
Submitted:
16 October 2023
Posted:
17 October 2023
You are already at the latest version
Abstract
Snakebite envenoming can be a life-threatening medical emergency that requires prompt medical intervention to neutralise the effects of venom toxins. Each year up to 138,000 people die from snakebite and 3-fold more victims suffer life-altering disabilities. Current treatment of snakebite relies solely on antivenom – polyclonal antibodies isolated from the plasma of hyperimmunised animals – which is associated with numerous deficiencies. The ADDovenom project seeks to deliver a novel snakebite therapy, through use of an innovative protein-based scaffold as a next-generation antivenom. The ADDomer is a megadalton-sized, thermostable synthetic nanoparticle derived from the adenovirus penton base protein with 60 high-avidity binding sites to neutralise venom toxins. Here, we outline our experimental strategies to achieve this goal using state-of-the-art protein engineering, expression technology, mass spectrometry, as well as in vitro and in vivo venom neutralisation assays. We anticipate that the approaches described here will produce antivenom with unparalleled efficacy, safety and affordability.
Keywords:
snakebite
; antivenom
; venom
; biologics
; ADDomer
1. Introduction
Snakebite envenoming is a Neglected Tropical Disease that is annually responsible for up to 138,000 deaths and 400,000 disabilities in surviving victims [1], disproportionately affecting the most economically and medically disadvantaged communities of Asia, sub-Saharan Africa and Latin America [2]. The first-choice treatment for snakebite is intravenously-delivered antivenom, which is produced by hyperimmunising equines or ovines with subtoxic doses of venom(s) and then purifying immunoglobulins from resulting sera/plasma samples. Consequently, the resulting antivenoms are associated with numerous deficiencies, including limited cross-snake species reactivity, poor dose-efficacy, and high incidence of adverse reactions [3]. The efficacy of current antivenom is often restricted to the snake venom(s) used as immunogens in the manufacturing process due to the significant variation in toxin composition observed between the venoms of different snake species, though venom components can even differ between different individuals of the same species or over the lifetime of a single snake [4,5]. Snake venoms typically contain mixtures of functionally distinct protein isoforms encoded by relatively few toxin families, and are biochemically diverse secretions that cause variable pathologies in snakebite victims (i.e. haemotoxicity, neurotoxicity and/or cytotoxicity) [1]. Venom toxin variation therefore underpins the restrictive cross-snake species efficacy of antivenom, which in turn translates into the limited geographic utility, reduced economies of scale and poor commercial manufacturing incentives associated with these life-saving therapeutics [6].
Most regions affected by snakebite envenoming are home to several species of medically-important snakes, and therefore most antivenom manufacturers use venom from multiple snake species to produce polyspecific (polyvalent) antivenoms for the target region [7]. This inherently results in a smaller proportion of antibodies in the antivenom directed against the toxins from any one species in comparison to antivenoms raised against only one venom (“monospecific” or “monovalent” antivenom), and therefore much higher doses of polyspecific antivenoms are required to effectively treat envenoming [8]. Further compounding the poor dose-efficacy of antivenoms is evidence that only 5-36% of immunoglobulins purified from the animal sera/plasma are specific to the venom proteins used for immunisation [9,10,11,12], and only a proportion of those are likely to be neutralising antibodies. The weak potency of antivenoms, which often require multiple vials to effect cure, increases the risk of adverse reactions to the large quantity (often grams) of intravenously-administered, non-human immunoglobulin, while also resulting in increased costs to the patient. Another consideration regarding the accessibility of antivenoms is cold-chain transport and stable low temperature storage requirements, which frequently prevents their effective distribution in peripheral rural health centres close to the populations that have the greatest need [13].
Snakebite was recognised as a priority neglected tropical disease by the World Health Organisation (WHO) in 2017 due to the significant public health problem it presents across most of the rural tropics. A number of novel snakebite treatments are under development: monoclonal human/humanised antibodies (or cocktails thereof), non-antibody toxin-binding proteins or peptides, small molecule drugs, and aptamers (for reviews on these therapeutic modalities for snakebite see [14,15,16,17]). These treatments aim to be better tolerated, more affordable, and possess broader snake species-utility than existing antivenoms; however only two are currently in clinical trials [18,19]. Thus, there remains a strong need to explore alternative biological therapeutics that may provide the next breakthrough in snakebite treatment. Here, we describe the proposed development of ADDomer - a novel protein-based nanoparticle therapeutic for snakebite - in our so named ‘ADDovenom’ project.
2. ADDomer – a versatile thermostable nanoparticle
ADDomer is a nanoparticle scaffold derived from the Adenovirus penton base protein protomer [20]. In Adenovirus, the protomer forms pentons at the vertices of the capsid, providing a base for fibre protein attachment [21]. When expressed recombinantly in isolation, 60 protomers spontaneously self-assemble into pentons that form a dodecahedron, known as ADDomer (for Adenovirus-Derived Dodecamer) (Figure 1a,b). ADDomer-based nanoparticles have been developed as efficient vaccines, e.g. against Chikungunya, SARS-CoV-2 or Foot-and-mouth disease [20,22,23,24]. The penton base protein protomer comprises two domains, a jelly roll fold and a crown domain (Fig 1c). The jelly roll fold mediates multimerisation into the penton and dodecamer assembly. The crown domain comprises two loops on its surface, the variable loop (VL) and the RGD loop (Figure 1c). The VL and RGD loops are highly variable in sequence and length among the penton base proteins found in different Adenoviruses. Immunogenic epitopes and other sequences can be inserted into these loops without compromising efficient folding of the penton base protein and assembly into the ADDomer [20]. The immunogenic epitopes are then displayed on the surface of the ADDomer in 60, 120 or even more copies, depending on the number of epitopes inserted into VL and RGD loops [20].
ADDomer nanoparticles have a number of key advantages that make them uniquely suitable for deployment in developing countries:
- Thermotolerance up to >45 °C [20,25]. It was shown that the ADDomer nanoparticle can be stored for one month at ~20°C, it can be frozen and thawed and/or heated to 45 °C for 1 hour, virtually without losing its structural integrity [20]. Consistently, ADDomer does not require cold-chain storage which constitutes a key logistic advance.
- ADDomers can be lyophilised [25]. This manufacturing step increases storage life, providing a commercial incentive for manufacturers.
- ADDomers can be produced as recombinant protein-based nanoparticles with exceptionally good yields using a baculovirus insect cell expression system, allowing establishment of good manufacturing practices with stringent quality control.
- Importantly, of all the Adenovirus capsid proteins, the penton base protein is the least immunogenic. Therefore, ADDomer presents a low immunogenicity scaffold.
The aforementioned characteristics of the ADDomer scaffold – thermotolerance, shelf-life, recombinant production, low immunogenicity – are equally desirable for antivenoms. We found that the architecture of the crown domain with its highly variable loops has remote similarities with complementarity determining regions (CDR loops) in antibodies, used to recognise cognate antigens. We hypothesised that the crown domain could be functionalised to bind toxins. Then, the ADDomer could act as a superbinder ‘sponge’ to neutralise toxins. A prerequisite for such a superbinder is the feasibility of a binder molecule representing the crown domain of the protomer. The bipartite architecture of the protomer suggested a possibility to separate crown from jelly roll fold. Thus, we designed ADDobody, comprising only the crown domain and produced the prototype in Escherichia coli with excellent yields as a monomeric protein (Figure 1d) [26]. Next, the VL and the RGD loops of the crown domain were randomised in length and sequence, mimicking the CDR loops of antibodies (Figure 1d). The resulting synthetic ADDobody library is used for ribosome display in vitro selections (Figure 1e) against native and recombinantly produced toxins. Selected specific binders are biophysically characterised and tested for toxin neutralisation using enzymatic or cell-based assays. In a next step, the selected toxin-neutralising ADDobody can be re-joined with the jelly roll fold multimerisation domain to produce the ADDomer-based superbinders [26] (Figure 1). The resulting antitoxin nanoparticle displays 60 binding domains against the toxin target, and therefore may ensure high-avidity binding and highly efficient neutralisation of the snake toxin targets.
3. The EU-funded ADDovenom project
In the ADDovenom project, we will focus on neutralising venoms from two medically important groups of African snakes that cause severe life threatening envenoming across the continent, specifically the saw-scaled vipers (Viperidae: Echis spp.) and the mambas (Elapidae: Dendroaspis spp.) [27]. Echis saw-scaled vipers are responsible for the largest numbers of bites and deaths in the northern half of Africa, and their venom mainly causes life-threatening haemorrhage and coagulopathy and debilitating local tissue necrosis [28]. These effects are caused by toxins such as the snake venom metalloproteinases (SVMPs), phospholipases A2 (PLA2s), serine proteases, C-type lectin-like proteins and disintegrins [29,30]. Contrastingly, envenoming by Dendroaspis mambas causes neuromuscular paralysis, which may become rapidly lethal if respiratory paralysis occurs [27]. Their venom contains several distinct neurotoxins [31], which are mostly members of the three-finger toxin (3FTx) and Kunitz-type toxin families [32,33].
We seek to develop ADDomers and ADDobodies with neutralising capabilities against Echis SVMPs, PLA2s, and disintegrins, and mamba 3FTx and Kunitz-type toxins, using approaches shown in Figure 2. First, venom mass spectrometry will be performed to define the venom toxin content of each species. Proteomics will identify the protein sequence of priority toxins to be targeted, their post-translational modifications, disulphide bonds, and relative abundance in each venom. Priority toxins will then be expressed as recombinant toxins using bacterial and eukaryotic expression systems. A naïve synthetic ADDobody library and recombinant toxins will be used to select for specific, high-affinity toxin-binders by ribosome display. Resulting ADDobodies and ADDomers (multimerised ADDobodies) will then be tested for their in vitro and in vivo neutralising capabilities against specific venom toxins and crude venom, and will also be assessed in vivo for their safety and pharmacokinetic characteristics. Finally, a good manufacturing practice (GMP)-compatible platform will be developed for the production of toxin-neutralising ADDomers.
