
Submitted:
02 November 2023
Posted:
03 November 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The interplay between thrombosis and inflammation, known as thromboinflammation, is a significant pathway driving cardiovascular and autoimmune diseases, as well as COVID-19. Key modulators of this process have emerged as innate immune cells. Strategically positioned to promote thromboinflammation are neutrophils, the most predominant white blood cells in humans. Neutrophils can trigger an organized cell death pathway by releasing decondensed chromatin structures known as neutrophil extracellular traps. These structures are decorated with histones, cytoplasmic, and granular proteins, and have cytotoxic, immunogenic, and prothrombotic effects, which can accelerate disease progression. The activities of PAD4, which catalyses the citrullination of histones, and the neutrophil inflammasome are required for distinct steps leading to extracellular DNA release (NETosis). PAD4 activity has important implications for understanding the processes that drive thromboinflammation by linking the immunological function of neutrophils with the procoagulant and proinflammatory activities of monocytes and platelets. We will discuss how vascular blockages in thromboinflammation occur due to the interaction between neutrophil extracellular traps and ultra-large VWF (von Willebrand Factor). PAD4 activity has important implications for understanding the processes that drive thromboinflammation by linking the immunological function of neutrophils with the procoagulant and proinflammatory activities of monocytes and platelets. It will also review the mechanisms whereby vaso-occlusive events in thrombo-inflammation depend on the interaction of neutrophil extracellular traps with von Willebrand factor and suggest the importance of PAD4 in neutrophil inflammasome assembly and neutrophil extracellular traps in thrombo-inflammatory diseases such as atherosclerosis and CVD.
Keywords:
Thromboinflammation
; atherosclerosis
; neutrophil extracellular traps
; NETosis
; von Willebrand Factor
; neutrophil inflammasome
1. Introduction
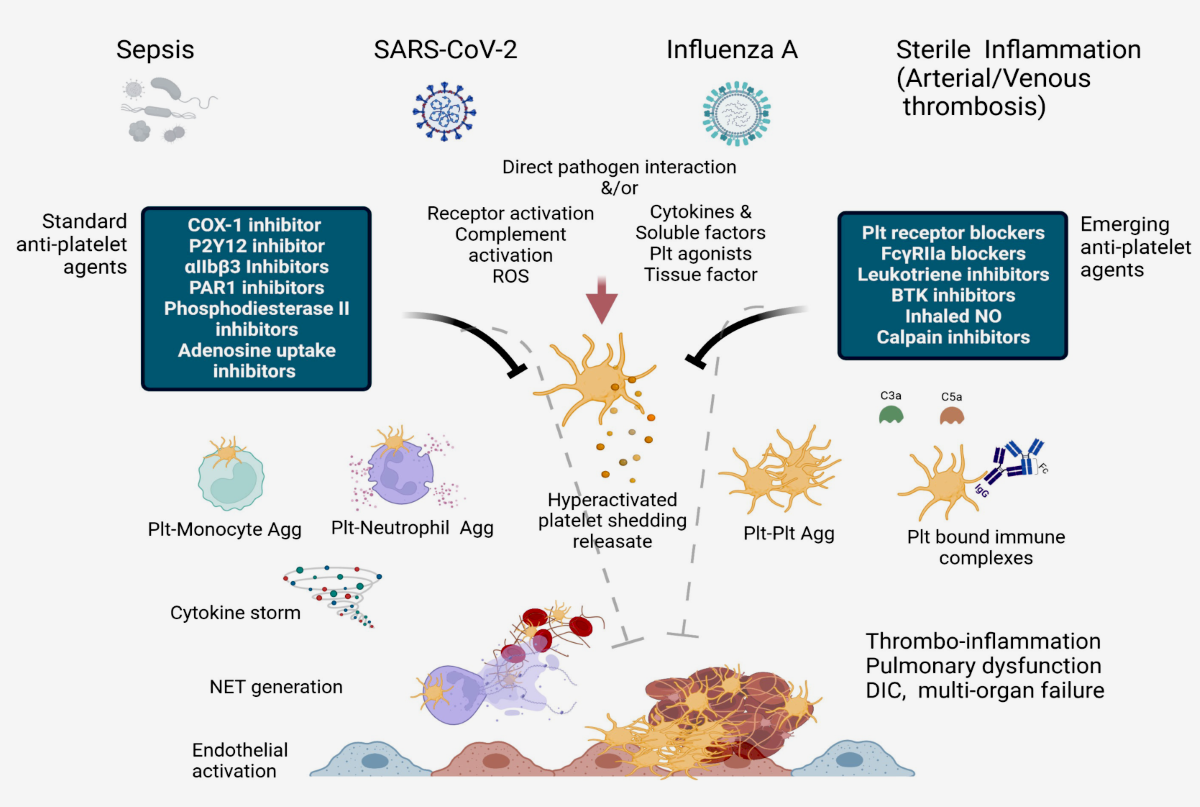
Our understanding of the impact of the inflammatory process in atherogenesis and other chronic diseases has increased significantly since the pioneering work dating back over 2 decades. (1) Thrombotic events, including myocardial infarction after plaque rupture, are now known to involve inflammation and innate immune cells. We previously suggested that platelet activation promotes a procoagulant state that drives inflammation and thrombosis in a vicious cycle. (2,3) Over the last decade, the idea that innate immune cells also contribute to thrombosis has gained recognition. Initially considered a safeguard response against the invasion of pathogens by means of local fibrin accumulation, it is now clear that innate immune cells can also make a significant contribution to sterile pathological thrombosis. (4,5) Thromboinflammation is now increasingly recognised as important therapeutic objective for a number of human disorders. The activation of platelets and immune cells, along with endothelial activation/dysfunction (as shown in the graphical abstract), is key to the concept of thromboinflammation. The result is microvascular thrombosis and ultimately organ dysfunction. (6)
2. Understanding the role of platelets and P-selectin as key actors in thromboinflammation
In thromboinflammation, platelets play an important role. (Figure 1). (7-9) Upon activation, they create heterotypic activation complexes with monocytes and neutrophils by binding to the adhesion molecule P-selectin (CD62P). P-selectin plays a critical role in leukocyte recruitment and activation and is stored and released by platelets and Weibel-Palade bodies in endothelial cells (10-13). The interplay between P-selectin and PSGL-1 results in P-selectin being cleaved to form soluble P-selectin (sP-selectin). The substance still possesses many of its procoagulant and stimulatory characteristics (14-16). High plasma levels of sP-selectin are linked to an elevated risk of cardiovascular disease, myocardial infarction, and stroke in both humans and mice. (17-20) Note that acute thromboembolism was a significant issue in Covid 19 patients referred for emergency cardiac surgery, with devastating post-operative complications that were difficult to manage. Thromboembolic episodes before disseminated intravascular coagulation led to severe complications in patients with acute aortic dissection and NSTEMI requiring emergency revascularization. (21-28)
On monocytes, P-selectin binding rapidly exposes tissue factor (TF), the coagulation initiator, to the surface. (29-34) Over time, the expression of the TF gene increases and TF is released from the surface of monocytes in extracellular vesicles. The primary source of TF in the blood is then the activated monocyte. (35) TF and Factor VIIa combine to aid in the formation of the prothrombinase complex on the exterior of triggered platelets, thereby producing high quantities of thrombin. Interestingly, researchers (3,9) have shown that platelet-specific P-selectin deficiency alters the initiation of atherosclerosis in a preclinical model of atherosclerosis based on the work of Russell Ross (1). This modification has the potential to decrease smooth muscle cell mobility, which may significantly impact lipid levels and the number of cells in the growing plaque. (36-39) Nevertheless, trials on patients presenting with acute coronary syndrome and administered inclacumab, an anti-P-selectin antibody, found that while the drug reduced troponin release, it did not have any effect on adverse effects during the SELECT-ACS trial.(40,41) This may be attributed to E-selectin, another type of selectin, being expressed as a result of endothelial activation through factors such as platelet factor-4 or other cytokines. (43,44) Perhaps in treating atherothrombosis, it is necessary to adopt a dual approach that inhibits both P- and E-selectin. Of these, 5-HT (serotonin) is of particular interest. In addition to inducing vasoconstriction, 5-HT also amplifies platelet and endothelial activation, as evidenced by its ability to facilitate Weibel-Palade body exocytosis. (15,45,46). Etulain and co-workers suggested that in aseptic inflammation, platelets induce neutrophils to release neutrophil extracellular traps (NETs) via P-selectin and PSGL-1 signals. (47)
These processes, together with platelet aggregation and fibrin deposition, lead to pathological vascular occlusion even in the absence of physical vascular injury. (48) However, it is important to note that platelets have a complex role in inflammation. They also act as protectors by preventing hemorrhaging in inflamed venules due to leukocyte transmigration through endothelial junction disruption. (49-51)
3. Interaction of NETs with Ultra-Large VWF in Thromboinflammatory Vasculopathy
3.1. Identifying the role played by NETs.
An organized cell death pathway, known as NETosis, takes place in a precise subset of neutrophils in response to different pathological stimuli, like ischemia. (53) Recently, the cell biology of NETosis has been further researched. (54,55) The enzyme PAD4 (protein arginine deiminase 4) plays a pivotal role in this process. (56-58) PAD4 possesses the sole nuclear localization signal among the PAD family. On entry into the nucleus, PAD4 converts positively charged arginine residues, common in histones, into citrulline, an uncharged amino acid. This process weakens histone interactions within nucleosomes and DNA, resulting in chromatin decondensation, histone proteolysis, and unwinding into NETs. H3 and H4 biomarkers are useful for identifying NETs in both animals and humans. They can be detected by analysing plasma samples and tissue sections. (56,58)
PAD4 is thought to have specific cytoplasmic targets that impact the cell biology of NETosis and neutrophil inflammasome composition. Neutrophils deficient in PAD4 function, either genetically or through inhibition, exhibit a marked impairment in NETosis. (58-61) The investigators used high-resolution time-lapse microscopy to prompt NETosis in stimulated mouse neutrophils and human neutrophil-like cells. They showed that PAD4 has the required enzymatic and nuclear localization capabilities for various stages, such as rupturing the nuclear envelope and releasing extracellular DNA. (61,62). Accordingly, researchers have provided evidence demonstrating a correlation between the elevated NETosis and heightened neutrophil PAD4 protein expression in type 1 diabetes patients. (58,61) This finding clarifies their pro-NETotic phenotype. Moreover, a critical aspect for neutrophils to undergo cell death and subsequently release chromatin involves the formation of gasdermin D-dependent membrane pores. (63,64) However, it appears that not all pathways leading to NETs result in neutrophil demise. Significantly, in the course of an infection, it was feasible to recognise well-operating cytoplasts (enucleated cells) with the ability to underpin phagocytosis (65,66).
