
Submitted:
08 October 2023
Posted:
09 October 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Mucosal vaccination appears to be suitable to protect against SARS-CoV-2 infection. In this study, we tested an intranasal mucosal vaccine candidate to COVID-19 constituted of a cationic liposome containing a trimeric SARS-CoV-2 spike protein and CpG-ODNs, a Toll-like receptor 9 agonist, as adjuvant. In vitro and in vivo experiments indicated the absence of toxicity following the intranasal administration of this vaccine formulation. First, we found that subcutaneous or intranasal vaccination protected hACE-2 transgenic mice from infection with the wild-type (Wuhan) SARS-CoV-2 strain, as shown by weight lost and mortality indicators. However, when compared to the subcutaneous administration, the intranasal route was more effective in the pulmonary clearance of the virus and induced higher neutralizing antibodies and anti-S IgA titers. In addition, the intranasal vaccination afforded protection against gamma, delta and omicron virus variants of concern. Furthermore, the intranasal vaccine formulation was superior to intramuscular vaccination with a recombinant, replication-deficient chimpanzee adenovirus vector encoding the SARS-CoV-2 spike glycoprotein (Oxford/AstraZeneca) in terms of virus lung clearance and production of neutralizing antibodies in serum and bronchial alveolar lavage (BAL). Finally, the intranasal liposomal formulation boosted heterologous immunity induced by previous intramuscular vaccination with the Oxford/AstraZeneca vaccine that was more robust than homologous immunity.
Keywords:
SARS-CoV-2
; vaccine
; hACE2 transgenic mice
; intranasal route
; spike protein
; cationic liposome
; CpG-ODNs
; heterologous immunity
Introduction
Infection by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has emerged as one of the major public health problems since 2019 due to the global spread of coronavirus disease (COVID-19) [1]. Paralleling the outbreak of the COVID-19 pandemic, was the extraordinary innovation and unprecedent development of highly effective vaccines [2]. For example, all vaccines approved by the European Medical Agency were highly efficacious against severe COVID-19 infection [3]. Indeed, in Brazil, the first model city vaccination program was performed with the CoronaVac vaccine (Sinovac Biotech) in the town of Serrana, São Paulo State, and the result of the efficient immunization campaign was a reduced death toll rate and related COVID-19 morbidity when compared to the rest of Brazil where the immunization rates were not as high [4]. However, systemic humoral immunity induced by vaccination wanes over time, as revealed by declining neutralizing antibody titers it became apparent that the [5]. Intramuscular vaccination, although safe and effective at inducing protective immunity, it might fail to induce optimal mucosal immunity in the airways, thus facilitating virus transmission. The emergence of viral variants of concern (VOC) has further complicated the pandemic. To address these challenges, the World Health Organization has recommended vaccine booster shots to enhance immunity [6]. It is anticipated that a mucosal vaccine might be advantageous in this scenario due to the potential to prevent infection and transmission; and may be more effective against VOCs, as they can induce local immune responses at the sites of viral entry. In this regard, the oral vaccine against poliovirus (Sabin vaccine) was the first vaccine to proof the concept of mucosal immunity: in contrast to the Salk intramuscular vaccine, the Sabin vaccine prevented the disease as well as its transmission by IgA neutralizing antibodies [7]. Consistently, in COVID-19 IgA antibodies dominated the early SARS-CoV-2-spexcific humoral responses, contributing to virus neutralization to a greater extent than IgG antibodies [8]. Pre-clinical studies in mice confirmed the superior protective immunity to SARS-CoV-2 achieved by intranasal adenovirus-vectored vaccine [9] or intranasal trivalent next-generation COVID-19 vaccines [10]. Besides adaptive humoral immunity, cellular immunity has also been shown to play a role in the control of COVID-19 [11]. Indeed, in murine models of COVID-19, both humoral and cellular adaptive immunity contribute to viral clearance, although the protection to infection appears to be largely mediated by the antibody response [12]. Altogether, vaccines against SARS-CoV-2 might need to be updated periodically and be administered preferentially by the mucosal route to avoid loss of clinical efficacy and prevent transmissibility. In addition, given the persistence of the COVID-19 pandemic worldwide, it is highly appropriate that vaccine formulations be versatile and adaptable to different VOC, stable and easily manufactured in different countries and under dissimilar conditions.
Here, we have used a vaccine platform consisting of a cationic liposome containing a recombinant trimeric SARS-CoV-2 spike protein to induce robust T-follicular helper cell and humoral responses [13], adjuvanted with CpG oligonucleotides to boost mucosal IgA antibody production [14] and Th1 cellular immunity, in a transgenic mouse model (K18-hACE2) of COVID-19 [15]. The study results show that our candidate COVID-19 vaccine is not toxic and that its intranasal administration induces a superior immunity compared to the subcutaneous or intramuscular routes. Also, the intranasal vaccine afforded protection against gamma, delta and Omicron virus variants. In addition, the intranasal liposomal formulation boosted heterologous immunity, which was better than the homologous immunity induced by previous vaccination with a recombinant, replication-deficient chimpanzee adenovirus vector encoding the SARS-CoV-2 Spike glycoprotein (Oxford/AstraZeneca vaccine).
Material and Methods
Vaccine Formulation
The vaccine formulation contained as antigen the trimeric spike protein of SARS-CoV-2 stabilized in the prefusion conformation was provided by the Cell Culture Engineering Laboratory of COPPE/UFRJ by means of serum-free cultivation of stably transfected HEK293 cells and purified by affinity chromatography, as described by Alvim et al [16]. In all experiments, the spike protein corresponded to the Wuhan strain (1208 aminoacids that form the spike ectodomain, as proposed by Wrapp et al [17] except for the toxicity study, where the Wuhan aminoacid sequence was slightly altered by one mutation (D614G). The adjuvant was a Class C CpG oligonucleotide (Human/Murine TLR9 ligand, ODN 2395, Invivogen, San Diego, CA, USA or Exxtend, Campinas, Brazil). Each 30L vaccine dose used in mice studies contained 5g of spike protein and 10g of CpG, entraped in DOTAP (ROCHE DOTAP Liposomal Transfection Reagent N-[1-(23-Dioleoyloxy) propyl]-NNN-trimethylammonium methyl-sulfate code:11202375001, Basel, Switzerland).
Physicochemical characterization: Size and polydispersity index (PDI) measurements of the vaccine formulation were performed using the Zetasizer Nano Ultra equipment (Malvern, United Kingdom) with a polystyrene cuvette at a backscatter angle (173°). To analyze the surface charge of the particle, specific cuvettes were used to measure the zeta potential. Vaccine formulations were diluted to a volume of 1mL before measurement. Results were measured in three replicates which are reported as mean ± standard deviation. To evaluate particle size, we used average hydrodynamic size weighted by particle intensity analyzed by dynamic light scattering (DLS) methodology.
Toxicity of Vaccine Formulation
In vivo toxicity: Female (n=15) and male (n=15) CD1 mice were intranasally vaccinated on days 0 and 14, according to the vaccination protocol described below (Vaccination, COVID-19 animal model). Blood samples were collected at days 0 (before vaccination), 2, 16, 29. Hemogram, albumin/globulin rate, aspartate aminotransferase (AST), alanine aminotransferase (ALT), phosphatase alkaline (FA), total bilirubin, urea, creatinine, calcium, phosphorus, total proteins, albumin, glucose, total cholesterol and triglycerides, alpha-1-glycoprotein were evaluated. Organs were weighted, macro and microscopically evaluated at day 16 (n=20) and day 29 (n=10): aorta, spleen, urinary bladder, brain, nasal cavity, tongue, heart, esophagus, stomach, liver, adrenal, mammary, pituitary and mandibular salivary glands, thyroid and parathyroid, small and large intestines, mesenteric lymph node, cervical lymph nodes (submandibular, near the site of administration), thymus, spinal cord, bone marrow, skeletal muscle, sciatic nerve, femur, ovaries, pancreas, skin, prostate, lungs, kidneys, testicles, epididymis, seminal vesicle, trachea, uterus, cervix, vagina, eyes, optic nerve. Body weight, food consumption, clinical score, morbidity, and mortality were evaluated at least once a day. Body temperature was measure before, 3h and 24h after vaccination. Mice were treated according to animal welfare guidelines of CIEnP (Ethic Protocol CEUA 308/00) under National Legislation-11.794 Law, which complies with the research complies with the commonly-accepted 3Rs: reduction, replacement and refinement.
In vitro toxicity: Vaccine cytotoxicity was evaluated according to [18]. Briefly, 3T3 fibroblasts, CALU-3 lung cells, primary human fibroblasts (HLF) and rat lung cells (pneumocytes) were distributed into flat-bottom 96-well plates (Corning, USA) at a concentration of 8,000 cells per well and incubated in a humid atmosphere and 5% CO2 at 37ºC for 48 hours. After this period, the vaccine formulation diluted in PBS pH 7.2 was added (0.1, 1.0 or 10 µl) in the final volume of 100 µl, for 24 or 48 h. Cell viability was evaluated by the colorimetric method of resazurin (7-hydroxy-3H-phenoxazine-3-one 10-oxide), based on the intracellular reduction of resazurin to resorufin by viable and metabolically active cells [19]. The plates were kept with the resazurin solution (0.02 mg/mL) (Sigma-Aldrich, USA - cod. M2003), in DMEM without phenol, for 2 h at 37°C. The absorbance of the samples was determined at 570 and 600 nm in a Synergy HT plate reader (Biotek, Winooski, VT, USA) using the Gen5 ™ software (BioTek, Winooski, VT, USA). The tests were performed in sextuplicate, and the results were expressed in terms of the mean ± standard error of two different experiments. Cells incubated with PBS were used as negative controls, while cells incubated in 10% dimethyl sulfoxide (DMSO) served as positive controls for cell death.
COVID-19 Animal Model
Animals: Transgenic C57BL/6 mice (K18-hACE2) expressing the human hACE2 receptor, lineage B6.Cg-Tg(K18-ACE2)2Prlmn/J HEMI Homozygous for Tg(K18-ACE2)2 Prlmn, [20] here purchased from Jackson laboratories and bred at ICTB, Rio de Janeiro, Brazil. Mice were kept in a Specific Pathogen Free (SPF) Biosafety Level (BSL)-2 animal house and transferred to a BSL-3 environment 2 days before virus infection. Food and water were provided ad libitum. Environment enrichment was provided in the BSL-2 animal house. Mice were treated according to animal welfare guidelines of ICB - USP (Ethic Protocol 4344010720) under National Legislation-11.794 Law.
