
Submitted:
14 October 2023
Posted:
17 October 2023
You are already at the latest version
Abstract
The agamid lizard Phrynocephalus melanurus, is restricted to the Northwest China (Dzungar Basin) and adjacent Eastern Kazakhstan (Zaisan and Alakol basins). To elucidate the phylogeography of P. melanurus, we obtained the mitochondrial DNA COI segments of 175 sampled lizards from 44 localities across the whole distribution range. Phylogenetic analyses revealed two main clades comprising six geographically structured lineages (I, IIa, IIb, IIc1, IIc2, and IIc3) that fits an isola-tion by distance (IBD) model. The divergence from the most recent ancestor was dated to ~1.86 million years ago (Ma). Demographic analyses demonstrated lineage-specific response to past climate change: stable population for Clade I, IIb and IIc2; while past population expansion for IIc1 and IIc3 since 0.02 Ma and 0.18 Ma, respectively. Bayesian phylogeographic diffusion anal-yses detected initial spreading at the Saur Mount vicinity approximately 1.8 Ma. Historical species distribution modeling (SDM) projected expansion of the suitable habitat in the last interglacial period and stepwise contraction in the Last Glacial Maximum and Holocene epochs. The SDM predicts a shrinkage and shift of suitable area eastward as a response to future climate change. Our findings suggest that the evolution of P. melanurus followed a parapatric divergence with subsequent dispersal and adaptation to cold and dry environments during the Quaternary. Moreover, future conservation strategies need to be taken to maintain the population of lineage IIc1.
Keywords:
Agama
; Aridification
; Colonization
; Mitochondrial DNA
; Northwest China
; Phylogeography
; Species Distribution Modeling
1. Introduction
The historical geological events and climate changes, coupled with the substantial environmental heterogeneity, have shaped the phylogeography of organisms across the globe. This complex interplay may have influenced significantly the processes of diversification and speciation of local biota in drylands [1,2]. It has been recognized that the climate fluctuations during the Quaternary have greatly impacted the distribution ranges of arid adapted organisms [3,4,5,6]. Thus, many species adapted to the cold and dry environments have recently attracted much attention in the field of biogeography and phylogeography [7,8,9,10,11].
Late Cenozoic progressive aridification in Central Asia is thought to be one of the most prominent climate changes in the Northern Hemisphere [12,13,14], resulting in the emergence of diverse arid landscapes, including sandy deserts, rocky deserts like the Gobi [15]. Meanwhile, the stepwise desertification of Northwest China contributed to the emergence of the Guerbantonggut Desert, the second largest sandy desert in Northwest China [16]. This desert is a major component of the Dzungar Basin, which is located in the Northern part of Xinjiang of China. Being an intermontane basin, Dzungar Basin is bounded by Dzungarian Alatau, Tarbagatay, and Saur mountains at the west, Tian Shan Mountains at the south, and Altai Mountains at the north. The formation of the sandy deserts and the Tianshan Mountains plays a crucial role in diversifying the regional environmental conditions [17]. This complex landscape has shaped rich biodiversity and makes the area an ideal system for examining species evolution under a phylogeographic perspective [18,19,20,21].
As such, plant phylogeographic studies in arid Northwest China have sprung up like mushrooms after rain [21,22,23]. However, animal phylogeographic studies, particularly those of lizards are still limited [8,11,25]. In fact, lizards are useful for studying the effects of environmental and geological conditions on phylogeographic structure due to their lower dispersal abilities, and susceptibility to climate fluctuations [26,27]. Nevertheless, a general conclusion has not been drawn on how the flora and fauna in Northwest China responded to the Quaternary climatic oscillations [23,28].
Phrynocephalus melanurus Eichwald, 1831, also known as the Zaisan toad-headed agama, is one of the highly adapted to sandy and gravel deserts species of the Phrynocephalus guttatus complex [29]. Its distribution range spans from the Zaisan and Alakol (Dzungar Gate vicinity) basins in the Eastern Kazakhstan to the Dzungar Basin in Northwest China [29,30,31]. Due to adaptation to the changing environments, the lizard has become specialized in accommodating the heterogeneous structure of the habitat and forming “substrate races” [32]. This intricate process has given rise to an array of morphological variations that display discernible phenotypic disparities, which have historically led to their classification under diverse taxonomic designations (reviewed by [33] and references therein). The species status of P. melanurus has been disputed, and was historically classified as a subspecies of P. guttatus [33,34,35]. Recent work suggested that the morphological differences between P. melanurus and P. guttatus may be corroborated by genetic data, with a high mean sequence divergence of 6.9 % in mtDNA ND2 gene [3], and of 2.48% in mtDNA COI gene [34]. Based on limited sampling (8 individuals restricted to two localities), Melville et al. [3] initially found that P. melanurus comprises a lineage from the Dzungar Basin (Xinjiang, China) and another from the Zaisan Lake area. Dunayev et al. [34] confirmed the differentiation of P. melanurus in Kazakhstan into two lineages inhabiting the Zaisan and Alakol depressions. However, one potential shortcoming of Dunayev et al. [34] lies in the lack of adequate sampling of P. melanurus from its whole distribution range. There are two passages connecting Dzungar with Kazakh Steppe (Great Dala) through the Dzungar Gate (western) and Irtysh valley (northwestern). Thus, agamas could migrate between Dzungar and adjacent Kazakhstan despite partial isolation of this region by mountains.
Thereby, our analyses were motivated by the goals to obtain a more detailed picture of the genetic structure and to trace the population history of P. melanurus across its distribution range. We used a phylogeographic approach complemented with species distribution modeling. Specifically, we aimed to (i) document the phylogeographic structure and the timing of genetic diversification within the Zaisan toad-headed agama; (ii) reconstruct the center of origin and colonization routes; and (iii) explore the relationship between historical demographic changes and past climate fluctuations. Analyses of the mtDNA sequences tell only one part of a potentially more complex story [36,37], yet they provide valuable insights into the evolutionary history of a species, including consequences of habitat changes, impacts from climate fluctuations, divergence and colonization. To incorporate the data published by Solovyeva et al. [38] and especially the data from the Zaisan Basin [34], we amplified the mitochondrial COI gene segment in this study.
2. Materials and Methods
2.1. Population Sampling
A total of 165 individuals of P. melanurus were collected from 36 sites across its whole distribution range from 2008‒2019 (Figure 1) including 1 sample from the type locality (Kyzylkum sands (previous name ‒ Bukon sands), of the Kurchum district, E KZ), which covers the Northern Xinjiang region of China, and the adjacent Eastern part of Kazakhstan. Additionally, 10 samples of P. melanurus from Kazakhstan (the Zaisan and Alakol basins) were taken from the previous studies [34,38]; two outgroup sequences were retrieved from GenBank – Phrynocephalus alpherakii (KF691705), Phrynocephalus guttatus guttatus (MK461381). Detailed sampling information is listed in Supplementary Materials, Table S1. Animals were euthanized with an overdose of sodium pentobarbital via intraperitoneal injection, and liver tissues were extracted and preserved in 95% ethanol following the animal-use protocols approved by Chengdu Institute of Biology (CIB), Chinese Academy of Sciences. Liver or tail tissue from specimens was fixed with 95% ethanol and stored at −20°C before DNA extraction. The voucher specimens for all populations are deposited in CIB.
2.2. DNA Isolation, PCR Amplification and Sequencing
Genomic DNA was extracted either from the 95% ethanol-preserved tail or liver tissue samples using the universal high-salt procedures [39]. Amplification of the COI gene fragment was implemented by using the pairs of primer: PhCOIf (5׳-AATTCAGCCATCTTACCATGTCAAC-3׳), PhCOIr (5׳-TATACTTCTGGGTGGCCAAAGAA- 3׳), which was designed particularly for this study. The length of amplified sequences was 680 bp. Each PCR reaction contained 25 μL of Taq PCR Master Mix (Omega Bio-Tek), 2 μL each primer (0.4 μM), 1−2 μL genomic DNA (~50 ng), and 19−20 μL double sterilized water for a total reaction volume of 50 μL. The PCR protocol involved an initial denaturation at 94°C for 4 min, followed by 35 cycles at 94°C for 30 s, 55°C for 30 s, and elongation at 72°C for 53 s, and a final extension at 72°C for 10 min. The PCR products were assessed using 1% agarose gel electrophoresis, purified, and sequenced for double strands with the PCR primers. All fragments were sequenced with ABI 3730 automated DNA Analyzer at Sangon Biotech (Shanghai, China).
2.3. Phylogenetic Reconstruction
All obtained nucleotide sequences were checked and assembled using SeqMan II program included in LASERGENE 7 .0 software package (DNAStR Inc., Madision, USA). In total, 177 sequences – which included those from [34,40] (available in GenBank) – were aligned using Clustal X v.1.81 [41] under default parameters. Subsequently, the aligned sequences were translated to amino acids with SeaView v.5.0.1 [42], and no stop codons were detected. Identical sequences were collapsed into a single haplotype using DnaSP v.6.0 [43].
Phylogenetic inference was used to establish the relationships among the observed haplotypes and their associated populations of P. melanurus. We used Bayesian inference (BI) and maximum likelihood (ML) approaches to reconstruct phylogenetic relationships among the mitochondrial haplotypes. Bayesian analyses were implemented using MrBayes v.3.2.6 [44] employing partition-specific modeling. The best-fit models of nucleotide substitution for each partition scheme were selected using PartitionFinder v.2.1.1 [45]. Three codon partitions and their corresponding substitution model for the COI gene sequences were proposed: first codon – K80+I; second codon – HKY; third codon – GTR+G. Two simultaneous parallel runs were performed with four heated Markov (using default heating values) chains per run (three heated and one cold) for 10 million generations with sampling frequency of every 1000 generations. Convergence of the runs was assessed by the effective sample sizes (ESS) (>200) using Tracer v.1.7 [46] and the average standard deviation of split frequencies < 0.01. The first 25% of trees were discarded as burn-in and a 50% majority-rule consensus tree was constructed to calculate the posterior probabilities (PPs) of nodes. Partitioned maximum likelihood (ML) analyses were carried out in RAxMLHPC v.8.2.4 [47] with the same partitioning strategy as for BI. The GTR+G model was used for all subsets, and 100 replicate ML inferences were performed for each analysis. Each inference was initiated with a random starting tree and nodal bootstrap support (BS) was assessed with 1000 pseudoreplicates [48].
2.4. Divergence Times Estimation
To estimate the divergence time for mitochondrial haplotypes of P. melanurus we conducted the Bayesian dating in BEAST v.1.8.4 [49]. Owing to lack of a reliable fossil record and substitution rate of COI for P. melanurus, we applied as secondary calibration points the estimated mean values of internal nodes of clades of P. guttatus-versicolor species complex (6.5 million years ago, Ma), and P. guttatus specie complex (5.0 Ma) from the previous study [40]. Additional sequences were retrieved from GenBank (Supplementary Materials, Table S1) and incorporated with our dataset in order to expand the dataset and re-calculate the divergence age of P. guttatus species group. The taxon sets were defined in two groups: P. guttatus-versicolor and P. guttatus group. The HKY+G site substitution model was used, under the uncorrelated lognormal clock model. The calibration was implemented as a normal prior with standard deviation equal to 1.0; mean values were set as 6.5 Ma and 5.0 Ma, respectively. A birth-death speciation prior was used for the tree. Non partitioned dataset was used for a single run. Analysis was run for 10 million generations, with random starting tree. Convergence of the four runs was assessed by the effective sample size (ESS) ≥ 200. The first 10% of generations were discarded as burn-in using LogCombiner, and TreeAnnotator was used to infer the ultrametric tree [49]. We interpreted PP ≥ 0.95 to be strongly supported [50]. Final trees were visualized and edited in FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) [51].
2.5. Genetic Diversity and Population Genetic Structure Analyses
Haplotypes were extracted using DnaSP v.6.0 [43]. Population genetic diversity was quantified using indices of polymorphic site, number of mutations (m), nucleotide diversity [52], haplotype diversity [53], number of haplotypes (Nh), and average number of nucleotide differences (k), which were calculated in DnaSP. Uncorrected pairwise sequence divergences (p-distances) between and within phylogroups were calculated using MEGA v.X [53]. To infer geographic distribution and relationships of the P. melanurus haplotypes, a median-joining haplotype network was constructed using PopART v.1.7 [54].
The grouping of the population was performed via spatial analysis of molecular variance (SAMOVA) using SAMOVA v.2.0 software [55]. The analysis was run for values of K ranging from 2 to 8. The number of groups was selected according to FCT value (the differentiation of groups) using the sum of squared differences between haplotypes, with 100 simulated annealing processes. The configuration of K that had one or more single-population groups were excluded due to the group disappearing [56,57]. To estimate genetic variation within populations, among populations within groups and between groups (phyloclades, and as identified by SAMOVA), AMOVA was carried out in the program Arlequin v.3.11 [58], with significance test based on 1000 permutations. Subdivision on two regions represented the Zaisan Basin population and the Dzungar Basin population, which is in accordance with Clade I and Clade II, respectively. Another AMOVA test for four groups, representing four geographic regions North (Clade I), Central (Subclade IIc3), Southwest (Subclade IIc1) and Southeast (Subclade IIc2), was also conducted.
To test the spatial genetic structure of populations, isolation by distance (IBD) [59] was determined by testing the correlation between geographic distance and pairwise FST/(1–FST) using the Mantel test with Mantel test with GenAlEX v.6.5. [60]. The procedure was implemented on Clade I and II separately with 999 number of permutations, under the default parameters. Geographic distances among populations were estimated in R package using package Vegan v.2. (https://CRAN.R-project.org/package=vegan) [61].