Adorned with histones, cytoplasmic, and granular proteins, NETs create a substantial framework that damages the nearby tissue and introduces neoantigens, ultimately causing autoimmune diseases. Recently, we demonstrated that the introduction of neutrophil extracellular traps (NETs) with G-CSF and collagen injections triggered arthritis and joint erosion in a mouse strain typically resistant to the disease. (67) In blood vessels, NETs, like VWF, act as a platform for platelet adhesion and initiation of coagulation. (54,55,57,58)
Active PAD4, which is released in conjunction with NETs, also facilitates the citrullination of ADAMTS13, thus impeding VWF scission and allowing platelet aggregates to remain close to the vessel wall in the coexistence of PAD4. (68,69)
Certain neutrophils release pieces of the inflammasome that include ASC (apoptosis-associated speck-like protein containing a CARD) tangled in NETs. (70) These inflammasome leftovers, when taken up by cells, have been observed to propagate inflammasome formation, similar to prion proteins. This cascade of events can increase IL-1β production and accelerate systemic inflammatory responses. (71) Studies have found TF-containing microparticles within NETs, consistent with their procoagulant function. Additionally, microparticles can originate from other cells such as malignant ones. Microparticles are derived from multiple sources including monocytes, which are the primary source of procoagulant microparticles. (72) NETosis and the increase in NET-associated TF have recently been linked to systemic inflammation and IL-1β levels, suggesting a common regulatory pathway (71). Furthermore, activation of both canonical and non-canonical inflammasomes stimulates TF secretion from activated macrophages and monocytes, as demonstrated by recent studies. (73)
Over time, a plethora of clinical and experimental evidence has linked NETs to various ischemia-related and thromboinflammatory ailments. (54,55,57,58,66,74-77) Inhibiting NET production or their cleavage through DNases has been suggested as a novel therapy, akin to ADAMTS13's VWF cleavage. (68)
Recently, Novotny and colleagues (78) conducted an in-depth histological examination of arterial thrombi, revealing differences in both thrombus architecture and leukocyte subset abundance among patients with acute ischemic stroke (AIS) and acute myocardial infarction (AMI). The researchers discovered that while leukocyte (p= 0.133) and neutrophil (p= 0.56) levels were similar between AIS and AMI thrombi, monocyte (p= 0.0052), eosinophil (p< 0.0001), B-cell (p< 0.0001), and T-cell (p< 0.0001) counts were higher in stroke patients compared to those with AMI thrombi. Moreover, there was an uneven distribution of NETs in terms of quantity and appearance. These were evident in all patients with AIS, but only in 20.8% of those with AMI. The abundance of NETs in thrombi correlated with inferior outcome scores among patients with AIS. Conversely, patients with AMI displayed reduced ejection fraction. This disparity in patient outcomes distinguishes the crucial influence of NETs on thrombus stability in both disorders.
Ducrox et al. (79) provided histological evidence from a controlled trial of 108 acute ischaemic stroke patients, from whom thrombi were retrieved. The study aimed to investigate the presence of NETs in thrombi extracted during endovascular therapy in AIS patients and to evaluate their impact on tPA-induced thrombolysis. The authors identified clusters of NETs in all thrombi, with a higher network density in the peripheral layers of the thrombus. The study has found that the presence of thrombus NET content causes resistance to reperfusion. The study investigated both mechanistic and pharmacological approaches, utilizing intravenous tPA. This was done irrespective of the underlying cause. Therefore, the combination of DNAse 1 with tPA should be considered a new strategy for exploration in the context of AIS. Novotny and Ducrox discuss the importance of NETs in thrombosis and their potential clinical benefits. The most significant finding is that recombinant DNAse 1 increased thrombolysis induced by tissue plasminogen activator ex vivo. However, DNAse 1 alone did not produce the same effect. (105)
Most notably, Blasco et al. (80) presented findings of NETs in coronary thrombi among patients with COVID-19 who had STEMI. The study reveals the fundamental process of coronary blockage in STEMI patients, emphasizing the crucial involvement of NETs in the development of COVID-19-associated coronary thrombosis. (77) Researchers found elevated levels of NETs in the blood clots of all COVID-19 patients. Specifically, patients with STEMI and COVID-19 had notably more NETs than those without, according to earlier findings from the same group. Immunohistochemical analysis demonstrated that all clots comprised a greater proportion of fibrin and polymorphonuclear cells. The complete lesion analysis, involving thrombi, NETs, and cellular infiltrate, demonstrated the absence of atheromatous plaques. In contrast, 65% of non-infected patients displayed STEMI with visible atheromatous plaques. It is also worth mentioning that the percentage of plaque fragments in the patient historical control closely resembled that of a previous series of 142 patients who did not experience STEMI (80,81). Furthermore, it is noteworthy that in the cohort studied by Blasco et al., patients with STEMI did not report significant changes in the coagulation parameters mentioned earlier, except for one patient who had a high concentration of D-dimer. Furthermore, this investigation furnishes a reliable explanation for the significant contribution of neutrophils and NETs to coronary thrombus formation in COVID-19 subjects, despite the constraint of a small sample size. (80)
Due to a lack of reliable evidence, it is unclear whether there is a causal relationship between circulating NETs and adverse clinical outcomes after STEMI. Langseth et al. analysed serum collected an average of 18 hours after PCI and correlated peripherally measured NET-specific components with clinical outcomes in STEMI. (81) The observational cohort study followed 956 patients who received PCI for STEMI for a median duration of 4.6 years. Patients' serum double-stranded DNA (dsDNA) was used to assess the more precise NETs markers, such as myeloperoxidase DNA and citrullinated histone. The authors did not find any significant differences in the levels of NETs markers between groups with or without a primary composite endpoint that encompassed reinfarction, stroke, heart failure rehospitalization, unscheduled revascularization post the initial infarction for more than three months, or all-cause mortality, regardless of their occurrence sequence. Despite this, there was a significant increase in dsDNA levels (p < 0.001) in patients who didn't survive (n = 76) compared to those who did. High dsDNA levels above the median were found to be associated with an increased mortality rate (54 vs 22, p < 0.001), and upper quartile levels of dsDNA were linked to a greater risk of mortality. Additionally, dsDNA showed a weak correlation with D-dimer (rs = 0.17, p < 0.001), while elevated dsDNA levels were linked to a higher risk of all-cause mortality. Similarly, in STEMI patients, elevated dsDNA levels were weakly associated with hypercoagulability. (82)
Blasco et al. (80,81) and Langseth et al. (82) conducted studies which validated the significance of NETs in the pathogenesis of SARS-CoV-2 infection. These findings endorse the notion that aiming for intravascular NETs is a pertinent goal in managing patients with STEMI and presents a practicable method to prevent coronary thrombosis among patients with severe COVID-19. (Figure 2) (77,80,81)
3.2. The role of VWF and ADAMTS13
Von Willebrand factor (VWF) is a multimeric glycoprotein that binds to platelet glycoprotein Ibα and plays a crucial role in the recruitment and activation of platelets. Von Willebrand factor (VWF) is located in the same area as P-selectin, found in Weibel-Palade bodies of ECs and α-granules. It plays a key role in supporting platelet tethering and leukocyte adhesion in a similar fashion. Moreover, if the ultra-large VWF stored in Weibel-Palade bodies is not cleaved, it forms long strings, which are temporarily anchored to the endothelial surface. (83) In artificial endothelial microchannels, von Willebrand factor (VWF) released from activated endothelial cells associates with itself to form elongated strands that can span across the vascular lumen. (84,85) These ultra-large VWF molecules fragment red blood cells, leading to the formation of schistocytes, as observed in thrombotic thrombocytopenic purpura. This is observed in thrombotic thrombocytopenic purpura, which impairs the activity of ADAMTS13 (a disintegrin-like metallopeptidase with thrombospondin motif type 1 member 13). (86,87) Fractured red blood cells release heme, which triggers NETosis, intensifying the thromboinflammatory process. (88,89) The uncut VWF, with its activated binding sites, stimulates the creation of platelet strings (see Figure 4) and microthrombi. (90,91) Increased levels of von Willebrand factor (VWF) are associated with increased disease severity in cardiovascular disease and stroke. (92-94) Studies in mice show that deficiency of ADAMTS13, an enzyme that converts VWF, results in increased thrombosis and inflammation. (95,96) Two reports from the same group demonstrate the potential anti-inflammatory benefits of recombinant ADAMTS13 in both stroke and myocardial ischaemia/reperfusion injury in mice. The investigators have shown that recombinant ADAMTS13 has a protective anti-inflammatory effect when administered in both scenarios. (97,98)
Interwoven fibrin, NETs and VWF, localised in the solid matrix of thrombi, are seen in histological examination of both animal and human thrombi. VWF acts as a bridge between the vessel wall and NETs, stabilizing thrombi. Histones released with NETs promote VWF release from ECs and stimulation of platelet activation (83,99). A direct interaction between VWF and NETs is evident (Figure 4). Both DNA and histones bind to VWF and keep NETs in place. (100-102) In a crucial study (103) predating the discovery of NETs, (57) a distinct binding between the A1 domain of VWF and histones had been identified, elucidating the purpose of this interaction site on VWF. It has been observed that recombinant ADAMTS13 therapy not only clears VWF from endothelial surfaces, but also removes NETs. (83, 104-106) Figure 3
Inflammasone to the direction of all actors
Excluding cellular interactions, the thromboinflammatory process is complex. It involves the interaction of the coagulation cascade, complement and cytokines, predominantly the IL-1 family. A variety of regulatory proteins are produced as inactive precursors and necessitate proteolytic processing in order to attain biological functionality. Initially reported in 1989 by Black et al (108,109), caspase 1, which is predominantly accountable for processing pro-IL-1β intracellularly, (110) was eventually unveiled as the primary component of the inflammasome. (111,112) Inflammasomes are complex protein structures composed of several components that gather in innate immune cells after activation. (113) Recent reports suggest that inflammasome assembly in neutrophils, which was previously mainly researched in monocytes/macrophages, also takes place (Figure 4). Although neutrophils are responsive to the same stimuli as monocytes, the latest studies have demonstrated that they do not require LPS priming in vitro, indicating a more rapid response time, aligning with their function as the host's primary immune defense (60). The main driving force for the assembly of the inflammasome in sterile thrombo-inflammation is the activated platelet. (114,115) Please refer to Figure for further details. It should be noted that there are numerous other intracellular and extracellular stimulators that activate different pathways of activation, which are discussed in more detail. (116,117) The importance of the inflammasome in pro-IL-1β processing renders it a focal point in the evolution of thromboinflammation, thereby presenting a plausible target for modifying inflammatory pathways. The most extensively studied inflammasome in IL-1β activation is the NLRP3 receptor (pyrin domain containing 3 of the NLR family). (118)
Upon activation and release, IL-1β functions as a prominent pro-inflammatory cytokine, triggering the generation of endothelial adhesion molecules that draw leukocytes, such as E-selectin, ICAM-1 (intercellular adhesion molecule-1), and VCAM-1 (vascular cell adhesion molecule-1).(119-123) Certain infrequent genetic disorders accentuate the significance of the inflammasome in regulating the excessive pro-inflammatory effects resulting from the release of IL-1β. Gain-of-function mutations in the NLRP3 inflammasome are responsible for cryopyrin-associated periodic syndromes (CAPS). Canakinumab, an anti–IL-1β antibody, is approved as an orphan medication for treating these conditions. A recent study showing that an exclusively neutrophil-expressed mutation that induces inflammasome assembly is sufficient to trigger cryopyrin-associated periodic syndromes. This discovery emphasizes the vital function of neutrophils in cryopyrin-associated periodic syndromes. (112) Thiam and colleagues (62) have recently discovered that the formation of ASC specks is a useful indicator of inflammasome activity in murine neutrophils, occurring before chromatin decondensation. In line with this, Münzer and colleagues (60) demonstrated that NLRP3 deficiency considerably decreases NETosis in vitro and leads to a lower density of NETs in thrombi created by a mouse model of deep vein thrombosis induced by stenosis. This is an intriguing discovery as it places the inflammasome upstream of yet another thromboinflammatory process known as NETosis. Additionally, citrullination promotes inflammasome assembly, (124) and in neutrophils, the citrullinating enzyme is PAD4. (125)
Pathological thromboinflammation is underpinned by the progression of the PAD4-mediated inflammasome in neutrophils and subsequent NETosis. PAD4-mediated inflammasome assembly and subsequent NETosis underpin pathological thromboinflammation, connecting the immunological role of neutrophils to the activation of platelets and monocytes. Figure 4
4. Focusing on the thromboinflammation process in atherosclerosis and COVID-19.
It is evident that the interaction between platelets, VWF, NETs, and inflammasomes plays a crucial role in the progression of thromboinflammation associated with various diseases. In this text, we will explore two distinct pathological processes where thromboinflammation is believed to be a contributing factor.
