Mumtaz, U.; et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci 2013, 56, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Wainger, B.J.; Kiskinis, E.; Mellin, C.; Wiskow, O.; Han, S.S.; Sandoe, J.; Perez, N.P.; Williams, L.A.; Lee, S.; Boulting, G.; et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep 2014, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McNeish, J.; Gardner, J.P.; Wainger, B.J.; Woolf, C.J.; Eggan, K. From Dish to Bedside: Lessons Learned While Translating Findings from a Stem Cell Model of Disease to a Clinical Trial. Cell Stem Cell 2015, 17, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat Rev Dis Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Sethi, K.D. The impact of levodopa on quality of life in patients with Parkinson disease. Neurologist 2010, 16, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Strecker, R.; Clarke, D.; Widner, H.; Nilsson, O.; Åstedt, B.; Lindvall, O.; Björklund, A. Chapter 57 Can human fetal dopamine neuron grafts provide a therapy for Parkinson's disease? In Progress in Brain Research, Elsevier, Ed. 1988; Vol. 78, pp. 441–448.
- Barker, R.A.; Parmar, M.; Kirkeby, A.; Björklund, A.; Thompson, L.; Brundin, P. Are Stem Cell-Based Therapies for Parkinson's Disease Ready for the Clinic in 2016? J Parkinsons Dis 2016, 6, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Kameda, M.; Sasaki, T.; Tajiri, N.; Date, I. Cell Therapy for Parkinson's Disease. Cell Transplant 2017, 26, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.W.; et al. Parkinson's disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat Commun 2011, 2, 440. [Google Scholar] [CrossRef]
- Woodard, C.M.; Campos, B.A.; Kuo, S.H.; Nirenberg, M.J.; Nestor, M.W.; Zimmer, M.; Mosharov, E.V.; Sulzer, D.; Zhou, H.; Paull, D.; et al. iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson's disease. Cell Rep 2014, 9, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson's Disease. N Engl J Med 2020, 382, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Stoddard-Bennett, T.; Reijo Pera, R. Treatment of Parkinson's Disease through Personalized Medicine and Induced Pluripotent Stem Cells. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Götz, J. Amyloid-β and tau complexity - towards improved biomarkers and targeted therapies. Nat Rev Neurol 2018, 14, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Vitorica, J. Toward a New Concept of Alzheimer's Disease Models: A Perspective from Neuroinflammation. J Alzheimers Dis 2018, 64, S329–S338. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer's disease with induced pluripotent stem cells. Hum Mol Genet 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer's disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum Mol Genet 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Imamura, K.; Funayama, M.; Tsukita, K.; Miyake, M.; Ohta, A.; Woltjen, K.; Nakagawa, M.; Asada, T.; Arai, T.; et al. iPSC-Based Compound Screening and In Vitro Trials Identify a Synergistic Anti-amyloid β Combination for Alzheimer's Disease. Cell Rep 2017, 21, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lei, Y.; Luo, J.; Wang, J.; Zhang, S.; Yang, X.J.; Sun, M.; Nuwaysir, E.; Fan, G.; Zhao, J.; et al. Prevention of β-amyloid induced toxicity in human iPS cell-derived neurons by inhibition of Cyclin-dependent kinases and associated cell cycle events. Stem Cell Res 2013, 10, 213–227. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef]
- Iovino, M.; Agathou, S.; González-Rueda, A.; Del Castillo Velasco-Herrera, M.; Borroni, B.; Alberici, A.; Lynch, T.; O'Dowd, S.; Geti, I.; Gaffney, D.; et al. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 2015, 138, 3345–3359. [Google Scholar] [CrossRef] [PubMed]
