Submitted:
28 September 2023
Posted:
29 September 2023
You are already at the latest version
Abstract
A sustainable, scalable source of energy‐dense chemical fuel is urgently needed to ensure the security of our energy supply for future generations. Solar energy is the only renewable energy source of sufficient scale to replace fossil fuels and meet rising environmental demand. Hydrogen is expected to play a key role as an energy carrier in future energy systems of the world. As fossil fuel supplies become scarcer and environmental concerns increase, hydrogen is likely to become an increasingly important chemical energy carrier and eventually may become the principal chemical energy carrier. When most of the world’s energy sources become non-fossil based, hydrogen and electricity are expected to be the two dominant energy carriers for the provision of end-use services. Photocatalytic hydrogen evolution from water by solar energy over semiconductors offers a promising way for clean and renewable production of hydrogen. The TiO2-based materials have received considerable attention in recent years due to their extensive application in photocatalysis for hydrogen evolution. Though TiO2 is the most widely investigated photocatalyst for hydrogen evolution, it still provides low photocatalytic activity due to the wide bandgap that results in photocatalytic activity only under ultraviolet (UV) irradiation. Modification of TiO2 photocatalysts with several transition metals has been extensively studied to extend the absorbance capacity of TiO2 into the visible range. The effect of different photocatalytic deposition and reaction parameters also play major roles in enhancing the photostability of photoanode and increasing the hydrogen gas output, respectively.
Keywords:
photocatalytic water splitting
; photoelectrochemical cell
; dye solar cell
; tandem solar cell
; hydrogen
1. Introduction
One of the vital global concerns of the twenty-first century is keeping up with the growth in global energy demand due to increasing population and rising standards of living. For instance, in 2019, 16 TW-energy was consumed by approximately seven billion people worldwide. By 2050, these numbers are expected to escalate to 30 TW and nine billion people, respectively [1]. Figure 1 illustrates that oil, natural gas, and coal continue to serve as the predominant sources of global energy, with their use for electricity generation showing consistent growth both in absolute terms and about other sources. In 2021, fossil fuels accounted for 61.42% of worldwide electricity generation, a slight increase from 61.26% in 2020 [2]. However, the finite nature and uneven distribution of fossil fuel resources make it unlikely for them to meet the rising energy demand. Additionally, fossil fuel reserves are becoming less accessible as the easily exploitable ones are depleted, and their prices are on the rise due to reduced accessibility and political uncertainties among nations holding these resources [3]. Beyond economic concerns, the emission of greenhouse gases, primarily carbon dioxide (CO2), resulting from fossil fuel usage and their contribution to global warming, has raised significant environmental issues. Therefore, transitioning to non-fossil fuel energy sources has the potential to substantially reduce CO2 emissions and mitigate their adverse impact on global warming.
The restrictions on the power generation research and development in renewable energy sources for developing countries of the world constrain the technology expertise in renewable energy resources and advances in the study. As the country’s power generation system, these main factors have limited under-propagated renewable energy sources [4]. Malaysia faces the same problem that many other developed countries face. Malaysia depends heavily on fossil fuels as a primary energy source. Fossil fuel power plants operate by burning coal or oil to produce heat, which is then utilized to generate steam that drives turbines, ultimately producing electricity. In the case of gas power plants, hot gases are employed to turn turbines for electricity generation. In contrast, combined cycle gas turbine (CCGT) plants enhance their efficiency by incorporating a steam generator to further increase electricity production. These power plants are known for their reliability over extended periods and are typically cost-effective to construct Nevertheless, the combustion of carbon-based fuels in these plants releases significant amounts of carbon dioxide (CO2), contributing to the accumulation of greenhouse gases in the atmosphere. This build-up of greenhouse gases is a primary driver of global warming and is directly linked to climate change issues [5]. Indeed, in addition to carbon dioxide (CO2), fossil fuel power plants release other harmful pollutants into the atmosphere. Two significant pollutants are sulfur dioxide (SO2) and nitrogen oxides (NOx). When these pollutants react with atmospheric moisture and other compounds, they can contribute to the formation of acid rain.
The global energy demand increase has led to discussions around clean, cheap, and sustainable energy generation sources [6,7]. The global population is projected to reach a potential 10 billion people by the year 2050, which is anticipated to result in a significant exponential increase in energy demands. The widespread use of fossil fuels for energy generation has played a pivotal role in driving economic growth worldwide. However, this reliance on fossil fuels has come at a considerable environmental cost. One of the major drawbacks of fossil fuel usage is its contribution to environmental issues, particularly the emission of greenhouse gases like carbon dioxide (CO2), which is a leading driver of global warming and climate change. Given these environmental concerns and other compelling reasons, researchers and scientists are actively exploring various alternative forms of energy generation that have either zero or significantly lower negative impacts on the environment [8,9].
The hydrogen atom is typically composed of a proton and an electron, and it possesses several distinctive characteristics. It is both colorless and odorless, and its density is lower than that of air. What makes hydrogen particularly intriguing for energy applications is its impressive energy gravimetric density, which is roughly seven times higher than that of fossil fuels [10]. To put it in perspective, 1.0 kilogram of hydrogen contains significantly more energy storage capacity than 2.75 kilograms of gasoline. Consequently, 1 liter (L) of hydrogen carries roughly the same energy content as 0.25 liters of gasoline [11]. The production of hydrogen is primarily achieved through the conversion of materials containing the hydrogen element, such as carbohydrates or water. It's important to note that a significant portion of the world's hydrogen, approximately 96%, is currently obtained from conventional fossil fuels. This distribution can be broken down into 30% from naphtha reforming, 48% from steam reforming of natural gas, and 18% from coal gasification [11]. However, these conventional methods of hydrogen production are associated with environmental pollution, contributing to global environmental challenges. For this reason, professionals in the energy and environmental sectors are advocating for more sustainable approaches to hydrogen production using renewable sources (RS). Shifting towards renewable hydrogen production methods can help reduce the environmental impact associated with hydrogen generation, aligning with efforts to address environmental pollution and promote a cleaner and more sustainable energy future.
Hydrogen is indeed a versatile resource with the potential to play a significant role in various sectors beyond its current industrial applications. While it's primarily used as a raw material in industries, particularly in the production of ammonia, and has a substantial role in oil refining, its versatility opens up numerous possibilities.35% [12]. Hydrogen can be used as a fuel for both freight and passenger transport. It can power internal combustion engines in vehicles, and it is a key component in fuel cell vehicles (FCVs), which are known for their efficiency and zero-emission operation [12,13]. Hydrogen can be burned in turbines or used in fuel cells to generate electricity. This provides a clean and efficient source of power, especially when combined with renewable energy sources [14]. Furthermore, power-to-gas technology involves converting excess electricity into hydrogen through electrolysis. This hydrogen can then be injected into natural gas pipelines or used for various applications, enhancing the integration of renewable energy into existing infrastructure. Realizing hydrogen's role as a flexible energy carrier (Figure 2) is essential for achieving a sustainable and low-carbon energy future. By expanding its applications across these sectors and integrating them into various energy systems, we can harness hydrogen's potential to reduce greenhouse gas emissions, enhance energy security, and contribute to a more sustainable energy landscape.
It's evident from the literature you've mentioned that there is a growing interest in hydrogen as an energy carrier, and researchers are actively exploring various aspects of hydrogen production, storage, safety, and its role in the energy transition. Yue et al. review recent developments in hydrogen technologies and their applications in power systems, covering hydrogen production, storage, and re-electrification [16]. Dawood et al. also review different hydrogen production pathways and assess their interrelationships within the hydrogen supply chain, providing insights into the broader context of hydrogen technology [17]. A study by Najjar focuses on the safety aspects of hydrogen throughout its lifecycle, particularly emphasizing issues related to hydrogen's ignition and combustion characteristics [18]. Parra et al. evaluate the progress of hydrogen production with an emphasis on cost considerations, highlighting the economic implications of hydrogen production from renewable sources [19]. Kovac discusses hydrogen's role in the energy transition and its importance in the context of technological advancements, recognizing the changing landscape of energy systems [20]. Maggio et al. explore how hydrogen production from renewable sources can impact fuel markets, particularly in the transport sector, and suggest that hydrogen electrolysis could lead to significant changes in the energy market [21]. Hanley et al. examine hydrogen production through low-carbon methods and identify policy scenarios and factors that could promote hydrogen as an energy carrier, while also considering competition from other low-carbon technologies and electric vehicles [12,12]. Additionally, Abe et al. give an overview of hydrogen as an energy carrier identify storage as a key challenge to hydrogen development, and provide recommendations for addressing this obstacle [22]. These studies collectively contribute to our understanding of hydrogen's potential and challenges as a clean energy carrier. They touch upon various aspects of hydrogen technology, safety, economics, and policy considerations, highlighting the multidisciplinary nature of hydrogen research in the context of the transition to more sustainable energy systems.
The studies referenced provide valuable insights into the trends, challenges, and various aspects of hydrogen production and storage technologies. Finally, Liu et al. examine trends and future challenges in hydrogen production and storage. It notes that the most researched hydrogen production mechanism during their study period was the photocatalytic decomposition of water to hydrogen [23]. Mengdi and Wang cover a wide range of hydrogen production technologies, including both renewable and non-renewable resources. It also includes a comparison of the life cycle environmental impact assessment for these technologies, providing insights into their environmental sustainability [24]. The research investigated by Hossein and Wahid on various hydrogen production technologies highlights that the high cost and low efficiency of photovoltaic cells are significant barriers to the commercialization of solar-based hydrogen production [25]. Similarly, El-Emam and Ozcan focus on the economic, technological, and environmental aspects of hydrogen production. It suggests that lower electricity costs associated with geothermal and nuclear energies make them ideal sources for low-cost hydrogen production [26]. Okonkwo et al. explore the possibility of producing, using, and exporting carbon-free hydrogen from Qatar. It finds that blue ammonia production and export are currently the best pathways, with green hydrogen potentially becoming more competitive in the mid-future [27]. In another study, Lane et al. forecast the technology shares of renewable hydrogen production. It indicates that biomass gasifiers dominate the early market, but electrolyzers may gain equal shares by the mid-term due to their higher learning rate and declining costs for renewable energy, ultimately becoming the leading technology for new facilities [28]. These studies collectively contribute to our understanding of the diverse landscape of hydrogen production technologies, their economic and environmental implications, and the factors influencing their adoption and commercialization. They highlight the importance of addressing challenges such as cost, efficiency, and environmental impact to promote the widespread use of hydrogen as a clean energy carrier.
It is clear that this paper contributes to the ongoing discussion on hydrogen production technologies, with a particular focus on clean methods. While previous studies have touched upon various aspects of hydrogen production processes, the unique contribution of this paper lies in its emphasis on clean technologies and its exploration of the limitations associated with these methods. Delving into both the strengths and weaknesses of clean hydrogen production technologies provides a more comprehensive understanding of the challenges and opportunities in this critical area of research and development. This holistic approach can help inform decision-making and policy development related to sustainable hydrogen production and utilization.
2. Hydrogen as Energy Carrier
The utilization of fossil fuels in modern industry has led to significant environmental problems and energy crises. As a response to these challenges, there is a growing interest in shifting from fossil fuels to solar energy as a promising strategy. Solar energy is considered a clean, safe, and virtually inexhaustible energy resource [29]. Various technologies have been developed to convert solar energy into different forms of usable energy, including electrical energy, heat energy, and chemical energy. Among these technologies, photocatalytic hydrogen production from water has gained significant attention since Fujishima's groundbreaking report in 1972, in which he demonstrated that titanium dioxide (TiO2) could act as a semiconductor for photocatalytic water splitting to produce hydrogen. This approach has several key advantages [30], 1) abundance of water; 2) environmentally friendly; 3) sustainable energy cycle. However, a critical challenge in realizing this potential is the need for high-efficiency catalysts to drive the process of splitting water into hydrogen and oxygen using sunlight. The development of such catalysts is a crucial factor in harnessing solar energy for clean and sustainable hydrogen production.
Green hydrogen is indeed an increasingly promising alternative to conventional forms of energy generation. It is called "green" hydrogen because its production process, when powered by renewable sources (RS), generates no greenhouse gas emissions (GHG). Green hydrogen is produced through a process called electrolysis, in which electricity generated from renewable sources is used to split water into hydrogen and oxygen. Since this process relies on clean, renewable energy, it does not contribute to GHG emissions. Indeed, one of the notable advantages of hydrogen is its ability to carry energy efficiently across geographical areas. This is primarily due to its electrochemical conversion capabilities, lightweight nature, and high mass-energy density. Here's how these features enable hydrogen to be used for energy transportation: [31].
a) Electrochemical potential: Hydrogen can be easily produced through the electrochemical process of water electrolysis, as mentioned earlier. This means that excess renewable energy, such as solar or wind power, can be used to produce hydrogen when it's abundant, and then the hydrogen can be stored for later use or transported to areas where energy is needed.
b) Lightweight: Hydrogen is the lightest element, which makes it an attractive option for transportation. When used as a fuel, it can power vehicles and ships while minimizing the weight of the fuel itself, improving overall energy efficiency.
c) High Mass-Energy Density: Hydrogen has a high energy content per unit mass, meaning a relatively small amount of hydrogen can carry a significant amount of energy. This high mass-energy density makes it an efficient energy carrier, especially when compared to some other forms of energy storage.
d) Transportation Methods: Hydrogen can be transported in various forms. Liquid hydrogen is one option, and it's used in some rocket propulsion systems. Additionally, hydrogen can be transported in the form of ammonia or other chemical compounds. It can also be injected into natural gas pipelines or transported using dedicated pipelines.
e) Freight Ships: Hydrogen can be used as a fuel for ships, including freight vessels. This can help reduce the carbon footprint of the shipping industry, which is a significant contributor to global emissions.
f) Pipelines: Dedicated hydrogen pipelines can transport hydrogen efficiently over long distances. These pipelines are being developed in various regions to support the growth of hydrogen as an energy carrier.
Hydrogen indeed boasts higher Higher Heating Value (HHV) and Lower Heating Value (LHV) compared to many conventional fossil fuels; as shown in Table 1. These high heating values indicate that hydrogen contains a substantial amount of energy per unit of mass or volume; making it an attractive energy carrier. Besides; Ball and Weeda mentioned that hydrogen can be used as a fuel in fuel cells. Fuel cells are electrochemical devices that efficiently convert hydrogen and oxygen into electricity and water through a chemical reaction. This electrochemical process is highly efficient and environmentally friendly since it generates electricity without combustion; producing only water vapor as a by-product. Fuel cells have the potential to provide clean and efficient power generation for a variety of applications; including vehicles; stationary power systems; and portable devices [32]. The combination of hydrogen's high energy content and its compatibility with fuel cells positions it as a promising option for clean and sustainable electricity generation; contributing to efforts to reduce greenhouse gas emissions and transition to a more environmentally friendly energy landscape
In summary, hydrogen's electrochemical conversion, lightweight nature, and high energy density make it a versatile and efficient means of carrying energy across geographical areas. This is especially valuable as we seek ways to transport and store energy from renewable sources and reduce greenhouse gas emissions in various sectors of the economy.
3. Hydrogen Production Methods
Hydrogen can indeed be sourced from various natural substances and feedstocks, and the method of extraction or production can significantly impact its environmental impact. Hydrogen can also be extracted from fossil hydrocarbons, biomass, hydrogen sulfide, or some other substances. The term "green hydrogen" is often used to describe hydrogen production methods that are environmentally friendly and produce minimal or no greenhouse gas emissions. Green hydrogen is typically produced through processes like water electrolysis using renewable energy sources like solar or wind power. These methods prioritize sustainability and contribute to reducing the carbon footprint associated with hydrogen production [34].
Various forms of energy can be utilized or hydrogen production from different natural resources. These energy sources can be classified into four categories: thermal, electrical, photonic, and biochemical. These kinds of energy can be obtained from green energy sources and it is proposed that green energy comprises renewable energy, nuclear energy, and recovered energy for example, recovered industrial heat, landfill gas, wastes that can be incinerated, etc. Thermal energy involves using heat as an energy source to drive hydrogen production processes. Thermal energy can be obtained from various green sources, including solar thermal energy, geothermal energy, and recovered industrial heat. For instance, solar thermal collectors can concentrate sunlight to provide the high temperatures needed for thermochemical hydrogen production. Electrical energy can come from a range of green energy sources, such as solar panels (photovoltaic cells), wind turbines, hydropower, and even nuclear power plants. It's commonly used in processes like water electrolysis, where electricity is used to split water into hydrogen and oxygen. Photonic energy refers specifically to energy derived from solar radiation. Solar panels, also known as photovoltaic cells, convert sunlight directly into electrical energy, which can be used for electrolysis or other hydrogen production methods. Biochemical energy is stored in organic matter, such as carbohydrates, glucose, and sugars. It can be harnessed through various biological processes, including dark fermentation and bio-photolysis. Microorganisms play a crucial role in extracting hydrogen from various substrates or converting organic matter into thermal energy. Table 2 likely provides an overview of hydrogen production methods based on the type of energy used to drive the processes. This classification helps in understanding the diversity of approaches available for sustainable hydrogen production, with a focus on utilizing green energy sources to minimize environmental impacts and reduce carbon emissions.
Renewable energy sources have emerged as highly promising alternatives to fossil fuels for hydrogen generation [51]. These sources offer numerous advantages, including their ability to mitigate the release of greenhouse gases into the environment. Strategies for hydrogen production that rely on renewable energy sources as their power input have gained traction. These approaches not only contribute to clean energy generation but also help prevent the dispersion of ozone-damaging materials. [52,53,54]. Hydrogen serves as a crucial link between renewable electricity generation and various applications, particularly in transportation [55]. Renewable energy initiators, such as wind, ocean, and solar power, have become integral to hydrogen production processes. This environmentally friendly approach to hydrogen generation is often referred to as "green hydrogen" [56,57]. In summary, green hydrogen production methods leverage renewable energy sources to produce hydrogen sustainably. These methods not only reduce the carbon footprint associated with hydrogen production but also promote the broader adoption of clean and renewable energy technologies.
Green hydrogen is indeed a compelling choice due to its economic feasibility and environmental friendliness with zero emissions. Its suitability as a transportation fuel makes it a valuable option for reducing carbon emissions in the transportation sector.[26,58]. Solar-based hydrogen systems are particularly noteworthy for their portability and sustainability. They harness the power of solar irradiance to produce hydrogen through a process known as water splitting, which separates water into its constituent elements, oxygen and hydrogen. This method is highly reliable because both water and solar irradiance are abundant resources on Earth, making it a sustainable and renewable approach to hydrogen production [59,60]. Table 3 highlights the various reviews conducted on different methods of green hydrogen production. These reviews provide insights into the technologies, processes, and advancements in green hydrogen production, further emphasizing the growing interest in sustainable hydrogen generation and its diverse applications.
The increasing awareness and concerns about greenhouse gas emissions and global warming led to a shift away from fossil fuels and hydrocarbons as feedstocks for hydrogen production. [74]. Instead, there is a growing focus on water electrolysis powered by renewable energy sources, which is considered a zero-emission method[75,76]. Water electrolysis is favored for its reducing cost trend, high purity of hydrogen produced, and absence of undesirable impurities like carbon oxides and sulfate oxides [77,78]. However, there are emerging technologies that aim to accelerate the transition to a hydrogen economy by reducing capital costs and increasing production efficiency. One of these emerging technologies is the photoelectrochemical (PEC) cell, which uses solar energy to split water into hydrogen and oxygen. It is considered renewable and environmentally friendly because it only requires water as a raw material [79]. The PEC technology is described as having a low environmental impact on both large and small scales, and it doesn't produce undesirable by-products [80]. Despite its potential, PEC hydrogen production faces challenges, including low efficiency, typically below 10%. Researchers are working to improve the efficiency of solar energy conversion systems to address these challenges and make PEC technology economically viable [81,82]. Therefore, efficient solar energy conversion systems are seen as essential for solving global energy issues and transitioning to cleaner and more sustainable energy sources. This passage underscores the importance of clean and renewable methods for hydrogen production and highlights the potential of PEC technology while acknowledging the need for continued research and development to overcome its challenges and increase efficiency.
4. Photocatalytic Semiconductor Materials
The efficiency of water photo-electrolysis is closely related to the properties of the materials used in the photoelectrode of a PEC cell. In PEC, Materials selected for the photoelectrode must serve two fundamental functions namely optical function that can absorb the maximum amount of solar energy, as this energy is used to drive the water-splitting reaction, and catalytic function that possesses catalytic properties that enable them to facilitate the decomposition of water molecules into hydrogen and oxygen [83]. In PEC cells, semiconductor materials are commonly used for photoelectrodes. Semiconductors have the electronic configuration necessary to absorb the solar spectrum required for water splitting. The bandgap energy (Eg) of a semiconductor is a crucial factor in its photoactivity. It is defined as the energy difference between the highest occupied valence band (VB) and the lowest empty conduction band (CB). The Eg determines which portion of the solar spectrum can be absorbed by the semiconductor. Figure 3 illustrates the general principle of photocatalysis in a semiconductor material. From the diagram, the photocatalyst will absorb photons with energy equal to or greater than its bandgap energy. When photons are absorbed, they generate electron-hole pairs within the semiconductor material. Electrons are promoted to the conduction band (CB), leaving behind positively charged holes in the valence band (VB). The photogenerated electrons and holes participate in surface chemical reactions, which include reducing an acceptor species (A → A-) and oxidizing a donor species (D → D+) [84]. This passage provides insights into the fundamental principles of photocatalysis and the role of semiconductor materials in the photoelectrode of a PEC cell, emphasizing the importance of bandgap energy for efficient solar-driven water splitting.
5. Tandem Cells
Using sunlight to split water into hydrogen and oxygen through photoelectrochemical (PEC) cells is indeed an attractive solution for addressing the intermittent nature of solar energy generation. This process, known as photoelectrolysis, offers several advantages [85]. One of the primary benefits of PEC-based hydrogen production is its ability to store energy effectively. Hydrogen can be stored in tanks or underground reservoirs for extended periods, making it a versatile energy carrier. This stored hydrogen can then be converted back into electricity or heat when needed, providing a continuous energy supply even during nighttime or cloudy days. Besides, hydrogen produced through photoelectrolysis is a clean and renewable energy source. It does not emit greenhouse gases or other pollutants when burned or used in fuel cells. This makes it an environmentally friendly option that can help reduce carbon emissions and combat climate change [86]. In addition, solar radiation is an abundant and virtually limitless source of energy. Unlike fossil fuels, which are finite resources, sunlight is always available as long as the sun is shining. This makes hydrogen production through PEC cells a sustainable and inexhaustible energy solution. However, it's essential to acknowledge that PEC technology is still in the research and development stage, and there are challenges to overcome. These challenges include improving the efficiency and durability of PEC cells, reducing production costs, and optimizing the materials used in the process. Additionally, the infrastructure for hydrogen storage and distribution needs to be developed to fully harness the potential of this technology.
PEC cells use semiconductor (SC) materials to harness the energy of sunlight and split water into hydrogen and oxygen. When photons with energy hυ higher than the semiconductor’s band gap strike the semiconductor surface, they create electron-hole pairs (+ ) in the conduction and valence bands, respectively [83]. These photoexcited electrons and holes play crucial roles in the subsequent water-splitting reactions.
At the semiconductor’s surface, holes oxidize water to oxygen,
while the photoexcited conduction band electrons, transferred to the counter-electrode of the PEC cell, reduce water to form hydrogen gas,
Many semiconductors used in PEC cells have relatively large energy band gaps, which can limit their efficiency in absorbing visible light. Additionally, the band edge potentials of these semiconductors may not be ideally suited for the water-splitting reactions. For efficient water cleavage, the conduction band of the semiconductor must be positioned at a more negative potential than the reduction potential of water (Ec < Ered), and the valence band must be positioned at a more positive potential than the oxidation reaction (Ev > Eox). To address the limitations of single semiconductors, researchers have explored tandem cell configurations. In a tandem cell, two semiconductors with different band gaps are coupled together. This arrangement allows for more efficient utilization of a broader range of the solar spectrum and can better match the energetics required for both oxygen and hydrogen evolution.[87]. Another promising approach is to couple a typical PEC cell with a photovoltaic (PV) cell. In this configuration, the photovoltaic compartment generates a photovoltage that compensates for any potential deficiencies in the PEC cell, ensuring that the required energetics for water splitting are met. This configuration is known as an "unbiased" solar water splitting system and is highly attractive for achieving efficient water photocleavage. It's important to note that ongoing research in materials science and device engineering is aimed at identifying and developing semiconductor materials and configurations that can improve the efficiency and practicality of PEC water splitting. Achieving high conversion efficiencies and scalability is crucial for realizing the full potential of this technology in the renewable energy landscape.
Tandem cells consist of two distinct photosystems connected (Figure 4). The first photosystem is the PEC cell, which is responsible for generating electron-hole pairs through band gap excitation of a semiconductor. The PEC cell primarily absorbs the blue part of the solar spectrum. In this process, holes migrate to the semiconductor's surface, where they participate in the oxygen evolution reaction, and electrons are injected into the conduction band of the semiconductor. The second photosystem in the tandem cell is the Dye Solar Cell (DSC). Unlike the PEC cell, the DSC is capable of absorbing a broader range of light in the visible and near-infrared spectrum. Its main function in this configuration is to collect the photoexcited electrons generated by the PEC cell. These electrons are then passed through the DSC. As the electrons pass through the DSC, their electrochemical potential is increased. This means that the DSC helps to elevate the energy state of the electrons, making them more suitable for the reduction of water to produce hydrogen. The increased electrochemical potential is used to drive the water-splitting reaction at the counter-electrode of the PEC cell [88]. In the described embodiment, the two photosystems, the PEC cell, and the DSC, are not physically connected, meaning they operate as independent devices. This setup has several advantages, including the ability to use the DSC to simultaneously provide the necessary anodic bias to multiple PEC cells. It also allows the DSC to generate electricity for other applications, effectively making use of the energy contained in photons that are absorbed by the DSC. In summary, tandem cells represent an innovative approach to improving the efficiency of solar-driven water splitting for hydrogen production. By combining the strengths of two independent photosystems, one optimized for specific light absorption and electron generation and the other for increasing electron energy, researchers can enhance the overall performance of PEC water-splitting systems. This concept has the potential to contribute to the development of more efficient and practical renewable energy solutions.
6. Limitations Associated with Current Technologies for Hydrogen Production
The potential of hydrogen as a clean and renewable source of energy is widely recognized, and it is often referred to as the "fuel of the future" due to its environmental benefits and versatility. However, as you mentioned, the challenge lies in the energy-intensive processes currently used for industrial-scale hydrogen production, which can be disadvantageous in terms of cost and environmental impact [8,89,90]. Photocatalytic water splitting, particularly through photoelectrochemical (PEC) methods, is seen as a promising and environmentally friendly alternative for hydrogen production where the photoanode in the PEC system absorbs sunlight and splits water directly into hydrogen and oxygen [91]. The main work in PEC water splitting still concerns increasing the efficiency and stability of the photoactive materials to achieve the required efficiency target of 10% that will be viable for commercialization.
In general, PEC water splitting is promising, however, the reported solar-to-hydrogen (STH) conversion efficiencies are still considered low for practical use. Over the past decade, many semiconductor materials, such as TiO2, WO3, CdS, Ta2O5, BiVO4, etc., have been extensively studied due to their suitable band gap, stability, and effective response for H2 production under solar irradiation. Among these, TiO2 is the most widely explored photocatalyst due to its global availability, low toxicity, stability, and near-visible light photocatalytic activity [92]. However, due to a large bandgap of 3.0-3.2 eV, TiO2 can only absorb light in the ultraviolet (UV) region. As UV light accounts for only 4% of solar energy, the theoretical STH efficiency of TiO2 photoanodes can only reach around 2% under solar irradiation, even when assuming 100% absorbed-photon-to-hydrogen conversion efficiency [93,94]. Shifting the active region of the photocatalyst from the UV region to the visible region of sunlight is needed and very relevant since the source is free and abundant. Therefore, doping TiO2 with non-noble metals like Fe and Ni is more practical and they have been used to decrease the band gap of TiO2 and to expand the photo response of the TiO2 into the visible region [95,96]. Cobalt (Co) and nickel (Ni) have received much attention as metal dopants as these metals are cheap and abundant in supply. A great deal of research has been conducted using Co and Ni impregnated on TiO2, but co-impregnated Co and Ni on TiO2 is rarely reported. Besides, the high electron-hole recombination process at the surface or within the bulk reduces the efficiency of photocatalytic water splitting. It means that the recombination of photogenerated charge carries happens very fast resulting in the low timescale of surface reaction [97]. And it happens in the range of picoseconds to femtoseconds. The use of metal doping in semiconductor materials, such as titanium dioxide (TiO2), is indeed a promising approach to enhance the performance of photocatalytic and photoelectrochemical processes. Metal doping can influence the electronic band structure of the semiconductor and improve charge carrier separation, which is crucial for efficient photocatalysis and water splitting. The Fermi levels of the metal dopants are often lower than that of TiO2. This energy level mismatch can facilitate the transfer of photoexcited electrons from TiO2 to the conduction band of the metal particles. This separation of charge carriers helps prevent fast recombination of electrons and holes, which can significantly enhance photocatalytic activity [98]. Ni et al. concluded that the promoting effect of bimetallic co-doping TiO2 can further improve charge carrier separation and trapping. Each metal can play a specific role: one metal can act as a collector of photogenerated electrons, inhibiting recombination, while the other metal can serve as a trap for photogenerated holes. This dual role helps to extend the lifetime of charge carriers and increase overall photocatalytic efficiency [99]. Therefore, the higher photo activities of TiO2 promoted with nickel or (and) cobalt will contribute to the improvement of light absorption and lower recombination rate of photogenerated electrons and holes [100]. Nickel is known as an effective catalyst for the hydrogen evolution reaction (HER) and can act as a collector of photogenerated electrons. By efficiently collecting electrons, it helps prevent their recombination with photogenerated holes [101]. Similarly, cobalt is an effective catalyst for the oxidation reaction and can trap photogenerated holes. This trapping of holes prevents their recombination with electrons. Both nickel and cobalt doping, therefore, contribute to lower recombination rates and improved charge carrier lifetimes, leading to enhanced photocatalytic activity [102].