4.1. Implication of atherosclerosis
In 2019, it will become possible to target inflammation in atherosclerosis using clinical intervention, as shown by CANTOS, which supports the thesis that the development and progression of atherosclerosis is mainly due to the inflammatory response (1,119-122,124,126). The outcomes of this significant study demonstrate that interventions aimed at inflammation can lead to encouraging clinical results. The CANTOS trial evidenced that administering an anti-IL-1β antibody to patients with cardiovascular stability after a myocardial infarction, following the guidelines, lowered the occurrence of renewed major adverse cardiovascular events. (113) Nevertheless, this constructive result was associated with a considerable increase in infections, some of which resulted in fatalities. (113) In this section, we examine the interconnection between atherogenesis and thromboinflammation while identifying significant contributors. The goal is to identify potential therapy targets. See other sources for a comprehensive overview of the condition. (126,127)
As with other thromboinflammatory illnesses, in atherosclerosis, the initial stages of leukocyte recruitment are reliant on the activation of endothelium and platelets. (128) Platelets secrete chemokines (such as CCL5) and cytokines (IL-1 beta) which promote monocyte/neutrophil adhesion to the endothelium. (129). Leukocyte adhesion and the progression of atherosclerotic lesions are supported by both platelet and endothelial P-selectin. (38) Soluble E-selectin increases the risk of cardiovascular disease. (130,131) The combined deficiency of P-selectin and E-selectin had the most detrimental effect on lesion progression in mice. (131,132) Molecular ultrasound imaging has revealed an increase in VWF-mediated platelet attachment to the endothelium prior to the appearance of atherosclerotic plaques, hinting at a possible role of VWF in initiating the platelet function within the lesion. (133) This finding explains that VWF-deficient mice fed with a high-fat diet developed atherosclerotic-prone sites later than their wild-type counterparts, and in unique locations. (134)
In turbulent flow where lesions tend to form, von Willebrand factor (VWF) is necessary to facilitate platelet adhesion, ultimately marking the site for monocyte recruitment. In addition, the fatty streaks observed in VWF-deficient mice were smaller in size and contained fewer monocytes. This finding is in line with the discovery that endothelial VWF, as opposed to platelet-derived VWF, is essential in the development of atherosclerosis in mice. (135) In all animal disease models where VWF is pathologically released, ADAMTS13 deficiency accelerates the progression of atherosclerotic lesions, in contrast to VWF deficiency. (136,137)
High plasma levels of sP-selectin are correlated with the severity of cardiovascular disease in humans, similar to VWF. Knock-in mice were created by deleting the cytoplasmic domain necessary for P-selectin storage, resulting in an enhanced procoagulant state due to overproduction of thrombin. These mice exhibited an increased susceptibility to atherosclerosis. (20) Although monocytes are widely considered the most critical factor in atherosclerosis induced by lipid, there is evidence to support the involvement of neutrophil-derived NETs in a process similar to endothelial erosion in mice. (126) Recent genetic research revealed the presence of such NETs in this disease, with PAD4 deficiency being an effective inhibitor. Reducing endothelial discontinuity and EC apoptosis in a mouse model was also possible with the administration of DNAse. (138) This treatment not only tackles the blood clotting effects caused by NETs but also their toxicity. Additionally, it has been discovered that histone H4 in neutrophil extracellular traps (NETs) can cause the death of smooth muscle cells in the arterial wall, which accelerates the destabilisation of atherosclerotic plaques. (139) This finding may explain how acute infections, causing an excessive amount of NETosis, contribute to cardiovascular risk. (126,140,141) Targeting NETs could be beneficial for enhancing plaque stability in secondary prevention therapy, such as for carotid disease and stable coronary artery disease. (126)
There is feedback from PAD4-mediated inflammasome assembly and NETosis to IL-1β activation, which further encourages NETosis and atherosclerosis (142). Experimental evidence shows that cholesterol crystals activate NLRP3 inflammasome, directly connecting it to the path of atherogenesis. Research has indicated that reducing atherosclerosis in mice can be accomplished by inhibiting NLRP3 or genetically deleting it (126,143,144). Additionally, as NLRP3 deficiency also decreases NETosis and the accumulation of NETs in thrombi (125), inhibition of PAD4 and NLRP3 targeting may present potential interventions for hindering atherothrombosis.
4.2. Implication of Thromboinflammation in COVID-19
Since the COVID-19 outbreak and the frequent occurrence of coagulopathy, thromboinflammation has been attracting attention. Blood clots in dialysis and ECMO circuits have been observed in COVID-19 patients despite adequate anticoagulation, in line with the definition of thromboinflammation. The principal pathogenic mechanisms underlying the pathology of COVID-19 are endothelial dysfunction (147-149) and thromboinflammation (125). Patients with the disease present an increase in platelet activation, formation of platelet-monocyte aggregates, and a significant rise in monocyte tissue factor expression (150). Despite the use of antiplatelet therapy, the outcome of hospitalized COVID-19 patients has not shown any significant improvement. (151-153) This suggests that severe COVID-19 is not solely driven by platelet aggregation but rather by the interaction between platelets and various factors, such as NETs. Patients with COVID-19 exhibit a specific coagulopathy marked by heightened levels of fibrinogen, D-dimer fibrin degradation products, and significantly increased levels of VWF in the bloodstream. (153-155) There is emerging evidence that that heightened activation of endothelial cells, causing the release of large von Willebrand factor (VWF) multimers, in combination with insufficient VWF cleavage due to ADAMTS13 consumption or disease pathophysiology related to COVID-19, might lead to escalated interactions between platelets and vessel walls, ultimately resulting in thrombotic microangiopathy. (156) Reports reveal COVID-19 to cause an excessive stimulation of the complement and coagulation systems. (157,158)
Given its procoagulant properties, it's not surprising that sP-selectin is associated with disease severity in COVID-19 patients, (159,160) but a placebo-controlled, randomised trial evaluating the effect of a single dose of crizanlizumab, a P-selectin inhibitor, in mild COVID-19 patients found no significant differences in clinical outcomes compared to placebo, despite evidence of reduced thrombin activation and sP-selectin levels. (161) It is planned to conduct a larger study with a larger number of random doses. Furthermore, neutrophil extracellular traps (NETs) are probably implicated in the thromboinflammation responsible for COVID-19 and the subsequent severe lung impairment. (162) In 2012, scientists revealed NETs' pathological involvement in acute respiratory distress syndrome associated with acute lung injury due to blood transfusion in mice and humans, with NETs found in the lungs. (163,164) Similarly, autopsy case reports of COVID-19 patients have identified NETs in the lung parenchyma and alveolar space. (165) Aymonnier et al (166) conducted a study of freshly isolated neutrophils from severe COVID-19 patients as part of a clinical trial on DNase 1 inhalation (https://www.clinicaltrials.gov; unique identifier: NCT04402944). Investigators discovered that a significant number of neutrophils were poised for NETosis, evidenced by 40% of nuclei testing positive for citrullinated histone H3. Additionally, 2% of neutrophils taken from either blood or tracheal aspirate (i.e. from the lung) were observed to be forming inflammasome, as detected by ASC speck assembly. Observation of the spots in the vicinity of multilobulated neutrophil nuclei before the standard nuclear rounding of NETosis indicates that inflammasome assembly takes place prior to NETosis in COVID-19 patients, (166) thereby highlighting its potential as a treatment target. In our study, though, neutrophils and monocytes depicted a comparable rate of inflammasome positivity (2%). However, it is imperative to note that the blood stream contains ten times more neutrophils than monocytes. Thus, the possible underestimation of the importance of neutrophil-produced IL-1β cannot be ignored. The same autocrine IL-1β feed-forward loop, activated by the inflammasome, could contribute to the development of acute adult respiratory distress syndrome, cytokine storm, and microvascular thrombosis, ultimately leading to multi-organ failure in severe COVID-19 cases. (166)
5. Conclusion and Next Steps
It is evident that thrombosis and inflammation do not occur in isolation during diseases. As further knowledge is acquired, the complexity of vascular processes is unveiled. For example, the formation of a blood clot can no longer be interpreted as being solely a matter of cleaved fibrinogen with plasma proteins, since the DNA content of the clot also has to be taken into account for clot lysis. Likewise, platelet clots are no longer just fibrinogen-crosslinked platelets. It was unexpected to discover that thrombi could still be produced in mice following an injury, despite their lack of both fibrinogen and VWF. (167)
Recent studies show that deep vein thrombosis formation is not only related to platelets and red blood cells, but also involves monocytes and neutrophils (168-170). Researchers have been aware of the role of platelets and platelet-derived factors in inflammation since the beginning with ongoing research supporting their impact. (3) The traditional cellular players of inflammation and thrombosis fully intertwine, validating the term thromboinflammation.
Neutrophils have been identified as central to thromboinflammation, owing to their newly-discovered ability to assemble inflammasomes and produce neutrophil extracellular traps (NETs). Given that NETs contribute equally to thrombosis, immune response, and tissue damage, they play a critical role in the development of this disease. NETosis sets off platelets, leads to the release of toxic histones (171-173) and active enzymes, hence modifying thrombus stability. (174,175) The assembly of the inflammasome, along with heightened PAD4 activity, leads to the discharge of cytokines that are both pro-inflammatory and pro-thrombotic. This also activates the endothelium, further increasing the recruitment of leukocytes as well as inflammasome assembly in adjacent cells. As such, PAD4 and inflammasome emerge as pertinent targets for antithromboinflammatory therapy. However, there are numerous outstanding mechanistic queries that remain unanswered. Identifying the specific protein targets of PAD4 citrullination necessary for inflammasome formation, and the intracellular substrates of caspase 1 (the enzyme produced by inflammasomes) that must be cleaved to trigger NETosis, could enhance our comprehension of how to regulate the behaviour of neutrophils.