Vaccination: Vaccinated animals were injected with i) the vaccine candidate was administered intranasally (i.n.) in a total volume of 30L (15L per nostril) or subcutaneously (s.c.), administered in the dorsal neck region (total of 100L, prepared by mixing 30L of vaccine formulation with 70L of sterile saline), or ii) intramuscularly (i.m.) in the thigh muscles of the hind limb (total of 70 L, 35L per leg) with the Oxford/AstraZeneca (AZ) Covid-19 vaccine (produced at FIOCRUZ/Bio-Manguinhos, Rio de Janeiro, Brazil). Animals were vaccinated on days 0 and 7 and for some experiments, a vaccination booster was administered on day 14. Non-vaccinated mice received only phosphate buffered saline (PBS). Details of the protocols performed in each experiment are indicated in the figures’ diagrams. Mice were anesthetized intra-peritoneally with ketamine (50 mg/kg, Syntec, SP, Brazil) and xylazine (20 mg/kg, Syntec, SP, Brazil) before each injection.
Virus infection: Four different strains of SARS-Cov-2 were used: Wuhan (wild type)
EPI_ISL_1557222, strain B.1.1.28 from nasopharyngeal swab taken from an infected patient from São Caetano do Sul, City, Brazil in April, 2020; Gamma, EPI_ISL_1060902, strain P.1 was obtained from a nasopharyngeal specimen of a patient from Manaus City, Brazil, in December, 2020 that was previously classified as belonging to the P.1 lineage by virus genome sequencing; Delta, EPI_ISL_2938096 – strain B.1.617.2, Instituto Butantan, and Ômicron (EPI_ISL_6901961 – strain B.1.1.529, Instituto de Ciências Biomédicas, University of São Paulo. K18-hACE2 C57Bl/6J mice were anesthetized as described for vaccinations and were inoculated i.n. with 10ˆ5 TCID50 of SARS-CoV-2 per mouse, in a total volume of 30L (15L per nostril). Infected mice were kept in Biosafety Level 3 (BSL-3) environment for a maximum of 7 days. Body weight and clinical score were recorded daily, and euthanasia was performed in mice reaching 20% of weight loss or showing signals of suffering, such as decreased activity, piloerection, un-groomed appearance, abnormal stance with ataxia, changes in eye brightness, or change in respiratory pattern. Euthanasia was performed intra-peritoneally with lethal doses of inhaled isoflurane (Cristalia, Itapira, Brazil). Mice were treated according to animal welfare guidelines of FCF - USP (Ethic Protocol CEUA 621) under National Legislation-11.794 Law.
Acid Nucleic Extraction and RT-qPCR (Quantitative Real Time PCR Based on REVERSE Transcriptase) Assay for SARS-CoV-2
Lung lobe tissue samples were macerated mechanically and, simultaneously, the viral inactivation and the digestion were performed in lysis buffer (containing guanidinium isothiocyanate + proteinases K), according to the manufacturer’s instructions for the Extract kit fast DNA and RNA viral reagents (Loccus®, Cotia, São Paulo, Brazil). The specimens underwent nucleic acid extraction and purification by using the automatized extractor EXTRACTA 32 with magnetic beads (Loccus®, Cotia, São Paulo, Brazil), according to the manufacturer’s instructions. For in house SARS-CoV-2 quantification by RT-qPCR, specific primers and probes for the E-gene of SARS-CoV2 were synthesized as described previously [21,22]. Standard curves were generated for the quantitative RT-qPCR for E-gene SARS-CoV-2 with known amounts of the synthetic oligos as previously reported [23] by using TaqMan™ Fast Virus 1-Step Master Mix system (Thermo Fisher Scientific®, Austin, USA). The data were analyzed using QuantStudio Design & Analysis Software v.1.4.1. The viral load was expressed as the log10 number of viral copies per ng of RNA. Samples were handled according to laboratory biosafety guidelines.
RNA Isolation, cDNA Synthesis and RT-qPCR for Cytokines
Olfactory bulb and hippocampus were dissected in PBS 1x, incubated in RNA Later overnight at 4oC (Sigma Aldrich, cat# R0901) and kept at -80oC. Tissues were lysed using QIAzol reagent (Qiagen, cat# 79306) and the RNeasy Plus Kit (Qiagen cat# 74134) was used for genomic DNA elimination and RNA isolation. RNA integrity was confirmed by electrophoresis on 1% agarose gel under denaturing conditions. cDNA was synthetized using the High-Capacity RNA-to-cDNA kit (Thermo Fisher Scientific, cat# 4388950), according to the manufacturer's instructions. RT-qPCR reactions were performed using SYBR green amplifications (PowerTrack SYBR Green Master Mix, Thermo Fisher Scientific, cat# A46109). Reactions consisted of 10 μL of SYBR mix, 2 μL of cDNA, 1 μL (10 μM) of each primer and 6 μL of UltraPure water (cat# AM9932). The cycling conditions were: 95ºC for 20s, 40 cycles of 95 ºC for 1s and 60ºC for 20s.Targets and primer sequences are listed below. Relative gene expression was determined by applying the 2-ΔΔCt method, using β-Actin expression for normalization and comparing treated mice to non-infected controls.
| Gene | Forward primer | Reverse primer |
| Actb | 5'-GAA GAT CAT TGC TCC TC-3' | 5'-CCT GCT TGC TGA TCC ACA TC-3' |
| Il1b | 5'-CAG GCA GGC AGT ATC ACT CA-3' | 5'-AGC TCA TAT GGG TCC GAC AG-3' |
| Il6 | 5'-TAG TCC TTC CTA CCC CAA TTT CC-3' | 5'-TTG GTC CTT AGC CAC TCC TTC-3' |
| Tnf | 5'-TGT AGC CCA CGT CGT AGC AAA-3' | 5'GGC TCA GCC ACT CCA GCT G-3' |
Virus Neutralization Test (VNT)
The Cytopathic Effect (CPE)- based Virus Neutralization Test (VNT) was adapted from Nurtop et al [24] and applied as previously described ([25,26,27]). The VNT was performed with SARS-CoV-2 Wuhan (wild type) EPI_ISL_1557222, strain B.1.1.28 from nasopharyngeal swab taken from an infected patient from São Caetano do Sul, City, Brazil in April, 2020) in 96-well microtiter plates containing 5 × 104 Vero cells/mL. Vero cells were seeded in a 96-well microtiter plate and allowed to grow for 24 h prior to infection. Sera and BAL to be tested were heat-inactivated for 30 min at 56°C. Then, 110 μL of two-fold serially diluted sera (from 1:20 to 1:2560) were added to mixed vol/vol with 10^3 TCID50/mL of SARS-CoV-2 and incubated at 37°C for 1h for virus neutralization. The sera-virus and BAL-virus mixture was transferred onto the confluent Vero cell monolayer and incubated for 72 h. Cultures at 37°C and 5% CO2 were observed daily for a CPE. After 72 h, the plates were analyzed by light microscopy (Nikkon, Japan), distinguishing the presence/absence of CPE-VNT. To verify the initial observations, after 72 h the monolayers were fixed and stained with Naphthol Blue Black (Sigma-Aldrich Co., Deisenhofen, Germany) dissolved in sodium acetate-acid acetic for 30 min. Dilutions of serum associated with CPE were considered a negative result. The absence of CPE or complete neutralization of SARS-CoV2 inoculum was considered a positive result. For each reaction, virus diluted in DMEM with 2.5% FBS was used as positive control, while DMEM with 2.5% FBS without added virus served as a negative control. As additional controls, a serum specimen taken from a patient with a SARS-CoV-2 infection was used as a positive control, and a sample from a patient without neutralizing antibodies, with known VNT results, was used as negative control. The antibody titer was calculated as the highest dilution where CPE was completely inhibited. Titers ≥1:20 were reported as positive. Virus isolation and VNT were performed in a Biosafety Level (BSL)-3 laboratory.
Enzyme-Linked Immunosorbent Assay (ELISA) for Antibodies
S-Protein-specific IgA was determined by adding serum or BAL (broncho-alveolar lavage) samples at multiple dilutions to 96-well plates previously coated with spike protein provided by the Cell Culture Engineering Laboratory of COPPE/UFRJ, followed by incubation with a goat anti-mouse IgA antibody conjugated to HRP (cat#1040-05, Southern Biotech, Birmingham AL, USA). Purified mouse IgA (cat#1040-01, Southern Biotech, Birmingham AL, USA) was used as standard. Serum assays were performed in 96-well plates (cat#442404, Thermo Fisher, Waltham, MA, USA) and BAL assays in 96-well plates (cat#3690, Corning, Corning NY, USA). Statistical analysis
The results were expressed as the mean ± standard error (SE) or standard deviation (SD), indicted in the figures. Statistically significant differences were determined using unpaired t-tests or ANOVA tests followed by the Bonferroni post-test, as applicable and according to test requirements. A p-value < 0.05 was considered statistically significant (* p < 0.05, ** p < 0.01, *** p < 0.001). GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA) was used for statistical analysis and graph generation. No statistical methods were used to predetermine sample size. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Results
Toxicity of Nasal Vaccine Formulation
The toxicity of the nasal vaccine formulation was tested using in vivo and in vitro models. In vivo toxicity tests were performed under good laboratory practices (GLP) by CIEnP (Center of Innovation and Pre-clinical studies) and accredited by the Brazilian Accreditation body (INMETRO). The parameters evaluated included clinical score, morbidity, mortality, body temperature, body weight, food consumption, hemogram, biochemical blood parameters, histopathology of several organs as detailed in M&M. No toxicity was recorded after intranasal vaccine administration (data not shown).
For in vitro toxicity tests, four different cell types were used. Firstly, 3T3 cells, a standardized cell lineage for cytotoxicity assays, served to verify that the vaccine formulation was not cytotoxic at any of the used concentrations and periods of incubation (Figure 1A,B). The viability of CALU-3 cells, a human lung cancer cell line used as respiratory model in preclinical applications [28] was then assessed in the presence or absence of the vaccine formulation, showing that the formulation was not cytotoxic (Figure 1C,D). Finally, human lung fibroblasts and primary rat pneumocytes remained viable after 24h of incubation with the vaccine formulation, even at the highest concentrations tested (Figure 1E,F).