3.1. Venom mass spectrometry and bioinformatics
One key challenge for the design of toxin-specific therapies is that it is often not apparent which specific isoforms within a family of toxins must be neutralised to prevent mortality and morbidity. The ADDovenom project will take advantage of the versality of mass spectrometry approaches [34,35,36,37] to determine the inventory of peptides and proteins present in the nine African snake venoms collected from saw-scaled vipers (E. ocellatus, E. pyramidum leakeyi, E. leucogaster, E. coloratus) and mambas (D. polylepis, D. angusticeps, D. viridis, D. jamesoni jamesoni and D. j. kaimosae) to be used in this project. From the peptide sequences, the prediction of toxin families and their related pharmacological activities will enable a ranking of toxins according to their toxicological importance to generate a list of ‘priority’ target toxins to be neutralised for each venom. A toxin database containing all this information will be generated to identify the target toxins for recombinant expression (see Section 3.2).
These objectives will be achieved through application of venomics, a methodology that integrates data generated from transcriptomics and proteomics [38,39,40]. This rational approach has already been demonstrated to be particularly powerful to explore the remarkable toxin arsenal of animal venoms. Snake venoms are typically composed of a combination of tens to hundreds of different components, mostly peptides and proteins (>90%), that vary between and within snake species [41,42]. Consequently, identifying the most potent toxins with proteomics can help identify targets for the production of recombinant antivenoms, which represent promising new approaches to treating snakebites.
In the ADDovenom project, we will characterise the crude venom complexity of nine medically important African snakes, as summarised in Figure 3. Each venom will be analysed by two-dimensional sodium dodecyl sulphate polyacrylamide gel electrophoresis, leading to a rough evaluation of their structural family. Accurate masses of the most concentrated toxins are obtained by analysing the venoms by Matrix-Assisted Laser Desorption/Ionization Time-Of-Flight Mass Spectrometry (MALDI-TOF MS). In addition to this visualisation of venom composition, the results will be exploited to roughly evaluate the abundance of each detected family of toxin aiming to extract information on the pharmacological activity in each venom that requires neutralising. Each venom is then subjected to the bottom-up proteomics strategy: peptides sourced from digested toxins are sequenced by liquid chromatography (LC)-MS/MS using Orbitrap analysers, exploiting their high resolution (70,000 at m/z 200), mass accuracy (less than 1 ppm at 2 m/z) and fragmentation efficacy (Higher Energy Collision Dissociation - HCD) for toxin sequencing and identification.
Several methodologies can be considered to increase the efficiency of the sequencing step. The first approach is based on bottom-up shotgun proteomics where each crude venom is chemically treated to reduce and alkylate the cysteines responsible for disulphide bonds formation. The samples are then digested by three enzymes (trypsin, chymotrypsin and GluC serine protease) to amplify the level of information gathered through the MS/MS process, by creating overlapping peptides [43]. The huge amount of proteomics data is then analysed by dedicated bioinformatics tools such as Peaks X (Bioinformatics Solutions) [44]. This software allows the prediction of sequences from MS/MS spectra (de novo sequencing), but also enables matching of protein annotations to the obtained mass spectra/sequences via protein databases extracted from the Uniprot or NCBI servers (e.g. “toxins” AND “snake”). In the case of ADDovenom, the transcriptomics data from mRNA sequencing of the snake venom glands are included to generate a more precise and complete protein database to match with the proteomics data [42,45].
Every toxin sequenced from the nine venoms is added to a home-made database containing; toxin name, Uniprot accession number (if known), sequence, mass, toxin family, relative toxin abundance and, where available, predicted/putative biologic activity. This database constitutes a critical tool essential for the identification and selection of the most potent toxins to be targeted by the ADDobodies/ADDomers produced in this project.
In addition, evaluation of what the produced antitoxin molecules are binding is of prime importance. A methodology allowing the determination at the molecular level of which families of toxins are captured by functionalised ADDomers (qualitative) and how many ADDomers are needed for neutralising a defined quantity of injected venom (quantitative) is desired. Using antivenomic principles [38,46], we will establish a high-throughput methodology based on magnetic beads coated, in the first instance, with antibodies sourced from gold standard antivenoms as a proof of concept, followed by specific ADDomers targeting toxins. After venom incubation, mass spectrometry will be applied to monitor which toxins are captured by the antibodies or ADDomers. This approach will provide rapid and robust evaluation of the potency of antitoxins to selectively and quantitatively bind to targeted toxins. Furthermore, it will provide an evidence base as to whether ADDomer constructs have higher avidities than classical antibody-based antivenoms.
3.2. Production of recombinant toxins as antigens for ribosome display selection
Current antivenoms are generated using crude or fractionated venom for immunisation. Recombinant toxin production has the advantage that no venomous snakes are required, tags can be added to facilitate downstream work, and high protein purity can be achieved, allowing the biochemical and biophysical characterisation of the recombinant toxins prior to their use as antigens in ribosome display selection experiments. Ribosome display is an in vitro selection and evolution method ideally suited for generating high-affinity binders from very large synthetic libraries (encoding antibody single-chain variable fragments, nanobodies or engineered proteins) [47,48,49,50,51,52]. While it is desirable to work with purified, well-characterised toxins, a key challenge is to establish expression protocols for the active toxins; in particular, SVMPs contain many disulphide bridges (some SVMPs comprise 40 cysteines) which make them difficult to express and fold correctly. Successful recombinant protein production, however, will remove the batch-to-batch biological variability in venoms and yield reliably pure protein with defined activity.
Only very few production protocols for toxins are available in the literature [53], and our objective is to develop new protocols, optimised for individual toxins, during this project using prokaryotic (E. coli) and eukaryotic (baculovirus insect cell expression) production systems. Moreover, to test the activity of recombinantly produced toxins, specific functional tests are required for each toxin family, confirming that the purified proteins are correctly folded and bioactive. For instance, the N-terminal and C-terminal residues of venom toxins can contribute to ligand binding and biological activity [54,55], and therefore functional tests must be performed to confirm that the addition of tags does not interfere with toxin activity.
In a first approach, all prioritised toxin types (see Section 3.1) will be produced in E. coli and purified following a high-throughput protocol that has been successfully validated for a previous EU project called VENOMICS [56] to purify thousands of toxins including 3FTxs, Kunitz-type toxins and disintegrins [56,57]. The toxins are produced in the periplasm of E. coli using an N-terminal hexahistidine-tagged redox-active DsbC as fusion tag. DsbC fusions increase solubility and contribute to oxidation of the cysteines, allowing efficient formation of correctly folded disulphide bridges [56]. The fusion protein can be cleaved off using TEV protease after affinity purification, restoring the native N-terminus in the toxin. For ribosome display selections, a non-cleavable C-terminal Avi-tag for biotinylation is fused to the toxins [58]. Importantly, folding of the toxins can be further improved by the co-expression of two cysteine chaperones, protein disulphide isomerase (PDI) and sulfhydryl oxidase Erv1p (the CyDisCo system, see [59]).
Larger toxins with enzymatic activity and composed of several domains, such as SVMPs, contain many disulphide bonds required for structural integrity and possibly also cysteines in the active centre for metal coordination. Due to this increased complexity, bacterial expression systems often fail to produce active enzymes. If the toxins cannot be produced in high yields in E. coli or are non-functional, our expression system of choice is MultiBac, a state-of-the-art baculovirus insect cell expression system [60]. The toxins are then expressed with a melittin signal sequence fused to the N-terminus to ensure that they are secreted into the medium [61] facilitating purification. Insect cell expression may lead to post-translational modification (PTM) of toxins, glycosylation and phosphorylation, and the impact of these PTMs on toxin activity and solubility are largely unknown to date.
A further strategy regarding toxin antigen production for selection experiments we will pursue is generation of ‘epitope strings’ comprising an N-terminal, highly produced fusion protein (e.g. maltose-binding protein) and the most conserved sequences from one toxin family fused via glycine-serine rich linker sequences [62] - the idea being that conserved regions are functionally important and binding these regions with ADDomers may interfere with their toxicity/function.
Our objective in ADDovenom by implementing these approaches is two-fold: to provide the toxin components as antigens for our selection studies, and to develop protocols and a knowledge base for how to comprehensively produce toxins of interest in the best suitable expression system.
3.3. Evaluation of the neutralising ability of ADDobodies and ADDomers
Ribosome display selected ADDobodies and corresponding ADDomers capable of binding recombinant toxins will be assessed for their ability to neutralise toxin-function using a panel of serological, phenotypic and functional in vitro assays against crude venom and recombinant toxins. First, serological methods such as end-point ELISA and immunoblots will be used to demonstrate recognition of the target toxin and venom proteins. Typically, an indirect ELISA format is used in which the venom/toxin of interest is coated on an ELISA plate, incubated with the test antibody (i.e., antivenom or other protein therapeutic), and a secondary antibody capable of binding the test antibody/protein used to detect binding. Although ELISAs generally show good correlation with in vivo neutralising ability, for some venoms a poor correlation is observed [63], highlighting the need for rigorous functional in vitro and in vivo testing for each candidate therapeutic beyond serological assays.
ADDobodies/ADDomers directed against Echis SVMP, PLA2 and disintegrin toxins will be tested using specific enzyme activity assays and phenotypic assays, as indicated in Figure 4. Neutralisation of SVMP activity will be measured using the fluorogenic assay previously described [64]. Briefly, the quenched fluorescent peptide Mca-KPLGL-Dpa-AR-NH2 (ES010, BioTechne) is cleaved by SVMP between the Gly and Leu residues, releasing the fluorescent Mca-containing fragment from the quenching Dpa-containing fragment (Figure 4a), resulting in increased fluorescence. Other assays of SVMP bioactivities, including enzymatic activity, cytotoxicity assays and endothelial cell tube formation assays are reviewed by Macedo and Fox (2016) [65]. The enzymatic activity (phospholipid cleavage) of Echis PLA2 toxins will be determined using a fluorescent liposome-based PLA2 assay (EnzCheck, ThermoFisher) [45]. The assay reagent is prepared by encapsulating a fluorescent dye (BODIPY PC-A2) in a liposome containing dioleoylphosphatidylcholine and dioleoylphosphatidylglycerol (Figure 4b). Liposomes cleaved by PLA2 release the fluorescent substrate. Lower enzymatic activity is reported for S49-containing snake venom PLA2s which exert cytotoxic activities in a calcium-independent manner [66] which can be measured in standard cell viability assays [67]. Echis venoms contain both D49 (enzymatically active) and S49 (enzymatically inactive) PLA2 variants.