2.6. Inference of Demographic History
Mismatch distributions of pairwise nucleotide differences were applied to test the sudden demographic expansion for the matrilines of P. melanurus using DnaSP [43], with 10,000 coalescent simulations. The goodness-of-fit between observed and expected distribution was tested by calculating Harpending’s Raggedness index (Rg) and the sum of squares deviation (SSD). Additionally, three types of neutrality test statistics were applied: Tajima’s D [62], Fu’s Fs [63] and R2 statistic [64] calculated in DnaSP as additional assessments of possible population expansion. Significantly positive R2 value was taken as evidence of population expansion.
Bayesian skyline plots (BSP) [65] were implemented in BEAST v.1.8.4 [49] to estimate the changes in effective population size on an evolutionary time scale. We applied a strict molecular clock with a mean substitution rate (0.012 per site per million years) obtained from the BEAST analysis described above. Owning to the small sample size (three sequences) for the Subclade IIa, BSP analysis was not conducted. We used HKY substitution model with empirical base frequencies, and three partitions into codon positions. Tree parameters were set to randomly-generated starting tree and piece-wise constant Skyline model with default number of groups (m = 10), except Subclade IIb where we reduced the number of groups to m = 5, due to its relatively small sample size — 15 sequences. This method allows to prevent over-parameterization of the model and get biased results [66]. The initial 10% of steps was discarded as burn-in. We obtained consistent demographic inferences across three replicates of the analysis visualized by Tracer v.1.7 [46].
2.7. Phylogeographic Diffusion in Continuous Space
Following Shi et al. [10], we reconstructed the spatiotemporal history of P. melanurus throughout their distribution range using the approach of Bayesian phylogeographic diffusion [67] in continuous space implemented in BEAST v.1.10.4 [49]. We analyzed a total of 175 individuals representing 44 sampling localities (Supplementary Materials, Table S1).
We applied a Yule speciation process as a tree prior, Cauchy Relaxed Random Walk (RRW) as a continuous trait model for spatial diffusion, and a strict molecular clock model with a substitution rate of 0.012 per site per million years estimated in this study. Geographic coordinates were provided for all sequences, adding a random jitter to tips with a ± 0.50° window size to create unique coordinates for individuals collected at identical sites. Analyses were run for 50 million generations, sampling every 5,000 generations. The convergence of the MCMC chains were checked using Tracer v.1.7.1 [46] to ensure adequate mixing and convergence. Finally, the sampled trees were annotated using TreeAnnotator v.1.10.4 and the final tree analysed in SpreaD3 v.1.0.7 [68] to visualize the ancestral area for each lineage.
2.8. Past, Present and Future Distribution Models
In order to reconstruct the potential species distribution loss-expansion scenarios of the P. melanurus driven by climate change, three-time projections were applied – past, present and future. Past scenarios followed the Last Interglacial (LIG; 0.14−0.12 Ma) [69], the Last Glacial Maximum (LGM; 0.026−0.019 Ma) [70], and the Late Holocene, Meghalayan period (LH; 0.0042−0.0003 Ma) [71]. Present-time constituted the time frames from 1979 to 2013 [72], and future period (2050s) was run under the three gas emission scenarios (RCP 2.6; RCP 4.5; RCP 8.5).
The SDM was executed in MAXENT v.3.3.3 using a maximum entropy algorithm [73,74]. We applied 19 bioclimatic variables for each period from the PaleoClim database (http://www.paleoclim.org) [71], and resolution of 2.5 arc-minutes was used for environmental layers. For the LGM and LIG, we used the predictions of general circulation model, i.e., community climate system model (CCSM4). For future periods, we used the projections of the model for interdisciplinary research on climate (MIROC5). All layers were clipped to span from 80˚E to 93˚E and 42˚N to 50˚N. To avoid strong correlation between environmental variables that may cause the overfitting [75], we discarded those variables with Pearson correlation coefficients r > 0.8 based on pairwise comparison of raster files in SDMtoolbox v.1.1c [76], whereas retained variables were used for subsequent analysis (see Supplementary Materials, Table S2). We used the species occurrence data from the resources of Global Biodiversity Information Facility (GBIF: http://www.gbif.org/) (accessed 24 April 2023) [77], the records from our field surveys, and the information of the published literatures. The distribution sites without coordinates, low precise (decimal < 2), and duplicate coordinates were removed from initial data. To ensure that input occurrence data are spatially not correlated and to reduce sampling bias, all sampling records were rarefied at a spatial distance of 10 km using SDMtoolbox in ArcGIS. The total amount of the occurrence points were 44 localities (including literature data) for P. melanurus, whereas 31 sampling sites were retained after rarefying (see Supplementary Materials, Table S3).
Maxent analyses were carried out with default parameters: 75% of the species records were used for training and 25% for testing the model, regularization multiplier = 1.0, 5000 iterations with the threshold for available presence data = 10 percentile that defines the minimum probability of suitable habitat. The area under the receiving operator characteristics curve (AUC) was used to evaluate the performance of the models. Finally, potential distribution ranges for the past, present and future time were projected in ArcMap 10.2 (ESRI, Redlands, CA, USA).
3. Results
3.1. Sequence Characteristics
The final alignment of COI gene fragment was 630 bp, in which 64 sites were variable across 165 individuals, with 15 singleton variables sites and 49 parsimony-informative sites. No indel (insertion or deletion) was observed in the alignment. The average frequencies of C, T, A, and G were 27.6%, 27.3%, 31.1%, and 14.0%, respectively. The A/T contents (58.4%) were significantly higher than the C/G contents (41.3%). A total of 50 haplotypes were identified. Among these, 11 haplotypes were shared by individuals from several sampling sites. Haplotype 6 and 3 were the most frequent one that comprised in H6 ‒ 7 sampling sites (1, 2, 3, 5, 6, 10, 11), and in H3 ‒ 5 sampling sites (20, 25, 27-29) respectively. Five haplotypes were shared by as many as 3 sampling sites: H2 - (sites 24, 26, 27), H9 ‒ (sites 32-34), H13 ‒ (sites 1, 2, 4), H16 ‒ (sites 13, 16, 23), H27 - (sites 17, 22, 25). Four haplotypes were shared by two sampling sites: H24 - (sites 19, 22), H30 ‒ (sites 17, 25); H38 ‒ (sites 6, 11), H49 ‒ (sites 35, 36). Haplotype diversity (Hd) among populations was very high, ranging from 0.182 to 1.000, while nucleotide diversity (π) was relatively low, ranging from 0.00029 to 0.01138 (Table 1). Total Hd for all populations was 0.950±0.00006, while π was 0.01443±0.00075 (Table 2). All new sequences were deposited in GenBank with accession numbers MW856918‒MW857082 (Supplementary Materials, Table S1).
3.2. Phylogenetic relationships
Bayesian inference and ML analyses produced highly congruent topology, with only minor conflicts on recent nodes. Thus, only the BI tree with both PP and BS from ML is presented (Figure 2). Two main clades were uncovered, with highly supported PP and BS values. Clade I (PP = 1.0, BS = 96%) covered the NW part of the Dzungar Basin, comprising the haplotypes of Altay Prefecture in NW China, and the Zaisan Basin in Eastern Kazakhstan. Clade II consisted of five matrilineal lineages, with IIa representing the haplotypes of Qitai (PP = 1.0, BS = 100%), IIb including the haplotypes of Jinghe (PP = 1.0, BS = 100%), IIc1 incorporating the haplotypes of W part of the Dzungar Basin and adjacent Dzungar Gate, and Alakol Basin area in Eastern Kazakhstan (PP = 0.53). Lineage IIc2 representing haplotypes of Fukang (PP = 0.99, BS = 59%), and IIc3 of Hoboksar (PP = 0.73). Overall, the genealogical structure was significant, which reflects a strong geographic association of each lineage.
3.3. Timeframe of the Diversification
As shown in Figure 3, the most rencent common ancestor (MRCA) of P. melanurus was dated in the Early Pleistocene (~1.87 Ma; 95% HPD: 1.04‒2.85 Ma). Subsequent divergences within P. melanurus occurred at various times throughout the Pleistocene. Within Clade I divergence was estimated to be ~0.39 Ma (95% HPD: 0.14‒0.74Ma). Within Clade II divergence occurred at approximately 1.3 Ma (95% HPD: 0.73‒2.01 Ma). Separation of Subclades IIb from IIc took place approximately 0.87 Ma (95% HPD: 0.48-1.34 Ma), whereas divergence within the Subclade IIc occurred at ~0.74 Ma (95% HPD: 0.43‒1.11 Ma). Divergence within Subclade IIb dated at ~0.09 Ma (95% HPD: 0‒0.24 Ma), and Subclade IIa at ~0.1 Ma (95% HPD: 0‒0.3 Ma) in the Late Quaternary.
3.4. Population Genetic Structure
A median-joining network demonstrated the relationships of the 50 haplotypes (Figure 4). The main feature of the haplotypes distribution was the occurrence of an apparent geographic structure, similar to the phylogenetic reconstruction with six structured haplogroups. A star-shaped network in IIc3 suggested that many haplotypes differed from H6 by only one or two mutations, which suggested that IIc3 experienced a population expansion event. Uncorrected genetic distances (p-distances) ranged from 2.5%‒3% between clades/subclades (Table 3).
The analysis of spatial genetic structure showed that the FCT values did not have the highest differentiation among groups when K = 5; however, one or more groups contained a single population when K ≥ 5. Therefore, we retained the configuration of K = 5 with overall FCT = 0.771 under the Tamura molecular distance model. Group 1 contained populations 1-12; Group 2 incorporatied populations 13-16, and 23; Group 3 comprised populations 17-22, 25, and 28-29; Group 4 incuded populations from 24, 26, and 27; Group 5 contained populations from 30 to 36, which is concordant with Clade I.
The AMOVA analyses were performed on two regions (the Zaisan and Dzungar basins) — Clade I and Clade II, and on four geographic regions (North, Central, Southwest, Southeast). The hierarchical analysis demonstrated that 73.94% or 73.11% of the variation was explained by the variation among two regions or four groups, respectively. Fixation index over all examined data showed significant differences (p ≤ 0.001) (Supplementary Materials, Table S4).
Mantel test for IBD was conducted to estimate the correlation between the genetic distance and geographic distance of P. melanurus. If the dispersal of P. melanurs is limited by distance, genetic and geographic distances should be positively correlated, producing a pattern of isolation by distance. Applying the Mantel test, the weak but significant positive correlation (r = 0.20, p = 0.03) between genetic and geographic distance was observed among populations in the Zaisan Basin (Clade I). Meanwhile, a moderate and signifciant IBD (r = 0.412, p = 0.01) was obeserved among populations in the Dzungar Basin (Clade II) (Supplementary Figure S1.). Overall, the correlation between genetic and geographic distance was confirmed by the positive slope of the first order regression line which is significantly different from zero (r2 = 0.6298, p = 0.01) for entire sample (Figure S1).
3.5. Historical Demography
Demographic analysis was conducted by applying different approaches to all groups except IIa, due to the small sample size. The neutrality tests resulted insignificant values of Tajima's D and Fu's Fs for all populations except Subclade IIc3 (Table 4). Notably, the Subclade IIc2 is characterized by statistically not significant positive value of D and a negative Fs statistic that indicates the lack of rare alleles and decrease in population size or balancing selection. On the contrary, Subclade IIc3 has observed the past population expansion based on large and negative D and Fs values. Significant small values of R2 statistics were captured for the Clade I and Subclade IIc3, which also supported the population growth.
Mismatch distribution analysis of Clade I and Subclade IIc3 produced a unimodal curves suggesting the rapid population expansion, which was additionally supported by the insignificant values of the Rg and SSD indices, as well as by small positive R2 statistics (Figure 5; Table 4). Meanwhile, subclades IIb and IIc2 with the same modality and non-significant Rg and SSD values, rejecting population stationary. A trimodal curves were observed in Subclade IIc1, which indicates the rejection of population expansion.
The BSP analysis demonstrated the population stability through time in Clade I, subclades IIb, and IIc2. The Subclade IIc1 has experienced a rapid population size growth starting about 0.02 Ma. Subclade IIc3 detected a past population expansion starting at approximately 0.18 Ma (Figure 6).
3.6. The Spatiotemporal Diffusion for P. melanurs
For the P. melanurus, the ancestral area was detected on the territory of current Hoboksar-Mongol Autonomous county which is in Tacheng prefecture of Xinjiang province of Northwest China. The intital colonization event started approximately 1.8 Ma (Figure 7A). The subsequent colonization route followed multiple directions and at 1.15 Ma the population reached Zaisan Basin, Karamay region, southwestward through Ebinur, Jinghe reached the Dzungar Gate territory, and southeastward Fukang-Qitai region (Figure 7B). At ~0.93 Ma local spreading was inferred throughout the main directions and reached the Ulungur territory, Kuytun, and Jimsar regions (Figure 7C). The final dispersal was detected throughout the vast territory of Zaisan and Dzungar basins, reaching the Alakol Basin in E KZ occured around 0.63 Ma (Figure 7D).
3.7. Potential Species Distribution Modeling
The reconstructed potential distributions of P. melanurus for past and present-day projections are presented in Figure 8. Under the selected models, the test of AUC value for the all periods of P. melanurus was r ≥ 0.9, which was considered as reliable for prediction. The Jackknife analysis of regularized training gained from retained environmental variables that highly contributed to the distribution model is shown in Figure S2. The projected LIG scenario showed the broad area of the potentially suitable habitat for P. melanurus, concentrated mostly on the southern part of its distribution range. Modelled LGM climate reconstruction showed the discontinuity of potentially suitable area at southern part, whereas apparently northern part experienced a noticeable expansion — from the Zaisan Basin toward to the Black Irtysh Valley. Late Holocene period simulation was characterized by drastic habitat diminishing, particularly, massive contraction occurring at the northern and southern periphery of Northern Xinjiang. Present-day scenario demonstrated a recovery of the southern habitat corridor approximately as it was in the LIG (Figure 8); the highly fitted habitat for P. melanurus is concentrated in the North, West and South part of its distribution range, in consistent with all known up today occurrence records.
