Submitted:
14 November 2023
Posted:
14 November 2023
You are already at the latest version
Abstract
Traumatic Brain Injury (TBI) remains a leading cause of morbidity and mortality among all ages; despite the advances, understanding pathophysiological responses after TBI is still complex, involving multiple mechanisms. Previous reviews have focused on potential targets; however, the research on potential targets has continuously grown in the last five years, bringing even more alternatives and elucidating previous mechanisms. Knowing the key and updated pathophysiology concepts is vital for adequate management and better outcomes. This article reviews the underlying molecular mechanisms, the latest updates, and future directions for pathophysiology-based TBI management.
Keywords:
Secondary TBI
; Traumatic Brain Injury
; Brain Injuries
; Physiopathology
1. Introduction
Traumatic brain injury (TBI) is a leading cause of disability and death among children and young adults, with an incidence of approximately 1.7 million per year in the USA, resulting in 52,000 deaths [1]. Survivors of the initial impact must still contend with the consequences of trauma, as not all injury occurs at the time of impact. The primary injury results from forces applied to the head and involve direct structural damage to the brain. This triggers a cascade of events leading to neurological damage that evolves (secondary injury) [2]. Several external brain insults, both intracranial and systemic, may complicate and worsen the secondary injury [3].
As was pointed out by Ng et al., current therapeutic strategies focus on preventing secondary injury through early surgical intervention, multiparameter monitoring, and targeted treatment in the intensive care setting [2,4]. Adherence to guidelines for managing severe TBI is associated with better outcomes [5]. However, treatment should be individualized to address underlying mechanisms following TBI and the role of secondary injury in recovery. This work focuses on the underlying pathophysiological mechanisms and their related treatments to systematize the thought process for TBI management, highlighting the role.
2. Materials and Methods
We conducted a literature search of PUBMED, SCOPUS, and Google Scholar from 1980 to 2023 using search terms related to the pathophysiology and treatment of traumatic brain injury. This review (Figure 1) aims to concisely evaluate the pathophysiology of TBI and provide guidance for understanding current and emerging treatment modalities. Recommendations in this review are based on the guidelines released by the Brain Trauma Foundation in 2016 [6].
Each section on the pathophysiological basis is followed by a discussion of the specific treatment evolved and its current status. Treatments with multiple action mechanisms are primarily discussed in the section with a more experimental or clinical background. We also searched the clinicaltrials.gov website to evaluate any ongoing clinical trials (status: “recruiting patients,” “not yet recruiting,” or “active, not recruiting”).
3. Literature Review
3.1. Pathophysiology of Secondary Brain Injury
Secondary traumatic brain injury is the neurological damage that evolves due to the primary injury (Figure 2). Within the first few hours following the injury, a series of events may trigger a cascade of secondary metabolic, inflammatory, and ischemic insults that can exacerbate the primary neuronal injury. These events are associated with gene changes that release excitatory amino acids, free radicals, and inflammatory cytokines, alter ion flux across membranes, upregulate neuroprotective cascades, and induce programmed cell death [7].
Alterations in cellular metabolism and signaling pathways (Figure 3) can lead to the depletion of energy reserves. As a result, neuronal cells may no longer be able to maintain depolarization of their membranes, causing alterations in ion fluxes and osmotic swelling of the cells. Therefore, following TBI, it is crucial to recognize, prevent, and treat secondary injury to ensure neuronal survival.
3.2. Sequelae from the Primary Impact
3.2.1. Cellular Events
Excitotoxicity and Calcium
Severe TBI cause an abrupt increase in extracellular excitatory amino acids such as glutamate and aspartate [2]. Evidence shows a 40% decline in the expression of astrocytic sodium-dependent glutamate transporters GLAST (EAAT1) and GLT-1 (EAAT2) within 24 h following TBI, leading to a significant decrease in the resorption of glutamate [2]. These excitatory amino acids activate N-methyl-D-Aspartate (NMDA) and non-NMDA receptors, leading to cell membrane depolarization and the influx of sodium (Na2+), potassium (K+), and calcium (Ca2+). High intracellular calcium concentrations activate phospholipase and calpain enzymes that alter membrane and cytoskeletal integrity, eventually resulting in neuronal cell damage and death. The specific mechanism of cell death will be explained later [2].
Magnesium (Mg2+) can regulate excitotoxic processes by blocking NMDA receptors and calcium channels. However, Mg2+ concentration decreases after TBI and persists for at least four days after the impact (Health), and its deficiency has been associated with poor neurological outcomes [8]; increasing extracellular magnesium concentration improves the recovery of hippocampal neuronal high-energy phosphates and accelerates regional cerebral flow to the ischemic brain area [9].
Excitotoxicity is complicated when cortical spreading depression (CSD) occurs [3], a depolarization wave in cerebral gray matter that propagates across the brain [10]. It can break down ion homeostasis, facilitating the release of excitatory amino acids. CSD is associated with poor outcomes in TBI patients [10]. In humans, microdialysis studies demonstrate an increased lactate-pyruvate ratio without consistent evidence of ischemia, and this finding is strongly correlated with outcome [11]. CSD and/or mitochondrial dysfunction may underlie this marker of disturbed metabolism since they are associated with lactate accumulation [7,12]
Specific Treatment
Until this point, statins have been proven to have the potential to protect cultured neurons from excitotoxic death caused by the glutamate receptor agonist NMDA[13]. Interestingly, when evaluating chronic TBI patients with prolonged disorders of consciousness, the use of amantadine, an assumed NMDA antagonist and dopamine agonist, accelerates functional recovery [14]
Multiple clinical trials in acute TBI targeting glutamate and specifically its NMDA receptor have shown contradictory results to demonstrate any beneficial effect [15,16,17], as well as treatment with nimodipine (a calcium channel blocker) [18]. Some authors argue that blocking synaptic transmission mediated by NMDA receptors hinders neuronal survival [16].
Despite promising experimental studies with the use of intravenous magnesium sulfate, its clinical efficiency has yet to be proven magnesium sulfate therapy is effective in the treatment of patients with DAI [19]
Regarding CSD, clinical management can focus on controlling factors that increase its incidence and duration, such as systemic hypotension, pyrexia, hypoxia, and low plasma glucose [10].
Free Radicals and Oxidative Stress in Traumatic Brain Injury
Free radicals are by-products of energy metabolism in cells and play a role in vascular tone and immune function. Signaling pathways can increase free radical formation after traumatic brain injury (TBI). Calcium activates pathways that release free radicals from mitochondria and increase nitric oxide (NO) production through inducible NO synthase (iNOS) [20]. Nicotinamide adenine dinucleotide (NADPH) oxidase produces reactive oxygen species (ROS) within the first hour after trauma [21]. ROS increases affinity to NO, forming peroxynitrites (ONOO-) [21], which destroy the cytoskeleton, cell membranes, and DNA [22]. NO inhibits cytochrome c oxidase, causing mitochondrial disruption and cell death [23].
NO formed by vascular endothelium (endothelial nitric oxide synthase – eNOS) exerts beneficial effects after TBI, including vasodilation and increased cerebral blood flow [23]. Peroxynitrites are the leading cause of oxidative stress, as indicated by increased nitrotyrosine in TBI patients with poor outcomes [24]. ONOO- inhibits potassium channels, increasing vascular tone and impairing vascular reactivity [23]. Iron compounds from hemoglobin degradation form reactive free radical oxidants, altering synaptic function and contributing to posttraumatic seizures [25].
Specific Treatment
Citicoline has multiple neuroprotective mechanisms, including inhibition of oxidative stress and apoptotic pathways [26]. However, a large randomized controlled trial (RCT) showed no benefit in TBI patients [27].
