
Submitted:
03 November 2023
Posted:
06 November 2023
You are already at the latest version
Abstract
Recent research has unveiled intriguing insights suggesting that the body's immune system may be implicated in Parkinson's disease (PD) development. Studies have observed disparities in pro-inflammatory and anti-inflammatory markers between PD patients and healthy individuals. This finding underscores the potential influence of immune system dysfunction in the genesis of this condition. A dysfunctional immune system can serve as a primary catalyst for systemic in-flammation in the body, which may contribute to the emergence of various brain disorders. The identification of several genes associated with PD, as well as their connection to neuroinflamma-tion, raises the likelihood of disease susceptibility. Moreover, advancing age and mitochondrial dysfunction can weaken the immune system, potentially implicating them in the onset of the dis-ease, particularly among older individuals. Compromised integrity of the blood-brain barrier could facilitate the immune system's access to brain tissue. This exposure may lead to encounters with native antigens or infections, potentially triggering an autoimmune response. Furthermore, there is mounting evidence supporting the notion that gut dysbiosis might represent an initial trigger for brain inflammation, ultimately promoting neurodegeneration. In this comprehensive review, we will delve into the numerous hypotheses surrounding the role of both innate and adaptive immunity in PD.
Keywords:
Parkinson’s disease
; immunity
; neuroinflammation
; mitochondria
; dysbiosis
; infections.
1. Introduction
Parkinson's disease (PD) is a multifaceted, progressive neurodegenerative disorder that impacts not only the central nervous system (CNS) but also peripheral organs [1]. It is believed that the development of PD is the result of a complex interplay between genetic and environmental factors, potentially involving an altered immune system [2]. Numerous clinical studies have revealed notable changes in inflammation markers and immune cell populations in both the peripheral blood and cerebrospinal fluid (CSF) of individuals with PD [3]. Additionally, evidence points to the presence of ongoing and end-stage neuroinflammatory processes within the brains of PD patients [4], suggesting a significant role for neuroinflammation in initiating or worsening the neurodegeneration of the dopaminergic nigrostriatal pathway.
PD is primarily characterized by the degeneration of dopaminergic neurons within the substantia nigra pars compacta [5]. Its distinctive pathological feature involves the presence of intracellular α-synuclein (α-syn) aggregates, commonly known as Lewy bodies [6]. A-syn, in this context, assumes a beta-sheet-rich conformation, facilitating the formation of noxious oligomeric clusters that amass within the nerve fibers of both the central and peripheral nervous systems [7]. These accumulations of α-syn can be expelled from cells and subsequently taken up by adjacent cellular structures like cell bodies, dendrites, or axons [8]. Recent research has hinted at the possibility that α-syn pathology may originate outside the CNS, implying an intricate interaction between brain-resident immune cells and the peripheral immune system [9]. Novel assays system, named immunoprecipitation-based real-time quaking-induced conversion (IP/RT-QuIC), can efficiently detect α-syn seeds in various tissues and blood of PD patients [6]. Histopathological evidence of phosphorylated α-syn has been detected not only in the brain but also in the spinal cord [10], cervical and thoracic ganglia [11], as well as various peripheral organs and the gastrointestinal tract [12]. Concerning the latter, it is conceivable that the gut-brain axis plays a significant role in PD's pathogenesis, with α-syn potentially generated in the intestinal tract, capable of migrating into the bloodstream or even reaching the brain by traversing along the vagus nerve or sympathetic nerves [13]. The fact that people with PD frequently show an increased occurrence of T-cells that identify α-syn peptides strongly implies the participation of autoimmune mechanisms in the disease development [14]. A-syn-reactive T-cells are most prevalent shortly after the diagnosis of motor PD and might exist years prior to the identification of motor PD [15].
In this review, we examine the relationship between genes associated with PD and the immune system, the blood-brain barrier changes in PD and the role of peripheral and CNS innate immunity. Finally, we explore the importance of the microbiota as well as the role of mitochondrial dysfunction in immunity and infections (Figure 1).
These factors collectively contribute to peripheral (involving activated innate cells and B/T cell signaling in the enteric nervous system and blood) and central nervous system inflammation (involving activated microglia and astrocyte).
2. PD genes associated with the function of the immune system
While most cases of PD are considered idiopathic, it's worth noting that mutations in over 20 genes have been identified, leading to monogenic forms of PD, which account for approximately 5-10% of all PD cases [16]. On the other hand, the vast majority of cases are attributed to polygenic inheritance, where a complex interplay of environmental factors, aging, and genetic predisposition contributes to the development of PD pathology [17].
Some of the genes associated with monogenic PD have potential roles in regulating immune responses, and there are shared genetic variants between PD patients and individuals with autoimmune and inflammatory diseases [18]. For instance, a common non-coding single nucleotide polymorphism known as rs3129882 in major histocompatibility complex, class II, DR alpha (HLA-DRA), located within the MHC-II locus, has been linked to an increased susceptibility to idiopathic PD [19]. This association may be related to the presentation of pathogenic, immunodominant antigens or a shift towards a more pro-inflammatory CD4 T-cell response, particularly in response to specific environmental exposures like pyrethroid exposure, possibly through genetic or epigenetic mechanisms that influence MHC-II gene expression [19].
Variations in genes related to mitochondrial function, which are a part of the genetic factors contributing to the hereditary component of PD, have been associated to the modulation of both innate and adaptive immune pathways [20]. Proteins encoded by autosomal recessive PD genes, such as E3 ubiquitin ligase PARKIN, PTEN-induced kinase 1 (PINK1), and DJ-1, play roles in mitochondrial pathways [21]. Further supporting the link to mitochondrial dysfunction, evidence emerges from models of autosomal-dominant PD associated with mutations in leucine-rich repeat kinase 2 (LRRK2) and alpha-synuclein (SNCA) [22].
PARKIN and PINK1 genes are vital for maintaining mitochondrial homeostasis and quality control by eliminating damaged mitochondria through a process known as mitophagy [23]. However, recent research, including our own investigations, has revealed intriguing connections between these proteins and mechanisms associated with both innate and adaptive immunity [24]. PARKIN and PINK1 actively suppress the formation of mitochondrial-derived vesicles and the presentation of mitochondrial antigens, identifying them as regulators of the immune system [25]. This underscores the potential involvement of autoimmune processes in the progression of PD. The systemic deletion of PARK2 and PINK1 exacerbates experimental autoimmune encephalomyelitis (EAE), an animal model of autoimmune inflammatory diseases of the CNS, in terms of disease severity [24,26]. This age-related exacerbation occurs through the modulation of both peripheral and CNS immune responses. The absence of PINK1 also alters glial innate immune responses and amplifies inflammation-induced neuron death mediated by nitric oxide [27]. Furthermore, intestinal infection with Gram-negative bacteria triggers PD-like symptoms in PINK1 knockout mice, leading to the development of cytotoxic mitochondria-specific T-cells in both the periphery and the brain [28]. These findings suggest a fascinating interplay between these PD-related genes and the immune system, shedding light on potential immunological mechanisms underlying the disease's progression.
PARK7, responsible for encoding the DJ-1 protein, is another gene that play a significant role in autosomal recessive early-onset PD and that has garnered attention concerning microglial function and neuroinflammation [29]. When DJ-1 function is impaired or lost, microglial cells exhibit an upregulation of pro-inflammatory cytokines while simultaneously downregulating anti-inflammatory pathways [30]. This alteration in microglial behavior may contribute to an increased risk of developing PD by amplifying neuroinflammatory processes within these cells. Interestingly, loss of DJ-1 in PD unexpectedly reduces signs of immunoaging, impacting T-cell compartments in humans and mice [31]. DJ-1 regulates immunoaging through hematopoietic-intrinsic and naïve-CD8-intrinsic mechanisms, presenting a potential target for aging-related diseases [31].
The expression of autosomal-dominant PD associated SNCA gene, that encodes α-syn, has been observed in various hematopoietic and immune cells [32]. This suggests a potential involvement of immune-mediated mechanisms regulated by α-syn in the pathogenesis of PD. It has been reported that alpha-syn exerts its pathogenic effects through non-cell-autonomous neurotoxic actions and induces inflammatory responses in microglia [33]. These activated microglia, in turn, release neurotoxic agents by activating toll-like receptor 2 (TLR2), which acts as a receptor for neuron-derived α-syn. This cascade ultimately contributes to neurodegeneration [33].