The haemostatic disturbances caused by Echis venoms can be measured using a phenotypic in vitro plasma clotting assay [68], in which the time for ‘normal’ plasma to clot is measured spectrophotometrically, and simultaneously disturbances to plasma clotting (either pro-coagulant or anti-coagulant) by venom toxins are detected by the differences in time to form a clot (Figure 4c). Disintegrin activity can be measured using several different methods that examine the effects of the toxins on platelet aggregation. Traditionally an aggregometer is used [65], but this approach requires specialist equipment and is low-throughput. Alternative approaches include a functional ELISA and flow cytometry, both employing antibodies that are selective for activated platelets, thus facilitating differentiation between activated and disintegrin-inhibited platelets (Figure 4d).
ADDobodies/ADDomers targeting the selected mamba toxins will be tested in cell-based assays for antagonism of muscle-type nicotinic acetylcholine receptors (nAChRs) and voltage-gated potassium channels, the targets of α-neurotoxins (a subclass of 3FTx) and Kunitz-type toxins respectively. To measure the effects of mamba long- and short-chain α-neurotoxins on nAChRs, we will use a functional assay that measures the ability of the receptors to activate in response to binding of the agonist acetylcholine (Figure 4e) [69]. This assay uses a cell line natively expressing muscle-type nAChR to measure the effects of α-neurotoxins, and complements an existing assay that measures the effects of neurotoxins on the neuronal α7-nAChR subtype [70], which are selectively targeted by long-chain 3FTx [71]. Traditional assays of the inhibitory activity of Kunitz-type toxins on voltage-gated potassium channels (Kv) use electrophysiological recording of Xenopus oocytes or mammalian cells engineered to express Kv channels [72]. Electrophysiology techniques are considered the gold-standard in ion channel research but require specialist equipment and training, and are low-throughput [73]. In the ADDovenom project, we are developing a higher-throughput (96-well format) assay to measure dendrotoxin-mediated block of Kv1 channels. HEK293 cells stably expressing tetrameric human Kv1.1 and 1.2 are stimulated by potassium sulphate, causing a change in membrane potential measured by a commercial membrane potential dye (FLIPR Membrane Potential Assay Kit, Molecular Devices), and antagonism of Kv channels by Kunitz-type toxins can therefore also be detected (Figure 4f).
Neutralising ADDobodies/ADDomers will subsequently be tested for venom neutralisation in vivo using murine models of envenoming. We will assess ADDobodies/ADDomers for their efficacy against the lethal systemic effects of Echis and Dendroaspis venoms using the WHO-approved ‘effective dose 50’ (ED50) murine model of snakebite envenoming, where venom challenge and treatment are preincubated together ahead of intravenous co-administration [74]. Evidence of efficacy in this ‘best case’ in vivo model of envenoming will provide a compelling justification to apply more challenging animal models that better recapitulate an envenoming scenario, specifically with therapeutic administration occurring after venom challenge [75].
3.4. Scalable bioprocess for ADDomer production
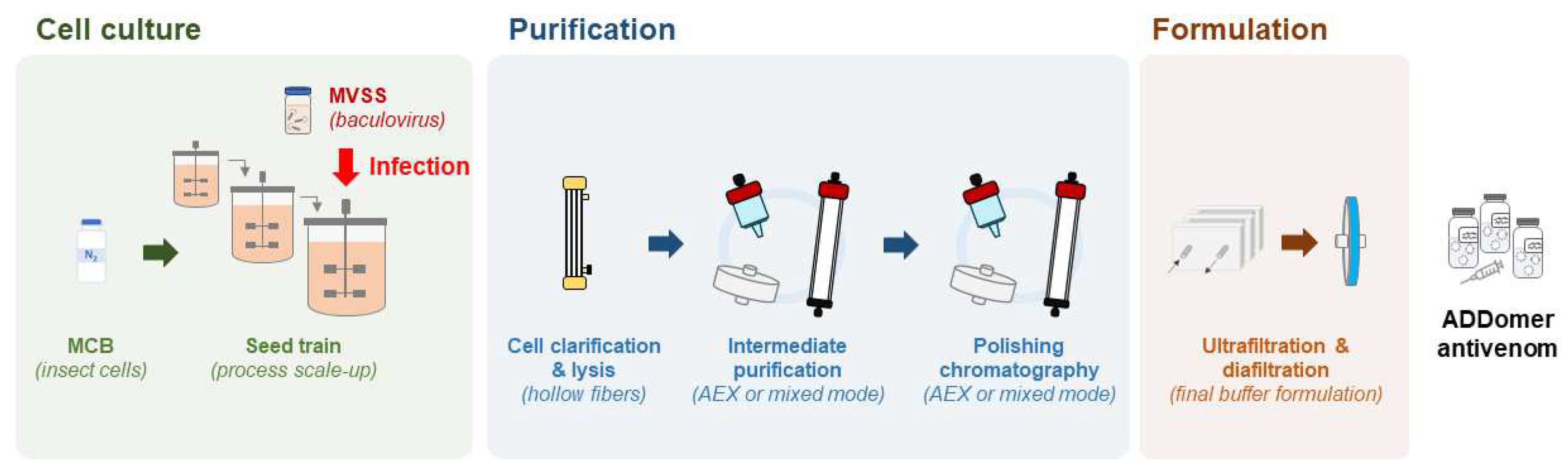
ADDomer nanoparticles are produced in insect cells, a very competitive manufacturing host, that enables expression at high level of exogenous proteins encoded in a baculovirus genome. As eukaryotes, insect cells can process complex secondary structures, are easy to culture and can grow in serum-free media, require less energy than mammalian cells and have low biosafety requirements [76]. The most frequently used insect cells derive from Spodoptera frugiperda and Trichoplusia ni and both offer GMP compatible cell lines [77,78,79]. By testing a variety of culture modes and culture medium supplementation strategies, production yields can be increased. Even though the most used culture mode is the batch system, protein-based nanoparticles and other insect cell derived products can also be produced in fed-batch or perfusion [80,81], both to be tested for ADDomer production (Figure 5). A hybrid perfusion process will also be tested, considering the cost and benefit of using larger media volumes and implementing cell-medium separation devices on the production phase [81]. Use of stainless steel or of single use bioreactors will be considered [82], with future transfer of the final process to a GMP facility in mind, and the latter providing easier technology transfer to GMP. The choice of upstream parameters will be combined with downstream optimisation, ensuring a good compromise between purity and product loss along the process. Once more, ensuring process scalability and GMP compatibility are the main focus. Moving towards a continuous bioprocess has been a major interest as the higher automation will lead to process intensification, reduce steps and shorten the production cycle, thus reducing the production cost [83,84]. Downstream process optimisation encompasses an array of techniques ranging from bench top to industrial scale, however, not all can easily be performed under GMP standards. Centrifugation steps and chromatography with low loading capacity will be avoided (e.g., size exclusion chromatography). The first step will be to optimise the clarification via microfiltration, using for example hollow fiber tangential flow filtration systems. As ADDomer nanoparticles are an intracellular product, this system will be used to separate the cells from the supernatant followed by in situ lysis of cells and recovery of the clarified ADDomer particles (Figure 5). Purification strategies will combine different types of chromatography in bind/elute or flow through modes with anion exchange membranes, these last ones removing DNA, baculovirus and charged host cell proteins. The process will be finalised by ultrafiltration/diafiltration in tangential flow mode and sterile filtration to ensure sterility and stability in the defined storage buffer.
Quality control protocols will be established ensuring reliable efficacy of the antivenom and robust, large-scale ADDomer nanoparticle production. These will cover from the production phase to the final product. During production, virus titre and cell growth rate will be analysed prior to infection. During downstream processing, the intermediate products will be screened by high-performance liquid chromatography (HPLC) for particles quantification and purity analysis. The final product will be screened for the main properties of the ADDomer nanoparticle as an antivenom: size and morphology will be screened by transmission electron microscopy, presence of aggregates will be evaluated by multi-angle dynamic light scattering (MADLS), and HPLC will enable accurate quantification. Impurities will also be controlled by measuring the amount of residual host cell protein, endotoxin levels, host cell DNA concentration and mass spectrometry to identify main contaminants.
For vaccines, cold-chain logistical issues account for up to one third of the final cost, and amounts for up to 50% of yearly dose wastage [85,86]. It is likely that similar numbers apply to antivenoms. The need to reduce costs and wastage has driven several studies to develop thermostable, cold-chain free solutions in a variety of systems [87,88,89]. As mentioned in Section 2, ADDomer-based proteins are inherently very stable (Tm>45 °C) which will translate in a wider range of storage and transport temperatures and consequently reduce the associated cost and reduce reliance on cold-chain logistics. Buffer formulation can play an important role in nanoparticle stability. Even though stability of ADDomer is not a main concern, different buffers (e.g., phosphate-, citrate-based) and stabilisers reported as “generally regarded as safe (GRAS)” will be considered and the cost-benefit of their introduction evaluated. Non-animal origin stabilisers such as amino acids and sugars have been reported to increase protein stability by increasing solubility, preventing protein aggregation, or reducing oxidation [90]. ADDomer antivenom final formulation will be tested under different temperature settings as well as freeze-thaw cycles to evaluate long term stability.