The prediction of species distribution in the near future (2050s) were predicted by three climatic scenarios of greenhouse gas concentration: optimistic (RCP 2.6), intermediate (RCP 4.5), and pessimistic (RCP 8.5) under the MIROC5 (Figure 9). All predicted future projections displayed the vast expansion of habitat range occupying the central and eastern regions. Also, the partial reduction of SW habitat area (the Dzungar Gate, Alatau) is observed. RCP 4.5 scenario demonstrated the discontinuity between W and SW sectors with the loss of corridor that was joining the SE and SW regions. RCP 8.5 scenario showed the irreversible loss of habitat, dividing the distribution area on two regions - SW (Dzungar Gate, Alatau) and E (eastern part of the Dzungar Basin), and N (the Zaisan Basin and adjacent area).
4. Discussion
The present study generates the substantive mtDNA data of P. melanurus covering its whole distribution range, and unravels the spatial genetic structure, demographic history and divergence date. Furthermore, the work provides the primary evidence for the species ancestral center and subsequent colonization trajectories emphasized by historical changes of the populations during the Pleistocene climatic oscillations. However, the results should be interpreted with caution, since the phylogeographic analyses are based on only one gene of mitochondrial DNA. Nevertheless, mtDNA is highly variable in natural populations because of its elevated mutation rate, which can generate a signal about population history over short time frames [36,37].
4.1. Phylogeographic Pattern and Diversification History
By utilizing mtDNA COI gene sequences in phylogenetic analysis, we revealed six lineages within two main clades of P. melanurus, where most of branches are highly supported (Figure 2). Clade I represent the Zaisan Basin lineage spanning from E KZ to the Altay Prefecture of N Xinjiang in China, while Clade II embodies the lineages of Dzungar Basin (Figure 1). Genetic distances between the Zaisan lineage and the lineages of Dzungar Basin ranged from 2.5%‒3%, which indicates a subspecies status due to an ongoing lineage sorting process and/or gene flow. Similar findings were captured by Solovyova et al. [38] and Dunayev et al. [34], where the Zaisan lineage diverged 2.6% and 2.48% from that of the Dzungar Gate, respectively. Later Solovyova et al. [40] contended the recognition of P. melanurus from the Zaisan and Dzungar basins as a distinct species based on p-distances of COI gene, morphology and geographic distribution. The population from Northwest China was examined by Wang and Fu [78] and Melville et al. [3] based on the analysis of ND2 gene sequences. Their results demonstrated that the population of Northwest China is sister to E KZ population, with 3.1%‒4.0% uncorrected sequence divergence between them.
SAMOVA divided all sampled populations into five groups with certain geographic distribution. Analysis of molecular variance (AMOVA) also suggested that the genetic variation in P. melanurus was high among groups (Supplementary Materials, Table S4). Haplotype 6 is dominate in the population of the northern Dzungar Basin and indicates a potential ancestral haplotype. The haplotype 3 was frequent for the Subclade IIc1, while haplotype 2 is shared between IIa and IIb, which might result from the secondary contact during last interglacial cycles (Figure 4). All these facts demonstrate that there is a distinct genetic differentiation among the geographic groups. Similar population structures were documented in previous studies of xerophyte plants [79,80].
The haplotype and nucleotide diversity indices of the Dzungar Basin population (Table 1) were detected to be significantly higher than those of the Zaisan Basin. This suggests that P. melanurus experienced an expansion and recent stepwise dispersal, which is also supported by the haplotype network analysis revealing a star-shaped topology of IIc3. Similar findings were reported for the desert scorpion Mesobutbus mongolicus [10], agamid lizards Phrynocephalus spp. [3] and the rapid racerunner Eremias velox [8].
The estimated divergence time from the MRCA of P. melanurus occurred approximately 1.86 Ma (Figure 3.), which falls into the early Pleistocene, the phase of intense uplift of Himalayas and Tianshan Mountains [81]. These findings are slightly older than the previous studies of Solovyova et al. [40] and Dunayev et al. [34], which might be due to the limited sampling size of P. melanurus analyzed by the mentioned authors. The Dzungar Basin lineage initiated divergence at ~1.3 Ma with subsequent intraspecific differentiation at Middle and Late Pleistocene epochs (Figure 3). Notably, Pleistocene climatic transitions placed a unique arid environment in Northwest China [82,83], which may have shaped the genetic diversity of vertebrate populations, particularly an isolation of desert-dwelling species [84,85].
4.2. Origin and Colonization
The origin of the P. melanurus has been debated for decades. Previously, researchers hypothesized that an ancestral form of the P. guttatus group originated in the eastern part of the Kazakh Upland and later spread to the Zaisan, Balkhash, and Ili basins, eventually settling in western Kazakhstan [32]. Melville et al. [3] assumed that the Irtysh valley could serve as a dispersal route for P. melanurus populations, while Golubev [29] proposed a hypothesis that P. melanurus may have penetrated into Kazakhstan from China through the Dzungar Gate in the Middle Pleistocene. However, our study highlights that the ancestral form of P. melanurus colonized Zaisan Basin from Chinese Dzungar. Bayesian phylogeographic diffusion analysis showed that the dispersal of the P. melanurus populations from its projected ancestral area occurred in the Middle Pleistocene epoch, which was conditioned by the mid-Pleistocene climatic transition (Figure 7A,B). The subsequent expansions of deserts in the Dzungar Basin (0.65 Ma and 0.5 Ma) [16] uncovered the vast territories for the settlement of P. melanurus and promoted the spatial diffusion of the population in the Dzungar Basin at 0.63 Ma (Figure 7D).
Subsequently, following the mountain and foothill trails of Saur-Tarbagatay and Alatau mountains, populations reached Ebi-Nur Lake (IIb) and spread eastward to the modern settlement of Qitai (IIa). During that period, the activity of shallow rivers and temporary streams along the northern slopes of the Dzungar Alatau could have contributed to the formation of sandy-pebble desert landscapes that eventually developed into a relatively integral piedmont plume [86,87]. This plume may have also served as a pathway for lizards from Ebi-Nur to penetrate the Alakol Basin via the Dzungar Gate, which stands with the Golubev’s assumption [29].
4.3. Lineage-Specific Response to Quaternary Climatic Oscillations
We suggest that the historical climate has greatly influenced the population dynamics of P. melanurus in Northwest China. Demographic analysis of the lineages examined in this study reveal that the population growth was captured in the lineages IIc1 and IIc3, matching glacial expansion model [88,89]. For the subclade IIc3 a star-like haplotype network, significant values of neutrality statistics, as well as unimodal mismatch distribution evidenced the population expansion event (Figure 4, Figure 5 and Figure 6). Moreover, BSP analysis detected the signal of the past population growth started at approximately 0.18 Ma and lasted during the LIG in IIc3 (Figures 6). SDM modelling of the LIG period demonstrated the habitat expansion at the south margins of the areal, which supports the possibility of the population expansion in the past (Figure 8).
Clade I and subclades IIb and IIc2 kept the constant population over the time, which also shows the stability of the distribution area during the LIG and the LGM epochs. While, subclade IIc1 likely experienced a recent population growth around 0.02 Ma, which falls with the LGM period (Figure 6). SDMs of the LGM and the Holocene identified that the southwest part of the distribution range of P. melanurus, which is attributed to the subclade IIc1, remained highly suitable in comparison to the eastern and northwestern habitat diminishment (Figure 8).
Species distribution modelling follows the glacial expansion of the overall distribution area of the P. melanurus. Interestingly, the area of suitability in northern part of the NW China expanded during the LGM, while that in the southern margins disconnected and decreased in the volume (Figure 8). The development of aridification in northwestern China in the late Pleistocene resulted in the enlargement of deserts in the LGM [90], which may in some cases have had the effect of forming broader suitable habitats (edges of deserts and arid piedmont) for expansion of the arid-adapted species. As expected, the potentially suitable habitat is dramatically diminished (Figure 8) in the Holocene epoch suitable due to the warm and humid episodes that have promoted the development of the mesic biota [91]. Similar suitable habitat expansions during the LGM were noticed in the other representatives of herpetofauna in arid Central Asia, such as the Turpan racerunner (Eremias roborowskii) [8] and sunwatcher toad-headed agama (Phrynocephalus helioscopus) [11].
It is widely accepted that glaciation in the Pleistocene generally led to the emergence of species refugia [92,93,94,95]. Refugia may be generally predicted as areas possessing high levels of genetic diversity, and may have a distinct characteristic, such as a presence of ancestral and unique haplotypes that have disappeared from other populations [93]. In our study, the high level of genetic diversity in populations of IIc3, IIa, and IIb is detected. Additionally, the potential ancestral haplotype (haplotype 6) comes from the Hoboksar region, which coincides with the projected ancestral area inferred from Bayesian phylogeographic analysis. All these facts indicate that the Hoboksar region may serve as a conditional refugium where the population survived during the interglacial-glacial periods. Nevertheless, it should be noted that Eastern Tianshan Mountain did not advance the glaciers/permafrost below 2400 m a.s.l. during the LGM due to the extra continental climate [81]. As such, multiple glacial refugia may have existed along the Saur-Tarbagatay Mountain system in the west, Alatau and Western Tianshan in the southwest, Eastern Tianshan (Bogda Mount) in southeast.
Future distribution modelling (for the 2050; Figure 9) proposes the scenario of habitat shift toward eastern part of the NW China, as a response to the climate change extensive aridification and urbanization growth. It may cause the different scenarios for the P. melanurus populations, such as extirpation, or reduction of the population size, which may require the future conservation strategies.
5. Conclusions
To the best of our knowledge, this work represents the first range-wide phylogeography of P. melanurus by integrating mtDNA and species distribution modeling. It sheds light on the lineage diversification and historical demography of the desert-dwelling species, which contributes to the dryland phylogeography of Eastern Palearctic. Our findings reveal that the population of P. melanurus is geographically structured into two main clades: the Zaisan lineage and the Dzungar lineage, with the latter being further sub-structured into several groups. Genetic distances among these lineages demonstrate their relatedness, and thus preclude recognition of as distinct species, due to ongoing diversification processes and incomplete lineage sorting. Furthermore, our results suggest that the ancestral form of P. melanurus migrated from the northwest of the Dzungar Basin during the Middle Pleistocene, and subsequently spread throughout Zaisan and Dzungar basins, with some populations accessing the Alakol Basin in Kazakhstan via the Dzungar Gate. Overall, our study provides a valuable contribution to understanding the importance of considering the geographic context of a species' distribution range, genetic structure and evolutionary history. Moreover, we acknowledge that further studies on agamid lizard P. melanurus using nuclear markers and morphological analysis are necessary to elucidate a complete picture of the evolution of P. melanurus.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Correlation analysis between the pairwise FST/(1–FST) values and the geographic distance based on mtDNA COI sequences: for (a) Clade I, Zaisan populations; (b) Clade II, Dzungar populations; (c) all data; Figure S2: The Jackknife analysis of regularized training gain from retained environmental variables that highly contributed to the distribution model; Table S1: List of analyzed specimens, geographic origin, clade assignment and GenBank accession number; Table S2: Environmental variables highly contributed to the SDM for P. melanurus with Pearson correlation coefficients r ≥ 0.8; Table S3: Sampling sites retained after rarefication for species distribution modeling; Table S4: Hierarchical analysis of AMOVA for testing the genetic subdivision of populations for P. melanurus using COI sequences. Statistical significance at p ≤ 0.001.
Author Contributions
Conceptualization, X.G., D.U. and X.Z.; methodology, D.U., J.L.L. and X.G.; validation, D.U., T.D. and X.G.; formal analysis, D.U., J.LL. and L.T.; investigation, D.U., J.L.L., X.G., B.C.; resources, X.G. and T.D.; data curation, X.G.; writing—original draft preparation, D.U.; writing—review and editing, D.U., X.G., T.D., J.L., L.T., X.Z. and B.C.; visualization, D.U. and X.G.; supervision, X.G. and X.Z.; project administration, X.G. and X.Z.; funding acquisition, X.G. and T.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Third Xinjiang Scientific Expedition Program (Grant No. 2021xjkk0600), the National Natural Science Foundation of China (Grant Nos. 32070433 and 32000288), and the Science Committee of the Ministry of Science and Higher Education of the Republic of Kazakhstan on the Red Data Book of Kazakhstan (Grant No. BR18574058). The first author (D.U.) was supported by the Chinese Academy of Sciences-The World Academy of Sciences (CAS-TWAS) President’s PhD Fellowship program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
The animal study protocol was approved by the Animal Protection and Utilization Committee of Chengdu Institute of Biology, Chinese Academy of Sciences (license numbers: CIB-20200767 and CIBDWLL2021001).
Data Availability Statement
All novel sequences obtained in this study were deposited in GenBank under accession numbers MW856918‒MW857082.