As a typical n-type semiconductor, TiO2 has especially attracted extensive attention due to the outstanding advantages of excellent optical and electronic properties. In contrast, the pristine TiO2 photoanode experienced a photocurrent decay of ~16.4% and this poor stability can be ascribed to the photo corrosion that was induced by the accumulation of photogenerated holes at the surface of the TiO2 nanotube arrays [103]. In addition, they partially undergo photoinduced dissolution in electrolytes and many photogenerated holes will accumulate on the surface of the photoanode, resulting in photocorrosion [104,105]. The part of the glass substrate in contact with the semiconductor material tends to crack, resulting in severe interfacial recombination and affecting the long-range stability of the device. This phenomenon leads to the instability of photoanodes which is still a major factor limiting PEC performance. For this problem, regardless of the type of photoanode material, some of the photocatalytic deposition parameters will be investigated to study the photo corrosion stability mechanism of photoanode.
Usually, in PEC water-splitting studies, the performance of the chosen material is arbitrated by observing its obtained photocurrent density value [106]. Hence, every PEC water splitting study using different materials always seeks to acquire higher photocurrent density because of having higher photocurrent density, there is a higher probability of hydrogen production which is directly proportional to solar to hydrogen efficiency. The enhancement in photocurrent density was also attributed to the improved photogeneration of electron–holes [107]. Furthermore, the backward reaction of hydrogen to oxygen to form water is thermodynamically favorable making the high production of hydrogen from pure water splitting difficult to achieve [108]. Therefore, we need to use electron donors to consume the photogenerated holes. It is also known as a sacrificial agent which can reduce the recombination tendency of electrons and holes and accelerate the rate of hydrogen generation. Owing to the less positive oxidation potential, it can react more quickly with holes than water, and this will lead to the accumulation of electrons on the TiO2 surface for the photoreduction of water [109].
7. Theory of Water Splitting
This section provided a concise description of the fundamental principles of water splitting in an electrochemical cell, which is a well-established process for generating hydrogen and oxygen gases from water. The electrochemical cell is the central component of the water-splitting process. It typically consists of two electrodes immersed in pure water or an electrolyte solution. The electrodes are usually made of conductive materials like platinum, graphite, or other metals. In the electrochemical cell, there are two electrodes: the cathode (-) and the anode (+). The cathode is where reduction reactions occur, leading to the production of hydrogen gas (H2). The anode is where oxidation reactions take place, resulting in the production of oxygen gas (O2). To drive the water-splitting reactions, a certain voltage difference (electromotive force or EMF) is applied between the cathode and anode. This voltage, often referred to as the cell voltage or overpotential, is necessary to overcome the activation energy barriers associated with the water-splitting reactions. The amount of hydrogen and oxygen gases produced per unit of time in the electrochemical cell is directly controlled by the electric current passing through the cell. The current is typically measured in amperes (A) and is a key parameter in determining the rate of gas production. In aqueous solutions like pure water, water molecules (H2O) can dissociate into ionic species, primarily hydronium ions (H3O+) and hydroxide ions (OH-). These ions are important in the overall electrochemical reactions, as they participate in the proton exchange and charge-balancing processes during water splitting. In addition, ionic species of water, including H+ (aq) and OH− (aq) have a certain percentage as represented in Equation 1.4.
Hydrogen and oxygen gas can be generated at the surface of the cathode and anode within Equations 1.5 and 1.6.
These reactions show that electrons (e-) are transferred through the external circuit from the anode to the cathode, creating an electric current, while hydrogen and oxygen gases are produced at their respective electrodes. Efficient water splitting for hydrogen production is an essential process in various applications, including renewable energy storage and clean fuel generation. Researchers continue to work on improving the efficiency and sustainability of water-splitting technologies to make them more practical and environmentally friendly. In summary, water splitting is a thermodynamically favorable process with a theoretical minimum cell voltage of approximately 1.23 volts. However, practical systems often require higher applied voltages in the range of 1.6 to 2 volts to overcome various losses and challenges associated with the kinetics of the electrochemical reactions and charge transfer processes. Achieving efficient and cost-effective water splitting at these practical voltages is a key goal in the development of hydrogen production technologies [22].
8. Principles of Photocatalytic Water Splitting in PEC Cell
Indeed, hydrogen production through solar-driven water splitting is a promising and environmentally friendly process, as it utilizes renewable and abundant resources. The generated hydrogen can be used as a clean fuel source and has a variety of applications, including in fuel cells for energy generation. The regeneration and consumption cycle of hydrogen in a fuel cell is a key aspect of the overall process. The regeneration and consumption cycle of hydrogen is displayed in Equations 1.7 and 1.8 [110].
In a fuel cell, hydrogen (H2) combines with oxygen (O2) from the air in the presence of a catalyst to produce electricity and water vapor (H2O). This is an electrochemical process that generates electrical energy as a byproduct of the reaction. The hydrogen-oxygen fuel cell is highly efficient and environmentally friendly, as it produces electricity without the release of harmful pollutants or greenhouse gases. The only byproduct of the reaction is pure water, which makes it a clean and sustainable energy conversion technology. The ability to store and transport hydrogen makes it a valuable energy carrier, and when combined with fuel cells, it can provide a reliable and efficient source of electricity for a wide range of applications, including transportation, stationary power generation, and more. The cycle of hydrogen production and consumption, as represented by Equations 2.4 and 2.5, plays a crucial role in enabling a sustainable and clean energy future.
Photovoltaic water splitting can be carried out using two types of systems: a photo-batch reactor and a PEC cell. Each system has its advantages and disadvantages, and the choice between them depends on various factors, including the specific application and desired outcomes. In the photo-batch reactor system, powdered photocatalysts are dispersed in water, and hydrogen is produced during a photocatalytic reaction under sunlight (Figure 5). This system has some disadvantages, namely the necessity to separate separate the generated hydrogen from oxygen. This separation process can be challenging and requires additional equipment [111,112,113]. It can be difficult to separate the inactive or spent catalyst particles from the reaction mixture after the photocatalytic process. Catalyst particles in powder form tend to agglomerate or clump together, reducing their surface area and overall efficiency. To address the drawbacks of the photo-batch reactor system, researchers have developed an alternative approach, which involves immobilizing the photocatalyst in the form of a thin film [111]. This approach is commonly used in PEC cells, where the photocatalyst is integrated into the cell structure, allowing for direct water splitting and efficient separation of hydrogen and oxygen. Both systems have their applications and advantages, and the choice between them depends on factors such as the scale of the operation, the desired level of efficiency, and the ease of catalyst recovery. Researchers continue to explore and develop various approaches to enhance the efficiency and practicality of photovoltaic water splitting for clean hydrogen production.
The pioneering work by Akira Fujishima and Kenichi Honda in 1972 marked a significant milestone in the field of photoelectrochemical (PEC) water splitting. Their discovery of photoelectrolysis of water on the surface of a titanium dioxide (TiO2) photoelectrode under ultraviolet (UV) irradiation laid the foundation for modern research in this area (Figure 6) [114]. A photoelectrochemical cell (PEC cell) is a device used for water splitting, where water is separated into oxygen (O2) and hydrogen (H2) using the energy of absorbed sunlight. The PEC cell typically consists of two electrodes immersed in an electrolyte. At least one of these electrodes should be made of a semiconductor material capable of absorbing sunlight to initiate the water-splitting reaction. To efficiently utilize sunlight and produce hydrogen (H2) in a PEC cell, the photoanode (the semiconductor electrode exposed to light) should meet specific criteria. It should have an optimal bandgap energy, approximately 2 electronvolts (eV), to absorb sunlight efficiently. Additionally, the conduction band potential of the semiconductor should be more negative than the hydrogen reduction potential to drive the reaction [83]. While the theoretical minimum cell voltage for water splitting is approximately 1.23 volts (the thermodynamic voltage), practical PEC cells typically require a higher potential to overcome overpotentials and losses within the system. [115]. Overpotential refers to the additional voltage needed to drive the reaction at a reasonable rate due to factors like reaction kinetics and charge transfer resistance. In a PEC cell, water reduction occurs at the cathode (often made of a material like platinum), where hydrogen ions (protons) and electrons combine to form hydrogen gas (H2). Water oxidation takes place at the anode, typically composed of a semiconductor material like TiO2, resulting in the production of oxygen gas (O2) [82,91].
9. PEC Water-splitting Process in Photoanode
An excellent photoanode device should satisfy the following criteria: (i) The photoelectrode has a suitable Eg (2.0–2.6 eV) to fully utilize the energy of the solar spectrum and generate enough photogenerated carriers; (ii) the conduction band (CB) position of the photoelectrode must be below the reaction potential of H+/H2 (0 V versus the reversible hydrogen electrode, 0 V vs. RHE); (iii) the valence band (VB) position of the photoelectrode must be higher than the reaction potential of O2/H2O (1.23 V vs. RHE); (iv) photogenerated carriers can rapidly separate and transfer in the photoelectrode; (v) photoelectrode materials are abundant and nontoxic [116].
In general, the water-splitting steps that take place on the photoanode are roughly described as follows: When sunlight illuminates the semiconductor material, photogenerated electrons, and holes appear at the CB and VB positions of the photoanode, respectively. Because of the upward energy band bending at the interface between the n-type semiconductor and the electrolyte, the photogenerated holes inside the semiconductor migrate to the electrolyte/photoanode interface and undergo an oxidation reaction to generate oxygen. At the same time, the photogenerated electrons are transferred to the Pt electrode through an external circuit with the assistance of an external voltage, and thus, a reduction reaction occurs to generate hydrogen [117]. The specific process on the photoanode can be roughly divided into three steps:
i. Generation of photogenerated electron–hole pairs (light absorption process): The energy of incident sunlight is absorbed by the semiconductor. When the energy of the incident photon is greater than Eg, the electrons are excited from the VB to the CB, leaving holes in the VB, which generates photogenerated electron–hole pairs in the bulk. This process can be expressed by the absorption efficiency (ηabs).
ii. Bulk transfer of photogenerated electron–hole pairs (a bulk separation process): The generated electron–hole pairs migrate inside the semiconductor, where the photogenerated holes move toward the semiconductor/electrolyte interface, and the photogenerated electrons move toward fluorine-doped tin oxide (FTO) and are transferred to the counter electrode through the external circuit. This process can be expressed by the bulk separation efficiency (ηsep).
iii. Photogenerated holes conduct a water oxidation reaction on the surface (surface catalytic process): The photogenerated electrons transferring to the counter electrode reduce water molecules, while the photogenerated holes transferring to the interface oxidize water molecules, realizing the conversion of solar energy to hydrogen energy. This process can be expressed by the surface injection efficiency (ηinj).
The above three factors jointly exert influence on the final efficiency of PEC water splitting, which can be expressed as [118].
where Jmax is the integral value of the spectral irradiance under a simulated air mass of 1.5 global (AM 1.5G) up to the absorption wavelength. In the overall process of PEC water splitting, the three processes of ηabs, ηsep, and ηinj together determine the final efficiency of the entire PEC water-splitting device. The above three processes have substantial effects on the STH efficiency of photoanodes because the photo corrosion phenomenon mainly results from the deactivation of the semiconductor material. On the whole, ηabs, ηsep, and ηinj have a certain influence on the long-range stability of the photoanode.
10. Photocatalysis for Water Splitting
The choice of a suitable photocatalyst for water splitting is crucial, and there are several functional requirements that a photocatalyst must meet to be effective in this process. First, the energy levels of the valence band (VB) and conduction band (CB) of the photocatalyst are critical. Specifically, the CB should be more negative (lower energy) than the reduction potential of water (H2O -> H2 + 1/2O2), while the VB should be more positive (higher energy) than the oxidation potential of water. This ensures that the photoexcited electrons can participate in the reduction of water to hydrogen (H2), while the photogenerated holes can be involved in the oxidation of water to oxygen (O2) [119,120]. Second, the bandgap energy of the photocatalyst is also important. It determines the range of the electromagnetic spectrum (i.e., the wavelength of light) that the material can absorb. A suitable bandgap energy allows the photocatalyst to absorb visible light effectively, as much of the solar spectrum is in the visible region. In Figure 7, different photocatalysts are compared in terms of their band level positions, bandgap energies, and their relation to the redox potential of water [112]. Third, photocatalysts must be stable in the presence of water and the harsh conditions of the water-splitting process. Some materials, like CdS and CdSe, have suitable band-level positions and bandgap energies for water splitting but are unstable in water. This is because S2- and Se2- ions can be more readily oxidized by photogenerated holes than water molecules, leading to photo corrosion and reduced photocatalyst durability [112,121].
WO3 has a bandgap energy of 2.8 eV, which limits its ability to efficiently absorb solar light. While it can utilize approximately 12% of solar light at a wavelength of 480 nm, the bandgap is still considered too large for effective absorption across the solar spectrum. The conduction band edge of bulk WO3 has been reported to be around +0.4 eV or +0.31 eV below the hydrogen redox potential. These positions are still too positive to facilitate hydrogen production, indicating that WO3 may not be an ideal photocatalyst for this purpose [30]. α-Fe2O3 has a bandgap energy of 2.3 eV, which allows it to absorb longer wavelengths of light, making it capable of utilizing approximately 40% of the solar spectrum. However, α-Fe2O3 faces several challenges in PEC cell applications. Its conduction band edge is still reported to be positive (+0.37 eV) relative to the hydrogen redox potential. This means that significant external bias is required for efficient hydrogen production. Additionally, α-Fe2O3 has limitations in terms of electrical conductivity, short hole diffusion length, and a relatively short excited-state lifetime [122,123]. These materials, including SrTiO3, TiO2, and NaTaO3, are known for their good photostability and resistance to photo-corrosion under light irradiation [112]. However, they all have relatively large bandgap energies, which can limit their efficiency in photocatalytic hydrogen production. Their bandgap energies may not allow for efficient absorption of a significant portion of the solar spectrum.
The bandgap of a semiconductor dictates the energy of photons that it can absorb effectively. A wider bandgap, such as that of titanium dioxide (TiO2) with a bandgap of around 3.2 eV, primarily absorbs ultraviolet (UV) light, which represents only a small fraction (about 4%) of the total solar energy. In contrast, semiconductors with narrower bandgaps, like cadmium sulfide (CdS) with a bandgap of approximately 2.4 eV, can absorb visible light, which constitutes a more significant portion of the solar spectrum. This broader absorption range is advantageous for harnessing solar energy effectively [124]. The positions of the conduction band (CB) and valence band (VB) edges of a semiconductor are critical for its performance in photocatalysis. These positions should be compatible with the redox potentials (electrochemical potentials) of the chemical species involved in the reactions. (Figure 8). For the evolution of hydrogen (H2) from water (H2O), the CB edge of the semiconductor should be at a higher (more negative) potential than the redox potential of the H2/H2O couple. This ensures that photoexcited electrons from the semiconductor can reduce water to produce hydrogen gas. In the reduction of carbon dioxide (CO2) to produce various carbon-based compounds such as methane (CH4), methanol (CH3OH), formaldehyde (HCHO), formic acid (HCOOH), or carbon monoxide (CO), the CB edge should also be positioned at a potential more negative than the relevant redox potentials, such as CH4/CO2 or H2O/CO2. Conversely, the VB edge should be at a lower (more positive) potential than the redox potential of the O2/H2O couple to enable the oxidation of water to oxygen gas.
Thus, one significant challenge in photocatalysis is to engineer the energy bands of semiconductor photocatalysts to efficiently utilize visible light. Since visible light constitutes a substantial portion of the solar spectrum, the ability to harness this light for photocatalytic reactions is essential for improving the efficiency of solar-driven processes. Researchers focus on designing and modifying semiconductor materials to achieve narrower bandgaps that allow for effective absorption of visible light. This engineering of energy bands is crucial to broadening the range of solar light that can be utilized in photocatalytic reactions. Another key challenge in photocatalysis is enhancing efficiency by minimizing charge recombination. One of the major limitations to achieving high efficiency in photocatalytic reactions is the recombination of photogenerated electrons and holes. This recombination process typically occurs on a very short timescale, on the order of 10^-9 seconds. [125]. Chemical interactions between photogenerated charge carriers and adsorbed species on the catalyst surface occur on a longer timescale, typically ranging from 10^-8 to 10^-3 seconds. To maximize the efficiency of photocatalysis, it is crucial to prevent or minimize the rapid recombination of electron-hole pairs before they can participate in the desired chemical reactions. Strategies to address this challenge include the development of materials and structures that promote efficient charge separation and migration, as well as the introduction of cocatalysts and surface modifications to suppress recombination processes [126].
In summary, finding a photocatalyst that satisfies all these requirements is a significant challenge. Researchers continue to investigate various materials and strategies to develop stable and efficient photocatalysts for water splitting. While these materials have certain advantages, such as photostability and non-photo-corrosiveness, their large bandgap energies remain a challenge for achieving efficient photocatalytic hydrogen production. The highlighted challenges are actively researched areas, and advancements in materials science and catalysis continue to drive progress in improving the efficiency and effectiveness of photocatalytic processes for applications such as hydrogen production and carbon dioxide reduction. These efforts are vital for advancing sustainable and renewable energy technologies.
a. TiO2 Photocatalyst in PEC Cell
Titanium dioxide (TiO2) is indeed one of the most widely used photocatalysts in photocatalytic applications, including hydrogen production and environmental remediation [127]. Titanium is an abundant element in nature, and TiO2 is readily available at a relatively low cost. This abundance makes TiO2 an economically viable choice for large-scale photocatalytic processes [128]. TiO2 can crystallize in different structures, with the anatase and rutile phases being the most commonly used for photocatalytic applications[97]. Both crystal structures consist of chains of distorted TiO6 octahedra, but they exhibit slight structural differences (Figure 9). Anatase TiO2 has a specific crystal structure characterized by distorted octahedra. It is known for its higher photocatalytic activity and has a relatively larger bandgap compared to rutile TiO2. While rutile TiO2 also consists of distorted octahedra but has a different structural arrangement compared to anatase. Rutile is known for its higher stability and durability. Because it has a smaller bandgap, it is less active as a photocatalyst compared to anatase. [125]. The structural differences between anatase and rutile TiO2 result in variations in their electronic band properties, which can impact their photocatalytic performance. Researchers choose between these phases based on the specific requirements of their photocatalytic applications, balancing factors such as activity, stability, and bandgap energy
The differences in the photocatalytic activity between anatase and rutile phases of TiO2 are indeed intriguing and have been the subject of extensive research and investigation. The electronic band structures of anatase and rutile TiO2 play a crucial role in their photocatalytic activity. While the valence band (VB) positions are similar in both phases, the conduction band (CB) potential of anatase is shifted cathodically (toward a more negative potential) by approximately 0.2 V compared to rutile. This small shift in CB potential results in a more favorable driving force for water reduction in anatase compared to rutile, making anatase a more active photocatalyst for hydrogen generation. In most investigations anatase is more active than rutile [130,131,132]. This activity difference can be explained by considering their different electronic band structures. The location of the VBs (mainly originating from O2p) of these two phases is almost the same (situated at ca. 3.0 V) [133], but their CBs (mainly comprising Ti3d) are slightly different (Figure 10). Another contributing factor is the light absorption capability. Rutile TiO2 has a limited ability to absorb light in the UV region, which may reduce its efficiency in photocatalytic reactions that rely on UV light absorption. Anatase TiO2, with its slightly larger bandgap, exhibits better light absorption properties in the UV region [130,132]. The mobility of photogenerated charge carriers (electrons and holes) is also a factor. Rutile TiO2 typically has lower charge carrier mobility than anatase, which can affect the efficiency of charge separation and migration in the material. [134].
To obtain high-activity TiO2 for photocatalytic applications, it is essential to control the fabrication conditions to favor the formation of the anatase phase without the coexistence of the rutile phase. This can be achieved by controlling the calcination temperature during the sol-gel process can favor the formation of the anatase phase [135]. The reaction temperature and solution pH during hydrothermal or solvothermal processes can also be optimized to promote anatase formation [136,137]. Furthermore, higher crystallinity, smaller particle size, and larger surface area of anatase TiO2 are favorable for hydrogen production. These factors can be controlled through various synthesis methods and conditions [138,139]. In summary, proper selection of the TiO2 crystal structure, crystallinity, particle size, and surface area can substantially improve its photocatalytic activity for hydrogen evolution and other photocatalytic reactions. Researchers continue to explore and optimize these factors to enhance the efficiency of TiO2-based photocatalysts for sustainable energy production.
The research and development of TiO2-based photoelectrodes for photoelectrochemical (PEC) water splitting have indeed been a subject of significant interest and investigation. As shown in Table 4, PEC systems with different types of TiO2 have attracted particular interest [117,140,141,142,143]. TiO2 is known for its remarkable chemical stability but presents challenges in PEC systems due to its wide bandgap (Eg = 3.2 eV). This wide bandgap limits its light absorption to the UV region of the solar spectrum and results in significant charge carrier recombination, which can hinder overall PEC performance. Researchers have made various efforts to enhance the light absorption capabilities of TiO2 materials, particularly extending their absorption into the visible region of the solar spectrum. This is often achieved through doping TiO2 with ions or modifying its structure to create heterojunctions. A comprehensive review by Yu et al. reports that TiO2-based photoelectrodes constructed in 1D/3D configurations have shown improved optical response and higher charge carrier separation compared to other configurations. This suggests that the choice of nanoarchitecture is crucial for optimizing PEC performance [140]. According to their survey, surface/interface properties of TiO2 nanotube array photoelectrodes can be enhanced through molecular monolayer modification. Different functional groups, such as alkane, amine, fluoroalkyl silane, and polymer molecules, have been employed for this purpose. Proper modification of these molecular monolayers can improve various aspects of PEC performance, including solid-liquid interface transfer, solid-gas interface transfer, and PEC oxidation reaction kinetics [117].
b. Modification of TiO2 Photocatalyst in PEC Cell
The modification of TiO2 surfaces to enhance its performance for hydrogen production in photoelectrochemical (PEC) cells is a common approach in photocatalysis research. These modifications aim to address limitations such as the absorption of light primarily in the UV region, charge carrier recombination, and fast backward reactions. One effective strategy is to introduce impurities or dopants into the TiO2 crystal structure, which can alter its conductivity and electronic properties[145]. Introducing impurities or dopants into the TiO2 crystal lattice is a way to modify its properties. These dopants can create new energy states within the TiO2 band structure, altering its physical, electronic, and light absorption properties. This modification is achieved by incorporating the dopant atoms into the host TiO2 lattice. Figure 11 illustrates the band diagram of TiO2 after dopant incorporation. The process creates donor levels located above the valence band (VB) and acceptor levels located below the conduction band (CB) of TiO2. These additional energy levels can influence the behavior of charge carriers in the material [146]. One of the primary goals of dopant incorporation is to extend the absorbance edge of TiO2 to the visible region of the electromagnetic spectrum. By doing so, TiO2 becomes capable of absorbing visible light, which is crucial for harnessing a broader range of solar energy in PEC water splitting. Dopants can also act as traps for photogenerated charge carriers (electrons and holes). When charge carriers migrate from the interior to the surface of the photocatalyst, dopants can capture and temporarily hold these carriers. This can help prevent rapid charge carrier recombination and promote more efficient charge separation and utilization in photocatalytic reactions.
Doping transition metals into TiO2 introduces several beneficial effects and mechanisms that can enhance the photocatalytic performance of TiO2-based materials for processes such as hydrogen production in photoelectrochemical (PEC) cells. Here's an overview of these mechanisms and the roles that metal dopants play [147].
1). Electron trap mechanism - Metal dopants can serve as electron or hole trapping sites within the TiO2 lattice. When photons are absorbed by the photocatalyst, they generate electron-hole pairs. These charge carriers need to be efficiently separated to participate in chemical reactions. Metal dopants can capture and temporarily hold electrons or holes, preventing rapid charge carrier recombination. This trapping mechanism prolongs the lifetime of charge carriers, increasing the chances of their participation in desired reactions, such as the reduction of water to produce hydrogen.
2). Creation of new energy levels - Doping transition metals introduce local energy states within the forbidden energy gap (bandgap) of TiO2. These newly created energy levels can facilitate charge transfer processes and narrow the bandgap. Narrowing the bandgap extends the absorbance of TiO2 into the visible light region, making it more effective for absorbing a broader range of solar energy. These local energy states can also influence the movement of charge carriers and enhance their mobility, improving overall photocatalytic activity.
3). Impact on particle size, surface area, and morphology - Metal doping can alter the morphology, crystallinity, particle size, and specific surface area of TiO2 nanoparticles. These changes can have a significant impact on the photocatalytic performance of TiO2. For example, smaller particle sizes and larger surface areas can enhance light absorption and increase the available surface area for photocatalytic reactions. Different morphologies (e.g., nanotubes, and nanorods) can provide more favorable structural configurations for charge carrier separation and migration.
c. Monometallic Doped TiO2
Doping transition metal ions and non-metals into TiO2 photocatalysts has been a successful strategy for improving their photocatalytic activity, especially in processes like hydrogen production [24]. Transition metals such as Fe, Cu, Ni, Cr, and Co have been effectively doped into TiO2 to enhance photocatalytic activity. These metal dopants help minimize electron-hole recombination and improve charge transfer at the interface, leading to higher hydrogen production rates.
The study by Rosseler et al. provides valuable insights into the effects of metal doping and other factors on the photocatalytic activity of TiO2 for hydrogen production by using Au and Pt-doped TiO2 [148]. They showed that the optimum metal loading value 3 wt% Au/TiO2 and Cu-coated TiO2 showed enhanced hydrogen production rates up to 120 μmol.min-1 in methanol (1 v/v%) solution under 150W metal halide lamp which emits a large portion of visible light. This indicates that certain metal dopants can extend the absorption edge of TiO2 into the visible region, allowing it to harness more of the solar spectrum for photocatalysis. The photocatalytic activity of Pt/TiO2 was lower compared to Au/TiO2 and Cu-coated TiO2. This difference may be attributed to Pt's limited ability to extend the absorption edge into the visible region and its high activity in the reverse reaction of hydrogen and oxygen (H2 + 1/2O2 → H2O), even at room temperature. The study highlights that the photocatalytic activity of a catalyst can be fine-tuned by considering several parameters, including:
1). The type and content of the metallic co-catalyst: The choice of metal dopant and its concentration significantly affect photocatalytic performance.
2). Metal-support interactions: The interactions between the metal dopant and the TiO2 support can influence charge transfer processes.
3). Surface properties of TiO2: Factors such as the anatase/rutile ratio and porosity of TiO2 can impact its photocatalytic activity.
4). Sacrificial reagent: The relative amount of methanol added as a sacrificial reagent can affect the overall hydrogen production rate [43].
The study by Alenzi et al. provides valuable insights into the photocatalytic water-splitting performance of Ag/TiO2 compared to pure TiO2 under UV irradiation with 10 mW/cm2 in an aqueous methanol solution [81]. Ag/TiO2 exhibited a significantly higher averaged hydrogen production rate (147.9 ± 35.5 μmol.h-1.g-1) compared to amorphous TiO2 when irradiated with UV light (4.65 ± 0.39 μmol.h-1.g-1). The better performance of Ag/TiO2, specifically the anatase phase, highlights the importance of the crystal structure of TiO2 in photocatalysis. Anatase TiO2 is known for its superior photocatalytic activity compared to the rutile phase, which may explain the higher hydrogen production rate observed [11]. Ag/TiO2 likely exhibited improved performance due to its lower bandgap energy. A lower bandgap allows the photocatalyst to absorb a broader range of light, including visible light, which can contribute to enhanced photocatalytic activity. Ag/TiO2 extended its absorbance edge into the visible region compared to amorphous TiO2. This is a crucial advantage, as visible light constitutes a significant portion of the solar spectrum. Extending the absorbance edge to visible light allows for more efficient utilization of sunlight in photocatalytic reactions.
Indeed, the high cost and limited availability of noble metals like gold (Au) and platinum (Pt) can pose significant challenges for their large-scale application in photocatalytic systems, despite their excellent photocatalytic properties. To address these limitations, researchers have been exploring the use of non-noble metals as alternatives for various photocatalytic applications [53,54,55,56]. The use of cobalt (Co) as a non-noble metal dopant in titanium dioxide (TiO2) photocatalysts has shown promise in various photocatalytic applications, particularly in the context of hydrogen production and other reactions. Sadandam et al. proposed a photocatalyst cobalt-doped TiO2 for efficient photocatalytic hydrogen production [96]. Sadanandam et al. demonstrated that cobalt-doped TiO2 (Co/TiO2) exhibited a remarkable hydrogen production rate of 11, 021 μmol.h-1.g-1in a glycerol aqueous solution under UV-Vis illumination. This high hydrogen production rate indicates the efficiency of co-doping for photocatalytic water splitting [96]. Coordinatively, cobalt sites in Co/TiO2 photocatalysts are effective in suppressing the recombination of photogenerated electron-hole pairs. This is a crucial factor for enhancing the overall photocatalytic efficiency. Kumaraswamy et al. reported cobalt-loaded TiO2 (Co/TiO2) to prepare 2-Aryl Benzimidazoles [149]. The Co-TiO2 catalysts exhibited excellent photocatalytic performance under solar light conditions. This suggests that co-doping can enable the utilization of solar energy for various photocatalytic reactions. The presence of cobalt species in the TiO2 lattice can prolong the lifetime of charge carriers (electrons and holes), allowing them more time to participate in surface reactions, such as the reduction of water to hydrogen and the oxidation of water to oxygen. In addition to cobalt, other non-noble metal dopants like copper (Cu) have also demonstrated high hydrogen evolution rates in conjunction with TiO2 photocatalysts. The study by Montoya et al. highlights the use of Cu-coated TiO2 for efficient hydrogen evolution as high as 8500μmol.h-1.g-1 using continuous monitoring of reactor headspace gases by portable mass spectrometry [150].
