
Submitted:
25 October 2023
Posted:
26 October 2023
You are already at the latest version
Abstract
Following ischemic stroke, the degradation of myelin and other cellular membranes surpasses the lipid-processing capabilities of resident microglia and infiltrating macrophages. This imbalance leads to foam cell formation in the infarct and areas of secondary neurodegeneration, instigating sustained inflammation and furthering neurological damage. Given that mitochondria are the primary sites of fatty acid metabolism, augmenting mitochondrial biogenesis (MB) may enhance lipid processing, curtailing foam cell formation and post-stroke chronic inflammation. Previous studies have shown that the pharmacological activation of the β2-adrenergic receptor (β2-AR) stimulates MB. Consequently, our study sought to discern the effects of intensified β2-AR signal-ing on MB, the processing of brain lipid debris, and neurological outcome using a mouse stroke model. To achieve this goal, aged mice were treated with formoterol, a long-acting β2-AR ago-nist, daily for two and eight weeks following stroke. Formoterol increased MB in the infarct re-gion, modified fatty acid metabolism, and reduced foam cell formation. However, it did not re-duce markers of post-stroke neurodegeneration or improve recovery. Our findings suggest that boosting MB in myeloid cells is one component of a therapeutic avenue for improving brain lipid debris processing after stroke.
Keywords:
stroke
; aging
; mitochondrial biogenesis
; beta-2-adrenergic activation
1. Introduction
Stroke is a leading cause of death and disability worldwide [1]. Following an ischemic stroke, the breakdown of myelin and other cell membranes exceeds the lipid processing abilities of both infiltrating macrophages and resident microglia. This results in the formation of foam cells within the infarct, which perpetuates a chronic inflammatory response and contributes to neurological damage (reviewed in: [2]).
In our prior research, we demonstrated that fatty acid β-oxidation, as evidenced by acyl carnitine levels, is notably elevated in myeloid cells in tissue affected by stroke for up to 4 weeks post-stroke [3]. This likely reflects the processing of myelin and other brain lipid debris by microglia and macrophages as they transition into foam cells.
Fatty acid oxidation primarily takes place in the mitochondria of a cell [4]. Therefore, increasing mitochondrial biogenesis (MB), could potentially help cells process fatty acids more efficiently and reduce foam cell accumulation and chronic inflammation after stroke.
Beta-2-adrenergic receptors (β2-ARs) are a type of G protein-coupled receptor found on the surface of various cell types including neurons, astrocytes, microglia, vascular cells, and immune cells in the periphery [5]. They play a pivotal role in the sympathetic nervous system’s response by mediating the actions of catecholamines, specifically epinephrine and norepinephrine. When activated by these catecholamines, they set off a series of intracellular events leading to a range of physiological responses, from relaxation of smooth muscles in the airways to vasodilation and increased heart rate [6]. Recent studies also indicate that β2-ARs have a role in mitochondrial biogenesis [7,8,9]. This is because β2-AR activation increases the activation of the PGC-1α (Peroxisome proliferator-activated receptor-gamma coactivator)/NRF1 (Nuclear respiratory factor 1)/TFAM (mitochondrial transcription factor A)- pathway, a key transcriptional regulator of MB [10]. This pathway is known to regulate the expression of genes involved in mitochondrial DNA replication, transcription, translation, and assembly [11].
Pharmacological stimulation of the β2-AR has previously been shown to induce mitochondrial biogenesis and improve outcome in the injured central nervous system, but not in the context of stroke [7,8]. Therefore, to build upon these previous studies, we aimed to determine how increasing β2-adrenergic signaling after stroke impacts mitochondrial biogenesis, the processing of fatty acids derived from brain lipid debris, and the formation of foam cells in the brain in a mouse model of stroke. We hypothesized that increasing β2-adrenergic signaling after stroke would promote mitochondrial biogenesis, enhance fatty acid processing, and reduce foam cell formation, thereby improving neurological outcome.
To test our hypothesis, we administered formoterol, a long-acting β2-AR agonist, daily for two and eight weeks after stroke in aged mice. We used aged mice to better represent the age of stroke patients, who are more likely to experience poorer outcomes after stroke. As clinically relevant primary endpoints, we assessed infarct size and brain atrophy using magnetic resonance imaging (MRI), neurofilament light (NF-L) release in the plasma as a biomarker of neurodegeneration, and the overall effect on recovery using a horizontal ladder test and a nest building test. We also measured cyclic adenosine monophosphate (cAMP) levels to validate that our assessment of formoterol was at a dose capable of sustaining β2-adrenergic signaling for at least two weeks after stroke.
2. Results
2.1. Study Design
The timeline of the experiment is presented in Figure 1. Briefly, for cohort 1, mice were treated with 0.3 or 1.0mg/kg of formoterol or vehicle daily, with treatment beginning 24 hours after stroke. Mice were sacrificed at 14 days and their brains were processed for western blot, immunostaining, Boron-dipyrromethene (BODIPY) staining, and measurement of cAMP and acyl carnitine levels. Their plasma was processed for NF-L quantitation. In cohort 2, mice were administered 0.3 mg/kg of formoterol or vehicle daily starting 24 hours post-stroke and were sacrificed at 8 weeks post-stroke. Behavioral assessments were conducted pre-stroke, 24 hours post-stroke, and then weekly. At 3 days post-stroke, infarct size was determined using MRI. Ventricle size was evaluated by MRI as a marker of brain atrophy at both 2 and 8 weeks post-stroke.
2.2. Formoterol administration induces MB in the infarct
Upon binding to the β2-AR, formoterol activates the Gα-subunit, which in turn stimulates adenylate cyclase [10]. This cascade leads to an accumulation of cyclic adenosine monophosphate (cAMP) as depicted in Figure 2a. Our metabolomic analysis revealed that when mice were administered formoterol at 0.3 mg/kg for two weeks after stroke, the concentration of cAMP in the infarct was increased compared to vehicle-treated mice (Figure 2b). However, this rise in the cAMP concentration wasn’t observed at a 1.0 mg/kg dose. Notably, while the activation of the Gα subunit isn’t required for MB, the release of the Gβγ heterodimer post formoterol-β2AR binding plays a crucial role [10] (Figure 2a).
The release of the Gβγ heterodimer stimulates phosphoinositide 3-kinase (PI3K), which activates the serine/threonine kinase Akt [10]. This sequence of events leads to the phosphorylation of endothelial nitric oxide synthase (eNOS), triggering nitric oxide (NO) production. Consequently, NO activates soluble guanylyl cyclase (sGC), causing an accumulation of cyclic guanosine monophosphate (cGMP). This cascade results in increased expression of TFAM and increased levels of NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial (NDUSF1) —a marker of MB [12] (Figure 2a).
Mice treated with 0.3 mg/kg of formoterol for 2 weeks exhibited significant increases in mitochondrial DNA (mtDNA) copy numbers and elevated levels of TFAM and NDUSF1 within the infarcted region (Figure 2c). Extending this treatment to 8 weeks saw a rise in mtDNA copy numbers and TFAM, with a noticeable trend towards higher NDUSF1 levels (Figure 2d). Yet, when the dosage was amplified to 1.0 mg/kg over a 2-week span, these enhancements were no longer statistically significant (Figure 2e). These data demonstrate that treating mice with 0.3mg/kg of formoterol induces mitochondrial biogenesis in the infarct, and that treatment with 1.0mg/kg has a diminished effect, perhaps due to β2-AR receptor desensitization in response to excessive stimulation.
2.3. Formoterol administration alters phagocytic cell morphology in the infarcted region
To evaluate the effect of 2 weeks of formoterol treatment on microglia and macrophage morphology, we used Iba-1 as a pan marker for microglia and macrophages, and CD68 to identify activated, phagocytotic microglia or macrophages. In the area of gliosis adjacent to the infarct core (depicted in Figure 3a), there was an increased number of Iba-1 positive cells in formoterol-treated mice compared to the vehicle-treated group (Figure 3b-c). Furthermore, the process length of these cells was shorter in the formoterol group (Figure 3d). The immunoreactive area associated with CD68 expression was significantly larger in mice treated with formoterol than in those receiving the vehicle (Figure 3e-f). This suggests that the microglia and macrophages in the infarcted area of the brains of formoterol-treated mice are more activated, more phagocytotic, and more numerous than in vehicle-treated mice. Meanwhile, GFAP staining, indicative of activated astrocytes, revealed neither morphological nor expression level differences between the two groups (Figure 3g-h).
2.4. Lipid droplets were significantly reduced in the infarcts of mice receiving formoterol
BODIPY 492/515, a green, fluorescent lipophilic dye, is frequently employed to label lipid droplets [13]. Given our observations suggesting enhanced phagocytic activity in the microglia and macrophages of formoterol-treated mice, we aimed to assess lipid droplet accumulation within the infarcted regions of these mice. To account for potential autofluorescence in stroke-infarcted regions, we imaged unstained brain sections in parallel with BODIPY-stained sections, using the same fluorescence channel (Figure 4a). No fluorescence was observed in the unstained sections at an exposure time of 1/150 seconds (first image in Figure 4a). Detectable autofluorescence in the unstained section necessitated an extended exposure of 1.2 seconds (second image in Figure 4a). However, the BODIPY-stained sections displayed robust fluorescence even at the shorter 1/150 seconds exposure (third image in Figure 4a). We noted a pronounced decrease in BODIPY fluorescence in the infarcted regions of formoterol-treated mice compared to controls (Figure 4b, with representative images in Figure 4c). Figure 4d presents a composite image of the infarcted region, highlighting Iba-1, CD68, and BODIPY staining, underscoring that the majority of the staining is intracellular and within microglia or macrophages. Collectively, these findings indicate that the microglia/macrophages in the infarcts of formoterol-treated mice contain fewer lipid droplets. This supports the hypothesis that they process brain lipid debris more efficiently than their counterparts in vehicle-treated mice.