Acknowledgments
We are indebted to Yongfei He and Feng Xu for their kind help during the fieldwork in Xinjiang, China. We are grateful to Dali Chen, Marina Chirikova, Xiong Gong, Tianhe Zhou, Qi Song, Hongfu Wan, Minli Chen, Na Wu, and Song Wang who participated in the fieldwork in Xinjiang, China. Special thanks go to Qi Song, Xingong Gong, and Minli Chen for their assistance in lab experiments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Byrne, M.; Yeates, D.K.; Joseph, L.; Kearney, M.; Bowler, J.; Williams, M.A.J.; Coper, S.; Donnellan, S.C.; Keogh, J.S.; Leys, R.; Melville, J.; Murphy, D.J.; Porch, N.; Wyrwoll, K.H. Birth of a biome: insights into the assembly and maintenance of the Australian arid zone biota. Mol. Ecol. 2008, 17, 4398–4417. [Google Scholar] [CrossRef]
- Byrne, M.P.; Pendergrass, A.G.; Rapp, A.D.; Wodzicki, K.R. Response of the intertropical convergence zone to climate change: Location, width, and strength. Curr. Clim. Change Rep. 2018, 4, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Melville, J.; Hale, J.; Mantziou, G.; Ananjeva, N.B.; Milto, K.; Clemann, N. Historical biogeography, phylogenetic relationships and intraspecific diversity of agamid lizards in the Central Asian deserts of Kazakhstan and Uzbekistan. Mol. Phylogenet. Evol. 2009, 53, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.M.; Ji, Y.J.; Liu, L.; Wang, L.; Zhang, D.X. Impact of climate changes from Middle Miocene onwards on evolutionary diversification in Eurasia: insights from the mesobuthid scorpions. Mol. Ecol. 2013, 22, 1700–1716. [Google Scholar] [CrossRef] [PubMed]
- Kearns, A.; Joseph, L.; Toon, A.; Cook, L.G. Australia’s arid-adapted butcherbirds experienced range expansions during Pleistocene glacial maxima. Nat. Commun. 2014, 5, 3994. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.D.; Zhang, L.J.; Lin, L.; Yu, S.; Chen, Z.; Wang, W. Insights into the historical assembly of global dryland floras: the diversification of Zygophyllaceae. BMC Evol. Biol. 2018, 18, 166. [Google Scholar] [CrossRef]
- Riddle, B.R.; Hafner, D.J. A step-wise approach to integrating phylogeographic and phylogenetic biogeographic perspectives on the history of a core North American warm deserts biota. J. Arid Environ. 2006, 66, 435–461. [Google Scholar] [CrossRef]
- Liu, J.; Guo, X.; Chen, D.; Li, J.; Yue, B.; Zeng, X. Diversification and historical demography of the rapid racerunner (Eremias velox) in relation to geological history and Pleistocene climatic oscillations in arid Central Asia. Mol. Phylogenet. Evol. 2019, 130, 244–258. [Google Scholar] [CrossRef]
- Maestre, F.T.; Benito, B.M.; Berdugo, M.; Concostrina-Zubiri, L.; Delgado-Baquerizo, M.; Eldridge, D.J.; Guirado, E.; Gross, N.; Kéfi, S.; Le Bagousse-Pinguet, Y.; Ochoa-Hueso, R.; Soliveres, S. Biogeography of global drylands. New Phytol. 2021, 231, 540–558. [Google Scholar] [CrossRef]
- Shi, C.M.; Zhang, X.S.; Liu, L.; Ji, Y.J.; Zhang, D.X. Phylogeography of the desert scorpion illuminates a route out of Central Asia. Curr. Zool. 2023, 69, 442–455. zoac061. [Google Scholar] [CrossRef]
- Wu, N.; Wang, S.; Dujsebayeva, T.; Chen, D.; Ali, A.; Guo, X. Geography and past climate changes have shaped the evolution of a widespread lizard in arid Central Asia. Mol. Phylogenet. Evol. 2023, 184, 107781. [Google Scholar] [CrossRef]
- An, Z.S.; Kutzbach, J.E.; Prell, W.L.; Porter, S.C. Evolution of Asian monsoons and phased uplift of the Himalaya-Tibetan plateau since Late Miocene times. Nature 2001, 411, 62–66. [Google Scholar] [CrossRef]
- Guo, Z.T.; Ruddiman, W.F.; Hao, Q.Z.; Wu, H.B.; Qiao, Y.S.; Zhu, R.X.; Peng, S.Z.; Wei, J. J.; Yuan, B.Y.; Liu, T.S. Onset of Asian desertification by 22 Myr ago inferred from loess deposits in China. Nature 2002, 416, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ye, J.; Wu, W.; Ni, X.; Bi, S.; Zhang, Z.; Liu, W.; Meng, J. Late Oligocene-Miocene mid-latitude aridification and wind patterns in the Asian interior. Geology 2010, 38, 515–518. [Google Scholar] [CrossRef]
- Lu, H.; Wang, X.; Wang, X.; Chang, X.; Zhang, H.; Xu, Z.; Zhang, W.; Wei, H.; Zhang, X.; Yi, S.; Zhang, W.; Feng, H.; Wang, Y.; Wang, Y.; Han, Z. Formation and evolution of Gobi Desert in central and eastern Asia. Earth Sci. Rev. 2019, 194, 251–263. [Google Scholar] [CrossRef]
- Fang, X.; Lu, L.; Yang, S.; Li, J.; An, Z.; Jiang, P.; Chen, X. Loess in Kunlun Mountains and its implications on desert development and Tibetan Plateau uplift in west China. Sci. China Ser. D 2002, 45, 289–299. [Google Scholar] [CrossRef]
- Miao, Y.; Herrmann, M.; Wu, F.; Yan, X.; Yang, S. What controlled Mid–Late Miocene long-term aridification in Central Asia? — Global cooling or Tibetan Plateau uplift: A review. Earth-Sci. Rev. 2012, 112, 155–172. [Google Scholar] [CrossRef]
- Li, Z.H.; Chen, J.; Zhao, G.F.; Guo, Y.P.; Kou, Y.X.; Ma, Y.Z.; Wang, G.; Ma, X.F. Response of a desert shrub to past geological and climatic change: a phylogeographic study of Reaumuria soongarica (Tamaricaceae) in western China. J. Syst. Evol. 2012, 50, 351–361. [Google Scholar] [CrossRef]
- Meng, H.H.; Zhang, M.L. Diversification of plant species in arid northwest China: species-level phylogeographic history of Lagochilus Bunge ex Bentham (Lamiaceae). Mol. Phylogenet. Evol. 2013, 68, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Su, S.H.; Zhang, M.L. A range wide geographic pattern of genetic diversity and population structure of Hexinia polydichotoma (Asteraceae) in Tarim Basin and adjacent areas. Biochem. Syst. Ecol. 2014, 56, 49–59. [Google Scholar] [CrossRef]
- Nanova, O.G.; Lebedev, V.S.; Matrosova, V.A.; Adiya, Y.; Undrakhbayar, E.; Surov, A.V.; Shenbrot, G.I. Phylogeography, phylogeny, and taxonomical revision of the Midday jird (Meriones meridianus) species complex from Dzungaria. J. Zool. Syst. Evol. Res. 2020, 58, 1335–1358. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, Y.; Li, W.; Fu, A.; Ma, X.; Gui, D.; Chen, Y. Distribution pattern of plant species diversity in the mountainous region of Ili River Valley, Xinjiang Environ. Monit. Assess. 2011, 177, 681–694. [Google Scholar] [CrossRef]
- Meng, H.H.; Gao, X.Y.; Huang, J.F.; Zhang, M.L. Plant phylogeography in arid Northwest China: Retrospectives and perspectives. J. Syst. Evol. 2015, 53, 33–46. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, H.-X. Population genetic structure and biodiversity conservation of a relict and medicinal subshrub Capparis spinosa in arid Central Asia. Diversity 2022, 14, 146. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, G.; Feng, Y.; Zhang, D.; Shi, C. Genetic analysis and ecological niche modeling delimit species boundary of the Przewalski’s scorpion (Scorpions: Buthidae) in arid Asian inland. Zool. Syst. 2020, 45, 81–96. [Google Scholar] [CrossRef]
- Camargo, A.; Werneck, F.P.; Morando, M.; Sites Jr, J.W.; Avila, L.J. Quaternary range and demographic expansion of Liolaemus darwinii (Squamata: Liolaemidae) in the Monte Desert of Central Argentina using Bayesian phylogeography and ecological niche modelling. Mol. Ecol. 2013, 22, 4038–4054. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.N.; Barley, A.J.; Poe, S.; Thomson, R.C.; Nieto-Montes de Oca, A.; Wang, I.J. Phylogeography of a widespread lizard complex reflects patterns of both geographic and ecological isolation. Mol. Ecol. 2019, 28, 644–657. [Google Scholar] [CrossRef]
- Fu, J.; Wen, L. Impacts of Quaternary glaciation, geological history and geography on animal species history in continental East Asia: A phylogeographic review. Mol. Ecol. 2023, 32, 4497–4514. [Google Scholar] [CrossRef]
- Golubev, M.L. Three controversial questions of systematics and nomenclature in Phrynocephalus of USSR fauna (Phrynocephalus, Agamidae). Questions of Herpetology: Abstracts of the 7th Conference; Kiev, 1989, pp. 44–45. [in Russian].
- Golubev, M.L. Variegated toad-head agama Phrynocephalus versicolor (Reptilia, Agamidae) in Dzungar gates (East Kazakhstan) with notes on the species systematics. Herald Zool. 1992, 2, 31–238, [In Russian]. [Google Scholar]
- Dunayev, E.A. Systematic condition, ecological and behavioral peculiarities of Zaisan toad-headed agama Phrynocephalus melanurus Eichwald, 1831 (Reptilia: Agamidae). Bull. Moscow Soc. Nat. Ser. Bio. 1989, 94, 41–53, [In Russian]. [Google Scholar]
- Dunayev, E.A. Systematics and Paleogeography: Conceptual Synthesis Using an Example of Phrynocephalus (superspecies guttatus) (Reptilia: Agamidae). Evolution and Systematics: Lamark and Darwin in Modern Research: Proceedings of the Zoological Museum of Moscow State University 2009, 50, 275–298, [In Russian]. [Google Scholar]
- Barabanov, A.V.; Ananjeva, N.B. Catalogue of the available scientific species-group names for lizards of the genus Phrynocephalus Kaup, 1825 (Reptilia, Sauria, Agamidae). Zootaxa 2007, 1399, 1–56. [Google Scholar] [CrossRef]
- Dunayev, E.A.; Solovyova, E.N.; Poyarkov, N.A. Taxonomy, phylogeny and distribution of Phrynocephalus (superspecies guttatus) (Reptilia: Agamidae). Curr. Stud. Herpetol. 2020, 20, 16–34, [In Russian]. [Google Scholar] [CrossRef]
- Dunayev, E.A. On the possible use of the ethological features in the taxonomy and phylogeny of toad agamas. Russ. J. Herpetol. 1996, 3, 32–38. [Google Scholar] [CrossRef]
- Ballard, J.W.O.; Whitlock, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Nahbolz, B.; Glemin, S.; Hurst, G.D. Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol. Ecol. 2009, 18, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Solovyeva, E.N.; Poyarkov, N.A.; Dunayev, E.A.; Nazarov, R.A.; Lebedev, V.S.; Bannikova, A.A. Phylogenetic relationships and subgeneric taxonomy of toad headed agamas Phrynocephalus (Reptilia, Squamata, Agamidae) as determined by mitochondrial DNA sequencing. Dokl. Biol. Sci. 2014, 455, 119–124. [Google Scholar] [CrossRef]
- Aljanabi, S.M.; Martinez, I. Universal and rapid salt-extraction of high-quality genomic DNA for PCR-based techniques. Nucl. Acids Res. 1997, 25, 4692–4693. [Google Scholar] [CrossRef] [PubMed]
- Solovyova, E.N.; Lebedev, V.S.; Dunayev, E.A.; Nazarov, R.A.; Bannikova, A.A.; Che, J.; Murphy, R.W.; Poyarkov, N.A. Cenozoic aridization in central Eurasia shaped diversification of toad-headed agamas (Phrynocephalus; Agamidae, Reptilia). PeerJ 2018, 6, 4543. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. 2018. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Rannala, B. Frequentist properties of Bayesian posterior probabilities of phylogenetic trees under simple and complex substitution models. Syst. Biol. 2004, 53, 904–913. [Google Scholar] [CrossRef] [PubMed]
- FigTree v1.4.4. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh. http://tree.bio.ed.ac.uk/software/figtree/.
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, 1987; 512 pp. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. PopART: full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Dupanloup, I.; Schneider, S.; Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 2002, 11, 2571–2581. [Google Scholar] [CrossRef]
- Godbout, J.; Jaramillo-Correa, J.P.; Beaulieu, J. A mitochondrial DNA minisatellite reveals the postglacial history of jack pine (Pinus banksiana), a broad range North American conifer. Mol. Ecol. 2005, 141, 3497–3512. [Google Scholar] [CrossRef]
- Magri, D.; Vendramin, G.G.; Comps, B.; Dupanloup, I.; Geburek, T.; Gömöry, D.; Latałowa, M.; Litt, T.; Paule, L.; Roure, J.M.; Tantau, I.; van der Knaap, W.O.; Petit, R.J.; de Beaulieu, J.L. A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytol. 2006, 171, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O`hara, R.B.; Simpson, L.; Solymos, P.; Henry, M.; Stevens, H.; Wagner, H. Package ‘vegan’: Community Ecology Package. R package version 2.4-3; 2017. https://CRAN.R-project.org/package=vegan.
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Ramos-Onsins, S.E.; Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 2002, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J. Bayesian Evolutionary analysis of viruses: A practical introduction to BEAST. 2009; 34 pp.