Statins up-regulate eNOS expression and inhibit inducible NO, interleukin1 β (IL1 β), and tumor necrosis factor α (TNF α) [27]. They reduce post-traumatic hypoperfusion and rebound hyperemia [27], protect neurons from excitotoxic death [13], and may reduce cerebral edema and intracranial hypertension [28]. Susanto et al. demonstrated that compared to nonusers, either simvastatin 40mg, atorvastatin 20mg, or rosuvastatin 20mg for ten days reduced mortality risk in TBI individuals. In contrast, statin discontinuation was associated with increased mortality [28].
Inflammatory Mediators and Cascades
Traumatic injuries cause disturbances in the normal cellular functioning of the brain due to the impact of direct, rotational, and shear forces [29]. Axonal injury leads to localized swelling, which hampers the transmission of signals. Traumatic injuries are also connected to alterations in cerebral blood flow, causing an initial decrease in blood circulation and subsequent unresponsive vasodilation, believed to be caused by the release of nitric oxide in the affected tissue [30].
Following a focal injury, the first component of the neuronal structure to be affected at the cellular level is the axonal membranes, given the rotational and direct forces. This axonal damage leads to the release of potassium from the intracellular environment. After membrane depolarization, calcium entry through voltage-dependent channels promotes the release of excitatory amino acids and neurotransmitters. At this point, potassium and calcium freely move between the intra- and extracellular spaces, disrupting intracellular homeostasis [29].
The impairment of intracellular functions leads the neuronal tissue into anaerobiosis(cite). Lactate levels rise, further contributing to local damage to the blood-brain barrier and cell death. This process can occur for 4 to 6 hours [30].
In brief, the disruption of cellular membranes resulting from the primary mechanical insult or secondary injury triggers the release of damage-associated molecular patterns (DAMPs). This prompts the rapid upregulation of tumor necrosis factor (TNF), IL-6, and IL-1β by local glial cells and infiltrating immune cells, acting as early mediators that drive the inflammatory response following traumatic injury [31].
The release of damage-associated molecular patterns (DAMPs) by injured neurons and proinflammatory and oxidative mediators from infiltrating immune cells leads to polarization towards an M1-like phenotype [32]. The expression of proinflammatory factors (Table 1) such as IL-1β, TNF, IL-6, NOS2, IL-12p40, and NOX2 characterizes M1-like cells.
In response to anti-inflammatory and neurotrophic signals, microglia and macrophages can shift towards an M2-like phenotype [33]. M2-like cells express proteins such as CD206, CD163, arginase-1, FCγR, Ym1, IL-10, and TGFβ. Molecular pathways involved in regulating M2-like phenotypic transitions include STAT6/3, IRF-4/7, NF-κB p50/p50, Nrf2, and miR-124. M2-like microglia and macrophages release anti-inflammatory and trophic factors, promoting the resolution of inflammation [34,35]. Microglia and macrophages possess remarkable plasticity and can transition between M1-like and M2-like phenotypes. Mixed phenotypes are present in the acute phase following TBI, eventually transitioning to an M1-like dominant phenotype in the chronic phase [35,36].
Treatments Based on the Pathway Level
The development of innovative anti-inflammatory drugs for managing traumatic brain injury (TBI) is facilitated by targeting various signaling pathways such as NF-κB, MAPKs, JAK/STAT, PI3K/Akt/mTOR, and TGF-β1[37]. Once inflammatory mediators are released, immune response and glial cells are recruited. Microglial cells form the first line of differentiation between the intact and injured tissue [33,38]. Microglial cells release oxidation metabolites and pro-inflammatory reactants and cytokines such as interferon-gamma, interleukins, and tumor necrosis factor-alpha, especially in the latter. All of this leads to the stimulation of astrocytes for the formation of glial scars at the sites of trauma [9].
Nuclear factor-kappa B (NF-κB) synthesizes inflammatory molecules, pro-inflammatory cytokines, and chemokines [39]. In glial cells, NF-κB promotes inflammation, while in neurons, it is associated with neuroplasticity and neuronal development. Inhibiting this factor could reduce apoptosis and secondary inflammation in TBI [40,41].
In TBI, the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathway decreases its expression, leading to increased cell death. In a study, recombinant erythropoietin was administered to cortical cells of rats with prior TBI, resulting in an increase in JAK and STAT and a reduction in apoptosis [42].
The MAPK (Mitogen-Activated Protein Kinase) pathway plays a crucial role in cell differentiation, proliferation, and survival. The path consists of c-Jun NH (2)-terminal kinase (JNK), extracellular signal-regulated protein kinase (ERK), and p38 [43]. Several studies have indicated that activating the p38 and JNK pathways contributes to increased neuronal damage in spinal cord injury and cerebral ischemia cases, activating a complex cascade in the mitochondria of brain cells and leading to apoptosis [44].
The dual nature of inflammation has been illustrated in experimental models exploring the involvement of TNF and inducible nitric oxide synthase (iNOS) following TBI [45]. TNF has been associated with brain edema, blood-brain barrier (BBB) disruption, and leukocyte recruitment [46]. Surprisingly, mice lacking TNF exhibited motor function impairment and larger lesions four weeks after the injury despite showing early neuroprotection [47]. Similarly, while TBI increased iNOS expression in the brain, which has multiple proinflammatory and neurotoxic effects, the genetic or chemical blockade of iNOS worsened spatial memory two to three weeks after the injury [48,49,50]. Cell death through programmed necrosis, such as necroptosis triggered by TNF-mediated RIP kinase activation, can initiate a detrimental cycle: necrosis leads to further membrane disruption, promoting the release of damage-associated molecular patterns (DAMPs), which, in turn, exacerbates necrosis and amplifies inflammation [48,49,50].
Other Neuroinflammatory Components
In response to TBI and glutamate toxicity, endogenous neuroprotectant adenosine levels are produced due to adenosine triphosphate and mRNA breakdown [51,52]. Following the ATP production and the mRNA, the activation of adenosine receptor A1 after TBI has been found to have anti-excitotoxic and anti-inflammatory effects in mice. However, it is essential to note that systemic administration of adenosine to patients can lead to bradycardia and hypotension [51,53].
Other components, including free radicals, lipid peroxidation, and direct impact, trigger the release of inflammatory mediators such as cytokines, chemokines, and complement. Cytokines are signaling molecules produced by immune system cells and brain cells, including microglia, astrocytes, and neurons. They act synergistically in cascades. After traumatic brain injury (TBI), tumor necrosis factor α and interleukin1 β trigger the formation of peptides, reactive oxygen species (ROS), and nitrogen species [54]. These mediators perpetuate secondary brain injury by activating arachidonic acid and coagulation cascades and disrupting the blood-brain barrier [55].
Specific Treatment
The activation of inflammasomes, which leads to the release of IL-1β, can be inhibited by targeting IL-1 receptors, which can inhibit the invasion of circulating immune cells into the CNS [59].
Treatments like intravenous immunoglobulin can inhibit the priming of T cells from entering the CNS [60]. Furthermore, alterations in the gut microbiome may influence the balance between proinflammatory and anti-inflammatory T lymphocytes [61]. Impaired glymphatic clearance after TBI may hinder the removal of proinflammatory mediators, and ongoing investigations aim to enhance glymphatic flow, with increased clearance observed during sleep [62].
Nonsteroidal anti-inflammatory drugs (NSAIDs) produce an inhibition of COX that significantly reduces the levels of IL-1β and hinders the synthesis of IL-6 by modulating the pathways involved in producing vasodilator prostaglandins [63]. Another critical aspect of NSAIDs is their ability to stimulate the proliferator-activated receptor (PPAR), which elicits transcriptional regulatory effects that reduce the levels of various proinflammatory substances and decrease microglial activity. Additionally, several NSAIDs exhibit antioxidant properties and can inhibit the activation of NF-kB [64].
Mitochondrial Dysfunction in Traumatic Brain Injury
The activation of inflammasomes, which leads to the release of IL-1β, can be inhibited by targeting IL-1 receptors, which can inhibit the invasion of circulating immune cells into the CNS [59].