Research has provided evidence of the significant role played by α-syn in the maturation and regulation of T-lymphocytes, as demonstrated in α-syn knockout mice (B6;129X1-Sncatm1Ros1/J) [34]. These mice exhibited an increase in the population of CD4 and CD8 double-negative thymocytes, along with notable decreases in the counts of CD4 single-positive and CD8 single-positive T-cells [34]. Furthermore, α-syn-deficient mice also displayed impaired B-cell lymphopoiesis and deficiencies in IgG production. These findings underscore the importance of α-syn in hematopoiesis, B-cell lymphopoiesis, and the adaptive immune response [35].
The VPS35 gene, an ortholog of Vacuolar Protein Sorting 35, has been associated with late-onset, autosomal dominant familial PD in various studies [36]. This gene appears to play a crucial role in regulating the functions and polarization of microglial cells by influencing the trafficking and recycling of immunomodulating receptors and mediators [36].
The glucocerebrosidase (GBA) gene, when mutated, significantly elevates the risk of developing PD by 5 to 30 times [37]. This gene is expressed in immune cells, including monocytes/macrophages and lymphocytes, and has been linked to an abnormal inflammatory response mediated by these immune cells [38]. Research has shown that non-cell-autonomous mechanisms play a role in the development of GBA1-linked PD [39]. This has been demonstrated in mouse astrocytes with heterozygous and homozygous GBA1 D409V knock in mutations. These astrocytes not only exhibited widespread issues with lysosomal structure and function but also displayed significant deficiencies in both basal and TLR4-dependent cytokine production [39]. Notably, inhibiting LRRK2 kinase activity normalized lysosomal function and reduced inflammatory responses, suggesting a functional interplay between glucocerebrosidase and LRRK2 activities in astrocytes [39]. Furthermore, positron emission tomography (PET) scans have detected neuroinflammation in brain regions vulnerable to Lewy pathology in individuals with glucocerebrosidase gene mutations who do not have PD [40]. This finding demonstrates an association with pronounced astrocytic and microglial activation, which can exacerbate neuroinflammatory responses.
Pathogenic mutations within the LRRK2 gene stand as the prevailing genetic culprits behind PD. Interestingly, LRRK2 exhibits heightened expression within immune cells, such as monocytes, neutrophils, and dendritic cells, when compared to its expression in neurons or glial cells [41]. However, intriguing evidence suggests a dynamic interplay between LRRK2 and α-syn in the context of neuroinflammation-driven PD progression [41]. The inhibition of LRRK2 can substantially mitigate the neuroinflammatory responses triggered by α-syn binding to TLR2 [42]. This inhibition effectively alleviates neuroinflammation-induced dopaminergic neuronal loss in various experimental models, including mouse glioma cells, primary rat microglia, and human microglia cell lines [42]. Furthermore, studies have demonstrated that LRRK2 kinase inhibition attenuates neuroinflammation, gliosis, and cytotoxicity in murine models receiving intracerebral injections of α-syn preformed fibrils, thereby reinforcing the anti-inflammatory potential of LRRK2 kinase inhibition in preclinical settings (PMID: 37443833). Recent investigations employing a chimeric mouse model revealed that replacing mutant LRRK2 with the wild-type variant of the protein in T- and B-lymphocytes exerts a marked reduction in lipopolysaccharide (LPS)-mediated inflammation [43]. Additionally, this genetic alteration rescues dopaminergic neuron loss within the substantia nigra pars compacta, underscoring the profound influence of LRRK2 mutations on the adaptive immune system and its significant role in shaping neuropathological outcomes [44]. Notably, the excessive production of interleukin-6 (IL-6) in the periphery emerges as a pivotal factor in driving LRRK2-mediated neurotoxicity. This suggests that signals originating from the peripheral adaptive immune system act as initiators in a cascade of events culminating in the development of PD pathology. Importantly, the neutralization of excessive peripheral IL-6 production has been demonstrated to prevent the loss of dopaminergic neurons within the substantia nigra pars compacta, highlighting the potential therapeutic significance of targeting these peripheral immune signals in PD [44]. Intriguingly, our ongoing research efforts have unveiled preliminary data indicating that the systemic absence of LRRK2 may have a preventive or ameliorative effect on the development of active-induced EAE. While these findings remain unpublished, they hint at a broader role for LRRK2 gene in immune-related neurological disorders.
Targeting gene-mediated immune dysregulation holds promise as a novel therapeutic avenue for mitigating the progression of PD and related neurodegenerative conditions. The existence of this immunological process could be closely linked to the incapacity of certain individuals with inherited mutations in these genes to efficiently remove abnormal α-syn inclusions from their bodies, thereby resulting in the formation of intracellular Lewy bodies and subsequent neuronal loss and worsening the advancement of the condition.
3. Blood bran-barrier and blood-cerebrospinal barrier in PD
The blood-brain barrier (BBB), composed of brain endothelial cells, and the blood- CSF barrier, formed by the tight junctions between choroid plexus epithelial cells, are two crucial anatomical barriers that constitute the primary interface between the extracellular fluids of the brain and the bloodstream [45]. These barriers serve as the foremost guardians, not only regulating the passage of various circulating substances between brain fluids and blood but also governing interactions between the peripheral immune system and the CNS [45]. The malfunction of the blood-CSF barrier and, particularly, the BBB has been associated with the initiation and progression of numerous neurological disorders, including PD [46]. Several clinical studies, including functional imaging of human patients, analysis of postmortem brain specimens, permeability assessments of drugs used for PD treatment, analysis of albumin/IgG levels in the CSF, and animal models induced with toxins, have identified diverse pathological mechanisms responsible for disrupting the BBB [47]. These mechanisms include the breakdown of intercellular junctions, the accumulation of toxic substances, vascular inflammation, and oxidative stress. Among the factors contributing to BBB disruption, notable attention has been directed toward phenotypic changes in astrocytes and endothelial cells, which, together with pericytes, constitute the neurovascular unit [47].
Mitochondrial oxidative stress within brain endothelial cells emerges as a critical factor in multiple pathological processes responsible for BBB disruption [48]. This oxidative stress harms intercellular junctions, contributes to abnormal cerebral angiogenesis, results in neurovascular decoupling, and actively participates in and exacerbates vascular inflammation [48].
Moreover, reactive astrogliosis, even its precise role in the pathogenesis of PD is not fully understood, it seems to be linked to alterations in BBB permeability [49]. Reactive astrogliosis, often accompanied by neuronal death, can result from various insults to the brain, including infection, inflammatory processes, trauma, α-syn accumulation, and ischemia [50]. Conversely, mutations in the PARKIN gene can lead to astrocytic alterations, suggesting the possibility of an astrocyte-related non-autonomous cell death mechanism [51]. Similarly, DJ-1 deficiency can disrupt astrocyte-mediated repair processes through the destabilization of Sox9 and impair the astrogliosis response [52]. In PD genetic animal models, dysfunctional astrocytes were observed. Specifically, in PARKIN and PINK1 knockout mice subjected to active EAE, an animal model of neuroinflammation characterized by disrupted BBB, there was a reduced number of astrocytes cells were observed during the later, chronic stages of the disease, especially in aged mice [24,26]. These findings indicate that under conditions of neuroinflammation and BBB dysfunction, the absence of these genes exacerbates both peripheral and CNS inflammation, highlighting the significance of the immune system and the crosstalk between the periphery and CNS in PD.
In a three-dimensional model replicating the BBB, it was noted that astrocytes carrying the LRRK2 G2019S mutation displayed a pro-inflammatory profile and induced changes in the morphology and function of brain blood vessels [46]. Further examination of postmortem human brain tissue confirmed that the vascular characteristics observed in the in vitro model closely corresponded to alterations seen in the brains of individuals with sporadic PD [46].
While it remains uncertain whether BBB dysfunction is an early event or a consequence of the primary insult in these diseases, it appears that astrocytes play a pivotal role in connecting the pathological processes occurring in the CNS and the periphery through a compromised BBB.
4. Peripheral and CNS immune responses in PD
The dysregulation of both cellular and humoral immune responses in peripheral tissues has been observed in patients with PD across various age groups, disease durations, and severity of motor or psychiatric symptoms. Research has shown that monocytes derived from the peripheral blood of individuals with PD exhibit heightened phagocytic activity when compared to monocytes from control subjects [53]. Additionally, the treatment of PD patient-derived red blood cell-derived extracellular vesicles has been associated with inflammatory sensitization of human monocytes and an increase in the expression of LRRK2 [54].
Interestingly, the expression of TLRs in peripheral immune cells also appears to be implicated in PD pathogenesis. In women with PD, there is a decrease in TLR4 expression, while the expression of TLR2 seems to correlate with the severity of motor symptoms [55]. Notably, monocytes possess the ability to degrade α-syn through their lysosomal system, and mutations in the GBA gene, which impair lysosomal function, are associated with an elevated risk of PD [56]. Furthermore, peripheral monocytes can act as antigen-presenting cells by expressing MHC class I and class II molecules, some of which have been linked to PD [53].