2.5. Formoterol reduced levels of acyl carnitines in the infarcted region
Following an injury to the CNS, such as a stroke, damaged, or apoptotic cells —including their myelin debris— are taken up by phagocytosis into phagolysosomes. Within these compartments, enzymes like lysosomal acid lipase degrade the ingested myelin lipids, releasing free fatty acids and other lipid derivatives [14]. Once liberated, these fatty acids must cross the lysosomal membrane to reach the cytosol. Here, they bind to fatty acid-binding proteins (FABPs), which facilitate their transport to various cellular destinations: the mitochondria for β-oxidation, the endoplasmic reticulum for lipid synthesis, or other sites for lipid-mediated signaling [15].
For β-oxidation in the mitochondria, the cytosolic fatty acids are first activated to fatty acyl-CoA molecules by the enzyme acyl-CoA synthetase (also known as fatty acid CoA ligase) [16]. Next, on the outer mitochondrial membrane, carnitine palmitoyltransferase I (CPT1) transfers the fatty acyl group from CoA to carnitine, producing acyl carnitine. This acyl-carnitine is shuttled across the inner mitochondrial membrane by a translocase protein, which, in the process, exports a free carnitine molecule from the mitochondria to the intermembrane space. Once within the mitochondrial matrix, carnitine palmitoyltransferase II (CPT2) reconverts acyl carnitine back to fatty acyl-CoA and a free carnitine molecule. The fatty acyl-CoA then undergoes β-oxidation, sequentially cleaving two-carbon units in the form of acetyl-CoA, while also producing NADH and FADH2. These acetyl-CoA units can subsequently enter the citric acid (Krebs) cycle, driving further oxidative metabolism and energy generation (Figure 5a).
Considering this pathway, we assessed acyl carnitine and carnitine levels as markers for fatty acid beta-oxidation and the metabolic breakdown of myelin and other brain lipid debris. As anticipated, and as we have shown previously [3], acyl carnitine levels in the infarcted region of the ipsilateral hemisphere of the vehicle-treated mice were markedly elevated compared to those in the equivalent region of the contralateral hemisphere. Contrastingly, formoterol-treated brains exhibited a significant reduction in acyl carnitine levels in the infarcted region compared to those treated with the vehicle (Figure 5b). Carnitine concentrations followed a similar pattern; they were notably augmented in the stroked tissue, but formoterol treatment aligned these levels more closely with those in the unaffected, contralateral cortex (Figure 5c).
Cumulatively, these results support that two weeks of formoterol treatment post-stroke optimizes fatty acid metabolism and enhances the clearance of brain lipid debris within the infarcted region.
2.6. Formoterol did not alter infarct size, ventricle size, or plasma NF-L release
Having observed changes in lipid processing after two weeks of treatment with 0.3mg/kg of formoterol, we sought to evaluate its impact on post-stroke neurodegeneration. Neurofilament light chain (NF-L) is one of the subunits of neurofilaments, which are intermediate filament proteins found in neurons. Elevated levels of NF-L in blood plasma are associated with neuronal damage or axonal injury [17]. Accordingly, we measured NF-L concentrations in the plasma of the mice that underwent vehicle- or formoterol treatment at 2 weeks post stroke. While stroke markedly raised NF-L plasma levels in comparison to age-matched naive mice, formoterol administration yielded no significant changes (Figure 6a). We then assessed infarct size at 3 days following stroke, and ventricle size at 2 weeks and 8 weeks after stroke as a marker of brain atrophy. No discernable differences were detected between the groups for any evaluated timepoint (Figure 6b-d). Together these results indicate that formoterol did not have a notable effect on post-stroke neurodegeneration.
2.7. Formoterol treatment did not alter motor recovery
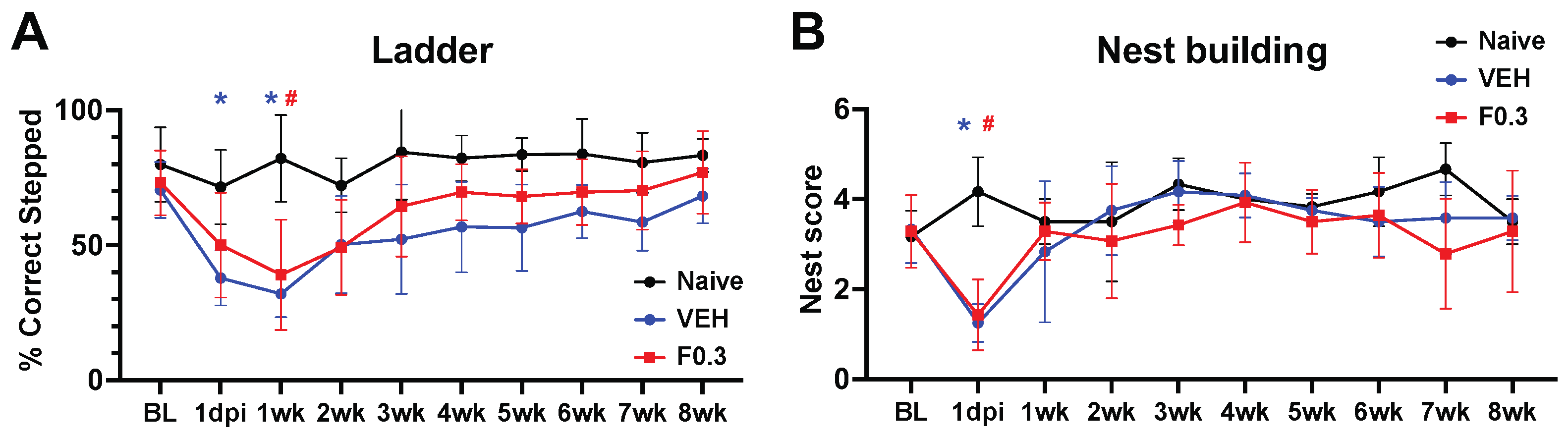
We assessed motor recovery of the mice using the ladder and nest building tests. In the ladder test, stroked mice performed notably worse than age-matched naïve controls. However, formoterol treatment didn’t enhance recovery compared to vehicle-treated mice, as determined by repeated measures ANOVA (Figure 7a). Similarly, in the nest building test, both the vehicle and formoterol-treated mice exhibited significant deficits on day 1 post-stroke compared to age-matched naïve mice. Yet, no discernible difference between the formoterol and vehicle groups was identified using repeated measures ANOVA (Figure 7b).
3. Discussion
The effect of β2-AR activation on stroke outcomes is complex and has yielded mixed results in the literature. This may be due to a number of factors, including the different stroke models used, the age of the animals, the method of activation or inactivation, and the starting time of the treatment in relation to the time of the ischemia.
Some studies have shown that β2-AR activation can be beneficial in stroke. For example, Culmsee et al. (1999) demonstrated that pretreatment with a β2-AR agonist could decrease infarct size in both mice and rats [18]. However, other studies have shown that β2-AR activation can be harmful in stroke. For example, Lechtenberg et al. (2019) observed that treatment with clenbuterol 3 hours post-photothrombotic stroke not only enlarged the lesion size but also altered microglial morphology and reduced TNFα and IL-10 expression in mice [19]. Similarly, Sun et al. (2017) discovered that β2-AR blockade can mitigate blood-brain barrier (BBB) damage during acute cerebral ischemia in mice, suggesting that β2-AR activation can contribute to BBB damage [20].
However, our research uniquely explores the chronic effects of β2-AR activation post-stroke on MB, lipid metabolism, and foam cell formation. Previous work from our team has demonstrated formoterol’s capacity to induce MB across diverse cell types, encompassing renal proximal tubule cells, cardiomyocytes, and skeletal muscle cells, as well as in various disease models [10,21,22]. For instance, formoterol treatment enhanced mitochondrial function and ATP production in a mouse model of acute kidney injury [23,24]. Furthermore, a 2018 publication from our group highlighted the benefits of formoterol in reducing neuronal death, augmenting motor function, and promoting MB in a mouse model of SCI, while also attenuating spinal cord inflammation [8]. Moreover, studies like the one by Lee et al. (2013) have identified formoterol’s potential to augment fatty acid oxidation in human tissue [25].
In this study, we administered formoterol to aged mice after experimental stroke and observed that the 0.3 mg/kg dose yielded positive results, including increased mitochondrial biogenesis, and reduced foam cell accumulation. Interestingly, the higher dose of 1.0 mg/kg did not replicate these effects, which raises the possibility of β2-AR desensitization at elevated doses [26].
Following two weeks of treatment, a decrease in acyl-carnitine levels within the infarcted brain region was evident, suggesting a modulation in lipid metabolism. While this could mean that formoterol accelerates lipid metabolism in microglia and macrophages, another interpretation revolves around the inherent metabolic dysfunction within foam cells [27]. This dysfunction could lead to incomplete beta-oxidation of fatty acids and result in acyl-carnitine accumulation in the infarcted region. Supporting this possibility, elevated acyl-carnitine levels have been associated with an incomplete fatty acid oxidation process [28]. In such a scenario, the intervention of formoterol might serve to re-establish mitochondrial functionality in microglia and macrophages, thereby facilitating the complete beta-oxidation of fatty acids.
Despite these potential therapeutic mechanisms, our MRI and NF-L assay analyses showed no neuroprotective effects of formoterol. Furthermore, there was no observable difference in motor recovery post-stroke between formoterol-treated mice and control mice. Given the known side effects of formoterol, such as tachycardia and tremors, and its association with worsened cardiac function, the adverse effects might overshadow its benefits, especially in aged mice [29]. It is also important to note that formoterol is a potent β2-AR agonist, and research by McCulloch et al has revealed that adrenergic signaling, triggered by the surge in sympathetic activity after stroke, decreases the number of B cells in the periphery and contributes to post stroke immunosuppression [30]. This immunosuppressive effect may be amplified by formoterol administration after stroke.
In conclusion, our study sheds light on the potential of mitochondrial function enhancement in microglia and macrophages as a therapeutic strategy post-stroke. While formoterol did not improve structural or motor recovery, it could be valuable in a multi-drug therapy. Further research is required to determine the optimal dose, timing, and safety of formoterol treatment after stroke, as well as to explore alternative methods of enhancing mitochondrial function in myeloid cells.