Treatments like intravenous immunoglobulin can inhibit the priming of T cells from entering the CNS [60]. Furthermore, alterations in the gut microbiome may influence the balance between proinflammatory and anti-inflammatory T lymphocytes [61]. Impaired glymphatic clearance after TBI may hinder the removal of proinflammatory mediators, and ongoing investigations aim to enhance glymphatic flow, with increased clearance observed during sleep [62].
Nonsteroidal anti-inflammatory drugs (NSAIDs) produce an inhibition of COX that significantly reduces the levels of IL-1β and hinders the synthesis of IL-6 by modulating the pathways involved in producing vasodilator prostaglandins [63]. Another critical aspect of NSAIDs is their ability to stimulate the proliferator-activated receptor (PPAR), which elicits transcriptional regulatory effects that reduce the levels of various proinflammatory substances and decrease microglial activity. Additionally, several NSAIDs exhibit antioxidant properties and can inhibit the activation of NF-kB [64].
Specific Treatment
Cyclosporine A (CsA) stabilizes the mitochondrial permeability transition pore (mPTP) and has demonstrated neuroprotection in pre-clinical TBI studies [74]. Sullivan et al. showed on animal models that animals receiving CsA demonstrated a reduction in the lesion volume, even up to 74% [74]. Another mechanism that has been explored is the reduction of brain metabolic activity through hypothermia [75]. However, a recent meta-analysis conducted by Chen et al., which included 23 trials involving 2796 patients, demonstrated that therapeutic hypothermia did not reduce, but surprisingly, can increase the mortality rate of patients with TBI in some high-quality studies. However, the therapy can benefit patients with demonstrated increased intracranial pressure, not as a prophylactic therapy but as a therapy within the first 24 hours [75].
Finally, barbiturates suppress cerebral metabolism and reduce cerebral blood volume and intracranial pressure (ICP) [76]. In a recent multicenter European trial, high-dose barbiturate treatment caused a decrease of 69% in ICP; however, this effect was also accompanied by hemodynamic instability, leading to more extended periods of mean arterial pressure <70mmHg despite the use of vasopressors [76]. All of these previously mentioned effects, without significantly impacting the outcome at any stage after injury [76].
Cell Death
Cell death after traumatic brain injury (TBI) can occur through necrosis or apoptosis. Both can occur in regions remote from the impact site within days and weeks after trauma. Necrosis occurs in response to severe mechanical or ischemic/hypoxic tissue damage, excessive excitatory amino acid neurotransmitter release, and metabolic failure [77]. Caspases and calpain are important mediators of programmed cell death (apoptosis), with calpain activation more associated with necrosis [78].
Caspases are activated through the extrinsic pathway initiated by cell surface death receptor ligation (receptor-linked caspase-8 pathway) and the intrinsic pathway arising from mitochondria (mitochondrial caspase-9 pathway) [78]. Caspase 3 compromises membrane permeability to Ca2+, leading to elevated intracellular Ca2+ levels. Caspase 3 also degrades calpastatin, facilitating calpain activation [78]. Endogenous inhibitors, such as the inhibitors of the apoptosis family, modulate caspase activity within these pathways [78].
Activation of caspase-3 via extrinsic and intrinsic apoptotic pathways after traumatic brain injury (TBI) has been documented, and this response is also mirrored in the retina [79]. However, experimental studies suggest that when sustained activation, calpain is more important than caspase-3 in mediating cell death after TBI [80]. The anti-apoptotic modulator B-cell lymphoma (Bcl-2) inhibits the mitochondrial permeability transition pore (mPTP), preserving mitochondrial homeostasis and preventing mitochondrial Ca2+ leak and programmed cell death [81]. Several in vivo studies with transgenic mice have shown promising results, at least with a reduced deficit in mice overexpressing the BCL-2 [82]. In clinical trials, a literature review published by Deng et al. showed reduced mortality and better outcomes in the Glasgow Coma Score (GOS) in the patients with increased Bcl-2 in the peritraumatic tissue, highlighting the importance of the peptide as a potential biomarker and therapeutic target [83].
Specific Treatment
Erythropoietin (Epo) and its receptor (Epor) are expressed throughout the central nervous system [84]. Epo modulates caspase 1, caspase 3, and caspase 8-like activities, maintaining genomic DNA integrity and preventing acute cellular injury and microglial activation [84]. Epo also modulates mitochondrial membrane permeability and cytochrome c release, reduces cerebral vasospasm, and improves cerebral blood flow when given early post-injury [84].
3.2.2. BBB Disruption and Neutrophil Invasion
The blood-brain barrier (BBB) regulates the exchange of substances between plasma and brain interstitium [85]. This barrier is formed by brain endothelial cells connected by tight junctions, with mechanical support from astrocytes critical for normal function [85]. Ion homeostasis and uptake of small molecules are conducted via specific endothelial membrane channels and solute carriers. Larger peptides and proteins are transported by endo- or transcytosis pathways within caveolae and clathrin-coated microvesicles. Paracellular diffusion is restricted by tight junctions between adjacent endothelial cells [86]. BBB disruption after injury is typically biphasic, with an immediate phase of hyperpermeability, temporary restoration of BBB function, and a delayed opening period [86]. BBB disruption leads to excess interstitial water (vasogenic edema) accumulation, contributing to cerebral swelling, brain shift, and herniation [85].
As previously mentioned, the BBB tight junction is based on the interaction between endothelial cells and astrocytes. Some studies have shown that the mechanical damage to astrocytes initiates oxidative-stress-mediated edema, altering astrocyte ionic gradients and increasing BBB permeability [85,87]. Reactive oxygen species (ROS) increase brain endothelium permeability and promote post-traumatic invasion of inflammatory cells by upregulating endothelial expression of cell adhesion molecules such as intercellular adhesion molecule-1 (ICAM1) [87]. Degradation of membrane proteins and increased permeability contribute to BBB opening, vasogenic edema formation, and increased intracranial pressure [88]
Inflammatory cells provide the primary source of matrix metalloproteinase (MMP) activity [89]. MMPs promote cell death, including apoptosis [89]. Other inflammatory mediators in BBB opening include substance P, kinins, and bradykinins [88]. Particularly, substance P and its receptor, neurokinin 1 (NK1R), have been promising [90]. MMPs are released in ischemic brain injury and contribute to BBB disruption by degrading basal lamina components and tight junctions [89]. Later, MMPs are involved in tissue remodeling and neurovascular recovery. Vascular endothelial growth factor (VEGF) alters BBB permeability by changing the distribution and downregulating the expression of tight junction proteins [89].
Specific Treatment
Substance P antagonists (SP, NK1 receptor antagonist) show promising results in limiting BBB opening and edema after TBI [90]. Vink et al., in their review article, showed that NK1 antagonists can reduce posttraumatic ICP and improve brain oxygenation after TBI [90]. In vitro studies have shown the potential for the NK1-R antagonist to reverse the compromise, integrity, and function of the BBB [91].
Another alternative is the Hyperbaric Oxygen Therapy (HBOT). HBOT reduces MMP-9 expression and inhibits neutrophilic infiltration. It may also counter capillary vasodilation within hypoxic tissues [92]. Hadanny et al. showed in their review that a search from 1969 to 2023 showed that HBOT should be recommended in acute moderate-severe TBI, specifically for patients suffering from prolonged post-concussion syndrome who have precise evidence of metabolic dysfunctional brain regions. However, further studies are needed to evaluate outcomes and determine the optimal treatment protocols [92].