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef]
- Otto-Bliesner, B.L.; Brady, E.C.; Clauzet, G.; Tomas, R.; Levis, S.; Kothavala, Z. Last Glacial Maximum and Holocene climate in CCSM3. J. Clim. 2006, 19, 2526–2544. [Google Scholar] [CrossRef]
- Karger, D.N.; Nobis, M.P.; Normand, S.; Graham, C.N.; Zimmermann, N.E. CHELSA-TraCE21k – high-resolution (1 km) downscaled transient temperature and precipitation data since the Last Glacial Maximum. Clim. Past 2023, 19, 439–456. [Google Scholar] [CrossRef]
- Brown, J.L.; Hill, D.J.; Dolan, A.M.; Carnaval, A.C.; Haywood, A.M. PaleoClim, high spatial resolution paleoclimate surfaces for global land areas. Sci. Data 2018, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Karger, D.N.; Conrad, O.; Böhner, J.; Kawohl, T.; Kreft, H.; Soria-Auza, R.W.; Zimmermann, N.E.; Kessler, M. Data Descriptor: Climatologies at high resolution for the earth’s land surface areas. Sci. Data 2017, 4, 170122. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.B.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Phillips, S.J.; Dudik, M. Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography 2008, 31, 161–175. [Google Scholar] [CrossRef]
- Synes, N.W.; Osborne, P.E. Choice of predictor variables as a source of uncertainty in continental-scale species distribution modelling under climate change. Global Ecol. Biogeogr. 2011, 20, 904–914. [Google Scholar] [CrossRef]
- Brown, J.L. SDMtoolbox: a python-based GIS toolkit for landscape genetic, biogeographic and species distribution model analyses. Methods Ecol. Evol. 2014, 5, 694–700. [Google Scholar] [CrossRef]
- GBIF.org (24 April 2023) GBIF Occurrence Download. [CrossRef]
- Wang, Y.; Fu, J. Cladogenesis and vicariance patterns in the toad-headed lizard Phrynocephalus versicolor species complex. Copeia 2004, 2004, 199–206. [Google Scholar] [CrossRef]
- Xu, Zh.; Zhang, M.-L. Phylogeography of the arid shrub Atrafaxis frutescens (Polygonacea) on Northwest China: Evidence from cpDNA sequences. J. Hered. 2015, 106, 184–195. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.-L.; Yin, L.-K. Phylogeographic structure of a Tethyan relict Capparis spinosa (Capparaceae) traces Pleistocene geologic and climatic changes in the Western Himalayas, Tianshan Mountains, and adjacent desert regions. Hindawi 2016, 2016, 1–13. [Google Scholar] [CrossRef]
- Shi, Y.-F.; Cui, Z.-J.; Su, Z. The Quaternary Glaciations and Environmental Variations in China; Hebei Science and Technology Press: Shijiazhuang, China, 2006; [In Chinese]. [Google Scholar]
- Liu, Y.Q.; Wang, Z.X.; Jin, X.C.; Li, T.; Li, Y. Evolution, chronology and depositional effect of uplifting in the eastern sector of the Tianshan Mountains. Acta Geol. Sin. 2004, 78, 319–331, [In Chinese with English Abstract]. [Google Scholar] [CrossRef]
- Sun, J.M.; Zhu, R.X. Cenozoic deposits in the Northern Tianshan mountains and its implications for neotectonics and environmental changes. Quatern. Sci. 2006, 1, 14–19, [In Chinese with English Abstract]. [Google Scholar] [CrossRef]
- Nicolas, V.; Mboumba, J.F.; Verheyen, E.; Denys, C.; Lecompte, E.; Olayemi, A.; Missoup, A.D.; Katuala, P.; Colyn, M. Phylogeographic structure and regional history of Lemniscomys striatus (Rodentia: Muridae) in tropical Africa. J. Biogeogr. 2008, 35, 2074–2089. [Google Scholar] [CrossRef]
- Brouat, C.; Tatard, C.; Bâ, K.; Cosson, J.F.; Dobigny, G.; Fichet-Calvet, E.; Granjon, L.; Lecompte, E.; Loiseau, A.; Mouline, K.; Piry, S.; Duplantier, J.M. Phylogeography of the Guinea multimammate mouse (Mastomys erythroleucus): a case study for Sahelian species in West Africa. J. Biogeogr. 2009, 36, 2237–2250. [Google Scholar] [CrossRef]
- Svarichevskaya, Z.A. Geomorphology of Kazakhstan and Middle Asia; Leningrad: LSU, 1965; 296 pp. [In Russian]. [Google Scholar]
- Shyukin, I.S. Geomorphology of Central Asia; Moscow: MSU, 1983; 432 pp. [Google Scholar]
- Willis KJ, Bennett KD, Walker D, Hewitt GM, 2004. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 183–195. [Google Scholar] [CrossRef]
- Galbreath, K.E.; Hafner, D.J.; Zamudio, K.R. When cold is better: Climate-driven elevation shifts yield complex patterns of diversification and demography in an alpine specialist (American pika, Ochotona princeps). Evoluiotn 2009, 63, 2848–2863. [Google Scholar] [CrossRef]
- Lu, H.; Yi, S.; Xu, Z.; Zhou, Y.; Zeng, L.; Zhu, F.; Feng, H.; Dong, L.; Zhuo, H.; Yu, K.; Mason, J.; Wang, X.; Chen, Y.; Lu, Q.; Wu, B.; Dong, Z.; Qu, J.; Wang, X.; Guo, Z. Chinese deserts and sand fields in Last Glacial Maximum and Holocene Optimum. Chin. Sci. Bull. 2013, 58, 2775–2783. [Google Scholar] [CrossRef]
- Taberlet, P.; Fumagalli, L.; Wust–Saucy, A.G.; Cosson, J.F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef]
- Hewitt, G.M. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Petit, R.J.; Aguinagalde, I.; de Beaulieu, J.L.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, M.; Mohanty, A.; Muller-Starck, G.; Demesure-Musch, B.; Palme, A.; Martin, J.P.; Rendell, S.; Vendramin, G.G. Glacial refugia: hotspots but not melting pots of genetic diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef] [PubMed]
- Provan, J.; Bennett, K.D. Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 2008, 23, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.R.; Lister, A.M.; Barnes, I.; Dalén, L. Refugia revisited: individualistic responses of species in space and time. Proc. Biol. Sci. 2009, 277, 661–671. [Google Scholar] [CrossRef]
Figure 1.
Collection sites for the samples of P. melanurus used in this study. Sites are numbered as in Table 1 and Table S1; phylogenetic lineages (clades/subclades) are highlighted by different color. Dashed lines represent the soft boundaries isolating the populations of IIc1, IIc2, and IIc3, respectively. The background outlines the current distribution of P. melanurus according to Dunayev et al. [34].
Figure 1.
Collection sites for the samples of P. melanurus used in this study. Sites are numbered as in Table 1 and Table S1; phylogenetic lineages (clades/subclades) are highlighted by different color. Dashed lines represent the soft boundaries isolating the populations of IIc1, IIc2, and IIc3, respectively. The background outlines the current distribution of P. melanurus according to Dunayev et al. [34].
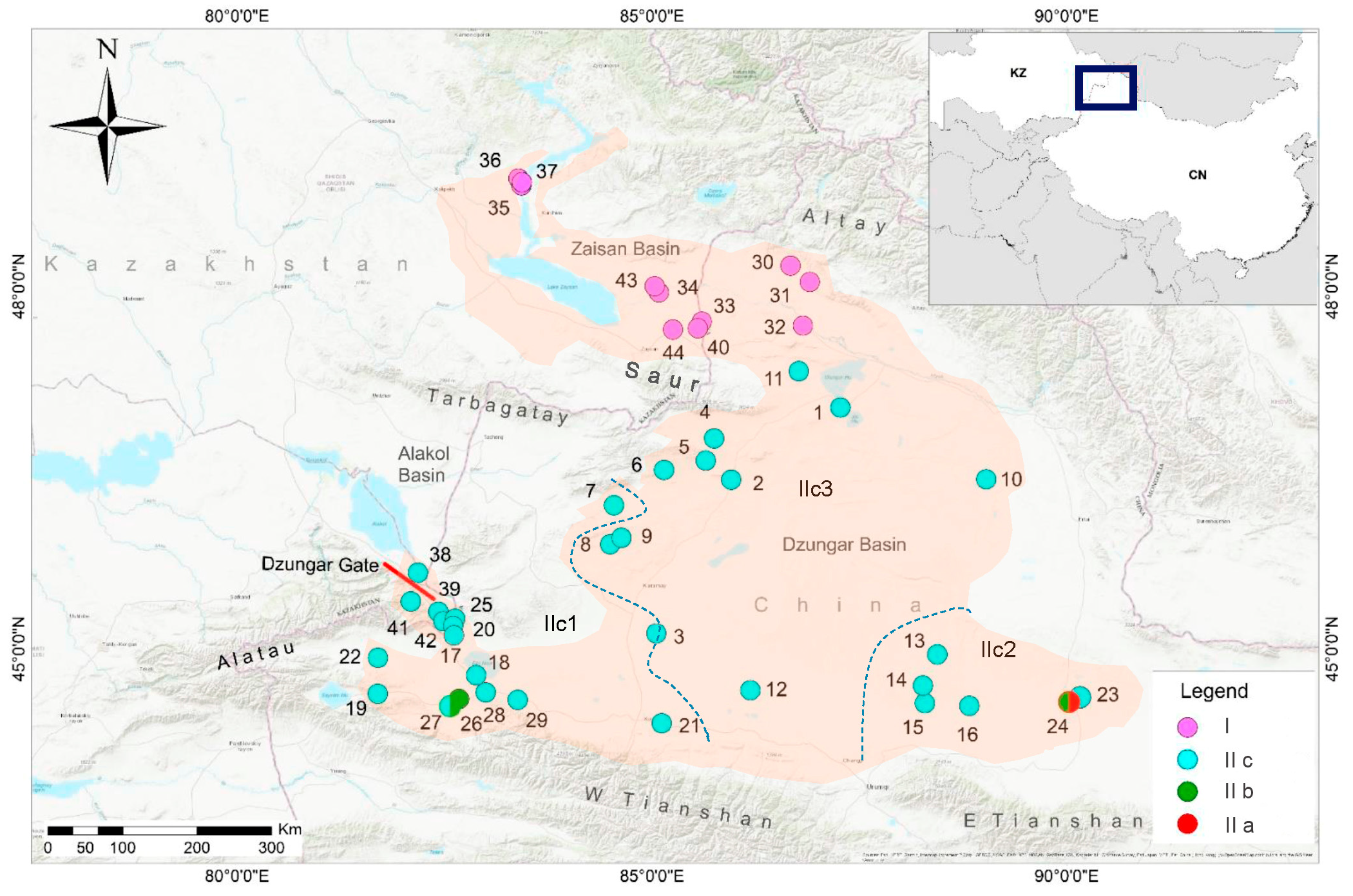
Figure 2.
The 50% majority-concensus tree for P. melanurus resulting from partitioned Bayesian analysis, associations with less than 0.5 posterior probability being collapsed. Bayeisan posterior probabilities and maximum likelihood bootstrap values are shown. Nodal support less than 50% was not shown in the tree. Dashes represent nodes with bootstrap support lower than 50% or represent nodes not existed. Photo of P. melanurus by X.G.
Figure 2.
The 50% majority-concensus tree for P. melanurus resulting from partitioned Bayesian analysis, associations with less than 0.5 posterior probability being collapsed. Bayeisan posterior probabilities and maximum likelihood bootstrap values are shown. Nodal support less than 50% was not shown in the tree. Dashes represent nodes with bootstrap support lower than 50% or represent nodes not existed. Photo of P. melanurus by X.G.
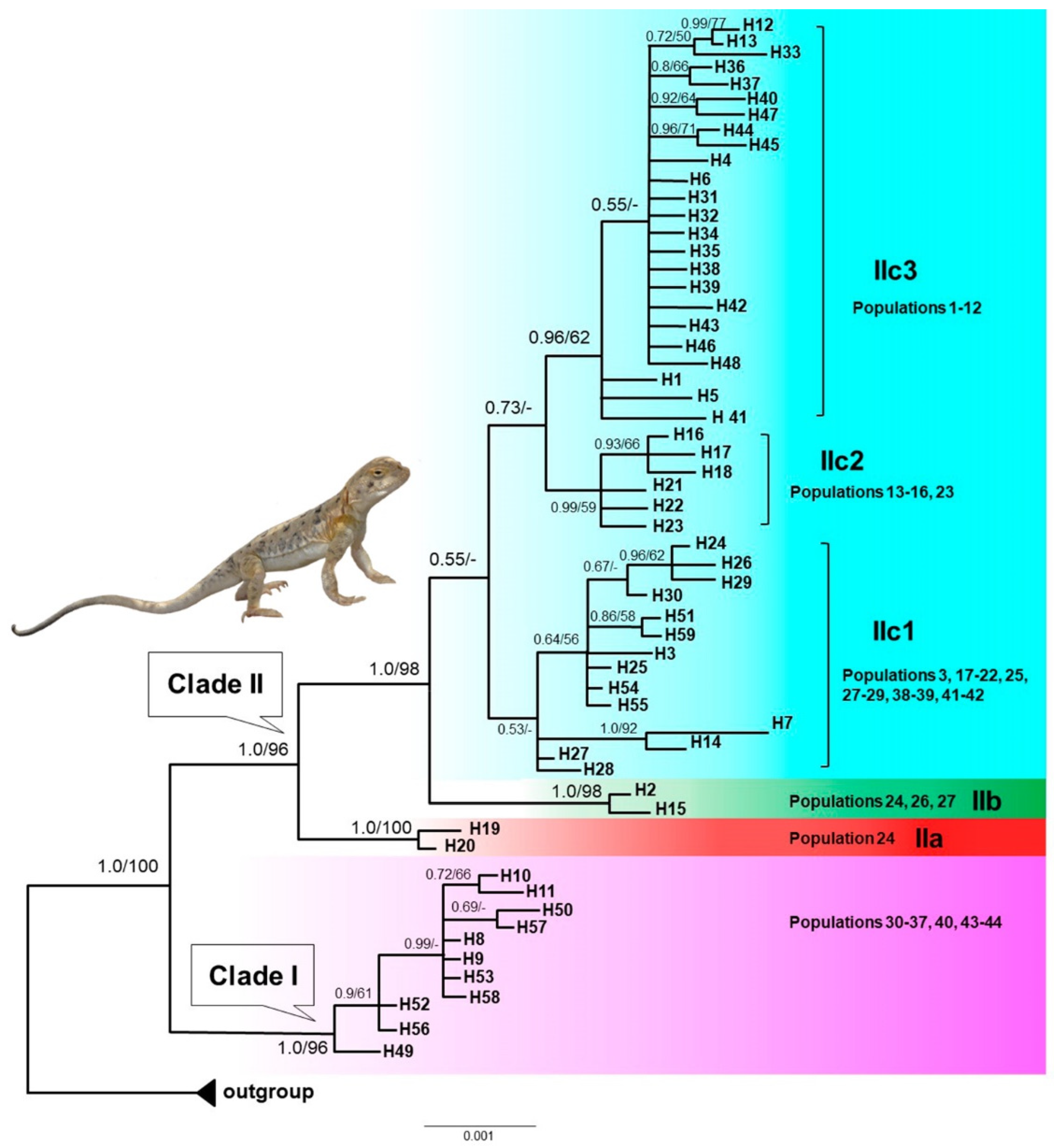
Figure 3.