Author Contributions
Conceptualization, F.N.; methodology, F.N..; software, F.N.; validation, F.N.; formal analysis, F.N; investigation, F.N.; data curation, F.N.; writing—original draft preparation, F.N.; writing—review and editing, F.N.; visualization, F.N.; supervision, F.N.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999 Jan 14;340(2):115-26. [CrossRef]
- Trivigno SMG, Guidetti GF, Barbieri SS, Zarà M. Blood Platelets in Infection: The Multiple Roles of the Platelet Signalling Machinery. Int J Mol Sci. 2023 Apr 18;24(8):7462. [CrossRef]
- Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. 2008; 111:5271–5281. [CrossRef]
- Yao M, Ma J, Wu D, Fang C, Wang Z, Guo T, Mo J. Neutrophil extracellular traps mediate deep vein thrombosis : from mechanism to therapy. Front Immunol. 2023 Aug 23 ;14:1198952. [CrossRef]
- Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013; 13:34–45. [CrossRef]
- Sharma S, Tyagi T, Antoniak S. Platelet in thrombo-inflammation: Unraveling new therapeutic targets. Front Immunol. 2022 Nov 14 ;13:1039843. [CrossRef]
- Krott KJ, Feige T, Elvers M. Flow Chamber Analyses in Cardiovascular Research : Impact of Platelets and the Intercellular Crosstalk with Endothelial Cells, Leukocytes, and Red Blood Cells. Hamostaseologie. 2023 Oct;43(5):338-347. [CrossRef]
- Thachil J. Semin Thromb Hemost. Platelets in Inflammatory Disorders: A Pathophysiological and Clinical Perspective 2015 Sep;41(6):572-81. [CrossRef]
- Wagner DD, Burger PC. Platelets in inflammation and thrombosis. Arterioscler Thromb Vasc Biol. 2003;23:2131–2137. [CrossRef]
- Solomon SD, Lowenstein CJ, Bhatt AS, Peikert A, Vardeny O, Kosiborod MN, Berger JS, Reynolds HR, Mavromichalis S, Barytol A, Althouse AD, Luther JF, Leifer ES, Kindzelski AL, Cushman M, Gong MN, Kornblith LZ, Khatri P, Kim KS, Baumann Kreuziger L, Wahid L, Kirwan BA, Geraci MW, Neal MD, Hochman JS; ACTIV4a Investigators. Effect of the P-Selectin Inhibitor Crizanlizumab on Survival Free of Organ Support in Patients Hospitalized for COVID-19: A Randomized Controlled Trial. Circulation. 2023 Aug;148(5):381-390. [CrossRef]
- Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte rolling and extravasation are severely compromised in P selectin-deficient mice. Cell. 1993; 74:541–554. [CrossRef]
- Hrachovinová I, Cambien B, Hafezi-Moghadam A, Kappelmayer J, Camphausen RT, Widom A, Xia L, Kazazian HH Jr, Schaub RG, McEver RP, Wagner DD. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat Med. 2003 Aug;9(8):1020-5. [CrossRef]
- Polgar J, Matuskova J, Wagner DD. The P-selectin, tissue factor, coagulation triad. J Thromb Haemost. 2005; 3:1590–1596. [CrossRef]
- Braun OO, Slotta JE, Menger MD, Erlinge D, Thorlacius H. Primary and secondary capture of platelets onto inflamed femoral artery endothelium is dependent on P-selectin and PSGL-1. Eur J Pharmacol. 2008 Sep 11;592(1-3):128-32. [CrossRef]
- Dole VS, Bergmeier W, Patten IS, Hirahashi J, Mayadas TN, Wagner DD. PSGL-1 regulates platelet P-selectin-mediated endothelial activation and shedding of P-selectin from activated platelets. Thromb Haemost. 2007; 98:806–812. [CrossRef]
- Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Acti- vated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. 2005; 106:2334–2339. [CrossRef]
- Blann AD, Nadar SK, Lip GY. The adhesion molecule P-selectin and cardiovascular disease. Eur Heart J. 2003 Dec;24(24):2166-79. [CrossRef]
- Ridker PM, Buring JE, Rifai N. Soluble P-selectin and the risk of future cardiovascular events. Circulation. 2001; 103:491–495. [CrossRef]
- Trivigno SMG, Guidetti GF, Barbieri SS, Zarà M. Blood Platelets in Infection: The Multiple Roles of the Platelet Signalling Machinery. Int J Mol Sci. 2023 Apr 18;24(8):7462. [CrossRef]
- Kisucka J, Chauhan AK, Zhao BQ, Patten IS, Yesilaltay A, Krieger M, Wagner DD. Elevated levels of soluble P-selectin in mice alter blood-brain barrier function, exacerbate stroke, and promote atherosclerosis. Blood. 2009; 113:6015–6022. [CrossRef]
- NIHR Global Health Unit on Global Surgery; Elective surgery system strengthening development, measurement, and validation of the surgical preparedness index across 1632 hospitals in 119 countries. COVIDSurg Collaborative. Lancet. 2022 Nov 5;400(10363):1607-1617. [CrossRef]
- COVIDSurg Collaborative; GlobalSurg Collaborative. SARS-CoV-2 infection and venous thromboembolism after surgery: an international prospective cohort study. Anaesthesia. 2022 Jan;77(1):28-39. Epub 2021 Aug 24. [CrossRef]
- COVIDSurg Collaborative, GlobalSurg Collaborative. SARS-CoV-2 vaccination modelling for safe surgery to save lives: data from an international prospective cohort study.Br J Surg. 2021 Sep 27;108(9):1056-1063. [CrossRef]
- COVIDSurg Collaborative; GlobalSurg Collaborative. Timing of surgery following SARS-CoV-2 infection: an international prospective cohort study. Anaesthesia.2021 Jun;76(6):748-758. [CrossRef]
- COVIDSurg Collaborative; GlobalSurg Collaborative. Effects of pre-operative isolation on postoperative pulmonary complications after elective surgery: an international prospective cohort study. Anaesthesia. 2021 Nov;76(11):1454-1464. [CrossRef]
- Nappi F, Giacinto O, Ellouze O, Nenna A, Avtaar Singh SS, Chello M, Bouzguenda A, Copie X. Association between COVID-19 Diagnosis and Coronary Artery Thrombosis: A Narrative Review. Biomedicines. 2022 Mar 18;10(3):702. [CrossRef]
- Fukuhara S, Rosati CM, El-Dalati S. Acute Type A Aortic Dissection During the COVID-19 Outbreak. Ann Thorac Surg. 2020 Nov;110(5):e405-e407. [CrossRef]
- Nappi F. Incertitude Pathophysiology and Management During the First Phase of the COVID-19 Pandemic. Ann Thorac Surg. 2022 Feb;113(2):693. [CrossRef]
- Troisi R, Balasco N, Autiero I, Sica F, Vitagliano L New insight into the traditional model of the coagulation cascade and its regulation: illustrated review of a three-dimensional view. Res Pract Thromb Haemost. 2023 Aug 7;7(6):102160. [CrossRef]
- Rolling CC, Barrett TJ, Berger JS. Platelet-monocyte aggregates: molecular mediators of thromboinflammation. Front Cardiovasc Med. 2023 May 15; 10:960398. [CrossRef]
- Grover SP, Mackman N. Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. 2018; 38:709–725. [CrossRef]
- Heestermans M, Poenou G, Duchez AC, Hamzeh-Cognasse H, Bertoletti L, Cognasse F. Immunothrombosis and the Role of Platelets in Venous Thromboembolic Diseases. Int J Mol Sci. 2022 Oct 29;23(21):13176. [CrossRef]
- Scholz T, Temmler U, Krause S, Heptinstall S, Lösche W. Transfer of tissue factor from platelets to monocytes: role of platelet-derived microvesicles and CD62P. Thromb Haemost. 2002 Dec;88(6):1033-8. [CrossRef]
- Ivanov II, Apta BHR, Bonna AM, Harper MT. Platelet P-selectin triggers rapid surface exposure of tissue factor in monocytes. Sci Rep. 2019; 9:13397. [CrossRef]
- Hrachovinová I, Cambien B, Hafezi-Moghadam A, Kappelmayer J, Camphausen RT, Widom A, Xia L, Kazazian HH Jr, Schaub RG, McEver RP, et al. Interaction of P-selectin and PSGL-1 generates microparticles that cor- rect hemostasis in a mouse model of hemophilia A. Nat Med. 2003;9:1020– 1025. [CrossRef]
- Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003; 101:2661–2666. [CrossRef]
- Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008; 28:812–819. [CrossRef]
- Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003; 101:2661–2666. [CrossRef]
- Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008; 28:812–819. [CrossRef]
- Libby P, Everett BM. Novel antiatherosclerotic therapies. Arterioscler Thromb Vasc Biol. 2019; 39:538–545. [CrossRef]
- Zhang W, Zhao J, Deng L, Ishimwe N, Pauli J, Wu W, Shan S, Kempf W, Ballantyne MD, Kim D, Lyu Q, Bennett M, Rodor J, Turner AW, Lu YW, Gao P, Choi M, Warthi G, Kim HW, Barroso MM, Bryant WB, Miller CL, Weintraub NL, Maegdefessel L, Miano JM, Baker AH, Long X. INKILN is a Novel Long Noncoding RNA Promoting Vascular Smooth Muscle Inflammation via Scaffolding MKL1 and USP10. Circulation. 2023 Jul 4;148(1):47-67. [CrossRef]
- Zhong M, Wang XH, Zhao Y. Platelet factor 4 (PF4) induces cluster of differentiation 40 (CD40) expression in human aortic endothelial cells (HAECs) through the SIRT1/NF-κB/p65 signaling pathway. In Vitro Cell Dev Biol Anim. 2023 Sep;59(8):624-635. [CrossRef]
- Yu G, Rux AH, Ma P, Bdeir K, Sachais BS. Endothelial expression of E-selec- tin is induced by the platelet-specific chemokine platelet factor 4 through LRP in an NF-kappaB-dependent manner. Blood. 2005; 105:3545–3551. [CrossRef]
- Stadtmann A, Brinkhaus L, Mueller H, Rossaint J, Bolomini-Vittori M, Bergmeier W, Van Aken H, Wagner DD, Laudanna C, Ley K, et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin- dependent slow leukocyte rolling. Eur J Immunol. 2011;41:2074–2085. [CrossRef]
- Yuan T, Aisan A, Maheshati T, Tian R, Li Y, Chen Y. Predictive value of combining leucocyte and platelet counts for mortality in ST-segment elevation myocardial infarction patients after percutaneous coronary intervention treatment in Chinese population: a retrospective cohort study. BMJ Open. 2023 Jul 18;13(7):e060756. [CrossRef]
- Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, Cifuni SM, Mauler M, Cicko S, Bader M, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood. 2013; 121:1008–1015. [CrossRef]
- Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015; 126:242–246. [CrossRef]
- Yun SH, Sim EH, Goh RY, Park JI, Han JY. Platelet activation: the mecha- nisms and potential biomarkers. Biomed Res Int. 2016; 2016:9060143. [CrossRef]
- Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O’Donnell E, Zhao BQ, Cifuni SM, Wagner DD. Inflammation induces hemorrhage in thrombocy- topenia. Blood. 2008; 111:4958–4964. [CrossRef]
- Ho-Tin-Noé B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008; 68:6851–6858. [CrossRef]
- Graca FA, Stephan A, Minden-Birkenmaier BA, Shirinifard A, Wang YD, Demontis F, Labelle M. Platelet-derived chemokines promote skeletal muscle regeneration by guiding neutrophil recruitment to injured muscles. Nat Commun. 2023 May 22;14(1):2900. [CrossRef]
- Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005 Dec ;115(12):3378-84. [CrossRef]
- Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutro- phil extracellular traps. J Cell Biol. 2007; 176:231–241. [CrossRef]
- Nappi F, Bellomo F, Avtaar Singh SS. Worsening Thrombotic Complication of Atherosclerotic Plaques Due to Neutrophils Extracellular Traps: A Systematic Review.Biomedicines. 2023 Jan 2;11(1):113. [CrossRef]
- Nappi F, Nappi P, Gambardella I, Avtaar Singh SS. Thromboembolic Disease and Cardiac Thrombotic Complication in COVID-19: A Systematic Review. Metabolites. 2022 Sep 22;12(10):889. [CrossRef]
- Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020; 36:191–218. [CrossRef]
- Nappi F, Bellomo F, Avtaar Singh SS. Insights into the Role of Neutrophils and Neutrophil Extracellular Traps in Causing Cardiovascular Complications in Patients with COVID-19: A Systematic Review. J Clin Med. 2022 Apr 27;11(9):2460. [CrossRef]
- Nappi F, Iervolino A, Avtaar Singh SS. Thromboembolic Complications of SARS-CoV-2 and Metabolic Derangements: Suggestions from Clinical Practice Evidence to Causative Agents. Metabolites. 2021 May 25;11(6):341. [CrossRef]
- Wong SL, Wagner DD. Peptidylarginine deiminase 4: a nuclear button trig- gering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018;32:fj201800691R. [CrossRef]
- Münzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, Cherpokova D, Gutch S, Sorvillo N, Shi L, et al. NLRP3 inflammasome assembly in neutrophils is supported by PAD4 and promotes NETosis under Sterile Conditions. Front Immunol. 2021; 12:683803. [CrossRef]
- Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015; 21:815–819. [CrossRef]
- Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, Goldman RD, Wagner DD, Waterman CM. Reply to Liu: the disassembly of the actin cytoskeleton is an early event during NETosis. Proc Natl Acad Sci USA. 2020; 117:22655–22656. [CrossRef]
- Sorvillo N, Cherpokova D, Martinod K, Wagner DD. Extracellular DNA NET- works with dire consequences for health. Circ Res. 2019; 125:470–88. [CrossRef]
- Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3: eaar6689. [CrossRef]
- Krishnamoorthy N, Douda DN, Brüggemann TR, Ricklefs I, Duvall MG, Abdulnour RE, Martinod K, Tavares L, Wang X, Cernadas M, et al; National Heart, Lung, and Blood Institute Severe Asthma Research Program-3 Inves- tigators. Neutrophil cytoplasts induce TH17 differentiation and skew inflam- mation toward neutrophilia in severe asthma. Sci Immunol. 2018;3: eaao4747. [CrossRef]
- Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013; 122:2784–2794. [CrossRef]
- Fukui S, Gutch S, Fukui S, Cherpokova D, Aymonnier K, Sheehy CE, Chu L, Wagner DD. The prominent role of hematopoietic peptidyl argi- nine deiminase 4 in arthritis: collagen- and granulocyte colony-stimulating factor-induced arthritis model in C57BL/6 MICE. Arthritis Rheumatol. 2022; 74:1139–1146. [CrossRef]
- Morrell CN, Hilt ZT, Pariser DN, Maurya P. PAD4 and von Willebrand Factor Link Inflammation and Thrombosis. Circ Res. 2019 Aug 16;125(5):520-522. [CrossRef]
- Sorvillo N, Mizurini DM, Coxon C, Martinod K, Tilvawala R, Cherpokova D, Salinger AJ, Seward RJ, Staudinger C, Weerapana E, et al. Plasma peptidy- larginine deiminase IV promotes VWF-platelet string formation and accel- erates thrombosis after vessel injury. Circ Res. 2019; 125:507–519. [CrossRef]
- Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al-Amoudi A, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014; 15:727–737. [CrossRef]
- Liberale L, Holy EW, Akhmedov A, Bonetti NR, Nietlispach F, Matter CM, Mach F, Montecucco F, Beer JH, Paneni F, et al. Interleukin-1β mediates arterial thrombus formation via NET-associated tissue factor. J Clin Med. 2019;8:E2072. [CrossRef]
- Liman TG, Bachelier-Walenta K, Neeb L, Rosinski J, Reuter U, Böhm M, Endres M. Circulating endothelial microparticles in female migraineurs with aura. Cephalalgia. 2015; 35:88–94. [CrossRef]
- Wu R, Wang N, Comish PB, Tang D, Kang R. Inflammasome-dependent coagulation activation in sepsis. Front Immunol. 2021; 12:641750. [CrossRef]
- Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014; 123:2768–2776. [CrossRef]
- Döring Y, Libby P, Soehnlein O. Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights. Circ Res. 2020; 126:1228–1241. [CrossRef]
- Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, Dentali F, Montecucco F, Massberg S, Levi M, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021;21:319–329. [CrossRef]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.P. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [CrossRef]
- Novotny,J.;Oberdieck,P.;Titova,A.;Pelisek,J.;Chandraratne,S.;Nicol,P.;Hapfelmeier,A.;Joner,M.;Maegdefessel,L.;Poppert, H.; et al. Thrombus NET content is associated with clinical outcome in stroke and myocardial infarction. Neurology 2020, 94, e2346–e2360. [CrossRef]
- Ducroux,C.;DiMeglio,L.;Loyau,S.;Delbosc,S.;Boisseau,W.;Deschildre,C.;BenMaacha,M.;Blanc,R.;Redjem,H.;Ciccio,G.; et al. Thrombus neutrophil extracellular traps content impair tPA-induced thrombolysis in acute ischemic stroke. Stroke 2018, 49, 754–757. [CrossRef]
- Blasco, A.; Coronado, M.-J.; Hernández-Terciado, F.; Martín, P.; Royuela, A.; Ramil, E.; García, D.; Goicolea, J.; Del Trigo, M.; Ortega, J.; et al. Assessment of Neutrophil Extracellular Traps in Coronary Thrombus of a Case Series of Patients With COVID-19 and Myocardial Infarction. JAMA Cardiol. 2021, 6, 469. [CrossRef]
- Blasco A, Bellas C, Goicolea L, Muñiz A, Abraira V, Royuela A, Mingo S, Oteo JF, García-Touchard A, Goicolea FJ. Immunohistological analysis of intracoronary thrombus aspirate in STEMI patients: clinical implications of pathological findings. Rev Esp Cardiol (Engl Ed). 2017;70(3):170-177. [CrossRef]
- Langseth, M.S.; Helseth, R.; Ritschel, V.; Hansen, C.H.; Andersen, G.; Eritsland, J.; Halvorsen, S.; Fagerland, M.W.; Solheim, S.; Arnesen, H.; et al. Double-Stranded DNA and NETs Components in Relation to Clinical Outcome After ST-Elevation Myocardial Infarction. Sci. Rep. 2020, 10, 5007. [CrossRef]
- Yang J, Wu Z, Long Q, Huang J, Hong T, Liu W, Lin J. Insights into immunothrombosis: the interplay among neutrophil extracellular trap, von Wil- lebrand Factor, and ADAMTS13. Front Immunol. 2020;11:610696. [CrossRef]
- Zheng Y, Chen J, López JA. Flow-driven assembly of VWF fibres and webs in in vitro microvessels. Nat Commun. 2015; 6:7858. [CrossRef]
- Chen J, Chung DW. Inflammation, von Willebrand factor, and ADAMTS13. Blood. 2018;132:141–147. [CrossRef]
- Chauhan AK, Walsh MT, Zhu G, Ginsburg D, Wagner DD, Motto DG. The combined roles of ADAMTS13 and VWF in murine models of TTP, endotox- emia, and thrombosis. Blood. 2008;111:3452–3457. [CrossRef]
- Sadler JE. Pathophysiology of thrombotic thrombocytopenic purpura. Blood. 2017;130:1181–1188. [CrossRef]
- Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new con- cepts, and future promise. J Clin Invest. 2007; 117:850–858. [CrossRef]
- Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS. Heme- induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood. 2014; 123:3818–3827. [CrossRef]
- Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124:1412–1425. [CrossRef]
- Arya M, Anvari B, Romo GM, Cruz MA, Dong JF, McIntire LV, Moake JL, López JA. Ultralarge multimers of von Willebrand factor form sponta- neous high-strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood. 2002; 99:3971–7. [CrossRef]
- Peetermans M, Meyers S, Liesenborghs L, Vanhoorelbeke K, De Meyer SF, Vandenbriele C, Lox M, Hoylaerts MF, Martinod K, Jacquemin M, et al. Von Willebrand factor and ADAMTS13 impact on the outcome of Staph- ylococcus aureus sepsis. J Thromb Haemost. 2020; 18:722–731. [CrossRef]
- Vischer UM. von Willebrand factor, endothelial dysfunction, and cardiovas- cular disease. J Thromb Haemost. 2006; 4:1186–1193. [CrossRef]
- Hanson E, Jood K, Karlsson S, Nilsson S, Blomstrand C, Jern C. Plasma levels of von Willebrand factor in the etiologic subtypes of ischemic stroke. J Thromb Haemost. 2011; 9:275–281. [CrossRef]
- Chauhan AK, Kisucka J, Brill A, Walsh MT, Scheiflinger F, Wagner DD. ADAMTS13: a new link between thrombosis and inflammation. J Exp Med. 2008; 205:2065–2074. [CrossRef]
- Gandhi C, Motto DG, Jensen M, Lentz SR, Chauhan AK. ADAMTS13 deficiency exacerbates VWF-dependent acute myocardial ischemia/ reperfusion injury in mice. Blood. 2012; 120:5224–5230. [CrossRef]
- De Meyer SF, Savchenko AS, Haas MS, Schatzberg D, Carroll MC, Schiviz A, Dietrich B, Rottensteiner H, Scheiflinger F, Wagner DD. Protective anti-inflammatory effect of ADAMTS13 on myocardial isch- emia/reperfusion injury in mice. Blood. 2012; 120:5217–5223. [CrossRef]
- Zhao BQ, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, Scheiflinger F, Wagner DD. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood. 2009; 114:3329–3334. [CrossRef]
- Michels A, Albánez S, Mewburn J, Nesbitt K, Gould TJ, Liaw PC, James PD, Swystun LL, Lillicrap D. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J Thromb Haemost. 2016; 14:2274–2286. [CrossRef]
- Matsui T, Hamako J. Structure and function of snake venom toxins interacting with human von Willebrand factor. Toxicon. 2005 Jun 15;45(8):1075-87. [CrossRef]
- Colicchia M, Perrella G, Gant P, Rayes J. Novel mechanisms of thrombo-inflammation during infection: spotlight on neutrophil extracellular trap-mediated platelet activation. Res Pract Thromb Haemost. 2023 Mar 11;7(2):100116. [CrossRef]
- Courson JA, Lam FW, Langlois KW, Rumbaut RE. Histone-stimulated platelet adhesion to mouse cremaster venules in vivo is dependent on von Willebrand factor. Microcirculation. 2022 Nov;29(8):e12782. [CrossRef]
- Ward CM, Tetaz TJ, Andrews RK, Berndt MC. Binding of the von Willebrand factor A1 domain to histone. Thromb Res. 1997; 86:469–477. [CrossRef]
- DeYoung V, Singh K, Kretz CA. Mechanisms of ADAMTS13 regulation. J Thromb Haemost. 2022 Dec;20(12):2722-2732. [CrossRef]
- Wong SL, Goverman J, Staudinger C, Wagner DD. Recombinant human ADAMTS13 treatment and anti-NET strategies enhance skin allograft sur- vival in mice. Am J Transplant. 2020; 20:1162–1169. [CrossRef]
- Gajendran C, Fukui S, Sadhu NM, Zainuddin M, Rajagopal S, Gosu R, Gutch S, Fukui S, Sheehy CE, Chu L, Vishwakarma S, Jeyaraj DA, Hallur G, Wagner DD, Sivanandhan D. Alleviation of arthritis through prevention of neutrophil extracellular traps by an orally available inhibitor of protein arginine deiminase 4. Sci Rep. 2023 Feb 23;13(1):3189. [CrossRef]
- Arisz RA, de Vries JJ, Schols SEM, Eikenboom JCJ, de Maat MPM. Interaction of von Willebrand factor with blood cells in flow models: a systematic review. Blood Adv. 2022 Jul 12;6(13):3979-3990. [CrossRef]
- Black RA, Kronheim SR, Sleath Activation of interleukin-1 beta by a co-induced protease. PR. FEBS Lett. 1989 Apr 24;247(2):386-90. [CrossRef]
- Black RA, Kronheim SR, Merriam JE, March CJ, Hopp TP. A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J Biol Chem. 1989 Apr 5;264(10):5323-6. [CrossRef]
- Krieger TJ, Hook VY. Purification and characterization of a novel thiol protease involved in processing the enkephalin precursor. J Biol Chem. 1991 May 5;266(13):8376-83. [CrossRef]
- Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009; 10:241–247. [CrossRef]
- Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med. 2020;217: e20190314. [CrossRef]
- Libby P. Targeting inflammatory pathways in cardiovascular disease: the inflammasome, Interleukin-1, Interleukin-6 and BEYOND. Cells. 2021; 10:951. [CrossRef]
- Kocatürk B, Lee Y, Nosaka N, Abe M, Martinon D, Lane ME, Moreira D, Chen S, Fishbein MC, Porritt RA, Franklin BS, Noval Rivas M, Arditi M. Platelets exacerbate cardiovascular inflammation in a murine model of Kawasaki disease vasculitis. JCI Insight. 2023 Jul 24;8(14):e169855. [CrossRef]
- Rolfes V, Ribeiro LS, Hawwari I, Böttcher L, Rosero N, Maasewerd S, Santos MLS, Próchnicki T, Silva CMS, Wanderley CWS, et al. Platelets fuel the inflammasome activation of innate immune cells. Cell Rep. 2020; 31:107615. [CrossRef]
- Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015 May;265(1):6-21. [CrossRef]
- Zheng D, Liwinski T, Elinav E. Inflammasome activation and regula- tion: toward a better understanding of complex mechanisms. Cell Discov. 2020; 6:36. [CrossRef]
- Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 pathway in Atherosclerosis. Circ Res. 2018; 122:1722–1740. [CrossRef]
- Wang X, Feuerstein GZ, Clark RK, Yue TL. Enhanced leucocyte adhesion to interleukin-1 beta stimulated vascular smooth muscle cells is mainly through intercellular adhesion molecule-1. Cardiovasc Res. 1994 Dec;28(12):1808-14. [CrossRef]
- Wang X, Feuerstein GZ, Gu JL, Lysko PG, Yue TL Interleukin-1 beta induces expression of adhesion molecules in human vascular smooth muscle cells and enhances adhesion of leukocytes to smooth muscle cells. Atherosclerosis. 1995; 115:89–98. [CrossRef]
- González-Carnicero Z, Hernanz R, Martínez-Casales M, Barrús MT, Martín Á, Alonso MJ. Regulation by Nrf2 of IL-1β-induced inflammatory and oxidative response in VSMC and its relationship with TLR4. Front Pharmacol. 2023 Mar 2; 14:1058488. eCollection 2023. [CrossRef]