4. Conclusions
Managing traumatic brain injury (TBI) patients includes specialized prehospital care, intensive clinical care, and long-term rehabilitation. However, neuroprotective agents to limit secondary injury or enhance repair lack clinical effectiveness. The complexity of TBI pathophysiology may reflect the difficulty of translating preclinical benefits into clinical practice. The current goal is to follow and identify the sequence of events of secondary lesions to avoid further neuronal damage. Knowledge of these concepts, the development of more efficient clinical trial designs, and the possibility of combination therapies may change the course of treatment in the acute phase in the near future.
Author Contributions
Conceptualization, RLOA.; methodology, RLOA, LJMMF, LFF, MCPC, GAVG; data curation, RLOA, LJMMF, LFFX.X.; writing—original draft preparation, RLOA, LJMMF, LFF, MCPC, GAVG; writing—review and editing, RLOA, LJMMF, LFF, MCPC, GAVG, GJH; supervision, RLOA; project administration, RLOA. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Langlois, J.A.; Rutland-Brown, W.; Wald, M.M. The Epidemiology and Impact of Traumatic Brain Injury: A Brief Overview. J Head Trauma Rehabil 2006, 21, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Sharma, S. Recent Advances in Pathophysiology of Traumatic Brain Injury. CN 2018, 16, 1224–1238. [Google Scholar] [CrossRef] [PubMed]
- Helmy, A.; Carpenter, K.L.; Menon, D.K.; Pickard, J.D.; Hutchinson, P.J. The Cytokine Response to Human Traumatic Brain Injury: Temporal Profiles and Evidence for Cerebral Parenchymal Production. J Cereb Blood Flow Metab 2011, 31, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, S.M.; Trask, A.L.; Waller, M.A.; Watts, D.D. Management of Brain-Injured Patients by an Evidence-Based Medicine Protocol Improves Outcomes and Decreases Hospital Charges. The Journal of Trauma: Injury, Infection, and Critical Care 2004, 56, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Hawryluk, G.W.J.; Rubiano, A.M.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; Shutter, L.; et al. Guidelines for the Management of Severe Traumatic Brain Injury: 2020 Update of the Decompressive Craniectomy Recommendations. Neurosurg. 2020, 87, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Raghupathi, R. Cell Death Mechanisms Following Traumatic Brain Injury. Brain Pathology 2004, 14, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Kahriman, A.; Bouley, J.; Smith, T.W.; Bosco, D.A.; Woerman, A.L.; Henninger, N. Mouse Closed Head Traumatic Brain Injury Replicates the Histological Tau Pathology Pattern of Human Disease: Characterization of a Novel Model and Systematic Review of the Literature. acta neuropathol commun 2021, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, W.; Zhong, J.; Li, Y.; Cheng, Y.; Su, Z.; Zheng, W.; Guan, X. The Effects of Magnesium Sulfate Therapy after Severe Diffuse Axonal Injury. TCRM 2016, 12, 1481–1486. [Google Scholar] [CrossRef]
- Lauritzen, M.; Dreier, J.P.; Fabricius, M.; Hartings, J.A.; Graf, R.; Strong, A.J. Clinical Relevance of Cortical Spreading Depression in Neurological Disorders: Migraine, Malignant Stroke, Subarachnoid and Intracranial Hemorrhage, and Traumatic Brain Injury. J Cereb Blood Flow Metab 2011, 31, 17–35. [Google Scholar] [CrossRef]
- Timofeev, I.; Carpenter, K.L.H.; Nortje, J.; Al-Rawi, P.G.; O’Connell, M.T.; Czosnyka, M.; Smielewski, P.; Pickard, J.D.; Menon, D.K.; Kirkpatrick, P.J.; et al. Cerebral Extracellular Chemistry and Outcome Following Traumatic Brain Injury: A Microdialysis Study of 223 Patients. Brain 2011, 134, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Verweij, B.H.; Muizelaar, J.P.; Vinas, F.C.; Peterson, P.L.; Xiong, Y.; Lee, C.P. Impaired Cerebral Mitochondrial Function after Traumatic Brain Injury in Humans. Journal of Neurosurgery 2000, 93, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Wible, E.F.; Laskowitz, D.T. Statins in Traumatic Brain Injury. Neurotherapeutics 2010, 7, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Rühl, L.; Kuramatsu, J.B.; Sembill, J.A.; Kallmünzer, B.; Madzar, D.; Gerner, S.T.; Giede-Jeppe, A.; Balk, S.; Mueller, T.; Jäger, J.; et al. Amantadine Treatment Is Associated with Improved Consciousness in Patients with Non-Traumatic Brain Injury. J Neurol Neurosurg Psychiatry 2022, 93, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Baracaldo-Santamaría, D.; Ariza-Salamanca, D.F.; Corrales-Hernández, M.G.; Pachón-Londoño, M.J.; Hernandez-Duarte, I.; Calderon-Ospina, C.-A. Revisiting Excitotoxicity in Traumatic Brain Injury: From Bench to Bedside. Pharmaceutics 2022, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Turski, L. Why Did NMDA Receptor Antagonists Fail Clinical Trials for Stroke and Traumatic Brain Injury? The Lancet Neurology 2002, 1, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Shohami, E.; Biegon, A. Novel Approach to the Role of NMDA Receptors in Traumatic Brain Injury. CNSNDDT 2014, 13, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Vergouwen, M.D.; Vermeulen, M.; Roos, Y.B. Effect of Nimodipine on Outcome in Patients with Traumatic Subarachnoid Haemorrhage: A Systematic Review. The Lancet Neurology 2006, 5, 1029–1032. [Google Scholar] [CrossRef] [PubMed]
- Temkin, N.; Machamer, J.; Dikmen, S.; Nelson, L.D.; Barber, J.; Hwang, P.H.; Boase, K.; Stein, M.B.; Sun, X.; Giacino, J.; et al. Risk Factors for High Symptom Burden Three Months after Traumatic Brain Injury and Implications for Clinical Trial Design: A Transforming Research and Clinical Knowledge in Traumatic Brain Injury Study. Journal of Neurotrauma 2022, 39, 1524–1532. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal Models of Traumatic Brain Injury. Nat Rev Neurosci 2013, 14, 128–142. [Google Scholar] [CrossRef]
- Zhang, Z.-D.; Fang, H.-Y.; Pang, C.; Yang, Y.; Li, S.-Z.; Zhou, L.-L.; Bai, G.-H.; Sheng, H.-S. Giant Pediatric Supratentorial Tumor: Clinical Feature and Surgical Strategy. Front. Pediatr. 2022, 10, 870951. [Google Scholar] [CrossRef]
- Besson, V.C. Drug Targets for Traumatic Brain Injury from Poly(ADP-Ribose)Polymerase Pathway Modulation. British Journal of Pharmacology 2009, 157, 695–704. [Google Scholar] [CrossRef]
- O’Connell, K.M.; Littleton-Kearney, M.T. The Role of Free Radicals in Traumatic Brain Injury. Biological Research For Nursing 2013, 15, 253–263. [Google Scholar] [CrossRef]
- Darwish, R.S.; Amiridze, N.; Aarabi, B. Nitrotyrosine as an Oxidative Stress Marker: Evidence for Involvement in Neurologic Outcome in Human Traumatic Brain Injury. Journal of Trauma: Injury, Infection & Critical Care 2007, 63, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Nisenbaum, E.J.; Novikov, D.S.; Lui, Y.W. The Presence and Role of Iron in Mild Traumatic Brain Injury: An Imaging Perspective. Journal of Neurotrauma 2014, 31, 301–307. [Google Scholar] [CrossRef]
- Secades, J.J. Role of Citicoline in the Management of Traumatic Brain Injury. Pharmaceuticals 2021, 14, 410. [Google Scholar] [CrossRef]
- Zafonte, R.D.; Bagiella, E.; Ansel, B.M.; Novack, T.A.; Friedewald, W.T.; Hesdorffer, D.C.; Timmons, S.D.; Jallo, J.; Eisenberg, H.; Hart, T.; et al. Effect of Citicoline on Functional and Cognitive Status Among Patients With Traumatic Brain Injury: Citicoline Brain Injury Treatment Trial (COBRIT). JAMA 2012, 308, 1993. [Google Scholar] [CrossRef] [PubMed]