Bayesian divergence times estimation for P. melanurus based on the sequences of mtDNA COI gene fragment. Median intervals of divergence (in millions of years ago) and 95% highest posterior densities (HPD) are shown above and below branches, respectively. Blue bars at nodes indicate 95% HPD. Branches with “*” indicate Bayesian posterior probabilities (PP) > 0.95. .
Figure 3.
Bayesian divergence times estimation for P. melanurus based on the sequences of mtDNA COI gene fragment. Median intervals of divergence (in millions of years ago) and 95% highest posterior densities (HPD) are shown above and below branches, respectively. Blue bars at nodes indicate 95% HPD. Branches with “*” indicate Bayesian posterior probabilities (PP) > 0.95. .
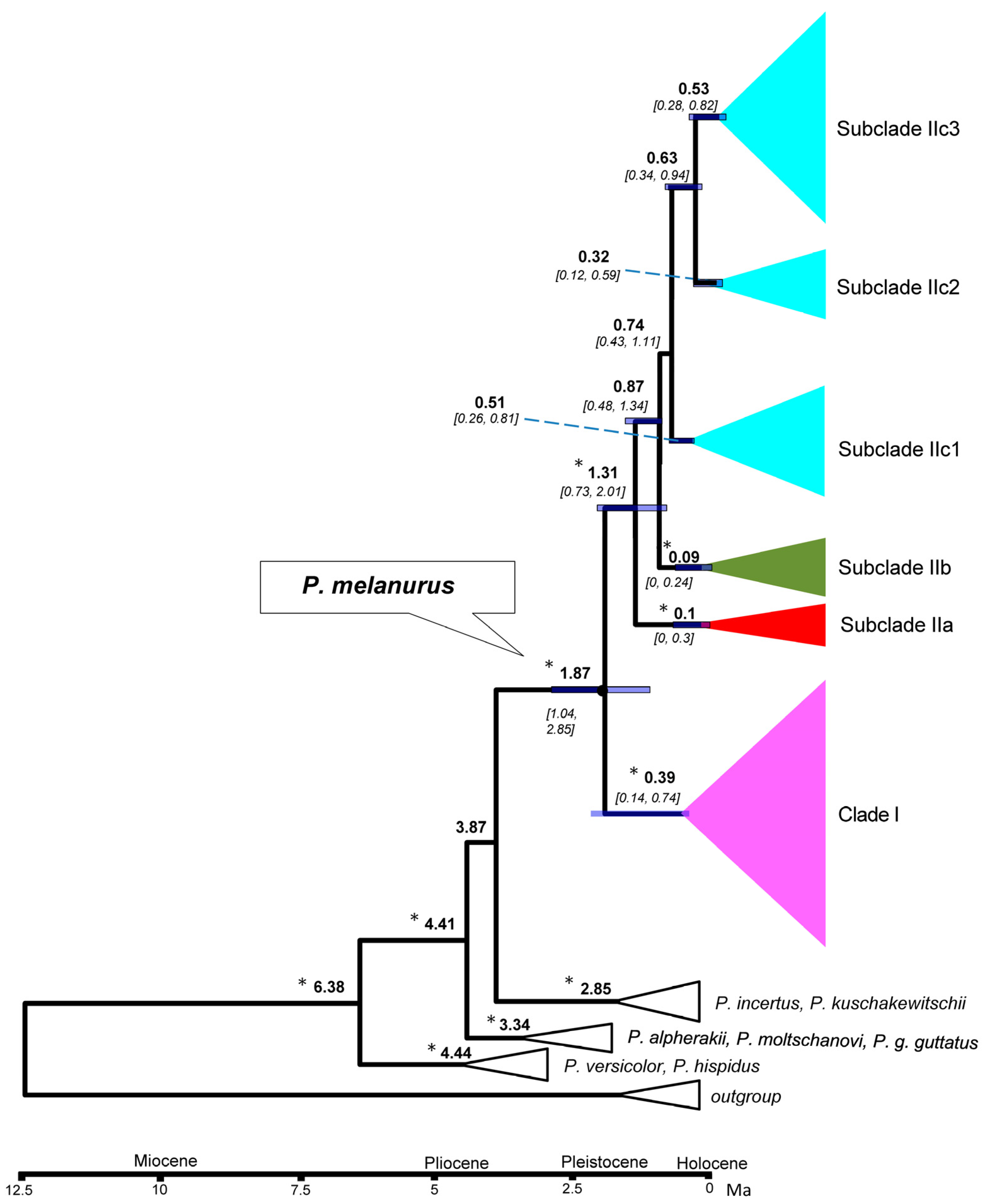
Figure 4.
Median-joining networks of mtDNA COI haplotypes for P. melanurus. Colors correspond to haplotypes of maternal lineages in Figure 2. Short bars crossing network branches indicate mutation steps; small dark circles indicate median vectors inferred by PopART software. Circle size corresponds to relative numbers of individuals sharing a particular haplotype.
Figure 4.
Median-joining networks of mtDNA COI haplotypes for P. melanurus. Colors correspond to haplotypes of maternal lineages in Figure 2. Short bars crossing network branches indicate mutation steps; small dark circles indicate median vectors inferred by PopART software. Circle size corresponds to relative numbers of individuals sharing a particular haplotype.
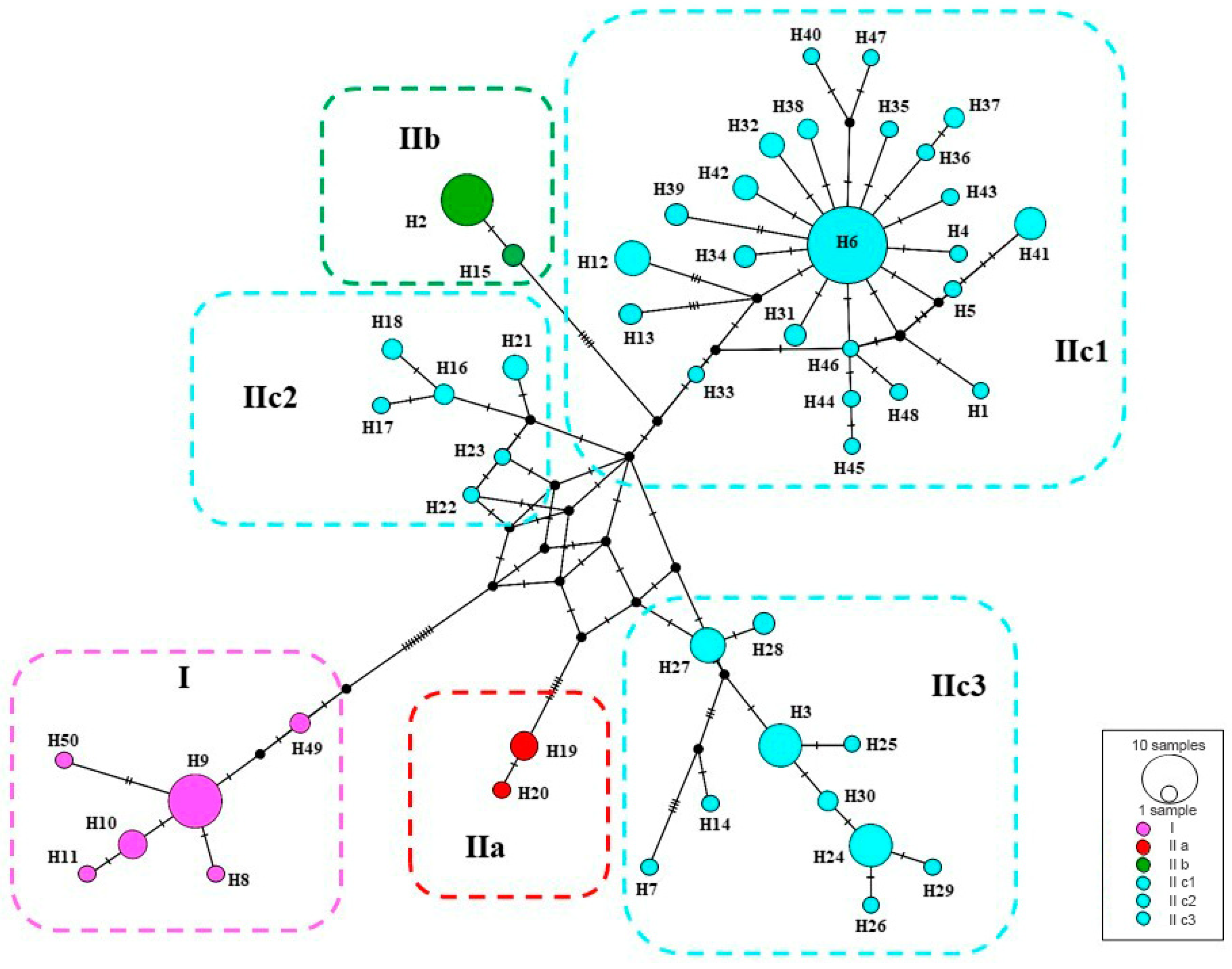
Figure 5.
Mismatch distribution plot for clade/subclade of P. melanurus. The red plots are the observed frequencies of pairwise divergences among sequences, while the green lines refer to the expeted values under a population growth scenario.
Figure 5.
Mismatch distribution plot for clade/subclade of P. melanurus. The red plots are the observed frequencies of pairwise divergences among sequences, while the green lines refer to the expeted values under a population growth scenario.
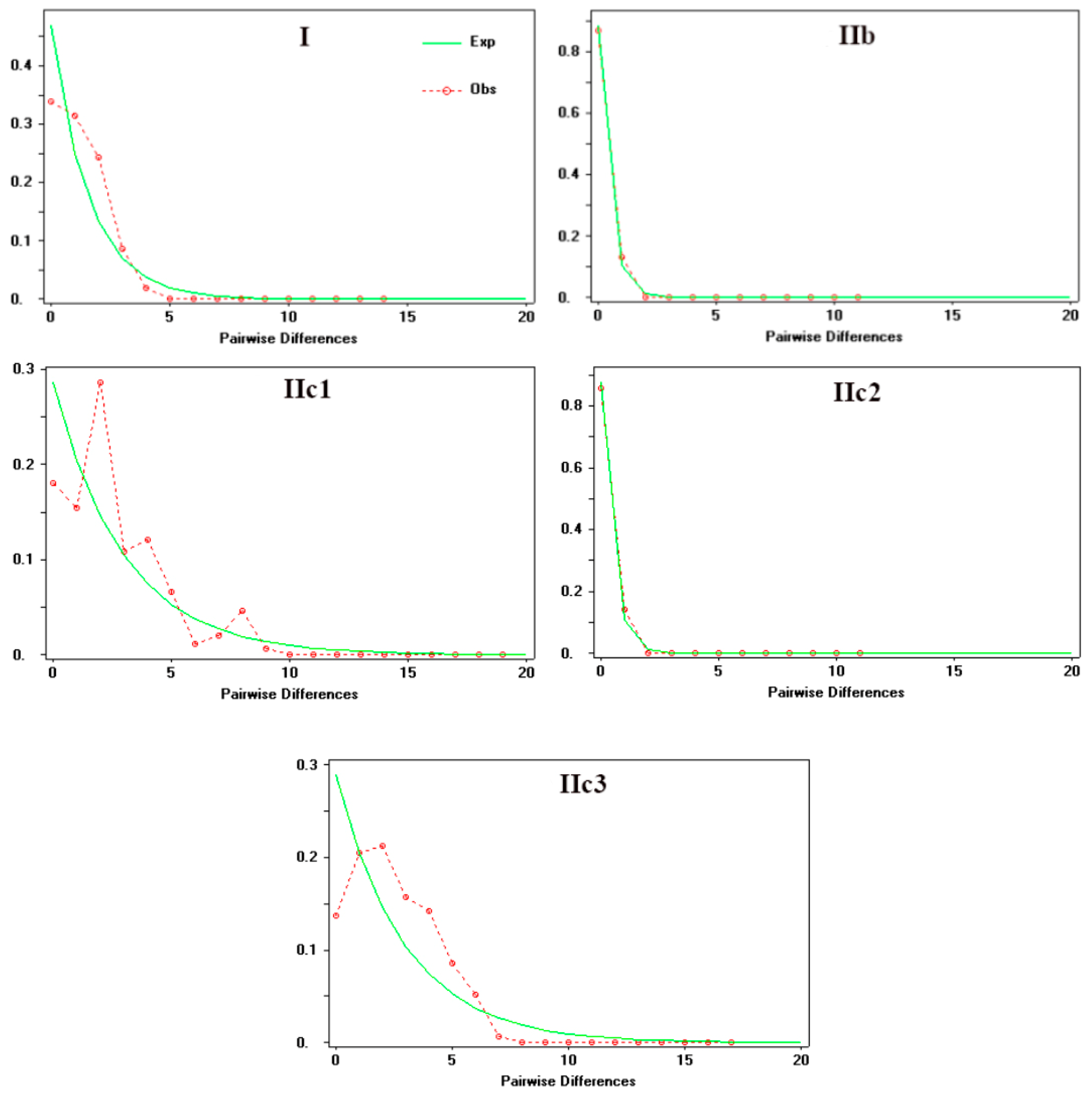
Figure 6.