4. Materials and Methods
4.1. Animals
In total, 86 aged (18- to 20-month-old) male C57BL/6J mice, sourced from the aged rodent colony at the National Institute on Aging, were used in this study. The mice were housed in a temperature-controlled facility with 12h light/dark cycles and free access to food and water. They were divided randomly (using GraphPad Prism Quick Calcs) into groups receiving either Formoterol (0.3 mg/kg or 1 mg/kg) or vehicle (DMSO) for 14 days following stroke, or into groups receiving Formoterol (0.3 mg/kg) or vehicle (DMSO) for 8 weeks after stroke. Six of these mice were harvested as naïve controls without any surgeries or treatments. The pre-set exclusion criteria of the study were (1) unsuccessful induction of ischemia (5 mice), (2) death of the animal during the experiment (8 mice), and (3) being a statistically significant outlier in any of the analyses (4 mice). All animal experiments followed the NIH guidelines and were approved by the University of Arizona Institutional Animal Care and Use Committee.
4.2. Stroke surgeries
Permanent distal middle cerebral artery occlusion (dMCAO) was performed following previously established methods [31]. Mice were anesthetized using 3% isoflurane (JD Medical, Phoenix, AZ, USA), which was maintained during surgery. Upon exposing and retracting the temporalis muscle, a 1 mm hole was drilled into the temporal bone to reveal the MCA. After dura removal, the artery was cauterized. The temporalis muscle was then repositioned, and the incision sealed with surgical glue. Throughout the operation, a heating pad with a rectal probe ensured the animals’ body temperature remained at 37 ± 0.5 °C. Both respiration and temperature were consistently monitored. Immediately after surgery, mice were placed in a hypoxia chamber (Coy Laboratory products, Grass Lake, MI, USA) containing 9% oxygen and 91% nitrogen for 45 minutes. The purpose of hypoxia in this model is to both increase infarct size and reduce variability in infarct size [31]. A single dose of buprenorphine hydrochloride (Buprenex® Injection 0.3 mg/mL, Henry Schein Medical, Melville, NY, USA; 0.1 mg/kg s.c.) was administered prior to surgery, and sustained release buprenorphine (Buprenorphine SR 1 mg/mL, Zoopharm LLC, Laramie, WY, USA; 1 mg/kg s.c.) was administered 24 hours after surgery as a post-operative analgesic. For pain management, buprenorphine hydrochloride (Buprenex® Injection 0.3 mg/mL, Henry Schein Medical, Melville, NY, USA; 0.1 mg/kg s.c.) was given before the procedure. A subsequent dose of sustained-release buprenorphine (Buprenorphine SR 1 mg/mL, Zoopharm LLC, Laramie, WY, USA; 1 mg/kg s.c.) was provided 24 hours post-surgery.
4.3. Perfusion and tissue collection
Fourteen days or eight weeks post-stroke, mice were anesthetized using 3% isoflurane. Blood samples were then drawn from the heart, followed by a transcardial perfusion using a 0.9% saline solution. For immunohistochemistry (IHC), whole brains were extracted and immediately fixed in 4% PFA in PBS for 22 hours at 4°C. Subsequently, they were immersed in a 30% sucrose solution at 4°C for a minimum of 48 hours before further processing. Using a Microm HM 450 sliding microtome (Thermo Fisher Scientific), coronal sections with a thickness of 40 µm were prepared spanning the entire brain. For biochemical evaluations, the infarct, peri-ischemic regions, and their equivalent areas on the contralateral side were excised and rapidly frozen in liquid nitrogen. Blood samples from control mice were collected in a similar fashion. Once drawn, the blood was centrifuged at 10,000 rpm for 15 minutes at 4ºC. The plasma layer was carefully collected, ensuring no disturbance to other layers, and immediately frozen in liquid nitrogen.
4.4. Immunostaining
Immunostaining was performed on free-floating or pre-mounted brain sections using standard protocols. Primary antibodies against CD68 (1:1000; Bio-Rad, cat. MCA1957GA), ionized calcium-binding adapter molecule 1 (IBA-1, 1:500; Wako cat. 019-19741), and glial fibrillary acidic protein (GFAP; 1:2000; Millipore Sigma, cat. AB5541) were used in conjunction with the appropriate secondary antibodies. Sections were imaged using a Leica DM6000B (Buffalo Grove, IL) light microscope coupled with a Leica DFC 7000 T camera, or confocal microscope (Zeiss NLO 880, San Diego, CA).
4.5. Microglia morphology analysis
For microglia morphology analysis, images were acquired on a confocal microscope (Zeiss NLO 880, San Diego, CA) using a 40X objective (236.16 x 236.16-micron area) from the area of gliosis proximal to the core of the infarct and corresponding area of the contralateral side. Microglia morphology was analyzed from 8-bit images, as described previously [32]. Microglia morphology parameters (process length and number of endpoints) were summed, and all data were divided by the cell soma count per image frame to calculate the summed microglia process length/cell. The average values from each region within each animal were used for statistical analysis.
4.6. Boron-dipyrromethene (BODIPY) 492/515 staining procedure
Tissue sections were allowed to air-dry overnight at room temperature (RT). Following the drying, sections underwent a series of washing steps in fresh 0.1M PBS at RT. Each wash lasted for 10 minutes, during which gentle agitation was applied, and any excess PBS was carefully removed after each cycle. For staining, a working solution of BODIPY 492/515 was formulated by diluting the stock (0.1 mg/ml in dimethyl sulfoxide, DMSO) to a final concentration of 1:25. The tissue sections were then submerged in this solution and incubated for 30 minutes in a dark environment at RT. Post-incubation, coverslips were applied using Vectashield HardSet Antifade mounting media to prepare the samples for subsequent fluorescence microscopy analysis.
4.7. Magnetic resonance imaging (MRI)
Infarct volumes and ventricle sizes were assessed by magnetic resonance imaging (MRI) at time points 3 days, 2 weeks, and 8 weeks after stroke using a Bruker Biospec 70/20 7.0T scanner with ParaVision-360.2.0 software and a 4-channel phase array mouse coil (Bruker Biospin GmbH, Germany). Mice were placed in a cradle equipped with a stereotaxic frame, an integrated heating system to maintain body temperature at 37±1°C, and a pressure probe to monitor respiration. During MRI acquisition, anesthesia was maintained by inhalation of 1.5-3% isoflurane. High-resolution structural images were acquired using a T2-weighted RARE multi-echo Bruker pulse sequence with the following parameters: repetition time (TR) = 2000 ms; flip angle = 180°; RARE factor = 12; matrix size = 128 x 128; averages = 2; field of view (FOV) = 1.92 cm x 1.92 cm; slice thickness = 0.8 mm; number of slices = 15; acquisition time = 8 min 53 s. Infarcts and hemispheric cross sections were manually delineated on T2-weighted images using Mango v4.1. Infarct volumes were calculated using the following equation: overall infarct volume × (1 − [ipsilateral hemisphere volume − contralateral hemisphere volume]/contralateral hemisphere volume) [33], after which they were normalized to vehicle. Ventricle sizes were measured with Mango and subsequently normalized to the contralateral ventricles. 12 slices were analyzed per mouse for each parameter.
4.8. Western Blot
Protein was extracted from the stroke infarcts using RIPA buffer with protease inhibitor cocktail (1:100), 1 mM sodium fluoride and 1 mM sodium orthovanadate (Sigma-Aldrich, St. Louis, MO) as described previously [8]. Protein was quantified using a bicinchoninic acid assay, and 10 μg of protein was separated via electrophoresis using 4–15% SDS-PAGE gels, then transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA). Membranes were blocked in 5% milk in TBST and incubated overnight with primary antibodies with constant agitation at 4 °C. Membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody and visualized using chemiluminescence (Thermo Scientific, Waltham, MA) on a GE ImageQuant LAS4000 (GE Life Sciences, Pittsburg, PA). Optical density was determined using Image Studio Lite software. Primary antibodies used were TFAM, NDUFS1 (1:1000) and α-tubulin (1:10,000, Abcam).
4.9. Ladder test
Motor recovery following stroke was evaluated using the Ladder test [34] on days 1 and 7 post-stroke and then weekly up to 8 weeks post-stroke. The Ladder test apparatus had plexiglass rungs, each 10.16 cm long and 0.32 cm in diameter, spaced 1.6 cm apart. These rungs were situated between two plexiglass walls, each 76.20 cm in length and 15.24 cm in height. The walls were set 3.18 cm apart, allowing ample room for the mice to traverse. The entire setup was elevated 18 cm above the table using blocks. Tests were conducted in a dimly lit room. A desk lamp lit the starting zone to motivate the mice to cross the ladder towards a small box at the end zone. At the commencement of the test, mice were positioned in a plastic chamber aligned with the ladder’s starting rungs. As each mouse navigated the ladder, its footfalls were captured by a camera, positioned beneath, and moved synchronously with the mouse along the table. Before official testing, each mouse underwent 8 training trials to attain a performance error rate of ≤10-12%. On actual test days, each mouse was evaluated twice, and the resultant scores were averaged. Between each trial, the ladder was cleaned to eliminate olfactory cues. Video footage of each mouse’s traversal was reviewed at playback speeds of 0.25-0.30x using standard video playback software. A skilled observer examined the front limb contralateral to the stroke infarct to tally correct and incorrect steps. Videos were scored independently by two trained examiners, with their scores averaged for final analysis. The percentage of accurate foot placements was determined as 100 x (number of correct steps / (number of correct steps + number of missteps)).
4.10. Nest building test
The nest building test was administered to evaluate the mice at 1 day and 1-week post-ischemia, followed by weekly assessments up to the 8-week mark. Each mouse was individually housed in a cage containing a cotton nestlet. They were permitted to engage with the nestlet overnight. The following morning, the degree of nestlet shredding was assessed. Scoring ranged from 0 (nestlet untouched) to 5 (nestlet fully shredded and nest constructed), as detailed in Deacon (2006) [35].
4.11. Neurofilament light -assay
Neurodegeneration levels were evaluated using terminal plasma samples at 2 weeks following stroke. These samples were forwarded to PBL Assay Science (Piscataway, NJ, USA) for analysis using the Simoa™ NF-Light® kit (Cat #103186, Quanterix, Billerica, MA, USA). Plasma from age-matched naïve mice (aligned in age with the formoterol and vehicle-treated groups) served as a control.