3. Future Perspective and Conclusions
We anticipate the resulting therapeutics arising from the ADDovenom project will feature numerous advantages over existing antivenoms in terms of efficacy, safety, affordability and manufacturing ability. Existing antivenom production uses animal immunisation with whole venom, which does not take into account the immunogenicity or toxicity of the numerous proteinaceous contents of venom, and thus many of the antibodies raised against venom are directed against non-toxic or low-abundance toxins. Our approach uses mass spectrometry and bioinformatics to rationally identify priority toxins to target. The low immunogenicity of the protein scaffold and the ability to manufacture ADDobodies and ADDomers to GMP-standards in bacterial and insect cells respectively will result in a reliable product with improved safety profiles over existing animal-derived antivenoms. The multi-modality format of ADDovenom (i.e., 38 kDa ADDobodies and 3.5 MDa ADDomers) lends itself to providing both a local treatment with characteristics suited to topical application and/or transcutaneous delivery, whilst the high-avidity ADDomers (60 binding sites) will be suited towards neutralising systemic toxins in the circulation. Finally, ADDobodies and ADDomers show impressive thermostability without the need for cold-chain storage, can be lyophilised to extend shelf-life, and can be produced recombinantly in exceptionally good yields at a competitive production cost. Collectively, these characteristics provide a strong rationale for the ADDovenom project and the future discovery of toxin-specific ADDomers as novel therapeutics for tropical snakebite.
Author Contributions
Writing—original draft preparation, S.K.M, R.A-G, F.G.A, I.A.C, A.R, R.V, I.B, L.Q, N.R.C, C.S.; writing—review and editing, all authors.; funding acquisition, A.R, P.M.A, R.V, I.B, L.Q, N.R.C, C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by a Horizon 2020 FET OPEN grant ‘ADDovenom’ (899670).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We are grateful to Dr. Rohit Patel (Liverpool School of Tropical Medicine) for critical review of the manuscript.
Conflicts of Interest
C.S. and I.B. report shareholding in Halo Therapeutics Ltd unrelated to this correspondence. I.B. reports shareholding in Geneva Biotech SARL, unrelated to this correspondence, and shareholding in Imophoron Ltd, related to this correspondence. Patents and patent applications have been filed related to ADDomer vaccines and therapeutics (WO2017167988A, EP22191583.8). The other authors do not declare competing interests. ADDomer and ADDobody are a registered trademark of Imophoron Ltd.
References
- Gutiérrez JM, Calvete JJ, Habib AG, Harrison RA, Williams DJ, Warrell DA. Snakebite envenoming. Nat Rev Dis Primer. 2017;3: 17063. [CrossRef]
- Harrison RA, Hargreaves A, Wagstaff SC, Faragher B, Lalloo DG. Snake Envenoming: A Disease of Poverty. PLoS Negl Trop Dis. 2009;3: e569. [CrossRef]
- Laustsen AH, María Gutiérrez J, Knudsen C, Johansen KH, Bermúdez-Méndez E, Cerni FA, et al. Pros and cons of different therapeutic antibody formats for recombinant antivenom development. Toxicon. 2018;146: 151–175. [CrossRef]
- Chippaux JP, Williams V, White J. Snake venom variability: methods of study, results and interpretation. Toxicon Off J Int Soc Toxinology. 1991;29: 1279–1303. [CrossRef]
- Casewell NR, Jackson TNW, Laustsen AH, Sunagar K. Causes and Consequences of Snake Venom Variation. Trends Pharmacol Sci. 2020;41: 570–581. [CrossRef]
- Harrison RA, Casewell NR, Ainsworth SA, Lalloo DG. The time is now: a call for action to translate recent momentum on tackling tropical snakebite into sustained benefit for victims. Trans R Soc Trop Med Hyg. 2019;113: 835–838. [CrossRef]
- World Health Organization. Guidelines for the Production, Control and Regulation of Snake Antivenom Immunoglobulins. 2017. Available: https://www.who.int/bloodproducts/AntivenomGLrevWHO_TRS_1004_web_Annex_5.pdf.
- Abubakar SB, Abubakar IS, Habib AG, Nasidi A, Durfa N, Yusuf PO, et al. Pre-clinical and preliminary dose-finding and safety studies to identify candidate antivenoms for treatment of envenoming by saw-scaled or carpet vipers (Echis ocellatus) in northern Nigeria. Toxicon Off J Int Soc Toxinology. 2010;55: 719–723. [CrossRef]
- Casewell NR, Cook DAN, Wagstaff SC, Nasidi A, Durfa N, Wüster W, et al. Pre-Clinical Assays Predict Pan-African Echis Viper Efficacy for a Species-Specific Antivenom. Williams DJ, editor. PLoS Negl Trop Dis. 2010;4: e851. [CrossRef]
- Segura Á, Herrera M, Villalta M, Vargas M, Gutiérrez JM, León G. Assessment of snake antivenom purity by comparing physicochemical and immunochemical methods. Biologicals. 2013;41: 93–97. [CrossRef]
- Rawat S, Laing G, Smith DC, Theakston D, Landon J. A new antivenom to treat eastern coral snake (Micrurus fulvius fulvius) envenoming. Toxicon. 1994;32: 185–190. [CrossRef]
- Herrera M, Paiva OK, Pagotto AH, Segura Á, Serrano SMT, Vargas M, et al. Antivenomic Characterization of Two Antivenoms Against the Venom of the Taipan, Oxyuranus scutellatus, from Papua New Guinea and Australia. Am J Trop Med Hyg. 2014;91: 887–894. [CrossRef]
- Harrison RA, Gutiérrez JM. Priority Actions and Progress to Substantially and Sustainably Reduce the Mortality, Morbidity and Socioeconomic Burden of Tropical Snakebite. Toxins. 2016;8: 351. [CrossRef]
- Pucca MB, Cerni FA, Janke R, Bermúdez-Méndez E, Ledsgaard L, Barbosa JE, et al. History of Envenoming Therapy and Current Perspectives. Front Immunol. 2019;10: 1598. [CrossRef]
- Gutiérrez JM, Albulescu L-O, Clare RH, Casewell NR, Abd El-Aziz TM, Escalante T, et al. The Search for Natural and Synthetic Inhibitors That Would Complement Antivenoms as Therapeutics for Snakebite Envenoming. Toxins. 2021;13: 451. [CrossRef]
- Clare RH, Hall SR, Patel RN, Casewell NR. Small Molecule Drug Discovery for Neglected Tropical Snakebite. Trends Pharmacol Sci. 2021;42: 340–353. [CrossRef]
- Jenkins TP, Fryer T, Dehli RI, Jürgensen JA, Fuglsang-Madsen A, Føns S, et al. Toxin Neutralization Using Alternative Binding Proteins. Toxins. 2019;11: 53. [CrossRef]
- Lewin MR, Carter RW, Matteo IA, Samuel SP, Rao S, Fry BG, et al. Varespladib in the Treatment of Snakebite Envenoming: Development History and Preclinical Evidence Supporting Advancement to Clinical Trials in Patients Bitten by Venomous Snakes. Toxins. 2022;14: 783. [CrossRef]
- Abouyannis M, FitzGerald R, Ngama M, Mwangudzah H, Nyambura Y, Ngome S, et al. TRUE-1: Trial of Repurposed Unithiol for snakebite Envenoming phase 1 (safety, tolerability, pharmacokinetics and pharmacodynamics in healthy Kenyan adults). Wellcome Open Res. 2022;7. [CrossRef]
- Vragniau C, Bufton JC, Garzoni F, Stermann E, Rabi F, Terrat C, et al. Synthetic self-assembling ADDomer platform for highly efficient vaccination by genetically encoded multiepitope display. Sci Adv. 2019;5: eaaw2853. [CrossRef]
- Besson S, Vragniau C, Vassal-Stermann E, Dagher MC, Fender P. The Adenovirus Dodecahedron: Beyond the Platonic Story. Viruses. 2020;12: 718. [CrossRef]
- Chevillard C, Amen A, Besson S, Hannani D, Bally I, Dettling V, et al. Elicitation of potent SARS-CoV-2 neutralizing antibody responses through immunization with a versatile adenovirus-inspired multimerization platform. Mol Ther. 2022;30: 1913–1925. [CrossRef]
- Luo C, Yan Q, Huang J, Liu J, Li Y, Wu K, et al. Using Self-Assembling ADDomer Platform to Display B and T Epitopes of Type O Foot-and-Mouth Disease Virus. Viruses. 2022;14: 1810. [CrossRef]
- Buzas D, Bunzel HA, Staufer O, Milodowski EJ, Edmunds GL, Bufton JC, et al. Antibodies generated in vitro and in vivo elucidate design of a thermostable ADDomer COVID-19 nasal nanoparticle vaccine. Biochemistry; 2023 Mar. [CrossRef]
- Zochowska M, Paca A, Schoehn G, Andrieu J-P, Chroboczek J, Dublet B, et al. Adenovirus Dodecahedron, as a Drug Delivery Vector. Jagetia GC, editor. PLoS ONE. 2009;4: e5569. [CrossRef]
- Buzas D, Sun H, Toelzer C, Yadav SKN, Borucu U, Gautam G, et al. Engineering the ADDobody protein scaffold for generation of high-avidity ADDomer super-binders. Synthetic Biology; 2023 Sep. [CrossRef]
- Gutiérrez JM, Theakston RDG, Warrell DA. Confronting the Neglected Problem of Snake Bite Envenoming: The Need for a Global Partnership. PLOS Med. 2006;3: e150. [CrossRef]
- Warrell DA, Davidson NMcD null, Greenwood BM, Ormerod LD, Pope HM, Watkins BJ, et al. Poisoning by bites of the saw-scaled or carpet viper (Echis carinatus) in Nigeria. Q J Med. 1977;46: 33–62.
- Casewell NR, Harrison RA, Wüster W, Wagstaff SC. Comparative venom gland transcriptome surveys of the saw-scaled vipers (Viperidae: Echis) reveal substantial intra-family gene diversity and novel venom transcripts. BMC Genomics. 2009;10: 564. [CrossRef]
- Dingwoke EJ, Adamude FA, Mohamed G, Klein A, Salihu A, Abubakar MS, et al. Venom proteomic analysis of medically important Nigerian viper Echis ocellatus and Bitis arietans snake species. Biochem Biophys Rep. 2021;28: 101164. [CrossRef]
- Rowan EG, Harvey AL. Snake toxins from mamba venoms: unique tools for the physiologist. Acta Chim Slov. 2011;58: 689–692.