Moreover, distinctions in peripheral immune cell populations, including CD8 T-cells and natural killer (NK) cells in peripheral blood, have been identified between individuals with early-onset PD and those with late-onset PD, defined by a disease duration of less or more than five years, respectively [57]. NK cells, which are recognized as the frontline responders within the immune system, play a pivotal role in promoting immune defense mechanisms through cytotoxicity and the secretion of cytokines. In post-mortem examinations of PD patients, NK cells were found in the substantia nigra [58], and there was an increase in the percentage of NK cells within the CNS parenchyma preceding dopaminergic neuronal degeneration. These findings suggest that NK cells migrate from the periphery into the brain, potentially targeting dysfunctional dopaminergic neurons and contributing to their demise during the progression of PD. The interplay between peripheral and CNS immune responses is a multifaceted aspect of PD pathophysiology. Dysregulated immune responses in the periphery, involving monocytes, TLRs, and specific immune cell populations such as NK cells, can impact the development and progression of PD.
Anomalies in the adaptive immune response affecting peripheral blood lymphocytes have been implicated in the pathogenesis of PD. Research has demonstrated notable differences in the immune profiles of PD patients when compared to healthy controls, shedding light on potential immune mechanisms underlying the disease. Specifically, individuals with PD exhibit a significantly reduced proportion of CD3 T-cells in their peripheral blood in comparison to control subjects, leading to an altered CD4 to CD8 T-cell ratio [59]. This observation highlights an abnormality in T-cell populations in PD patients, which is of considerable interest in understanding the disease's immune aspect. Furthermore, the peripheral immune profile seen in PD patients deviates from what is typically observed in an older population. Unlike the expected CD8 T-cell replicative senescence associated with normal aging, this phenomenon is notably absent in PD patients [60]. This atypical immune profile suggests that there may be unique immune mechanisms at play in PD, distinct from the immunological changes seen in the aging process. Intriguingly, peripheral CD4 and CD8 T-cells in PD patients have been found to produce Th1/Th2 cytokines in response to α-syn, suggesting the presence of a chronic memory T-cell response in PD, potentially contributing to the ongoing neuroinflammation [14]. Moreover, a recent animal study utilizing an adeno-associated viral model to induce α-syn overexpression in the substantia nigra pars compacta of mice has revealed that α-synuclein overexpression in the mouse midbrain leads to the infiltration of both CD8 and CD4 T-cells into the CNS. Notably, CD4 T cells play a particularly significant role in mediating α-syn-induced myeloid inflammation and neurodegeneration in this context [61]. Additional preclinical animal models have also indicated dysregulation within the CD8 T cell compartment. For instance, in PARKIN and PINK1 knockout mice exposed to peripheral inflammation induced by conditions such as EAE or infection with gram-negative bacteria, a substantial increase in peripheral CD8 T-cell numbers was observed when compared to control animals [24,26,28].
Regarding the CNS immune responses, studies have identified activated microglia in post-mortem brain tissue from patients diagnosed with PD [62]. Brain PET imaging studies have further revealed the presence of activated microglia, which were observed bilaterally in the midbrain, with a more pronounced presence on the side most affected by the disease [63]. In pathological conditions such as neurodegeneration, microglia, which are the resident macrophages of the CNS, respond promptly to disruptions in brain homeostasis. They migrate to the site of injury and release both pro-inflammatory and anti-inflammatory factors. Microglia also play a crucial role in activating astrocytes [64]. In vitro studies have shown that microglia phagocytose oligomeric α-syn, resulting in the activation of a pro-inflammatory phenotype in these cells. α-syn appears to influence microglial activation through TLRs and other proteins, such as integrin CD11b [65].
Recent research utilizing a seeding/spreading model of α-syn, which involves the injection of α-syn preformed fibrils into the mouse brain, has demonstrated the significant role of microglia in the pathogenesis of PD. Notably, it has been shown that neurodegeneration and microgliosis can occur independently of the formation of α-syn inclusions [66]. It appears that the early stages of PD progression are more likely driven by microglia, activated by oligomeric α-syn [66]. In a genetic mouse model using PARKIN knockout mice, neuroinflammation induced by active EAE leads to a robust activation of M1 microglia in the brains of knockout mice compared to wild-type controls [26]. It's worth noting that the autosomal recessive form of PD is characterized by the absence of Lewy bodies [67], suggesting that mitochondrial impairment can also trigger a microglial inflammatory response. Moreover, in the same genetic model, it was detected a higher number of CD8 T-cells in the brain of knockout mice compared to controls. In PD, there is an increased presence of CD8 T-cells in regions of the brain associated with the disease. Furthermore, there is a notable expansion of clonally distinct terminal effector CD8 T-cells within the CSF of individuals with PD [68].
4. Gut microbiota, infections, and mitochondrial dysfunction in PD
Although the origin of Lewy bodies remains unclear, data from human and animal studies have demonstrated the involvement of numerous neurotropic and non-neurotropic bacterial and viral agents in PD [69], indicating the importance of peripheral and CNS inflammation in the onset or progression of the disease.
One of the most relevant theories, the Braak’s hypothesis, suggested that an enteric nervous system lesion, triggered by pathogens, can promote α-syn aggregation and subsequently spread from the gut to the CNS via the vagus nerve [70]. According to this model, sporadic PD could be caused by a pathogen entering the CNS via the gut and anterior olfactory structures.
Clinical and neuropathological evidence has reported that alterations in intestinal permeability, also known as leaky gut syndrome, may be associated with changes in the composition of the intestinal microbiota and microbial metabolites, and are related to the accumulation of intestinal α-syn [71].
Increased intestinal permeability and translocation of bacteria and inflammatory bacterial products may lead to an overstimulation of the innate immune system, inflammation, and oxidative stress in the gastrointestinal tract, thus initiating the aggregation and accumulation of α-syn in the enteric nervous system [72]. Consequently, overstimulation of the innate immune system in the gut can produce systemic inflammation and neuroinflammation.
The prodromal phase of PD occurs several years before the typical motor symptoms and begins in extranigral structures, including the olfactory bulb or brainstem nuclei [73]. Indeed, hyposmia, an important non-motor symptom of PD that predicts disease progression, it thought to be related to α-syn aggregates in central olfactory structures [74]. It is possible that during this prodromal phase an infectious agent continues to replicate, triggering the immune response that breaks immune tolerance years later.
Age is the main risk factor for the development and progression of PD; is it known that age-related changes in central thymic tolerance can cause a breakdown of self-tolerance [75], with a T-cell compartment in older individuals mounting weak innate and adaptive immune responses to newly encountered pathogens. Indeed, not only has the role of infection become plausible due to Braak’s hypothesis, but cases of PD and parkinsonism have been described following virus infections, including the more recent severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [76].
Viral and bacterial infections interfere with peripheral tolerance through the induction of CD8 T- cells, whose functionality decreases with age [77]. Animal models have demonstrated age-associated changes in CD8 and DCs in mice lacking mitophagy-related genes [24]. Furthermore, age-related increased oxidative stress and impaired energy production may make neurons vulnerable to toxicity from infectious agents [78].
In addition, inflammation driven by dysfunctional mitochondria can occur in the context of aging, as mitochondria play a critical role during the proinflammatory response against pathogenic infections [79], and neuroinflammation-related mitochondrial dysfunction occurs in different neurodegenerative disorders such as PD [80].
Mitochondrial dysfunction is acknowledged as a contributor to both idiopathic and monogenic forms of PD. While it's still debated whether mitochondrial dysfunction is an initial cause or a consequence of PD, dysfunctional lysosomal degradation, and the resulting accumulation of α-syn are key pathological features [81]. Moreover, the activation of neuroinflammatory processes through the recognition of mitochondrial-derived damage-associated molecular patterns (mtDAMPs) by microglia plays a significant role in this context [82]. Mitochondria play diverse roles in immune responses, influencing metabolic pathways that can modulate immune cell activity, triggering neuroinflammatory responses.
For instance, the absence of mitophagy-related proteins worsens the acute inflammation caused by several stimuli including bacterial infection and LPS. Intestinal infection with Gram-negative bacteria in PINK1 knockout mice elicits the establishment of cytotoxic CD8 T-cells in the periphery and in the brain, highlighting the relevance of the gut–brain axis in the PD [28].