4.12. Measurement of cAMP and acyl carnitines
Analysis of cAMP and acyl carnitine levels in brain tissue was performed by Metabolon Inc as previously described [3]. Briefly, Brain tissue underwent cAMP and acyl carnitine profiling by Metabolon Inc. using UPLC-MS/MS. Upon receipt, samples were stored at −80°C and prepared with the MicroLab STAR® system. Proteins were precipitated with methanol to recover diverse metabolites, followed by fractionation for different analysis modes. Organic solvents were evaporated using TurboVap®, and sample extracts were stored under nitrogen before analysis.
Multiple controls were employed: a pooled sample for technical replication, water samples as process blanks, and QC standards spiked into every sample for instrument performance and chromatographic alignment. Instrument and process variabilities were determined by median RSD calculations. The UPLC-MS/MS utilized Waters ACQUITY UPLC and Thermo Scientific Q-Exactive spectrometer with a HESI-II source.
Data extraction involved Metabolon’s systems to peak-identify and QC process. cAMP and the acyl carnitines were identified against Metabolon’s library of authenticated standards, considering retention time/index, mass-to-charge ratio, and MS/MS spectral data.
4.13. Statistics
Statistical analyses were performed using GraphPad Prism software 9.3.1 (GraphPad Software, LaJolla, CA, USA), and normality was assessed using the Kolmogorov–Smirnov test. Statistical tests and sample sizes are provided in each figure legend, and p values less than 0.05 were considered to be statistically significant. Statistically significant outliers, calculated using GraphPad Prism QuickCalcs, were excluded from the datasets. Data are presented as mean ± SD. Median-scaled raw data was used to generate the heat maps in Figure 5 and Welch’s two-sample t-test was used to test whether two unknown means were different between two independent populations.
Author Contributions
Conceptualization, Rick Schnellmann and Kristian Doyle; Funding acquisition, Rick Schnellmann and Kristian Doyle; Investigation, Sanna Loppi, Marco Tavera-Garcia, Natalie Scholpa and Boaz Maiyo; Methodology, Danielle Becktel, Helena Morrison, Rick Schnellmann and Kristian Doyle; Project administration, Kristian Doyle; Resources, Helena Morrison and Kristian Doyle; Supervision, Rick Schnellmann and Kristian Doyle; Visualization, Sanna Loppi, Boaz Maiyo and Helena Morrison; Writing – original draft, Sanna Loppi and Kristian Doyle; Writing – review & editing, Sanna Loppi, Helena Morrison, Rick Schnellmann and Kristian Doyle. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NINDS RF1NS131110, NIA R01AG063808, United States Department of Veterans Affairs I01RX003224, NIH S10 OD025016, and the Leducq Foundation Transatlantic Network of Excellence Stroke-IMPaCT.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care & Use Committee (IACUC) of the University of Arizona.
Data Availability Statement
The data used in this research are available upon request from the corresponding author, in compliance with the ethical and legal considerations governing data sharing and confidentiality.
Acknowledgments
Confocal and MRI imaging was performed with the use of the Office of Research, Innovation and Impact’s Optical and MRI Imaging Core Facilities at the University of Arizona. We thank the core managers and technicians for their help with all data collection. We are also grateful to Metabolon Inc. for their expertise and assistance throughout all aspects of our study. Schematics created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Katan M, Luft A. Issue Theme Global Health Neurology. Neurol. 2018 [accessed 2023 Oct 12];38:208–211. [CrossRef]
- Zbesko JC, Stokes J, Becktel DA, Doyle KP. Targeting foam cell formation to improve recovery from ischemic stroke. Neurobiology of Disease. 2023 [accessed 2023 Oct 13];181:106130. [CrossRef]
- Loppi SH, Tavera-Garcia MA, Becktel DA, Maiyo BK, Johnson KE, Nguyen TV V., Schnellmann RG, Doyle KP. Increased fatty acid metabolism and decreased glycolysis are hallmarks of metabolic reprogramming within microglia in degenerating white matter during recovery from experimental stroke. Journal of Cerebral Blood Flow and Metabolism. 2023 [accessed 2023 Oct 13];43(7):1099–1114. [CrossRef]
- Talley JT, Mohiuddin SS. Biochemistry, Fatty Acid Oxidation. StatPearls. 2023 Jan 16 [accessed 2023 Oct 13]. https://www.ncbi.nlm.nih.gov/books/NBK556002/.
- Kumar Velmurugan B, Baskaran R, Huang C-Y. Detailed insight on β-adrenoceptors as therapeutic targets. 2019 [accessed 2023 Oct 14]. [CrossRef]
- Abosamak NR, Shahin MH. Beta2 receptor agonists and antagonists. 2023.
- Vekaria HJ, Hubbard WB, Scholpa NE, Spry ML, Gooch JL, Prince SJ, Schnellmann RG, Sullivan PG. Formoterol, a β2-adrenoreceptor agonist, induces mitochondrial biogenesis and promotes cognitive recovery after traumatic brain injury. Neurobiology of disease. 2020 [accessed 2023 Oct 14];140. https://pubmed.ncbi.nlm.nih.gov/32289370/. [CrossRef]
- Scholpa NE, Simmons EC, Tilley DG, Schnellmann RG. β2-adrenergic receptor-mediated mitochondrial biogenesis improves skeletal muscle recovery following spinal cord injury. Experimental neurology. 2019 [accessed 2023 Oct 14];322. https://pubmed.ncbi.nlm.nih.gov/31525347/. [CrossRef]
- Cameron RB, Beeson CC, Schnellmann RG. Development of Therapeutics That Induce Mitochondrial Biogenesis for the Treatment of Acute and Chronic Degenerative Diseases HHS Public Access. J Med Chem. 2016 [accessed 2023 Oct 13];59(23):10411–10434. [CrossRef]
- Wills LP, Trager RE, Beeson GC, Lindsey CC, Peterson YK, Beeson CC, Schnellmann RG. The 2-Adrenoceptor Agonist Formoterol Stimulates Mitochondrial Biogenesis. The Journal of Pharmacology and Experimental Therapeutics. 2012 [accessed 2023 Oct 15];342:106–118. [CrossRef]
- Taherzadeh-Fard E, Saft C, Akkad DA, Wieczorek S, Haghikia A, Chan A, Epplen JT, Arning L. PGC-1alpha downstream transcription factors NRF-1 and TFAM are genetic modifiers of Huntington disease. 2011 [accessed 2023 Oct 14]. http://www.molecularneurodegeneration.com/content/6/1/32. [CrossRef]
- UCSC Genome Browser at genome.ucsc.edu. Accessed. 2023 Oct 13.
- Kyong Fam T, Klymchenko AS, Collot M. Recent Advances in Fluorescent Probes for Lipid Droplets. Materials. 2018 [accessed 2023 Oct 15];11. www.mdpi.com/journal/materials. [CrossRef]
- Li F, Zhang H. Lysosomal Acid Lipase in Lipid Metabolism and Beyond. Arterioscler Thromb Vasc Biol. 2019 [accessed 2023 Oct 14];39:850–856. https://www.ahajournals.org/journal/atvb. [CrossRef]
- Longo N, Frigeni M, Pasquali M. Carnitine transport and fatty acid oxidation ☆. 2016 [accessed 2023 Oct 14]. [CrossRef]
- Houten SM, Violante S, Ventura F V, Wanders RJA. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. The Annual Review of Physiology is online at. 2016 [accessed 2023 Oct 15];78:23–44. www.annualreviews.org. [CrossRef]
- Shanker A, Narayanan S, Khera T, Subramaniam B. Faculty Opinions Neurofilament light: a narrative review on biomarker utility. Faculty Reviews. 2021 [accessed 2023 Oct 14];10(46). [CrossRef]