- Lauridsen LP, Laustsen AH, Lomonte B, Gutiérrez JM. Toxicovenomics and antivenom profiling of the Eastern green mamba snake (Dendroaspis angusticeps). J Proteomics. 2016;136: 248–261. [CrossRef]
- Laustsen AH, Lomonte B, Lohse B, Fernández J, Gutiérrez JM. Unveiling the nature of black mamba (Dendroaspis polylepis) venom through venomics and antivenom immunoprofiling: Identification of key toxin targets for antivenom development. J Proteomics. 2015;119: 126–142. [CrossRef]
- Degueldre M, Echterbille J, Smargiasso N, Damblon C, Gouin C, Mourier G, et al. In-Depth Glyco-Peptidomics Approach Reveals Unexpected Diversity of Glycosylated Peptides and Atypical Post-Translational Modifications in Dendroaspis angusticeps Snake Venom. Int J Mol Sci. 2017;18: 2453. [CrossRef]
- Quinton L, Demeure K, Dobson R, Gilles N, Gabelica V, De Pauw E. New Method for Characterizing Highly Disulfide-Bridged Peptides in Complex Mixtures: Application to Toxin Identification from Crude Venoms. J Proteome Res. 2007;6: 3216–3223. [CrossRef]
- Quinton L, Girard E, Maiga A, Rekik M, Lluel P, Masuyer G, et al. Isolation and pharmacological characterization of AdTx1, a natural peptide displaying specific insurmountable antagonism of the alpha1A-adrenoceptor. Br J Pharmacol. 2010;159: 316–325. [CrossRef]
- Dunbar JP, Fort A, Redureau D, Sulpice R, Dugon MM, Quinton L. Venomics Approach Reveals a High Proportion of Lactrodectus-Like Toxins in the Venom of the Noble False Widow Spider Steatoda nobilis. Toxins. 2020;12: 402. [CrossRef]
- Calvete JJ, Sanz L, Angulo Y, Lomonte B, Gutiérrez JM. Venoms, venomics, antivenomics. FEBS Lett. 2009;583: 1736–1743. [CrossRef]
- Wilson D, Daly NL. Venomics: A Mini-Review. High-Throughput. 2018;7: 19. [CrossRef]
- Oldrati V, Arrell M, Violette A, Perret F, Sprüngli X, Wolfender J-L, et al. Advances in venomics. Mol Biosyst. 2016;12: 3530–3543. [CrossRef]
- Oliveira AL, Viegas MF, da Silva SL, Soares AM, Ramos MJ, Fernandes PA. The chemistry of snake venom and its medicinal potential. Nat Rev Chem. 2022;6: 451–469. [CrossRef]
- Casewell NR, Wagstaff SC, Wüster W, Cook DAN, Bolton FMS, King SI, et al. Medically important differences in snake venom composition are dictated by distinct postgenomic mechanisms. Proc Natl Acad Sci. 2014;111: 9205–9210. [CrossRef]
- Morsa D, Baiwir D, La Rocca R, Zimmerman TA, Hanozin E, Grifnée E, et al. Multi-Enzymatic Limited Digestion: The Next-Generation Sequencing for Proteomics? J Proteome Res. 2019;18: 2501–2513. [CrossRef]
- Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, et al. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom RCM. 2003;17: 2337–2342. [CrossRef]
- Ainsworth S, Petras D, Engmark M, Süssmuth RD, Whiteley G, Albulescu L-O, et al. The medical threat of mamba envenoming in sub-Saharan Africa revealed by genus-wide analysis of venom composition, toxicity and antivenomics profiling of available antivenoms. J Proteomics. 2018;172: 173–189. [CrossRef]
- Gutiérrez JM, Lomonte B, León G, Alape-Girón A, Flores-Díaz M, Sanz L, et al. Snake venomics and antivenomics: Proteomic tools in the design and control of antivenoms for the treatment of snakebite envenoming. J Proteomics. 2009;72: 165–182. [CrossRef]
- Schaffitzel C, Hanes J, Jermutus L, Plückthun A. Ribosome display: an in vitro method for selection and evolution of antibodies from libraries. J Immunol Methods. 1999;231: 119–135. [CrossRef]
- Hanes J, Schaffitzel C, Knappik A, Plückthun A. Picomolar affinity antibodies from a fully synthetic naive library selected and evolved by ribosome display. Nat Biotechnol. 2000;18: 1287–1292. [CrossRef]
- Dreier B, Plückthun A. Rapid Selection of High-Affinity Antibody scFv Fragments Using Ribosome Display. Methods Mol Biol Clifton NJ. 2018;1827: 235–268. [CrossRef]
- Zimmermann I, Egloff P, Hutter CAJ, Kuhn BT, Bräuer P, Newstead S, et al. Generation of synthetic nanobodies against delicate proteins. Nat Protoc. 2020;15: 1707–1741. [CrossRef]
- Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004;22: 575–582. [CrossRef]
- Zahnd C, Spinelli S, Luginbühl B, Amstutz P, Cambillau C, Plückthun A. Directed in Vitro Evolution and Crystallographic Analysis of a Peptide-binding Single Chain Antibody Fragment (scFv) with Low Picomolar Affinity. J Biol Chem. 2004;279: 18870–18877. [CrossRef]
- Rivera-de-Torre E, Rimbault C, Jenkins TP, Sørensen CV, Damsbo A, Saez NJ, et al. Strategies for Heterologous Expression, Synthesis, and Purification of Animal Venom Toxins. Front Bioeng Biotechnol. 2022;9: 811905. [CrossRef]
- Simonato M, Morbiato L, Zorzi V, Caccin P, Fernández J, Massimino ML, et al. Production in Escherichia coli, folding, purification and characterization of notexin with wild type sequence and with N-terminal and catalytic site mutations. Toxicon. 2014;88: 11–20. [CrossRef]
- Chiou Y-L, Lin S-R, Chang L-S. Mutations on N-terminal region of Taiwan cobra phospholipase A2 result in structurally distorted effects. J Pept Sci. 2008;14: 890–897. [CrossRef]
- Turchetto J, Sequeira AF, Ramond L, Peysson F, Brás JLA, Saez NJ, et al. High-throughput expression of animal venom toxins in Escherichia coli to generate a large library of oxidized disulphide-reticulated peptides for drug discovery. Microb Cell Factories. 2017;16: 6. [CrossRef]
- Sequeira AF, Turchetto J, Saez NJ, Peysson F, Ramond L, Duhoo Y, et al. Gene design, fusion technology and TEV cleavage conditions influence the purification of oxidized disulphide-rich venom peptides in Escherichia coli. Microb Cell Factories. 2017;16: 4. [CrossRef]
- Fairhead M, Howarth M. Site-specific biotinylation of purified proteins using BirA. Methods Mol Biol Clifton NJ. 2015;1266: 171–184. [CrossRef]
- Nguyen VD, Hatahet F, Salo KE, Enlund E, Zhang C, Ruddock LW. Pre-expression of a sulfhydryl oxidase significantly increases the yields of eukaryotic disulfide bond containing proteins expressed in the cytoplasm of E.coli. Microb Cell Factories. 2011;10: 1. [CrossRef]
- Fitzgerald DJ, Berger P, Schaffitzel C, Yamada K, Richmond TJ, Berger I. Protein complex expression by using multigene baculoviral vectors. Nat Methods. 2006;3: 1021–1032. [CrossRef]
- Tessier DC, Thomas DY, Khouri HE, Laliberté F, Vernet T. Enhanced secretion from insect cells of a foreign protein fused to the honeybee melittin signal peptide. Gene. 1991;98: 177–183. [CrossRef]
- Wagstaff SC, Laing GD, Theakston RDG, Papaspyridis C, Harrison RA. Bioinformatics and Multiepitope DNA Immunization to Design Rational Snake Antivenom. PLOS Med. 2006;3: e184. [CrossRef]
- Gutiérrez JM, Vargas M, Segura Á, Herrera M, Villalta M, Solano G, et al. In Vitro Tests for Assessing the Neutralizing Ability of Snake Antivenoms: Toward the 3Rs Principles. Front Immunol. 2021;11. Available: https://www.frontiersin.org/articles/10.3389/fimmu.2020.617429.
- Albulescu L-O, Xie C, Ainsworth S, Alsolaiss J, Crittenden E, Dawson CA, et al. A therapeutic combination of two small molecule toxin inhibitors provides broad preclinical efficacy against viper snakebite. Nat Commun. 2020;11: 6094. [CrossRef]
- Macêdo JKA, Fox JW. Biological Activities and Assays of the Snake Venom Metalloproteinases (SVMPs). In: Gopalakrishnakone P, Calvete JJ, editors. Venom Genomics and Proteomics. Dordrecht: Springer Netherlands; 2016. pp. 211–238. [CrossRef]
- Hiu JJ, Yap MKK. Cytotoxicity of snake venom enzymatic toxins: phospholipase A2 and l-amino acid oxidase. Biochem Soc Trans. 2020;48: 719–731. [CrossRef]
- Conlon JM, Attoub S, Arafat H, Mechkarska M, Casewell NR, Harrison RA, et al. Cytotoxic activities of [Ser49]phospholipase A2 from the venom of the saw-scaled vipers Echis ocellatus, Echis pyramidum leakeyi, Echis carinatus sochureki, and Echis coloratus. Toxicon. 2013;71: 96–104. [CrossRef]
- Still KBM, Nandlal RSS, Slagboom J, Somsen GW, Casewell NR, Kool J. Multipurpose HTS Coagulation Analysis: Assay Development and Assessment of Coagulopathic Snake Venoms. Toxins. 2017;9: 382. [CrossRef]
- Patel RN, Clare RH, Ledsgaard L, Nys M, Kool J, Laustsen AH, et al. An in vitro assay to investigate venom neurotoxin activity on muscle-type nicotinic acetylcholine receptor activation and for the discovery of toxin-inhibitory molecules. Biochem Pharmacol. 2023; 115758. [CrossRef]
- Slagboom J, Otvos RA, Cardoso FC, Iyer J, Visser JC, van Doodewaerd BR, et al. Neurotoxicity fingerprinting of venoms using on-line microfluidic AChBP profiling. Toxicon. 2018;148: 213–222. [CrossRef]
- Servent D, Winckler-Dietrich V, Hu H-Y, Kessler P, Drevet P, Bertrand D, et al. Only Snake Curaremimetic Toxins with a Fifth Disulfide Bond Have High Affinity for the Neuronal α7 Nicotinic Receptor. J Biol Chem. 1997;272: 24279–24286. [CrossRef]
- Harvey AL. Twenty years of dendrotoxins. Toxicon. 2001;39: 15–26. [CrossRef]
- Yajuan X, Xin L, Zhiyuan L. A Comparison of the Performance and Application Differences Between Manual and Automated Patch-Clamp Techniques. Curr Chem Genomics. 2012;6: 87–92. [CrossRef]
- Theakston RD, Reid HA. Development of simple standard assay procedures for the characterization of snake venom. Bull World Health Organ. 1983;61: 949–956.