The physicochemical characterization of the vaccine formulation, evaluated at different stages, demonstrated that concerning size, there is a significant increase in the formulation when only the S protein is added to the DOTAP liposome. In this context, when CpG is subsequently added to this mixture, the size is reduced. When we compare the DOTAP liposome with and without the addition of CpG, without protein, we do not observe a significant modification in size (Figure 1G). The same effect is observed in the polydispersity, with a decrease in the polydispersity index (PDI) upon adding CpG compared to samples without the adjuvant, suggesting that the addition of the adjuvant is beneficial for the formulation in terms of physicochemical parameters (Figure 1H) . Regarding the charge of the vaccine formulation, we found that without the adjuvant the formulation has a positive charge and only with its presence does it becomes negative (Figure 1I).
The Effect of Intranasal versus Subcutaneous Vaccine Administration
Transgenic K18-hACE2 mice received the vaccine formulation intranasally (i.n.) or subcutaneously (s.c.) on days 0 and 7. Two weeks after the second dose, vaccinated (i.n. or s.c.) or non-vacinated (non-vac) animals were challenged i.n. with wild type (Wuhan) SARS-CoV-2 virus, (Figure 2A). All infected non-vaccinated mice died after 5 to 7 days post infection (DPI 5-7), while no mortality was observed in i.n. or s.c. vaccinated groups (Figure 2B). Non-vaccinated mice started to lose weight 4 days after receiving SARS-CoV-2, reaching 20% of weight loss at DPI-7 (Figure 2C). According to mortality data, both i.n. and s.c. vaccinated groups maintained stable body weight during 7 consecutive days after SARS-CoV-2 infection. These results indicate that i.n. or s.c. vaccination was equally protective against the SARS-CoV-2 infection.
The viral load in the lung (Figure 2D) and in the brain (Figure 2E) was measured by RT-PCR on different days after viral infection. Two days after viral infection (DPI-2) 40% of the vaccinated animals (n=2) (i.n. and s.c.) had no virus detected in the lung tissue (Figure 2D). However, at DPI-2, there was no statistically significant difference between vaccinated and non-vaccinated groups. At DPI-4, there was a significant decrease in the lung viral load only in the i.n. vaccinated group when compared to non-vaccinated mice (Figure 2D). Moreover, at DPI-6, i.n. vaccinated group SARS-CoV-2 mRNA could not be detected in the lung while in non-vaccinated animals a high viral load was detected (Supplementary Figure S1A).
Next, we determined viral load in proximal and distal sites of the nervous system (i.e., closer to and further away from the site of infection, respectively). Interestingly, all vaccinated and non-vaccinated groups were equally positive for the virus in the olfactory bulb (proximal site), but only non-vaccinated animals had the virus in distal areas such as the hippocampus (Figure 2E). As expected, in the lung and brain, SARS-CoV-2 was not detected in naive animals that did not receive the virus (Figure 2D,E). These results indicate that both vaccination routes are equally effective in preventing the virus spread to the hippocampus, but the i.n. vaccination was more effective than the s.c. vaccination in the clearance of the virus from the lung tissue. We further assessed brain inflammation to test whether the i.n. vaccination could prevent neuroinflammation. Following infection, the proinflammatory cytokines IL-1β, TNFα and IL-6 were upregulated in the olfactory bulb and hippocampus at DPI-4. The i.n. vaccination prevented the upregulation of these cytokines in both brain regions. (Supplementary Figure S1D,E).
We also measured the levels of anti-spike protein (S) antibodies in the serum of vaccinated and non-vaccinated K18-hACE2 mice. The levels of anti-S IgG1 at DPI-2 were significatively higher in the s.c. vaccinated mice when compared to non-vaccinated mice (Figure 2F). Although mice vaccinated by the i.n. route had detectable anti-S IgG1 in the serum, there was no statistically significant difference compared with the non-vaccinated animals. However, at DPI-4 mice in both vaccinated groups presented significatively higher levels of anti-S IgG1 than the animals from the non-vaccinated group (Figure 2F). The levels of IgG1 antibodies were generally higher at DPI-4 when compared to DPI-2 (Figure 2F). The levels of anti-S IgG2c at DPI-2 and DPI-4 were significantly higher in both groups of vaccinated animals (s.c. and i.n.) when compared with the non-vaccinated group (Figure 2G), with antibody levels generally higher at DPI-4 when compared to DPI-2 (Figure 2G). Importantly, serum anti-S IgA was only found in i.n. vaccinated mice that showed significantly higher IgA titers than non-vaccinated or s.c. vaccinated animals (Figure 2H). Similar anti-S IgA results were found in the broncho-alveolar lavage (BAL) (Supplementary Figure S1B). As expected, no anti-S immunoglobulins were found in non-vaccinated animals (data not shown).
For all antibodies measured, the levels were higher at DPI-4 than at DPI-2, indicating that the antibody production increased after viral infection. Altogether, these results indicate that the vaccine formulation was effective to induce the production of anti-S antibodies and that the i.n. route induced higher levels of specific IgG2c and IgA antibodies when compared to the s.c. administration.
Finally, the titers of SARS-CoV-2 neutralizing antibodies in the serum of vaccinated and non-vaccinated mice were measured by VNT (Figure 2I). At DPI-2, neutralizing antibodies were found only in i.n. vaccinated animals. At DPI-4, although neutralizing antibodies were detected in the serum of s.c. vaccinated animals, this was not significatively different from non-vaccinated animals. In contrast, the titers of neutralizing antibodies found in i.n. vaccinated mice were highly increased compared to the non-vaccinated group. The antibody titers were also increased in DPI-4 in relation to DPI-2, confirming the progressive increase in antibody production in i.n. vaccinated mice after the contact with the virus.
In conclusion, both vaccine administration routes were highly effective to protect mice against SARS-CoV-2 infection, but the i.n. route was more efficient in clearing SARS-CoV-2 from the lung and in inducing the production of S-specific IgG2c and IgA antibodies and neutralizing antibodies in the serum than the s.c. route. Therefore, we selected the i.n. vaccination to perform our next experiments.
Intranasal Vaccine Protects against SARS-CoV-2 Variants
Having established the effectiveness of i.n. vaccination, we investigated whether the the i.n. vaccination could protect mice against infection with different variants of of concerns (VOC) isolated from patients with COVID-19. For this, transgenic hACE2 mice received i.n. vaccination or PBS on days 0 and 7 and two weeks later the animals were challenged i.n. with different variants of active SARS-CoV-2 virus as depicted in Figure 3A. As shown in Figure 3B, mice from the non-vaccinated group infected with the gamma variant started to lose weight 3 days after viral infection, reaching 20% of weight loss at DPI-5. In addition, 60% of mortality was observed in this group of non-vaccinated animals at DPI-5, and by DPI-6 all animals from the group had died (Figure 3D). In contrast, intranasally vaccinated mice did not show weight loss nor mortality after the infection with the gamma variant (Figure 3B,D). Similar results were obtained when mice were infected with the delta variant of SARS-CoV-2 (Figure 3C,E). Again, non-vaccinated animals presented significant weight loss and high mortality (80% by DPI-6), while i.n. vaccinated mice did not present weight loss or mortality after the infection with the delta variant. Regarding the Omicron strain, we only measured the viral load in the lung of vaccinated and non-vaccinated animals since this variant does not induce weight loss or mortality in hACE2 mice [29]. We found that vaccinated mice presented significantly lower viral load in the lung than non-vaccinated animals at DPI-4 (Supplementary Figure S1C). We conclude that i.n. vaccination afforded protections against all VOC tested.
Intranasal Vaccine Boosts Heterologous Immunity and Is More Effective Than Homologous Oxford/AstraZeneca (AZ) Vaccine Boost
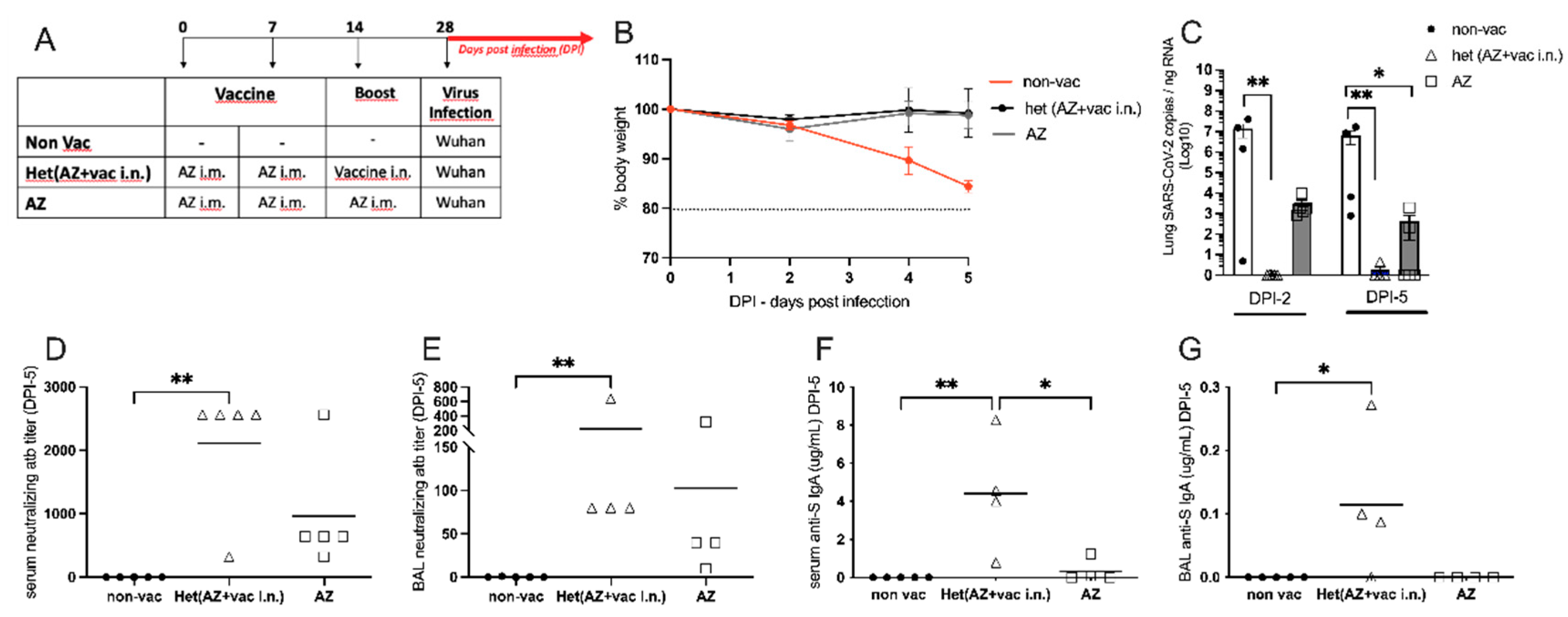
Considering that most of the world population has been vaccinated for COVID-19, we were interested in testing the efficiency of our nasal vaccine formulation to boost previous immunity (heterologous immunity) induced by Oxford/AstraZeneca i.m. vaccination when compared with an AZ i.m. homologous booster. For this, hACE2 transgenic mice received 2 i.m. doses of AZ vaccine on days 0 and 7 and were boosted with the i.n. vaccine formulation or with the i.m. AZ vaccine on day 14. One week after the boost, animals were infected with SARS-CoV-2 as depicted in Figure 5A. We found that the non-vaccinated group lost weight after viral infection, while both vaccinated groups maintained stable body weight after the infection with SARS-CoV-2 (Figure 5B), confirming that vaccination with the AZ formulation protects against infection. Next, we determined the viral load in the lungs 2 days or 5 days after virus infection. We found that at DPI-2 no virus was detected in mice that received the i.n. vaccine boost, being significantly different from the non-vaccinated group in Figure 5C. Conversely, all animals that received the AZ boost presented virus in the lung at DPI-2, at levels of viral load that were not significantly different from the non-vaccinated group (Figure 5C left). At DPI-5, although AZ-boosted mice had significantly less virus than the non-vaccinated group, 40% of the mice in the AZ-boosted group still had SARS-CoV-2 mRNA present in the lungs (Figure 5C right). These results indicate that the heterologous i.n. boost is more efficient than the homologous i.m. boost in virus clearance from the lungs. In the same vein, the titers of SARS-CoV-2 neutralizing antibodies in the serum and BAL were significantly higher in animals that received the i.n. boost when compared to the non-vaccinated group (Figure5D,E). Of note, although neutralizing antibodies were detected in AZ-boosted animals, they were not statistically different from the non-vaccinated group (Figure 5D,E). Finally, animals that received the i.n. boost produced significantly higher levels of anti-S IgA antibodies when compared to the AZ-vaccinated or non-vaccinated groups (Figure 5F,G).