- Pomeshchik, Y.; Klementieva, O.; Gil, J.; Martinsson, I.; Hansen, M.G.; de Vries, T.; Sancho-Balsells, A.; Russ, K.; Savchenko, E.; Collin, A.; et al. Human iPSC-Derived Hippocampal Spheroids: An Innovative Tool for Stratifying Alzheimer Disease Patient-Specific Cellular Phenotypes and Developing Therapies. Stem Cell Reports 2020, 15, 256–273. [Google Scholar] [CrossRef] [PubMed]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Gubert Olivé, M.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer's Disease. Stem Cell Reports 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, H.; Cabral-da-Silva, M.E.C.; Ohtonen, S.; Wojciechowski, S.; Shakirzyanova, A.; Caligola, S.; Giugno, R.; Ishchenko, Y.; Hernández, D.; Fazaludeen, M.F.; et al. PSEN1ΔE9, APPswe, and APOE4 Confer Disparate Phenotypes in Human iPSC-Derived Microglia. Stem Cell Reports 2019, 13, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D'Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer's Disease Phenotypes. PLoS One 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: from genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med 2012, 4, 145ra104. [Google Scholar] [CrossRef]
- Sareen, D.; O'Rourke, J.G.; Meera, P.; Muhammad, A.K.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 2013, 5, 208ra149. [Google Scholar] [CrossRef]
- Kiskinis, E.; Kralj, J.M.; Zou, P.; Weinstein, E.N.; Zhang, H.; Tsioras, K.; Wiskow, O.; Ortega, J.A.; Eggan, K.; Cohen, A.E. All-Optical Electrophysiology for High-Throughput Functional Characterization of a Human iPSC-Derived Motor Neuron Model of ALS. Stem Cell Reports 2018, 10, 1991–2004. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.P.; De Calbiac, H.; Kabashi, E.; Barmada, S.J. Autophagy and ALS: mechanistic insights and therapeutic implications. Autophagy 2022, 18, 254–282. [Google Scholar] [CrossRef] [PubMed]
- Madill, M.; McDonagh, K.; Ma, J.; Vajda, A.; McLoughlin, P.; O'Brien, T.; Hardiman, O.; Shen, S. Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol Brain 2017, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Meyer, K.; Sherwood, T.W.; Vick, J.; Likhite, S.; Frakes, A.; Miranda, C.J.; Braun, L.; Heath, P.R.; Pineda, R.; et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc Natl Acad Sci U S A 2016, 113, E6496–E6505. [Google Scholar] [CrossRef] [PubMed]
- Yamout, B.I.; Alroughani, R. Multiple Sclerosis. Semin Neurol 2018, 38, 212–225. [Google Scholar] [CrossRef] [PubMed]
- García-León, J.A.; Kumar, M.; Boon, R.; Chau, D.; One, J.; Wolfs, E.; Eggermont, K.; Berckmans, P.; Gunhanlar, N.; de Vrij, F.; et al. SOX10 Single Transcription Factor-Based Fast and Efficient Generation of Oligodendrocytes from Human Pluripotent Stem Cells. Stem Cell Reports 2018, 10, 655–672. [Google Scholar] [CrossRef]
- Najm, F.J.; Madhavan, M.; Zaremba, A.; Shick, E.; Karl, R.T.; Factor, D.C.; Miller, T.E.; Nevin, Z.S.; Kantor, C.; Sargent, A.; et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 2015, 522, 216–220. [Google Scholar] [CrossRef]
- Daviaud, N.; Chen, E.; Edwards, T.; Sadiq, S.A. Cerebral organoids in primary progressive multiple sclerosis reveal stem cell and oligodendrocyte differentiation defect. Biol Open 2023, 12. [Google Scholar] [CrossRef]
- Stoker, T.B.; Mason, S.L.; Greenland, J.C.; Holden, S.T.; Santini, H.; Barker, R.A. Huntington's disease: diagnosis and management. Pract Neurol 2022, 22, 32–41. [Google Scholar] [CrossRef]
- Consortium, H.i. Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 2012, 11, 264–278. [Google Scholar] [CrossRef]