- Susanto, M.; Pangihutan Siahaan, A.M.; Wirjomartani, B.A.; Setiawan, H.; Aryanti, C. ; Michael The Neuroprotective Effect of Statin in Traumatic Brain Injury: A Systematic Review. World Neurosurgery: X 2023, 19, 100211. [Google Scholar] [CrossRef]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury. Medical Clinics of North America 2020, 104, 213–238. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.J. Pathophysiology of Traumatic Brain Injury. Physical Medicine and Rehabilitation Clinics of North America 2017, 28, 215–225. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.-S. Microglia and Inflammation-Mediated Neurodegeneration: Multiple Triggers with a Common Mechanism. Progress in Neurobiology 2005, 76, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and Neuroprotection in Traumatic Brain Injury. JAMA Neurol 2015, 72, 355. [Google Scholar] [CrossRef] [PubMed]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory Response in Acute Traumatic Brain Injury: A Double-Edged Sword. Current Opinion in Critical Care, 2002; 8, 101–105. [Google Scholar] [CrossRef]
- David, S.; Kroner, A. Repertoire of Microglial and Macrophage Responses after Spinal Cord Injury. Nat Rev Neurosci 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Loane, D.J. Neuroinflammation after Traumatic Brain Injury: Opportunities for Therapeutic Intervention. Brain, Behavior, and Immunity 2012, 26, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Kumar, A.; Stoica, B.A.; Cabatbat, R.; Faden, A.I. Progressive Neurodegeneration After Experimental Brain Trauma: Association With Chronic Microglial Activation. Journal of Neuropathology & Experimental Neurology 2014, 73, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Malik, R.; Singh, G.; Bhatia, S.; Al-Harrasi, A.; Mohan, S.; Albratty, M.; Albarrati, A.; Tambuwala, M.M. Pathogenesis and Management of Traumatic Brain Injury (TBI): Role of Neuroinflammation and Anti-Inflammatory Drugs. Inflammopharmacol 2022, 30, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP Mediates Rapid Microglial Response to Local Brain Injury in Vivo. Nat Neurosci 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.P.-Y.; Tung, J.Y.-L.; Ku, D.T.-L.; Luk, C.-W.; Ling, A.S.-C.; Kwong, D.L.-W.; Cheng, K.K.-F.; Ho, W.W.-S.; Shing, M.M.-K.; Chan, G.C.-F. Outcome of Chinese Children with Craniopharyngioma: A 20-Year Population-Based Study by the Hong Kong Pediatric Hematology/Oncology Study Group. Childs Nerv Syst 2020, 36, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Camandola, S. NF-κB in Neuronal Plasticity and Neurodegenerative Disorders. J. Clin. Invest. 2001, 107, 247–254. [Google Scholar] [CrossRef]
- Yang, K.; Mu, X.S.; Hayes, R.L. Increased Cortical Nuclear Factor-κB (NF-κB) DNA Binding Activity after Traumatic Brain Injury in Rats. Neuroscience Letters 1995, 197, 101–104. [Google Scholar] [CrossRef]
- Oliva, A.A.; Kang, Y.; Sanchez-Molano, J.; Furones, C.; Atkins, C.M. STAT3 Signaling after Traumatic Brain Injury: STAT3 Activation after Traumatic Brain Injury. Journal of Neurochemistry 2012, 120, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, W.S.; Mandell, J.W. Transient Neuronal but Persistent Astroglial Activation of ERK/MAP Kinase after Focal Brain Injury in Mice. Journal of Neurotrauma 2003, 20, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, W.D.; Bramlett, H.M. Therapeutic Hypothermia and Targeted Temperature Management in Traumatic Brain Injury: Clinical Challenges for Successful Translation. Brain Research 2016, 1640, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, J.M.; Morganti-Kossmann, M.C. Involvement of Pro- and Anti-Inflammatory Cytokines and Chemokines in the Pathophysiology of Traumatic Brain Injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Scherbel, U.; Raghupathi, R.; Nakamura, M.; Saatman, K.E.; Trojanowski, J.Q.; Neugebauer, E.; Marino, M.W.; McIntosh, T.K. Differential Acute and Chronic Responses of Tumor Necrosis Factor-Deficient Mice to Experimental Brain Injury. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 8721–8726. [Google Scholar] [CrossRef]
- Sinz, E.H.; Kochanek, P.M.; Dixon, C.E.; Clark, R.S.B.; Carcillo, J.A.; Schiding, J.K.; Chen, M.; Wisniewski, S.R.; Carlos, T.M.; Williams, D.; et al. Inducible Nitric Oxide Synthase Is an Endogenous Neuroprotectant after Traumatic Brain Injury in Rats and Mice. J. Clin. Invest. 1999, 104, 647–656. [Google Scholar] [CrossRef]
- Au, A.K.; Aneja, R.K.; Bell, M.J.; Bayir, H.; Feldman, K.; Adelson, P.D.; Fink, E.L.; Kochanek, P.M.; Clark, R.S.B. Cerebrospinal Fluid Levels of High-Mobility Group Box 1 and Cytochrome C Predict Outcome after Pediatric Traumatic Brain Injury. Journal of Neurotrauma 2012, 29, 2013–2021. [Google Scholar] [CrossRef]
- Laird, M.D.; Shields, J.S.; Sukumari-Ramesh, S.; Kimbler, D.E.; Fessler, R.D.; Shakir, B.; Youssef, P.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. High Mobility Group Box Protein-1 Promotes Cerebral Edema after Traumatic Brain Injury via Activation of Toll-like Receptor 4. Glia 2014, 62, 26–38. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Savitz, S.I.; Yang, J.; Degterev, A.; Yuan, J.; Cuny, G.D.; Moskowitz, M.A.; Whalen, M.J. Necrostatin-1 Reduces Histopathology and Improves Functional Outcome after Controlled Cortical Impact in Mice. J Cereb Blood Flow Metab 2008, 28, 1564–1573. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Clark, R.S.B.; Ruppel, R.A.; Adelson, P.D.; Bell, M.J.; Whalen, M.J.; Robertson, C.L.; Satchell, M.A.; Seidberg, N.A.; Marion, D.W.; et al. Biochemical, Cellular, and Molecular Mechanisms in the Evolution of Secondary Damage after Severe Traumatic Brain Injury in Infants and Children: Lessons Learned from the Bedside. Pediatric Critical Care Medicine, 2000; 1, 4–19. [Google Scholar] [CrossRef]
- Viviani, B.; Boraso, M.; Marchetti, N.; Marinovich, M. Perspectives on Neuroinflammation and Excitotoxicity: A Neurotoxic Conspiracy? NeuroToxicology 2014, 43, 10–20. [Google Scholar] [CrossRef]
- Verrier, J.D.; Exo, J.L.; Jackson, T.C.; Ren, J.; Gillespie, D.G.; Dubey, R.K.; Kochanek, P.M.; Jackson, E.K. Expression of the 2′,3′-cAMP-Adenosine Pathway in Astrocytes and Microglia: 2′,3′-cAMP Metabolism in Astrocytes and Microglia. Journal of Neurochemistry 2011, 118, 979–987. [Google Scholar] [CrossRef]
- Cederberg, D.; Siesjö, P. What Has Inflammation to Do with Traumatic Brain Injury? Childs Nerv Syst 2010, 26, 221–226. [Google Scholar] [CrossRef]
- Dohi, K.; Kraemer, B.C.; Erickson, M.A.; McMillan, P.J.; Kovac, A.; Flachbartova, Z.; Hansen, K.M.; Shah, G.N.; Sheibani, N.; Salameh, T.; et al. Molecular Hydrogen in Drinking Water Protects against Neurodegenerative Changes Induced by Traumatic Brain Injury. PLoS ONE 2014, 9, e108034. [Google Scholar] [CrossRef]
- Chen, S.-F.; Hsu, C.-W.; Huang, W.-H.; Wang, J.-Y. Post-Injury Baicalein Improves Histological and Functional Outcomes and Reduces Inflammatory Cytokines after Experimental Traumatic Brain Injury: Baicalein Reduces Cytokine Expression in TBI. British Journal of Pharmacology 2008, 155, 1279–1296. [Google Scholar] [CrossRef]