The utilization of nickel (Ni) in various forms and as a dopant in titanium dioxide (TiO2) photocatalysts has shown significant potential for enhancing photocatalytic hydrogen production and other related reactions. Nickel particles, particularly at step edges, exhibit a high capacity for breaking C-C bonds. This property is beneficial for hydrogenation and hydrogenolysis reactions of hydrocarbons and plays a vital role in determining the overall catalytic activity of Ni-containing catalysts [151]. Nickel has demonstrated the capability to significantly improve hydrogen evolution rates in TiO2-based photocatalysts. For example, Xia et al. decorated a Ni-imidazole framework on TiO2 nanorods, leading to a remarkable hydrogen evolution rate of 21,712 μmol.h-1.g-1under visible light irradiation. This rate was substantially higher than that achieved with pure TiO2[152]. Ni can also be used as the cocatalyst to improve the hydrogen evolution performance. Su et al. atomically dispersed Ni in zinc sphalerite cadmium-zinc sulfide quantum dots (ZCS QDs) [153]. Nickel-doped TiO2 photocatalysts have shown the ability to utilize visible light (λ > 420 nm), expanding their range of effective light absorption and utilization. This visible light activity is crucial for harnessing sunlight for photocatalytic reactions. The finely tuned Ni SAs dispersed in ZCS QDs exhibit a photocatalytic H2 production activity of 18.87 mmol.h-1.g-1. Nickel dopants have been found to enhance surface engineering, leading to the creation of highly active sites. Additionally, they can contribute to the formation of surface heterojunctions that help reinforce the separation of photogenerated charge carriers. This improved carrier separation is vital for efficient photocatalysis. Furthermore, they suggested that The incorporation of Ni can stabilize the anatase phase of TiO2, preventing the phase transformation from anatase to rutile. This phase stability is essential for maintaining the desired photocatalytic properties. Nickel also has been proposed as an efficient dopant to improve the photocatalytic activity of some semiconductors but it has been rarely studied on TiO2 photocatalyst [154]. Jing et al. studied the photocatalytic hydrogen production of Ni/TiO2 under irradiation of a 300W high-pressure Hg lamp in 20-vol% methanol solution [155]. 1 wt% Ni/TiO2 photocatalyst had maximum hydrogen production rate of 185 μmol.h-1 compared to pure TiO2 due to incorporation of Ni2+ in TiO2 framework and trapping holes. Finally, they concluded that the presence of Ni2+ in the TiO2 framework can increase the thermal stability of the material, ensuring its suitability for prolonged photocatalytic reactions. Ni/TiO2 photocatalysts have been reported to produce mesoporous materials with high surface area and narrower pore distribution. These characteristics facilitate better diffusion of reactants and provide easier access to reactive sites, further enhancing photocatalytic performance.
The research conducted by Dholam et al. on Cr and Fe-doped TiO2 photocatalysts for use in a photoelectrochemical (PEC) cell provides valuable insights into the impact of different metal dopants on photocatalytic activity [156]. The photocatalytic performance of Cr/TiO2 and Fe/TiO2 photoanodes was evaluated in a PEC cell under irradiation from a 250 W tungsten halogen lamp. The study found that the hydrogen production rate for Fe/TiO2 was significantly higher, at 15.5 μmol/h, compared to Cr/TiO2, which had a hydrogen production rate of 5.3 μmol/h. The observed difference in photocatalytic activity between Cr/TiO2 and Fe/TiO2 suggests that the type of metal dopant plays a crucial role in determining photocatalytic performance. In this case, Fe was more effective in promoting hydrogen production compared to Cr. One of the reasons for the variation in photocatalytic activity between Cr and Fe dopants is their ability to trap photogenerated charge carriers. Fe dopants were found to have the capability to trap both photogenerated electrons and holes, whereas Cr dopants primarily trapped holes. Table 5 summarizes the key findings of metal-doped TiO2 developed in recent years for H2 production. The results showed that maximum H2 production is achieved when Cu, Co, and Ni are used as dopants. The photodeposited 3d metal species on the TiO2 surface likely act to reduce electron−hole recombination by facilitating the transfer of photoinduced TiO2 conduction band electrons to protons in solution that are reduced to H2.
d. Bimetallic Doped TiO2
Bimetallic catalysts have shown promise in improving the photocatalytic performance of TiO2 compared to single-doped TiO2. This improvement is attributed to the synergistic effect resulting from the presence of two different metals. The composition, structure, and preparation processes of bimetallic nanoparticles play a crucial role in their catalytic properties. Recently, Bimetallic cocatalysts composed of non-noble metals offer several advantages, including affordability, high stability, non-toxicity, and unique properties that differ from monometallic cocatalysts [157]. The introduction of one metal, such as selenium (Se), can have a significant impact on the Fermi level position of another semiconductor, like cadmium sulfide (CdS). In this case, the Fermi level position of CdS is elevated to a higher position due to the introduction of Se. This change in Fermi level position is crucial because it affects the capture of photogenerated electrons by trapping sites, effectively inhibiting the recombination of electron-hole pairs and prolonging the lifetime of charge carriers. The introduction of Se into CdS also results in a substantial increase in the density of charge carriers in CdS. This higher charge carrier density is advantageous because it promotes the production of more photogenerated electrons and holes, which are essential for photocatalytic reactions. As a result of the elevated Fermi level position and increased charge carrier density, the photocatalytic performance of the resulting bimetallic CdS0.9Se0.1 quantum dots (QDs) is significantly enhanced. These QDs exhibit a remarkable hydrogen evolution rate of 29.12 mmol.h-1.g-1under simulated solar irradiation (320–780 nm) without the need for additional co-catalysts. This rate is approximately 23.5 times higher than that of pristine CdS QDs under the same conditions.
For instance, Zhang et al. prepared a novel photocatalyst by modifying graphitic carbon nitride (g-C3N4) with NiMo bimetallic cocatalysts. This modified photocatalyst exhibited an impressive visible light photocatalytic H2 evolution rate, comparable to Pt/g-C3N4. The NiMo/g-C3N4 photocatalyst was synthesized using a simple one-pot method. The optimized composition of Ni0.4Mo0.6 with a content of 10 wt%, showed the highest photocatalytic H2 generation rate of 1785 μmol.h-1.g-1. This rate was approximately 37 times higher than that of pure g-C3N4 and comparable to the optimized Pt/g-C3N4. [158]. Furthermore, Liu et al. developed a photocatalyst by modifying CdS nanorods with NiCd bimetallic nanoparticles. [159]. The NiCd bimetallic cocatalyst effectively improved the separation and transfer rate of photoinduced carriers, leading to a significantly enhanced photocatalytic H2 production rate. This low-cost and efficient photocatalyst was designed using an ingenious in situ deposition method. The NiCd/CdS nanorods exhibited excellent photocatalytic H2 evolution activity under visible light irradiation (λ > 410 nm), with a corresponding H2 evolution rate of 11.57 mmol.h-1.g-1. This rate was approximately 64.8, 17.2, and 2.3 times higher than that of pristine CdS nanorods, Cd/CdS, and Ni/CdS, respectively.[160]. Altomare et al. fabricated NiCu bilayers on a TiO2 nanotube composite photocatalyst and found that the inclusion of NiCu bilayers significantly enhanced the H2 evolution performance of TiO2 nanotubes [160]. The photocatalytic experiments were conducted under both unfiltered and filtered (420 nm cut-off filter) simulated solar light (AM1.5G). The results indicated that under filtered illumination (λ > 420 nm), the NiCu-TiO2 photocatalyst exhibited negligible H2 generation (0.02 μL.h-1 cm-2). However, under full lamp illumination, the activity significantly increased to 0.28 μL.h-1 cm-2).
The study by Kim et al. highlights the synthesis of an Al-Cu/TiO2 photocatalyst using a solvothermal method for water splitting under UV irradiation in the presence of methanol. This photocatalyst demonstrated significantly enhanced hydrogen production rates compared to other photocatalysts, including Al/TiO2, Cu/TiO2, and pure TiO2 [161]. The Al-Cu/TiO2 photocatalyst exhibited a markedly increased hydrogen production rate of 1093 μmol.h-1.g-1. This rate was significantly higher compared to other photocatalysts, such as Al/TiO2 (312 μmol.h-1.g-1), Cu/TiO2 (900 μmol.h-1.g-1), and pure TiO2 (200 μmol.h-1.g-1) [161]. The study proposed that Cu or Ag components within the TiO2 framework have a greater reduction potential compared to pure TiO2. As a result, these components can capture excited electrons from the valence band of TiO2, thereby hindering the recombination of electrons and holes. This process increases the number of available holes in the valence band, facilitating the continued decomposition of methanol.
The studies conducted by Sun et al. explored the synthesis and photocatalytic properties of doped and bimetallic-doped TiO2 photocatalysts for hydrogen production [162]. Single (Fe and Ni) and bimetallic (Fe-Ni) doped TiO2 photocatalysts were synthesized using the alcohol thermal method. Among the various metal loading ratios, 5% Fe-4% Ni/TiO2 exhibited the highest hydrogen production rate of 361.64 μmol.h-1.g-1. This enhanced photocatalytic performance was attributed to several factors, including improved charge carrier separation, reduced recombination of charge carriers, and a red-shifted absorption edge to 514 nm compared to pure TiO2. Sun et al. successfully synthesized Ag and Fe-doped TiO2 using the solvothermal method [163]. The water photo splitting was conducted in an aqueous ethanol solution using two light sources: a 254 nm Hg lamp with an intensity of 11.7 mW/cm2 and a 500W Xenon lamp equipped with a 400 nm cut-off glass as a visible light source. The Fe-Ag/TiO2 photocatalyst demonstrated a significantly higher hydrogen production rate of 515.45 μmol.h-1.g-1 compared to monometallic doped and undoped TiO2. The enhanced photocatalytic activity was attributed to the interaction of Fe and Ag with TiO2, resulting in a reduction in particle size (approximately 12 nm), a shift in the absorption edge into the visible region (λ ≤ 367 nm), and improved charge carrier separation. These factors collectively contributed to the improved photocatalytic performance.
The research described the synthesis and characterization of a novel CoNi-ZnIn2S4 (CoNi-ZIS) photocatalyst composed of 2D ZnIn2S4 nanosheets and 2D CoNi bimetal nanosheets [164]. The photocatalytic hydrogen production rate of CoNi-ZIS was significantly improved compared to ZIS under visible light irradiation (λ > 420 nm) in the presence of 0.1 M ascorbic acid (AA). CoNi-ZIS photocatalyst was fabricated using a simple hydrothermal and chemical reduction process and the optimized CoNi-ZIS photocatalyst exhibited a photocatalytic hydrogen production rate of 100.1 μmol·h−1, which was approximately 4.3 times higher than that of ZIS. Detailed characterization results indicated that the introduction of CoNi bimetal was the primary factor responsible for the improved photocatalytic performance of CoNi-ZIS. This enhancement was attributed to its ability to accelerate charge migration and inhibit the recombination of photoinduced charge carriers (electrons and holes). The research highlighted the potential of bimetal cocatalysts, specifically CoNi, to enhance the activity of photocatalysts such as ZnIn2S4. CoNi's good electron conductivity and low overpotential, similar to noble-metal Pt, make it a promising cocatalyst for improving photocatalytic performance. This research contributes to the understanding of how bi-metal cocatalysts can play a crucial role in enhancing the efficiency of photocatalysts for hydrogen production, offering potential applications in renewable energy technologies.
To simplify these points, the photocatalytic activity of single-doped TiO2 nanoparticles is affected by electron-hole pair recombination and that of co-doped TiO2 nanoparticles is influenced by both recombination and shielding effect. In summary, the incorporation of dopants into TiO2 is a powerful strategy to tailor its properties for improved PEC performance. By creating new energy states, extending light absorption into the visible region, and mitigating charge carrier recombination, researchers aim to enhance the efficiency of TiO2-based photocatalysts for hydrogen production and other photocatalytic applications.
11. Factors Influencing the Photocatalytic Water Splitting
a. Recombination of Photogenerated Charge Carriers
The recombination of photogenerated electron-hole pairs is believed to be the reason for the low photocatalytic activity of TiO2. Structural imperfections in the TiO2 lattice generate trap sites which may act as recombination centers, leading to a decrease in the levels of electrons and holes. To enhance the TiO2 photocatalysis as well as the response to the visible spectrum of solar light, TiO2 has been impregnated with certain transition metals. Impregnated ions can also act as charge-trapping sites and thus reduce electron-hole recombination. The effect of impregnation on the activity of photocatalysts is governed by several factors, e.g., the type and concentration of dopant, the structure and the initial concentration of the pollutants, and the physicochemical properties of the photocatalyst. Both positive and negative results have been reported from impregnation with metal ions. The increase in charge separation efficiency will enhance the formation of both free hydroxyl radicals and active oxygen species [165]. In the photocatalytic of water splitting, the operating parameters that affect the hydrogen production process include calcination temperature, calcination duration, metal loading, and Co:Ni mass composition. These parameters will be considered one after the other in the processes of photocatalytic water splitting for hydrogen production.
i. Calcination Temperature and Duration
These studies highlight the importance of optimizing calcination conditions to achieve desired photocatalytic performance and underscore the versatility of photocatalytic materials for various applications, including water purification, air pollution control, and hydrogen production. The calcination temperature and duration of TiO2 or impregnated TiO2 photocatalyst could be a prominent factor that could highly influence the properties and structure of the photocatalyst depending on the preparation method. It is well known from the literature that calcination temperature is an important factor that probably influences the crystalline sizes, morphology, and surface area of TiO2 which can clearly affect the photocatalytic activity.
Generally, higher calcination temperature produces larger sizes of TiO2 particles [166,167]. It was reported by Chen et al. that anatase TiO2 was observed at 400°C, while rutile appeared as the main TiO2 phase at 600°C and above [168]. As the temperature increased from 400 to 700°C, the particle sizes also increased causing the specific surface area to decrease dramatically from 106.9 to 26.9 m2g-1. The reported decrease in BET surface area of C-doped TiO2 200 m2g-1 at 200°C to 80 m2g-1 at 400°C is a common phenomenon observed in materials science, particularly in the field of nanomaterials and catalysts. This decrease in specific surface area can be attributed to several factors [169]. At higher temperatures, the material undergoes crystallization, which means that the atoms or molecules in the material arrange themselves in a more ordered and structured manner. In the case of TiO2, it typically exists in an amorphous or disordered state at lower temperatures, and as the temperature increases, it transforms into a more crystalline structure. This transition from amorphous to crystalline reduces the surface area available for adsorption, resulting in a decrease in BET surface area.
The study by Peng et al. on La-doped TiO2 highlights the importance of calcination temperature in the photocatalytic activity of the material. Peng et al. found that at an optimum calcination temperature of 300°C, La-doped TiO2 exhibited the highest degradation of Rhodamine B (RhB) compared to the bare sample (without lanthanum doping) and P25 (a commercial TiO2 material). This enhancement in photocatalytic activity was attributed to the presence of lanthanum ions, which led to the formation of high-thermal-stability mesostructures in TiO2 [168]. Lanthanum doping was reported to increase the surface area of the TiO2 material. A larger surface area generally means more active sites available for catalytic reactions, which can lead to improved photocatalytic performance. The mesostructures formed due to lanthanum doping likely contributed to this increase in surface area. Interestingly, the researchers observed that as the calcination temperature increased from 300°C to 700°C, the photocatalytic activity decreased. This decrease in activity was correlated with a significant reduction in specific surface area, which dropped from 460.6 to 102.1 m2g-1. This decline in surface area was likely due to the processes mentioned earlier, such as crystallite growth and agglomeration. At higher calcination temperatures, particularly at 900°C, the anatase crystallites in the material began to grow extensively and transform into the rutile phase. This phase transformation is known to reduce the photocatalytic activity of TiO2 because rutile is less active than anatase in photocatalytic reactions. Additionally, the mesostructures in the material started to segregate, and, ultimately, the entire mesostructure was destroyed [168]
In the production of hydrogen (H2) by rutile TiO2, Amano et al. observed the deactivation of rutile TiO2 when it was calcined above 500°C [167]. Without calcination, the evolution rate of H2 was 98.9 μmolh-1 and the rate was similar for the samples calcined at different temperatures until 500°C. However, even though an insignificant reduction of specific surface area was observed for the photocatalysts calcined between 500 to 900°C, the evolution rate of H2 monotonically decreased, and further decreased for the sample calcined at 1100°C. They suggested the reduction of oxygen vacancy in TiO2 structure when calcined at higher temperatures as the reason for the low photocatalytic activity of the photocatalyst, which was confirmed by their study when they recalcined TiO2 at 500°C under H2 flow. The photocatalytic activity of the treated sample was significantly improved and the concentration of oxygen vacancies in the TiO2 structure increased with H2 treatment.
The study on the photocatalytic activity of Zn-TiO2 as affected by calcination temperature and duration provides valuable insights into the optimization of photocatalytic materials. The researchers investigated the photocatalytic degradation of Rhodamine B (RhB) using Zn-TiO2 and found that the photocatalytic activity exhibited a significant response to both calcination temperature and duration [170]. The photocatalytic activity rapidly increased and reached an optimum at 500°C with a 1-hour duration of calcination. When the calcination temperature was increased from 300°C to 500°C or the calcination duration was extended from 30 minutes to 2 hours, the photocatalytic activity improved. Interestingly, when the calcination temperature was further increased beyond 500°C or the calcination duration was extended beyond 1 hour, the photocatalytic activity of the Zn-TiO2 catalyst decreased. The researchers attributed this decrease in photocatalytic activity to the transformation of the anatase phase of TiO2 to the rutile phase. Rutile TiO2 has been found to have lower photocatalytic activity compared to anatase TiO2. The transformation of TiO2 from anatase to rutile is a well-known phenomenon. Anatase is the more photocatalytically active phase, while rutile is less active. This phase transition can reduce the number of active sites available for photocatalysis and thus decrease the overall photocatalytic activity.
The study by Zhiyong et al. on the effect of calcination temperature and duration in ZnSO4-TiO2 photocatalysts provides additional insights into how these parameters can impact photocatalytic performance. [171]. In this study, the researchers kept the calcination temperature constant at 500°C, and the variable parameter was the duration of calcination. This approach allowed them to isolate the effect of calcination time on the photocatalytic properties of the ZnSO4-TiO2 catalyst. The results showed that the catalyst treated at 500°C for 1 hour exhibited the best enhancement in the discoloration of Rhodamine B (RhB). This suggests that a specific calcination duration at this fixed temperature was particularly effective in improving photocatalytic activity. They attributed the trend of improved photocatalytic activity to the diffusion of zinc (Zn) ions into the TiO2 lattice. The incorporation of Zn ions into the TiO2 structure can introduce defects and alter the electronic structure, potentially enhancing the photocatalytic properties of the material.
ii. Metal Loading
The photocatalytic water splitting is highly related to the photocatalytic activity of TiO2. It is demonstrated that impregnation of ions onto TiO2 is one way to enhance the photocatalytic activity of TiO2. An important factor that governs the efficiency of the metal-incorporated photocatalyst is the metal content. Thus, the influence of the metal loading on the photocatalytic activity of the photocatalysts was investigated [172].
The optimum metal loading, either mol% or wt% varies depending on the preparation method and the application of the photocatalysts. Fe loading showed an increase in efficiency compared to bare material in the elimination of thiacloprid (TCL), with the optimum Fe content at 7.2 wt% giving complete degradation of TCL within 25 min [172]. The incorporation of 7.2 wt% Fe metal onto the TiO2 increased the surface area thus increasing the adsorption of TCL molecules and absorption of photon for an enhanced process.
Appreciable enhancement in photocatalytic efficiency could be observed with the incorporation of the appropriate amount of metal loading onto TiO2 nanoparticles. Photocatalytic activity increased with the increasing amount of metal loading which could be attributed to the formation of a space charge layer, which can separate the photoinduced electron-hole pairs. As the concentration of metal loading increases, the surface barrier on the surface of TiO2 becomes higher and the electron-hole pairs within the region are efficiently separated by the large electric field. Due to the difference in electron negativity between Ti and the metal, the Ti metal oxide (Ti-O-M) formed via M2+ entering the shallow surface of TiO2 could promote the charges to transfer, increasing photocatalytic activity.
The incorporation of metal ions as dopants into TiO2 photocatalysts indeed plays a crucial role in influencing their properties and photocatalytic activities. The introduction of metal ions can cause lattice deformation and produce defects in the TiO2 crystal structure. These defects can act as active sites for photocatalytic reactions by inhibiting the recombination of electron-hole pairs, which is a key process that can reduce photocatalytic efficiency. [173]. Incorporation of metal ion dopant can also reduce the band gap energy of TiO2 making the photocatalyst more active under visible light irradiation [174,175]. There is typically an optimum loading of metal ions in TiO2 beyond which photocatalytic performance may deteriorate. Excessive metal ion loading can lead to agglomeration during the activation process, resulting in a photocatalyst with a lower surface area. Reduced surface area can limit the availability of active sites for catalytic reactions, leading to decreased photocatalytic activity.[176,177]. When in excess, the existence of a dopant on the particle surface of TiO2 lessens the specific area of TiO2, impedes the adsorption of reactant, and thus, inhibits the photocatalytic activity [170]. Excess dopant loading on the surface of TiO2 particles can not only reduce the specific surface area but also screen the TiO2 from UV light. This screening effect can hinder the efficient transfer of interfacial electrons and holes, which are essential for photocatalysis. Consequently, an excess of surface dopant can result in reduced photocatalytic activity.] [178]. On the other hand, Xin et al. reported that Excessive oxygen vacancies and metal dopant species can become recombination centers for photoinduced electrons and holes. Instead of facilitating photocatalytic reactions, these excess defects may promote recombination, thereby reducing photocatalytic efficiency. Certain dopants, like copper (Cu), when present in excess as P-type CuO species, can cover the surface of TiO2 particles. This coverage can limit the accessibility of reactants to the TiO2 surface, leading to decreased photocatalytic activity [179].
The choice of dopants in photocatalysis and electrocatalysis can significantly impact the performance of materials for various reactions, including the photoelectrochemical (PEC) water-splitting reaction. In this context, Ni and Co dopants have extremely positive effects on PEC water splitting and are good catalysts for electrocatalytic water splitting [162]. Nickel (Ni) and cobalt (Co) are known to be effective dopants for improving the catalytic activity of materials in water-splitting reactions. These transition metals can introduce additional active sites on the surface of the photocatalyst or electrocatalyst, which can accelerate the rate of the water-splitting reaction. Ni and Co are known for their electrocatalytic properties, especially in the context of the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER), which are the two half-reactions involved in water splitting. They can lower the energy barrier for these reactions, making the overall water-splitting process more efficient. The fact that Ni and Co have negative H+ binding energy means that they have a favorable interaction with protons (H+ ions). This favorable interaction can facilitate the adsorption of reactants (such as H2O molecules) onto the catalyst's surface and the subsequent release of reaction products (H2 and O2) from the surface. This dynamic adsorption-desorption behavior is crucial for efficient catalysis. Because Ni and Co dopants promote the efficient adsorption and desorption of reactants and products, they can lead to a higher water-splitting efficiency. This means that a larger fraction of incident light or electrical energy is used to drive the desired water-splitting reaction, rather than being lost to inefficient processes.
Moreover, compared to Cu with positive H+ binding energy, it is less efficient at proton adsorption and subsequent water-splitting reactions [180]. This can result in a lower affinity for protons (H+ ions), making it less efficient in facilitating the initial step of the water-splitting reaction, which involves the adsorption of H+ ions. In contrast, Ni and Co have negative H+ binding energy, indicating that they have a more favorable interaction with H+ ions. This favorable interaction allows them to readily adsorb H+ ions on their surface, creating active sites for subsequent reactions. With Cu, because of its less favorable H+ binding energy, the adsorption of reactants (H2O molecules) and the release of products (H2 and O2) on the catalyst's surface may not occur as efficiently. This can lead to a lower water splitting efficiency as reactants may not readily attach to the catalyst or products may not easily detach from the surface. Overall, metal dopants play a crucial role in tailoring the properties of photocatalytic and electrocatalytic materials, making them more efficient for a wide range of applications, including water splitting. The choice of metal dopants and their loading levels should be carefully considered to optimize the performance of these materials for specific catalytic reactions.
iii. Metal Mass Composition
There have been several literatures that report on the beneficial effects of co-modification (bimetallic) of metals on TiO2 nanoparticles [181,182,183]. Wongwisate and Liu prepared Ag/TiO2 co-modified with Au and Ce, respectively, via sol-gel method [181,184]. A similar amount of Ag precursor (0.1%) was added during the synthesis of the photocatalyst. In their study, Liu fixed the amount of Ag and varied the amount of Ce to investigate the effect of different Ce:Ag ratios. The optimum amount of Ce was observed at 0.1% for photocatalytic removal of ammonium and nitrite in water. On the other hand, Wongwisate added 0.1% of Au to produce an Au-Ag/TiO2 photocatalyst for the degradation of 4-chlorophenol. In contrast to the encouraging enhancement observed for Ce-Ag/TiO, bimetallic photocatalyst, Au-Ag/TiO2 showed insignificant enhancement for photocatalytic activity compared to monometallic Ag/TiO2. This may be due to the inappropriate amount of Au added during the synthesis. The BET surface areas of both photocatalysts were similar, giving a similar performance. Monometallic Au/TiO2 with a lower amount of Au (0.05%) gave a similar performance as 0.1% Au/TiO2, but the lower Au content produced smaller particles. Thus, it could be suggested that a smaller amount of Au dopant for bimetallic Au-Ag/TiO2 would enhance the photocatalytic activity.
Production of hydrogen from methanol over Au-Cu/TiO2 photocatalyst was investigated by [185]. Bimetallic photocatalyst (1.0 wt% Au, 1.0 wt% Cu) displayed the best performance compared to 2.0 wt% Au or 2.0 wt% Cu. The synergistic effect of the co-modification was the reason for the enhancement. The particle size of Au in 2.0 wt% Au photocatalyst was 3.1 nm, larger compared to that in the bimetallic photocatalyst (2.6 nm). Thus, it could be concluded that a higher amount of Au in the photocatalyst produced a larger particle-size photocatalyst. This observation agrees with the lower performance of the Au-Ag/TiO2 photocatalyst prepared by Wongwisate [184].
The study conducted by Gao et al. on bimetallic photocatalysts for the reduction of nitrate highlights the potential advantages of combining multiple metals with TiO2 to enhance photocatalytic activity [186]. The researchers investigated the use of various metals, including Pt, Pd, Cu, and Ni, as monometallic dopants on the surface of TiO2, as well as the combination of Pt and Cu or Cu and Ni in bimetallic photocatalysts. Among the tested photocatalysts, Cu-Ni/TiO2 exhibited the highest photocatalytic activity for the reduction of nitrate. This was determined based on the highest yield of nitrite and ammonia, indicating an effective reduction of nitrate ions. The superior photocatalytic performance of Cu-Ni/TiO2 was attributed to the synergetic effects of having two different metals present. These metals effectively trapped both photo-generated electrons and holes, preventing their recombination. The separation of electrons and holes is crucial for promoting catalytic reactions. One of the critical aspects of photocatalysis is the efficient separation of photo-generated electron-hole pairs. The presence of Cu and Ni on the dispersion of metal dopants on the surface of TiO2 was noted as a beneficial factor in improving the photocatalytic activity of the bimetallic photocatalyst. Well-dispersed metal ions provide more active sites for catalytic reactions, enhancing the overall efficiency. surface facilitated this separation, leading to a higher yield of nitrite and ammonia.
Photoluminescence (PL) analysis conducted by Zhang et al. revealed that upon Cu and Fe co-modified onto TiO2, the PL emission spectra significantly decreased [182]. The PL emission spectra described results from the recombination of free carriers, thus higher spectra showed that the recombination rate is faster. During the study, several photocatalysts with different Cu:Fe molar ratio was analyzed. The lowest PL spectrum was obtained from 1.2%Cu-1.8% Fe/TiO2 photocatalyst, which also gave the best performance in photocatalytic degradation of methyl orange. Thus, the "appropriate amount" of co-dopants is crucial. Too little may not effectively improve charge separation, while too much may lead to undesirable effects, such as charge trapping or increased recombination. Researchers often optimize the dopant concentration to achieve the best photocatalytic performance.
b.Photoanode Instability Mechanism
A comprehensive understanding of the factor that contributes to the photo corrosion of semiconductor photoanode materials in PEC water-splitting applications is provided for designing and fabricating photoanodes with excellent performance. Photoanodes are typically made from semiconductor materials, including metal oxides, metal sulfides, metal nitrides, and organic materials. The stability of the photoanode largely depends on the intrinsic properties of the semiconductor material used. [187,188]. Therefore, the stability of the photoanode depends on the semiconductor material. If the semiconductor material corrodes during PEC water splitting, the stability of the photoanode will be poor. In other words, no matter what the material is, the water-splitting process of the semiconductor device as a photoanode is completed by the above three steps (light absorption process, bulk separation process, and surface catalytic process). Under this circumstance, the reasons for the degradation of photoanode materials have common points, and here, we mainly summarize the common causes of photoanode corrosion. Regardless of the photoanode semiconductor material, the main causes of photoanode instability under the PEC test are as follows.
First, the dissolution rate of the photoanode during PEC testing is determined by photogenerated holes. Holes weaken the chemical bonds between adjacent atoms, making them susceptible to dissolution when interacting with affinity reagents in the solution. Narrow-bandgap semiconductor materials, which have higher concentrations of photogenerated carriers, are more susceptible to photo corrosion [189,190]. This susceptibility is why narrow-bandgap photoanodes are very unstable, while wide-bandgap photoanodes are relatively stable [191,192]. Consequently, the choice of energy band gap (Eg) and the light absorption ability of the semiconductor material are critical for photoanode stability. Wide-bandgap photoanodes tend to be more stable than narrow-bandgap ones.
Second, the choice of electrolyte solution is crucial for the chemical stability of the photoanode. Many photoanode materials are unstable in acidic electrolyte solutions, so selecting the appropriate electrolyte can impact stability [193]. The choice of electrolyte can significantly affect the chemical stability of the photoanode. Different electrolytes, such as Na2SO4, Na2SO3, KOH, HCl, and others, can have varying impacts on photoanode stability.
Third, photoanode stability is also closely related to the relationship between the self-oxidation potential of the semiconductor material and the oxidation potential (Φox) of the photoanode [194]. In general, photochemical corrosion of semiconductors depends on the band alignment of the reduction potential (Φre) relative to the conduction band minimum (CBM) or the Φox relative to the valence band maximum (VBM). Therefore, the photoanode has a suitable energy band structure position and can have better stability performance.
Fourth, photogenerated holes generated by light are transferred to the semiconductor surface and undergo an oxidation reaction with water during PEC testing. If the photogenerated holes accumulate for various reasons (such as interface recombination or excessive surface states) at this time, the holes photogenerated too late to transfer will generate oxidation reactions with the semiconductor material, causing deactivation of the photoanode [195]. Regardless of whether the photoanode material is a metal oxide or a metal sulfide, photo corrosion is due to several of the above causes. Except for the above reasons, since each semiconductor material has unique properties, the deactivation mechanism of each photoanode has a certain anisotropy. The instability of BiVO4 during the PEC process is not limited to the above common causes, and the dissolution of V is also an important factor in photo corrosion [196].