Altogether these results show that AZ-vaccinated animals that received a heterologous boost with the i.n. vaccine formulation cleared the virus more rapidly and had higher titers of neutralizing antibodies and S-specific IgA in serum and the BAL than mice that received a homologous AZ vaccine boost. Therefore, the heterologous i.n. boost induces a more effective anti-SARS-CoV-2 immunity than the homologous AZ boost.
Discussion
Here we have developed a COVID-19 vaccine formulation using the SARS-CoV-2 trimeric spike protein and CpG type C (2395) entrapped in a cationic liposome. We designed this type of formulation to boost both humoral and cellular immunity, based on our previous works with allergen-specific immunotherapy with CpG using the OVA asthma model [28,29]. Also, it was shown that mice immunized with two doses of recombinant virus antigens mixed with cationic adjuvants showed higher serum IgG titers than animals treated with anionic adjuvants [30]. Moreover, lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust follicular T-helper cell and humoral responses [13]. Also, our formulation contained CpG type C, which is a potent inducer of IFN-α, a strong B-cell activator in human and mice [33] and a potent Th1 adjuvant [34].
We found that our vaccine formulation was devoid of toxicity either in cell cultures with different cell types, including human bronchial cells, or in preclinical experiments in CD1 mice (data not shown). We tested the efficacy of our candidate vaccine in hACE2 transgenic mice infected with the wild-type Wuhan strain of SARS-CoV-2, and compared the results obtained with s.c. versus i.n. administration. Regarding survival and body weight lost, non-vaccinated animals succumbed to infection while vaccination by both routes prevented viral spread into distal areas of the brain and protected all infected animals. These results are in line with other studies showing the effectiveness of different vaccines formulations administered by different routes [35] and indicate that our vaccine candidate is effective irrespective of the administration route. However, major differences between s.c. and i.n. vaccination were observed regarding lung viral load, anti-S IgA and IgG2c titers and levels of neutralizing antibodies in serum. All these parameters of humoral immunity were superior in animals vaccinated by the i.n. route. The enhanced IgA production might reflect the increased antibody neutralizing activity since it was shown that IgA dominates the early neutralizing antibody response to SARS-CoV-2 [8] while the increased IgG2c production might be associated with enhanced Th1 immunity [36]. It was expected that i.n. vaccination would increase the IgA production since mucosal delivery of vaccines targets the inductive sites for IgA responses in mucosal surfaces [37,38,39]. In addition, CpG activates the adaptor protein MyD88 that boosts IgA production (Suzuki et al., 2010). In the same vein, CpG also boosts IgG2c production [31]. However, it remains to be determined why i.n. vaccination induced higher titers of IgG2c than s.c. vaccination.
Experimental evidence indicates that vaccine-induced immunity to SARS-CoV-2 infection in hACE2 transgenic mice relies on humoral immunity and/or on T cell-mediated immunity [11]. Since our nasal vaccination also afforded protection against infections with VOC that are known to be more resistant to neutralizing antibodies [38], it is likely that our vaccine also induced T cell immunity. In line with this assumption, it was shown that an intranasal COVID-19 vaccine induced respiratory memory T cells and protected K18-hACE mice against SARS-CoV-2 infection [39].
We found that intranasal vaccination was superior to the intramuscular vaccination with the Oxford/AstraZeneca vaccine regarding virus clearance from the lung and production of neutralizing antibodies in serum and BAL. Finally, the intranasal liposomal formulation prevented neuroinflammation and boosted heterologous immunity induced by previous vaccination with the AZ vaccine.
The durability and breadth of our candidate vaccine remains to be determined, but it has been shown previously that nanoparticle-conjugated TLR9 agonists improve the potency, durability, and breadth of COVID-19 vaccines [40].
We conclude that our vaccine formulation is easy to be manufactured worldwide. This type of vaccine formulation could be adapted to be used against VOC or different infectious agents that cause pulmonary disease or to boost immunity induced by other vaccines.
Authors Contribution
Luciana Mirotti and Momtchilo Russo: conception, coordination, analysis, writing and review; Maria Cássia Mendes-Corrêa: coordination of virology unit at IMT-FMUSP; Mario H. Hirata: coordination of animal care at BS3 unit at FCF-USP; Bruna B. Lins, Victor Kersten, Livia Dati, Brisa M. Gomes: performed experiments at the animal care facility at ICB-USP and at BSL-3 unit at FCF-USP and ICB-USP, Paulo C. A. Pernambuco Filho ELISAs for antibodies at ICB-USP; Toni Ricardo Martins, Tânia R. Tozetto-Mendoza, Lucy S. Vilas Boas, Anderson V. de Paula: generation of virus SARS CoV2 strains, determination of neutralizing antibodies and virus titers at IMT-FMUSP. Amaro Duarte Neto: detection of virus antigens by immunochemistry at FMUSP; Gustavo R. Reigado, Felipe S. Chambergo, Viviane A. N. C. Dantas, in vitro cytotoxic test at EACH-USP; Ana P. Ano Bom, Ana B. T. Frederico, Danielle R. de A. de Brito e Cunha: physicochemical characterization of vaccine formulation at Bio-Manguinhos, Fiocruz; Rodrigo A. P. Martins, José I. G. da Silva, Carlos F. M. Vasconcelos: studies in the brain at ICB-UFRJ; Leda R. Castilho: production of Spike protein at COPPE-UFRJ. All authors discussed the results, contributed to the article and approved the submitted version.