Swine fever virus infection stimulates PARKIN and PINK1 expression and mitochondrial translocation, leading to mitochondrial fission and increased mitophagy [83]; hepatitis B protein recruits PARKIN to destroy depolarized mitochondria by regulating PINK1 expression [84]; SARS-CoV2 impairs intracellular signaling and affects mitochondrial biogenesis [85]. Several intracellular bacteria, such as mycobacteria, can act as virulence factors by modulating mitochondrial physiology for bacterial survival and immune evasion within host cells by inducing macrophage apoptosis via a mitochondrial pathway in macrophages [86].
All this evidence shows that deranged mitochondrial activity is involved in inflammation and the dysfunctional response to infection; however, very little is known about the molecular mechanism underlying the change in mitochondrial function in response to pathogens when inflammation occurs in the brain (Figure 2).
Mitochondrial DAMPs, owing to their resemblance to ancient prokaryotes and the similarity they share with pathogen-associated molecular patterns (PAMPs) presented by infectious bacteria, pose a set of challenges to the host immune system. These challenges may result in the immune system responding similarly to mitochondrial DAMPs as it would to a bacterial infection. When mitochondrial quality control pathways fail, it leads to the accumulation of DAMPs, subsequently activating multiple response pathways [87]. These include the TLR9 receptor pathway, the AIM2 and NLRP3 inflammasomes, as well as the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway[87]. This pro-inflammatory stimulus promotes the expression of antimicrobial peptides and virus-targeting interferon (IFN) proteins. It is noteworthy that a-syn exhibit several characteristics akin to antimicrobial peptides [88].
Regarding the association between bacteria and PD, polymorphisms in the LRRK2 and PARK7 genes lead to increased susceptibility to PD and confer shared effects on the risk of Crohn's disease, a human inflammatory bowel disease [89]. These same genetic defects are associated with susceptibility to mycobacteria, including Mycobacteria tuberculosis and Mycobacterium avium subsp. paratuberculosis [90]. M. tuberculosis is the causative agent of human tuberculosis and could induce neuroinflammation in astrocytes of PD-related brain regions in an LRRK2-dependent manner [91]. M. paratuberculosis is the causative agent of paratuberculosis in ruminants and a suspected causative agent of Crohn's disease in humans [92]. It has been hypothesized that PD-associated LRRK2 and PARK2 defects result in a permissive environment for M. paratuberculosis infection and ineffective xerophagy [93], suggesting that, starting as an enteric infection, M. paratuberculosis, via the vagus nerve, initiates a pathological process that results in targeted CNS neuroinvasion. On the other hand, studies in animal models have shown that different types of Bacillus Calmette-Guerin vaccine can confer non-specific neuroprotection and therapeutic benefits in PD by inducing specific regulatory T (Treg) responses and reducing microglia proliferation and activation [94]. Interestingly, rifampin, an antibiotic used for the treatment mycobacterial infections, can inhibit microglial inflammation and neurodegeneration induced by α-syn fibrillary aggregates [95].
Delving into the interactions among infections, immune reactions, and PD has the potential to unveil underlying mechanisms that contribute to the initiation and advancement of the disease.
5. Future perspective
Understanding the potential role of the immune system in the neurodegenerative processes observed in individuals with PD presents a wide range of opportunities for developing novel treatment approaches. There is significant interest in exploring various strategies, including immunomodulatory drugs and therapies targeting α-syn, to address the complex interplay between neuroinflammation, immunity, and PD pathogenesis.
One avenue of investigation involves examining drugs capable of dampening neuroinflammation, such as non-steroidal anti-inflammatory drugs (NSAIDs) and molecules designed to target specific proinflammatory pathways, such as TNF-α inhibitors. These compounds have shown promise in preclinical and clinical studies for their potential to mitigate neuroinflammation in PD. However, their efficacy remains a subject of ongoing debate. The challenge lies in striking the delicate balance between reducing neuroinflammation and preserving essential immune functions. Fully suppressing the immune response could leave PD patients vulnerable to infections, emphasizing the need for careful consideration when employing such treatments.
Another potential therapeutic strategy involves bolstering the function of Tregs. This can be achieved, for instance, through co-transplantation of autologous Treg cells as a part of cell therapy [96]. This approach holds promise in controlling uncontrolled innate and adaptive immune responses associated with PD.
Additionally, research efforts are focused on developing therapies aimed at reducing the accumulation of α-syn. Vaccines designed to stimulate an immune response against aggregated α-syn, such as α-synuclein mimetic vaccines PD01A and PD03A, are currently in development [97]. These vaccines have the potential to target and clear α-synuclein aggregates, potentially slowing disease progression.
The emerging concept of the gut-brain axis has garnered attention in PD research. Probiotics with neuroprotective properties are undergoing clinical trials [98] [99],. These probiotics are designed to modulate the gut microbiome in ways that influence immune responses, potentially impacting the neuroinflammatory processes that contribute to PD pathology.
Considering the heterogeneity of PD, with variations in clinical presentations and underlying mechanisms among patients, determining the optimal timing for intervention is a critical consideration. Initiating anti-inflammatory treatments either too early or too late in the disease course could yield different outcomes. Therefore, a personalized medicine approach tailored to individual patients based on their specific immune profiles and disease characteristics is a complex but essential endeavor in the pursuit of effective PD treatments.
Author Contributions
Conceptualization, D.C and N.H; writing, D.C.; supervision, T.H. and N.H.; funding acquisition, D.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI (JP23K14675) and PRIN 2022 (202238WEHT) to D.C.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lauritsen, J.; Romero-Ramos, M. The systemic immune response in Parkinson's disease: focus on the peripheral immune component. Trends Neurosci. 2023, 46, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.; Ho, P.W.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson's disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef]
- Abdi, I.Y.; Ghanem, S.S.; El-Agnaf, O.M. Immune-related biomarkers for Parkinson's disease. Neurobiol. Dis. 2022, 170, 105771. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Arakawa, S.; Sato, S.; Ishikawa, K.I.; Taniguchi, D.; Sakurai, H.T.; Honda, S.; Hiraoka, Y.; Ono, M.; Akamatsu, W.; et al. Involvement of casein kinase 1 epsilon/delta (Csnk1e/d) in the pathogenesis of familial Parkinson's disease caused by CHCHD2. EMBO Mol. Med. 2023, 15, e17451. [Google Scholar] [CrossRef] [PubMed]