- Stackowicz J, Gaudenzio N, Serhan N, Conde E, Godon O, Marichal T, Starkl P, Balbino B, Roers A, Bruhns P, et al. Neutrophil-specific gain- of-function mutations in Nlrp3 promote development of cryopyrin- associated periodic syndrome. J Exp Med. 2021;218: e20201466. [CrossRef]
- Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015 May;25(5):308-15. [CrossRef]
- Mishra N, Schwerdtner L, Sams K, Mondal S, Ahmad F, Schmidt RE, Coonrod SA, Thompson PR, Lerch MM, Bossaller L. Cutting edge: protein arginine deiminase 2 and 4 regulate NLRP3 inflammasome-dependent IL-1β maturation and ASC speck formation in macrophages. J Immunol. 2019; 203:795–800. [CrossRef]
- Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, Dentali F, Montecucco F, Massberg S, Levi M, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021; 21:319–329. [CrossRef]
- Soehnlein O, Libby P. Targeting inflammation in atherosclerosis - from experimental insights to the clinic. Nat Rev Drug Discov. 2021; 20:589–610. [CrossRef]
- Libby P. The changing landscape of atherosclerosis. Nature. 2021; 592:524– 533. [CrossRef]
- Nording HM, Seizer P, Langer HF. Platelets in inflammation and atherogenesis. Front Immunol. 2015 Mar 6;6:98. eCollection 2015. [CrossRef]
- Lievens D, von Hundelshausen P. Platelets in atherosclerosis. Thromb Haemost. 2011 Nov;106(5):827-38. [CrossRef]
- Blann AD, Nadar SK, Lip GY. The adhesion molecule P-selectin and cardiovascular disease. Eur Heart J. 2003 Dec;24(24):2166-79. [CrossRef]
- Wagner DD, Heger LA. Thromboinflammation: From Atherosclerosis to COVID-19. Arterioscler Thromb Vasc Biol. 2022 Sep;42(9):1103-1112. [CrossRef]
- Woollard KJ, Chin-Dusting J. Therapeutic targeting of p-selectin in atherosclerosis. Inflamm Allergy Drug Targets. 2007 Mar;6(1):69-74. [CrossRef]
- Shim CY, Liu YN, Atkinson T, Xie A, Foster T, Davidson BP, Treible M, Qi Y, López JA, Munday A, Ruggeri Z, Lindner JR. Molecular Imaging of Platelet-Endothelial Interactions and Endothelial von Willebrand Factor in Early and Mid-Stage Atherosclerosis. Circ Cardiovasc Imaging. 2015 Jul;8(7):e002765. [CrossRef]
- Methia N, André P, Denis CV, Economopoulos M, Wagner DD. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood. 2001;98:1424–1428. [CrossRef]
- Doddapattar P, Dev R, Jain M, Dhanesha N, Chauhan AK. Differential Roles of Endothelial Cell-Derived and Smooth Muscle Cell-Derived Fibronectin Containing Extra Domain A in Early and Late Atherosclerosis. Arterioscler Thromb Vasc Biol. 2020 Jul;40(7):1738-1747. [CrossRef]
- Gandhi C, Khan MM, Lentz SR, Chauhan AK. ADAMTS13 reduces vascular inflammation and the development of early atherosclerosis in mice. Blood. 2012; 119:2385–2391. [CrossRef]
- Jin SY, Tohyama J, Bauer RC, Cao NN, Rader DJ, Zheng XL. Genetic ablation of Adamts13 gene dramatically accelerates the formation of early atherosclerosis in a murine model. Arterioscler Thromb Vasc Biol. 2012; 32:1817–1823. [CrossRef]
- Franck G, Mawson TL, Folco EJ, Molinaro R, Ruvkun V, Engelbertsen D, Liu X, Tesmenitsky Y, Shvartz E, Sukhova GK, et al. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: implica- tions for superficial erosion. Circ Res. 2018; 123:33–42. [CrossRef]
- Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, Winter J, Adrover JM, Santos GS, Froese A, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. 2019; 569:236–240. [CrossRef]
- Violi F, Cangemi R, Calvieri C. Hospitalization for pneumonia and risk of cardiovascular disease. JAMA. 2015 May 5;313(17):1753. [CrossRef]
- Dalager-Pedersen M, Søgaard M, Schønheyder HC, Nielsen H, Thomsen RW. Risk for myocardial infarction and stroke after community-acquired bacteremia: a 20-year population-based cohort study. Circulation. 2014; 129:1387–1396. [CrossRef]
- Johnston WF, Salmon M, Su G, Lu G, Stone ML, Zhao Y, Owens GK, Upchurch GR Jr, Ailawadi G. Genetic and pharmacologic disruption of interleukin-1β signaling inhibits experimental aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2013 Feb;33(2):294-304. [CrossRef]
- Popa-Fotea NM, Ferdoschi CE, Micheu MM. Molecular and cellular mechanisms of inflammation in atherosclerosis. Front Cardiovasc Med. 2023 Aug 3;10:1200341. eCollection 2023. [CrossRef]
- Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, Wang W, Abramowicz S, la Bastide-van Gemert S, Wang N, Welch CL, Reilly MP, Stroes ES, Moore KJ, Tall AR. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation. 2018 Aug 28;138(9):898-912. [CrossRef]
- Parzy G, Daviet F, Puech B, Sylvestre A, Guervilly C, Porto A, Hraiech S, Chaumoitre K, Papazian L, Forel JM. Venous Thromboembolism Events Following Venovenous Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Syndrome Coronavirus 2 Based on CT Scans.Crit Care Med. 2020 Oct;48(10):e971-e975. [CrossRef]
- Rajagopal K, Keller SP, Akkanti B, Bime C, Loyalka P, Cheema FH, Zwischenberger JB, El Banayosy A, Pappalardo F, Slaughter MS, Slepian MJ. Advanced Pulmonary and Cardiac Support of COVID-19 Patients: Emerging Recommendations From ASAIO-a Living Working Document. Circ Heart Fail. 2020 May;13(5):e007175. [CrossRef]
- Khoo BZE, Lim RS, See YP, Yeo SC. Dialysis circuit clotting in critically ill patients with COVID-19 infection. BMC Nephrol. 2021 Apr 20;22(1):141. [CrossRef]
- Zhang J, Tecson KM, McCullough PA. Endothelial dysfunction contributes to COVID-19-associated vascular inflammation and coagulopathy. Rev Cardiovasc Med. 2020 Sep 30;21(3):315-319. [CrossRef]
- Levy JH, Iba T, Olson LB, Corey KM, Ghadimi K, Connors JM. COVID-19: Thrombosis, thromboinflammation, and anticoagulation considerations. Int J Lab Hematol. 2021 Jul;43 Suppl 1(Suppl 1):29-35. [CrossRef]
- Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, Muenchhoff M, Hellmuth JC, Ledderose S, Schulz H, Scherer C, Rudelius M, Zoller M, Höchter D, Keppler O, Teupser D, Zwißler B, von Bergwelt-Baildon M, Kääb S, Massberg S, Pekayvaz K, Stark K. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation. 2020 Sep 22;142(12):1176-1189. Epub 2020 Jul 28. [CrossRef]
- Group RC. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2022; 399:143–51. [CrossRef]
- Investigators R-CWCftR-C, Bradbury CA, Lawler PR, Stanworth SJ, McVerry BJ, McQuilten Z, Higgins AM, Mouncey SJ, Al-Beidh F, Rowan KM, Berry LR, et al. Effect of antiplatelet therapy on survival and organ support-free days in critically ill patients with COVID-19: a random- ized clinical trial. JAMA. 2022; 327:1247–59. [CrossRef]
- Kander T. Coagulation disorder in COVID-19. Lancet Haematol. 2020;7:e630–e632. [CrossRef]
- Sinkovits G, Réti M, Müller V, Iványi Z, Gál J, Gopcsa L, Reményi P, Szathmáry B, Lakatos B, Szlávik J, Bobek I, Prohászka ZZ, Förhécz Z, Mező B, Csuka D, Hurler L, Kajdácsi E, Cervenak L, Kiszel P, Masszi T, Vályi-Nagy I, Prohászka Z. Associations between the von Willebrand Factor-ADAMTS13 Axis, Complement Activation, and COVID-19 Severity and Mortality. Thromb Haemost. 2022 Feb;122(2):240-256. [CrossRef]
- Cugno M, Meroni PL, Gualtierotti R, Griffini S, Grovetti E, Torri A, Lonati P, Grossi C, Borghi MO, Novembrino C, Boscolo M, Uceda Renteria SC, Valenti L, Lamorte G, Manunta M, Prati D, Pesenti A, Blasi F, Costantino G, Gori A, Bandera A, Tedesco F, Peyvandi F. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J Autoimmun. 2021 Jan; 116:102560. [CrossRef]
- Pascreau T, Zia-Chahabi S, Zuber B, Tcherakian C, Farfour E, Vasse M. ADAMTS 13 deficiency is associated with abnormal distribution of von Willebrand factor multimers in patients with COVID-19. Thromb Res. 2021; 204:138–140. [CrossRef]
- Afzali B, Noris M, Lambrecht BN, Kemper C. The state of comple- ment in COVID-19. Nat Rev Immunol. 2022; 22:77–84. [CrossRef]
- The Lancet Haematology. COVID-19 coagulopathy: an evolving story. Lancet Haematol. 2020 Jun;7(6):e425. [CrossRef]
- Comer SP, Cullivan S, Szklanna PB, Weiss L, Cullen S, Kelliher S, Smolenski A, Murphy C, Altaie H, Curran J, O'Reilly K, Cotter AG, Marsh B, Gaine S, Mallon P, McCullagh B, Moran N, Ní Áinle F, Kevane B, Maguire PB; COCOON Study investigators. COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol. 2021 Feb 17;19(2):e3001109. eCollection 2021 Feb. [CrossRef]
- Yatim N, Boussier J, Chocron R, Hadjadj J, Philippe A, Gendron N, Barnabei L, Charbit B, Szwebel TA, Carlier N, et al. Platelet activation in criti- cally ill COVID-19 patients. Ann Intensive Care. 2021;11:113. [CrossRef]
- Gue YX, Pula G, Lip GYH. Crizanlizumab: A CRITICAL Drug During a CRITICAL Time? JACC Basic Transl Sci. 2021 Dec 27;6(12):946-947. eCollection 2021 Dec. [CrossRef]
- Barnes BJ, Adrover JM, Baxter-Stoltzfus A, et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J Exp Med. 2020;217(6):e20200652. [CrossRef]
- Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR. Platelets induce neutrophil ex- tracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012; 122:2661–2671. [CrossRef]
- Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, Cifuni SM, Fuchs TA, von Andrian UH, Hartwig JH, et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood. 2012; 119:6335–6343. [CrossRef]
- Radermecker C, Detrembleur N, Guiot J, Cavalier E, Henket M, d’Emal C, Vanwinge C, Cataldo D, Oury C, Delvenne P, et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular com- partments in severe COVID-19. J Exp Med. 2020;217:e20201012. [CrossRef]
- Aymonnier K, Ng J, Fredenburgh LE, Zambrano-Vera K, Münzer P, Gutch S, Fukui S, Desjardins M, Subramaniam M, Baron RM, Raby BA, Perrella MA, Lederer JA, Wagner DD.. Inflammasome activation in neutrophils of patients with severe COVID-19. Blood Adv. 2022; 6:2001–2013. [CrossRef]
- Ni H, Denis CV, Subbarao S, Degen JL, Sato TN, Hynes RO, Wagner DD. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest. 2000; 106:385–392. [CrossRef]
- Ammollo C.T., Semeraro F., Xu J., Esmon N.L., Esmon C.T. 2011. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 9:1795–1803. [CrossRef]
- Brill A., Fuchs T.A., Chauhan A.K., Yang J.J., De Meyer S.F., Köllnberger M., Wakefield T.W., Lämmle B., Massberg S., Wagner D.D. 2011. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 117:1400–1407. [CrossRef]
- von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, et al. Monocytes, neu- trophils, and platelets cooperate to initiate and propagate venous throm- bosis in mice in vivo. J Exp Med. 2012; 209:819–835. [CrossRef]
- Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, Hagemann JH, Rupanagudi KV, Ryu M, Schwarzenberger C, Hohenstein B, Hugo C, Uhl B, Reichel CA, Krombach F, Monestier M, Liapis H, Moreth K, Schaefer L, Anders HJ. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. 2012 Aug;23(8):1375-88. [CrossRef]
- Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011; 187:2626–2631. [CrossRef]
- Colicchia M, Perrella G, Gant P, Rayes Novel mechanisms of thrombo-inflammation during infection: spotlight on neutrophil extracellular trap-mediated platelet activation. J.Res Pract Thromb Haemost. 2023 Mar 11;7(2):100116. eCollection 2023 Feb. [CrossRef]
- Damiana T, Damgaard D, Sidelmann JJ, Nielsen CH, de Maat MPM, Münster AB, Palarasah Y. Citrullination of fibrinogen by peptidylarginine deiminase 2 impairs fibrin clot structure. Clin Chim Acta. 2020 Feb; 501:6-11. [CrossRef]
- Osca-Verdegal R, Beltrán-García J, Paes AB, Nacher-Sendra E, Novella S, Hermenegildo C, Carbonell N, García-Giménez JL, Pallardó FV. Histone Citrullination Mediates a Protective Role in Endothelium and Modulates Inflammation. Cells. 2022 Dec 15;11(24):4070. [CrossRef]
Figure 1.