In summary, understanding the factors contributing to photo corrosion in semiconductor photoanode materials is essential for designing and fabricating photoanodes with excellent performance in PEC water splitting and other photoelectrochemical applications. Researchers must carefully consider the choice of materials, energy band structure, electrolyte, and other parameters to optimize photoanode stability for specific applications. Optimization of PEC cells for hydrogen production involves fine-tuning various parameters. Some of the critical parameters mentioned include sintering temperature, photoanode thickness, and geometrical area. These parameters can significantly affect the efficiency of hydrogen production in a PEC cell. The seminal report on PEC water splitting, in 1972, used a thin layer of TiO2 (Eg ~3.2eV) as a photoanode for a PEC half-cell. Since then, photoanodes have been studied extensively with several material modifications and fabrication techniques. In a PEC cell, the photoanode plays a crucial role in the oxygen (O2) evolution, while the hydrogen (H2) evolution takes place on the counter electrode [114].
N-type semiconductors are preferred for photoanode applications because they are good conductors of heat and have a high concentration of free electrons. These free electrons can efficiently carry away heat and participate in photocatalytic reactions, making them suitable for PEC cells. The n-type semiconductors, such as TiO2 (3.2–3.4 eV) [197] BiVO4(2.2–2.4 eV) [198], WO3 (2.6–3 eV), g-C3N4 (2.5–2.8 eV) [199], CdS (2.2–2.4 eV) [200], SrTiO3 (3.2–3.4 eV), and Fe2O3 (2–2.2 eV) have been widely applied for the photoanode application in the PEC cell. Among all of these materials, TiO2 is noted for its stability, which is an important characteristic for long-term PEC cell operation. On the other hand, BiVO4 is known for its abundance and effective light absorption in the visible region of the electromagnetic spectrum.
The choice of photoanode material depends on the specific requirements of the PEC cell and the balance between stability and light absorption. In summary, optimizing PEC cells for photocatalytic hydrogen production involves a careful selection of photoanode materials and the fine-tuning of various parameters. The choice of material depends on factors like stability and light-absorbing capacity. Additionally, n-type semiconductors are commonly used as photoanodes due to their favorable electronic properties. These advancements in PEC cell technology are critical for the development of sustainable and clean energy sources.
i. Sintering Temperature of Photoanode
The optimization of sintering temperature in the fabrication of photoanodes for photoelectrochemical (PEC) water splitting is a critical factor that influences the performance and stability of the photoanode. Sintering is a process that electrically connects the components of the photoanode paste composition. It ensures that electrons injected into the film can be collected efficiently at the underlying conductive substrate. This connection is crucial for the transport of charge carriers (electrons) within the photoanode. Optimizing the sintering temperature is desirable to achieve good adherence of the photoanode to the substrate. This adherence enhances the photostability of the photoanode, ensuring that it remains intact during PEC water splitting. The sintering process also plays a role in eliminating organic residues present in the paste composition. Removing these residues is essential for improving the connection between nanocrystallites in the thin film, which, in turn, facilitates efficient charge transport [201]. Impedance spectra analysis demonstrates that charge transfer resistance (Rct) increases with higher sintering temperatures. This increase in Rct can have implications for charge transport and recombination processes in the photoanode. [202]. Sintering temperature influences the density of bulk trap and surface states within the TiO2 material. These states can affect the recombination process of electron-hole pairs. The collapse of the porous structure at temperatures beyond 400°C can introduce new surface states, leading to higher charge transfer resistance [203].
Robabeh et al show that the increasing sintering temperature had a negative effect on the photocurrent density and reaction rate on the electrode and electrolyte interface due to high Rct and higher recombination rate [204]. This also can be explained by the destruction of the crystal structure of the photocatalysts, which led to an acceleration of electron-hole pair recombination and a decrease in the rate of electron transfer between the molecules, resulting in high photo corrosion of the photoanode [205]. A study conducted by Basu et al. shows that CuO PPy calcined at 300°C for 3 hours can generate a maximum photocurrent density of 2.76 mA/cm2 at an applied potential of 0.1 V vs. RHE whereas CuO PPy calcined at 400°C and 500°C for 3 hours can generate photocurrent density of 2.05 and 1.87 mA/cm2, respectively [206]. It is clear that with an increase in the calcination temperature, the PEC activity of CuO PPy decreases and the CuO PPy calcined at 300°C for 3 hours is the most efficient one; thus the optimum condition for calcination is determined.
A study concluded that several photoanodes exhibited relatively stable photocurrent densities during long-term operation. This stability is crucial for the practical application of PEC cells. Increasing the calcination temperature during photoanode preparation had a notable impact on average photocurrent densities. Initially, as the calcination temperature increased, average photocurrent densities increased. This increase can be attributed to specific factors related to the photoanode's structure and properties. The average photocurrent densities of about 46.4 μA/cm2 for the TNT-450 photoanode were superior to that of about 26.5 μA/cm2 for the TNT-350 photoanode [127]. The study identified a synergetic effect between the plentiful pore structure and improved crystallinity of the TNT-450 photoanode. This synergy likely contributed to the higher average photocurrent densities observed in this photoanode compared to the TNT-350 photoanode. The presence of abundant pores can enhance the specific active surface area, potentially increasing the number of active sites for PEC reactions. While factors like pore structure and crystallinity can enhance PEC performance, the study also emphasized the importance of charge carrier transport. In the case of the TNT-350 photoanode, despite its abundant paths for VO2+ and proton transfer, limited by poor crystallinity, the transport of photo-excited electrons was inhibited [207]. The inhibited electron transfer in the TNT-350 photoanode suggested an aggravated recombination of electron-hole pairs. Efficient charge carrier transport is essential for preventing the recombination of these pairs, as recombination leads to a reduction in PEC efficiency. The findings underscore the need to strike a balance between various properties in photoanode design. While factors like pore structure and crystallinity can enhance performance, they must be optimized to avoid hindering charge carrier transport and increasing recombination rates.
Ling et al. also studied about stability of the photoanode using the sintering temperature parameter of the photoanode for photoelectrochemical water splitting [208]. The sudden drop of photocurrent for the Sn-doped hematite nanowires sintered at 850°C can be ascribed to the considerably increased resistivity of the FTO substrate. The hematite nanowires sintered at 800°C exhibited a pronounced photocurrent density of 1.24 mA/cm2 at 1.23 V vs. RHE and reached a maximum photocurrent density of 1.95 mA/cm2 at 1.6 V vs. RHE. This indicates that the choice of sintering temperature must consider the electrical properties of the substrate. In summary, optimizing the sintering temperature in the fabrication of photoanodes is a critical step in achieving efficient and stable photoelectrochemical water splitting. The choice of temperature affects charge transport, recombination rates, and the overall performance of the photoanode in PEC cells.
ii. Photoanode Thickness
The influence of photoanode thickness on the performance of TiO2 and other materials in photodegradation and water-splitting applications is a crucial aspect of photoelectrochemical (PEC) cell design [209]. Increasing the thickness of the photoanode from 12 to 24 μm can lead to a reduction in charge transfer resistance (Rct). This reduction in Rct suggests that thicker photoanodes can enhance interfacial adhesion between the photocatalyst nanoparticles (e.g., Cu-Ni/TiO2) and the substrate (e.g., FTO) while also improving stability in an alkaline solution. Lower Rct is generally desirable in PEC cells as it facilitates efficient charge transfer [210]. Thicker photoanodes can generate more excited electrons due to improved light absorption. The increased thickness allows for more photons to be absorbed, resulting in a greater number of photo-excited electrons available for the photocatalytic reaction. This can lead to higher photocurrent and improved PEC efficiency [211] There appears to be an optimal thickness for photoanodes beyond which performance starts to deteriorate. Bennani's conclusion about thicker photocatalyst layers increasing resistance to electron transport and electron-hole recombination aligns with this observation [212]. Beyond the optimal thickness, longer path lengths for photogenerated electrons, increased electron diffusion distances, and more grain boundaries (potential sites for electron-hole recombination) can negatively impact PEC performance.
Miller et al. also discussed the photocurrent density as a function of film thickness [213]. They found that as the film thickness continues to increase, film resistivity can also increase. This higher resistivity can impede charge transport, making it more difficult for electrons to move efficiently within the photoanode material. Excessive charge carriers and longer equilibration times can further hinder performance [214,215]. The increase in the volume of grain boundaries, which are often regions where electron-hole recombination can occur, can be a limiting factor in thicker photoanodes. While grain boundaries are important for charge separation in some cases, an excessive volume can lead to increased recombination rates and reduced photocurrent density [216]. In summary, optimizing the thickness of a photoanode is crucial for achieving the best performance in PEC cells. Thicker photoanodes can enhance light absorption and generate more excited electrons, but there is an optimal thickness range where these benefits are maximized. Beyond this range, increased film resistivity, longer electron diffusion distances, and greater grain boundary volume can lead to decreased performance. Therefore, the choice of photoanode thickness should consider a trade-off between enhanced light absorption and efficient charge transport to achieve optimal PEC efficiency.
iii. Photoanode Geometric Area
The geometric area of the photoanode is a crucial factor in the performance of a PEC cell for hydrogen production. It directly affects the amount of hydrogen generated, making it an essential parameter to study and optimize. A study found that as the photoanode's geometric area increased from 0.25 cm² to 1 cm², hydrogen production increased from 3.8 to 10 mL. This indicates that a larger photoanode surface area can capture more light and facilitate higher hydrogen production [217]. Interestingly, when the geometric area was further increased from 1 cm² to 2 cm², there was a decrease in hydrogen production from 10 to 9.5 mL. This suggests that there is an optimum geometric area beyond which increasing it does not lead to a proportional increase in hydrogen production, and in fact, it can have a negative impact. Besides, Mishra and Lee also mentioned that increasing the geometric area beyond the optimum can have negative effects [218,219]. One possible reason cited is that it can result in an increased number of defect states at grain boundaries or surface defects, which, in turn, leads to higher charge carrier recombination rates and lower photocurrent density. Essentially, the excess surface area may introduce defects that hinder the efficiency of the PEC cell. On the other hand, at very small geometric areas (0.25 cm²), the low hydrogen production is attributed to factors such as insufficient light absorption, limited migration of excited electrons to the photoanode/electrolyte interface, and the instability of the photoanode. These issues can limit the effectiveness of the PEC cell at small scales [145]. In summary, determining the optimum geometric area for the photoanode in a PEC cell is a critical aspect of optimizing hydrogen production. Too small an area may result in inefficiencies due to limited light absorption and electron migration, while too large an area can introduce defects and reduce overall performance. Finding the right balance is essential for maximizing the efficiency of the PEC cell.
c. Photocurrent Density Affects the Hydrogen Production
In the design of photoelectrodes for tandem PEC devices, there is a dilemma between achieving high transparency and high photocurrent density. The front cell of tandem devices needs to efficiently absorb sunlight but also be transparent enough to allow light to reach the rear cell for unaided operation. This balance is crucial for achieving high solar-to-hydrogen (STH) efficiency beyond 10%. In analysis, potential systematic errors have been discussed in typical, laboratory-scale PEC measurements, their impact on reported performance figures, and the implications on research strategy [60]. The focus of the previous study is tandem devices should have the prospect for superior STH efficiency and greater complexity.
Döscher et al. proposed a standardized PEC characterization to provide crucial insights and guidance for developing tandem devices [220]. Photocurrent density is a critical parameter in PEC characterization. It is complementary to the measurement of incident photon-to-current efficiency (IPCE) and provides insights into the solar generation potential of a photoelectrode. Proper consideration of photocurrent density is essential for assessing the performance of photoelectrodes.
To improve the efficiency of the PEC process, it is essential to increase the conductivity and reduce the resistance of the photoelectrodes. High conductivity facilitates the flow of charge carriers and can lead to higher photocurrent densities, contributing to improved efficiency in solar-to-hydrogen conversion [213]. Meanwhile, Chiang et. al said that the result of 2wt.% and 0.5 at.% doped CuO film photocathodes are also included [221]. Fitting these results, a correlation between the photocurrent density and conductivity of the film electrode is found to be y = 11.48x0.2478 where y is the photocurrent density and x is the conductivity, over a range of conductivity ca. 109–104 S/cm and a photocurrent density range of ca. 0.1–1.5 mA/cm2. As conductivity increases or decreases, there is a corresponding change in photocurrent density. However, beyond a certain conductivity threshold, further increases in conductivity do not linearly increase photocurrent density. This suggests that other factors, such as surface morphology and the availability of surface area for the water-splitting reaction, become limiting factors for efficiency. This is because once the conductivity reaches a certain level where it is no longer the limiting factor, surface morphology becomes more critical. The efficiency of the PEC process depends on the availability of surface area for photon-generated electrons to react with water. Inadequate surface area can limit efficiency, even in highly conductive photoelectrodes. In summary, achieving high STH efficiency in tandem PEC devices requires careful consideration of factors such as transparency, photocurrent density, conductivity, and surface morphology. Standardized testing methods and a deeper understanding of these parameters are essential for the development of efficient solar water-splitting devices for hydrogen generation. In the photocatalytic of water splitting, the reaction parameters that affect the hydrogen production process include the distance between two electrodes, depth of electrode immersion, illumination intensity, and type of sacrificial agent. These parameters will be considered one after the other in the processes of photocatalytic water splitting for hydrogen production.
i. Distance Between Two Electrodes
This section discusses the importance of the distance between electrodes in various electrochemical and photoelectrochemical processes, particularly in the context of water electrolysis and dye-sensitized solar cells. Equation (2.7) shows that electrical resistance (R) is inversely proportional to the cross-sectional area (A) and directly proportional to the length of the current path (l) through the material. Therefore, reducing the distance between electrodes can lower electrical resistance, which is beneficial for efficient electron transfer. Nagai et al. explored the effects of the space between electrodes in water electrolysis.[222]. They found that reducing the distance too much can lead to void fracture caused by gas bubble formation. This effect is more pronounced at higher current densities and can decrease the efficiency of the electrolysis process.
where R is the electrical resistance, ρ is the material resistivity, A is the cross-section area and l is the length of the current path. Electrons start their travel from the surface of an electrode, move through the electrolyte and finally end their journey to the surface of the other electrode.
In the context of hydrogen production through photoelectrochemical processes, reducing the distance between electrodes can lead to higher current densities at lower voltages. This improvement is attributed to the decrease in electrolyte resistivity with reduced electrode distance. Controlling the distance between electrodes can also help reduce overpotential, which can restrict the electrolysis process [223]. Yuzer et al reported that the higher current densities are obtained with lower voltages when the electrode distance is reduced. This improvement is caused by the decrease of electrolyte resistivity by decreasing the distance [224]. Hernandez et al also concluded that the overpotential could be reduced by controlling the distance between the two electrodes of the PEC water-splitting cell and the high overpotential anode restricts the electrolysis process in the cell [225]. The longer the distance between the electrode, the tougher the situation should be for OH- ions to move from one electrode to another and releases their electrons [226]. When the space between the pair of electrodes decreases, the performance of the cell increases due to the resulting decreases in electrical resistance between the electrodes which in turn leads to an increase in the electrical current.
Recently, Riegel et al. Riegel et al. conducted a detailed examination of bubble diffusion, convection, and transportation between electrodes in the context of water electrolysis. Bubbles are a common byproduct of the electrolysis process, and their behavior can impact the overall efficiency of the process [227]. Previous research has successfully explained the effects of bubbles on water electrolysis efficiency, but primarily at low current densities or in cases where the electrode spacing is relatively large. This suggests that under such conditions, the impact of bubbles on efficiency is more straightforward [228]. It is easily postulated, however, the researcher proposed that there is likely an optimum electrode spacing under high current density conditions. In scenarios where the current density is high and the electrode spacing is small, there is a tendency for the void fraction (the volume of space occupied by gas bubbles) between the electrodes to increase. As the void fraction increases, it leads to higher electrical resistance between the electrodes, which can decrease the efficiency of the water electrolysis process. When the electrode spacing is reduced to a small distance, it can create conditions where gas bubbles accumulate and occupy a significant portion of the space between the electrodes. This accumulation of bubbles can hinder the flow of electrons and ions, increasing electrical resistance and, subsequently, reducing the overall efficiency of the electrolysis process [222].
If the distance between the electrodes becomes long, namely, a large amount of electrolyte exists in the dye-sensitized solar cell, the amount of the impurities adsorbed on the TiO2 surface and in the electrolyte close to the TiO2 surface are expected to decrease relatively. Thus not many of the photoinjected electrons are consumed by the recombination reaction with impurities, leading to a longer effective electron lifetime. The longer electron lifetime in TiO2 anticipated an increase in photocurrent and photovoltage of cells [229]. Silvia also mentioned that by reducing the distance between the electrodes, the effective contact and transmission between the produced micro-bulbs in the cathode (H2, as a factor for floating the formed flocks), and anode (coagulant agent), would become more efficient and the release of the anode surface would be made in a better manner [230]. In summary, the distance between electrodes is a crucial parameter in various electrochemical and photoelectrochemical processes. Finding the right electrode spacing is essential to optimize the efficiency of these processes, and the optimal distance can vary depending on the specific application and conditions.
ii. Depth of Electrode Immersion
It is found that the higher efficiency of water electrolysis is observed when the height of the electrodes is smaller, especially at high current densities. [222]. This finding can be explained by the fact that, with smaller electrode heights, the average void fraction between the electrodes is reduced. In simpler terms, there is less space for gas bubbles (hydrogen and oxygen) to accumulate between the electrodes. This is beneficial for reducing electrical resistance and increasing the efficiency of the electrolysis process. Fouad et al. also noted that increasing the height of electrodes exposes more electrode area for the reaction to occur, which can lead to a higher rate of mass transfer. In other words, a larger electrode surface area allows for more efficient interaction between the electrolyte and the electrodes, promoting the transport of ions and gases involved in the electrolysis process [231]. Reducing the depth of immersion of the cathode rod (the depth to which the electrode is submerged in the electrolyte) reduces the surface area in contact with the electrolyte. As a result, this reduction in surface area increases the effective resistance of the cell [232]. To maintain a constant current in the electrolysis cell (as described by Ohm's law, V = IR), an increase in potential (voltage) is required when the effective resistance rises. In summary, the height of the electrodes in a water electrolysis system plays a crucial role in determining the efficiency of the process, particularly at high current densities. Smaller electrode heights reduce the void fraction between electrodes, minimizing the accumulation of gas bubbles and reducing electrical resistance, leading to higher efficiency. Additionally, increasing the height of the electrodes can expose more surface area for reactions, facilitating mass transfer, but it may also increase effective resistance, necessitating a higher voltage to maintain a constant current. The optimal electrode height depends on the specific conditions and goals of the electrolysis system.
iii. Illumination Intensity
Light intensity is a crucial parameter in photocatalytic water-splitting systems and PEC cells. It directly affects the rate of photon absorption, which in turn impacts the generation and behavior of charge carriers (electrons and holes) in the photoactive materials. To date, there has been minimal research conducted to date on the effect of light intensity on photocatalytic water-splitting reactions. This suggests that the impact of light intensity on such reactions is not yet well understood [233].
Robabeh's research findings indicate that the relationship between hydrogen evolution and light intensity is not linear. Up to a certain point (100 mW/cm²), increasing light intensity has a positive linear effect on hydrogen production. This suggests that at lower light intensities, charge carrier consumption through chemical reactions on the surface of the photoanode is faster than charge carrier recombination [234]. However, beyond 100 mW/cm², increasing light intensity has an antagonistic effect on hydrogen production due to factors such as increased charge carrier density, recombination rate, charge transfer resistance, and saturation of photocurrent density [235].
In addition, the limitation of the charge transfer rate from the photocatalyst to the electrolyte can lead to insufficient reactants at the electrolyte/photocatalyst interface, high resistance at this point, and non-linear hydrogen reduction with increasing light intensity. This suggests that there is an optimal range of light intensities for efficient charge transfer and hydrogen production [259]. In another perspective, light intensity also affects DSCs. Increasing light intensity above 100 mW/cm² can reduce the voltage and efficiency of DSCs. This reduction may be attributed to factors such as increased charge carrier recombination and reduced average electron lifetime. Adachi et al. reported that increasing light intensity to more than 100 mW/cm2 reduces the voltage and efficiency of DSC [207]. In addition, Kim et al. reported that higher cell operating temperatures can reduce electrolyte viscosity and charge transfer resistance between the counter electrode and electrolyte. However, this may also lead to increased charge carrier recombination and reduced DSC performance due to larger ion mobility in the liquid electrolyte [236]. In summary, the relationship between light intensity and the efficiency of photocatalytic water-splitting reactions and DSCs is complex. While lower light intensities may promote efficient charge carrier utilization, higher intensities can lead to various limiting factors, including charge carrier recombination and increased resistance, impacting overall performance. The optimal operating conditions depend on the specific system and the balance of these factors.
iv. Sacrificial Agent
This section discusses the use of sacrificial agents or hole scavengers in the context of photocatalytic hydrogen production from water. It also touches upon the influence of pH and the choice of reaction conditions on the efficiency of hydrogen production. Sacrificial agents are compounds added to the electrolyte in photocatalytic systems to reduce charge carrier recombination, thus improving the efficiency of hydrogen production. These agents react irreversibly with photogenerated holes, preventing them from recombining with electrons and facilitating the conversion of water to hydrogen. Both inorganic and organic compounds, such as Na2CO3, NaOH, formic acid, acetic acid, methanol, ethanol, 2-propanol, formaldehyde, and glycerol, have been reported as attractive sacrificial agents for enhancing photocatalytic hydrogen production from water [237].
Studies have shown that adding alkaline hydroxides like KOH and NaOH to the aqueous solution can enhance the efficiency of photo-splitting water to produce hydrogen and oxygen. This enhancement is observed over specific photocatalysts, such as Ni-doped K4Nb6O17 [238]. Arakawa and colleagues observed that the addition of Na2CO3 to the reaction system resulted in an enhancement of the hydrogen production rate. This enhancement is an important finding because it suggests a way to improve the efficiency of hydrogen generation in photocatalytic or photoelectrochemical processes. They identified an optimum amount of Na2CO3 for maximum hydrogen production rate. This optimal amount was determined to be 0.73 moles of Na2CO3 added to the reaction system and the pH conditions of the reaction system were adjusted to pH=11. This pH level appears to be favorable for the hydrogen production process in the presence of Na2CO3. They concluded that under the specified conditions (optimal amount of Na2CO3 and pH=11) over the Pt/TiO2 catalyst, the rate of hydrogen production reached 568 moles per hour. This indicates a relatively high efficiency in generating hydrogen gas from water through photocatalysis [239].
Many researchers have attempted to produce hydrogen simultaneously with the degradation of organic waste or pollutants instead of the expensive sacrificial agent. Patsoura et al. reported a single photocatalytic system of the photocatalytic reduction of water to hydrogen and oxidation of azo-dyes as a pollutant over Pt/TiO2 which enhanced the hydrogen production rate together with significant dye degradation [240]. Sadanandam et al. reported a comparison of photocatalytic hydrogen production over cobalt (Co) doped TiO2 in pure water and the presence of different alcohols such as methanol, ethanol, glycerol, ethylene glycol, and isopropanol under solar irradiation [149]. The amount of produced hydrogen showed that the presence of glycerol enhanced photocatalytic hydrogen production compared to pure water and other alcohols. It was concluded that the hydrogen production rate in the presence of alcohol depends on the alcohol polarity.
The pH of the solution plays a crucial role in hydrogen production [241]. This aspect is especially important in the presence of a sacrificial reagent. In general, weak alkaline solutions (pH > 10) are found to be more efficient for hydrogen production than strongly acidic or strongly alkaline ones. However, the specific pH conditions may depend on the photocatalyst and the reaction system. The concentration of protons (H+) in the solution affects the reaction rate, with more acidic conditions generally leading to higher reaction rates. Moreover, the pH level can also influence the corrosion stability of photocatalysts. For example, the efficiency of hydrogen production on a CuOx/TiO2 heterostructure was found to be highest in a slightly alkaline environment (pH 10) and lowest at pH 2. This is due to the stability of Cu(I) on the surface of TiO2 in different pH conditions [242]. Changes in pH can lead to modifications in the bandgap of the catalyst, which can further impact its photocatalytic activity. The bandgap determines the energy levels of the catalyst and influences the absorption and utilization of photons for the water-splitting reaction.
In summary, the choice of sacrificial agents, the pH of the solution, and the specific photocatalyst used all play significant roles in optimizing the efficiency of photocatalytic hydrogen production from water. Understanding these factors and their interactions is crucial for the development of efficient and sustainable hydrogen production methods.
12. Dye Solar Cell (DSC)
This section provides detailed information about dye-sensitized solar cells (DSCs) and their characteristics, including their structure, operation principles, and challenges associated with their scaling up and commercialization. It also discusses the potential of coupling typical photoelectrochemical (PEC) cells with photovoltaic (PV) cells to achieve unbiased solar water splitting. In this configuration, the PV compartment is expected to provide the necessary photovoltage to compensate for any deficiencies in the PEC cell, resulting in efficient hydrogen production from water. PV technologies are described as a means to convert abundant and clean solar energy into electrical energy. PV systems have gained significant attention as a sustainable source of electricity [243,244]. Of the PV technologies, Si-based PV systems, particularly those based on crystalline silicon, have dominated the global PV market for several decades. This dominance is attributed to several beneficial features of Si-based PV systems, including their efficiency in electricity generation under full sunlight and stability in various climatic conditions [243,244]. Despite their advantages, Si-based PV systems have certain drawbacks. These include energy-intensive production processes, which can impact their environmental footprint. Additionally, Si-based PV systems are often criticized for their aesthetics, which may limit their integration into building designs. Moreover, these systems tend to have lower photovoltaic performance in low-light conditions. The limitations of Si-based PV systems have restricted their widespread use in various applications. For example, they may not be the ideal choice for building integrated photovoltaics (BIPV), portable electronics, or indoor applications, where aesthetics and performance under low light intensities are important considerations.
Dye-sensitized solar cells are contrasted with traditional photovoltaic (PV) technologies, particularly silicon-based PV systems. DSCs are noted for their use of low-cost and readily available materials, as well as their scalable fabrication methods [245,246,247]. While DSCs offer advantages in terms of materials and fabrication, the passage acknowledges that they currently exhibit lower solar-to-electrical energy conversion efficiency and photovoltaic performance stability compared to existing commercial PV technologies. This has limited their competitiveness in bulk electricity generation outdoors [248]. DSCs are described as being well-suited for portable electronics due to their ability to be manufactured as thin and lightweight flexible solar modules. This characteristic makes them ideal for applications like charging portable electronic devices. One of the strengths of DSCs mentioned in the passage is their high efficiency under low-light conditions. They are noted for outperforming other technologies when used indoors or in environments with typical indoor lighting conditions. This makes them promising for applications such as powering wireless sensors in the Internet of Things (IoT) devices [243,249].
Dye-sensitized solar cells (DSCs) were significantly improved by O’Regan and Grätzel in 1991 [245]. Unlike traditional systems where semiconductors handle both light absorption and charge carrier transport, DSCs separate these functions. Light absorption is accomplished by a monolayer of dye molecules attached to a mesoporous layer of a wide-band-gap semiconductor, typically titanium dioxide (TiO2). Charge separation takes place at the semiconductor/dye interface [250]. Nanocrystalline DSCs have garnered significant attention as promising low-cost alternatives to conventional solar cells [251,252]. DSCs have achieved efficiencies of up to 13% under standard test conditions (1 sun irradiation, AM 1.5). This indicates that DSCs have made notable progress in improving their energy conversion efficiency [253]. These DSCs are characterized by their use of nanometer-sized particles and are known for their low production cost. TiO2 is commonly used as a semiconductor material, and it is noted for its low cost and wide use in pigments. DSCs are praised for their ease of fabrication, lightweight nature, and flexibility in design. These attributes contribute to their potential for various applications.[251].
Figure 12 provides a schematic presentation of DSC including nano-crystalline TiO2 thin film as a working electrode (WE) or photoanode which serves as the site where light absorption occurs and where the photogenerated electrons are injected into the TiO2. The monolayer of sensitizer molecules is in contact with the TiO2 photoanode. The sensitizer absorbs light and becomes excited, initiating the process of electron injection into the TiO2. Sensitizers are crucial for light absorption in DSCs. The CE, as a crucial component of DSSCs, performs two critical functions: it collects the electrons flowing from the external circuit and catalysis the reduction of I3− to I−, thereby realizing the regeneration of the sensitizer. The substrate on which the components are mounted is typically made of F-doped tin oxide (FTO) glass. This substrate provides support and electrical conductivity to the DSC. Different types of sensitizer dyes, such as ruthenium bipyridyl dyes like N3, N719, and black dye, can be used in DSCs. These dyes are chosen for their ability to efficiently absorb light and initiate the photoelectrochemical processes [254].
The operating principles of a Dye-sensitized Solar Cell (DSC) are explained in Figure 13, providing an overview of how the cell generates electrical power from light without undergoing any permanent chemical transformation. t the core of the DSC is a mesoporous oxide layer made up of nanometer-sized particles. This layer is designed to have a high surface area and is typically composed of materials like titanium dioxide (TiO2) in its anatase phase although alternative wide-band gap oxides such as ZnO [255], and Nb2O5 [238] have also been investigated. The mesoporous structure allows for efficient electronic conduction within the material. Attached to the surface of the nanocrystalline oxide film is a monolayer of a charge transfer dye. This dye plays a crucial role in the absorption of light energy. When it absorbs photons, it becomes excited. The excitation of the charge transfer dye results in the injection of an electron into the conduction band of the oxide material, typically TiO2. This electron injection is a key step in the conversion of light energy to electrical energy. After electron injection, the charge transfer dye is left in an oxidized state. To return to its original state, it needs to regain an electron. This electron is donated by the electrolyte, which is usually an organic solvent containing a redox system, such as the iodide/triiodide couple. The electrolyte plays a crucial role in regenerating the dye. The regeneration of the charge transfer dye by iodide ions (I-) intercepts the recapture of the conduction band electron by the oxidized dye. This process allows the dye to be reused for further light absorption. Iodide ions (I-) are regenerated through the reduction of triiodide ions (I3-) at the counter electrode (CE). This reduction process is catalyzed by the platinum (Pt) electrode at the CE. The electrons generated in the photoexcitation and injection process migrate through the external circuit, creating an electric current. This current can be harnessed to do work, such as powering electronic devices or charging batteries. The voltage generated in the DSC corresponds to the difference between the Fermi level of the electron in the solid material and the redox potential of the electrolyte. This voltage is what drives the flow of electrons through the external circuit and generates electrical power.