- Culmsee C, Stumm RK, Schäfer MKH, Weihe E, Krieglstein J. Clenbuterol induces growth factor mRNA, activates astrocytes, and protects rat brain tissue against ischemic damage. European Journal of Pharmacology. 1999 [accessed 2023 Oct 14];379(1):33–45. https://pubmed.ncbi.nlm.nih.gov/10499369/. [CrossRef]
- Lechtenberg KJ, Meyer ST, Doyle JB, Peterson TC, Buckwalter MS. Augmented β2-adrenergic signaling dampens the neuroinflammatory response following ischemic stroke and increases stroke size. Journal of Neuroinflammation. 2019 [accessed 2023 Oct 14];16(112). [CrossRef]
- Sun Y, Chen X, Zhang X, Shen X, Wang M, Wang X, Liu W-C, Liu C-F, Liu J, Liu W, et al. β2-Adrenergic Receptor-Mediated HIF-1α Upregulation Mediates Blood Brain Barrier Damage in Acute Cerebral Ischemia. 2017 [accessed 2023 Oct 14]. www.frontiersin.org. [CrossRef]
- Jesinkey SR, Korrapati MC, Rasbach KA, Beeson CC, Schnellmann RG, Johnson RH. Atomoxetine Prevents Dexamethasone-Induced Skeletal Muscle Atrophy in Mice. THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther. 2014 [accessed 2023 Oct 15];351:663–673. [CrossRef]
- Cameron RB, Gibbs WS, Miller SR, Dupre T V, Megyesi J, Beeson CC, Schnellmann RG. Proximal Tubule b 2-Adrenergic Receptor Mediates Formoterol-Induced Recovery of Mitochondrial and Renal Function after Ischemia-Reperfusion Injury s. THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther. 2019 [accessed 2023 Oct 15];369:173–180. [CrossRef]
- Arif E, Solanki AK, Srivastava P, Rahman B, Fitzgibbon WR, Deng P, Budisavljevic MN, Baicu CF, Zile MR, Megyesi J, et al. b2-adrenergic receptor agonist formoterol accelerates podocyte recovery from glomerular injury. Kidney International. 2019 [accessed 2023 Oct 15];96:656–673. www.kidney-international.org. [CrossRef]
- Vallorz EL, Janda J, Mansour HM, Schnellmann RG. Kidney targeting of formoterol containing polymeric nanoparticles improves recovery from ischemia reperfusion-induced acute kidney injury in mice. Kidney International. 2022;102(5):1073–1089. [CrossRef]
- Lee P, Day RO, Greenfield JR, Ho K. Formoterol, a highly beta2-selective agonist, increases energy expenditure and fat utilisation in men. International Journal of Obesity. 2013 [accessed 2023 Oct 14];37:593–597. [CrossRef]
- Goral V, Jin Y, Sun H, Ferrie AM, Wu Q, Fang Y. Agonist-Directed Desensitization of the b 2-Adrenergic Receptor. 2011 [accessed 2023 Oct 14]. www.plosone.org. [CrossRef]
- Bedse G, Corti A, Sorrentino R, Xiao H, Liu A, Xu W, Wei Z, Dong J, Duan F, Chen K, et al. Global Metabolomics Reveals the Metabolic Dysfunction in Ox-LDL Induced Macrophage-Derived Foam Cells. Front. Pharmacol. 2017 [accessed 2023 Oct 15];8:586. www.frontiersin.org. [CrossRef]
- Mccann MR, Vet M, De La Rosa G, Rosania GR, Stringer KA. L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine. Metabolites. 2021 [accessed 2023 Oct 15];11:51. [CrossRef]
- Guhan AR, Cooper S, Oborne J, Lewis S, Bennett J, Tattersfield AE. Systemic effects of formoterol and salmeterol: a dose-response comparison in healthy subjects. Thorax. 2000 [accessed 2023 Oct 15];55(8):650–656. www.thoraxjnl.com. [CrossRef]
- Mcculloch L, Smith CJ, Mccoll BW. Adrenergic-mediated loss of splenic marginal zone B cells contributes to infection susceptibility after stroke. Nature Communications. 2017. [CrossRef]
- Doyle KP, Fathali N, Siddiqui MR, Buckwalter MS. Distal hypoxic stroke: A new mouse model of stroke with high throughput, low variability and a quantifiable functional deficit. Journal of Neuroscience Methods. 2012 [accessed 2020 May 27];207(1):31–40. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3348433/. [CrossRef]
- Young K, Morrison H. Quantifying Microglia Morphology from Photomicrographs of Immunohistochemistry Prepared Tissue Using ImageJ. J. Vis. Exp. 2018 [accessed 2023 Oct 14];(136):57648. URL:https://www.jove.com/video/57648. [CrossRef]
- Xu W, Zhang Y, Su J, Liu A, Wang K, Li C, Liu Y, Zhang Y, Lv J, Jiang W. Ischemia Reperfusion Injury after Gradual versus Rapid Flow Restoration for Middle Cerebral Artery Occlusion Rats. Scientific Reports. 2018 [accessed 2019 Jul 12];8(1):1638. http://www.nature.com/articles/s41598-018-20095-9. [CrossRef]
- Metz GA, Whishaw IQ. The Ladder Rung Walking Task: A Scoring System and its Practical Application. 2009 [accessed 2023 Oct 14]:28. [CrossRef]
- Deacon RM. Assessing nest building in mice. Nature Protocols. 2006 [accessed 2023 Oct 14]. http://www.natureprotocols.com. [CrossRef]
Figure 1.
Study design. Cohort 1 received vehicle, 0.3 mg/kg formoterol, or 1.0 mg/kg formoterol for 2 weeks after stroke. Based on the results of Cohort 1, the 0.3 mg/kg dose was selected for Cohort 2, which received formoterol for 8 weeks. The 8-week study was designed to evaluate the long-term effects of formoterol treatment on stroke recovery. Outcomes included infarct size and brain atrophy assessment by MRI, and behavioral testing. WB=Western Blot, IHC=Immunohistochemistry, cAMP=cyclic adenosinemonophosphate, Acylcarn=Acyl carnitines, NF-L=Neurofilament Light Assay.
Figure 1.
Study design. Cohort 1 received vehicle, 0.3 mg/kg formoterol, or 1.0 mg/kg formoterol for 2 weeks after stroke. Based on the results of Cohort 1, the 0.3 mg/kg dose was selected for Cohort 2, which received formoterol for 8 weeks. The 8-week study was designed to evaluate the long-term effects of formoterol treatment on stroke recovery. Outcomes included infarct size and brain atrophy assessment by MRI, and behavioral testing. WB=Western Blot, IHC=Immunohistochemistry, cAMP=cyclic adenosinemonophosphate, Acylcarn=Acyl carnitines, NF-L=Neurofilament Light Assay.
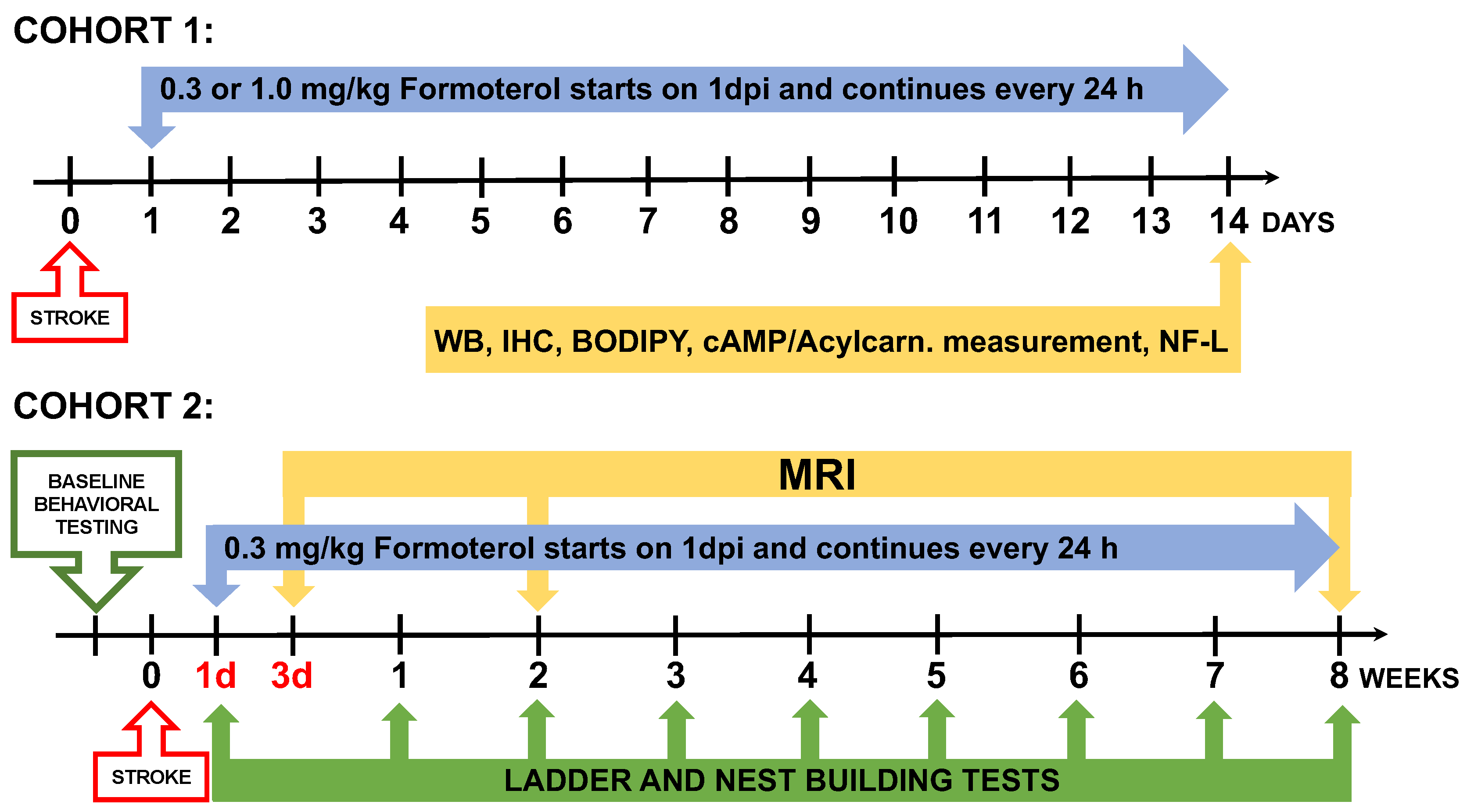
Figure 2.
Formoterol administered at 0.3 mg/kg, but not at 1.0 mg/kg, increased cAMP levels and MB in the infarcted area. (a) A schematic illustration of the proposed mechanism for how formoterol induces MB (according to [10]). cAMP= cyclic adenosinemonophosphate, PI3K=phosphoinositide 3-kinase, AKT=serine/threonine kinase, eNOS =endothelial nitric oxide synthase, sGC=soluble guanylyl cyclase, cGMP=cyclic guanosine monophosphate, TFAM=mitochondrial transcription factor A, NDUSF1=mitochondrial NADH-ubiquinone oxidoreductase 75 kDa subunit, mtDNA=mitochondrial DNA (created with BioRender.com). (b) Formoterol administered at 0.3 mg/kg, but not 1.0 mg/kg, elevated cAMP levels in the infarcted area of the brain; F*(3, 17.31)=6.887 Brown-Forsythe ANOVA test followed by Dunnett’s T3 multiple comparison test, ** adjusted p=0.0026, n=6, VEH=Vehicle, F0.3=Formoterol 0.3 mg/kg, F1.0=Formoterol 1.0 mg/kg, CL=contralateral. This indicates that 0.3 mg/kg formoterol is able to activate β2-AR signaling in the brain. (c) Formoterol 0.3 mg/kg elevated mitochondrial DNA copynumber (t(10)=3.190, **p=0.0097 unpaired two-tailed t-test), TFAM (t(10)=2.967, *p=0.0141 unpaired two-tailed t-test) and NDUSF1 (t(10)=2.283, *p=0.0456 unpaired two-tailed t-test, n=6) after 2 weeks of treatment, and this effect was still visible with mtDNA copynumber (t(6)=3.993, **p=0.0072 unpaired two-tailed t-test) and TFAM (*p=0.0286 two-tailed Mann-Whitney test, n=4) at 8 weeks (d). The 1.0 mg/kg dose of formoterol failed to induce these markers of MB (e). Data presented as mean ± SD.