- Xu, X.; Tay, Y.; Sim, B.; Yoon, S.I.; Huang, Y.; Ooi, J.; Utami, K.H.; Ziaei, A.; Ng, B.; Radulescu, C.; et al. Reversal of Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in Huntington Disease Patient-Derived Induced Pluripotent Stem Cells. Stem Cell Reports 2017, 8, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev 2010, 90, 905–981. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington's disease pathology in human induced pluripotent stem cell-derived neurons. Mol Neurodegener 2016, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Linville, R.M.; Nerenberg, R.F.; Grifno, G.; Arevalo, D.; Guo, Z.; Searson, P.C. Brain microvascular endothelial cell dysfunction in an isogenic juvenile iPSC model of Huntington's disease. Fluids Barriers CNS 2022, 19, 54. [Google Scholar] [CrossRef] [PubMed]
- Antonov, S.A.; Novosadova, E.V. Current State-of-the-Art and Unresolved Problems in Using Human Induced Pluripotent Stem Cell-Derived Dopamine Neurons for Parkinson's Disease Drug Development. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, G.; Comi, G.P.; Corti, S. Advancing Drug Discovery for Neurological Disorders Using iPSC-Derived Neural Organoids. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Balusu, S.; Praschberger, R.; Lauwers, E.; De Strooper, B.; Verstreken, P. Neurodegeneration cell per cell. Neuron 2023, 111, 767–786. [Google Scholar] [CrossRef] [PubMed]
- Baxi, E.G.; Thompson, T.; Li, J.; Kaye, J.A.; Lim, R.G.; Wu, J.; Ramamoorthy, D.; Lima, L.; Vaibhav, V.; Matlock, A.; et al. Answer ALS, a large-scale resource for sporadic and familial ALS combining clinical and multi-omics data from induced pluripotent cell lines. Nat Neurosci 2022, 25, 226–237. [Google Scholar] [CrossRef]
- Chang, C.Y.; Ting, H.C.; Liu, C.A.; Su, H.L.; Chiou, T.W.; Lin, S.Z.; Harn, H.J.; Ho, T.J. Induced Pluripotent Stem Cell (iPSC)-Based Neurodegenerative Disease Models for Phenotype Recapitulation and Drug Screening. Molecules 2020, 25. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of iPSC application. Somatic cells (a) can be obtained from patients with neurodegenerative diseases and induced into iPSC by pluripotency factors (b). Then, they can be genetically manipulated to undergo gene correction (c) and differentiated (f) into cell types implicated in disease onset. The iPSC can be differentiated into healthy neural cells for correct cell function and validation for transplant (d) or autologous transplantation in the donor patient (e). Alternatively, not genetically corrected iPSC (b), can be differentiated into neural cells implicated in disease onset (f) to model the cellular pathogenic phenotype in vitro (g). These cellular cultures or organoids can be studied in the laboratory to validate the iPSC-derived models (h) and be used to screen drugs, for example (i). The selected drugs can be tested in animal models in preclinical trials (j) and, in the case of beneficial results pointing to drug repositioning or combination (k), clinical trials in patients can be proposed.
Figure 1.
Schematic representation of iPSC application. Somatic cells (a) can be obtained from patients with neurodegenerative diseases and induced into iPSC by pluripotency factors (b). Then, they can be genetically manipulated to undergo gene correction (c) and differentiated (f) into cell types implicated in disease onset. The iPSC can be differentiated into healthy neural cells for correct cell function and validation for transplant (d) or autologous transplantation in the donor patient (e). Alternatively, not genetically corrected iPSC (b), can be differentiated into neural cells implicated in disease onset (f) to model the cellular pathogenic phenotype in vitro (g). These cellular cultures or organoids can be studied in the laboratory to validate the iPSC-derived models (h) and be used to screen drugs, for example (i). The selected drugs can be tested in animal models in preclinical trials (j) and, in the case of beneficial results pointing to drug repositioning or combination (k), clinical trials in patients can be proposed.