Funding
This study received funds from: Programa Inova Fiocruz (INOVA VPPCB-005-FIO-20-2-54); Verba parlamentar Davi Miranda (39540021); PIPAE-USP-2021 2021.1.10424.1.9.; Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (E-26/211.711/2021 to R.A.P.M.); International Retinal Research Foundation/2021 (to R.A.P.M.); Grant Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP) 2021/12502-7
Acknowledgments
We would like to thank the great support received from Christoph Milewski, Wanise Barroso, Isabel M. A. Freire and Fabienne P. de Paiva from ICTB, Fiocruz. Also thank João Calixto and his team from CIEnP, Prof. Carsten Wrenger and his team from ICB- USP; Felipe Rodrigues da Silva and Carlos Eduardo de Andrade Lima da Rocha from Plataforma Internacional de Ciência, Tecnologia e Inovação em Saúde (PICTIS), Fiocruz, and Jessica Santana, INOVA/Fiocruz scholarship holder.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Seyed Hosseini, E.; Riahi Kashani, N.; Nikzad, H.; Azadbakht, J.; Hassani Bafrani, H.; Haddad Kashani, H. The Novel Coronavirus Disease-2019 (COVID-19): Mechanism of Action, Detection and Recent Therapeutic Strategies. Virology 2020, 551, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Schwetz, T.A.; Tabak, L.A.; Lander, E.S. ARPA-H: Accelerating Biomedical Breakthroughs. Science (1979) 2021, 373, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Harder, T.; Külper-Schiek, W.; Reda, S.; Treskova-Schwarzbach, M.; Koch, J.; Vygen-Bonnet, S.; Wichmann, O. Effectiveness of COVID-19 Vaccines against SARS-CoV-2 Infection with the Delta (B.1.617.2) Variant: Second Interim Results of a Living Systematic Review and Meta-Analysis, 1 January to Literature Search. 1. 25 August. [CrossRef]
- Slavov, S.N.; de La-Roque, D.G.L.; da Costa, P.N.M.; Rodrigues, E.S.; Santos, E.V.; Borges, J.S.; Evaristo, M.; de Matos Maçonetto, J.; Marques, A.A.; Milhomens, J.; et al. Dynamics of SARS-CoV-2 Variants of Concern in Vaccination Model City in the State of Sao Paulo, Brazil. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.G.; Lustig, Y.; Cohen, C.; Fluss, R.; Indenbaum, V.; Amit, S.; Doolman, R.; Asraf, K.; Mendelson, E.; Ziv, A.; et al. Waning Immune Humoral Response to BNT162b2 Covid-19 Vaccine over 6 Months. N. Engl. J. Med. 2021, 385, e84. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Muecksch, F.; Muenn, F.; Cho, A.; Zong, S.; Raspe, R.; Ramos, V.; Johnson, B.; Tanfous, T. Ben; Dasilva, J.; et al. Humoral Immunity to SARS-CoV-2 Elicited by Combination COVID-19 Vaccination Regimens. J. Exp. Med. 2022, 219. [Google Scholar] [CrossRef]
- Ogra, P.L. Mucosal Immune Response to Poliovirus Vaccines in Childhood. Rev. Infect. Dis. 1984, 6, S361–S368. [Google Scholar] [CrossRef]
- Sterlin, D.; Mathian, A.; Miyara, M.; Mohr, A.; Anna, F.; Claër, L.; Quentric, P.; Fadlallah, J.; Devilliers, H.; Ghillani, P.; et al. IgA Dominates the Early Neutralizing Antibody Response to SARS-CoV-2. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Hassan, A.O.; Shrihari, S.; Gorman, M.J.; Ying, B.; Yaun, D.; Raju, S.; Chen, R.E.; Dmitriev, I.P.; Kashentseva, E.; Adams, L.J.; et al. An Intranasal Vaccine Durably Protects against SARS-CoV-2 Variants in Mice. Cell Rep. 2021, 36. [Google Scholar] [CrossRef]
- Afkhami, S.; D’Agostino, M.R.; Zhang, A.; Stacey, H.D.; Marzok, A.; Kang, A.; Singh, R.; Bavananthasivam, J.; Ye, G.; Luo, X.; et al. Respiratory Mucosal Delivery of Next-Generation COVID-19 Vaccine Provides Robust Protection against Both Ancestral and Variant Strains of SARS-CoV-2. Cell 2022, 185, 896–915. [Google Scholar] [CrossRef]
- Castro, J.T.; Azevedo, P.; Fumagalli, M.J.; Hojo-Souza, N.S.; Salazar, N.; Almeida, G.G.; Oliveira, L.I.; Faustino, L.; Antonelli, L.R.; Marçal, T.G.; et al. Promotion of Neutralizing Antibody-Independent Immunity to Wild-Type and SARS-CoV-2 Variants of Concern Using an RBD-Nucleocapsid Fusion Protein. Nat. Commun. 2022, 13. [Google Scholar] [CrossRef]
- Israelow, B.; Mao, T.; Klein, J.; Song, E.; Menasche, B.; Omer, S.B.; Iwasaki, A. Adaptive Immune Determinants of Viral Clearance and Protection in Mouse Models of SARS-CoV-2. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Alameh, M.G.; Tombácz, I.; Bettini, E.; Lederer, K.; Sittplangkoon, C.; Wilmore, J.R.; Gaudette, B.T.; Soliman, O.Y.; Pine, M.; Hicks, P.; et al. Lipid Nanoparticles Enhance the Efficacy of MRNA and Protein Subunit Vaccines by Inducing Robust T Follicular Helper Cell and Humoral Responses. Immunity 2021, 54, 2877–2892. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Maruya, M.; Kawamoto, S.; Sitnik, K.; Kitamura, H.; Agace, W.W.; Fagarasan, S. The Sensing of Environmental Stimuli by Follicular Dendritic Cells Promotes Immunoglobulin A Generation in the Gut. Immunity 2010, 33, 71–83. [Google Scholar] [CrossRef]
- Bao, L.; Deng, W.; Huang, B.; Gao, H.; Liu, J.; Ren, L.; Wei, Q.; Yu, P.; Xu, Y.; Qi, F.; et al. The Pathogenicity of SARS-CoV-2 in HACE2 Transgenic Mice. Nature 2020, 583. [Google Scholar] [CrossRef] [PubMed]
- Alvim, R.G.F.; Lima, T.M.; Rodrigues, D.A.S.; Marsili, F.F.; Bozza, V.B.T.; Higa, L.M.; Monteiro, F.L.; Abreu, D.P.B.; Leitão, I.C.; Carvalho, R.S.; et al. From a Recombinant Key Antigen to an Accurate, Affordable Serological Test: Lessons Learnt from COVID-19 for Future Pandemics. Biochem. Eng. J. 2022, 186, 108537. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; Mclellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020. [Google Scholar] [CrossRef]
- Reigado, G.R.; Adriani, P.P.; dos Santos, J.F.; Freitas, B.L.; Fernandes, M.T.P.; Chambergo Alcalde, F.S.; Leo, P.; Nunes, V.A. Delivery of Superoxide Dismutase by TAT and Abalone Peptides for the Protection of Skin Cells against Oxidative Stress. Biotechnol. Appl. Biochem. 2022, 69, 2673–2685. [Google Scholar] [CrossRef]
- Präbst, K.; Engelhardt, H.; Ringgeler, S.; Hübner, H. Basic Colorimetric Proliferation Assays: MTT, WST, and Resazurin. In Methods in Molecular Biology; Humana Press Inc., 2017; Vol. 1601, pp. 1–17. [CrossRef]
- McCray, P.B.; Pewe, L.; Wohlford-Lenane, C.; Hickey, M.; Manzel, L.; Shi, L.; Netland, J.; Jia, H.P.; Halabi, C.; Sigmund, C.D.; et al. Lethal Infection of K18- HACE2 Mice Infected with Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2007, 81, 813–821. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Pan, Y.; Cheng, S.M.S.; Hui, K.P.Y.; Krishnan, P.; Liu, Y.; Ng, D.Y.M.; Wan, C.K.C.; Yang, P.; Wang, Q.; et al. Molecular Diagnosis of a Novel Coronavirus (2019-NCoV) Causing an Outbreak of Pneumonia. Clin Chem 2020, 66, 549–555. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.W.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 Novel Coronavirus (2019-NCoV) by Real-Time RT-PCR. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef]
- Mendes-Correa, M.C.; Salomão, M.C.; Ghilardi, F.; Tozetto-Mendoza, T.R.; Santos Villas-Boas, L.; de Paula, A.V.; Paiao, H.G.O.; da Costa, A.C.; Leal, F.E.; Ferraz, A. de B.C.; et al. SARS-CoV-2 Detection and Culture in Different Biological Specimens from Immunocompetent and Immunosuppressed COVID-19 Patients Infected with Two Different Viral Strains. Viruses 2023, 15, 1270. [Google Scholar] [CrossRef] [PubMed]
- Nurtop, E.; Villarroel, P.M.S.; Pastorino, B.; Ninove, L.; Drexler, J.F.; Roca, Y.; Gake, B.; Dubot-Peres, A.; Grard, G.; Peyrefitte, C.; et al. Combination of ELISA Screening and Seroneutralisation Tests to Expedite Zika Virus Seroprevalence Studies. Virol J 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- Wendel, S.; Kutner, J.M.; Machado, R.; Fontão-Wendel, R.; Bub, C.; Fachini, R.; Yokoyama, A.; Candelaria, G.; Sakashita, A.; Achkar, R.; et al. Screening for SARS-CoV-2 Antibodies in Convalescent Plasma in Brazil: Preliminary Lessons from a Voluntary Convalescent Donor Program. Transfusion (Paris) 2020, 60, 2938–2951. [Google Scholar] [CrossRef] [PubMed]