- Kokiko-Cochran, O.N.; Godbout, J.P. The Inflammatory Continuum of Traumatic Brain Injury and Alzheimer’s Disease. Front. Immunol. 2018, 9, 672. [Google Scholar] [CrossRef] [PubMed]
- Thelin, E.P.; Hall, C.E.; Gupta, K.; Carpenter, K.L.H.; Chandran, S.; Hutchinson, P.J.; Patani, R.; Helmy, A. Elucidating Pro-Inflammatory Cytokine Responses after Traumatic Brain Injury in a Human Stem Cell Model. Journal of Neurotrauma 2018, 35, 341–352. [Google Scholar] [CrossRef]
- Anderson, G.D.; Peterson, T.C.; Vonder Haar, C.; Kantor, E.D.; Farin, F.M.; Bammler, T.K.; MacDonald, J.W.; Hoane, M.R. Comparison of the Effects of Erythropoietin and Anakinra on Functional Recovery and Gene Expression in a Traumatic Brain Injury Model. Front. Pharmacol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The Far-Reaching Scope of Neuroinflammation after Traumatic Brain Injury. Nat Rev Neurol 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Mazzeo, A.T.; Kunene, N.K.; Gilman, C.B.; Hamm, R.J.; Hafez, N.; Bullock, M.R. Severe Human Traumatic Brain Injury, but Not Cyclosporin A Treatment, Depresses Activated T Lymphocytes Early after Injury. Journal of Neurotrauma 2006, 23, 962–975. [Google Scholar] [CrossRef]
- Jin, X.; Ishii, H.; Bai, Z.; Itokazu, T.; Yamashita, T. Temporal Changes in Cell Marker Expression and Cellular Infiltration in a Controlled Cortical Impact Model in Adult Male C57BL/6 Mice. PLoS ONE 2012, 7, e41892. [Google Scholar] [CrossRef]
- Breitner, J.C.S. Inflammatory Processes and Antiinflammatory Drugs in Alzheimer’s Disease: A Current Appraisal. Neurobiology of Aging 1996, 17, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Grilli, M.; Pizzi, M.; Memo, M.; Spano, P. Neuroprotection by Aspirin and Sodium Salicylate Through Blockade of NF-κB Activation. Science 1996, 274, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol Rev 2016, 68, 20–48. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin Externalization to the Outer Mitochondrial Membrane Acts as an Elimination Signal for Mitophagy in Neuronal Cells. Nat Cell Biol 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Schurr, A.; Payne, R.S. Lactate, Not Pyruvate, Is Neuronal Aerobic Glycolysis End Product: An in Vitro Electrophysiological Study. Neuroscience 2007, 147, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-L.; Lin, T.-Y.; Chen, P.-L.; Guo, T.-N.; Huang, S.-Y.; Chen, C.-H.; Lin, C.-H.; Chan, C.-C. Mitochondrial Function and Parkinson’s Disease: From the Perspective of the Electron Transport Chain. Front. Mol. Neurosci. 2021, 14, 797833. [Google Scholar] [CrossRef] [PubMed]
- Hiebert, J.B.; Shen, Q.; Thimmesch, A.R.; Pierce, J.D. Traumatic Brain Injury and Mitochondrial Dysfunction. The American Journal of the Medical Sciences 2015, 350, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.G.; Alshboul, O.A. Pathophysiology of Traumatic Brain Injury. Neurosciences (Riyadh) 2013, 18, 222–234. [Google Scholar]
- Ahluwalia, M.; Kumar, M.; Ahluwalia, P.; Rahimi, S.; Vender, J.R.; Raju, R.P.; Hess, D.C.; Baban, B.; Vale, F.L.; Dhandapani, K.M.; et al. Rescuing Mitochondria in Traumatic Brain Injury and Intracerebral Hemorrhages - A Potential Therapeutic Approach. Neurochemistry International 2021, 150, 105192. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal Pathology in Traumatic Brain Injury. Experimental Neurology 2013, 246, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain Tumor Cells in Circulation Are Enriched for Mesenchymal Gene Expression. Cancer Discovery 2014, 4, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, F.; Yang, P.; Shao, J.; Chen, Q.; Zheng, R. A Meta-Analysis of the Effects of Therapeutic Hypothermia in Adult Patients with Traumatic Brain Injury. Crit Care 2019, 23, 396. [Google Scholar] [CrossRef] [PubMed]
- Majdan, M.; Mauritz, W.; Wilbacher, I.; Brazinova, A.; Rusnak, M.; Leitgeb, J. Barbiturates Use and Its Effects in Patients with Severe Traumatic Brain Injury in Five European Countries. Journal of Neurotrauma 2013, 30, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Engelhard, K. Pathophysiology of Traumatic Brain Injury. British Journal of Anaesthesia 2007, 99, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Eldadah, B.A.; Faden, A.I. Caspase Pathways, Neuronal Apoptosis, and CNS Injury. Journal of Neurotrauma 2000, 17, 811–829. [Google Scholar] [CrossRef] [PubMed]
- Kovács-Öller, T.; Zempléni, R.; Balogh, B.; Szarka, G.; Fazekas, B.; Tengölics, Á.J.; Amrein, K.; Czeiter, E.; Hernádi, I.; Büki, A.; et al. Traumatic Brain Injury Induces Microglial and Caspase3 Activation in the Retina. IJMS 2023, 24, 4451. [Google Scholar] [CrossRef]
- Saatman, K.E.; Creed, J.; Raghupathi, R. Calpain as a Therapeutic Target in Traumatic Brain Injury. Neurotherapeutics 2010, 7, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled Demolition at the Cellular Level. Nat Rev Mol Cell Biol 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Raghupathi, R.; Fernandez, S.C.; Murai, H.; Trusko, S.P.; Scott, R.W.; Nishioka, W.K.; McIntosh, T.K. BCL-2 Overexpression Attenuates Cortical Cell Loss after Traumatic Brain Injury in Transgenic Mice. J Cereb Blood Flow Metab 1998, 18, 1259–1269. [Google Scholar] [CrossRef]
- Deng, H.; Yue, J.K.; Zusman, B.E.; Nwachuku, E.L.; Abou-Al-Shaar, H.; Upadhyayula, P.S.; Okonkwo, D.O.; Puccio, A.M. B-Cell Lymphoma 2 (Bcl-2) and Regulation of Apoptosis after Traumatic Brain Injury: A Clinical Perspective. Medicina 2020, 56, 300. [Google Scholar] [CrossRef]
- Liu, M.; Wang, A.J.; Chen, Y.; Zhao, G.; Jiang, Z.; Wang, X.; Shi, D.; Zhang, T.; Sun, B.; He, H.; et al. Efficacy and Safety of Erythropoietin for Traumatic Brain Injury. BMC Neurol 2020, 20, 399. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and Treatment of Cerebral Edema in Traumatic Brain Injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The Blood–Brain Barrier: An Overview. Neurobiology of Disease 2004, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mira, R.G.; Lira, M.; Cerpa, W. Traumatic Brain Injury: Mechanisms of Glial Response. Front. Physiol. 2021, 12, 740939. [Google Scholar] [CrossRef]
- Donkin, J.J.; Vink, R. Mechanisms of Cerebral Edema in Traumatic Brain Injury: Therapeutic Developments. Current Opinion in Neurology, 2010; 23, 293–299. [Google Scholar] [CrossRef]
- Minta, K.; Brinkmalm, G.; Al Nimer, F.; Thelin, E.P.; Piehl, F.; Tullberg, M.; Jeppsson, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; et al. Dynamics of Cerebrospinal Fluid Levels of Matrix Metalloproteinases in Human Traumatic Brain Injury. Sci Rep 2020, 10, 18075. [Google Scholar] [CrossRef]
- Vink, R.; Gabrielian, L.; Thornton, E. The Role of Substance P in Secondary Pathophysiology after Traumatic Brain Injury. Front. Neurol. 2017, 8, 304. [Google Scholar] [CrossRef]
- Gao, X.; Bayraktutan, U. Substance P Reversibly Compromises the Integrity and Function of Blood-Brain Barrier. Peptides 2023, 167, 171048. [Google Scholar] [CrossRef]
- Hadanny, A.; Maroon, J.; Efrati, S. The Efficacy of Hyperbaric Oxygen Therapy in Traumatic Brain Injury Patients: Literature Review and Clinical Guidelines. MRAJ 2023, 11. [Google Scholar] [CrossRef]
Figure 1.