Figure 2.
Formoterol administered at 0.3 mg/kg, but not at 1.0 mg/kg, increased cAMP levels and MB in the infarcted area. (a) A schematic illustration of the proposed mechanism for how formoterol induces MB (according to [10]). cAMP= cyclic adenosinemonophosphate, PI3K=phosphoinositide 3-kinase, AKT=serine/threonine kinase, eNOS =endothelial nitric oxide synthase, sGC=soluble guanylyl cyclase, cGMP=cyclic guanosine monophosphate, TFAM=mitochondrial transcription factor A, NDUSF1=mitochondrial NADH-ubiquinone oxidoreductase 75 kDa subunit, mtDNA=mitochondrial DNA (created with BioRender.com). (b) Formoterol administered at 0.3 mg/kg, but not 1.0 mg/kg, elevated cAMP levels in the infarcted area of the brain; F*(3, 17.31)=6.887 Brown-Forsythe ANOVA test followed by Dunnett’s T3 multiple comparison test, ** adjusted p=0.0026, n=6, VEH=Vehicle, F0.3=Formoterol 0.3 mg/kg, F1.0=Formoterol 1.0 mg/kg, CL=contralateral. This indicates that 0.3 mg/kg formoterol is able to activate β2-AR signaling in the brain. (c) Formoterol 0.3 mg/kg elevated mitochondrial DNA copynumber (t(10)=3.190, **p=0.0097 unpaired two-tailed t-test), TFAM (t(10)=2.967, *p=0.0141 unpaired two-tailed t-test) and NDUSF1 (t(10)=2.283, *p=0.0456 unpaired two-tailed t-test, n=6) after 2 weeks of treatment, and this effect was still visible with mtDNA copynumber (t(6)=3.993, **p=0.0072 unpaired two-tailed t-test) and TFAM (*p=0.0286 two-tailed Mann-Whitney test, n=4) at 8 weeks (d). The 1.0 mg/kg dose of formoterol failed to induce these markers of MB (e). Data presented as mean ± SD.
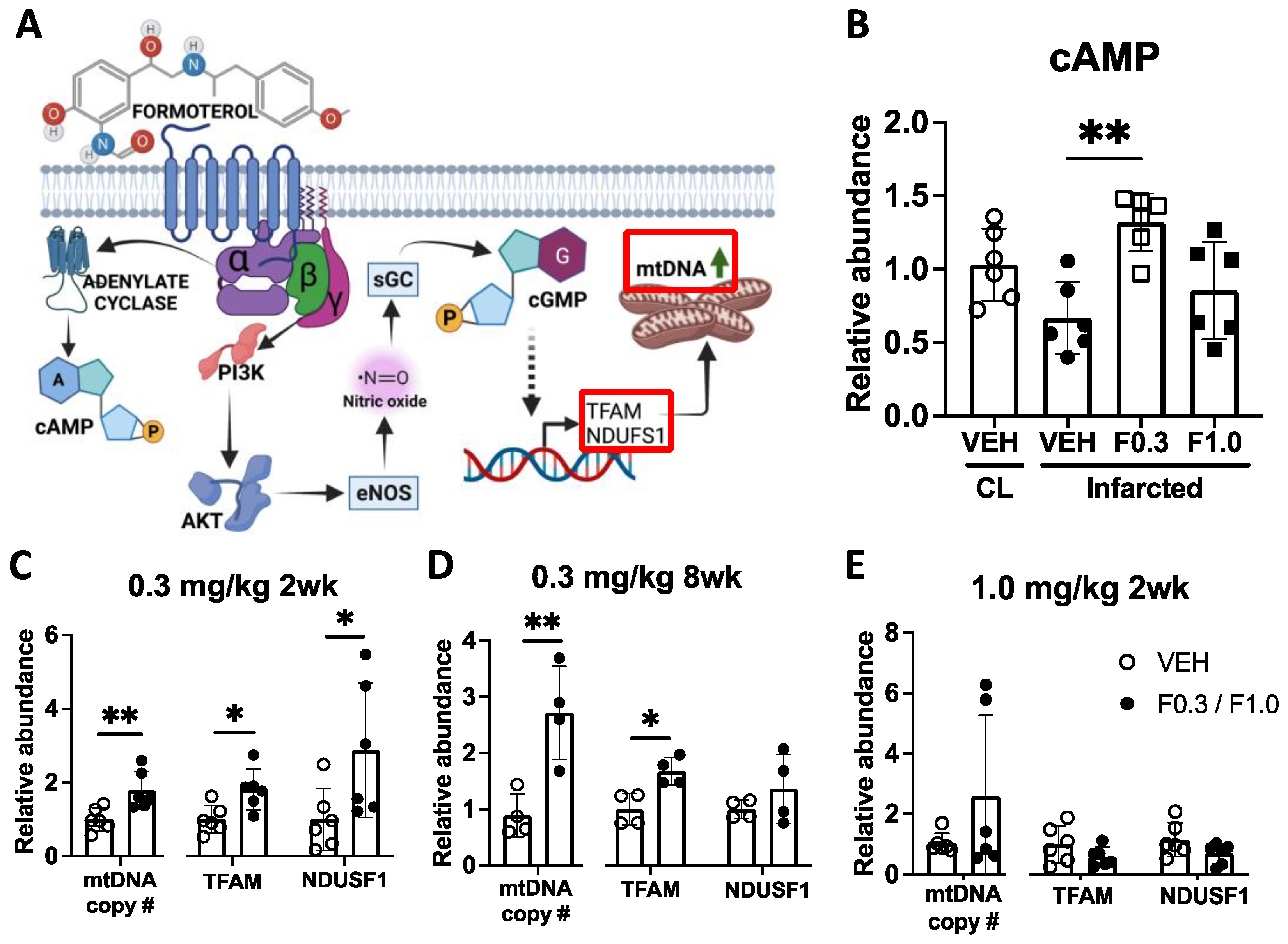
Figure 3.
Formoterol 0.3 mg/kg for 2 weeks increased microglia but not astrocyte responses to ischemic stroke. (a) Microglia and astrocytes were imaged in the indicated regions after ischemic stroke (created with BioRender.com). (b) Representative images of Iba-1 positive microglia and corresponding summary data of increased microglia cell soma counts per frame (c) in formoterol treated mice post-stroke; F(1,26)=9.174 p=0.0055 interaction effect, F(1,26)=96.08 p<0.0001 stroke effect, F(1,26)=3.924 p=0.0583 treatment effect of Two-way ANOVA followed by Sidak’s post hoc, **adj. p=0.0030. (d) Morphology analysis of Iba-1 positive cells. Mice treated with formoterol showed decreased microglia process length/cell in the infarcted area; F(1,24)=105.9 p<0.0001 stroke effect, F(1,24)=4.737 p=0.0396 treatment effect of Two-way ANOVA followed by Sidak’s post hoc, *adj. p= 0.0388. (e) Representative images of CD68 and corresponding summary data of increased area of positive immunoreactivity in formoterol treated mice post-stroke (f); F(1,26)=4.327 p=0.0475 interaction effect, F(1,26)=22.65 p<0.0001 stroke effect of Two-way ANOVA followed by Sidak’s post hoc, *adj. p=0.0284. (g) Representative images of GFAP and corresponding summary data of GFAP immunoreactivity area, increased post-stroke but unchanged with formoterol treatment (h). n=7-8 per group. Contra = contralateral side, Ipsi = ipsilateral side, VEH = vehicle, F0.3 = Formoterol 0.3 mg/kg for 2 weeks. Scale bar = 50 µm. Data presented as mean ± SD.
Figure 3.
Formoterol 0.3 mg/kg for 2 weeks increased microglia but not astrocyte responses to ischemic stroke. (a) Microglia and astrocytes were imaged in the indicated regions after ischemic stroke (created with BioRender.com). (b) Representative images of Iba-1 positive microglia and corresponding summary data of increased microglia cell soma counts per frame (c) in formoterol treated mice post-stroke; F(1,26)=9.174 p=0.0055 interaction effect, F(1,26)=96.08 p<0.0001 stroke effect, F(1,26)=3.924 p=0.0583 treatment effect of Two-way ANOVA followed by Sidak’s post hoc, **adj. p=0.0030. (d) Morphology analysis of Iba-1 positive cells. Mice treated with formoterol showed decreased microglia process length/cell in the infarcted area; F(1,24)=105.9 p<0.0001 stroke effect, F(1,24)=4.737 p=0.0396 treatment effect of Two-way ANOVA followed by Sidak’s post hoc, *adj. p= 0.0388. (e) Representative images of CD68 and corresponding summary data of increased area of positive immunoreactivity in formoterol treated mice post-stroke (f); F(1,26)=4.327 p=0.0475 interaction effect, F(1,26)=22.65 p<0.0001 stroke effect of Two-way ANOVA followed by Sidak’s post hoc, *adj. p=0.0284. (g) Representative images of GFAP and corresponding summary data of GFAP immunoreactivity area, increased post-stroke but unchanged with formoterol treatment (h). n=7-8 per group. Contra = contralateral side, Ipsi = ipsilateral side, VEH = vehicle, F0.3 = Formoterol 0.3 mg/kg for 2 weeks. Scale bar = 50 µm. Data presented as mean ± SD.