- Mendrone-Junior, A.; Dinardo, C.L.; Ferreira, S.C.; Nishya, A.; Salles, N.A.; de Almeida Neto, C.; Hamasaki, D.T.; Facincani, T.; de Oliveira Alves, L.B.; Machado, R.R.G.; et al. Correlation between SARS-COV-2 Antibody Screening by Immunoassay and Neutralizing Antibody Testing. Transfusion (Paris) 2021, 61, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Villas-Boas, L.S.; Paula, A.V.; Silva, A.R.D.; Jr, A.; Paiao, H.G.O.; Tozetto-Mendoza, T.R.; Manuli, E.R.; Leal, F.E.; Ferraz, A.B.C.; Sabino, E.C.; et al. Absence of Neutralizing Antibodies against the Omicron SARS-CoV-2 Variant in Convalescent Sera from Individuals Infected with the Ancestral SARS-CoV-2 Virus or Its Gamma Variant. Clinics 2022. [Google Scholar] [CrossRef]
- Zhu, Y.; Chidekel, A.; Shaffer, T.H. 3. Cultured Human Airway Epithelial Cells (Calu-3): A Model of Human Respiratory Function, Structure, and Inflammatory Responses. Crit Care Res Pract 2010, 2010. [Google Scholar] [CrossRef]
- Halfmann, P.J.; Iida, S.; Iwatsuki-Horimoto, K.; Maemura, T.; Kiso, M.; Scheaffer, S.M.; Darling, T.L.; Joshi, A.; Loeber, S.; Singh, G.; et al. SARS-CoV-2 Omicron Virus Causes Attenuated Disease in Mice and Hamsters. Nature 2022, 603, 687–692. [Google Scholar] [CrossRef]
- Alberca-Custodio, R.W.; Faustino, L.D.; Gomes, E.; Nunes, F.P.B.; Siqueira, M.K.; Labrada, A.; Almeida, R.R.; Camara, N.O.S.; Fonseca, D.M.; Russo, M. Allergen-Specific Immunotherapy With Liposome Containing CpG-ODN in Murine Model of Asthma Relies on MyD88 Signaling in Dendritic Cells Allergen-Specific Immunotherapy With Liposome Containing CpG-ODN in Murine Model of Asthma Relies on MyD88 Signaling in Dendritic Cells. Front. Immunol 2020, 11, 692. [Google Scholar] [CrossRef]
- Mirotti, L.; Custódio, R.W. A.; Gomes, E.; Rammauro, F.; de Araujo, E.F.; Calich, V.L.G.; Russo,M. CPG-ODN Shapes Alum Adjuvant Activity Signaling via MyD88 and Il-10. Front Immunol 2017, 8, 47. [Google Scholar] [CrossRef]
- Sengupta, A.; Azharuddin, M.; Cardona, M.E.; Devito, C.; von Castelmur, E.; Wehlin, A.; Pietras, Z.; Sunnerhagen, M.; Selegård, R.; Aili, D.; et al. Intranasal Coronavirus SARS-CoV-2 Immunization with Lipid Adjuvants Provides Systemic and Mucosal Immune Response against SARS-CoV-2 S1 Spike and Nucleocapsid Protein. Vaccines 2022, 10, 504. [Google Scholar] [CrossRef]
- Hartmann, G.; Battiany, J.; Poeck, H.; Wagner, M.; Kerkmann, M.; Lubenow, N.; Rothenfusser, S.; Endres, S. Rational Design of New CpG Oligonucleotides That Combine B Cell Activation with High IFN-α Induction in Plasmacytoid Dendritic Cells. Eur J Immunol 2003, 33, 1633–1641. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, J.; Weeratna, R.; Payette, P.; Jurk, M.; Schetter, C.; Laucht, M.; Wader, T.; Tluk, S.; Liu, M.; Davis, H.L.; et al. Characterization of Three CpG Oligodeoxynucleotide Classes with Distinct Immunostimulatory Activities. Eur J Immunol 2004, 34, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Firmino-Cruz, L.; dos-Santos, J.S.; da Fonseca-Martins, A.M.; Oliveira-Maciel, D.; Guadagnini-Perez, G.; Roncaglia-Pereira, V.A.; Dumard, C.H.; Guedes-da-Silva, F.H.; Vicente Santos, A.C.; Alvim, R.G.F.; et al. Intradermal Immunization of SARS-CoV-2 Original Strain Trimeric Spike Protein Associated to CpG and AddaS03 Adjuvants, but Not MPL, Provide Strong Humoral and Cellular Response in Mice. Vaccines (Basel) 2022, 10. [Google Scholar] [CrossRef]
- Rivera-Hernandez, T.; Rhyme, M.S.; Cork, A.J.; Jones, S.; Segui-Perez, C.; Brunner, L.; Richter, J.; Petrovsky, N.; Lawrenz, M.; Goldblatt, D.; et al. Vaccine-Induced Th1-Type Response Protects against Invasive Group a Streptococcus Infection in the Absence of Opsonizing Antibodies. mBio 2020, 11. [Google Scholar] [CrossRef]
- Boyaka, P.N. Inducing Mucosal IgA: A Challenge for Vaccine Adjuvants and Delivery Systems. J. Immunol. 2017, 199, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. Mucosal Immunity: Induction, Dissemination, and Effector Functions. Scand J Immunol 2009, 70, 505–515. [Google Scholar] [CrossRef]
- Hand, T.W.; Reboldi, A. Annual Review of Immunology Production and Function of Immunoglobulin A. Annu. Rev. Immunol. 2021 2021, 39, 695–718. [Google Scholar] [CrossRef]
- Suzuki, K.; Maruya, M.; Kawamoto, S.; Sitnik, K.; Kitamura, H.; Agace, W.W.; Fagarasan, S. The Sensing of Environmental Stimuli by Follicular Dendritic Cells Promotes Immunoglobulin A Generation in the Gut. Immunity 2010, 33, 71–83. [Google Scholar] [CrossRef]
- Castro, J.T.; Azevedo, P.; Fumagalli, M.J.; Hojo-Souza, N.S.; Salazar, N.; Almeida, G.G.; Oliveira, L.I.; Faustino, L.; Antonelli, L.R.; Marçal, T.G.; et al. Promotion of Neutralizing Antibody-Independent Immunity to Wild-Type and SARS-CoV-2 Variants of Concern Using an RBD-Nucleocapsid Fusion Protein. Nat Commun 2022, 13. [Google Scholar] [CrossRef]
- Zhang, G.F.; Meng, W.; Chen, L.; Ding, L.; Feng, J.; Perez, J.; Ali, A.; Sun, S.; Liu, Z.; Huang, Y.; et al. Neutralizing Antibodies to SARS-CoV-2 Variants of Concern Including Delta and Omicron in Subjects Receiving MRNA-1273, BNT162b2, and Ad26.COV2.S Vaccines. J Med Virol 2022, 94, 5678–5690. [Google Scholar] [CrossRef]
- Diallo, B.K.; Chasaide, C.N.; Wong, T.Y.; Schmitt, P.; Lee, K.S.; Weaver, K.; Miller, O.; Cooper, M.; Jazayeri, S.D.; Damron, F.H.; et al. Intranasal COVID-19 Vaccine Induces Respiratory Memory T Cells and Protects K18-HACE Mice against SARS-CoV-2 Infection. Vaccines (Basel) 2023. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.S.; Picece, V.C.T.M.; Baillet, J.; Gale, E.C.; Powelll, A.E.; Saouaf, O.M.; Yan, J.; Lopez Hernandez, H.; Appel, E.A. Nanoparticle-Conjugated TLR9 Agonists Improve the Potency, Durability, and Breadth of COVID-19 Vaccines. bioRxiv 2023. [Google Scholar] [CrossRef]
Figure 1.
- Cytotoxicity assays for vaccine formulation. (A-F) Incubation of different cell types with nasal vaccine formulation (grey bars), PBS – negative controls (white bars) or DMSO – positive controls (black bars). Axis X indicates de percentage of vaccine formulation, PBS or DMSO present in the wells. (A) Fibroblasts 3T3 incubated for 24h; (B) Fibroblasts 3T3 incubated for 48h; (C) CALU-3 – human lung adenocarcinoma cells - incubated for 24h, (D) CALU-3 – human lung adenocarcinoma cells - incubated for 48h; (E) Human lung fibroblasts incubated for 24h; (F) Rat pneumocytes were incubated with vaccine formulation for 24 h. Cytotoxicity assays were performed by resazurin method. Cultures incubate with 10% DMSO were positive controls for cell death. Evaluation of the physicochemical characteristics of different formulations, using the dynamic light scattering (DLS) technique (G) Hydrodynamic size (nm), (H) Charge (zeta potential - mV), (I) Polydispersity index (PDI). (A-F) The data are presented as the mean ± SE of four individual experiments (n=3) in sextuplicates. (G-I) Results were measured in three replicates which are reported as mean ± SE. * p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 1.
- Cytotoxicity assays for vaccine formulation. (A-F) Incubation of different cell types with nasal vaccine formulation (grey bars), PBS – negative controls (white bars) or DMSO – positive controls (black bars). Axis X indicates de percentage of vaccine formulation, PBS or DMSO present in the wells. (A) Fibroblasts 3T3 incubated for 24h; (B) Fibroblasts 3T3 incubated for 48h; (C) CALU-3 – human lung adenocarcinoma cells - incubated for 24h, (D) CALU-3 – human lung adenocarcinoma cells - incubated for 48h; (E) Human lung fibroblasts incubated for 24h; (F) Rat pneumocytes were incubated with vaccine formulation for 24 h. Cytotoxicity assays were performed by resazurin method. Cultures incubate with 10% DMSO were positive controls for cell death. Evaluation of the physicochemical characteristics of different formulations, using the dynamic light scattering (DLS) technique (G) Hydrodynamic size (nm), (H) Charge (zeta potential - mV), (I) Polydispersity index (PDI). (A-F) The data are presented as the mean ± SE of four individual experiments (n=3) in sextuplicates. (G-I) Results were measured in three replicates which are reported as mean ± SE. * p < 0.05, ** p < 0.01, *** p < 0.001.
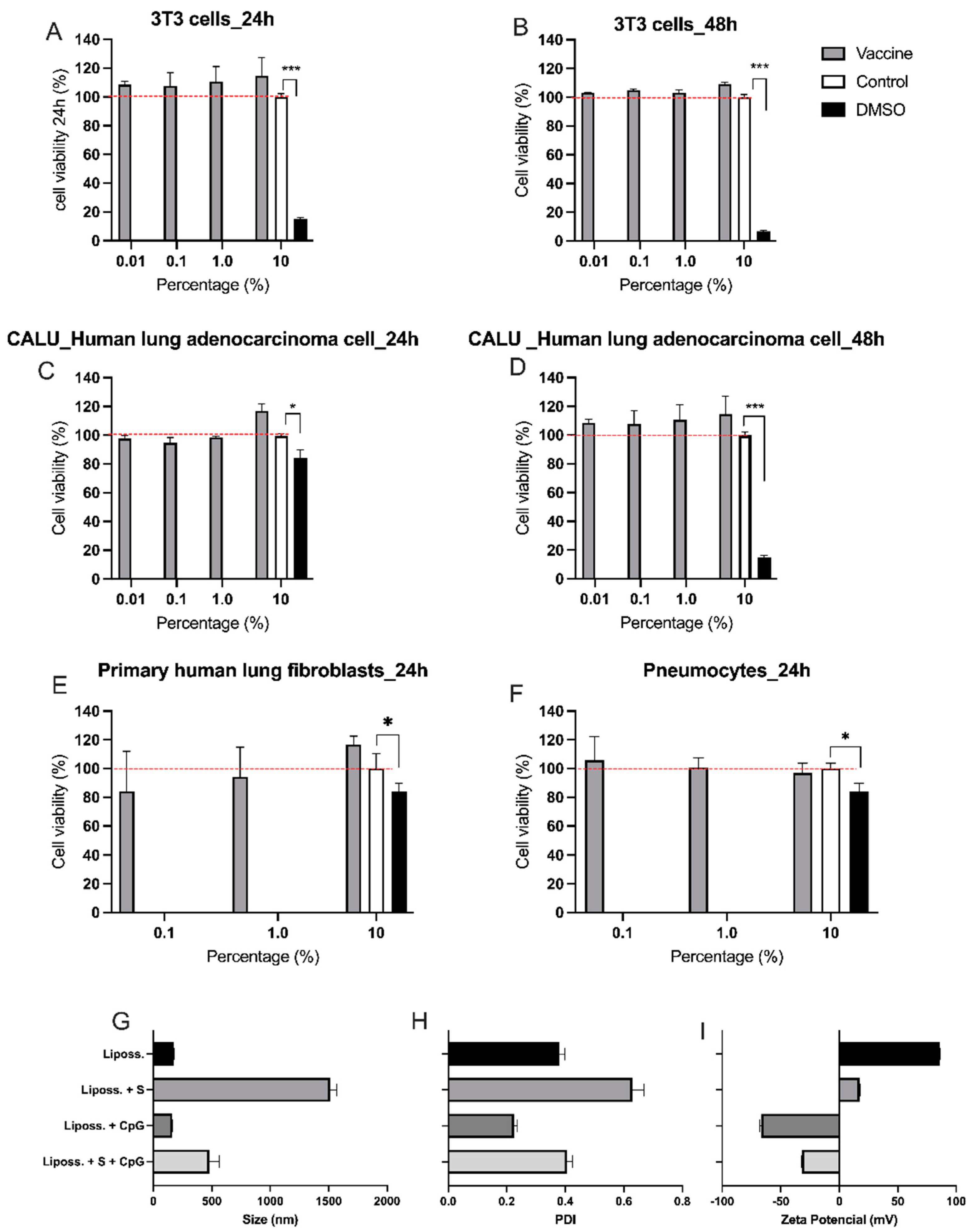
Figure 2.