- Okuzumi, A.; Hatano, T.; Matsumoto, G.; Nojiri, S.; Ueno, S.I.; Imamichi-Tatano, Y.; Kimura, H.; Kakuta, S.; Kondo, A.; Fukuhara, T.; et al. Propagative alpha-synuclein seeds as serum biomarkers for synucleinopathies. Nat. Med. 2023, 29, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Marotta, N.P.; Ara, J.; Uemura, N.; Lougee, M.G.; Meymand, E.S.; Zhang, B.; Petersson, E.J.; Trojanowski, J.Q.; Lee, V.M. Alpha-synuclein from patient Lewy bodies exhibits distinct pathological activity that can be propagated in vitro. Acta Neuropathol. Commun. 2021, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernandez, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Arguero-Sanchez, R.; Schule, B.; Guerra-Crespo, M. Alpha-Synuclein Physiology and Pathology: A Perspective on Cellular Structures and Organelles. Front. Neurosci. 2019, 13, 1399. [Google Scholar] [CrossRef]
- Reyes, J.F.; Ekmark-Lewen, S.; Perdiki, M.; Klingstedt, T.; Hoffmann, A.; Wiechec, E.; Nilsson, P.; Nilsson, K.P.R.; Alafuzoff, I.; Ingelsson, M.; et al. Accumulation of alpha-synuclein within the liver, potential role in the clearance of brain pathology associated with Parkinson's disease. Acta Neuropathol. Commun. 2021, 9, 46. [Google Scholar] [CrossRef]
- Mendritzki, S.; Schmidt, S.; Sczepan, T.; Zhu, X.R.; Segelcke, D.; Lubbert, H. Spinal cord pathology in alpha-synuclein transgenic mice. Parkinsons Dis. 2010, 2010, 375462. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Mikkelsen, T.W.; Alstrup, A.K.O.; Tamguney, G.; Karlsson, P.; Terkelsen, A.J.; Nyengaard, J.R.; Jensen, P.H.; Borghammer, P. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain 2021, 144, 1853–1868. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic alpha-Synuclein from the Gut to the Brain Models Parkinson's Disease. Neuron 2019, 103, 627–641.e627. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, X.; Zeng, W.; Zhang, X.; Yang, X.; Xu, Y.; Liu, K.; Zhang, Z.; Xu, Y.; Cao, X. Dual Effects: Intrastriatal Injection of alpha-syn N103/tau N368 Preformed Fibrils Promotes Endogenous alpha-synuclein Aggregates in the Proximal Colon. J. Parkinsons Dis. 2022, 12, 2097–2116. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson's disease recognize alpha-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. alpha-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson's disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef]
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular genetics of Parkinson's disease: Contributions and global trends. J. Hum. Genet. 2023, 68, 125–130. [Google Scholar] [CrossRef]
- Dulski, J.; Uitti, R.J.; Ross, O.A.; Wszolek, Z.K. Genetic architecture of Parkinson's disease subtypes - Review of the literature. Front. Aging Neurosci. 2022, 14, 1023574. [Google Scholar] [CrossRef]
- Garretti, F.; Monahan, C.; Sloan, N.; Bergen, J.; Shahriar, S.; Kim, S.W.; Sette, A.; Cutforth, T.; Kanter, E.; Agalliu, D.; et al. Interaction of an alpha-synuclein epitope with HLA-DRB1( *)15:01 triggers enteric features in mice reminiscent of prodromal Parkinson's disease. Neuron 2023. [Google Scholar] [CrossRef] [PubMed]
- Kannarkat, G.T.; Cook, D.A.; Lee, J.K.; Chang, J.; Chung, J.; Sandy, E.; Paul, K.C.; Ritz, B.; Bronstein, J.; Factor, S.A.; et al. Common Genetic Variant Association with Altered HLA Expression, Synergy with Pyrethroid Exposure, and Risk for Parkinson's Disease: An Observational and Case-Control Study. NPJ Parkinsons Dis. 2015, 1, 15002. [Google Scholar] [CrossRef]
- Magalhaes, J.D.; Cardoso, S.M. Mitochondrial signaling on innate immunity activation in Parkinson disease. Curr. Opin. Neurobiol. 2023, 78, 102664. [Google Scholar] [CrossRef]
- Imberechts, D.; Kinnart, I.; Wauters, F.; Terbeek, J.; Manders, L.; Wierda, K.; Eggermont, K.; Madeiro, R.F.; Sue, C.; Verfaillie, C.; et al. DJ-1 is an essential downstream mediator in PINK1/parkin-dependent mitophagy. Brain 2022, 145, 4368–4384. [Google Scholar] [CrossRef]
- Guedes, B.F.S.; Cardoso, S.M.; Esteves, A.R. The Impact of microRNAs on Mitochondrial Function and Immunity: Relevance to Parkinson's Disease. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Cossu, D.; Yokoyama, K.; Sechi, L.A.; Hattori, N. Potential of PINK1 and PARKIN Proteins as Biomarkers for Active Multiple Sclerosis: A Japanese Cohort Study. Front. Immunol. 2021, 12, 681386. [Google Scholar] [CrossRef] [PubMed]
- Cossu, D.; Yokoyama, K.; Sato, S.; Noda, S.; Sakanishi, T.; Sechi, L.A.; Hattori, N. Age related immune modulation of experimental autoimmune encephalomyelitis in PINK1 knockout mice. Front. Immunol. 2022, 13, 1036680. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson's Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef]
- Cossu, D.; Yokoyama, K.; Sato, S.; Noda, S.; Sechi, L.A.; Hattori, N. PARKIN modifies peripheral immune response and increases neuroinflammation in active experimental autoimmune encephalomyelitis (EAE). J. Neuroimmunol. 2021, 359, 577694. [Google Scholar] [CrossRef]
- Sun, L.; Shen, R.; Agnihotri, S.K.; Chen, Y.; Huang, Z.; Bueler, H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci. Rep. 2018, 8, 383. [Google Scholar] [CrossRef]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson's disease-like symptoms in Pink1(-/-) mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef]
- Zhao, N.; Li, Y.; Wang, C.; Xue, Y.; Peng, L.; Wang, T.; Zhao, Y.; Xu, G.; Yu, S. DJ-1 activates the Atg5-Atg12-Atg16L1 complex via Sirt1 to influence microglial polarization and alleviate cerebral ischemia/reperfusion-induced inflammatory injury. Neurochem. Int. 2022, 157, 105341. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Wang, J.; Yang, B.; He, Q.; Weng, Q. Role of DJ-1 in Immune and Inflammatory Diseases. Front. Immunol. 2020, 11, 994. [Google Scholar] [CrossRef]
- Zeng, N.; Capelle, C.M.; Baron, A.; Kobayashi, T.; Cire, S.; Tslaf, V.; Leonard, C.; Coowar, D.; Koseki, H.; Westendorf, A.M.; et al. DJ-1 depletion prevents immunoaging in T-cell compartments. EMBO Rep. 2022, 23, e53302. [Google Scholar] [CrossRef]
- Booms, A.; Coetzee, G.A. Functions of Intracellular Alpha-Synuclein in Microglia: Implications for Parkinson's Disease Risk. Front. Cell Neurosci. 2021, 15, 759571. [Google Scholar] [CrossRef]
- Kim, C.; Lee, H.J.; Masliah, E.; Lee, S.J. Non-cell-autonomous Neurotoxicity of alpha-synuclein Through Microglial Toll-like Receptor 2. Exp. Neurobiol. 2016, 25, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Shameli, A.; Xiao, W.; Zheng, Y.; Shyu, S.; Sumodi, J.; Meyerson, H.J.; Harding, C.V.; Maitta, R.W. A critical role for alpha-synuclein in development and function of T lymphocytes. Immunobiology 2016, 221, 333–340. [Google Scholar] [CrossRef]
- Xiao, W.; Shameli, A.; Harding, C.V.; Meyerson, H.J.; Maitta, R.W. Late stages of hematopoiesis and B cell lymphopoiesis are regulated by alpha-synuclein, a key player in Parkinson's disease. Immunobiology 2014, 219, 836–844. [Google Scholar] [CrossRef]
- Ye, S.Y.; Apple, J.E.; Ren, X.; Tang, F.L.; Yao, L.L.; Wang, Y.G.; Mei, L.; Zhou, Y.G.; Xiong, W.C. Microglial VPS35 deficiency regulates microglial polarization and decreases ischemic stroke-induced damage in the cortex. J. Neuroinflammation 2019, 16, 235. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Atashrazm, F.; Hammond, D.; Perera, G.; Dobson-Stone, C.; Mueller, N.; Pickford, R.; Kim, W.S.; Kwok, J.B.; Lewis, S.J.G.; Halliday, G.M.; et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson's disease. Sci. Rep. 2018, 8, 15446. [Google Scholar] [CrossRef]
- Sanyal, A.; DeAndrade, M.P.; Novis, H.S.; Lin, S.; Chang, J.; Lengacher, N.; Tomlinson, J.J.; Tansey, M.G.; LaVoie, M.J. Lysosome and Inflammatory Defects in GBA1-Mutant Astrocytes Are Normalized by LRRK2 Inhibition. Mov. Disord. 2020, 35, 760–773. [Google Scholar] [CrossRef]
- Mullin, S.; Stokholm, M.G.; Hughes, D.; Mehta, A.; Parbo, P.; Hinz, R.; Pavese, N.; Brooks, D.J.; Schapira, A.H.V. Brain Microglial Activation Increased in Glucocerebrosidase (GBA) Mutation Carriers without Parkinson's disease. Mov. Disord. 2021, 36, 774–779. [Google Scholar] [CrossRef]
- Wallings, R.L.; Herrick, M.K.; Tansey, M.G. LRRK2 at the Interface Between Peripheral and Central Immune Function in Parkinson's. Front. Neurosci. 2020, 14, 443. [Google Scholar] [CrossRef]