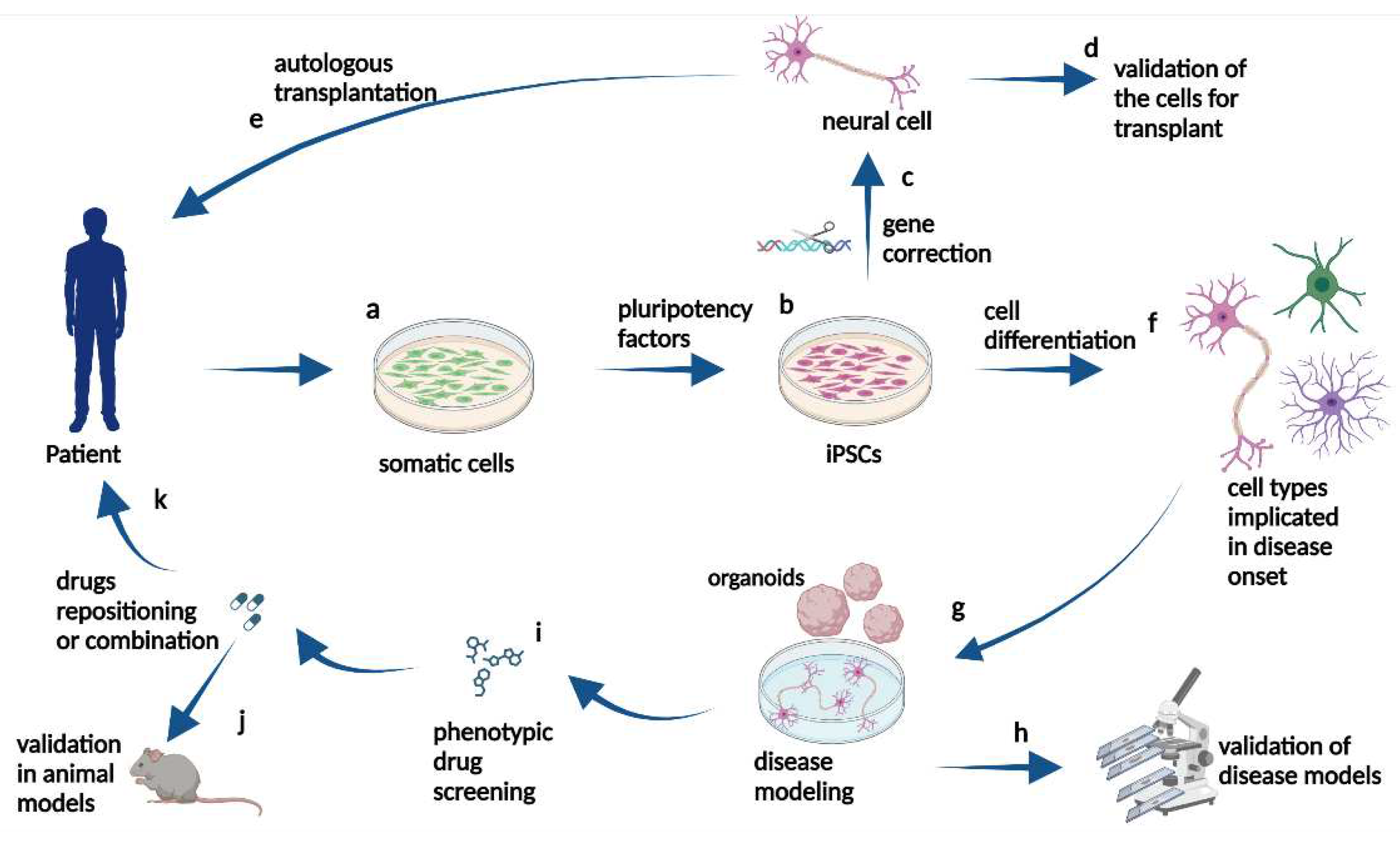
Figure 2.
Representative figure of the potential of iPSC obtained from a patient or a healthy adult. The neural cells obtained from the patient can be corrected by gene editing to obtain healthy neural cells, which will be used as an isogenic control in modeling the disease. Healthy neural cells obtained from a healthy adult can undergo gene editing, and neural cells carrying a mutation from a neurodegenerative disease can be obtained for disease modeling.
Figure 2.
Representative figure of the potential of iPSC obtained from a patient or a healthy adult. The neural cells obtained from the patient can be corrected by gene editing to obtain healthy neural cells, which will be used as an isogenic control in modeling the disease. Healthy neural cells obtained from a healthy adult can undergo gene editing, and neural cells carrying a mutation from a neurodegenerative disease can be obtained for disease modeling.