The study by Asagoe et al. highlights the importance of optimizing the composition and structure of the porous semiconductor layer in Dye-sensitized Solar Cells (DSSCs) to enhance their energy conversion efficiency [256]. In their experiment, the researchers used a porous semiconductor layer composed of a mixture of TiO2 nanoparticles and TiO2 nanowires. Specifically, they used a composition of 90% TiO2 nanoparticles and 10% TiO2 nanowires. The DSSC with this specific composition demonstrated a significantly higher energy conversion efficiency compared to a DSSC with a porous semiconductor layer made entirely of TiO2 nanoparticles (100% TiO2 nanoparticles). The increase in efficiency was measured at approximately 17%. The key finding was that a 10% concentration of TiO2 nanowires within the TiO2 nanoparticles was optimal for enhancing electron transport in the DSSC. This suggests that the presence of nanowires facilitated the movement of electrons within the semiconductor layer, improving overall device performance. Interestingly, the study also noted that as the concentration of TiO2 nanowires in the TiO2 nanoparticles increased beyond the optimal 10%, the energy conversion efficiency of the DSSC started to decrease. This decrease was attributed to the formation of an entangled structure at higher nanowire concentrations, which obstructed electron transport.
When DSCs are connected in series, each cell must be enclosed within a closed compartment. This design ensures that mass transport of the electrolyte between neighboring cells is prevented [257]. If there were a possibility for ion exchange between the redox electrolytes of neighboring cells, a process called photophoresis could occur. Photophoresis can lead to the separation of the redox couple and result in a drop in the electrical parameters of the DSC module [258]. To prevention exchange and photophoresis, a sealing material, such as glass frit, is used to create a barrier between neighboring cells. This barrier ensures that the electrolyte in each cell remains isolated from that of adjacent cells. Each cell compartment typically requires two openings—one for coloration (exposing the DSC to light) and one for electrolyte filling. The design of these compartments is critical to the proper functioning of the DSC module. In a strip pattern configuration, an optimal cell width for DSCs is suggested to be around 7–8 mm. This width is influenced by factors such as the sheet resistance of the transparent conductive oxide (TCO) substrate and the width of the dead area, which includes components like the protecting glass frit and Z-contact. To avoid the need for drilling and sealing a large number of filling holes in the glass substrate, one approach is to increase the width of an individual cell in the series connection. This can be achieved by screen printing a silver grid on both the front and counter electrodes. This reduces the effective sheet resistance of the TCO substrate and allows for wider cells. In summary, proper compartmentalization and sealing of individual cells in a series connection of DSCs are essential to prevention exchange and maintain the integrity of the redox electrolyte. The design of cell compartments, including their width and the use of materials like glass frit and silver grids, plays a crucial role in optimizing DSC modules for efficient photovoltaic performance.
As DSCs are scaled up, there is a higher likelihood of electrons recombining with electron acceptors within the nanocrystalline network of the cell. This recombination leads to current losses, reducing the overall efficiency of the DSC [251]. With up-scaling, the probability of issues like leaking holes, scratches on the TiO2 anode, and impurity defects also increases. These issues can further contribute to reduced efficiency and performance problems in DSCs. An increase in shunt current, which can be caused by the aforementioned issues, can lead to a rapid decrease in open circuit voltage and fill factor in DSCs. These parameters are critical for the overall performance of the cell. Without appropriate optimization of design methods and techniques, the efficiency of large-scale DSCs can drop significantly. In some cases, the efficiency may even fall below 1% when the cell size reaches 10 cm × 10 cm, making them less practical for widespread use [259]. DSCs typically use liquid electrolytes, which can introduce challenges related to encapsulation and long-term stability. Ensuring the integrity of the cell and preventing electrolyte leakage are important aspects of DSC design. Given these challenges, research institutes and commercial companies around the world have increasingly focused on developing design methods and conducting stability research for large-scale DSC modules. The goal is to overcome these obstacles and make DSC technology more viable for practical applications.
In summary, Dye-sensitized Solar Cells (DSCs) offer unique advantages in terms of materials, flexibility, and performance under low-light conditions. While they face challenges related to scaling up and stability, ongoing research aims to overcome these obstacles, making DSCs a promising technology for various applications in the renewable energy sector.
13. Conclusion
The combination of solar energy and photocatalytic hydrogen generation using materials like TiO2 holds promise for sustainable and environmentally friendly hydrogen production. Research efforts are focused on improving the efficiency of these photocatalysts to make them more practical for large-scale applications in the transition toward cleaner energy sources.
References
- Energy. Agency and International Energy Agency., World energy outlook 2013. International Energy Agency, 2013.
- bp, “Statistical Review of World Energy 2022.
- Dincer, I.; Acar, C. Review and evaluation of hydrogen production methods for better sustainability. Int. J. Hydrogen Energy 2015, 40, 11094–11111. [Google Scholar] [CrossRef]
- Peng, T.; Kellens, K.; Tang, R.; Chen, C.; Chen, G. Sustainability of additive manufacturing: An overview on its energy demand and environmental impact. Addit. Manuf. 2018, 21, 694–704. [Google Scholar] [CrossRef]
- Abdullah, W.M.Z.B.W.; Zainudin, W.N.R.A.B.; Ishak, W.W.B.M.; Sulong, F.B.; Haq, H.M.Z.U. Public Participation of Renewable Energy (PPRED) Model in Malaysia: An Instrument Development. Int. J. Renew. Energy Dev. 2020, 10, 119–137. [Google Scholar] [CrossRef]
- Agyekum, E.B. Energy poverty in energy rich Ghana: A SWOT analytical approach for the development of Ghana’s renewable energy. Sustain. Energy Technol. Assessments 2020, 40, 100760. [Google Scholar] [CrossRef]
- Gyamfi, B.A.; Adebayo, T.S.; Bekun, F.V.; Agyekum, E.B.; Kumar, N.M.; Alhelou, H.H.; Al-Hinai, A. Beyond environmental Kuznets curve and policy implications to promote sustainable development in Mediterranean. Energy Rep. 2021, 7, 6119–6129. [Google Scholar] [CrossRef]
- Tarhan, C.; Çil, M.A. A study on hydrogen, the clean energy of the future: Hydrogen storage methods. J. Energy Storage 2021, 40, 102676. [Google Scholar] [CrossRef]
- Yaqoob, S.J.; Motahhir, S.; Agyekum, E.B. A new model for a photovoltaic panel using Proteus software tool under arbitrary environmental conditions. J. Clean. Prod. 2021, 333, 130074. [Google Scholar] [CrossRef]
- Yang, J.; Sudik, A.; Wolverton, C.; Siegel, D.J. High capacity hydrogenstorage materials: attributes for automotive applications and techniques for materials discovery. Chem. Soc. Rev. 2009, 39, 656–675. [Google Scholar] [CrossRef]
- Da Silva Veras, T.; Mozer, T.S.; da Costa Rubim Messeder dos Santos, D.; da Silva César, A. Hydrogen: Trends, production and characterization of the main process worldwide. Int. J. Hydrog. Energy 2017, 42, 2018–2033. [Google Scholar] [CrossRef]
- Hanley, E.S.; Deane, J.P.; Gallachóir, B.P.Ó. The role of hydrogen in low carbon energy futures—A review of existing perspectives. Renew. Sustain. Energy Rev. 2018, 82, 3027–3045. [Google Scholar] [CrossRef]
- Niaz, S.; Manzoor, T.; Pandith, A.H. Hydrogen storage: Materials, methods and perspectives. Renew. Sustain. Energy Rev. 2015, 50, 457–469. [Google Scholar] [CrossRef]
- Bakenne, A.; Nuttall, W.; Kazantzis, N. Sankey-Diagram-based insights into the hydrogen economy of today. Int. J. Hydrogen Energy 2016, 41, 7744–7753. [Google Scholar] [CrossRef]
- Safari, F.; Dincer, I. A review and comparative evaluation of thermochemical water splitting cycles for hydrogen production. Energy Convers. Manag. 2020, 205, 112182. [Google Scholar] [CrossRef]
- Yue, M.; Lambert, H.; Pahon, E.; Roche, R.; Jemei, S.; Hissel, D. Hydrogen energy systems: A critical review of technologies, applications, trends and challenges. Renew. Sustain. Energy Rev. 2021, 146, 111180. [Google Scholar] [CrossRef]
- Dawood, F.; Anda, M.; Shafiullah, G.M. Hydrogen Production for Energy: An Overview. Int. J. Hydrog. Energy 2020, 45, 3847–3869. [Google Scholar] [CrossRef]
- Najjar, Y.S. Hydrogen safety: The road toward green technology. Int. J. Hydrogen Energy 2013, 38, 10716–10728. [Google Scholar] [CrossRef]
- Parra, D.; Valverde, L.; Pino, F.J.; Patel, M.K. A review on the role, cost and value of hydrogen energy systems for deep decarbonisation. Renew. Sustain. Energy Rev. 2019, 101, 279–294. [Google Scholar] [CrossRef]
- Kovač, A.; Paranos, M.; Marciuš, D. Hydrogen in energy transition: A review. 14th Conference on Sustainable Development of Energy, Water and Environment Systems (SDEWES). LOCATION OF CONFERENCE, COUNTRYDATE OF CONFERENCE; pp. 10016–10035.
- Maggio, G.; Nicita, A.; Squadrito, G. How the hydrogen production from RES could change energy and fuel markets: A review of recent literature. Int. J. Hydrogen Energy 2019, 44, 11371–11384. [Google Scholar] [CrossRef]
- Abe, J.O.; Popoola, A.P.I.; Ajenifuja, E.; Popoola, O.M. Hydrogen energy, economy and storage: Review and recommendation. Int. J. Hydrog. Energy 2019, 44, 15072–15086. [Google Scholar] [CrossRef]
- Liu, W.; Sun, L.; Li, Z.; Fujii, M.; Geng, Y.; Dong, L.; Fujita, T. Trends and future challenges in hydrogen production and storage research. Environ. Sci. Pollut. Res. 2020, 27, 31092–31104. [Google Scholar] [CrossRef]
- Ji, M.; Wang, J. Review and comparison of various hydrogen production methods based on costs and life cycle impact assessment indicators. Int. J. Hydrogen Energy 2021, 46, 38612–38635. [Google Scholar] [CrossRef]
- Hosseini, S.E.; Wahid, M.A. Hydrogen production from renewable and sustainable energy resources: Promising green energy carrier for clean development. Renew. Sustain. Energy Rev. 2016, 57, 850–866. [Google Scholar] [CrossRef]
- El-Emam, R.S.; Özcan, H. Comprehensive review on the techno-economics of sustainable large-scale clean hydrogen production. J. Clean. Prod. 2019, 220, 593–609. [Google Scholar] [CrossRef]
- Okonkwo, E.C.; Al-Breiki, M.; Bicer, Y.; Al-Ansari, T. Sustainable hydrogen roadmap: A holistic review and decision-making methodology for production, utilisation and exportation using Qatar as a case study. Int. J. Hydrogen Energy 2021, 46, 35525–35549. [Google Scholar] [CrossRef]
- Lane, B.; Reed, J.; Shaffer, B.; Samuelsen, S. Forecasting renewable hydrogen production technology shares under cost uncertainty. Int. J. Hydrogen Energy 2021, 46, 27293–27306. [Google Scholar] [CrossRef]
- Song, H.; Luo, S.; Huang, H.; Deng, B.; Ye, J. Solar-Driven Hydrogen Production: Recent Advances, Challenges, and Future Perspectives. ACS Energy Lett. 2022, 7, 1043–1065. [Google Scholar] [CrossRef]
- Fujishima Akira, Sugiyama Eiichi, and Honda Kenichi, “Photosensitized Electrolytic Oxidation of Iodide Ions on Cadmium Sulfide Single Crystal Electrode”.
- Oliveira, A.M.; Beswick, R.R.; Yan, Y. A green hydrogen economy for a renewable energy society. Curr. Opin. Chem. Eng. 2021, 33. [Google Scholar] [CrossRef]
- Ball, M.; Weeda, M. The hydrogen economy—Vision or reality? Int. J. Hydrogen Energy 2015, 40, 7903–7919. [Google Scholar] [CrossRef]
- Dincer, I. Green methods for hydrogen production. Int. J. Hydrogen Energy 2011, 37, 1954–1971. [Google Scholar] [CrossRef]
- Dincer, I. Green methods for hydrogen production. Int. J. Hydrogen Energy 2011, 37, 1954–1971. [Google Scholar] [CrossRef]
- Chi, J.; Yu, H. Water electrolysis based on renewable energy for hydrogen production. Chin. J. Catal. 2018, 39, 390–394. [Google Scholar] [CrossRef]
- Schmidt, O.; Gambhir, A.; Staffell, I.; Hawkes, A.; Nelson, J.; Few, S. Future cost and performance of water electrolysis: An expert elicitation study. Int. J. Hydrogen Energy 2017, 42, 30470–30492. [Google Scholar] [CrossRef]
- Kheirollahivash, M.; Rashidi, F.; Moshrefi, M.M. Hydrogen Production from Methane Decomposition Using a Mobile and Elongating Arc Plasma Reactor. Plasma Chem. Plasma Process. 2019, 39, 445–459. [Google Scholar] [CrossRef]
- Wang, M.; Wang, G.; Sun, Z.; Zhang, Y.; Xu, D. Review of renewable energy-based hydrogen production processes for sustainable energy innovation. Glob. Energy Interconnect. 2019, 2, 436–443. [Google Scholar] [CrossRef]
- Cong, T.Y.; Raj, A.; Chanaphet, J.; Mohammed, S.; Ibrahim, S.; Al Shoaibi, A. A detailed reaction mechanism for hydrogen production via hydrogen sulphide (H2S) thermolysis and oxidation. Int. J. Hydrogen Energy 2016, 41, 6662–6675. [Google Scholar] [CrossRef]
- Li, S.; Zheng, H.; Zheng, Y.; Tian, J.; Jing, T.; Chang, J.-S.; Ho, S.-H. Recent advances in hydrogen production by thermo-catalytic conversion of biomass. Int. J. Hydrogen Energy 2019, 44, 14266–14278. [Google Scholar] [CrossRef]
- Safari, F.; Dincer, I. A review and comparative evaluation of thermochemical water splitting cycles for hydrogen production. Energy Convers. Manag. 2020, 205, 112182. [Google Scholar] [CrossRef]
- Parthasarathy, P.; Narayanan, K.S. Hydrogen production from steam gasification of biomass: Influence of process parameters on hydrogen yield – A review. Renew. Energy 2014, 66, 570–579. [Google Scholar] [CrossRef]
- Pashchenko, D. First law energy analysis of thermochemical waste-heat recuperation by steam methane reforming. Energy 2018, 143, 478–487. [Google Scholar] [CrossRef]
- Gillis, R.J.; Al-Ali, K.; Green, W.H. Thermochemical production of hydrogen from hydrogen sulfide with iodine thermochemical cycles. Int. J. Hydrogen Energy 2018, 43, 12939–12947. [Google Scholar] [CrossRef]
- Pathak, A.K.; Kothari, R.; Tyagi, V.; Anand, S. Integrated approach for textile industry wastewater for efficient hydrogen production and treatment through solar PV electrolysis. Int. J. Hydrogen Energy 2020, 45, 25768–25782. [Google Scholar] [CrossRef]
- Ni, M.; Leung, M.K.; Leung, D.Y.; Sumathy, K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew. Sustain. Energy Rev. 2007, 11, 401–425. [Google Scholar] [CrossRef]
- Tayebi, M.; Lee, B.-K. Recent advances in BiVO4 semiconductor materials for hydrogen production using photoelectrochemical water splitting. Renew. Sustain. Energy Rev. 2019, 111, 332–343. [Google Scholar] [CrossRef]
- Hussain Rather, A. Kumar Srivastav, and A. Abdul, “A Study on Biohydrogen Production based on Biophotolysis from Cyanobacteria,” 2021. [Online]. Available: http://annalsofrscb.
- Łukajtis, R.; Hołowacz, I.; Kucharska, K.; Glinka, M.; Rybarczyk, P.; Przyjazny, A.; Kamiński, M. Hydrogen production from biomass using dark fermentation. Renew. Sustain. Energy Rev. 2018, 91, 665–694. [Google Scholar] [CrossRef]
- Zhang, Y.-H.P.; Evans, B.R.; Mielenz, J.R.; Hopkins, R.C.; Adams, M.W. High-Yield Hydrogen Production from Starch and Water by a Synthetic Enzymatic Pathway. PLOS ONE 2007, 2, e456. [Google Scholar] [CrossRef]
- González, M.S.; Marigorta, A.B.; Quintana, J.P. Review on hydrogen production technologies from solar energy. Renew. Energy Power Qual. J. 2011, 347–351. [Google Scholar] [CrossRef]
- Mehrpooya, M.; Valizadeh, F.; Askarimoghadam, R.; Sadeghi, S.; Pourfayaz, F.; Mousavi, S.A. Fabrication of nano-platinum alloy electrocatalysts and their performance in a micro-direct methanol fuel cell. Eur. Phys. J. Plus 2020, 135, 1–17. [Google Scholar] [CrossRef]
- Levene, J.I.; Mann, M.K.; Margolis, R.M.; Milbrandt, A. An analysis of hydrogen production from renewable electricity sources. Sol. Energy 2007, 81, 773–780. [Google Scholar] [CrossRef]
- Kuo, P.-C.; Illathukandy, B.; Wu, W.; Chang, J.-S. Energy, exergy, and environmental analyses of renewable hydrogen production through plasma gasification of microalgal biomass. Energy 2021, 223, 120025. [Google Scholar] [CrossRef]
- Apostolou, D. Optimisation of a hydrogen production – storage – re-powering system participating in electricity and transportation markets. A case study for Denmark. Appl. Energy 2020, 265, 114800. [Google Scholar] [CrossRef]
- Yilanci, A.; Dincer, I.; Ozturk, H.K. A review on solar-hydrogen/fuel cell hybrid energy systems for stationary applications. Prog. Energy Combust. Sci. 2009, 35, 231–244. [Google Scholar] [CrossRef]
- Ishaq, H.; Dincer, I. A comparative evaluation of OTEC, solar and wind energy based systems for clean hydrogen production. J. Clean. Prod. 2019, 246, 118736. [Google Scholar] [CrossRef]
- Razi, F.; Dincer, I. A critical evaluation of potential routes of solar hydrogen production for sustainable development. J. Clean. Prod. 2020, 264, 121582. [Google Scholar] [CrossRef]
- Spasiano, D.; Marotta, R.; Malato, S.; Fernandez-Ibañez, P.; Di Somma, I. Solar photocatalysis: Materials, reactors, some commercial, and pre-industrialized applications. A comprehensive approach. Appl. Catal. B: Environ. 2015, 170-171, 90–123. [Google Scholar] [CrossRef]
- Burton, N.; Padilla, R.; Rose, A.; Habibullah, H. Increasing the efficiency of hydrogen production from solar powered water electrolysis. Renew. Sustain. Energy Rev. 2020, 135, 110255. [Google Scholar] [CrossRef]
- Steinfeld, A. Solar thermochemical production of hydrogen–A review. Sol. Energy 2005, 78, 603–615. [Google Scholar] [CrossRef]
- Ngoh, S.K.; Njomo, D. An overview of hydrogen gas production from solar energy. Renew. Sustain. Energy Rev. 2012, 16, 6782–6792. [Google Scholar] [CrossRef]
- Xiao, L.; Wu, S.-Y.; Li, Y.-R. Advances in solar hydrogen production via two-step water-splitting thermochemical cycles based on metal redox reactions. Renew. Energy 2012, 41, 1–12. [Google Scholar] [CrossRef]
- Chaubey, R.; Sahu, S.; James, O.O.; Maity, S. A review on development of industrial processes and emerging techniques for production of hydrogen from renewable and sustainable sources. Renew. Sustain. Energy Rev. 2013, 23, 443–462. [Google Scholar] [CrossRef]
- Agrafiotis, C.; Roeb, M.; Sattler, C. A review on solar thermal syngas production via redox pair-based water/carbon dioxide splitting thermochemical cycles. Renew. Sustain. Energy Rev. 2015, 42, 254–285. [Google Scholar] [CrossRef]
- Villafán-Vidales, H.; Arancibia-Bulnes, C.; Riveros-Rosas, D.; Romero-Paredes, H.; Estrada, C. An overview of the solar thermochemical processes for hydrogen and syngas production: Reactors, and facilities. Renew. Sustain. Energy Rev. 2017, 75, 894–908. [Google Scholar] [CrossRef]
- Wang, B.; Shen, S.; Mao, S.S. Black TiO 2 for solar hydrogen conversion. J. Materiomics 2017, 3, 96–111. [Google Scholar] [CrossRef]
- Show, K.-Y.; Yan, Y.; Zong, C.; Guo, N.; Chang, J.-S.; Lee, D.-J. State of the art and challenges of biohydrogen from microalgae. Bioresour. Technol. 2019, 289, 121747. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Shi, R.; Waterhouse, G.I.; Zhang, T. Recent advances in niobium-based semiconductors for solar hydrogen production. Co-ord. Chem. Rev. 2020, 419, 213399. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, H. High visible transmittance of VO2 film prepared by DC magnetron sputtering with situ annealing. J. Opt. 2021, 50, 508–511. [Google Scholar] [CrossRef]
- Lee, J.E.; Shafiq, I.; Hussain, M.; Lam, S.S.; Rhee, G.H.; Park, Y.-K. A review on integrated thermochemical hydrogen production from water. Int. J. Hydrogen Energy 2021, 47, 4346–4356. [Google Scholar] [CrossRef]
- Li, R.; Li, C. Scalable solar water splitting using particulate photocatalysts. Curr. Opin. Green Sustain. Chem. 2021, 33, 100577. [Google Scholar] [CrossRef]
- Usman, M.; Zeb, Z.; Ullah, H.; Suliman, M.H.; Humayun, M.; Ullah, L.; Shah, S.N.A.; Ahmed, U.; Saeed, M. A review of metal-organic frameworks/graphitic carbon nitride composites for solar-driven green H2 production, CO2 reduction, and water purification. J. Environ. Chem. Eng. 2022, 10, 107548. [Google Scholar] [CrossRef]
- Gielen, D.; Boshell, F.; Saygin, D.; Bazilian, M.D.; Wagner, N.; Gorini, R. The role of renewable energy in the global energy transformation. Energy Strategy Rev. 2019, 24, 38–50. [Google Scholar] [CrossRef]
- Bidin, N.; Azni, S.R.; Abu Bakar, M.A.; Johari, A.R.; Munap, D.H.F.A.; Salebi, M.F.; Razak, S.N.A.; Sahidan, N.S.; Sulaiman, S.N.A. The effect of sunlight in hydrogen production from water electrolysis. Int. J. Hydrogen Energy 2017, 42, 133–142. [Google Scholar] [CrossRef]
- Parra, D.; Valverde, L.; Pino, F.J.; Patel, M.K. A review on the role, cost and value of hydrogen energy systems for deep decarbonisation. Renew. Sustain. Energy Rev. 2019, 101, 279–294. [Google Scholar] [CrossRef]
- Saba, S.M.; Müller, M.; Robinius, M.; Stolten, D. The investment costs of electrolysis – A comparison of cost studies from the past 30 years. Int. J. Hydrogen Energy 2018, 43, 1209–1223. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Nowotny, J.; Sorrell, C.; Sheppard, L.; Bak, T. Solar-hydrogen: Environmentally safe fuel for the future. Int. J. Hydrogen Energy 2005, 30, 521–544. [Google Scholar] [CrossRef]
- Liao, C.-H.; Huang, C.-W.; Wu, J.C.S. Hydrogen Production from Semiconductor-based Photocatalysis via Water Splitting. Catalysts 2012, 2, 490–516. [Google Scholar] [CrossRef]
- Alenzi, N.; Liao, W.-S.; Cremer, P.S.; Sanchez-Torres, V.; Wood, T.K.; Ehlig-Economides, C.; Cheng, Z. Photoelectrochemical hydrogen production from water/methanol decomposition using Ag/TiO2 nanocomposite thin films. Int. J. Hydrogen Energy 2010, 35, 11768–11775. [Google Scholar] [CrossRef]
- Arifin, K.; Majlan, E.H.; Daud, W.R.W.; Kassim, M.B. Bimetallic complexes in artificial photosynthesis for hydrogen production: A review. Int. J. Hydrogen Energy 2012, 37, 3066–3087. [Google Scholar] [CrossRef]
- T. Bak, J. T. Bak, J. Nowotny, M. Rekas, and C. C. Sorrell, “Photo-electrochemical hydrogen generation from water using solar energy. Materials-related aspects,” 2002. [Online]. Available: www.elsevier.
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2008, 38, 253–278. [Google Scholar] [CrossRef]
- van de Krol, R.; Liang, Y.; Schoonman, J. Solar hydrogen production with nanostructured metal oxides. J. Mater. Chem. 2008, 18, 2311–2320. [Google Scholar] [CrossRef]
- Nowotny, J.; Sorrell, C.; Sheppard, L.; Bak, T. Solar-hydrogen: Environmentally safe fuel for the future. Int. J. Hydrogen Energy 2005, 30, 521–544. [Google Scholar] [CrossRef]
- Andrade, L.; Cruz, R.; Ribeiro, H.A.; Mendes, A. Impedance characterization of dye-sensitized solar cells in a tandem arrangement for hydrogen production by water splitting. Int. J. Hydrogen Energy 2010, 35, 8876–8883. [Google Scholar] [CrossRef]
- Dusastre and Vincent, “Materials for Sustainable Energy,” 2010. [Online]. Available: www.worldscientific.
- Muradov, N.; Veziroǧlu, T. From hydrocarbon to hydrogen–carbon to hydrogen economy. Int. J. Hydrogen Energy 2005, 30, 225–237. [Google Scholar] [CrossRef]
- Lubitz, W.; Tumas, W. Hydrogen: An Overview. Chem. Rev. 2007, 107, 3900–3903. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, H.; Song, W.; Li, G.; Yi, B.; Shao, Z. A novel photoelectrochemical cell with self-organized TiO2 nanotubes as photoanodes for hydrogen generation. Int. J. Hydrogen Energy 2011, 36, 14374–14380. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef]
- Prévot, M.S.; Sivula, K. Photoelectrochemical Tandem Cells for Solar Water Splitting. J. Phys. Chem. C 2013, 117, 17879–17893. [Google Scholar] [CrossRef]
- Chen, Z.; Jaramillo, T.F.; Deutsch, T.G.; Kleiman-Shwarsctein, A.; Forman, A.J.; Gaillard, N.; Garland, R.; Takanabe, K.; Heske, C.; Sunkara, M.; et al. Accelerating materials development for photoelectrochemical hydrogen production: Standards for methods, definitions, and reporting protocols. J. Mater. Res. 2010, 25, 3–16. [Google Scholar] [CrossRef]
- Kerkez-Kuyumcu. ; Kibar, E.; Dayıoğlu, K.; Gedik, F.; Akın, A.N.; Özkara-Aydınoğlu,. A comparative study for removal of different dyes over M/TiO 2 (M = Cu, Ni, Co, Fe, Mn and Cr) photocatalysts under visible light irradiation. J. Photochem. Photobiol. A: Chem. 2015, 311, 176–185. [Google Scholar] [CrossRef]
- Sadanandam, G.; Lalitha, K.; Kumari, V.D.; Shankar, M.V.; Subrahmanyam, M. Cobalt doped TiO2: A stable and efficient photocatalyst for continuous hydrogen production from glycerol: Water mixtures under solar light irradiation. Int. J. Hydrogen Energy 2013, 38, 9655–9664. [Google Scholar] [CrossRef]
- Leung, D.Y.C.; Fu, X.; Wang, C.; Ni, M.; Leung, M.K.H.; Wang, X.; Fu, X. Hydrogen Production over Titania-Based Photocatalysts. ChemSusChem 2010, 3, 681–694. [Google Scholar] [CrossRef]
- Zhang, H.; Lv, X.; Li, Y.; Wang, Y.; Li, J. P25-Graphene Composite as a High Performance Photocatalyst. ACS Nano 2009, 4, 380–386. [Google Scholar] [CrossRef]
- Cao, H.; Lin, X.; Zhan, H.; Zhang, H.; Lin, J. Photocatalytic degradation kinetics and mechanism of phenobarbital in TiO2 aqueous solution. Chemosphere 2013, 90, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-D.; Yan, S.; Huang, Q.-D.; Fan, M.-T.; Yuan, Y.-Z.; Tan, T.T.-Y.; Liao, D.-W. TiO2 promoted by two different non-noble metal cocatalysts for enhanced photocatalytic H2 evolution. Appl. Surf. Sci. 2014, 309, 188–193. [Google Scholar] [CrossRef]
- Sreethawong, T.; Suzuki, Y.; Yoshikawa, S. Photocatalytic evolution of hydrogen over mesoporous TiO2 supported NiO photocatalyst prepared by single-step sol–gel process with surfactant template. Int. J. Hydrogen Energy 2005, 30, 1053–1062. [Google Scholar] [CrossRef]
- Harriman, A.; Pickering, I.J.; Thomas, J.M.; Christensen, P.A. Metal oxides as heterogeneous catalysts for oxygen evolution under photochemical conditions. J. Chem. Soc. Faraday Trans. 1: Phys. Chem. Condens. Phases 1988, 84. [Google Scholar] [CrossRef]
- Sheng, P.; Li, W.; Cai, J.; Wang, X.; Tong, X.; Cai, Q.; Grimes, C.A. A novel method for the preparation of a photocorrosion stable core/shell CdTe/CdS quantum dot TiO2 nanotube array photoelectrode demonstrating an AM 1.5G photoconversion efficiency of 6.12%. J. Mater. Chem. A 2013, 1, 7806–7815. [Google Scholar] [CrossRef]
- Meng, L.; Li, L. Recent research progress on operational stability of metal oxide/sulfide photoanodes in photoelectrochemical cells. Nano Res. Energy 2022, 1, e9120020. [Google Scholar] [CrossRef]
- Y. Nakato, H. Y. Nakato, H. Akanuma, J.-I. Shimizu, and Y. Magari, “Photo-oxidation reaction of water on an n-TiO 2 electrode. Improvement in efficiency through formation of surface micropores by photo-etching in H2SO 4 1,” 1995.