- Ho, D.H.; Nam, D.; Seo, M.; Park, S.W.; Seol, W.; Son, I. LRRK2 Inhibition Mitigates the Neuroinflammation Caused by TLR2-Specific alpha-Synuclein and Alleviates Neuroinflammation-Derived Dopaminergic Neuronal Loss. Cells 2022, 11. [Google Scholar] [CrossRef]
- Mutti, V.; Carini, G.; Filippini, A.; Castrezzati, S.; Giugno, L.; Gennarelli, M.; Russo, I. LRRK2 Kinase Inhibition Attenuates Neuroinflammation and Cytotoxicity in Animal Models of Alzheimer's and Parkinson's Disease-Related Neuroinflammation. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Kozina, E.; Byrne, M.; Smeyne, R.J. Mutant LRRK2 in lymphocytes regulates neurodegeneration via IL-6 in an inflammatory model of Parkinson's disease. NPJ Parkinsons Dis. 2022, 8, 24. [Google Scholar] [CrossRef]
- Zhang, S.; Gan, L.; Cao, F.; Wang, H.; Gong, P.; Ma, C.; Ren, L.; Lin, Y.; Lin, X. The barrier and interface mechanisms of the brain barrier, and brain drug delivery. Brain Res. Bull. 2022, 190, 69–83. [Google Scholar] [CrossRef]
- de Rus Jacquet, A.; Alpaugh, M.; Denis, H.L.; Tancredi, J.L.; Boutin, M.; Decaestecker, J.; Beauparlant, C.; Herrmann, L.; Saint-Pierre, M.; Parent, M.; et al. The contribution of inflammatory astrocytes to BBB impairments in a brain-chip model of Parkinson's disease. Nat. Commun. 2023, 14, 3651. [Google Scholar] [CrossRef]
- Wu, Y.C.; Sonninen, T.M.; Peltonen, S.; Koistinaho, J.; Lehtonen, S. Blood-Brain Barrier and Neurodegenerative Diseases-Modeling with iPSC-Derived Brain Cells. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J.; Wang, J.; He, L.; Lai, H.; Zhang, T.; Wang, X.; Li, W. Mitochondrial oxidative stress in brain microvascular endothelial cells: Triggering blood-brain barrier disruption. Mitochondrion 2023, 69, 71–82. [Google Scholar] [CrossRef]
- Kim, H.; Leng, K.; Park, J.; Sorets, A.G.; Kim, S.; Shostak, A.; Embalabala, R.J.; Mlouk, K.; Katdare, K.A.; Rose, I.V.L.; et al. Reactive astrocytes transduce inflammation in a blood-brain barrier model through a TNF-STAT3 signaling axis and secretion of alpha 1-antichymotrypsin. Nat. Commun. 2022, 13, 6581. [Google Scholar] [CrossRef] [PubMed]
- Chavarria, C.; Ivagnes, R.; Souza, J.M. Extracellular Alpha-Synuclein: Mechanisms for Glial Cell Internalization and Activation. Biomolecules 2022, 12. [Google Scholar] [CrossRef]
- Kano, M.; Takanashi, M.; Oyama, G.; Yoritaka, A.; Hatano, T.; Shiba-Fukushima, K.; Nagai, M.; Nishiyama, K.; Hasegawa, K.; Inoshita, T.; et al. Reduced astrocytic reactivity in human brains and midbrain organoids with PRKN mutations. NPJ Parkinsons Dis. 2020, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.J.; Eun, J.H.; Kim, B.G.; Jou, I.; Park, S.M.; Joe, E.H. A Parkinson's disease gene, DJ-1, repairs brain injury through Sox9 stabilization and astrogliosis. Glia 2018, 66, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Wijeyekoon, R.S.; Kronenberg-Versteeg, D.; Scott, K.M.; Hayat, S.; Jones, J.L.; Clatworthy, M.R.; Floto, R.A.; Barker, R.A.; Williams-Gray, C.H. Monocyte Function in Parkinson's Disease and the Impact of Autologous Serum on Phagocytosis. Front. Neurol. 2018, 9, 870. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chan, R.B.; Cai, Z.; Liu, X.; Wu, Y.; Yu, Z.; Feng, T.; Yang, Y.; Zhang, J. alpha-Synuclein-containing erythrocytic extracellular vesicles: essential contributors to hyperactivation of monocytes in Parkinson's disease. J. Neuroinflammation 2022, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Konstantin Nissen, S.; Farmen, K.; Carstensen, M.; Schulte, C.; Goldeck, D.; Brockmann, K.; Romero-Ramos, M. Changes in CD163+, CD11b+, and CCR2+ peripheral monocytes relate to Parkinson's disease and cognition. Brain Behav. Immun. 2022, 101, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, D.; Cisternas-Olmedo, M.; Arcos, J.; Nassif, M.; Vidal, R.L. Contribution of Autophagy-Lysosomal Pathway in the Exosomal Secretion of Alpha-Synuclein and Its Impact in the Progression of Parkinson's Disease. Front. Mol. Neurosci. 2022, 15, 805087. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Dai, S.B.; Jiang, S.S.; Yang, W.Y.; Yan, Y.Q.; Lin, Z.H.; Dong, J.X.; Liu, Y.; Zheng, R.; Chen, Y.; et al. Specific immune status in Parkinson's disease at different ages of onset. NPJ Parkinsons Dis. 2022, 8, 5. [Google Scholar] [CrossRef]
- Earls, R.H.; Lee, J.K. The role of natural killer cells in Parkinson's disease. Exp. Mol. Med. 2020, 52, 1517–1525. [Google Scholar] [CrossRef]
- Sun, C.; Zhao, Z.; Yu, W.; Mo, M.; Song, C.; Si, Y.; Liu, Y. Abnormal subpopulations of peripheral blood lymphocytes are involved in Parkinson's disease. Ann. Transl. Med. 2019, 7, 637. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Wijeyekoon, R.S.; Scott, K.M.; Hayat, S.; Barker, R.A.; Jones, J.L. Abnormalities of age-related T cell senescence in Parkinson's disease. J. Neuroinflammation 2018, 15, 166. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.P.; Schonhoff, A.M.; Jurkuvenaite, A.; Gallups, N.J.; Standaert, D.G.; Harms, A.S. CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson's disease. Brain 2021, 144, 2047–2059. [Google Scholar] [CrossRef]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson's Disease. Cells 2023, 12. [Google Scholar] [CrossRef]
- Lavisse, S.; Goutal, S.; Wimberley, C.; Tonietto, M.; Bottlaender, M.; Gervais, P.; Kuhnast, B.; Peyronneau, M.A.; Barret, O.; Lagarde, J.; et al. Increased microglial activation in patients with Parkinson disease using [(18)F]-DPA714 TSPO PET imaging. Parkinsonism Relat. Disord. 2021, 82, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and astrocyte dysfunction in parkinson's disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates alpha-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.; Jurgens-Wemheuer, W.; Uriarte Huarte, O.; Michelucci, A.; Masuch, A.; Brioschi, S.; Weihofen, A.; Koncina, E.; Coowar, D.; Heurtaux, T.; et al. Neurodegeneration and neuroinflammation are linked, but independent of alpha-synuclein inclusions, in a seeding/spreading mouse model of Parkinson's disease. Glia 2022, 70, 935–960. [Google Scholar] [CrossRef]
- Nishioka, K.; Imai, Y.; Yoshino, H.; Li, Y.; Funayama, M.; Hattori, N. Clinical Manifestations and Molecular Backgrounds of Parkinson's Disease Regarding Genes Identified From Familial and Population Studies. Front. Neurol. 2022, 13, 764917. [Google Scholar] [CrossRef]
- Wang, P.; Yao, L.; Luo, M.; Zhou, W.; Jin, X.; Xu, Z.; Yan, S.; Li, Y.; Xu, C.; Cheng, R.; et al. Single-cell transcriptome and TCR profiling reveal activated and expanded T cell populations in Parkinson's disease. Cell Discov. 2021, 7, 52. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Noyce, A.J.; Byrne, M.; Savica, R.; Marras, C. Infection and Risk of Parkinson's Disease. J. Parkinsons Dis. 2021, 11, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna) 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Claudino Dos Santos, J.C.; Lima, M.P.P.; Brito, G.A.C.; Viana, G.S.B. Role of enteric glia and microbiota-gut-brain axis in parkinson disease pathogenesis. Ageing Res. Rev. 2023, 84, 101812. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.L.; Huang, K.X.; Zheng, D.Z.; Lin, D.Y.; Chen, Y.; Jing, X.N.; Liang, Y.R.; Tao, E.X. Alpha-Synuclein Accumulation and Its Phosphorylation in the Enteric Nervous System of Patients Without Neurodegeneration: An Explorative Study. Front. Aging Neurosci. 2020, 12, 575481. [Google Scholar] [CrossRef] [PubMed]
- Simonet, C.; Schrag, A.; Lees, A.J.; Noyce, A.J. The motor prodromes of parkinson's disease: from bedside observation to large-scale application. J. Neurol. 2021, 268, 2099–2108. [Google Scholar] [CrossRef]
- Chen, F.; Liu, W.; Liu, P.; Wang, Z.; Zhou, Y.; Liu, X.; Li, A. alpha-Synuclein aggregation in the olfactory bulb induces olfactory deficits by perturbing granule cells and granular-mitral synaptic transmission. NPJ Parkinsons Dis. 2021, 7, 114. [Google Scholar] [CrossRef]