Platelet-endothelium adhesion occurs when activated endothelial surfaces express P-selectin, which interacts with platelet surface receptors GPIbα and PSGL-1 to mediate platelet rolling. Beta-3 integrins subsequently mediate firm adhesion. From Gawaz M et al J Clin Invest. 2005 Dec;115(12):3378-84. Ref 52.
Figure 1.
Platelet-endothelium adhesion occurs when activated endothelial surfaces express P-selectin, which interacts with platelet surface receptors GPIbα and PSGL-1 to mediate platelet rolling. Beta-3 integrins subsequently mediate firm adhesion. From Gawaz M et al J Clin Invest. 2005 Dec;115(12):3378-84. Ref 52.
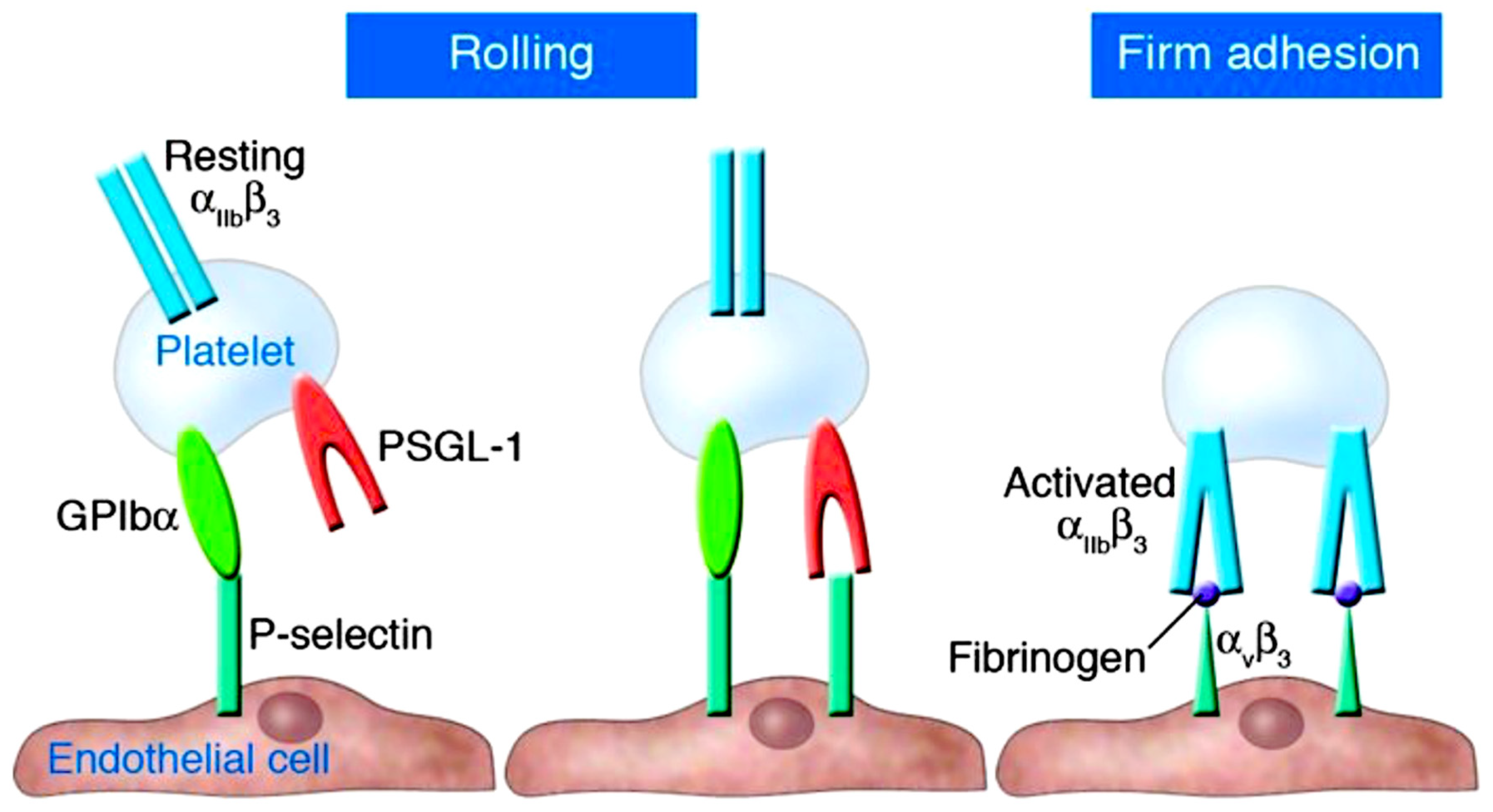
Figure 2.
The formation of Neutrophil Extracellular Traps (NETs) in severe COVID-19 patients leads to cardiac injury caused by vascular inflammation, thrombogenesis, and NETOSIS, which arise from the unstable atherosclerotic plaque. Abbreviations used: HMGB1, High Mobility Group Box 1; ISG-15, Interferon-Stimulated Gene 15; LDG, Low-Density Granulocytes; NDG, Normal Density Granulocytes; NAD, Nicotinamide Adenine Dinucleotide; ROS, Reactive Oxygen Species; SIRT3, Sirtuin 3. The same abbreviations are used as in the previous figure. The symbol ↑ represents an increase, while ↓ represents a decrease. From Nappi et al J Clin Med. 2022 Apr 27;11(9):2460. Ref 57.
Figure 2.
The formation of Neutrophil Extracellular Traps (NETs) in severe COVID-19 patients leads to cardiac injury caused by vascular inflammation, thrombogenesis, and NETOSIS, which arise from the unstable atherosclerotic plaque. Abbreviations used: HMGB1, High Mobility Group Box 1; ISG-15, Interferon-Stimulated Gene 15; LDG, Low-Density Granulocytes; NDG, Normal Density Granulocytes; NAD, Nicotinamide Adenine Dinucleotide; ROS, Reactive Oxygen Species; SIRT3, Sirtuin 3. The same abbreviations are used as in the previous figure. The symbol ↑ represents an increase, while ↓ represents a decrease. From Nappi et al J Clin Med. 2022 Apr 27;11(9):2460. Ref 57.
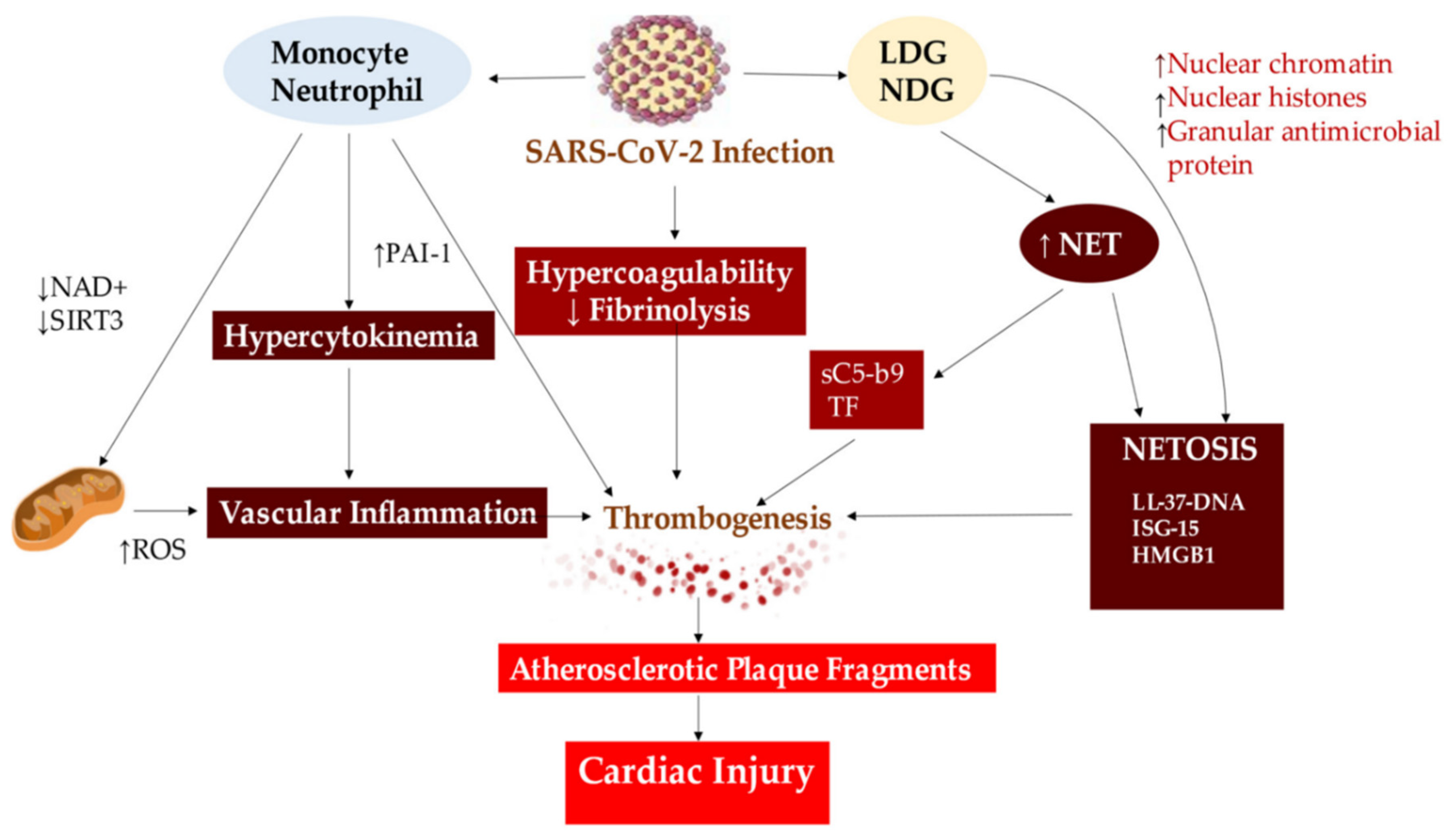
Figure 3.