- Koyale, P.A.; Delekar, S.D. A review on practical aspects of CeO2 and its composites for photoelectrochemical water splitting. Int. J. Hydrogen Energy 2023. [Google Scholar] [CrossRef]
- Lee, W.J.; Shinde, P.S.; Go, G.H.; Ramasamy, E. Ag grid induced photocurrent enhancement in WO3 photoanodes and their scale-up performance toward photoelectrochemical H2 generation. Int. J. Hydrogen Energy 2011, 36, 5262–5270. [Google Scholar] [CrossRef]
- Mishra, P.; Shukla, P.; Singh, A.; Srivastava, O. Investigation and optimization of nanostructured TiO2 photoelectrode in regard to hydrogen production through photoelectrochemical process. Int. J. Hydrogen Energy 2003. [Google Scholar] [CrossRef]
- Bashiri, R.; Mohamed, N.M.; Kait, C.F.; Sufian, S.; Kakooei, S.; Khatani, M.; Gholami, Z. Optimization hydrogen production over visible light-driven titania-supported bimetallic photocatalyst from water photosplitting in tandem photoelectrochemical cell. Renew. Energy 2016, 99, 960–970. [Google Scholar] [CrossRef]
- Minggu, L.J.; Daud, W.R.W.; Kassim, M.B. An overview of photocells and photoreactors for photoelectrochemical water splitting. Int. J. Hydrogen Energy 2010, 35, 5233–5244. [Google Scholar] [CrossRef]
- Alenzi, N.; Liao, W.-S.; Cremer, P.S.; Sanchez-Torres, V.; Wood, T.K.; Ehlig-Economides, C.; Cheng, Z. Photoelectrochemical hydrogen production from water/methanol decomposition using Ag/TiO2 nanocomposite thin films. Int. J. Hydrogen Energy 2010, 35, 11768–11775. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2008, 38, 253–278. [Google Scholar] [CrossRef]
- Lopes, T.; Andrade, L.; Ribeiro, H.A.; Mendes, A. Characterization of photoelectrochemical cells for water splitting by electrochemical impedance spectroscopy. Int. J. Hydrogen Energy 2010, 35, 11601–11608. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Minggu, L.J.; Daud, W.R.W.; Kassim, M.B. An overview of photocells and photoreactors for photoelectrochemical water splitting. Int. J. Hydrogen Energy 2010, 35, 5233–5244. [Google Scholar] [CrossRef]
- Ding, C.; Shi, J.; Wang, Z.; Li, C. Photoelectrocatalytic Water Splitting: Significance of Cocatalysts, Electrolyte, and Interfaces. ACS Catal. 2016, 7, 675–688. [Google Scholar] [CrossRef]
- Zhang, T.; Lin, P.; Wei, N.; Wang, D. Enhanced Photoelectrochemical Water-Splitting Property on TiO2 Nanotubes by Surface Chemical Modification and Wettability Control. ACS Appl. Mater. Interfaces 2020, 12, 20110–20118. [Google Scholar] [CrossRef]
- Meng, L.; Rao, D.; Tian, W.; Cao, F.; Yan, X.; Li, L. Simultaneous Manipulation of O-Doping and Metal Vacancy in Atomically Thin Zn 10 In 16 S 34 Nanosheet Arrays toward Improved Photoelectrochemical Performance. Angew. Chem. Int. Ed. 2018, 57, 16882–16887. [Google Scholar] [CrossRef]
- Maeda, K. Photocatalytic water splitting using semiconductor particles: History and recent developments. J. Photochem. Photobiol. C: Photochem. Rev. 2011, 12, 237–268. [Google Scholar] [CrossRef]
- Kudo, A. Photocatalysis and solar hydrogen production. Pure Appl. Chem. 2007, 79, 1917–1927. [Google Scholar] [CrossRef]
- Zhu, J.; Zäch, M. Nanostructured materials for photocatalytic hydrogen production. Curr. Opin. Colloid Interface Sci. 2009, 14, 260–269. [Google Scholar] [CrossRef]
- Moniz, S.J.A.; Shevlin, S.A.; Martin, D.J.; Guo, Z.-X.; Tang, J. Visible-light driven heterojunction photocatalysts for water splitting – a critical review. Energy Environ. Sci. 2015, 8, 731–759. [Google Scholar] [CrossRef]
- Gan, J.; Lu, X.; Tong, Y. Towards highly efficient photoanodes: boosting sunlight-driven semiconductor nanomaterials for water oxidation. Nanoscale 2014, 6, 7142–7164. [Google Scholar] [CrossRef] [PubMed]
- Dette, C.; Pérez-Osorio, M.A.; Kley, C.S.; Punke, P.; Patrick, C.E.; Jacobson, P.; Giustino, F.; Jung, S.J.; Kern, K. TiO2 Anatase with a Bandgap in the Visible Region. Nano Lett. 2014, 14, 6533–6538. [Google Scholar] [CrossRef]
- L. Linsebigler, G. L. Linsebigler, G. Lu, and J. T. Yates, “Photocatalysis on TiOn Surfaces: Principles, Mechanisms, and Selected Results,” 1995.
- Fan, W.; Zhang, Q.; Wang, Y. Semiconductor-based nanocomposites for photocatalytic H2 production and CO2 conversion. Phys. Chem. Chem. Phys. 2012, 15, 2632–2649. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Liu, J.; Zhang, Y.; Liu, D. Solar Energy Storage in an All-Vanadium Photoelectrochemical Cell: Structural Effect of Titania Nanocatalyst in Photoanode. Energies 2022, 15, 4508. [Google Scholar] [CrossRef]
- Dholam, R.; Patel, N.; Adami, M.; Miotello, A. Physically and chemically synthesized TiO2 composite thin films for hydrogen production by photocatalytic water splitting. Int. J. Hydrogen Energy 2008, 33, 6896–6903. [Google Scholar] [CrossRef]
- Bourikas, K.; Kordulis, C.; Lycourghiotis, A. Titanium Dioxide (Anatase and Rutile): Surface Chemistry, Liquid–Solid Interface Chemistry, and Scientific Synthesis of Supported Catalysts. Chem. Rev. 2014, 114, 9754–9823. [Google Scholar] [CrossRef]
- Jitputti, J.; Pavasupree, S.; Suzuki, Y.; Yoshikawa, S. Synthesis and photocatalytic activity for water-splitting reaction of nanocrystalline mesoporous titania prepared by hydrothermal method. J. Solid State Chem. 2007, 180, 1743–1749. [Google Scholar] [CrossRef]
- Chiarello, G.L.; Selli, E.; Forni, L. Photocatalytic hydrogen production over flame spray pyrolysis-synthesised TiO2 and Au/TiO2. Appl. Catal. B: Environ. 2008, 84, 332–339. [Google Scholar] [CrossRef]
- K. E. Karakitsou and X. E. Verykios’, “Effects of Altervalent Cation Doping of Ti02 on Its Performance as a Photocatalyst for Water Cleavage,” 1993.
- M. V Rao, K. M. V Rao, K. Rajeshwar, V. R. Pal Verneker, and J. Dubow, “Photosyntlhetic Production of H2 and H202 on Semiconducting Oxide Grains in Aqueous Solutlons,” 1980.
- Kolen’ko, Y.; Churagulov, B.; Kunst, M.; Mazerolles, L.; Colbeau-Justin, C. Photocatalytic properties of titania powders prepared by hydrothermal method. Appl. Catal. B: Environ. 2004, 54, 51–58. [Google Scholar] [CrossRef]
- Sreethawong, T.; Suzuki, Y.; Yoshikawa, S. Synthesis, characterization, and photocatalytic activity for hydrogen evolution of nanocrystalline mesoporous titania prepared by surfactant-assisted templating sol–gel process. J. Solid State Chem. 2005, 178, 329–338. [Google Scholar] [CrossRef]
- H. Kominami and B. Ohtani, “Preparation of Titanium Oxide-Based Powders and Thin Films of High Photocatalytic Activities Using Solvothermal Methods,” 2010, pp. 113–132. [CrossRef]
- Wang, C.-C.; Ying, J.Y. Sol−Gel Synthesis and Hydrothermal Processing of Anatase and Rutile Titania Nanocrystals. Chem. Mater. 1999, 11, 3113–3120. [Google Scholar] [CrossRef]
- Sreethawong, T.; Suzuki, Y.; Yoshikawa, S. Photocatalytic evolution of hydrogen over nanocrystalline mesoporous titania prepared by surfactant-assisted templating sol–gel process. Catal. Commun. 2005, 6, 119–124. [Google Scholar] [CrossRef]
- Korzhak, A.V.; Ermokhina, N.I.; Stroyuk, A.L.; Bukhtiyarov, V.K.; Raevskaya, A.E.; Litvin, V.I.; Kuchmiy, S.Y.; Ilyin, V.G.; Manorik, P.A. Photocatalytic hydrogen evolution over mesoporous TiO2/metal nanocomposites. J. Photochem. Photobiol. A: Chem. 2008, 198, 126–134. [Google Scholar] [CrossRef]
- Yu, Z.; Liu, H.; Zhu, M.; Li, Y.; Li, W. Interfacial Charge Transport in 1D TiO2 Based Photoelectrodes for Photoelectrochemical Water Splitting. Small 2019, 17, e1903378. [Google Scholar] [CrossRef]
- Jaramillo-Gutiérrez, M.; Rivero, E.; Cruz-Díaz, M.; Niño-Gómez, M.; Pedraza-Avella, J. Photoelectrocatalytic hydrogen production from oilfield-produced wastewater in a filter-press reactor using TiO2-based photoanodes. Catal. Today 2016, 266, 17–26. [Google Scholar] [CrossRef]
- Bahramian, A.; Rezaeivala, M.; He, K.; Dionysiou, D.D. Enhanced visible-light photoelectrochemical hydrogen evolution through degradation of methyl orange in a cell based on coral-like Pt-deposited TiO2 thin film with sub-2 nm pores. Catal. Today 2018, 335, 333–344. [Google Scholar] [CrossRef]
- Guaraldo, T.; Gonçales, V.; Silva, B.; de Torresi, S.; Zanoni, M. Hydrogen production and simultaneous photoelectrocatalytic pollutant oxidation using a TiO2/WO3 nanostructured photoanode under visible light irradiation. J. Electroanal. Chem. 2015, 765, 188–196. [Google Scholar] [CrossRef]
- Zhang, B.; Chou, L.; Bi, Y. Tuning surface electronegativity of BiVO4 photoanodes toward high-performance water splitting. Appl. Catal. B: Environ. 2019, 262, 118267. [Google Scholar] [CrossRef]
- Akshay, V.R.; Arun, B.; Mandal, G.; Mutta, G.R.; Chanda, A.; Vasundhara, M. Observation of Optical Band-Gap Narrowing and Enhanced Magnetic Moment in Co-Doped Sol–Gel-Derived Anatase TiO2 Nanocrystals. J. Phys. Chem. C 2018, 122, 26592–26604. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, Q. Mechanism of higher photocatalytic activity of anatase TiO2doped with nitrogen under visible-light irradiation from density functional theory calculation. J. Phys. D: Appl. Phys. 2007, 41. [Google Scholar] [CrossRef]
- Rafique, M.; Hajra, S.; Irshad, M.; Usman, M.; Imran, M.; Assiri, M.A.; Ashraf, W.M. Hydrogen Production Using TiO2-Based Photocatalysts: A Comprehensive Review. ACS Omega 2023, 8, 25640–25648. [Google Scholar] [CrossRef]
- Rosseler, O.; Shankar, M.V.; Karkmaz-Le Du, M.; Schmidlin, L.; Keller, N.; Keller, V. Solar light photocatalytic hydrogen production from water over Pt and Au/TiO2(anatase/rutile) photocatalysts: Influence of noble metal and porogen promotion. J. Catal. 2010, 269, 179–190. [Google Scholar] [CrossRef]
- Kumaraswamy, G.; Sadanandam, G.; Ledwaba, K.; Maroju, R. An efficient photocatalytic synthesis of benzimidazole over cobalt-loaded TiO2 catalysts under solar light irradiation. J. Photochem. Photobiol. A: Chem. 2022, 429. [Google Scholar] [CrossRef]
- Montoya, A.T.; Gillan, E.G. Enhanced Photocatalytic Hydrogen Evolution from Transition-Metal Surface-Modified TiO2. ACS Omega 2018, 3, 2947–2955. [Google Scholar] [CrossRef]
- Zanchet, D.; Santos, J.B.O.; Damyanova, S.; Gallo, J.M.R.; Bueno, J.M.C. Toward Understanding Metal-Catalyzed Ethanol Reforming. ACS Catal. 2015, 5, 3841–3863. [Google Scholar] [CrossRef]
- Xia, B.; Yang, Y.; Zhang, Y.; Xia, Y.; Jaroniec, M.; Yu, J.; Ran, J.; Qiao, S.-Z. Metal–organic framework with atomically dispersed Ni–N4 sites for greatly-raised visible-light photocatalytic H2 production. Chem. Eng. J. 2022, 431. [Google Scholar] [CrossRef]
- Su, D.W.; Ran, J.; Zhuang, Z.W.; Chen, C.; Qiao, S.Z.; Li, Y.D.; Wang, G.X. Atomically dispersed Ni in cadmium-zinc sulfide quantum dots for high-performance visible-light photocatalytic hydrogen production. Sci. Adv. 2020, 6, eaaz8447. [Google Scholar] [CrossRef]
- Tseng, Y.-H.; Huang, B.-K. Photocatalytic Degradation of Using Ni-Containing TiO2. Int. J. Photoenergy 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Jing, D.; Zhang, Y.; Guo, L. Study on the synthesis of Ni doped mesoporous TiO2 and its photocatalytic activity for hydrogen evolution in aqueous methanol solution. Chem. Phys. Lett. 2005, 415, 74–78. [Google Scholar] [CrossRef]
- Dholam, R.; Patel, N.; Adami, M.; Miotello, A. Hydrogen production by photocatalytic water-splitting using Cr- or Fe-doped TiO2 composite thin films photocatalyst. Int. J. Hydrogen Energy 2009, 34, 5337–5346. [Google Scholar] [CrossRef]
- Shi, J.-W.; Sun, D.; Zou, Y.; Ma, D.; He, C.; Ji, X.; Niu, C. Trap-level-tunable Se doped CdS quantum dots with excellent hydrogen evolution performance without co-catalyst. Chem. Eng. J. 2019, 364, 11–19. [Google Scholar] [CrossRef]
- Han, X.; Xu, D.; An, L.; Hou, C.; Li, Y.; Zhang, Q.; Wang, H. Ni-Mo nanoparticles as co-catalyst for drastically enhanced photocatalytic hydrogen production activity over g-C3N4. Appl. Catal. B: Environ. 2018, 243, 136–144. [Google Scholar] [CrossRef]
- Wang, B.; He, S.; Zhang, L.; Huang, X.; Gao, F.; Feng, W.; Liu, P. CdS nanorods decorated with inexpensive NiCd bimetallic nanoparticles as efficient photocatalysts for visible-light-driven photocatalytic hydrogen evolution. Appl. Catal. B: Environ. 2018, 243, 229–235. [Google Scholar] [CrossRef]
- Spanu, D.; Recchia, S.; Mohajernia, S.; Tomanec, O.; Kment. ; Zboril, R.; Schmuki, P.; Altomare, M. Templated Dewetting–Alloying of NiCu Bilayers on TiO2 Nanotubes Enables Efficient Noble-Metal-Free Photocatalytic H2 Evolution. ACS Catal. 2018, 8, 5298–5305. [Google Scholar] [CrossRef]
- Kim, A.-Y.; Kang, M. Effect of Al-Cu Bimetallic Components in a TiO2Framework for High Hydrogen Production on Methanol/Water Photo-Splitting. Int. J. Photoenergy 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Sun, T.; Fan, J.; Liu, E.; Liu, L.; Wang, Y.; Dai, H.; Yang, Y.; Hou, W.; Hu, X.; Jiang, Z. Fe and Ni co-doped TiO2 nanoparticles prepared by alcohol-thermal method: Application in hydrogen evolution by water splitting under visible light irradiation. Powder Technol. 2012, 228, 210–218. [Google Scholar] [CrossRef]
- Sun, T.; Liu, E.; Fan, J.; Hu, X.; Wu, F.; Hou, W.; Yang, Y.; Kang, L. High photocatalytic activity of hydrogen production from water over Fe doped and Ag deposited anatase TiO2 catalyst synthesized by solvothermal method. Chem. Eng. J. 2013, 228, 896–906. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Tian, W.; Meng, A.; Yang, L. CoNi Bimetal Cocatalyst Modifying a Hierarchical ZnIn2S4 Nanosheet-Based Microsphere Noble-Metal-Free Photocatalyst for Efficient Visible-Light-Driven Photocatalytic Hydrogen Production. ACS Sustain. Chem. Eng. 2019, 7, 20190–20201. [Google Scholar] [CrossRef]
- Ahmed, A.Y.; Kandiel, T.A.; Oekermann, T.; Bahnemann, D. Photocatalytic Activities of Different Well-defined Single Crystal TiO2 Surfaces: Anatase versus Rutile. J. Phys. Chem. Lett. 2011, 2, 2461–2465. [Google Scholar] [CrossRef]
- Riaz, N.; Chong, F.; Dutta, B.K.; Man, Z.B.; Khan, M.S.; Nurlaela, E. Photodegradation of Orange II under visible light using Cu–Ni/TiO2: Effect of calcination temperature. Chem. Eng. J. 2012, 185-186, 108–119. [Google Scholar] [CrossRef]
- Amano, F.; Nakata, M.; Asami, K.; Yamakata, A. Photocatalytic activity of titania particles calcined at high temperature: Investigating deactivation. Chem. Phys. Lett. 2013, 579, 111–113. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Lee, C.-Y.; Yeng, M.-Y.; Chiu, H.-T. The effect of calcination temperature on the crystallinity of TiO2 nanopowders. J. Cryst. Growth 2003, 247, 363–370. [Google Scholar] [CrossRef]
- Lim, J.-M.; Yi, G.-R.; Moon, J.H.; Heo, C.-J.; Yang, S.-M. Superhydrophobic Films of Electrospun Fibers with Multiple-Scale Surface Morphology. Langmuir 2007, 23, 7981–7989. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, X.; Xu, Y.; Niu, X.; Zheng, L.; Ding, X. The preparation of Zn2+-doped TiO2 nanoparticles by sol–gel and solid phase reaction methods respectively and their photocatalytic activities. Chemosphere 2005, 59, 1367–1371. [Google Scholar] [CrossRef]
- Zhiyong, Y.; Bensimon, M.; Sarria, V.; Stolitchnov, I.; Jardim, W.; Laub, D.; Mielczarski, E.; Mielczarski, J.; Kiwi-Minsker, L.; Kiwi, J. ZnSO4–TiO2 doped catalyst with higher activity in photocatalytic processes. Appl. Catal. B: Environ. 2007, 76, 185–195. [Google Scholar] [CrossRef]
- Babić, B.; Gulicovski, J.; Dohčević-Mitrović, Z.; Bučevac, D.; Prekajski, M.; Zagorac, J.; Matović, B. Synthesis and characterization of Fe3+ doped titanium dioxide nanopowders. Ceram. Int. 2012, 38, 635–640. [Google Scholar] [CrossRef]
- Padikkaparambil, S.; Yaakob, Z.; Narayanan, B.N.; Ramakrishnan, R.; Viswanathan, S. Novel preparation method of nanosilver doped sol gel TiO2 photocatalysts for dye pollutant degradation. J. Sol-Gel Sci. Technol. 2012, 63, 108–115. [Google Scholar] [CrossRef]
- Yoong, L.; Chong, F.; Dutta, B.K. Development of copper-doped TiO2 photocatalyst for hydrogen production under visible light. Energy 2009, 34, 1652–1661. [Google Scholar] [CrossRef]
- Angkaew, S.; Limsuwan, P. Preparation of silver-titanium dioxide core-shell (Ag@TiO2) nanoparticles: Effect of Ti-Ag mole ratio. Procedia Eng. 2012, 32, 649–655. [Google Scholar] [CrossRef]
- Gümüş, D.; Akbal, F. Photocatalytic Degradation of Textile Dye and Wastewater. Water, Air, Soil Pollut. 2010, 216, 117–124. [Google Scholar] [CrossRef]
- Adán, C.; Bahamonde, A.; Fernández-García, M.; Martínez-Arias, A. Structure and activity of nanosized iron-doped anatase TiO2 catalysts for phenol photocatalytic degradation. Appl. Catal. B: Environ. 2007, 72, 11–17. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Huo, Y.; Zhu, J. Supercritical Preparation of a Highly Active S-Doped TiO2 Photocatalyst for Methylene Blue Mineralization. Environ. Sci. Technol. 2007, 41, 4410–4414. [Google Scholar] [CrossRef]
- Xin, B.; Wang, P.; Ding, D.; Liu, J.; Ren, Z.; Fu, H. Effect of surface species on Cu-TiO2 photocatalytic activity. Appl. Surf. Sci. 2008, 254, 2569–2574. [Google Scholar] [CrossRef]
- Mohamed, N.M.; Bashiri, R.; Kait, C.F.; Sufian, S. Photocatalytic water splitting over titania supported copper and nickel oxide in photoelectrochemical cell; optimization of photoconversion efficiency. 348, 0120; 07. [Google Scholar] [CrossRef]
- Liu, L.-F.; Zhang, Y.; Yang, F.-L.; Chen, G.; Yu, J.C. Simultaneous photocatalytic removal of ammonium and nitrite in water using Ce3+–Ag+ modified TiO2. Sep. Purif. Technol. 2009, 67, 244–248. [Google Scholar] [CrossRef]
- Zhang, D. Enhanced photocatalytic activity for titanium dioxide by co-modification with copper and iron. Transit. Met. Chem. 2010, 35, 933–938. [Google Scholar] [CrossRef]
- Nogawa, T.; Isobe, T.; Matsushita, S.; Nakajima, A. Preparation and visible-light photocatalytic activity of Au- and Cu-modified TiO2 powders. Mater. Lett. 2012, 82, 174–177. [Google Scholar] [CrossRef]
- Wongwisate, P.; Chavadej, S.; Gulari, E.; Sreethawong, T.; Rangsunvigit, P. Effects of monometallic and bimetallic Au–Ag supported on sol–gel TiO2 on photocatalytic degradation of 4-chlorophenol and its intermediates. Desalination 2011, 272, 154–163. [Google Scholar] [CrossRef]
- Ou, T.-C.; Chang, F.-W.; Roselin, L.S. Production of hydrogen via partial oxidation of methanol over bimetallic Au–Cu/TiO2 catalysts. J. Mol. Catal. A: Chem. 2008, 293, 8–16. [Google Scholar] [CrossRef]
- Gao, W.; Jin, R.; Chen, J.; Guan, X.; Zeng, H.; Zhang, F.; Guan, N. Titania-supported bimetallic catalysts for photocatalytic reduction of nitrate. Catal. Today 2004, 90, 331–336. [Google Scholar] [CrossRef]
- Moss, B.; Babacan, O.; Kafizas, A.; Hankin, A. A Review of Inorganic Photoelectrode Developments and Reactor Scale-Up Challenges for Solar Hydrogen Production. Adv. Energy Mater. 2021, 11, 2003286. [Google Scholar] [CrossRef]
- Yang, Y.; Niu, S.; Han, D.; Liu, T.; Wang, G.; Li, Y. Progress in Developing Metal Oxide Nanomaterials for Photoelectrochemical Water Splitting. Adv. Energy Mater. 2017, 7. [Google Scholar] [CrossRef]
- Chu, S.; Vanka, S.; Wang, Y.; Gim, J.; Wang, Y.; Ra, Y.-H.; Hovden, R.; Guo, H.; Shih, I.; Mi, Z. Solar Water Oxidation by an InGaN Nanowire Photoanode with a Bandgap of 1.7 eV. ACS Energy Lett. 2018, 3, 307–314. [Google Scholar] [CrossRef]
- Chen, X.; Shen, X.; Shen, S.; Reese, M.O.; Hu, S. Stable CdTe Photoanodes with Energetics Matching Those of a Coating Intermediate Band. ACS Energy Lett. 2020, 5, 1865–1871. [Google Scholar] [CrossRef]
- Wang, G.; Yang, Y.; Ling, Y.; Wang, H.; Lu, X.; Pu, Y.-C.; Zhang, J.Z.; Tong, Y.; Li, Y. An electrochemical method to enhance the performance of metal oxides for photoelectrochemical water oxidation. J. Mater. Chem. A 2016, 4, 2849–2855. [Google Scholar] [CrossRef]
- Zhong, M.; Hisatomi, T.; Sasaki, Y.; Suzuki, S.; Teshima, K.; Nakabayashi, M.; Shibata, N.; Nishiyama, H.; Katayama, M.; Yamada, T.; et al. Highly Active GaN-Stabilized Ta3 N5 Thin-Film Photoanode for Solar Water Oxidation. Angew. Chem. Int. Ed. 2017, 56, 4739–4743. [Google Scholar] [CrossRef]
- Chen, S.; Huang, D.; Xu, P.; Xue, W.; Lei, L.; Cheng, M.; Wang, R.; Liu, X.; Deng, R. Semiconductor-based photocatalysts for photocatalytic and photoelectrochemical water splitting: will we stop with photocorrosion? J. Mater. Chem. A 2020, 8, 2286–2322. [Google Scholar] [CrossRef]
- Salvador, P. Mechanisms of water photooxidation at n-TiO2 rutile single crystal oriented electrodes under UV illumination in competition with photocorrosion. Prog. Surf. Sci. 2011, 86, 41–58. [Google Scholar] [CrossRef]
- Fu, J.; Fan, Z.; Nakabayashi, M.; Ju, H.; Pastukhova, N.; Xiao, Y.; Feng, C.; Shibata, N.; Domen, K.; Li, Y. Interface engineering of Ta3N5 thin film photoanode for highly efficient photoelectrochemical water splitting. Nat. Commun. 2022, 13, 1–9. [Google Scholar] [CrossRef]
- He, B.; Jia, S.; Zhao, M.; Wang, Y.; Chen, T.; Zhao, S.; Li, Z.; Lin, Z.; Zhao, Y.; Liu, X. General and Robust Photothermal-Heating-Enabled High-Efficiency Photoelectrochemical Water Splitting. Adv. Mater. 2021, 33, 2004406. [Google Scholar] [CrossRef] [PubMed]
- Subramanyam, P.; Kumar, P.N.; Deepa, M.; Subrahmanyam, C.; Ghosal, P. Bismuth sulfide nanocrystals and gold nanorods increase the photovoltaic response of a TiO2/CdS based cell. Sol. Energy Mater. Sol. Cells 2017, 159, 296–306. [Google Scholar] [CrossRef]
- Subramanyam, P.; Vinodkumar, T.; Nepak, D.; Deepa, M.; Subrahmanyam, C. Mo-doped BiVO4@reduced graphene oxide composite as an efficient photoanode for photoelectrochemical water splitting. 9th International Symposium on Group Five Elements. LOCATION OF CONFERENCE, COUNTRYDATE OF CONFERENCE; pp. 73–80.