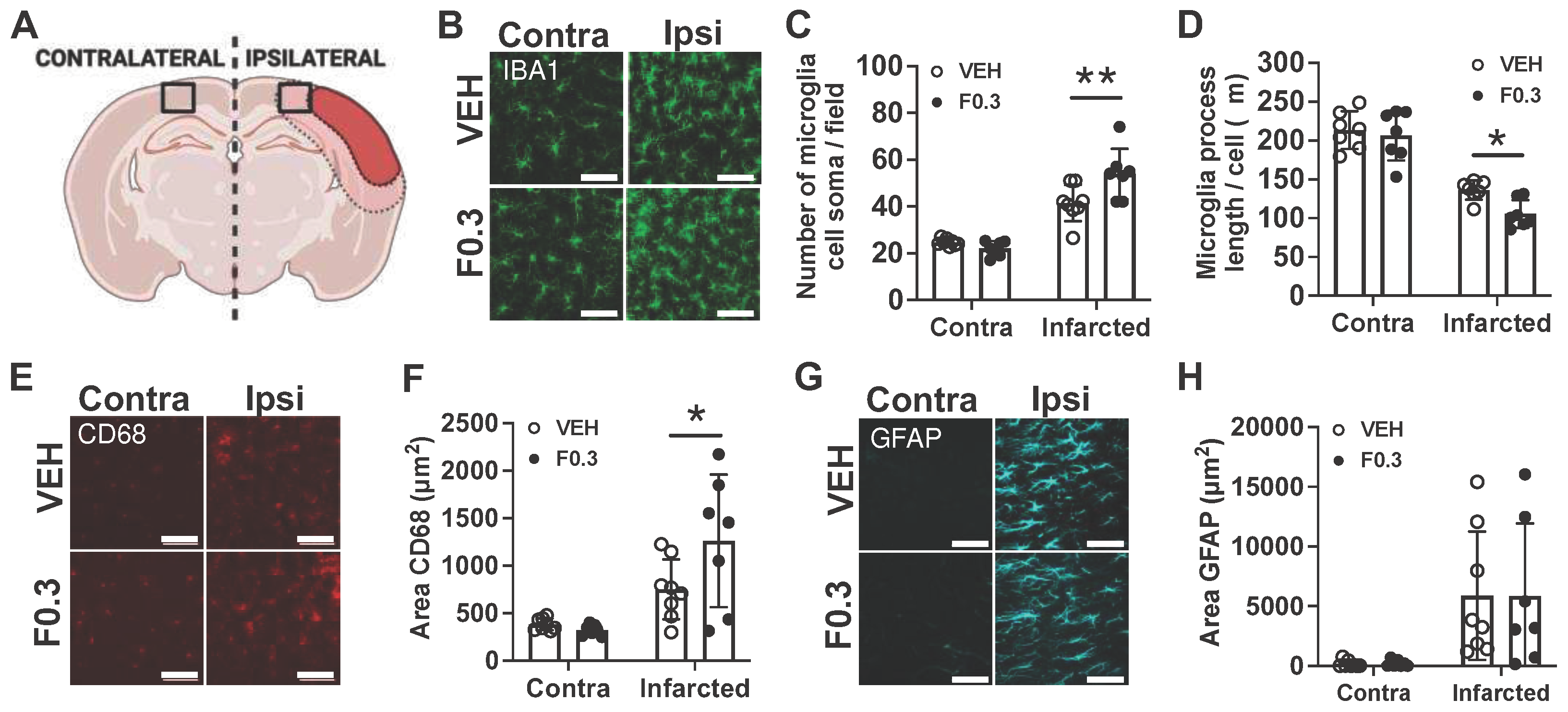
Figure 4.
BODIPY staining was reduced in the infarcts of mice treated with formoterol 0.3 mg/kg for 2 weeks after stroke. (a) Representative images showing the difference between autofluorescence and BODIPY staining. Images were captured at 20X magnification, with inserts highlighting the specificity of the staining. The first autofluorescence (AF) image and the BODIPY were taken with an exposure time (Exp.) of 1/150 second, while the second AF image was taken with an exposure time of 1.2 seconds. Scale bars: 100 µm for the entire infarct and 30 µm for the inserts. (b) Quantification of BODIPY staining in stroke infarcts at 2 weeks post stroke. BODIPY staining was significantly reduced in the formoterol treated group; t(14)=5.19, ***p=0.0001 unpaired two-tailed t-test, n=8. Data presented as mean ± SD. (c) Representative immunofluorescent images of BODIPY staining in the infarct. Images at 2 weeks post stroke were captured at 20X magnification, with inserts highlighting the infarct region. Scale bars: 100 µm for the entire infarct and 20 µm for the inserts. VEH = vehicle, F0.3 = formoterol 0.3 mg/kg for 2 weeks. (d) Immunofluorescent staining demonstrates individual as well as colocalized staining of IBA1 (cyan), CD68 (red), and BODIPY (green) in the infarct. Images were captured at 40X magnification (zoomed 2X), showcasing cellular colocalization within the infarct. Scale bars: 20 µm for the main images and 10 µm for the inserts.
Figure 4.
BODIPY staining was reduced in the infarcts of mice treated with formoterol 0.3 mg/kg for 2 weeks after stroke. (a) Representative images showing the difference between autofluorescence and BODIPY staining. Images were captured at 20X magnification, with inserts highlighting the specificity of the staining. The first autofluorescence (AF) image and the BODIPY were taken with an exposure time (Exp.) of 1/150 second, while the second AF image was taken with an exposure time of 1.2 seconds. Scale bars: 100 µm for the entire infarct and 30 µm for the inserts. (b) Quantification of BODIPY staining in stroke infarcts at 2 weeks post stroke. BODIPY staining was significantly reduced in the formoterol treated group; t(14)=5.19, ***p=0.0001 unpaired two-tailed t-test, n=8. Data presented as mean ± SD. (c) Representative immunofluorescent images of BODIPY staining in the infarct. Images at 2 weeks post stroke were captured at 20X magnification, with inserts highlighting the infarct region. Scale bars: 100 µm for the entire infarct and 20 µm for the inserts. VEH = vehicle, F0.3 = formoterol 0.3 mg/kg for 2 weeks. (d) Immunofluorescent staining demonstrates individual as well as colocalized staining of IBA1 (cyan), CD68 (red), and BODIPY (green) in the infarct. Images were captured at 40X magnification (zoomed 2X), showcasing cellular colocalization within the infarct. Scale bars: 20 µm for the main images and 10 µm for the inserts.
Figure 5.
Formoterol treatment reduced levels of acyl carnitines in the stroked brain. (a) A schematic illustration of transportation of fatty acids into mitochondria for beta-oxidation (created with BioRender.com). (b) A heatmap showing that the levels of almost all of the detected acyl carnitines were elevated in the ipsilateral (IL) side of the vehicle treated brains when compared to the contralateral side. However, the acyl carnitines in the ipsilateral side of the formoterol treated brains were markedly reduced when compared to the ipsilateral side of brains receiving vehicle. Red shaded cells indicate significant increase (p<0.05) of the metabolite in that comparison, and yellow shaded cells indicate a significant decrease. Light red and light orange shaded cells indicate a trend (0.05<p<0.1) towards the respective direction, and the values represent fold change. (c) Carnitine levels increased after stroke, similar to the acyl carnitines, and formoterol treatment reduced them towards contralateral (CL) side levels. F*(2,17.12) = 14.14 p=0.0002 Brown-Forsythe ANOVA test followed by Dunnett’s multiple comparisons test, adj. p(VEH CL vs. VEH IL) = 0.0019; adj. p(VEH IL vs. F0.3 IL) = 0.0059, n=9, data presented as mean ± SD.
Figure 5.
Formoterol treatment reduced levels of acyl carnitines in the stroked brain. (a) A schematic illustration of transportation of fatty acids into mitochondria for beta-oxidation (created with BioRender.com). (b) A heatmap showing that the levels of almost all of the detected acyl carnitines were elevated in the ipsilateral (IL) side of the vehicle treated brains when compared to the contralateral side. However, the acyl carnitines in the ipsilateral side of the formoterol treated brains were markedly reduced when compared to the ipsilateral side of brains receiving vehicle. Red shaded cells indicate significant increase (p<0.05) of the metabolite in that comparison, and yellow shaded cells indicate a significant decrease. Light red and light orange shaded cells indicate a trend (0.05<p<0.1) towards the respective direction, and the values represent fold change. (c) Carnitine levels increased after stroke, similar to the acyl carnitines, and formoterol treatment reduced them towards contralateral (CL) side levels. F*(2,17.12) = 14.14 p=0.0002 Brown-Forsythe ANOVA test followed by Dunnett’s multiple comparisons test, adj. p(VEH CL vs. VEH IL) = 0.0019; adj. p(VEH IL vs. F0.3 IL) = 0.0059, n=9, data presented as mean ± SD.
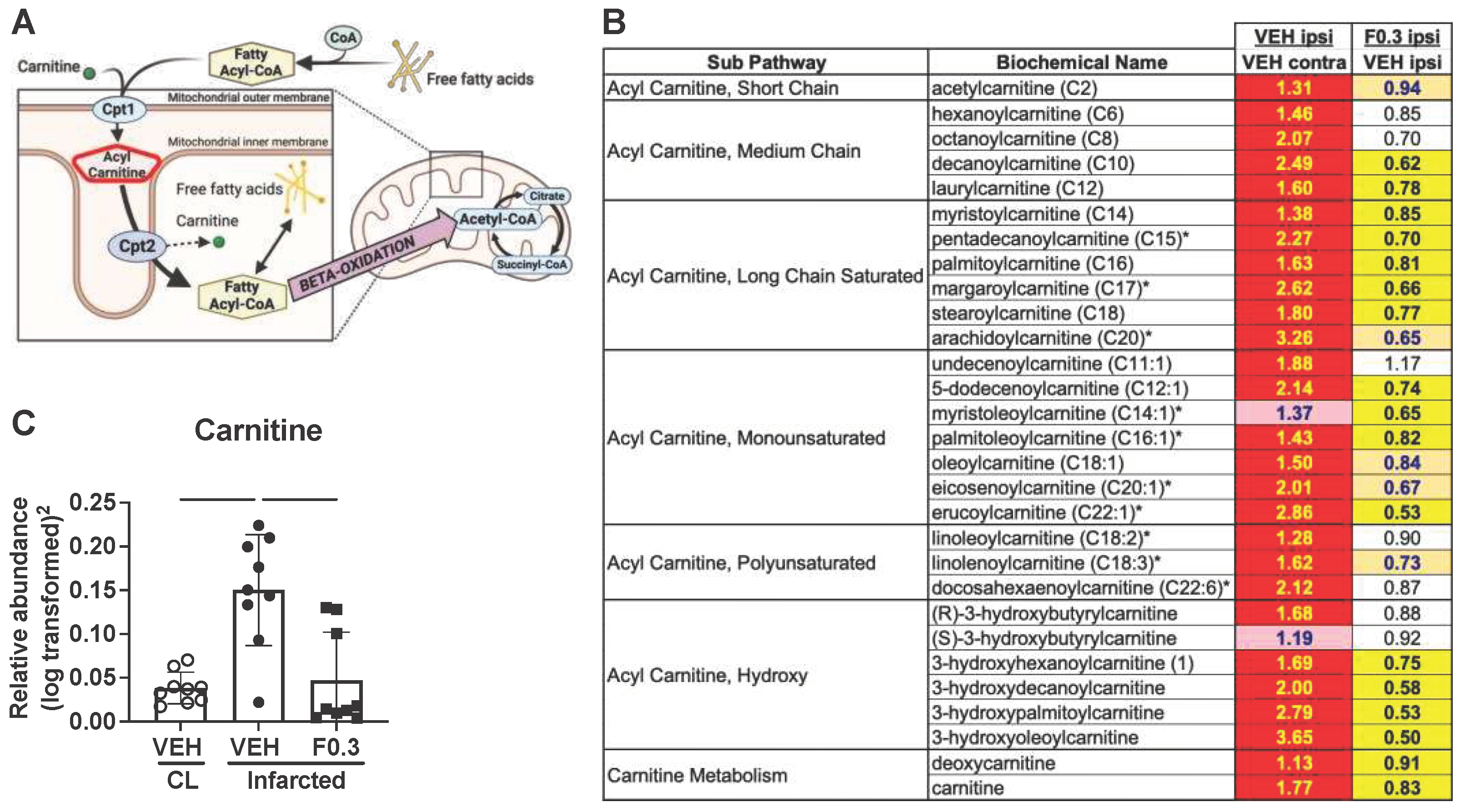
Figure 6.