- Intranasal versus subcutaneous vaccinations (A) Protocol design: K18-hACE2 mice were vaccinated via intra-nasal (vac i.n.) or via sub-cutaneous (vac s.c.) with the nasal vaccine formulation on days 0 and 7. Non-vaccinated mice (non-vac) received only PBS. All mice were infected intra-nasally with SARS-CoV-2 (Wuhan strain) 21 days after the first dose; (B) Survival rate following the virus infection (DPI- days after infection) represented by a Kaplan-Meier survival curve; (C) Body weight during the course of infection plotted as percent (100% representing the body weight on DPI-0, before the virus infection). (B and C) Red line represents non-vaccinated mice, gray line represents subcutaneously vaccinated mice and black line represents mice vaccinated intranasally; (D) RNA was isolated from the lungs and SARS-CoV-2 was quantitated by RT-qPCR measuring E-gene log copies number per ng RNA at two and four days after viral infection, DPI-2 and DPI-4 respectively (E) RNA was isolated from distinct brain regions (olfactory bulb and hippocampus) and SARS-CoV-2 was quantitated by RT-PCR measuring E-gene log copies number per ng RNA four days after viral infection (DPI-4); (F) Concentration of spike protein (S)-specific IgG1 in serum measured by ELISA; (G) Concentration of S-specific IgG2c in serum measured by ELISA; (H) Concentration of S-specific IgA in serum measured by ELISA; (I) Titers of SARS-CoV-2 neutralizing antibodies in serum measured by VNT, two and four days after virus infection (DPI-2 an DPI-4). N=5 per group; * p < 0.05, ** p < 0.01.
Figure 2.
- Intranasal versus subcutaneous vaccinations (A) Protocol design: K18-hACE2 mice were vaccinated via intra-nasal (vac i.n.) or via sub-cutaneous (vac s.c.) with the nasal vaccine formulation on days 0 and 7. Non-vaccinated mice (non-vac) received only PBS. All mice were infected intra-nasally with SARS-CoV-2 (Wuhan strain) 21 days after the first dose; (B) Survival rate following the virus infection (DPI- days after infection) represented by a Kaplan-Meier survival curve; (C) Body weight during the course of infection plotted as percent (100% representing the body weight on DPI-0, before the virus infection). (B and C) Red line represents non-vaccinated mice, gray line represents subcutaneously vaccinated mice and black line represents mice vaccinated intranasally; (D) RNA was isolated from the lungs and SARS-CoV-2 was quantitated by RT-qPCR measuring E-gene log copies number per ng RNA at two and four days after viral infection, DPI-2 and DPI-4 respectively (E) RNA was isolated from distinct brain regions (olfactory bulb and hippocampus) and SARS-CoV-2 was quantitated by RT-PCR measuring E-gene log copies number per ng RNA four days after viral infection (DPI-4); (F) Concentration of spike protein (S)-specific IgG1 in serum measured by ELISA; (G) Concentration of S-specific IgG2c in serum measured by ELISA; (H) Concentration of S-specific IgA in serum measured by ELISA; (I) Titers of SARS-CoV-2 neutralizing antibodies in serum measured by VNT, two and four days after virus infection (DPI-2 an DPI-4). N=5 per group; * p < 0.05, ** p < 0.01.
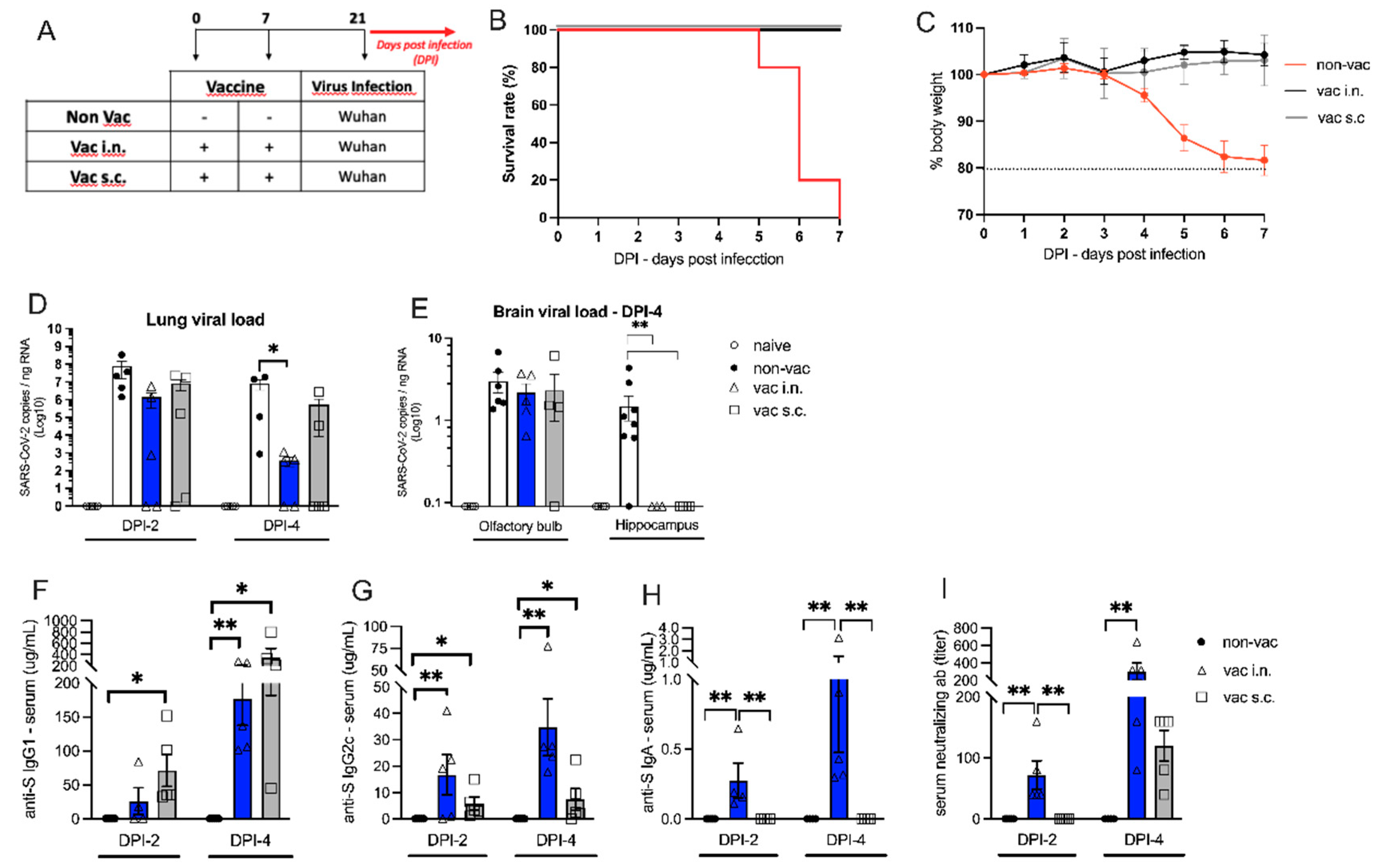
Figure 3.
- Different SARS-CoV-2 variants. (A) Protocol design: K18-hACE2 mice were vaccinated via intra-nasal (vac i.n.) with the nasal vaccine formulation on days 0 and 7. Non-vaccinated mice (non-vac) received PBS. All mice were infected intra-nasally with different SARS-CoV-2 VOC (Gamma or Delta strains) 21 days after the first dose. (B and C) Body weight during the course of infection plotted as percent (100% representing the body weight on DPI-0, before the virus infection), (B) animals infected with Gamma variant of concern (VOC), (C) animals infected with Delta VOC. (D and E) Survival rate following the with virus infection (DPI – days after infection) represented by a Kaplan-Meier survival curve: (D) animals infected Gamma VOC, (E) animals infected Delta VOC. N=5 per group.
Figure 3.
- Different SARS-CoV-2 variants. (A) Protocol design: K18-hACE2 mice were vaccinated via intra-nasal (vac i.n.) with the nasal vaccine formulation on days 0 and 7. Non-vaccinated mice (non-vac) received PBS. All mice were infected intra-nasally with different SARS-CoV-2 VOC (Gamma or Delta strains) 21 days after the first dose. (B and C) Body weight during the course of infection plotted as percent (100% representing the body weight on DPI-0, before the virus infection), (B) animals infected with Gamma variant of concern (VOC), (C) animals infected with Delta VOC. (D and E) Survival rate following the with virus infection (DPI – days after infection) represented by a Kaplan-Meier survival curve: (D) animals infected Gamma VOC, (E) animals infected Delta VOC. N=5 per group.