The summarized pathophysiology of traumatic brain injury. This review will be separated into two parts. Part I will focus on the cellular and BBB that evolve following the injury. Part II will focus on the consequence of these molecular mechanisms and its repair.
Figure 1.
The summarized pathophysiology of traumatic brain injury. This review will be separated into two parts. Part I will focus on the cellular and BBB that evolve following the injury. Part II will focus on the consequence of these molecular mechanisms and its repair.
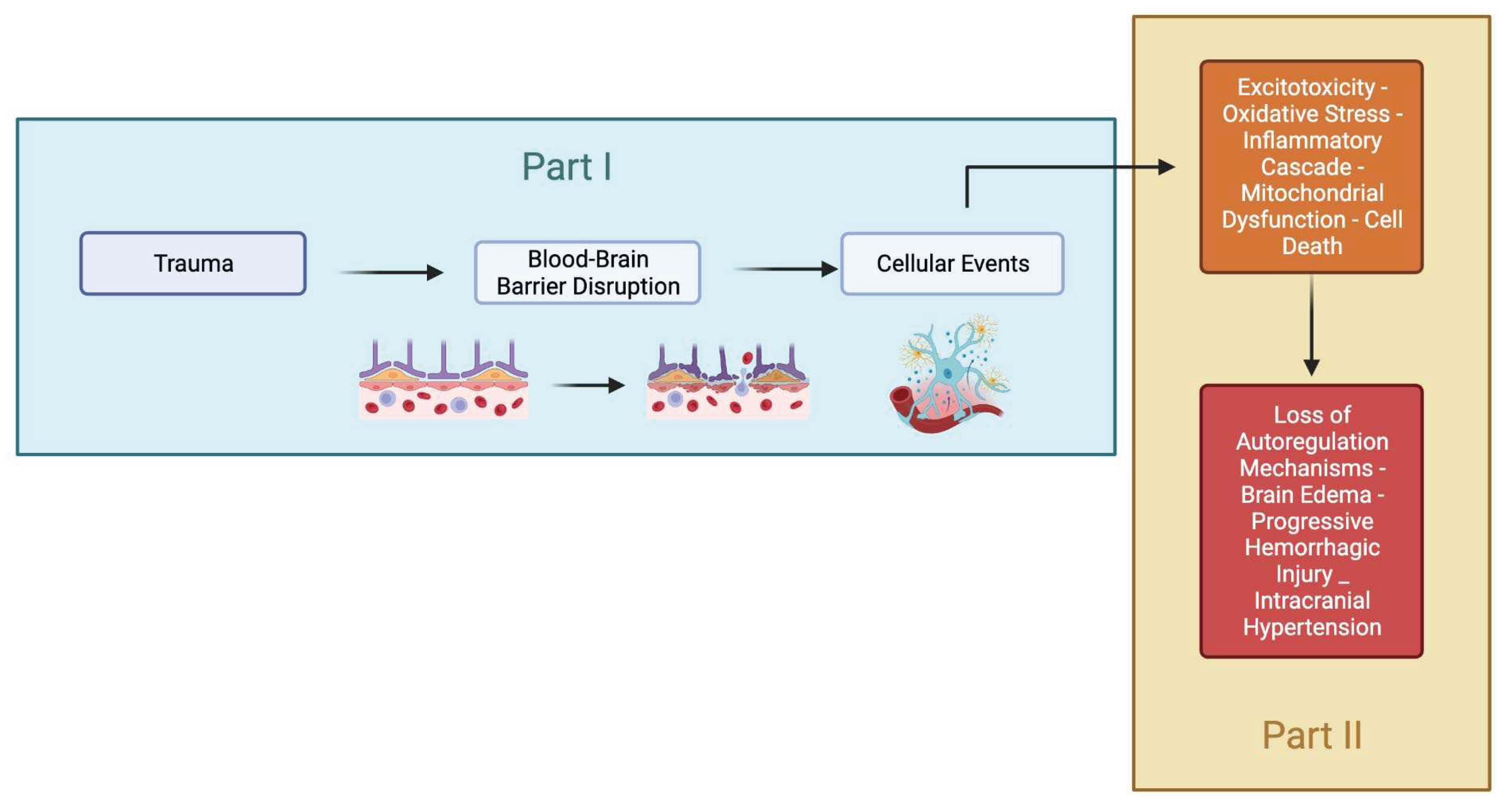
Figure 2.
Schematic view of the primary, secondary and tertiary injuries after TBI.
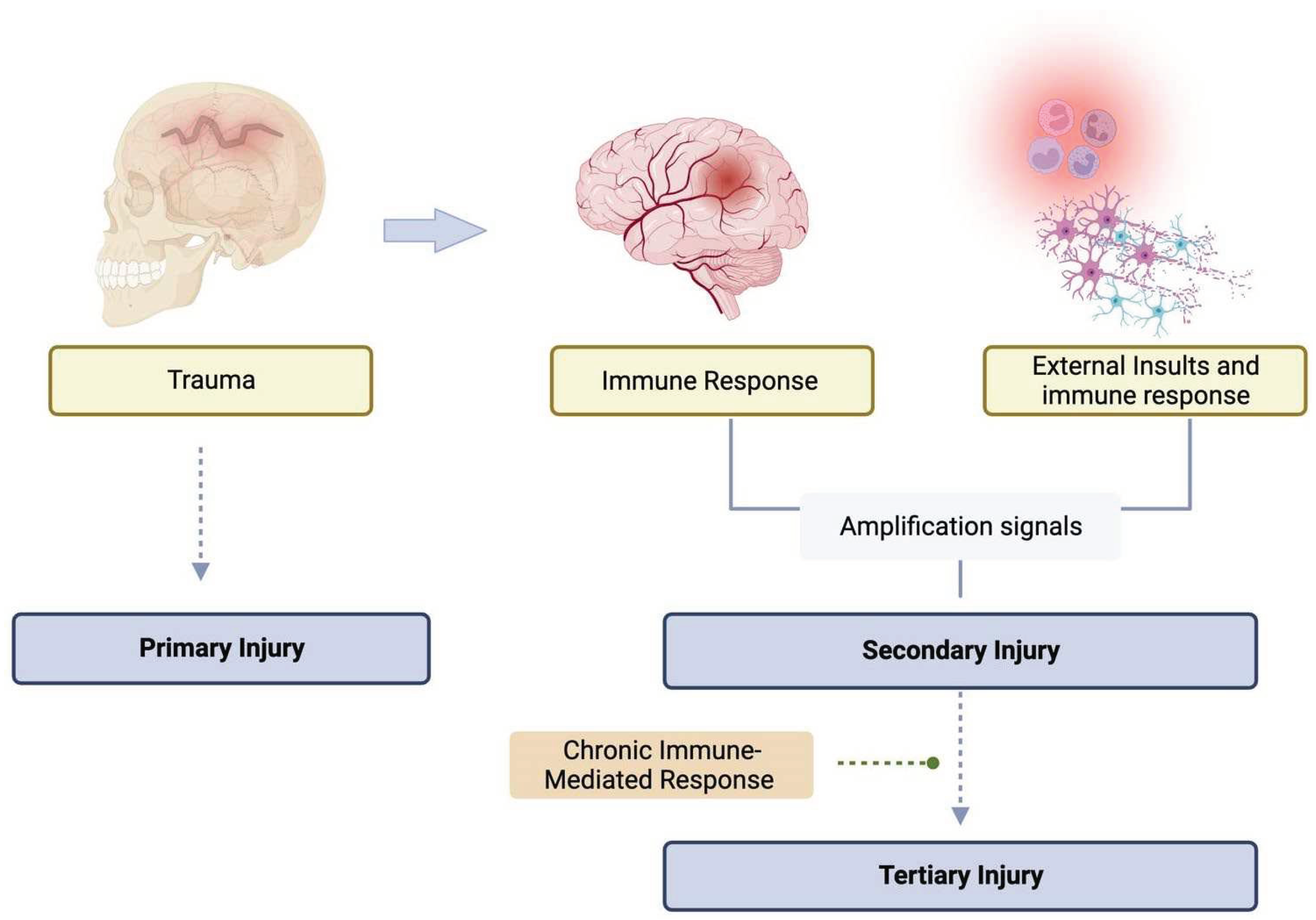
Figure 3.