- Subramanyam, P.; Meena, B.; Suryakala, D.; Deepa, M.; Subrahmanyam, C. Plasmonic nanometal decorated photoanodes for efficient photoelectrochemical water splitting. Catal. Today 2020, 379, 1–6. [Google Scholar] [CrossRef]
- Li, J.; Cushing, S.K.; Zheng, P.; Senty, T.; Meng, F.; Bristow, A.D.; Manivannan, A.; Wu, N. Solar Hydrogen Generation by a CdS-Au-TiO2 Sandwich Nanorod Array Enhanced with Au Nanoparticle as Electron Relay and Plasmonic Photosensitizer. J. Am. Chem. Soc. 2014, 136, 8438–8449. [Google Scholar] [CrossRef]
- Al Dahoudi, N.; Zhang, Q.; Cao, G. Low-Temperature Processing of Titanium Oxide Nanoparticles Photoanodes for Dye-Sensitized Solar Cells. J. Renew. Energy 2012, 2013, 1–8. [Google Scholar] [CrossRef]
- Zhao, D.; Peng, T.; Lu, L.; Cai, P.; Jiang, P.; Bian, Z. Effect of Annealing Temperature on the Photoelectrochemical Properties of Dye-Sensitized Solar Cells Made with Mesoporous TiO2 Nanoparticles. J. Phys. Chem. C 2008, 112, 8486–8494. [Google Scholar] [CrossRef]
- Lu, L.; Li, R.; Fan, K.; Peng, T. Effects of annealing conditions on the photoelectrochemical properties of dye-sensitized solar cells made with ZnO nanoparticles. Sol. Energy 2010, 84, 844–853. [Google Scholar] [CrossRef]
- Bashiri, R.; Mohamed, N.M.; Kait, C.F.; Sufian, S. Effect of heat treatment on the physical properties of bimetallic doped catalyst, Cu-Ni/TiO2. PROCEEDINGS OF THE 23RD SCIENTIFIC CONFERENCE OF MICROSCOPY SOCIETY MALAYSIA (SCMSM 2014). LOCATION OF CONFERENCE, MalaysiaDATE OF CONFERENCE;
- Zhang, L.J.; Zheng, R.; Li, S.; Liu, B.K.; Wang, D.J.; Wang, L.L.; Xie, T.F. Enhanced Photocatalytic H2 Generation on Cadmium Sulfide Nanorods with Cobalt Hydroxide as Cocatalyst and Insights into Their Photogenerated Charge Transfer Properties. ACS Appl. Mater. Interfaces 2014, 6, 13406–13412. [Google Scholar] [CrossRef]
- Basu, M. Porous Cupric Oxide: Efficient Photocathode for Photoelectrochemical Water Splitting. ChemPhotoChem 2019, 3, 1254–1262. [Google Scholar] [CrossRef]
- Adachi, M.; Sakamoto, M.; Jiu, J.; Ogata, Y.; Isoda, S. Determination of Parameters of Electron Transport in Dye-Sensitized Solar Cells Using Electrochemical Impedance Spectroscopy. J. Phys. Chem. B 2006, 110, 13872–13880. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Wang, G.; Wheeler, D.A.; Zhang, J.Z.; Li, Y. Sn-Doped Hematite Nanostructures for Photoelectrochemical Water Splitting. Nano Lett. 2011, 11, 2119–2125. [Google Scholar] [CrossRef]
- Heikkilä, M.; Puukilainen, E.; Ritala, M.; Leskelä, M. Effect of thickness of ALD grown TiO2 films on photoelectrocatalysis. J. Photochem. Photobiol. A: Chem. 2009, 204, 200–208. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, W.; Liu, H.; Wang, R.; Chen, Y.; Wang, Z. Effects of TiO2 film thickness on photovoltaic properties of dye-sensitized solar cell and its enhanced performance by graphene combination. Mater. Res. Bull. 2013, 49, 126–131. [Google Scholar] [CrossRef]
- Song, X.-M.; Wu, J.-M.; Yan, M. Photocatalytic and photoelectrocatalytic degradation of aqueous Rhodamine B by low-temperature deposited anatase thin films. Mater. Chem. Phys. 2008, 112, 510–515. [Google Scholar] [CrossRef]
- Bennani, Y.; Appel, P.; Rietveld, L.C. Optimisation of parameters in a solar light-induced photoelectrocatalytic process with a TiO2/Ti composite electrode prepared by paint-thermal decomposition. J. Photochem. Photobiol. A: Chem. 2015, 305, 83–92. [Google Scholar] [CrossRef]
- Miller, E.L.; Marsen, B.; Paluselli, D.; Rocheleau, R. Optimization of Hybrid Photoelectrodes for Solar Water-Splitting. Electrochem. Solid-state Lett. 2005, 8, A247–A249. [Google Scholar] [CrossRef]
- Yang, B.; Barnes, P.R.F.; Bertram, W.; Luca, V. Strong photoresponse of nanostructured tungsten trioxide films prepared via a sol–gel route. J. Mater. Chem. 2007, 17, 2722–2729. [Google Scholar] [CrossRef]
- Vidyarthi, V.S.; Hofmann, M.; Savan, A.; Sliozberg, K.; König, D.; Beranek, R.; Schuhmann, W.; Ludwig, A. Enhanced photoelectrochemical properties of WO3 thin films fabricated by reactive magnetron sputtering. Int. J. Hydrogen Energy 2011, 36, 4724–4731. [Google Scholar] [CrossRef]
- Hong, S.J.; Jun, H.; Lee, J.S. Nanocrystalline WO3 film with high photo-electrochemical activity prepared by polymer-assisted direct deposition. Scr. Mater. 2010, 63, 757–760. [Google Scholar] [CrossRef]
- Mohamed, N.M.; Bashiri, R.; Kait, C.F.; Sufian, S.; Khatani, M. Investigation of photoconversion efficiency of Cu and Ni doped TiO2 thin film in photoelectrochemical cell. 4TH INTERNATIONAL CONFERENCE ON FUNDAMENTAL AND APPLIED SCIENCES (ICFAS2016). LOCATION OF CONFERENCE, MalaysiaDATE OF CONFERENCE; p. 050022.
- Mishra, P.; Shukla, P.; Srivastava, O. Study of modular PEC solar cells for photoelectrochemical splitting of water employing nanostructured TiO2 photoelectrodes. Int. J. Hydrogen Energy 2007, 32, 1680–1685. [Google Scholar] [CrossRef]
- Lee, W.J.; Shinde, P.S.; Go, G.H.; Ramasamy, E. Ag grid induced photocurrent enhancement in WO3 photoanodes and their scale-up performance toward photoelectrochemical H2 generation. Int. J. Hydrogen Energy 2011, 36, 5262–5270. [Google Scholar] [CrossRef]
- H. Döscher et al., “Solar-to-hydrogen efficiency: shining light on photoelectrochemical device performance,” in AIChE Annual Meeting, Conference Proceedings, American Institute of Chemical Engineers, 2019. [CrossRef]
- Bashiri, R.; Mohamed, N.M.; Kait, C.F.; Sufian, S. Optimization of hydrogen production over TiO2 supported copper and nickel oxides: effect of photoelectrochemical features. J. Appl. Electrochem. 2018, 49, 27–38. [Google Scholar] [CrossRef]
- Nagai, N.; Takeuchi, M.; Kimura, T.; Oka, T. Existence of optimum space between electrodes on hydrogen production by water electrolysis. Int. J. Hydrogen Energy 2003, 28, 35–41. [Google Scholar] [CrossRef]
- Asl, M.A.; Tahvildari, K.; Bigdeli, T. Eco-friendly synthesis of biodiesel from WCO by using electrolysis technique with graphite electrodes. Fuel 2020, 270, 117582. [Google Scholar] [CrossRef]
- Yuzer, B.; Selcuk, H.; Chehade, G.; Demir, M.; Dincer, I. Evaluation of hydrogen production via electrolysis with ion exchange membranes. Energy 2019, 190, 116420. [Google Scholar] [CrossRef]
- Hernández, S.; Saracco, G.; Alexe-Ionescu, A.; Barbero, G. Electric investigation of a photo-electrochemical water splitting device based on a proton exchange membrane within drilled FTO-covered quartz electrodes: under dark and light conditions. Electrochimica Acta 2014, 144, 352–360. [Google Scholar] [CrossRef]
- Effect of Operating Parameters on Performance of Alkaline Water Electrolysis. 01-902-664-9706 2015, 09. [CrossRef]
- Riegel, H.; Mitrovic, J.; Stephan, K. Role of mass transfer on hydrogen evolution in aqueous media. J. Appl. Electrochem. 1997, 28, 10–17. [Google Scholar] [CrossRef]
- Funk, J.E.; Thorpe, J.F. Void Fraction and Current Density Distributions in a Water Electrolysis Cell. J. Electrochem. Soc. 1969, 116, 48–54. [Google Scholar] [CrossRef]
- Hoshikawa, T.; Yamada, M.; Kikuchi, R.; Eguchi, K. Impedance Analysis of Internal Resistance Affecting the Photoelectrochemical Performance of Dye-Sensitized Solar Cells. J. Electrochem. Soc. 2005, 152, E68–E73. [Google Scholar] [CrossRef]
- Curteanu, S.; Piuleac, C.G.; Godini, K.; Azaryan, G. Modeling of electrolysis process in wastewater treatment using different types of neural networks. Chem. Eng. J. 2011, 172, 267–276. [Google Scholar] [CrossRef]
- Fouad, M.; Sedahmed, G. Effect of gas evolution on the rate of mass transfer at vertical electrodes. Electrochimica Acta 1972, 17, 665–672. [Google Scholar] [CrossRef]
- Hsu, W.; Terrones, M.; Hare, J.; Terrones, H.; Kroto, H.; Walton, D. Electrolytic formation of carbon nanostructures. Chem. Phys. Lett. 1996, 262, 161–166. [Google Scholar] [CrossRef]
- Bell, S.; Will, G.; Bell, J. Light intensity effects on photocatalytic water splitting with a titania catalyst. Int. J. Hydrogen Energy 2013, 38, 6938–6947. [Google Scholar] [CrossRef]
- Mohamed, N.M.; Bashiri, R.; Chong, F.K.; Sufian, S.; Kakooei, S. Photoelectrochemical behavior of bimetallic Cu–Ni and monometallic Cu, Ni doped TiO2 for hydrogen production. Int. J. Hydrogen Energy 2015, 40, 14031–14038. [Google Scholar] [CrossRef]
- Lim, T.H.; Jeong, S.M.; Kim, S.D.; Gyenis, J. Photocatalytic decomposition of NO by TiO2 particles. J. Photochem. Photobiol. A: Chem. 2000, 134, 209–217. [Google Scholar] [CrossRef]
- Kim, J.H.; Moon, K.J.; Kim, J.M.; Lee, D.; Kim, S.H. Effects of various light-intensity and temperature environments on the photovoltaic performance of dye-sensitized solar cells. Sol. Energy 2015, 113, 251–257. [Google Scholar] [CrossRef]
- Liu, J.; Wan, J.; Liu, L.; Yang, W.; Low, J.; Gao, X.; Fu, F. Synergistic effect of oxygen defect and doping engineering on S-scheme O-ZnIn2S4/TiO2-x heterojunction for effective photocatalytic hydrogen production by water reduction coupled with oxidative dehydrogenation. Chem. Eng. J. 2021, 430, 133125. [Google Scholar] [CrossRef]
- Sayama, K.; Sugihara, H.; Arakawa, H. Photoelectrochemical Properties of a Porous Nb2O5 Electrode Sensitized by a Ruthenium Dye. Chem. Mater. 1998, 10, 3825–3832. [Google Scholar] [CrossRef]
- K. Sayama and H. Arakawa+, “Effect of Na,CO, addition on photocatalytic decomposition of liquid water over various semiconductor catalysts,” 1994.
- Patsoura, A.; Kondarides, D.I.; Verykios, X.E. Enhancement of photoinduced hydrogen production from irradiated Pt/TiO2 suspensions with simultaneous degradation of azo-dyes. Appl. Catal. B: Environ. 2006, 64, 171–179. [Google Scholar] [CrossRef]
- Dutta, A.; DuBois, D.L.; Roberts, J.A.S.; Shaw, W.J. Amino acid modified Ni catalyst exhibits reversible H 2 oxidation/production over a broad pH range at elevated temperatures. 111, 1628. [Google Scholar] [CrossRef]
- Seadira, T.; Sadanandam, G.; Ntho, T.A.; Lu, X.; Masuku, C.M.; Scurrell, M. Hydrogen production from glycerol reforming: conventional and green production. Rev. Chem. Eng. 2017, 34, 695–726. [Google Scholar] [CrossRef]
- Michaels, H.; Rinderle, M.; Freitag, R.; Benesperi, I.; Edvinsson, T.; Socher, R.; Gagliardi, A.; Freitag, M. Dye-sensitized solar cells under ambient light powering machine learning: towards autonomous smart sensors for the internet of things. Chem. Sci. 2020, 11, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Zhao, C.; You, S.; Zhang, Y.; Li, W. Recent progress of thin-film photovoltaics for indoor application. Chin. Chem. Lett. 2019, 31, 643–653. [Google Scholar] [CrossRef]
- Brian O’Regan and Michael Grätzel, “A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films,” Nature, vol. 353, pp. 737–740, 1991.
- Liu, S.; Yuan, J.; Deng, W.; Luo, M.; Xie, Y.; Liang, Q.; Zou, Y.; He, Z.; Wu, H.; Cao, Y. High-efficiency organic solar cells with low non-radiative recombination loss and low energetic disorder. Nat. Photon- 2020, 14, 300–305. [Google Scholar] [CrossRef]
- Burschka, J.; Pellet, N.; Moon, S.-J.; Humphry-Baker, R.; Gao, P.; Nazeeruddin, M.K.; Grätzel, M. Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature 2013, 499, 316–319. [Google Scholar] [CrossRef]
- Kokkonen, M.; Talebi, P.; Zhou, J.; Asgari, S.; Soomro, S.A.; Elsehrawy, F.; Halme, J.; Ahmad, S.; Hagfeldt, A.; Hashmi, S.G. Advanced research trends in dye-sensitized solar cells. J. Mater. Chem. A 2021, 9, 10527–10545. [Google Scholar] [CrossRef]
- Freitag, M.; Teuscher, J.; Saygili, Y.; Zhang, X.; Giordano, F.; Liska, P.; Hua, J.; Zakeeruddin, S.M.; Moser, J.-E.; Grätzel, M.; et al. Dye-sensitized solar cells for efficient power generation under ambient lighting. Nat. Photon- 2017, 11, 372–378. [Google Scholar] [CrossRef]
- Grätzel, M. Dye-sensitized solar cells. J. Photochem. Photobiol. C: Photochem. Rev. 2003, 4, 145–153. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Sun, L. Iodine-free redox couples for dye-sensitized solar cells. J. Mater. Chem. 2011, 21, 10592–10601. [Google Scholar] [CrossRef]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.E.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, K.; Grätzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Nazeeruddin, Mohammad K., Filippo De Angelis, Simona Fantacci, Bessho Takeru, and Michael Grätzel., “Combined Experimental and DFT-TDDFT Computational Study of Photoelectrochemical Cell Ruthenium Sensitizers,” J Am Chem Soc, vol. 127, no. 48, pp. 16835–16847, 2005.
- Tennakone, K.; Senadeera, G.K.R.; Perera, V.P.S.; Kottegoda, I.R.M.; De Silva, L.A.A. Dye-Sensitized Photoelectrochemical Cells Based on Porous SnO2/ZnO Composite and TiO2 Films with a Polymer Electrolyte. Chem. Mater. 1999, 11, 2474–2477. [Google Scholar] [CrossRef]
- Asagoe, K.; Suzuki, Y.; Ngamsinlapasathian, S.; Yoshikawa, S. TiO2-Anatase Nanowire Dispersed Composite Electrode for Dye-Sensitized Solar Cells. 61, 1112. [Google Scholar] [CrossRef]
- Wang, L.; Fang, X.; Zhang, Z. Design methods for large scale dye-sensitized solar modules and the progress of stability research. Renew. Sustain. Energy Rev. 2010, 14, 3178–3184. [Google Scholar] [CrossRef]
- Sastrawan, R.; Beier, J.; Belledin, U.; Hemming, S.; Hinsch, A.; Kern, R.; Vetter, C.; Petrat, F.M.; Prodi-Schwab, A.; Lechner, P.; et al. New interdigital design for large area dye solar modules using a lead-free glass frit sealing. Prog. Photovoltaics: Res. Appl. 2006, 14, 697–709. [Google Scholar] [CrossRef]
- Späth, M.; Sommeling, P.M.; van Roosmalen, J.A.M.; Smit, H.J.P.; van der Burg, N.P.G.; Mahieu, D.R.; Bakker, N.J.; Kroon, J.M. Reproducible manufacturing of dye-sensitized solar cells on a semi-automated baseline. Prog. Photovoltaics: Res. Appl. 2003, 11, 207–220. [Google Scholar] [CrossRef]
Figure 1.
World's fuel shares of electricity generation in 2020 and 2021 (Other includes geothermal, solar, wind, heat, waste, etc. [2].
Figure 1.
World's fuel shares of electricity generation in 2020 and 2021 (Other includes geothermal, solar, wind, heat, waste, etc. [2].
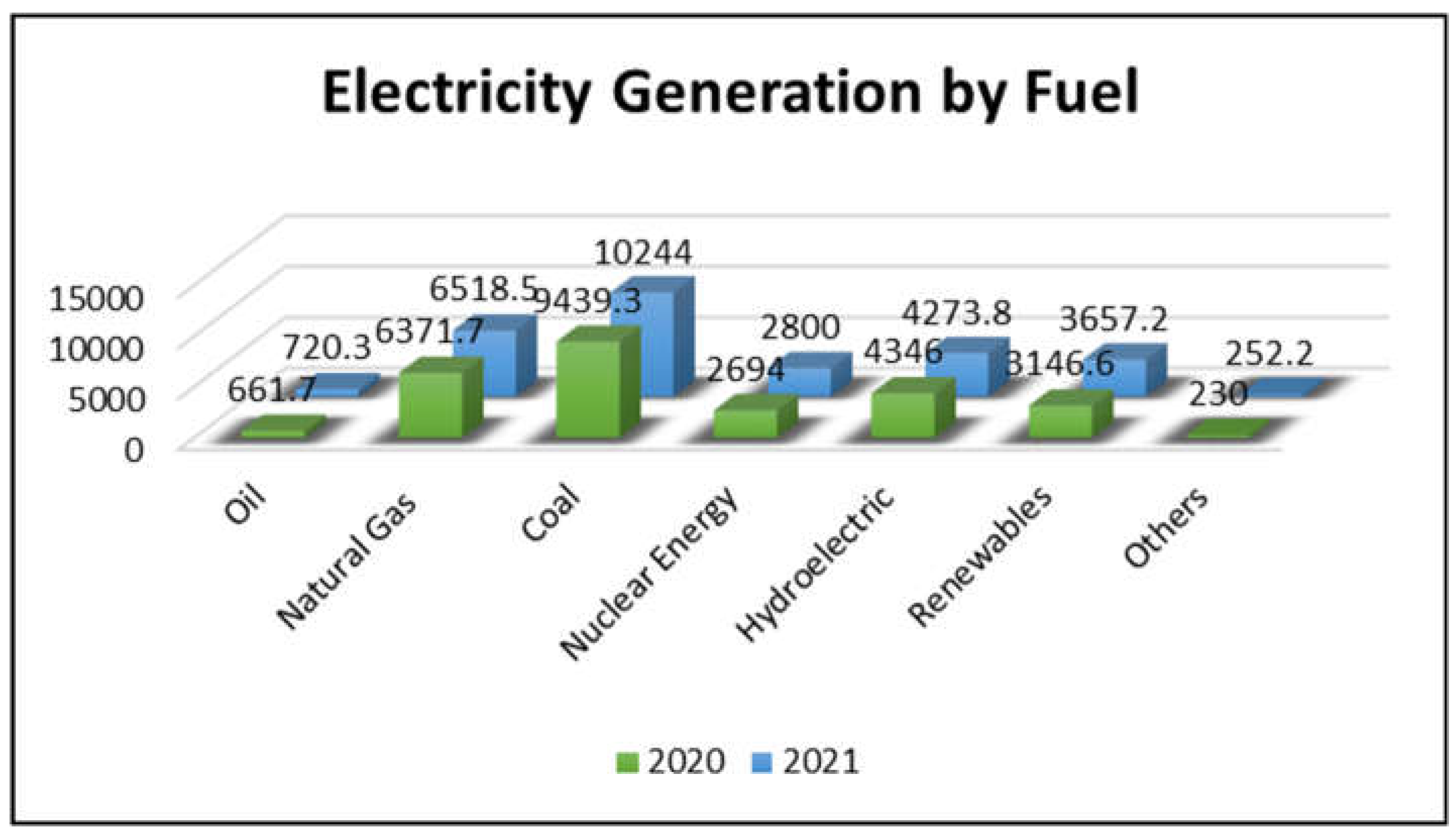
Figure 2.
Effect of hydrogen production on other sectors [15].
Figure 2.
Effect of hydrogen production on other sectors [15].
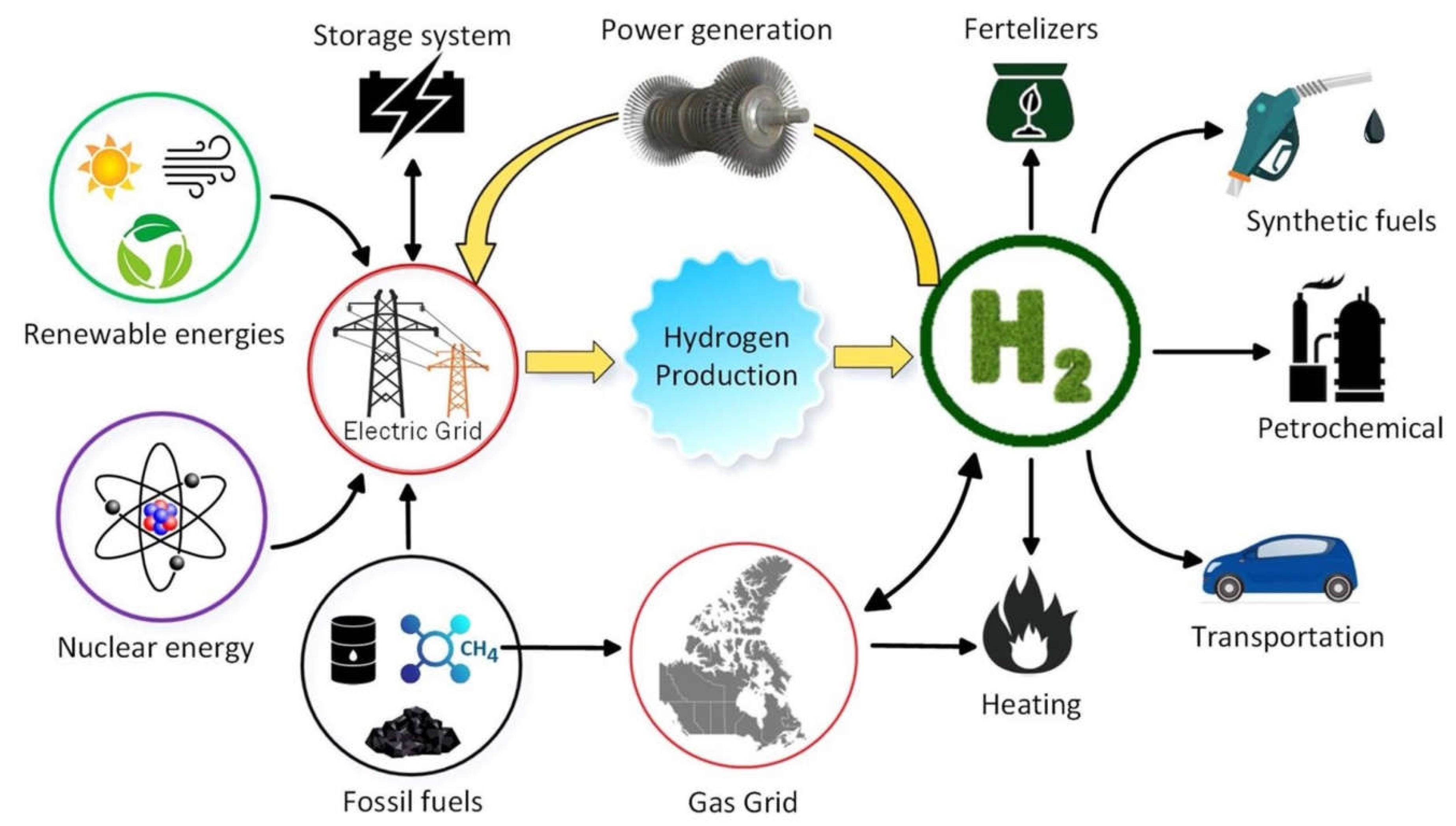
Figure 3.
A schematic diagram of general principle of photocatalysis.
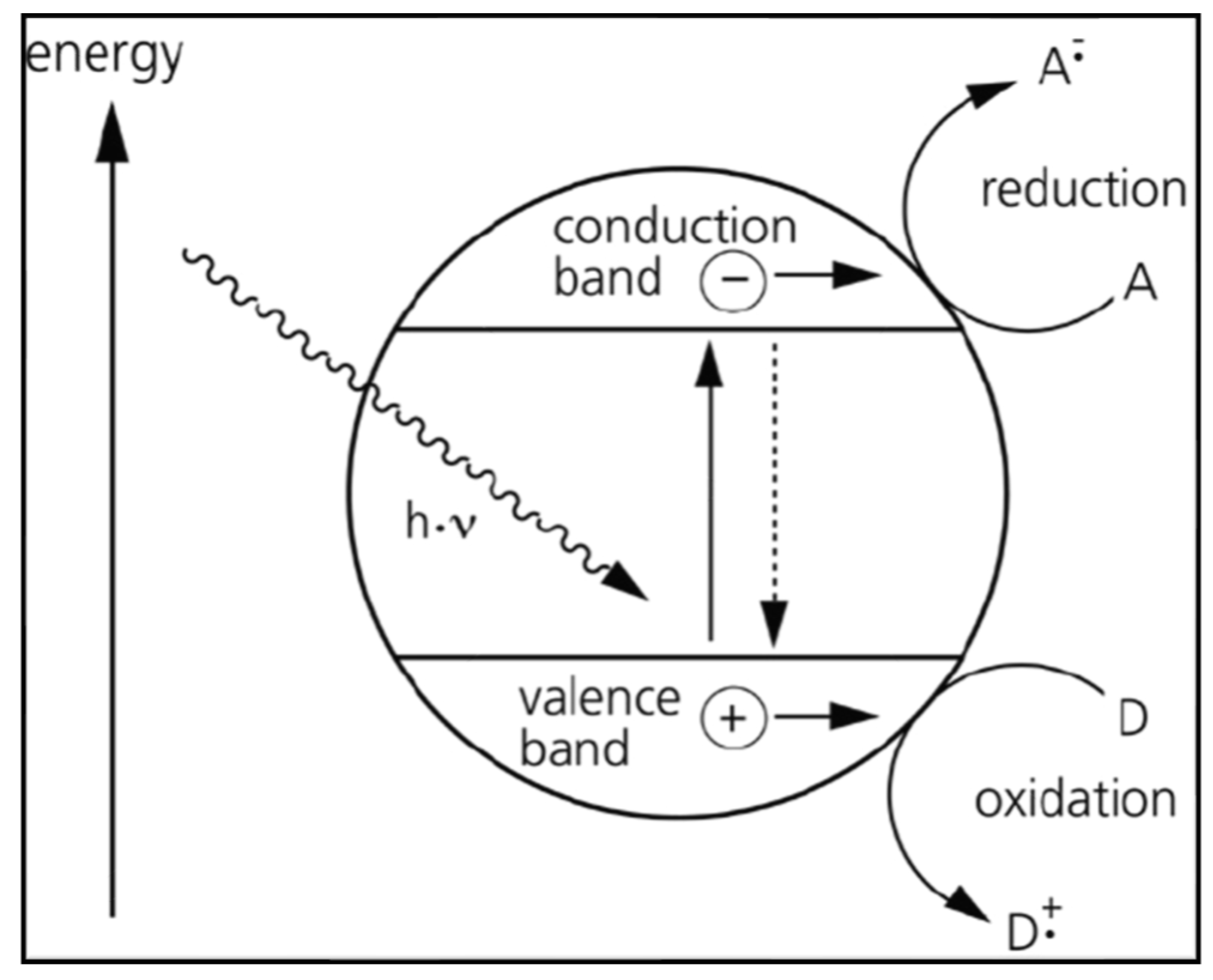
Figure 4.
Scheme of a standard DSC/PEC tandem cell configuration.
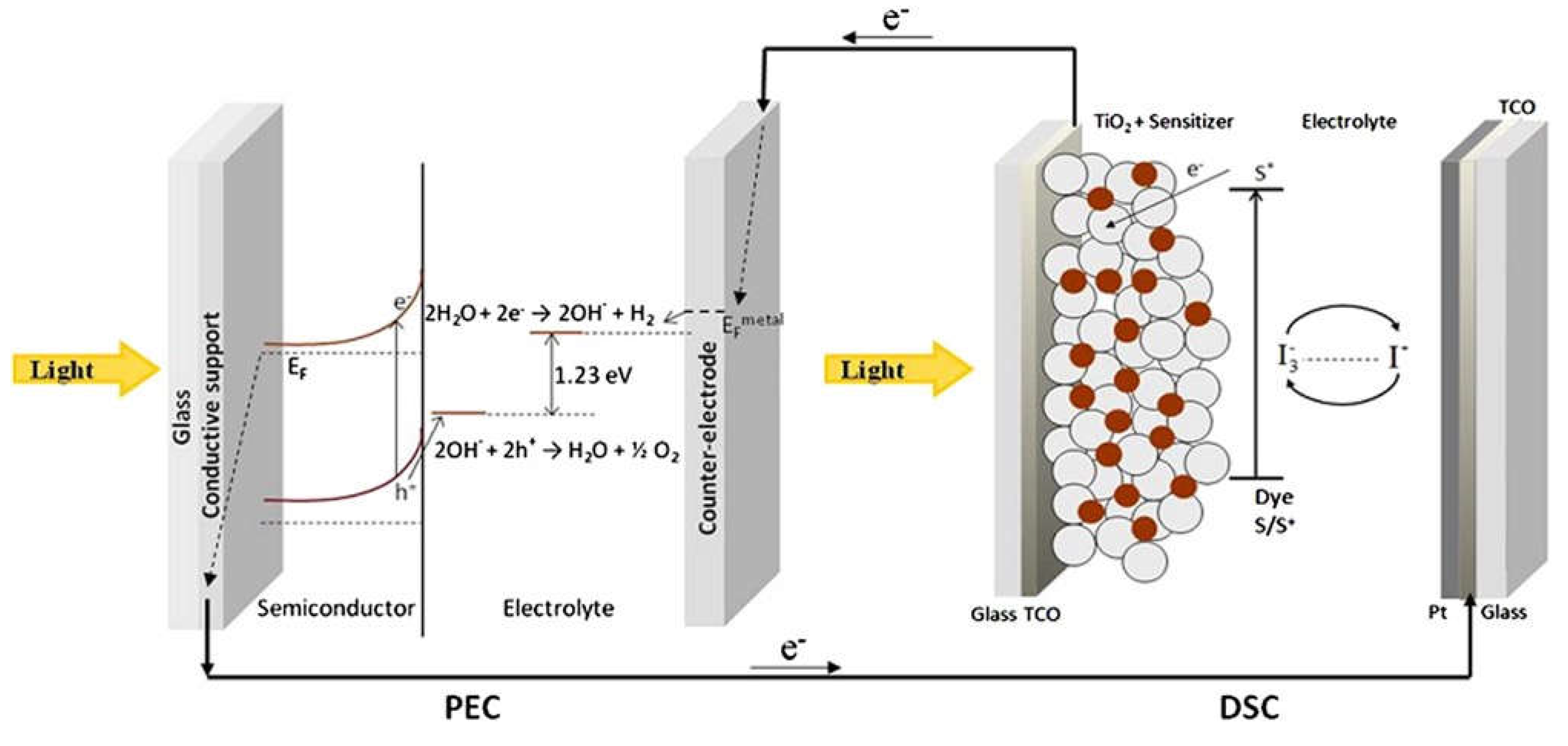
Figure 5.
Hydrogen production from water using powder photocatalyst.
Figure 6.
Schematic representation of a PEC reported by Fujishima and Honda [24].
Figure 6.
Schematic representation of a PEC reported by Fujishima and Honda [24].