Flow models were used to investigate interactions between von Willebrand factor (VWF) and neutrophils, providing insights into the relationships between the A1 domain of VWF multimers, platelets, neutrophils, and NETs under conditions of high shear flow (indicated by red arrows) and low shear flow (indicated by blue arrows). Abbreviations; ds, double strand; GP, glycoprotein.
Figure 3.
Flow models were used to investigate interactions between von Willebrand factor (VWF) and neutrophils, providing insights into the relationships between the A1 domain of VWF multimers, platelets, neutrophils, and NETs under conditions of high shear flow (indicated by red arrows) and low shear flow (indicated by blue arrows). Abbreviations; ds, double strand; GP, glycoprotein.
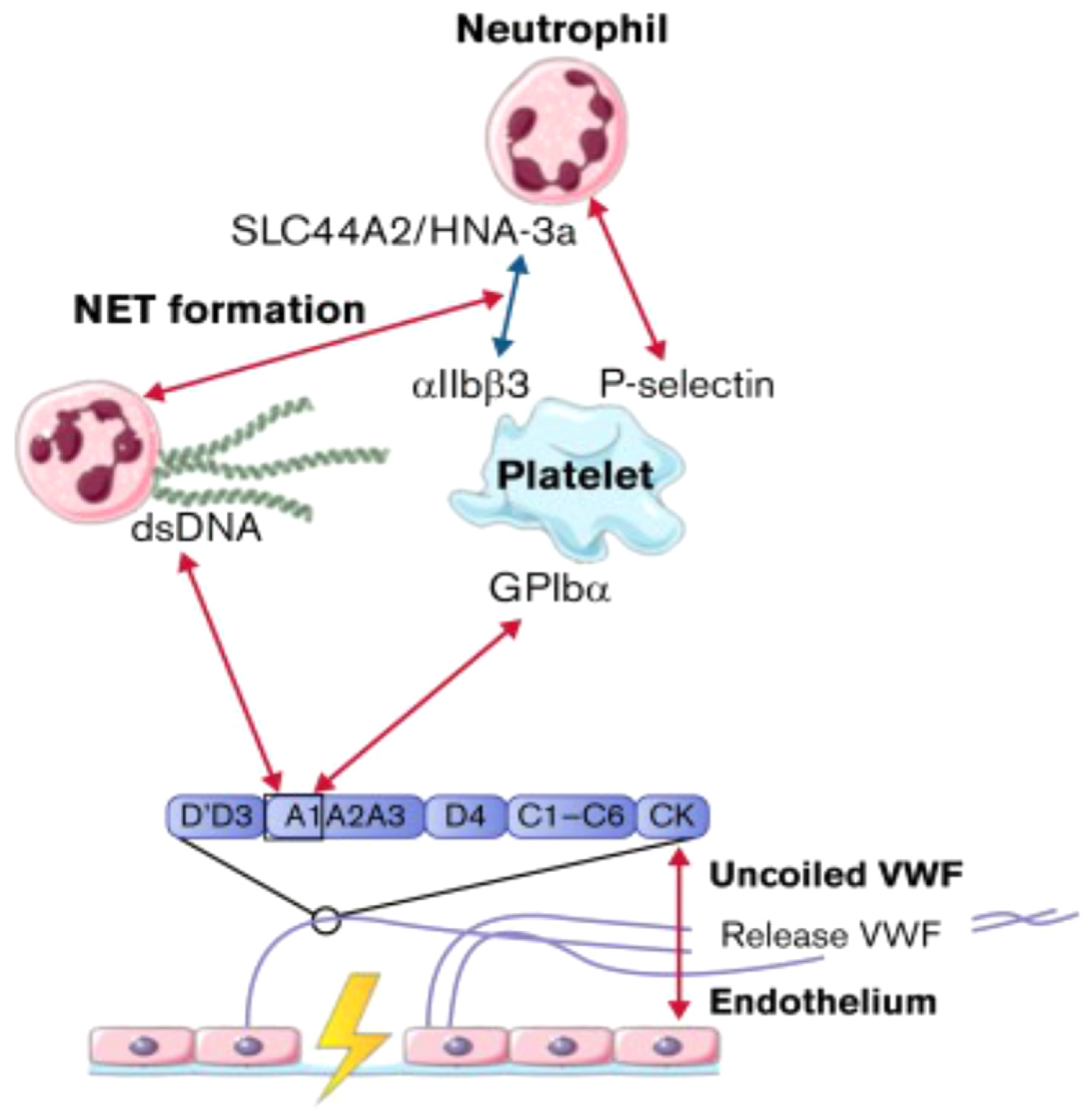
Figure 4.
Canonical inflammasomes consist of sensors from the NLR or ALR family. NLRC4 is triggered by bacterial flagellin and T3SS components, NLRP1b by anthrax lethal toxin, and AIM2 by cytosolic dsDNA. NLRP3 is stimulated by various signals, including pore-forming cytotoxins, ATP, uric acid, and alum. Once activated, the receptors produce an inflammasome complex, with or without the adaptor ASC, which attracts procaspase-1. Following that, procaspase-1 is cleaved into active caspase-1. Abbreviations: AIM2, absent in melanoma 2; ALR, AIM2-like receptor; ASC, apoptosis-associated speck-like protein containing a CARD; IL, interleukin; NLR, nucleotide-binding domain (NBD) and leucine-rich-repeat-(LRR)-containing family; T3SS, type III secretion system. From Vanaja SK et al Trends Cell Biol. 2015 May;25(5):308-15. Ref 123.
Figure 4.
Canonical inflammasomes consist of sensors from the NLR or ALR family. NLRC4 is triggered by bacterial flagellin and T3SS components, NLRP1b by anthrax lethal toxin, and AIM2 by cytosolic dsDNA. NLRP3 is stimulated by various signals, including pore-forming cytotoxins, ATP, uric acid, and alum. Once activated, the receptors produce an inflammasome complex, with or without the adaptor ASC, which attracts procaspase-1. Following that, procaspase-1 is cleaved into active caspase-1. Abbreviations: AIM2, absent in melanoma 2; ALR, AIM2-like receptor; ASC, apoptosis-associated speck-like protein containing a CARD; IL, interleukin; NLR, nucleotide-binding domain (NBD) and leucine-rich-repeat-(LRR)-containing family; T3SS, type III secretion system. From Vanaja SK et al Trends Cell Biol. 2015 May;25(5):308-15. Ref 123.
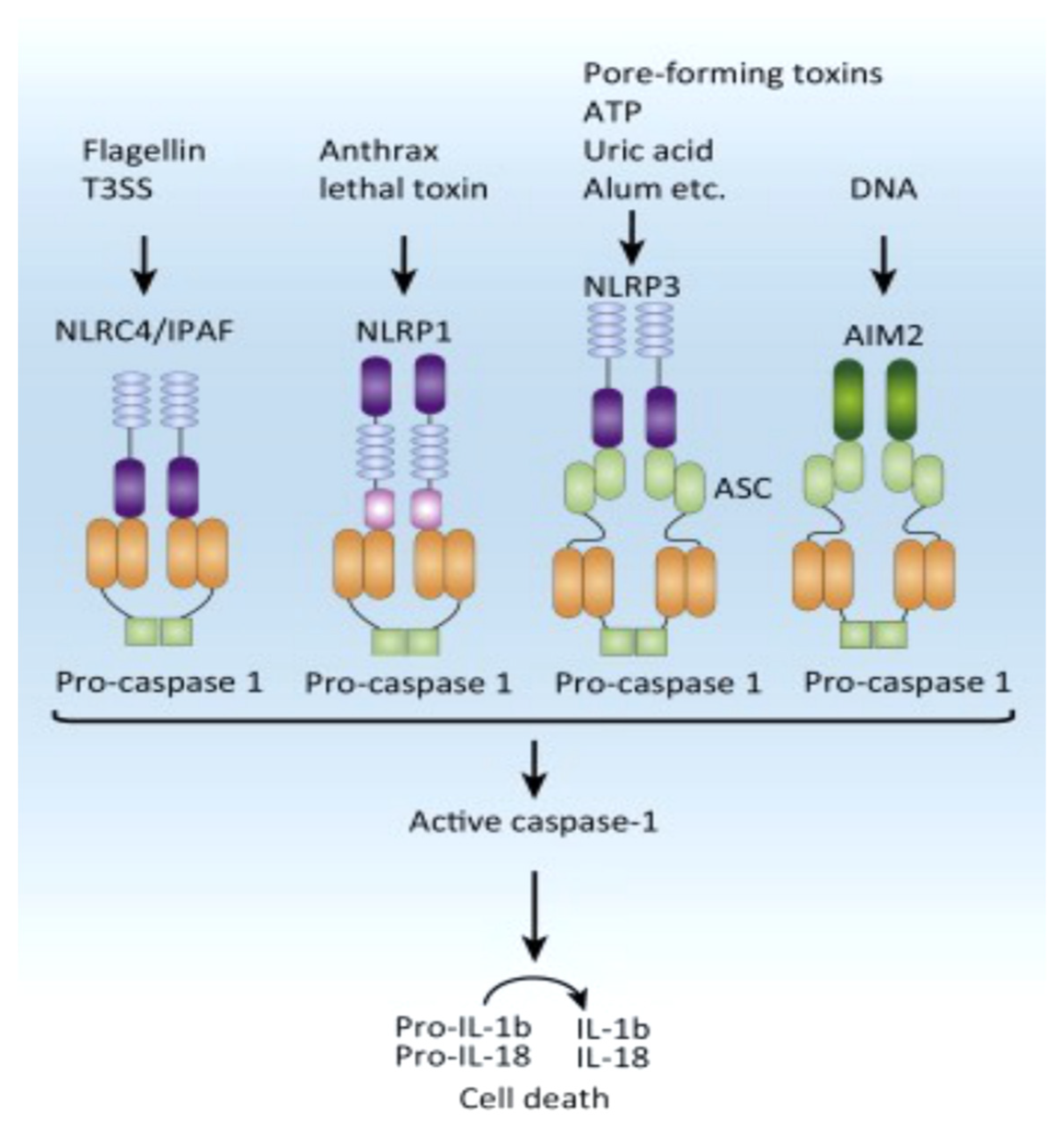
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