Bayesian skyline plots (BSP) for the clade/subclade of P. melanurus. The y-axes represent the estimated effective population size on a log scale (Ne*τ/106, the product of the female effective population size and generation length in years); x-axes represent time in millions of years ago (Ma).
Figure 6.
Bayesian skyline plots (BSP) for the clade/subclade of P. melanurus. The y-axes represent the estimated effective population size on a log scale (Ne*τ/106, the product of the female effective population size and generation length in years); x-axes represent time in millions of years ago (Ma).
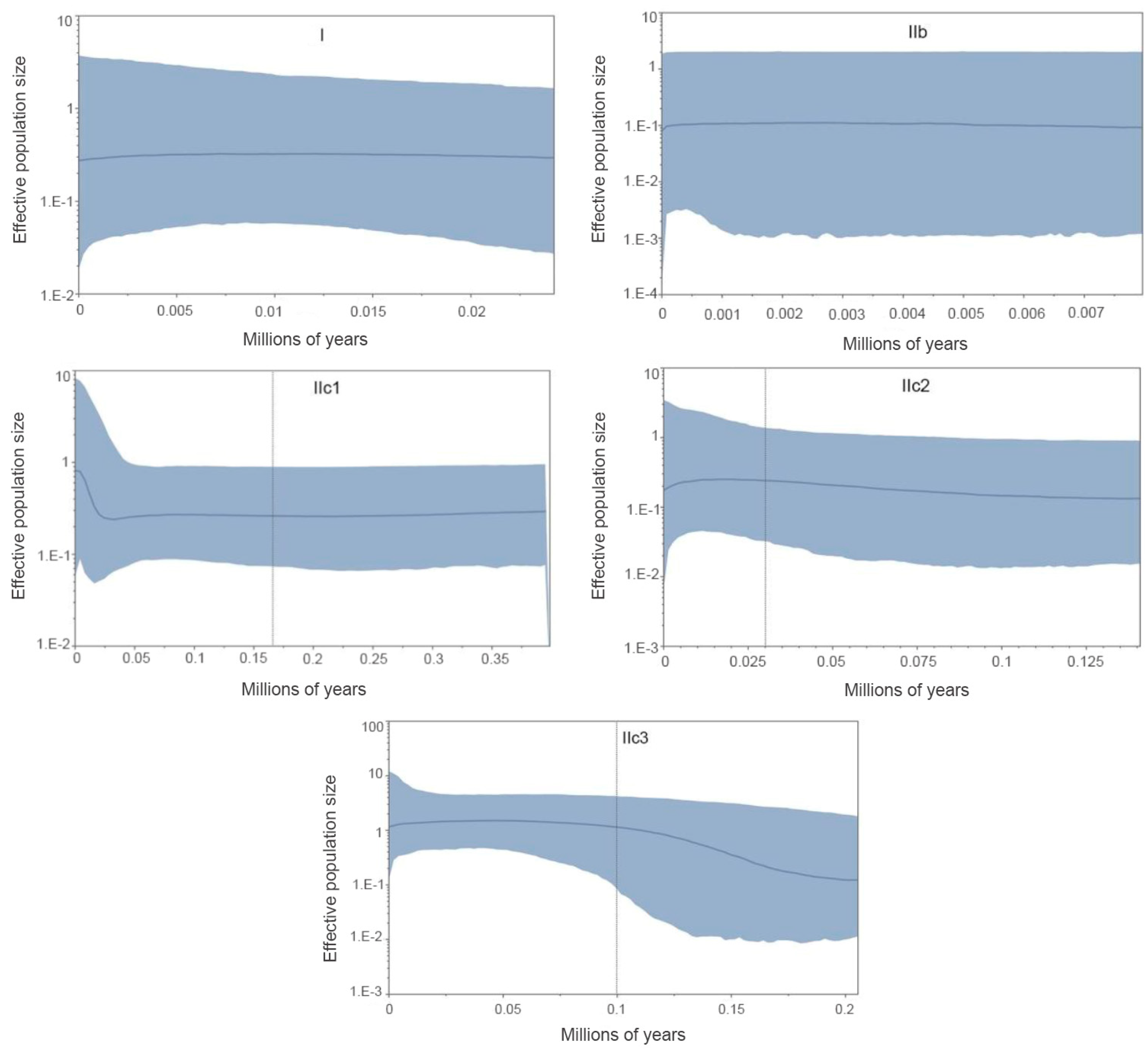
Figure 7.
Spatiotemporal diffusion for P. melanurus from the potential ancestarl area inferred from Bayesian phylogeographic analysis. Four snapshots of colonization events throughout the time are shown: A — the Dzungar Basin origin; B — subsequent multiple spreading northward to E KZ (Zaisan Basin), southward to Karamay region, and southeast to Qitai-Fukang, and southwest through Jinghe and Ebinur reaching Dzungar Gate area. C — the west to east spreading from Zaisan Basin in E KZ to Altay Prefecture in NW CN, and from Bortala Autonomous county to Kuytun area. D — the full event of dispersal with accession to Alakol Basin in E KZ. Colored plogons represent the 80% HPD intervals which indicate the uncertainity of phylogeographic estimates for the nodes.
Figure 7.
Spatiotemporal diffusion for P. melanurus from the potential ancestarl area inferred from Bayesian phylogeographic analysis. Four snapshots of colonization events throughout the time are shown: A — the Dzungar Basin origin; B — subsequent multiple spreading northward to E KZ (Zaisan Basin), southward to Karamay region, and southeast to Qitai-Fukang, and southwest through Jinghe and Ebinur reaching Dzungar Gate area. C — the west to east spreading from Zaisan Basin in E KZ to Altay Prefecture in NW CN, and from Bortala Autonomous county to Kuytun area. D — the full event of dispersal with accession to Alakol Basin in E KZ. Colored plogons represent the 80% HPD intervals which indicate the uncertainity of phylogeographic estimates for the nodes.
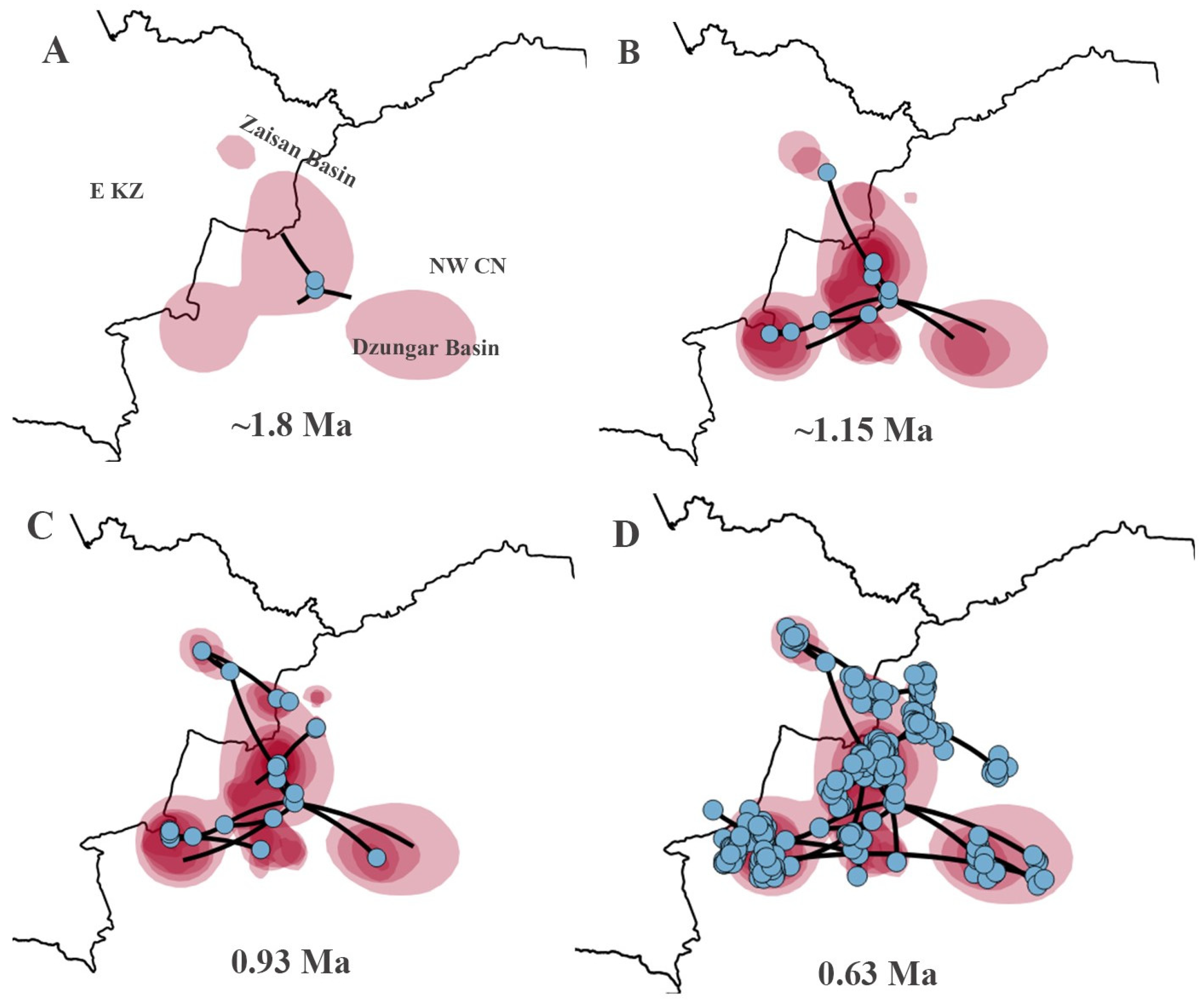
Figure 8.
Species distribution modeling for P. melanurus at the LIG, LGM, Holocene, and current periods.
Figure 8.
Species distribution modeling for P. melanurus at the LIG, LGM, Holocene, and current periods.
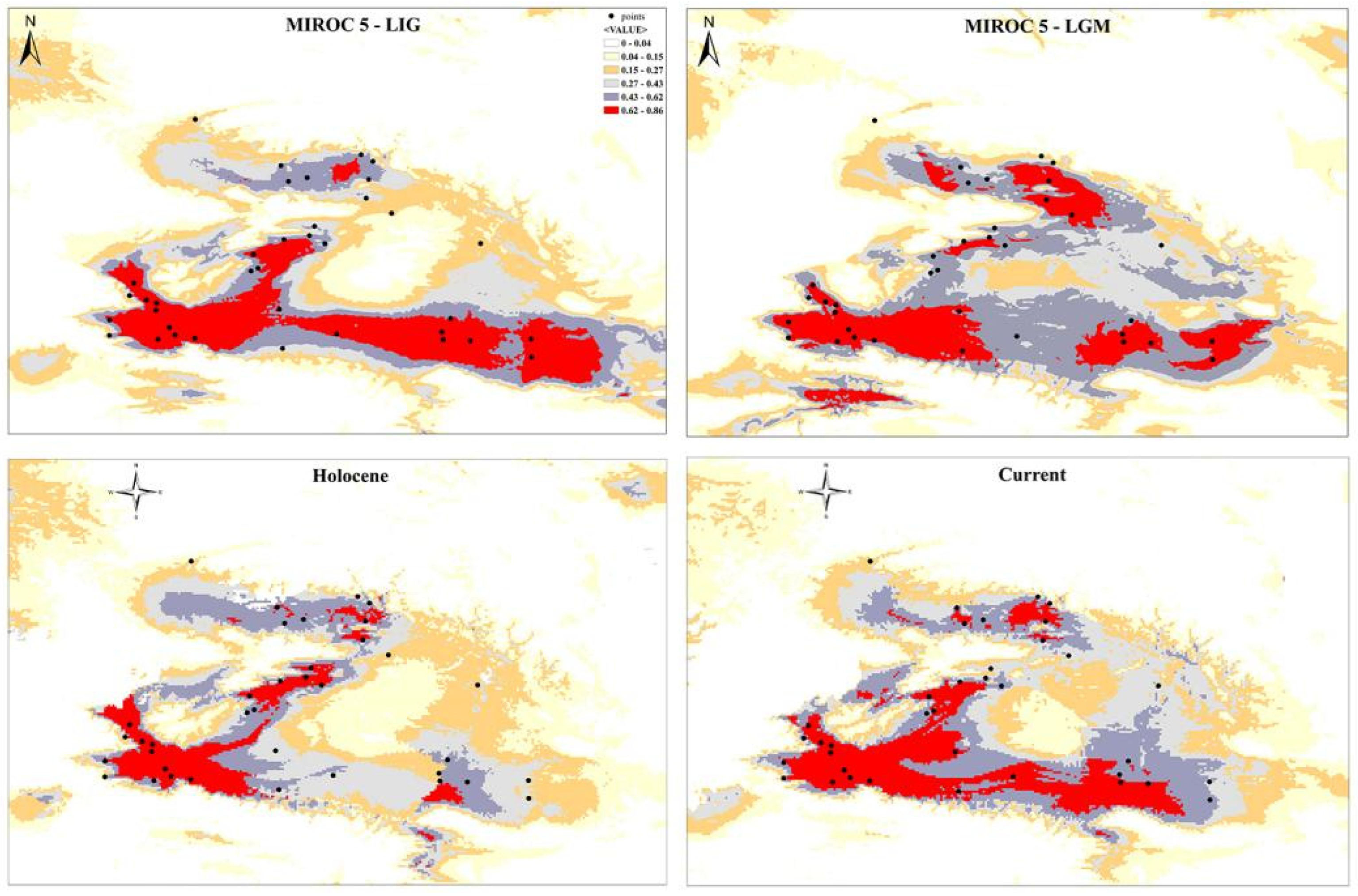
Figure 9.