- Knudsen C, Casewell NR, Lomonte B, Gutiérrez JM, Vaiyapuri S, Laustsen AH. Novel Snakebite Therapeutics Must Be Tested in Appropriate Rescue Models to Robustly Assess Their Preclinical Efficacy. Toxins. 2020;12: 528. [CrossRef]
- Ikonomou L, Schneider Y-J, Agathos SN. Insect cell culture for industrial production of recombinant proteins. Appl Microbiol Biotechnol. 2003;62: 1–20. [CrossRef]
- Yee CM, Zak AJ, Hill BD, Wen F. The Coming Age of Insect Cells for Manufacturing and Development of Protein Therapeutics. Ind Eng Chem Res. 2018;57: 10061–10070. [CrossRef]
- Maghodia AB, Geisler C, Jarvis DL. Characterization of an Sf-rhabdovirus-negative Spodoptera frugiperda cell line as an alternative host for recombinant protein production in the baculovirus-insect cell system. Protein Expr Purif. 2016;122: 45–55. [CrossRef]
- Maghodia AB, Geisler C, Jarvis DL. A new nodavirus-negative Trichoplusia ni cell line for baculovirus-mediated protein production. Biotechnol Bioeng. 2020;117: 3248–3264. [CrossRef]
- Fuenmayor J, Gòdia F, Cervera L. Production of virus-like particles for vaccines. New Biotechnol. 2017;39: 174–180. [CrossRef]
- Roldão A, Mellado MCM, Castilho LR, Carrondo MJ, Alves PM. Virus-like particles in vaccine development. Expert Rev Vaccines. 2010;9: 1149–1176. [CrossRef]
- Junne S, Neubauer P. How scalable and suitable are single-use bioreactors? Curr Opin Biotechnol. 2018;53: 240–247. [CrossRef]
- Gerstweiler L, Bi J, Middelberg APJ. Continuous downstream bioprocessing for intensified manufacture of biopharmaceuticals and antibodies. Chem Eng Sci. 2021;231: 116272. [CrossRef]
- Gerstweiler L, Billakanti J, Bi J, Middelberg APJ. An integrated and continuous downstream process for microbial virus-like particle vaccine biomanufacture. Biotechnol Bioeng. 2022;119: 2122–2133. [CrossRef]
- Mvundura M, Kien VD, Nga NT, Robertson J, Van Cuong N, Tung HT, et al. How much does it cost to get a dose of vaccine to the service delivery location? Empirical evidence from Vietnam’s Expanded Program on Immunization. Vaccine. 2014;32: 834–838. [CrossRef]
- Mvundura M, Lorenson K, Chweya A, Kigadye R, Bartholomew K, Makame M, et al. Estimating the costs of the vaccine supply chain and service delivery for selected districts in Kenya and Tanzania. Vaccine. 2015;33: 2697–2703. [CrossRef]
- Kumar R, Srivastava V, Baindara P, Ahmad A. Thermostable vaccines: an innovative concept in vaccine development. Expert Rev Vaccines. 2022;21: 811–824. [CrossRef]
- Daniell H, Rai V, Xiao Y. Cold chain and virus-free oral polio booster vaccine made in lettuce chloroplasts confers protection against all three poliovirus serotypes. Plant Biotechnol J. 2019;17: 1357–1368. [CrossRef]
- Guo M, Li J, Teng Z, Ren M, Dong H, Zhang Y, et al. Four Simple Biomimetic Mineralization Methods to Improve the Thermostability and Immunogenicity of Virus-like Particles as a Vaccine against Foot-and-Mouth Disease. Vaccines. 2021;9: 891. [CrossRef]
- Cardoso FMC, Petrovajová D, Horňáková T. Viral vaccine stabilizers: status and trends. Acta Virol. 2017;61: 231–239. [CrossRef]
Figure 1.
Structure-based design of ADDobody. (a) Twelve pentons self-assemble into the ADDomer particle (PDB ID 6HCR [20]). ADDomer is shown in grey, one penton is highlighted in colour. (b) The penton consists of five penton base proteins (depicted in magenta, purple, light blue, blue, dark blue). (c) Penton base protein comprises a jelly roll fold multimerisation domain and a crown domain with hypervariable RGD and VL loops. (d) ADDobody is the engineered crown domain. RGD and VL loops were randomised in sequence and length to generate the ADDobody library. (e) Scheme of ribosome display in vitro selection and evolution. The DNA library encoding the ADDobody library (up to 1012 members) is transcribed using T7 RNA polymerase and then translated in vitro. Ribosome-mRNA-ADDobody complexes form and ADDobodies (magenta) can fold outside the ribosomal tunnel due to an unstructured ‘spacer’ fused to the C-terminus of ADDobody. The complexes are used for selection against immobilised antigens/toxins. Non-binders are eliminated during stringent washing steps. mRNA is recovered by dissociating the ribosomal complexes with EDTA, reverse-transcribed and PCR amplified (RT-PCR). During PCR, the ribosome-binding site and T7 promoter sequence are reintroduced into the construct. Error-prone PCR introduces mutations into selected binders allowing in vitro evolution. The resulting DNA library is enriched for binders and can be used for a new ribosome display round or cloned into a plasmid to clonally isolate and express selected ADDobodies.
Figure 1.
Structure-based design of ADDobody. (a) Twelve pentons self-assemble into the ADDomer particle (PDB ID 6HCR [20]). ADDomer is shown in grey, one penton is highlighted in colour. (b) The penton consists of five penton base proteins (depicted in magenta, purple, light blue, blue, dark blue). (c) Penton base protein comprises a jelly roll fold multimerisation domain and a crown domain with hypervariable RGD and VL loops. (d) ADDobody is the engineered crown domain. RGD and VL loops were randomised in sequence and length to generate the ADDobody library. (e) Scheme of ribosome display in vitro selection and evolution. The DNA library encoding the ADDobody library (up to 1012 members) is transcribed using T7 RNA polymerase and then translated in vitro. Ribosome-mRNA-ADDobody complexes form and ADDobodies (magenta) can fold outside the ribosomal tunnel due to an unstructured ‘spacer’ fused to the C-terminus of ADDobody. The complexes are used for selection against immobilised antigens/toxins. Non-binders are eliminated during stringent washing steps. mRNA is recovered by dissociating the ribosomal complexes with EDTA, reverse-transcribed and PCR amplified (RT-PCR). During PCR, the ribosome-binding site and T7 promoter sequence are reintroduced into the construct. Error-prone PCR introduces mutations into selected binders allowing in vitro evolution. The resulting DNA library is enriched for binders and can be used for a new ribosome display round or cloned into a plasmid to clonally isolate and express selected ADDobodies.
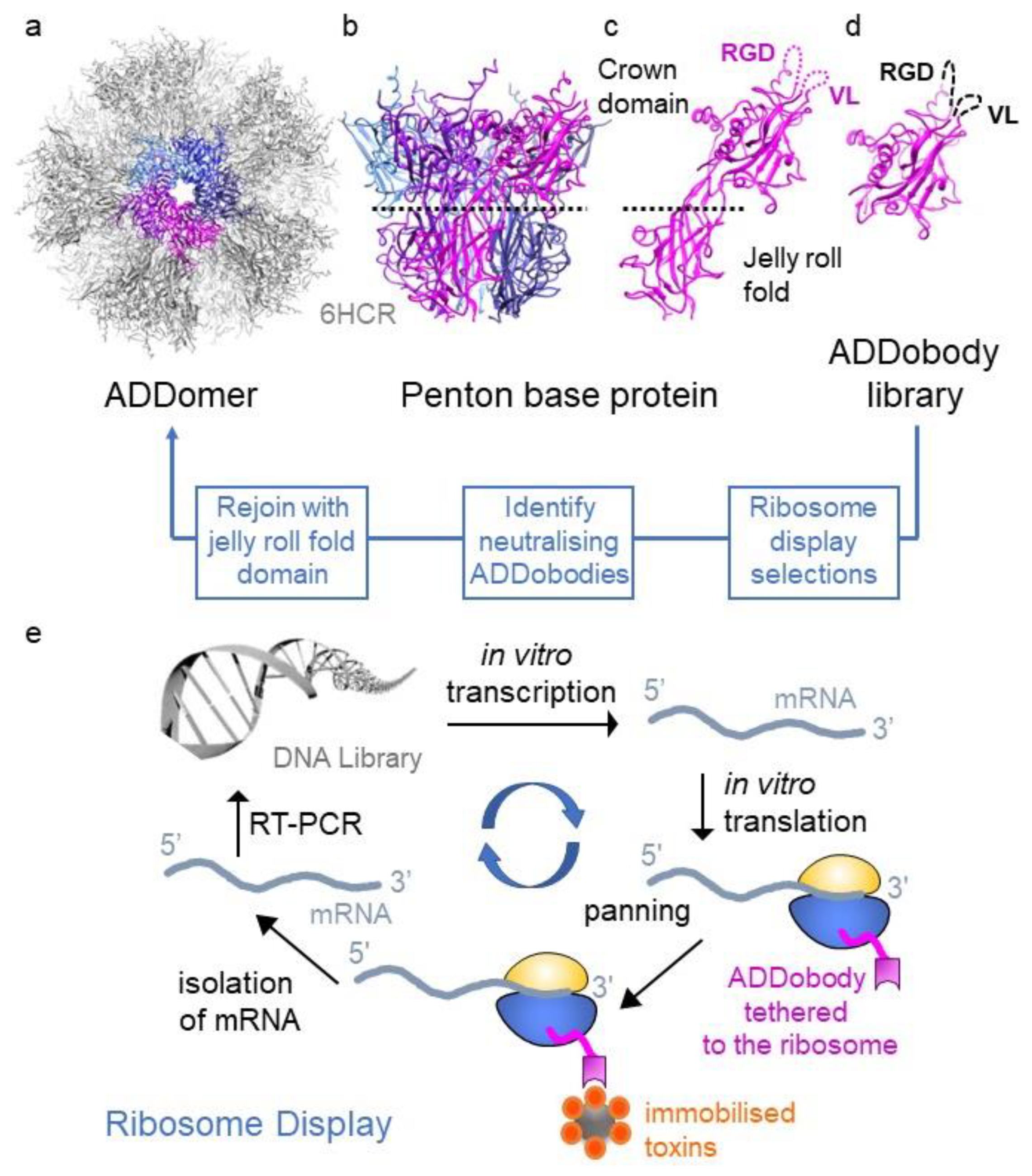
Figure 2.