Formoterol did not affect neurodegeneration. (a) NF-L levels in plasma from vehicle (VEH)- and formoterol 0.3 mg/kg (F0.3)-treated mice were significantly elevated after stroke compared to age-matched naïve mice. Formoterol treatment did not further affect NF-L levels; F*(2,7.266)=14.21 Brown-Forsythe ANOVA followed by Dunnett’s T3 multiple comparisons test, *adj.p=0.0192, n=5. (b) There was no difference in the size of the lesion at 3 days after ischemia. (c) Ventricle size at 2 weeks after stroke was significantly larger in the ipsilateral (IL) than the contralateral (CL) side, but there was no difference between the treatment groups; F*(2,10.65)=17.21 Brown-Forsythe ANOVA followed by Dunnett’s T3 multiple comparisons test, adj.p(VEH CL vs. VEH IL)=0.0031, adj.p(VEH CL vs. F0.3 IL)=0.0054, n=6. (d) Ventricle sizes did not differ between the groups at the 8-week timepoint. .
Figure 6.
Formoterol did not affect neurodegeneration. (a) NF-L levels in plasma from vehicle (VEH)- and formoterol 0.3 mg/kg (F0.3)-treated mice were significantly elevated after stroke compared to age-matched naïve mice. Formoterol treatment did not further affect NF-L levels; F*(2,7.266)=14.21 Brown-Forsythe ANOVA followed by Dunnett’s T3 multiple comparisons test, *adj.p=0.0192, n=5. (b) There was no difference in the size of the lesion at 3 days after ischemia. (c) Ventricle size at 2 weeks after stroke was significantly larger in the ipsilateral (IL) than the contralateral (CL) side, but there was no difference between the treatment groups; F*(2,10.65)=17.21 Brown-Forsythe ANOVA followed by Dunnett’s T3 multiple comparisons test, adj.p(VEH CL vs. VEH IL)=0.0031, adj.p(VEH CL vs. F0.3 IL)=0.0054, n=6. (d) Ventricle sizes did not differ between the groups at the 8-week timepoint. .
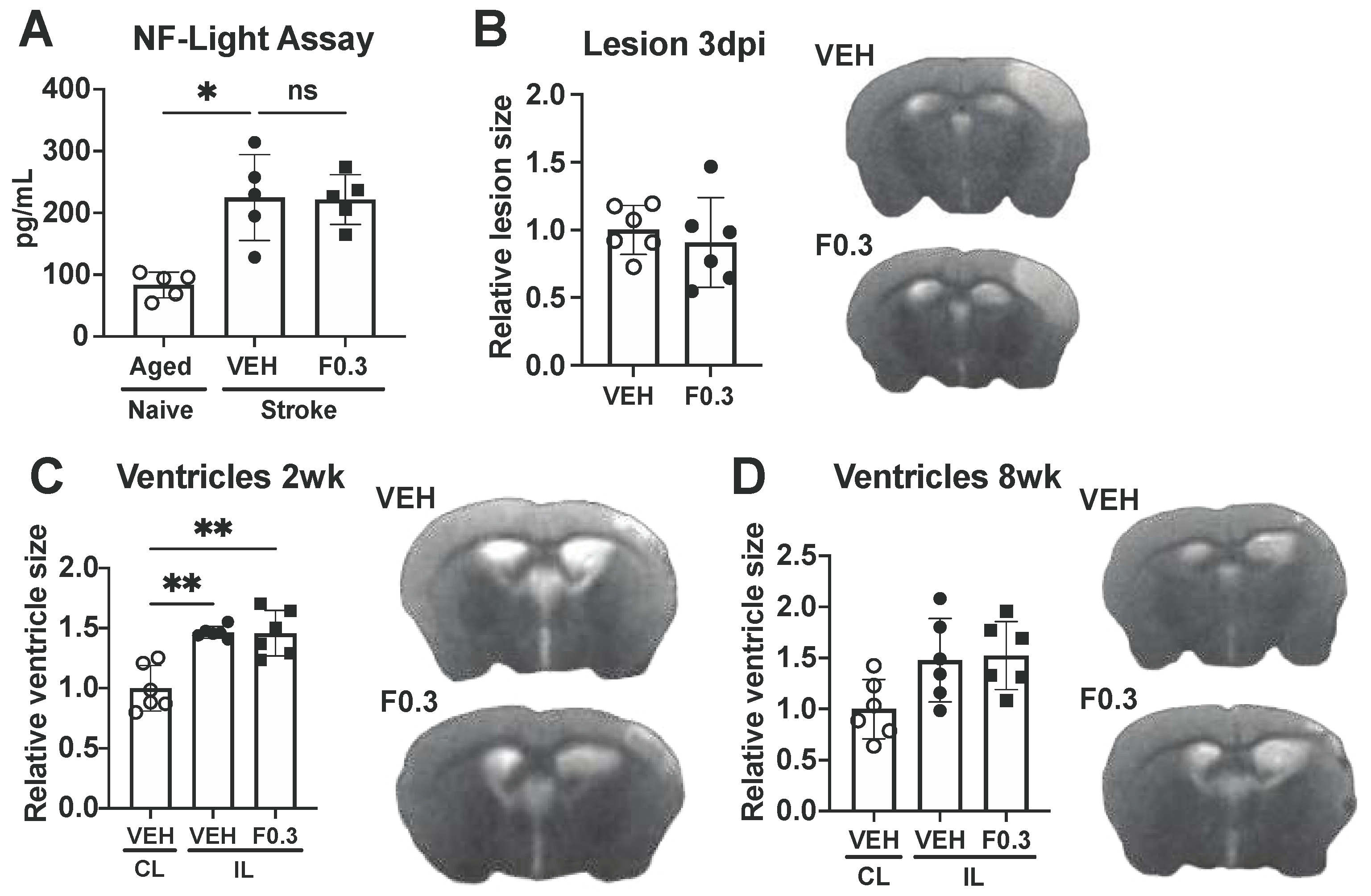
Figure 7.
Formoterol did not affect motor recovery. (a) Both formoterol- and vehicle treated mice showed similar stroke deficits during week 1 of the ladder test when compared to their age matched naive counterparts; Blue * = VEH = vehicle, Red # = F0.3 = formoterol, F(18, 144)=1.622 p=0.0616 time x treatment, F(3.201, 51.21)=10.05 p<0.0001 time, F(2, 16)=11.81 p=0.0007 treatment, F(16, 144)=5.477 p<0.0001 subject effect of Two-way Repeated Measures ANOVA followed by Tukey’s multiple comparisons test, 1dpi: blue * adj.p=0.0190, 1 wk: blue * adj.p=0.0102, red # adj.p=0.0124. (b) The same applied to the nest building test. There was a significant stroke effect on day 1 after stroke, but the mice quickly recovered to the level of age-matched naïve animals; F(18, 117)=2.518 p=0.0016 time x treatment, F(3.697, 48.06)=6.873 p=0.0003 time, F(13, 117)=5.374 p<0.0001 subject effect of Two-way Repeated Measures ANOVA followed by Tukey’s multiple comparisons test, blue * adj.p=0.0246, red # adj.p=0.0151. n=4-8. Data presented as mean ± SD.
Figure 7.
Formoterol did not affect motor recovery. (a) Both formoterol- and vehicle treated mice showed similar stroke deficits during week 1 of the ladder test when compared to their age matched naive counterparts; Blue * = VEH = vehicle, Red # = F0.3 = formoterol, F(18, 144)=1.622 p=0.0616 time x treatment, F(3.201, 51.21)=10.05 p<0.0001 time, F(2, 16)=11.81 p=0.0007 treatment, F(16, 144)=5.477 p<0.0001 subject effect of Two-way Repeated Measures ANOVA followed by Tukey’s multiple comparisons test, 1dpi: blue * adj.p=0.0190, 1 wk: blue * adj.p=0.0102, red # adj.p=0.0124. (b) The same applied to the nest building test. There was a significant stroke effect on day 1 after stroke, but the mice quickly recovered to the level of age-matched naïve animals; F(18, 117)=2.518 p=0.0016 time x treatment, F(3.697, 48.06)=6.873 p=0.0003 time, F(13, 117)=5.374 p<0.0001 subject effect of Two-way Repeated Measures ANOVA followed by Tukey’s multiple comparisons test, blue * adj.p=0.0246, red # adj.p=0.0151. n=4-8. Data presented as mean ± SD.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