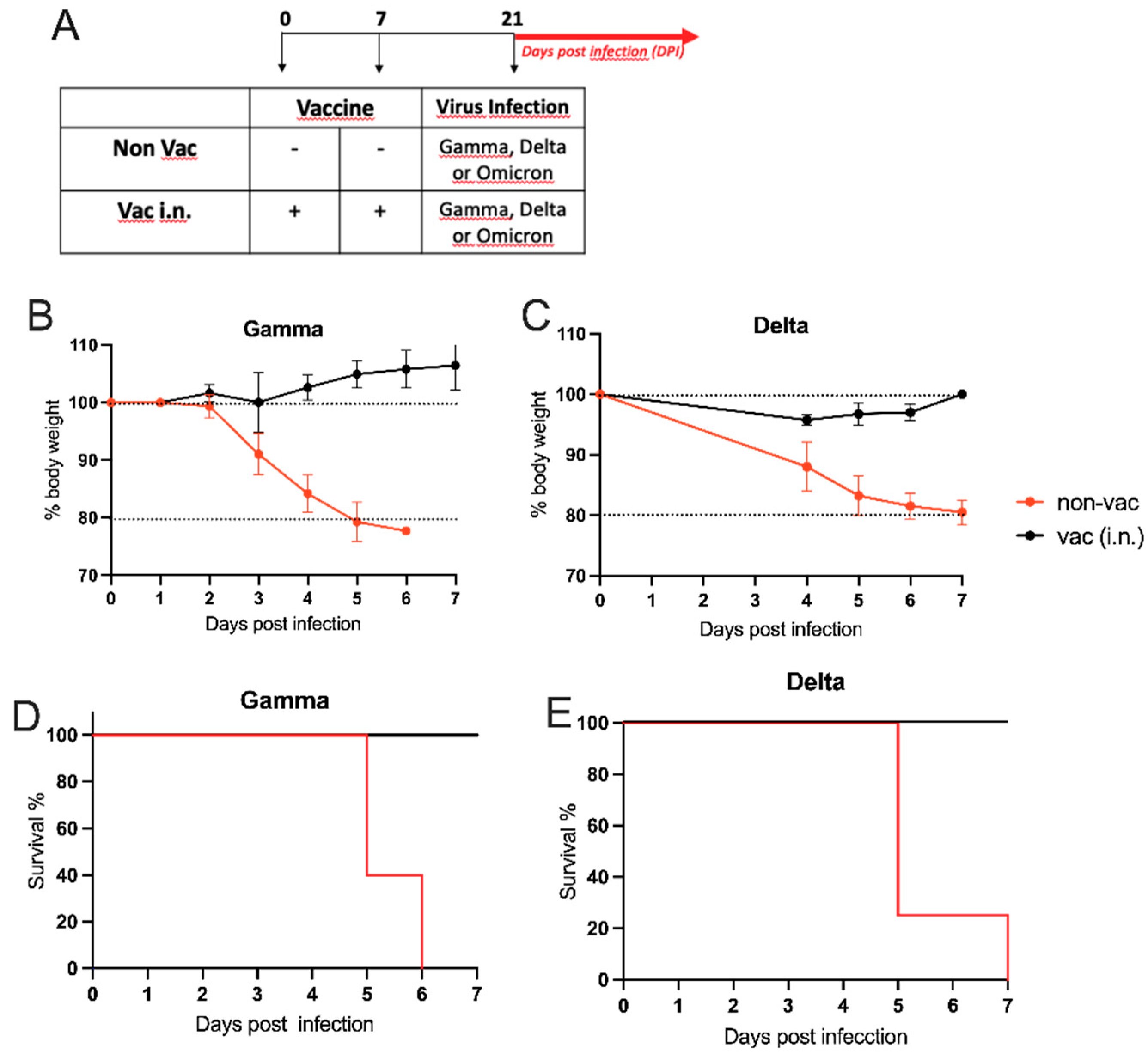
Figure 4.
- Nasal formulation versus Oxford/AstraZeneca vaccination (A) Protocol design: K18-hACE2 mice were vaccinated on days 0 and 7 with nasal vaccine formulation via intra-nasal (vac i.n.) or vaccinated with Oxford/AstraZeneca vaccine via intramuscular (AZ). Non-vaccinated mice (non-vac) received PBS. All mice were infected intra-nasally with SARS-CoV-2 (Wuhan strain) 21 days following the first dose. All samples collected four days after infection (DPI-4). (B) Body weight during the course of infection plotted as percent change (100% representing the body weight on DPI-0, before the virus infection); (C) RNA was isolated from the lungs and SARS-CoV-2 was quantitated by RT-qPCR measuring E-gene log copies number per ng RNA; (D) Titers of SARS-CoV-2 neutralizing antibodies in serum measured by VNT; (E) Concentration of Spike (S)-specific IgA in serum measured by ELISA; (F) Titers of SARS-CoV-2 neutralizing antibodies in BAL (broncho-alveolar lavage) measured by VNT; (G) Concentration of S-specific IgA in BAL measured by ELISA. N=5 per group; ** p < 0.05, ** p < 0.01.
Figure 4.
- Nasal formulation versus Oxford/AstraZeneca vaccination (A) Protocol design: K18-hACE2 mice were vaccinated on days 0 and 7 with nasal vaccine formulation via intra-nasal (vac i.n.) or vaccinated with Oxford/AstraZeneca vaccine via intramuscular (AZ). Non-vaccinated mice (non-vac) received PBS. All mice were infected intra-nasally with SARS-CoV-2 (Wuhan strain) 21 days following the first dose. All samples collected four days after infection (DPI-4). (B) Body weight during the course of infection plotted as percent change (100% representing the body weight on DPI-0, before the virus infection); (C) RNA was isolated from the lungs and SARS-CoV-2 was quantitated by RT-qPCR measuring E-gene log copies number per ng RNA; (D) Titers of SARS-CoV-2 neutralizing antibodies in serum measured by VNT; (E) Concentration of Spike (S)-specific IgA in serum measured by ELISA; (F) Titers of SARS-CoV-2 neutralizing antibodies in BAL (broncho-alveolar lavage) measured by VNT; (G) Concentration of S-specific IgA in BAL measured by ELISA. N=5 per group; ** p < 0.05, ** p < 0.01.
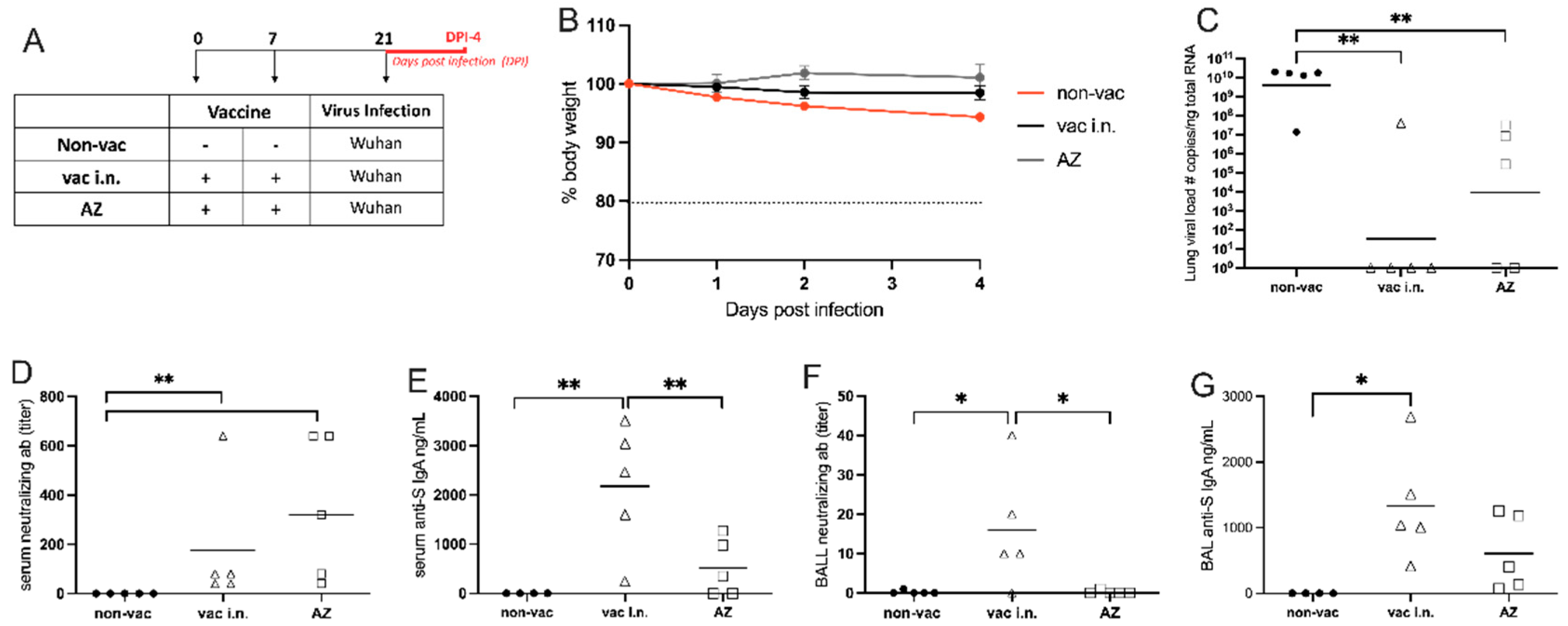
Figure 5.
- Heterologous vaccination: Booster with nasal formulation versus Oxford/AstraZeneca. (A) Protocol design: all K18-hACE2 mice were vaccinated with Oxford/AstraZeneca via intramuscular on days 0 and 7. On 14th day, mice were boosted either with the nasal formulation via intra-nasal: Het(AZ+vaci.n) or with Oxford/AstraZeneca via intramuscular (AZ). Non-vaccinated animals (non-vac) received only PBS. All mice were infected intra-nasally with SARS-CoV-2 (Wuhan strain) 28 days after the first dose; (B) Body weight during the course of infection plotted as percent change (100% representing the body weight on DPI-0, before the virus infection). (C) RNA was isolated from the lungs and SARS-CoV-2 was quantitated by RT-qPCR measuring E-gene log copies number per ng RNA at two (DPI-2) and five (DPI-5) days after viral infection; (D-G) Samples collected five days after viral infection (DPI-5); (D) Titers of SARS-CoV-2 neutralizing antibodies in serum measured by VNT, (E) Titers of SARS-CoV-2 neutralizing antibody in BAL (broncho-alveolar lavage) measured by ELISA; (F) Concentration of S-specific IgA in serum measured by ELISA; (G) Concentration of S-specific IgA in BAL measured by ELISA. N=5 per group; * p < 0.05, ** p < 0.01.
Figure 5.
- Heterologous vaccination: Booster with nasal formulation versus Oxford/AstraZeneca. (A) Protocol design: all K18-hACE2 mice were vaccinated with Oxford/AstraZeneca via intramuscular on days 0 and 7. On 14th day, mice were boosted either with the nasal formulation via intra-nasal: Het(AZ+vaci.n) or with Oxford/AstraZeneca via intramuscular (AZ). Non-vaccinated animals (non-vac) received only PBS. All mice were infected intra-nasally with SARS-CoV-2 (Wuhan strain) 28 days after the first dose; (B) Body weight during the course of infection plotted as percent change (100% representing the body weight on DPI-0, before the virus infection). (C) RNA was isolated from the lungs and SARS-CoV-2 was quantitated by RT-qPCR measuring E-gene log copies number per ng RNA at two (DPI-2) and five (DPI-5) days after viral infection; (D-G) Samples collected five days after viral infection (DPI-5); (D) Titers of SARS-CoV-2 neutralizing antibodies in serum measured by VNT, (E) Titers of SARS-CoV-2 neutralizing antibody in BAL (broncho-alveolar lavage) measured by ELISA; (F) Concentration of S-specific IgA in serum measured by ELISA; (G) Concentration of S-specific IgA in BAL measured by ELISA. N=5 per group; * p < 0.05, ** p < 0.01.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