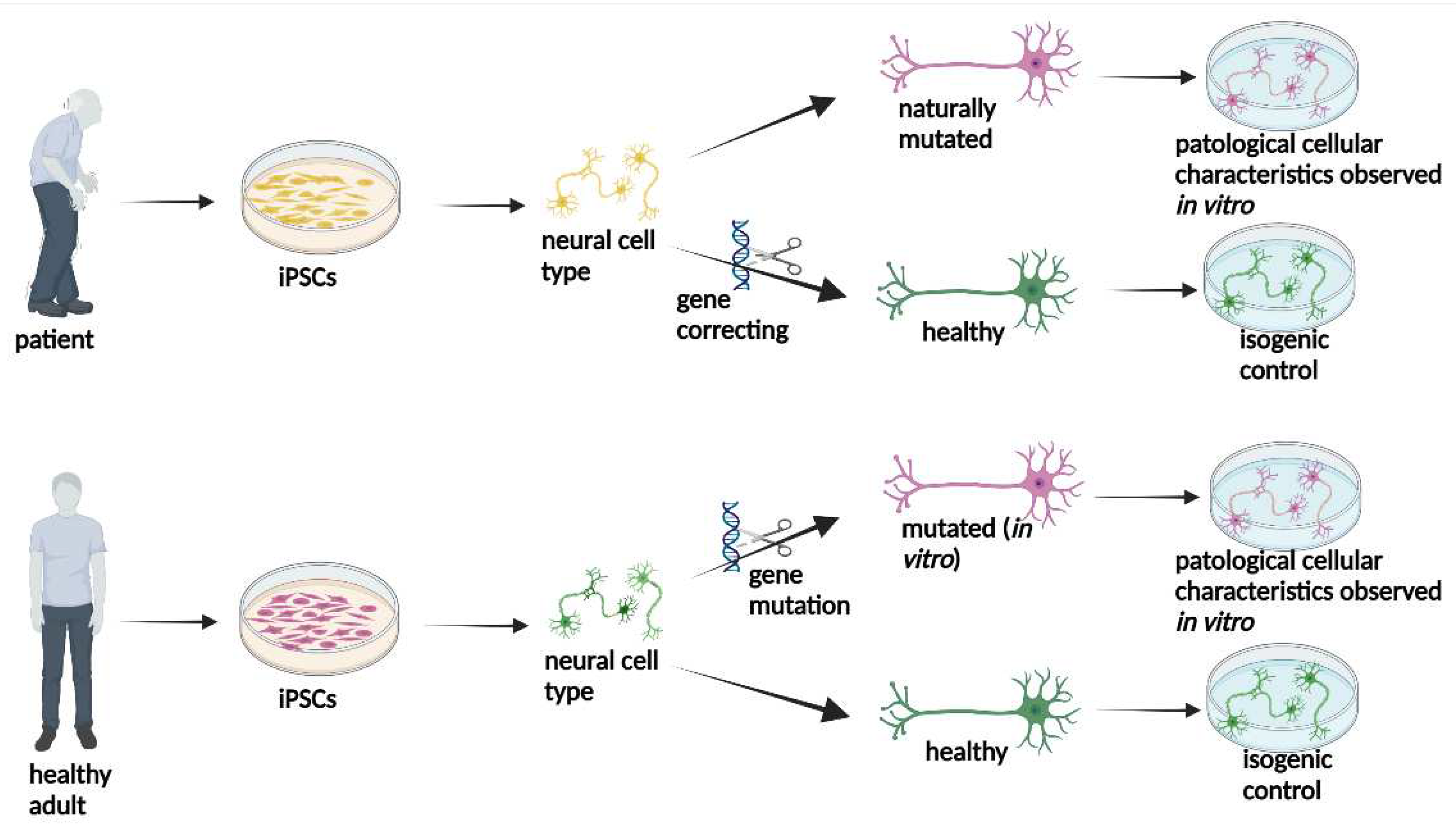
Figure 3.
Table showing the relevance of each type of neural cell obtained from iPSC for each neurodegenerative disease. ALS: Amyotrophic Lateral Sclerosis, PD: Parkinson Disease, AD: Alzheimer Disease, HD: Huntington’s disease, MS: Multiple sclerosis.
Figure 3.
Table showing the relevance of each type of neural cell obtained from iPSC for each neurodegenerative disease. ALS: Amyotrophic Lateral Sclerosis, PD: Parkinson Disease, AD: Alzheimer Disease, HD: Huntington’s disease, MS: Multiple sclerosis.