Overview of the ADDovenom project. Mass spectrometry will be used to define the venom composition of nine snake venoms and identify toxins. Toxins will then be produced as recombinant proteins in bacterial and insect cell expression systems. A synthetic library of ADDobodies will be created and screened using ribosome display to identify specific, high-affinity binders to the recombinant toxins. ADDobodies and ADDomers will then be assessed for toxin neutralising ability using in vitro assays and in vivo models of venom bioactivity. Finally, a GMP-compatible platform will be developed to produce toxin-neutralising ADDomers at scale.
Figure 2.
Overview of the ADDovenom project. Mass spectrometry will be used to define the venom composition of nine snake venoms and identify toxins. Toxins will then be produced as recombinant proteins in bacterial and insect cell expression systems. A synthetic library of ADDobodies will be created and screened using ribosome display to identify specific, high-affinity binders to the recombinant toxins. ADDobodies and ADDomers will then be assessed for toxin neutralising ability using in vitro assays and in vivo models of venom bioactivity. Finally, a GMP-compatible platform will be developed to produce toxin-neutralising ADDomers at scale.
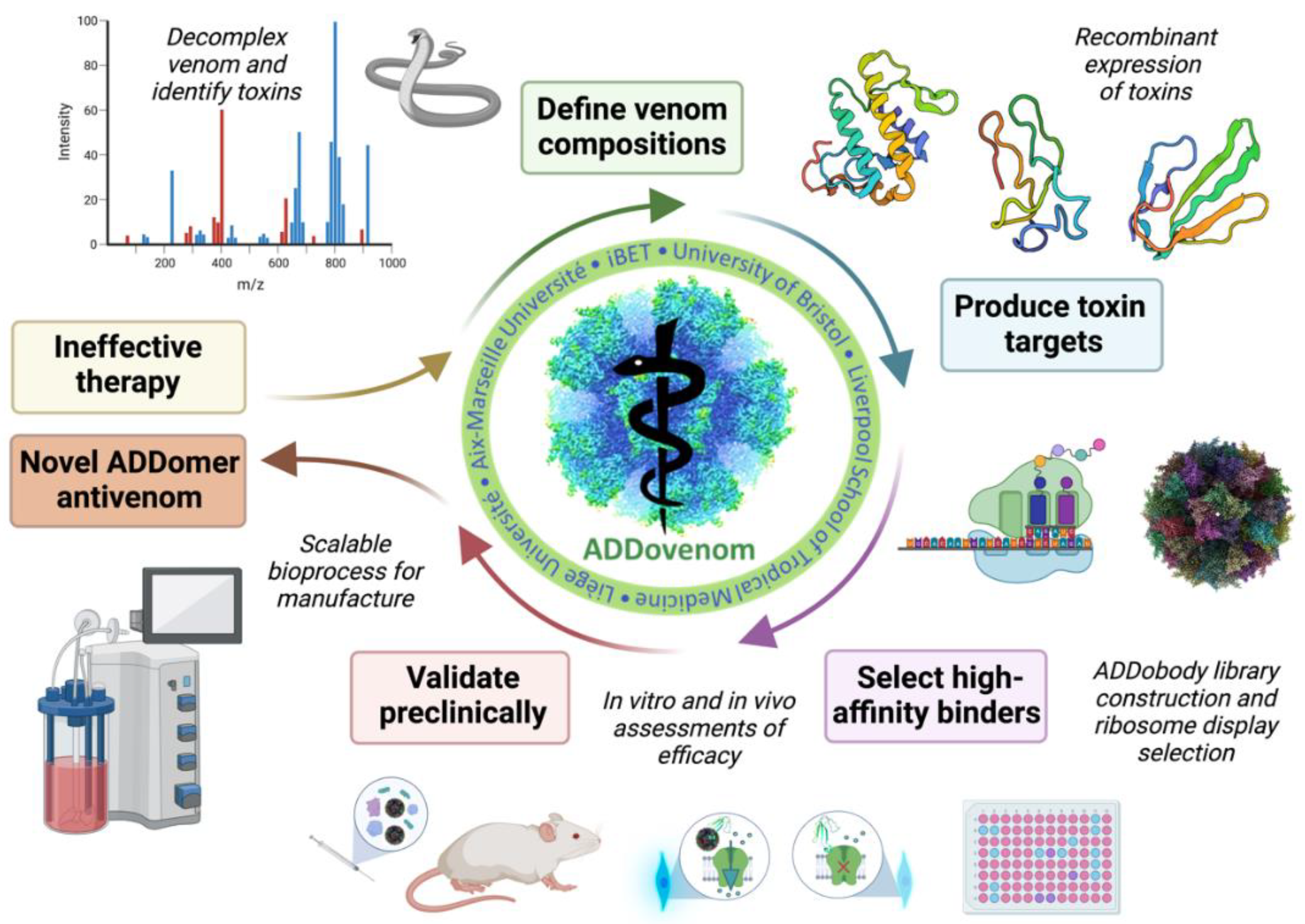
Figure 3.
A brief overview to illustrate the aims of the mass spectrometry work package. Nine of the deadliest snakes found in Sub-Saharan Africa are selected for this study, including five species of Dendroaspis and four species of Echis. Shotgun proteomics, along with the use of a database repository and transcriptome data, will be used to generate a toxin database. Bioinformatic tools will then be utilised to predict the biochemical and structural features of the toxins and, when combined with the symptoms of envenoming, help to identify and select the most potent toxins in the venom for use in ADDomer production.
Figure 3.
A brief overview to illustrate the aims of the mass spectrometry work package. Nine of the deadliest snakes found in Sub-Saharan Africa are selected for this study, including five species of Dendroaspis and four species of Echis. Shotgun proteomics, along with the use of a database repository and transcriptome data, will be used to generate a toxin database. Bioinformatic tools will then be utilised to predict the biochemical and structural features of the toxins and, when combined with the symptoms of envenoming, help to identify and select the most potent toxins in the venom for use in ADDomer production.
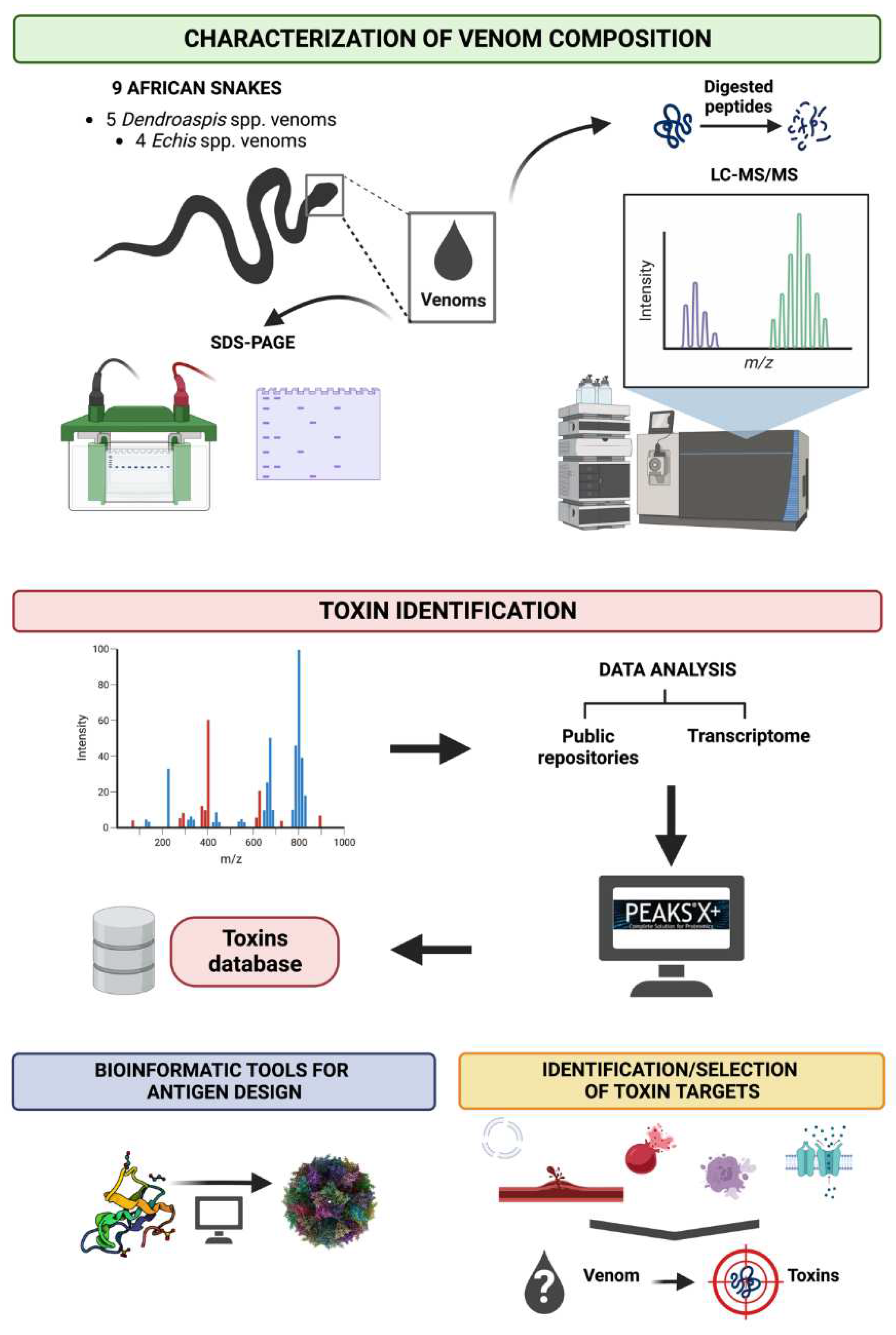
Figure 4.