Schematic evolution of the major mechanisms involved in the secondary injury of TBI. The sequence of events can occur simultaneously and interact to exacerbate injury and initiate neuronal and vascular repair. Potential drugs are placed in red-doted arrows directed to their leading site of action.
Figure 3.
Schematic evolution of the major mechanisms involved in the secondary injury of TBI. The sequence of events can occur simultaneously and interact to exacerbate injury and initiate neuronal and vascular repair. Potential drugs are placed in red-doted arrows directed to their leading site of action.
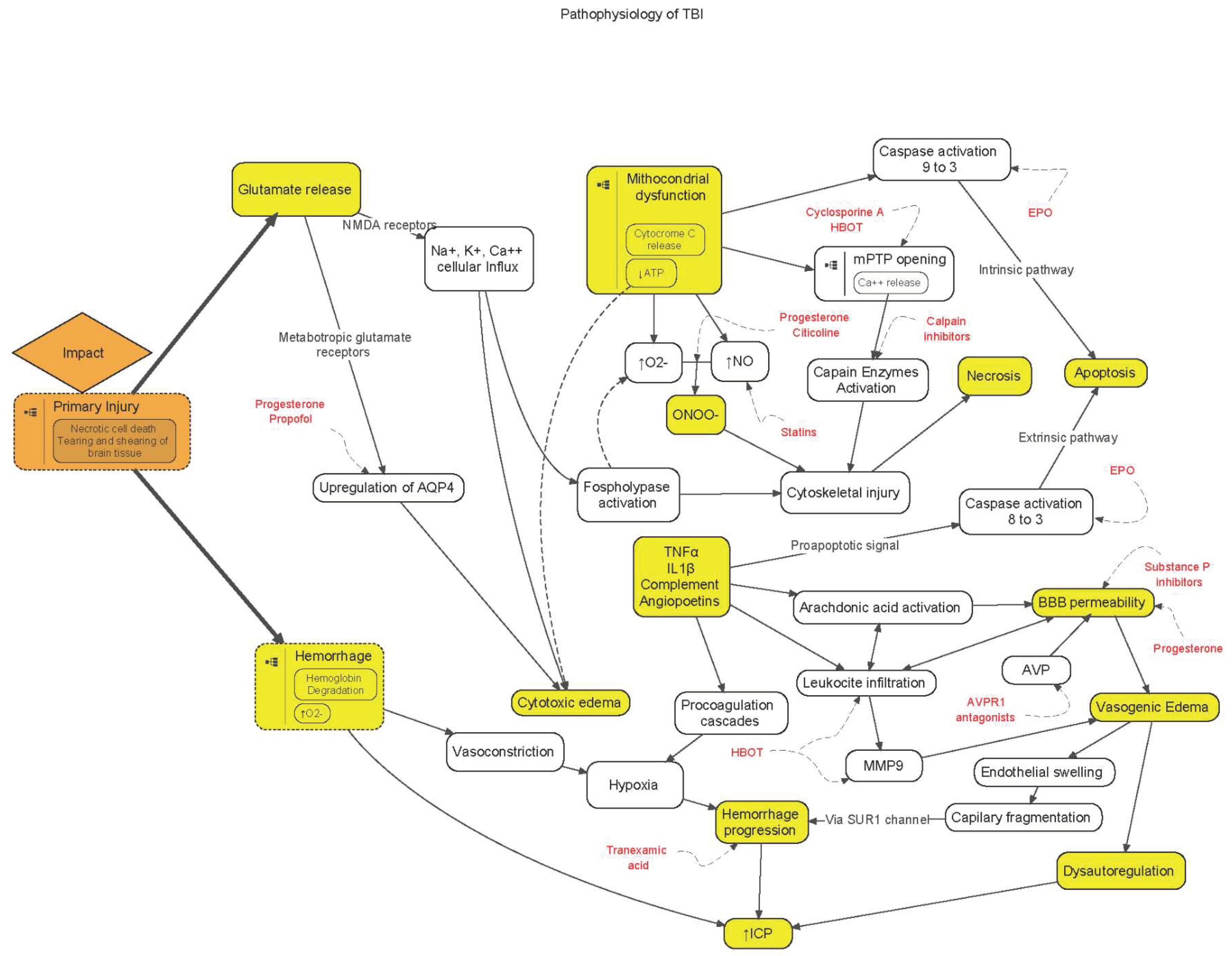

Table 1.
Main cytokines1 involved in traumatic brain injury.
| Main Effects in TBI | Highlights | |
|---|---|---|
| IL1β | Detrimental | Raised CSF IL1β correlates with both raised ICP and poorer outcome. The balance between members of the IL1 cytokine family, in particular between IL1β and its endogenous inhibitor IL1ra, is an important determinant of the degree of inflammatory response, rather than the absolute concentration of IL1β. In TBI patients, high microdialysate IL1ra/IL1b ratio is associated to favourable outcomes. |
| TNFα | Detrimental in acute phase and beneficial during the healing process | Upregulated in the injured brain early after trauma, reaching a peak within a few hours following the initial injury. This cytokine triggers the apoptotic cascade but also, pathways resulting in activation of pro-survival genes. |
| IL6 | Beneficial | It is a VEGF, a trophic factor that is upregulated in the CNS after injury and promotes neuronal survival and brain repair through astroglia and vascular remodelling. Following TBI, its concentration rises dramatically. |
| Complement | Detrimental | Complement is the major trigger of inflammatory molecules. That ultimately lead to increase BBB permeability, induction of cytokines, chemokines, and adhesion molecules and induction of apoptosis and cell death by creation of pores on cell membranes. |
| Angiopoietins | Beneficial (Ang1) and detrimental (Ang2) | They are family of growth factors that are important in regulating angiogenesis and vascular permeability, and also have been implicated in BBB disruption. TBIs models show acute decrease in Angiopoetin-1 expression and concomitant increase in Angiopoetin-2 which is associated with endothelial apoptosis and BBB permeability. |
1 IL1β: interleukin1β; TNFα: tumor necrosis factor α; IL6: interleukin6.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