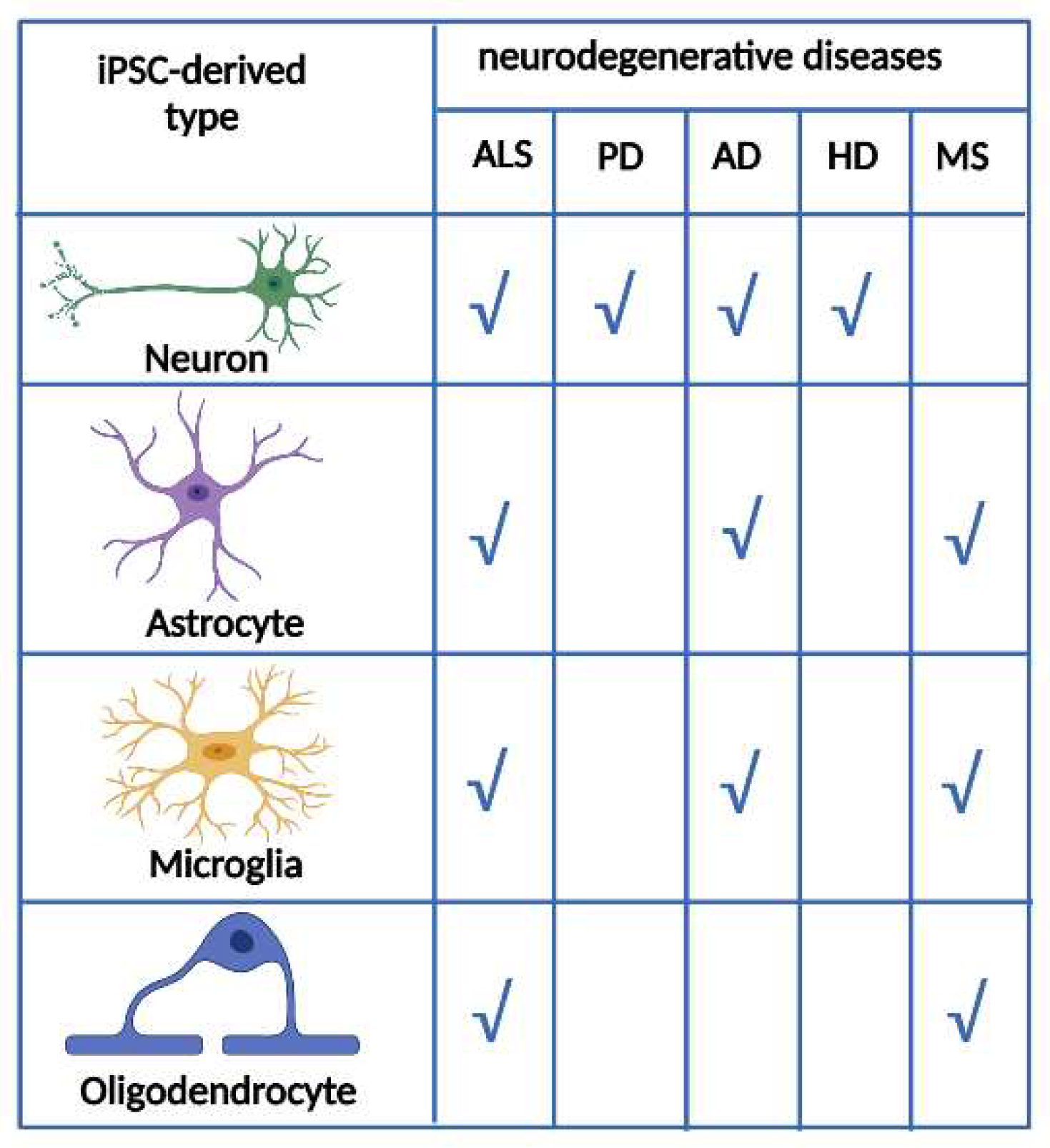
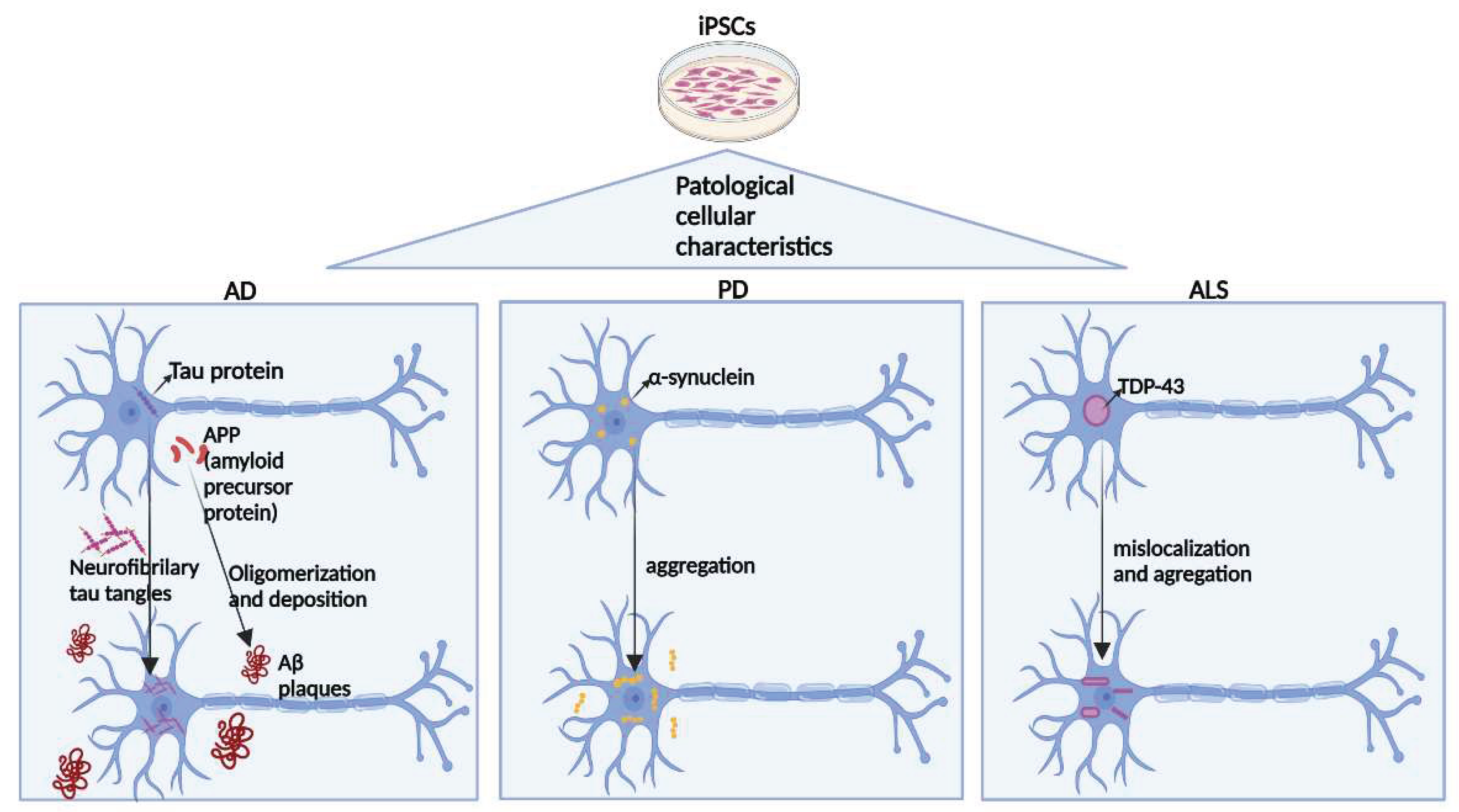
Figure 4.
Common general pathogenic mechanisms of the three main neurodegenerative diseases can be obtained for disease modeling from iPSCs. In AD, Tau protein and APP (amyloid precursor protein) present in the healthy brain undergo aggregation in the disease. Tau protein forms neurofibrillary tau tangles inside neurons, and the APP protein undergoes oligomerization and deposition, forming extracellular AB plaques. α-Synuclein present within cells in the healthy brain undergoes aggregation in PD. Finally, in ALS, the TDP-43 protein inside the nucleus in the healthy brain migrates to the cytoplasm and undergoes aggregation in the disease.
Figure 4.
Common general pathogenic mechanisms of the three main neurodegenerative diseases can be obtained for disease modeling from iPSCs. In AD, Tau protein and APP (amyloid precursor protein) present in the healthy brain undergo aggregation in the disease. Tau protein forms neurofibrillary tau tangles inside neurons, and the APP protein undergoes oligomerization and deposition, forming extracellular AB plaques. α-Synuclein present within cells in the healthy brain undergoes aggregation in PD. Finally, in ALS, the TDP-43 protein inside the nucleus in the healthy brain migrates to the cytoplasm and undergoes aggregation in the disease.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