In vitro assays that will be used to measure venom activity and neutralisation by ADDomers. (a) The SVMP assay uses a quenched fluorogenic peptide substrate (ES010, BioTechne) which is cleaved by SVMPs, resulting in a free unquenched fragment. Structural model shown (green-pink-cyan) is an E. ocellatus SVMP (AlphaFold AF-Q2UXQ5-F1). (b) Enzymatically active PLA2s are measured using a fluorescent assay (EnzCheck, ThermoFisher) in which a fluorescent substrate (BODIPY PC-A2) is encapsulated in liposomes. PLA2s hydrolyse the phospholipids, resulting in increased fluorescence. Structural model shown (blue) is a PLA2 from E. ocellatus (AlphaFold AF-P59171-F1). (c) The plasma clotting assay measures the optical density (OD) of plasma in a spectrophotometer to detect clot formation where a higher OD indicates clotting. A normal plasma clotting control is concurrently run, and compared to venom samples to indicate whether venom is causing pro-coagulant or anti-coagulant effects. (d) Flow cytometry and sandwich ELISA assays of disintegrin assays use antibodies to detect aggregated platelets following ADP-activation. (e) The neurotoxic activity of α-neurotoxins (a subclass of 3FTx) can be measured in a cell-based assay using cells natively expressing muscle-type nAChRs incubated with a fluorescent dye (FLIPR Membrane Potential Assay, Molecular Devices) that measures the change in membrane potential upon acetylcholine-induced nAChR activation. Protein shown (cyan-green) is α-elapitoxin-Dpp2d from D. polylepis (PDB ID: 4LFT). (f) The neurotoxic activity of Kunitz-type toxins on voltage-gated potassium channels will be measured in a cell-based assay using transfected HEK293 cells expressing human Kv1.1 or Kv1.2 channels. Cells are incubated with the fluorescent FLIPR Membrane Potential Assay dye (Molecular Devices) that measures the change in membrane potential upon application of extracellular potassium sulphate which causes cell membrane depolarisation and therefore activation of Kv channels. Protein shown (red-yellow) is dendrotoxin-I from D. polylepis (PDB ID: 1DEM).
Figure 4.
In vitro assays that will be used to measure venom activity and neutralisation by ADDomers. (a) The SVMP assay uses a quenched fluorogenic peptide substrate (ES010, BioTechne) which is cleaved by SVMPs, resulting in a free unquenched fragment. Structural model shown (green-pink-cyan) is an E. ocellatus SVMP (AlphaFold AF-Q2UXQ5-F1). (b) Enzymatically active PLA2s are measured using a fluorescent assay (EnzCheck, ThermoFisher) in which a fluorescent substrate (BODIPY PC-A2) is encapsulated in liposomes. PLA2s hydrolyse the phospholipids, resulting in increased fluorescence. Structural model shown (blue) is a PLA2 from E. ocellatus (AlphaFold AF-P59171-F1). (c) The plasma clotting assay measures the optical density (OD) of plasma in a spectrophotometer to detect clot formation where a higher OD indicates clotting. A normal plasma clotting control is concurrently run, and compared to venom samples to indicate whether venom is causing pro-coagulant or anti-coagulant effects. (d) Flow cytometry and sandwich ELISA assays of disintegrin assays use antibodies to detect aggregated platelets following ADP-activation. (e) The neurotoxic activity of α-neurotoxins (a subclass of 3FTx) can be measured in a cell-based assay using cells natively expressing muscle-type nAChRs incubated with a fluorescent dye (FLIPR Membrane Potential Assay, Molecular Devices) that measures the change in membrane potential upon acetylcholine-induced nAChR activation. Protein shown (cyan-green) is α-elapitoxin-Dpp2d from D. polylepis (PDB ID: 4LFT). (f) The neurotoxic activity of Kunitz-type toxins on voltage-gated potassium channels will be measured in a cell-based assay using transfected HEK293 cells expressing human Kv1.1 or Kv1.2 channels. Cells are incubated with the fluorescent FLIPR Membrane Potential Assay dye (Molecular Devices) that measures the change in membrane potential upon application of extracellular potassium sulphate which causes cell membrane depolarisation and therefore activation of Kv channels. Protein shown (red-yellow) is dendrotoxin-I from D. polylepis (PDB ID: 1DEM).
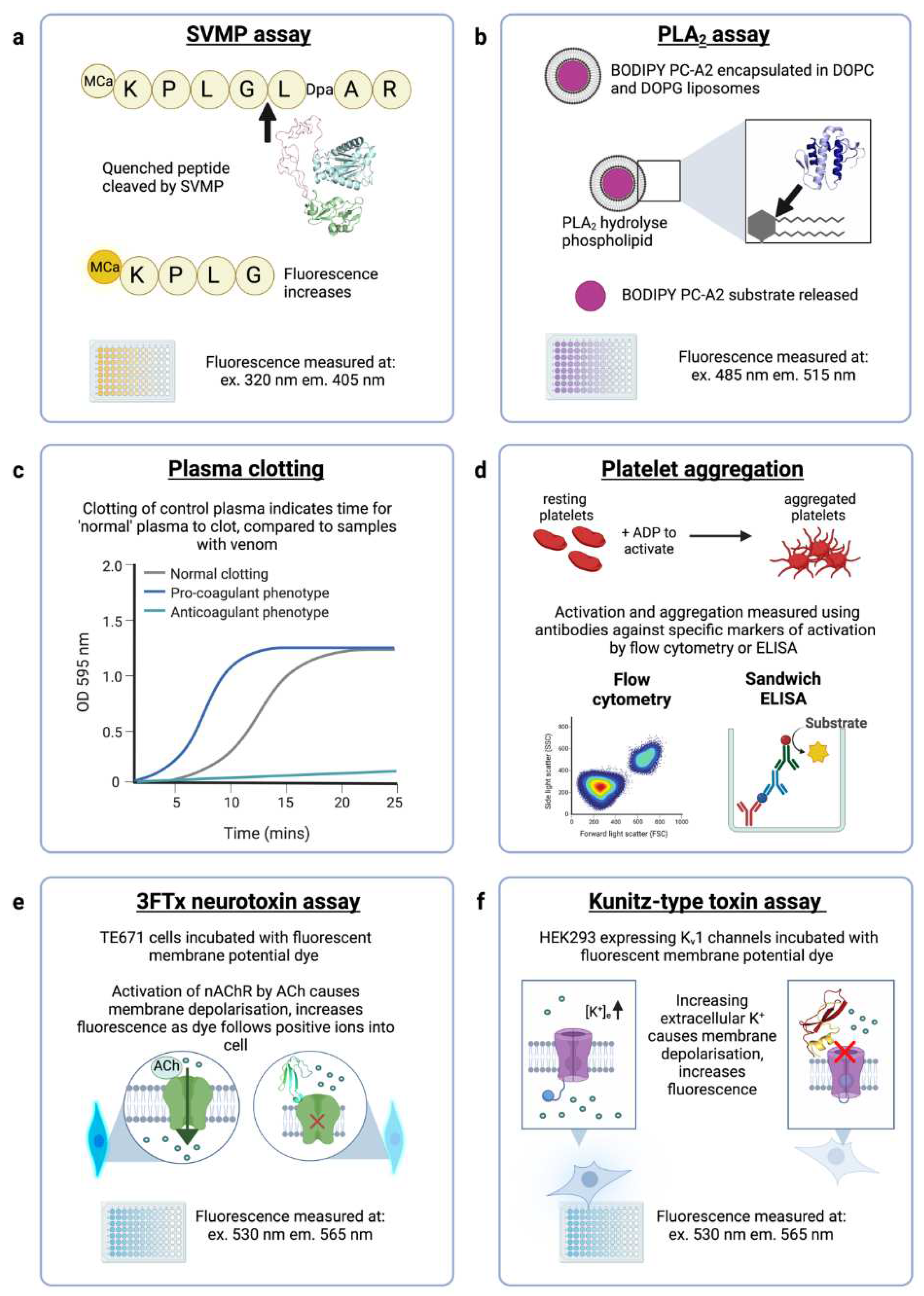
Figure 4.
Scalable, GMP-compliant process for ADDomer production. MCB: master cell bank; MVSS: master virus seed stock. See main text for further details.
Figure 4.
Scalable, GMP-compliant process for ADDomer production. MCB: master cell bank; MVSS: master virus seed stock. See main text for further details.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