- Srinivasan, J.; Lancaster, J.N.; Singarapu, N.; Hale, L.P.; Ehrlich, L.I.R.; Richie, E.R. Age-Related Changes in Thymic Central Tolerance. Front. Immunol. 2021, 12, 676236. [Google Scholar] [CrossRef] [PubMed]
- Iacono, S.; Schiro, G.; Davi, C.; Mastrilli, S.; Abbott, M.; Guajana, F.; Arnao, V.; Aridon, P.; Ragonese, P.; Gagliardo, C.; et al. COVID-19 and neurological disorders: what might connect Parkinson's disease to SARS-CoV-2 infection. Front. Neurol. 2023, 14, 1172416. [Google Scholar] [CrossRef]
- Quinn, K.M.; Fox, A.; Harland, K.L.; Russ, B.E.; Li, J.; Nguyen, T.H.O.; Loh, L.; Olshanksy, M.; Naeem, H.; Tsyganov, K.; et al. Age-Related Decline in Primary CD8(+) T Cell Responses Is Associated with the Development of Senescence in Virtual Memory CD8(+) T Cells. Cell Rep. 2018, 23, 3512–3524. [Google Scholar] [CrossRef]
- Olufunmilayo, E.O.; Gerke-Duncan, M.B.; Holsinger, R.M.D. Oxidative Stress and Antioxidants in Neurodegenerative Disorders. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef]
- Pizarro-Galleguillos, B.M.; Kunert, L.; Bruggemann, N.; Prasuhn, J. Neuroinflammation and Mitochondrial Dysfunction in Parkinson's Disease: Connecting Neuroimaging with Pathophysiology. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Xu, J.; Ao, Y.L.; Huang, C.; Song, X.; Zhang, G.; Cui, W.; Wang, Y.; Zhang, X.Q.; Zhang, Z. Harmol promotes alpha-synuclein degradation and improves motor impairment in Parkinson's models via regulating autophagy-lysosome pathway. NPJ Parkinsons Dis. 2022, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Tavares, H.; Beatriz, M.; Mota, S.; Lopes, C. Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders. Cells 2022, 11. [Google Scholar] [CrossRef]
- Gou, H.; Zhao, M.; Xu, H.; Yuan, J.; He, W.; Zhu, M.; Ding, H.; Yi, L.; Chen, J. CSFV induced mitochondrial fission and mitophagy to inhibit apoptosis. Oncotarget 2017, 8, 39382–39400. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Mozzi, A.; Oldani, M.; Forcella, M.E.; Vantaggiato, C.; Cappelletti, G.; Pontremoli, C.; Valenti, F.; Forni, D.; Saresella, M.; Biasin, M.; et al. SARS-CoV-2 ORF3c impairs mitochondrial respiratory metabolism, oxidative stress, and autophagic flux. iScience 2023, 26, 107118. [Google Scholar] [CrossRef]
- Lee, K.I.; Whang, J.; Choi, H.G.; Son, Y.J.; Jeon, H.S.; Back, Y.W.; Park, H.S.; Paik, S.; Park, J.K.; Choi, C.H.; et al. Mycobacterium avium MAV2054 protein induces macrophage apoptosis by targeting mitochondria and reduces intracellular bacterial growth. Sci. Rep. 2016, 6, 37804. [Google Scholar] [CrossRef]
- Singh, B.; Avula, K.; Sufi, S.A.; Parwin, N.; Das, S.; Alam, M.F.; Samantaray, S.; Bankapalli, L.; Rani, A.; Poornima, K.; et al. Defective Mitochondrial Quality Control during Dengue Infection Contributes to Disease Pathogenesis. J. Virol. 2022, 96, e0082822. [Google Scholar] [CrossRef]
- Park, S.C.; Moon, J.C.; Shin, S.Y.; Son, H.; Jung, Y.J.; Kim, N.H.; Kim, Y.M.; Jang, M.K.; Lee, J.R. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem. Biophys. Res. Commun. 2016, 478, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Cabezudo, D.; Baekelandt, V.; Lobbestael, E. Multiple-Hit Hypothesis in Parkinson's Disease: LRRK2 and Inflammation. Front. Neurosci. 2020, 14, 376. [Google Scholar] [CrossRef]
- Manzanillo, P.S.; Ayres, J.S.; Watson, R.O.; Collins, A.C.; Souza, G.; Rae, C.S.; Schneider, D.S.; Nakamura, K.; Shiloh, M.U.; Cox, J.S. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 2013, 501, 512–516. [Google Scholar] [CrossRef]
- Weindel, C.G.; Bell, S.L.; Vail, K.J.; West, K.O.; Patrick, K.L.; Watson, R.O. LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. Elife 2020, 9. [Google Scholar] [CrossRef]
- Cossu, D.; Masala, S.; Frau, J.; Mameli, G.; Marrosu, M.G.; Cocco, E.; Sechi, L.A. Antigenic epitopes of MAP2694 homologous to T-cell receptor gamma-chain are highly recognized in multiple sclerosis Sardinian patients. Mol. Immunol. 2014, 57, 138–140. [Google Scholar] [CrossRef]
- Arru, G.; Caggiu, E.; Paulus, K.; Sechi, G.P.; Mameli, G.; Sechi, L.A. Is there a role for Mycobacterium avium subspecies paratuberculosis in Parkinson's disease? J. Neuroimmunol. 2016, 293, 86–90. [Google Scholar] [CrossRef]
- Cossu, D.; Yokoyama, K.; Sakanishi, T.; Sechi, L.A.; Hattori, N. Bacillus Calmette-Guerin Tokyo-172 vaccine provides age-related neuroprotection in actively induced and spontaneous experimental autoimmune encephalomyelitis models. Clin. Exp. Immunol. 2023, 212, 70–80. [Google Scholar] [CrossRef]
- Cossu, D.; Ruberto, S.; Yokoyama, K.; Hattori, N.; Sechi, L.A. Efficacy of BCG vaccine in animal models of neurological disorders. Vaccine 2022, 40, 432–436. [Google Scholar] [CrossRef]
- Park, T.Y.; Jeon, J.; Lee, N.; Kim, J.; Song, B.; Kim, J.H.; Lee, S.K.; Liu, D.; Cha, Y.; Kim, M.; et al. Co-transplantation of autologous T(reg) cells in a cell therapy for Parkinson's disease. Nature 2023, 619, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Meissner, W.G.; Traon, A.P.; Foubert-Samier, A.; Galabova, G.; Galitzky, M.; Kutzelnigg, A.; Laurens, B.; Luhrs, P.; Medori, R.; Peran, P.; et al. A Phase 1 Randomized Trial of Specific Active alpha-Synuclein Immunotherapies PD01A and PD03A in Multiple System Atrophy. Mov. Disord. 2020, 35, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Tamtaji, O.R.; Taghizadeh, M.; Daneshvar Kakhaki, R.; Kouchaki, E.; Bahmani, F.; Borzabadi, S.; Oryan, S.; Mafi, A.; Asemi, Z. Clinical and metabolic response to probiotic administration in people with Parkinson's disease: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2019, 38, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, H.; Jin, Y.; Yu, J.; Mao, S.; Su, K.P.; Ling, Z.; Liu, J. Probiotic Clostridium butyricum ameliorated motor deficits in a mouse model of Parkinson's disease via gut microbiota-GLP-1 pathway. Brain Behav. Immun. 2021, 91, 703–715. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
potential contributors to immune system dysfunction in Parkinson’s Disease (PD). 1) Genetic variations associated with an increased risk of PD may also influence immune functions. 2) Dysfunction in mitochondria has been linked to both immune system activation and neural degeneration in PD. 3) According to the Braak hypothesis, Lewy body pathologies may initiate in the peripheral regions such as the nose and in the gut. 4) Epidemiological evidence suggests that during the prodromal stage, a combination of factors, including infections, may contribute to the risk of developing PD. 5) Increased leakage of Blood brain barrier (BBB) and Cerebrospinal Fluid Barrier (CSFB) in PD patients and animal models points to potential involvement of peripheral immune response and altered drug efficacy in disease progression. 6) Age-associated changes in the immune system may increase susceptibility to infection and age-acquired autoimmunity.
Figure 1.
potential contributors to immune system dysfunction in Parkinson’s Disease (PD). 1) Genetic variations associated with an increased risk of PD may also influence immune functions. 2) Dysfunction in mitochondria has been linked to both immune system activation and neural degeneration in PD. 3) According to the Braak hypothesis, Lewy body pathologies may initiate in the peripheral regions such as the nose and in the gut. 4) Epidemiological evidence suggests that during the prodromal stage, a combination of factors, including infections, may contribute to the risk of developing PD. 5) Increased leakage of Blood brain barrier (BBB) and Cerebrospinal Fluid Barrier (CSFB) in PD patients and animal models points to potential involvement of peripheral immune response and altered drug efficacy in disease progression. 6) Age-associated changes in the immune system may increase susceptibility to infection and age-acquired autoimmunity.