Species distribution modeling for P. melanurus for future projections under the RCP 2.5, RCP 4.5, and RCP 8.5 gas emissions models.
Figure 9.
Species distribution modeling for P. melanurus for future projections under the RCP 2.5, RCP 4.5, and RCP 8.5 gas emissions models.
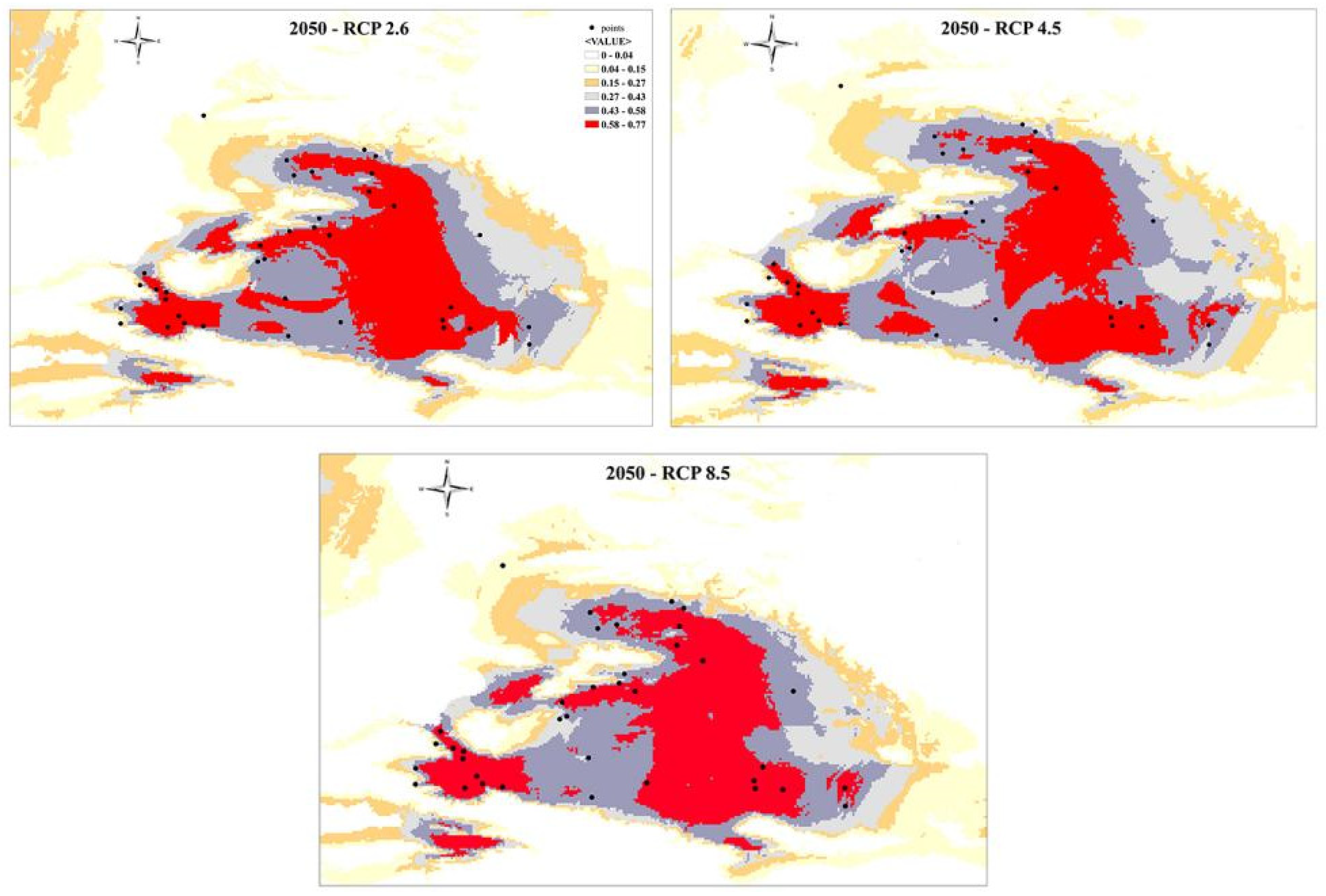

Table 1.
Sampling, sample size, haplotype and nucleotide diversity for 36 populations for P. melanurus. Abbreviation KZ representing Kazakhstan, and CN – China. N – number of samples, X – longitude, Y – latitude, Hd – haplotype diversity, π – nucleotide diversity, SD – standard deviation.
Table 1.
Sampling, sample size, haplotype and nucleotide diversity for 36 populations for P. melanurus. Abbreviation KZ representing Kazakhstan, and CN – China. N – number of samples, X – longitude, Y – latitude, Hd – haplotype diversity, π – nucleotide diversity, SD – standard deviation.
| Site Number | Locality | N | X | Y | Genetic Diversity | ||
|---|---|---|---|---|---|---|---|
| Haplotypes | Hd±SD | ||||||
| 1 | Ulungur, CN | 5 | 87.26 | 47.01 | H6, H12-13 | 0.700±0.218 | 0.00254±0.00079 |
| 2 | Karamay 1, CN | 9 | 85.95 | 46.41 | H6, H13, H32-35 | 0.889±0.091 | 0.00300±0.00073 |
| 3 | Karamay 2, CN | 4 | 85.05 | 45.13 | H6-7 | 0.500±0.07031 | 0.00873±0.00463 |
| 4 | Hoboksar 1, CN | 11 | 85.74 | 46.76 | H13, H42 | 0.509±0.101 | 0.00485±0.00096 |
| 5 | Hoboksar 2, CN | 6 | 85.64 | 46.57 | H6, H36-37 | 0.733±0.155 | 0.00180±0.00039 |
| 6 | Hoboksar 3, CN | 10 | 85.14 | 46.50 | H6, H38-40, H48 | 0.844±0.080 | 0.00317±0.00052 |
| 7 | Emin, CN | 1 | 84.53 | 46.20 | H7 | ‒ | ‒ |
| 8 | Toli 1, CN | 3 | 84.49 | 45.88 | H4-5 | 0.667±0.314 | 0.00529±0.00249 |
| 9 | Toli 2, CN | 2 | 84.62 | 45.93 | H4 | 0.000±0.000 | 0.00000±0.00000 |
| 10 | Fuyun, CN | 6 | 89.02 | 46.42 | H6 | 0.000±0.000 | 0.00000±0.00000 |
| 11 | Jeminay, CN | 13 | 86.76 | 47.31 | H6, H39. H43-47, H49 | 0.808±0.113 | 0.00265±0.00061 |
| 12 | Shihezi, CN | 1 | 86.18 | 44.64 | H1 | ‒ | ‒ |
| 13 | Fukang 1, CN | 4 | 88.43 | 44.95 | H16-18 | 0.833±0.222 | 0.00185±0.00058 |
| 14 | Fukang 2, CN | 3 | 88.28 | 44.53 | H17, H21 | 0.667±0.314 | 0.00317±0.00150 |
| 15 | Fukang 3, CN | 2 | 88.26 | 44.68 | H21 | ‒ | ‒ |
| 16 | Jimsar, CN | 4 | 88.82 | 44.51 | H16, H22-23 | 0.833±0.222 | 0.00265±0.00071 |
| 17 | Ebinur 1, CN | 2 | 82.61 | 45.11 | H28, H31 | 1.000±0.500 | 0.00476±0.00238 |
| 18 | Ebinur 2, CN | 1 | 82.87 | 44.77 | H25 | ‒ | ‒ |
| 19 | Bortala 1, CN | 7 | 81.69 | 44.61 | H24, H26 | 0.286±0.196 | 0.00045±0.00031 |
| 20 | Bortala 2, CN | 1 | 82.60 | 45.19 | H3 | ‒ | ‒ |
| 21 | Kuytun, CN | 1 | 85.11 | 44.36 | H14 | ‒ | ‒ |
| 22 | Bole, CN | 11 | 81.69 | 44.92 | H24, H27-30 | 0.709±0.099 | 0.00404±0.00056 |
| 23 | Qitai 1, CN | 1 | 90.03 | 44.19 | H16 | ‒ | ‒ |
| 24 | Qitai 2, CN | 4 | 90.02 | 44.54 | H2, H19-20 | 0.833±0.222 | 0.01138±0.00554 |
| 25 | Alashankou, CN | 9 | 82.62 | 45.25 | H3, H28, H31 | 0.556±0.165 | 0.00132±0.00055 |
| 26 | Jinghe 1, CN | 11 | 82.65 | 44.54 | H2, H15 | 0.182±0.144 | 0.00029±0.00023 |
| 27 | Jinghe 2, CN | 4 | 82.58 | 44.54 | H2, H3 | 0.500±0.265 | 0.00635±0.00337 |
| 28 | Jinghe 3, CN | 1 | 82.99 | 44.62 | H3 | ‒ | ‒ |
| 29 | Jinghe 4, CN | 1 | 83.37 | 44.56 | H3 | ‒ | ‒ |
| 30 | Bolade, CN | 1 | 86.66 | 48.16 | H11 | ‒ | ‒ |
| 31 | Buerjin 1, CN | 5 | 86.90 | 48.03 | H10 | 0.000±0.000 | 0.00000±0.00000 |
| 32 | Buerjin 2, CN | 9 | 86.81 | 47.69 | H9 | 0.000±0.000 | 0.00000±0.00000 |
| 33 | Zaisan, KZ | 4 | 85.59 | 47.71 | H8, H9 | 0.667±0.204 | 0.00106±0.00032 |
| 34 | Kurchum, KZ | 5 | 85.08 | 47.94 | H9, H50 | 0.400±0.237 | 0.00127±0.00075 |
| 35 | Kokpekty 1, KZ | 1 | 83.38 | 48.85 | H50 | ‒ | ‒ |
| 36 | Kokpekty 2, KZ | 2 | 83.42 | 48.80 | H50 | 0.000±0.000 | 0.00000±0.00000 |
Table 2.
Molecular diversity indices of P. melanurus lineages. N – number of individuals, Nh –number of haplotypes, S – number of polymorphic sites, m – number of mutations, Hd – haplotype diversity, π - nucleotide diversity.
Table 2.
Molecular diversity indices of P. melanurus lineages. N – number of individuals, Nh –number of haplotypes, S – number of polymorphic sites, m – number of mutations, Hd – haplotype diversity, π - nucleotide diversity.
| Clade/Subclade | N | Nh | S | m | k | Hd ±SD | π ±SD |
|---|---|---|---|---|---|---|---|
| I | 27 | 6 | 7 | 7 | 1.13390 | 0.661±0.086 | 0.00180±0.00038 |
| IIa | 3 | 2 | 1 | 1 | 0.66667 | 0.667±0.314 | 0.00106±0.00050 |
| IIb | 15 | 2 | 1 | 1 | 0.13333 | 0.133±0.112 | 0.00021±0.00018 |
| IIc1 | 36 | 10 | 15 | 15 | 2.50159 | 0.819±0.036 | 0.00397±0.00063 |
| IIc2 | 14 | 6 | 6 | 6 | 1.89011 | 0.857±0.056 | 0.00300±0.00029 |
| IIc3 | 70 | 24 | 29 | 31 | 2.46253 | 0.863±0.034 | 0.00391±0.00040 |
| All | 165 | 50 | 64 | 69 | 9.08914 | 0.950±0.00006 | 0.01443±0.00075 |
Table 3.
Uncorrected p-distances between groups of P. melanurus are shown below the diagonal. Standard error estimate(s) are shown above the diagonal and were obtained by a bootstrap procedure (1000 replicates).
Table 3.
Uncorrected p-distances between groups of P. melanurus are shown below the diagonal. Standard error estimate(s) are shown above the diagonal and were obtained by a bootstrap procedure (1000 replicates).
| Clade/Subclade | I | IIa | IIb | IIc1 | IIc2 | IIc3 |
| I | 0.00550 | 0.00626 | 0.00555 | 0.00570 | 0.00585 | |
| IIa | 0.02534 | 0.00562 | 0.00490 | 0.00519 | 0.00517 | |
| IIb | 0.03017 | 0.02180 | 0.00414 | 0.00418 | 0.00432 | |
| IIc1 | 0.02727 | 0.01963 | 0.01451 | 0.00305 | 0.00311 | |
| IIc2 | 0.02699 | 0.02033 | 0.01463 | 0.01011 | 0.00264 | |
| IIc3 | 0.02874 | 0.02165 | 0.01539 | 0.01095 | 0.00846 |
Table 4.
Neutrality tests and mismatch distribution analyses of P. melanurus.
| Clade/Subclade | Tajim’s D | Fu’s Fs | R2 | Rg | SSD |
| I | ‒1.13891 | ‒1.418 | 0.0881** | 0.035 | 0.001 |
| IIb | ‒1.15945 | ‒0.649 | 0.2494 | 0.427 | 0.012 |
| IIc1 | ‒1.00244 | ‒1.722 | 0.0788 | 0.058 | 0.012 |
| IIc2 | 0.00646 | ‒1.120 | 0.1459 | 0.188 | 0.043*** |
| IIc3 | ‒1.96737* | ‒15.870* | 0.0397** | 0.014 | 0.001 |
| All | ‒0.77779 | ‒13.991 | 0.0696 | 0.006 | 0.010 |
SSD, sum of squared distribution; Rg, Harpending’s raggedness index. *p < 0.05;**p < 0.01;***p < 0.001
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