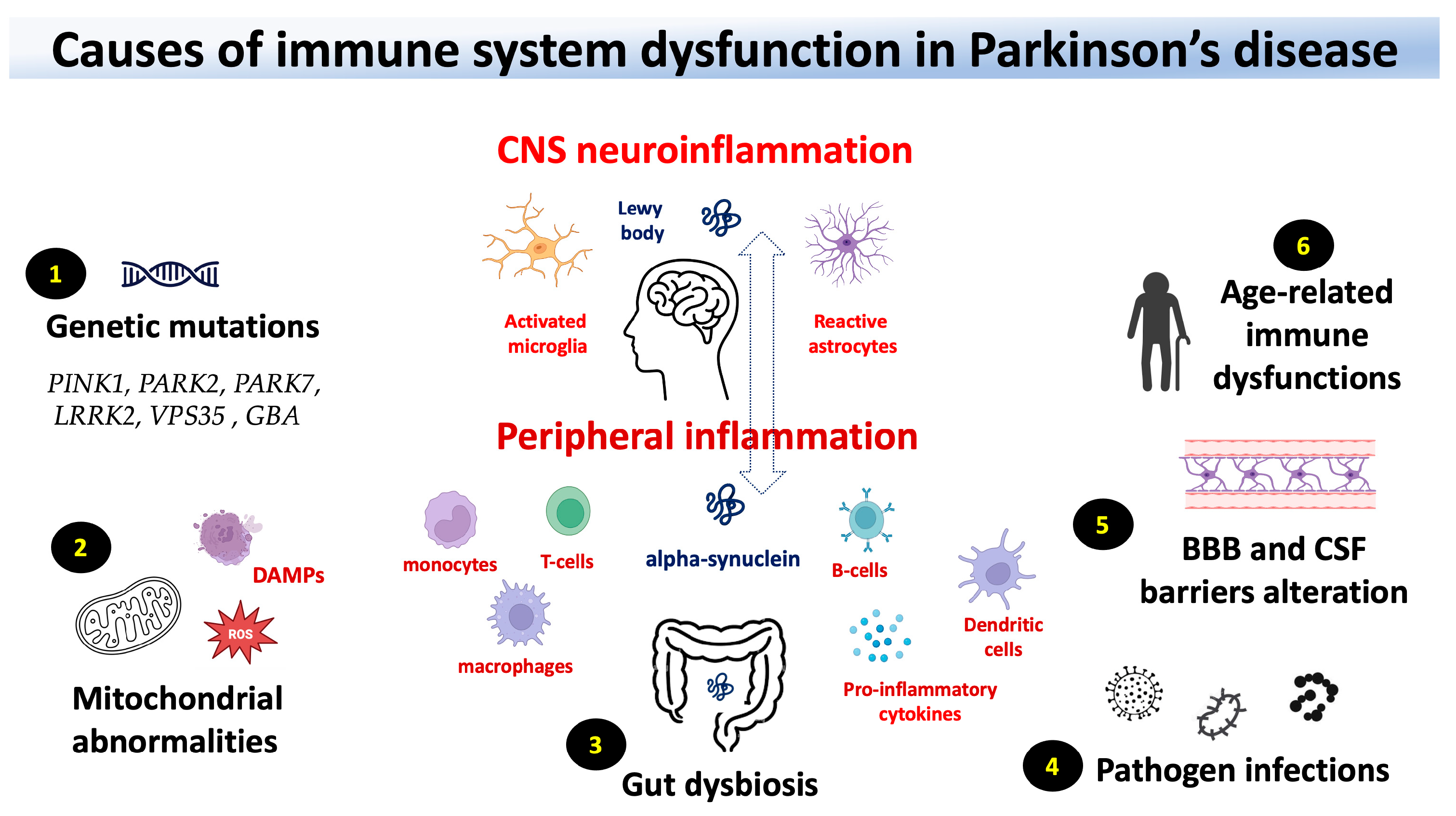
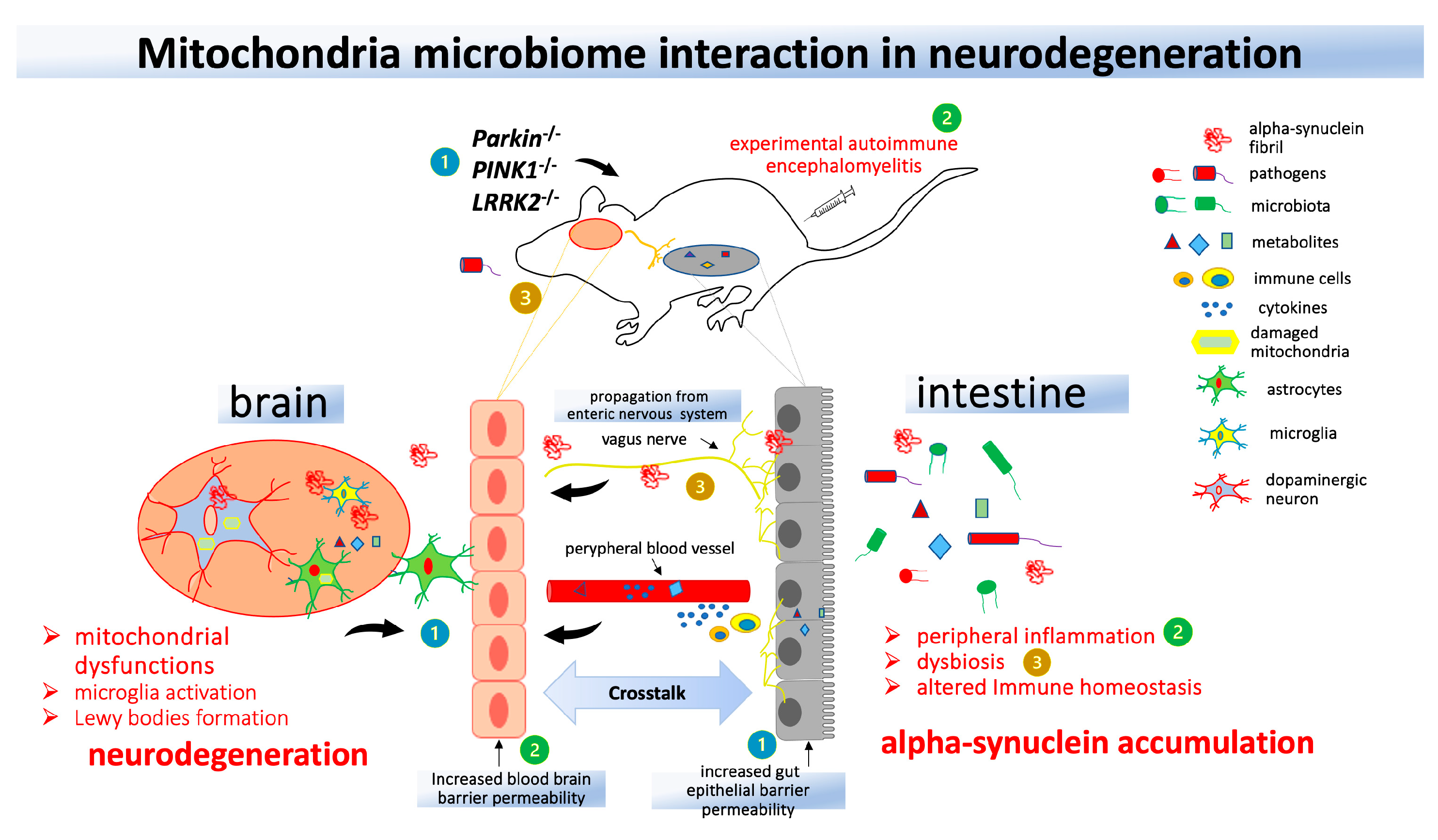
Figure 2.
Interaction between mitochondria and gut in animal models of Parkinson’s Disease (PD). Genetic models of mitochondria knockout animals (1) demonstrate an increase of central nervous system neuroinflammation following the induction of experimental autoimmune encephalomyelitis (2) or administration of lipopolysaccharide or Gram-negative bacteria (3). Mitochondrial dysfunction and peripheral inflammation may contribute to dysbiosis and alpha-synuclein accumulation, which can reach the brain through the vagus nerve and lead to neurodegeneration.
Figure 2.
Interaction between mitochondria and gut in animal models of Parkinson’s Disease (PD). Genetic models of mitochondria knockout animals (1) demonstrate an increase of central nervous system neuroinflammation following the induction of experimental autoimmune encephalomyelitis (2) or administration of lipopolysaccharide or Gram-negative bacteria (3). Mitochondrial dysfunction and peripheral inflammation may contribute to dysbiosis and alpha-synuclein accumulation, which can reach the brain through the vagus nerve and lead to neurodegeneration.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