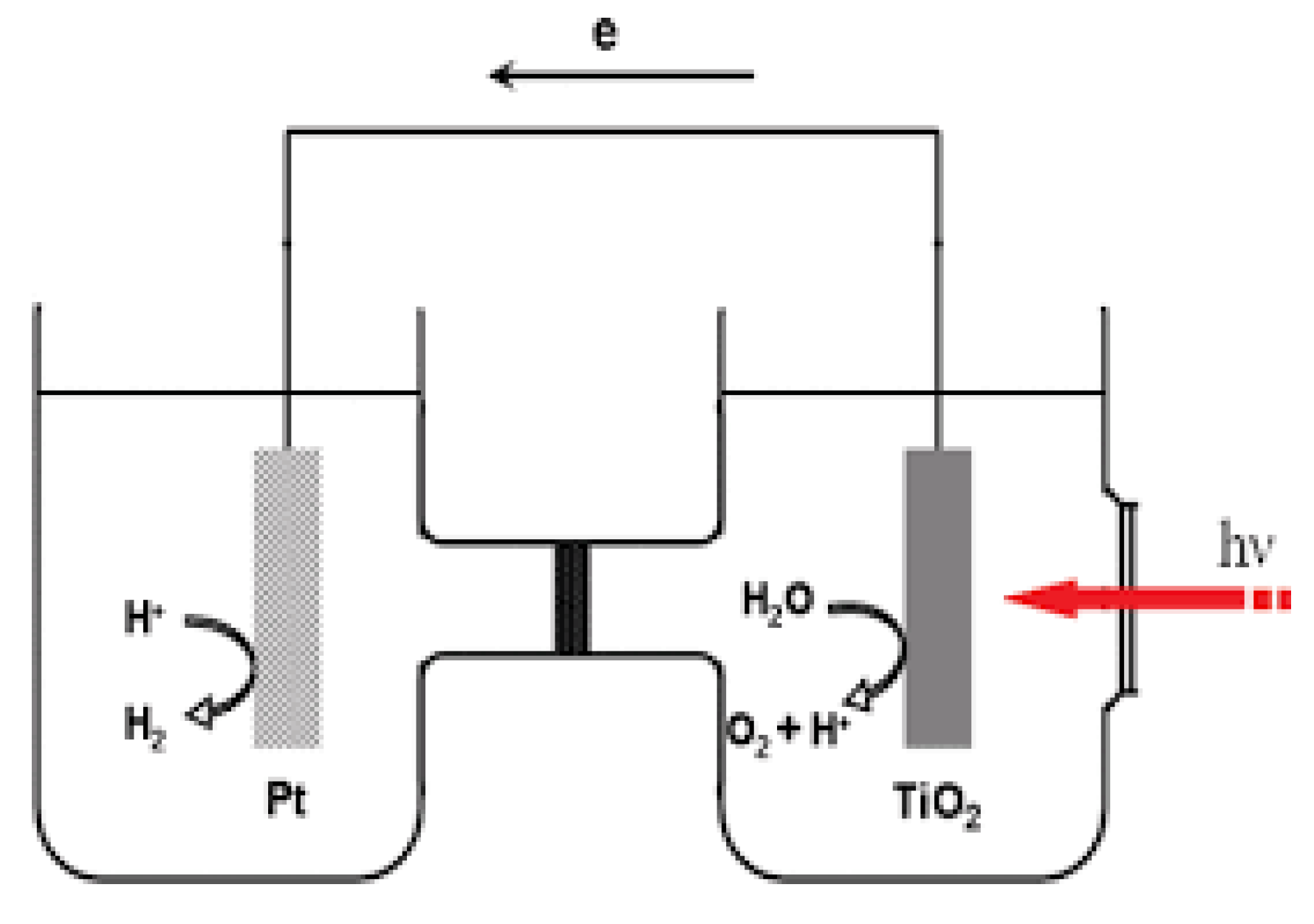
Figure 7.
The desirable CB and VB level position for water splitting in a photocatalyst.
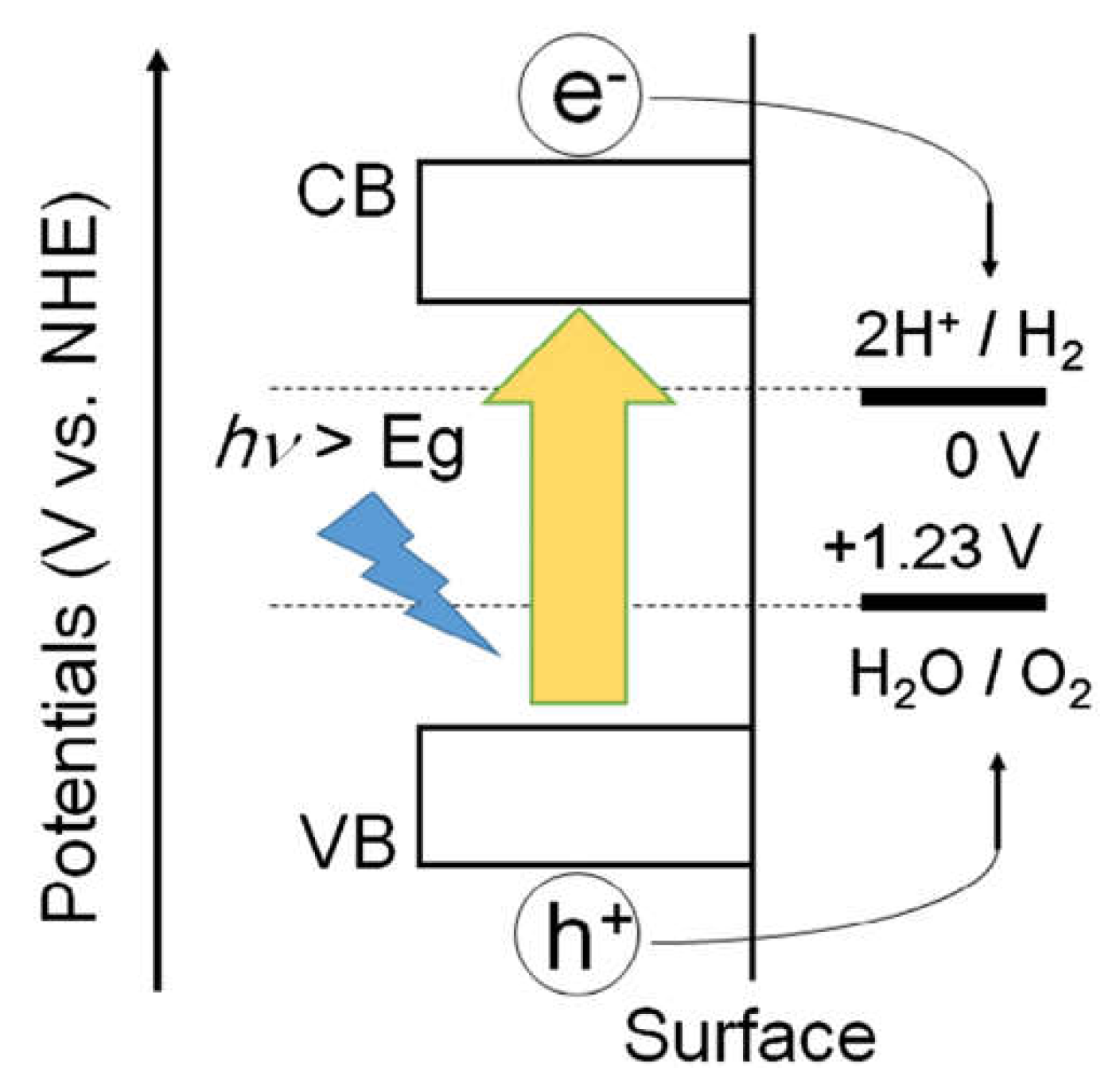
Figure 8.
Schematic of the band-level positions of different photocatalysts.
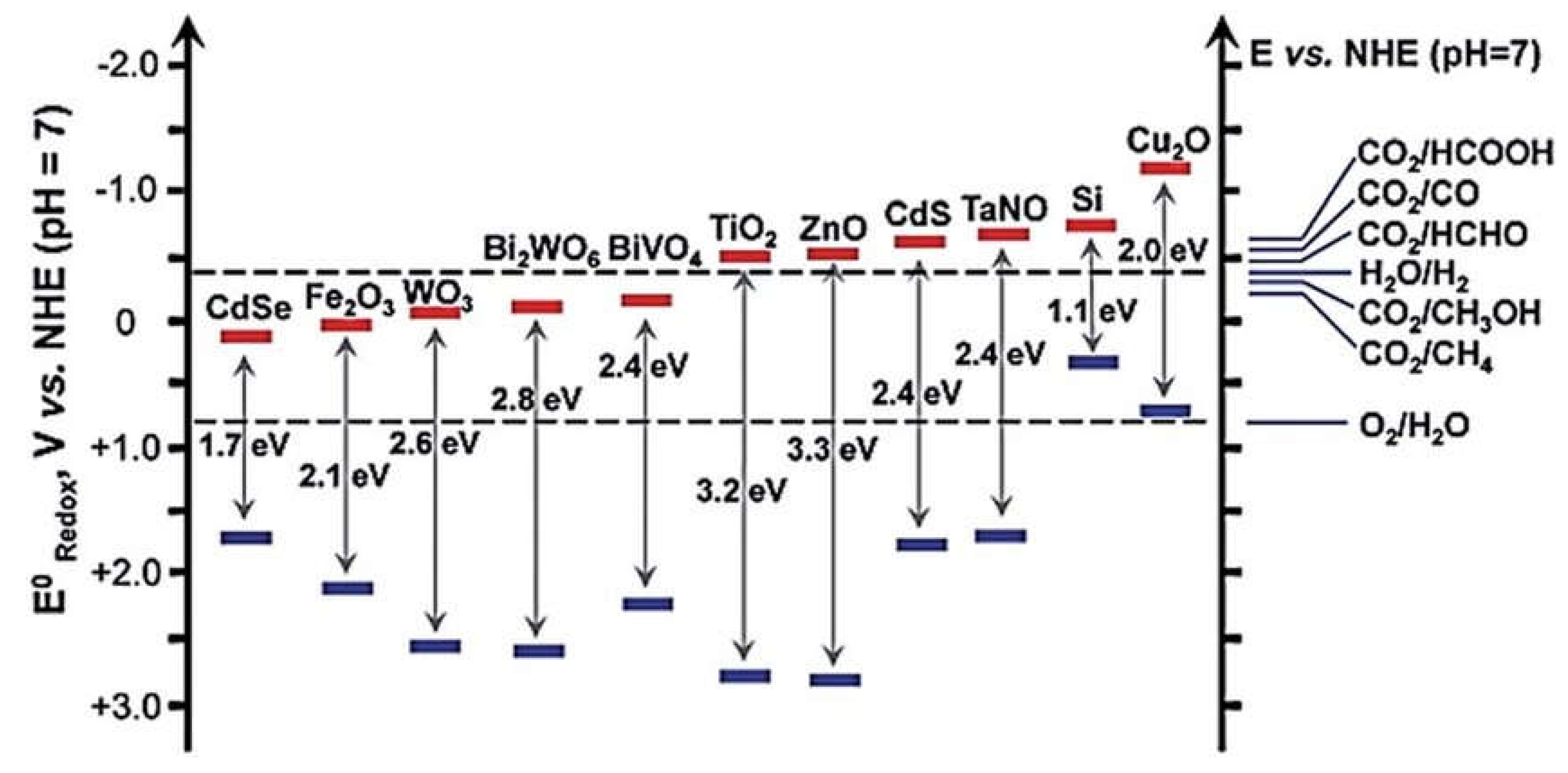
Figure 9.
Schematic representation of the distorted TiO6 octahedron of TiO2 (anatase and rutile). Each Ti atom (grey ball) is coordinated with the six neighboring oxygen atoms (red balls) [129].
Figure 9.
Schematic representation of the distorted TiO6 octahedron of TiO2 (anatase and rutile). Each Ti atom (grey ball) is coordinated with the six neighboring oxygen atoms (red balls) [129].

Figure 10.
Band structures of anatase and rutile TiO2.
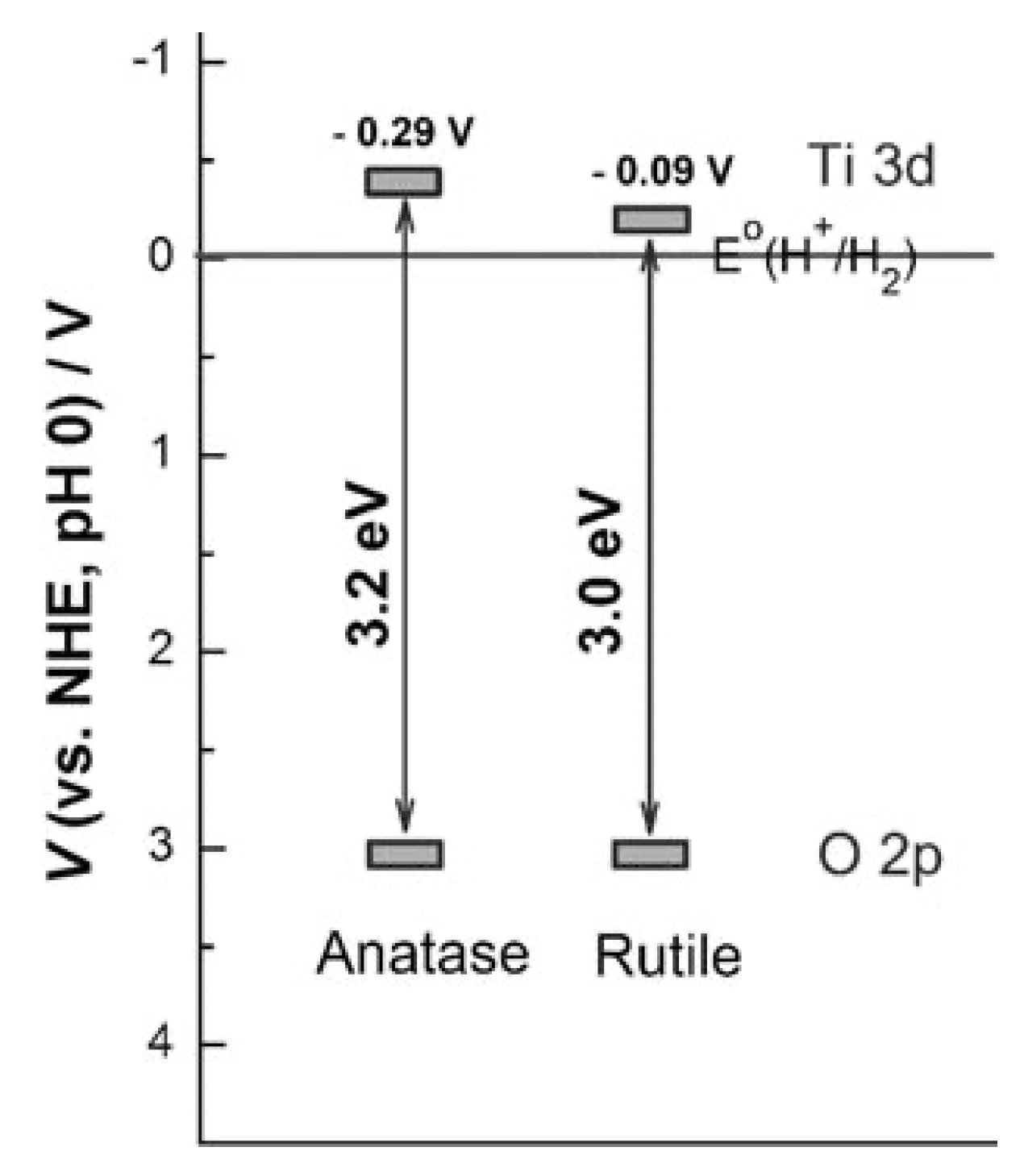
Figure 11.
Formation of (a) donor and (b) acceptor level by metal dopant.
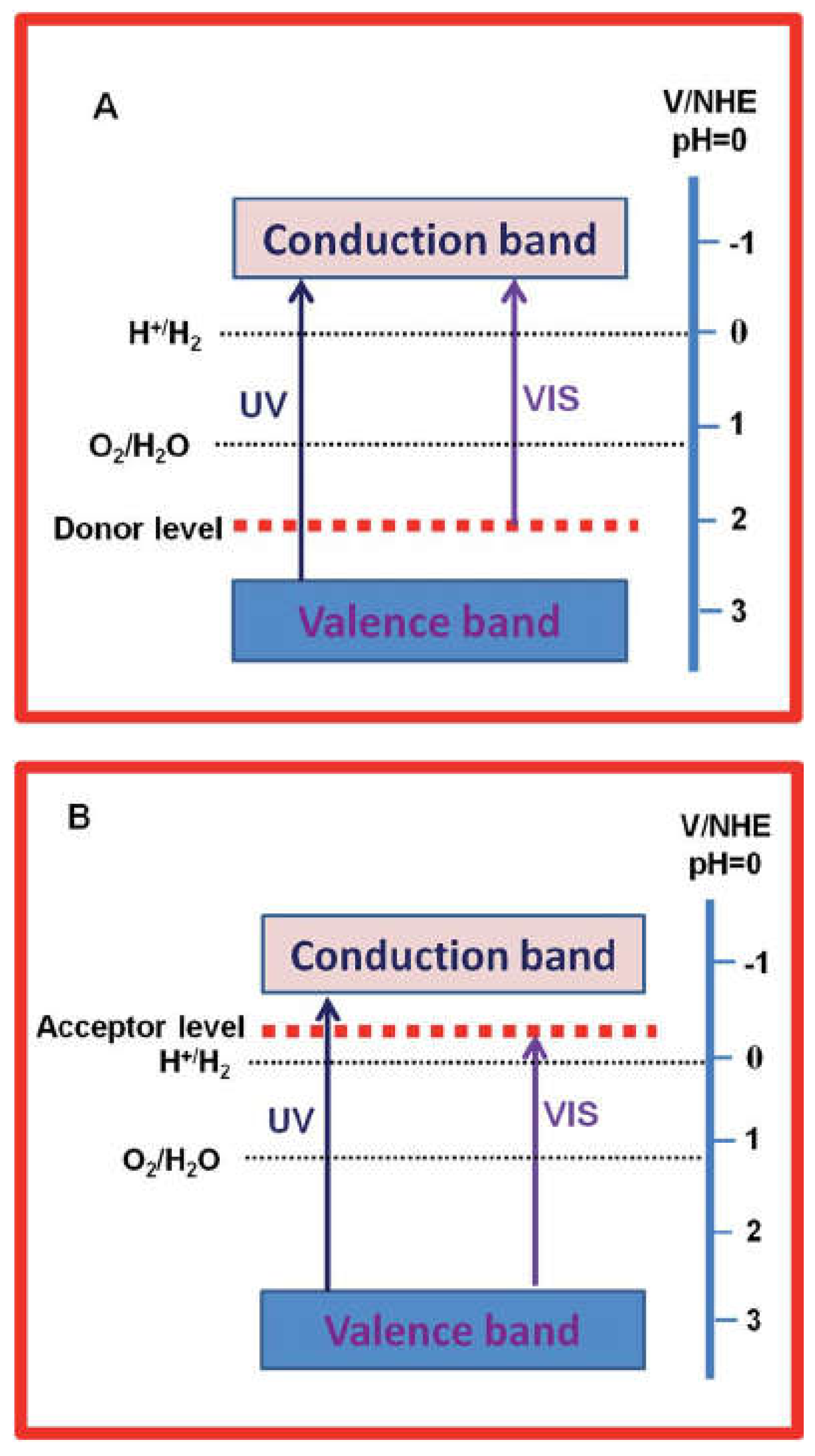
Figure 12.
Schematic diagram of dye solar cell.
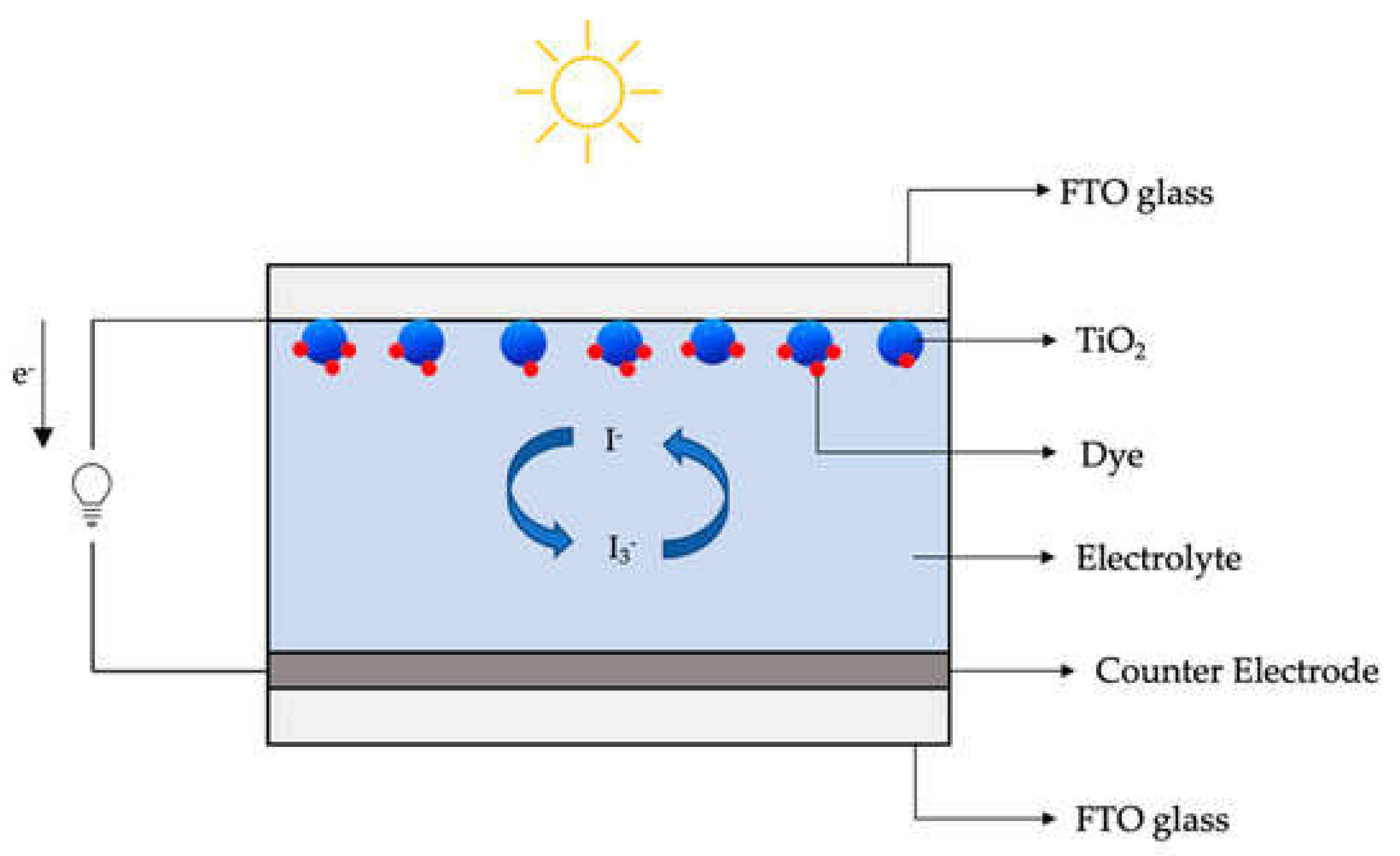
Figure 13.
Principle of operation and energy level of DSC.
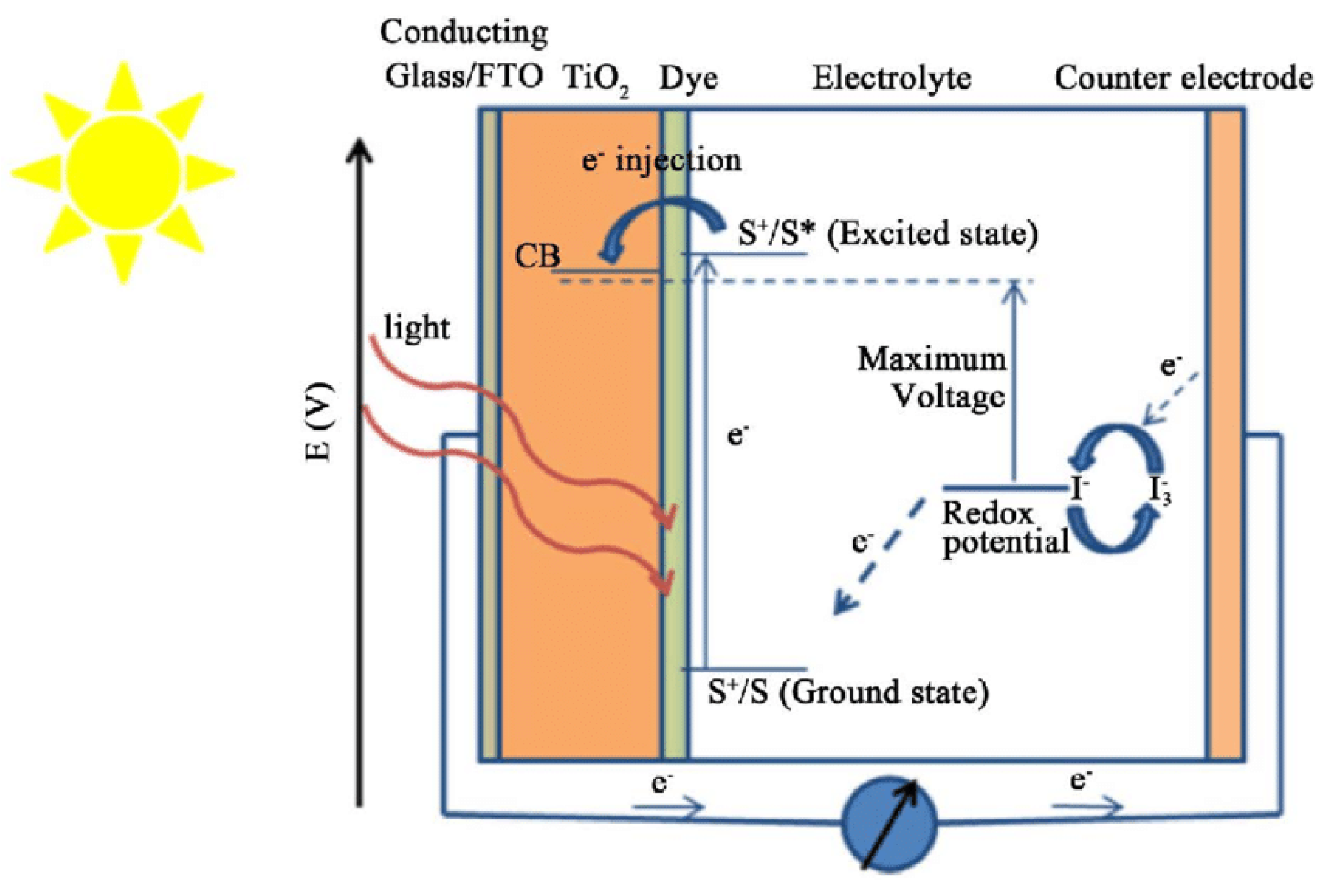

Table 1.
Higher and lower heating values of hydrogen and common fossil fuels at 25°C and 1 atm [33].
Table 1.
Higher and lower heating values of hydrogen and common fossil fuels at 25°C and 1 atm [33].
| Fuel | HHV (kJ/g) | LHV (kJ/g) |
|---|---|---|
| Hydrogen | 141.9 | 119.9 |
| Methane | 55.5 | 50.0 |
| Gasoline | 47.5 | 44.5 |
| Diesel | 44.8 | 42.5 |
| Methanol | 20.0 | 18.1 |
Table 2.
Summary of different hydrogen production.
| Process | Hydrogen Production Method | Feedstocks | Brief description | Ref. |
|---|---|---|---|---|
| Electrical energy | Electrolysis | Water | Conversion of electric power to chemical energy in the form of H2 and O2 as a by-product of water with two reactions in each electrode; anode and cathode | [11] [12] |
| Plasma arc decomposition | Natural gas | H2 and carbon soot were produced through methane decomposition in a plasma reactor with a parallel set of screw type helix and rod-like electrodes | [37] |
|
| Thermal energy | Thermolysis | Water | Steam is brought to temperature of over 2500K at which water molecule decomposes thermally into H2 and O2 | [38] |
| Thermocatalysis H2S cracking |
Hydrogen sulfide | Sufficient heat is supplied to break down H2S into H2 and S2 | [39] | |
| Biomass conversion |
Biomass | Thermocatalytic conversion of biomass to H2 | [40] | |
| Thermochemical processes Water splitting |
Water | Water decomposition through a repetitive series of chemical reactions using intermediate reactions and substances which are all recycled during the process so that the overall reactions is equivalent to the dissociation of the water molecule into H2 and O2 | [41] | |
| Gasification | Biomass | Biomass steam gasification converts carbonaceous material to permanent gases (H2, CO, CO2, CH4 and light hydrocarbon), char and tar | [42] | |
| Reforming | Biofuels | Transformation of exhaust heat into the chemical energy of a new synthetic fuel that has higher calorimetric properties such as low heating value | [43] | |
| H2S splitting | Hydrogen sulfide | Decompose H2S into H2S and S2 over two reaction steps; creating and then decomposing hydroiodic acid. | [44] | |
| Photonic energy | PV-electrolysis | Water | The system is equipped with 20 W photovoltaic panel to generate electricity and drive electrolyzer which is exposed to direct solar radiation till mid noon | [45] |
| Photocatalysis | Water | For H2 production, the conduction band level should be more negative than hydrogen production level while the valence band should be more positive than water oxidation level for efficient O2 production from water by photocatalysis | [46] | |
| Photoelectrochemical | Water | H2 and O2 are generated from water dissociation on the surface of PEC materials with enough solar energy absorption and proper use of electrolysis | [47] | |
| Bio-photolysis | Water | Under anaerobic condition, the cyanobacterium carries out the biological fixation of atmospheric nitrogen (N2) into ammonia (NH3), concurrently producing H2 as by-product | [48] | |
| Biochemical energy | Dark fermentation | Biomass | In dark fermentation, substrates are converted by anaerobic bacteria grown in the dark | [49] |
| Enzymatic | Water | A synthetic enzymatic pathway consisting of 13 enzymes for producing hydrogen from starch and water | [50] |
Table 3.
Recorded data of reviews of solar-powered hydrogen generation.
| No | Year | Description | Ref |
|---|---|---|---|
| 1. | 2004 | Dr. Aldo Steinfeld's work in the field of solar thermochemical hydrogen generation aligns with the broader efforts to harness solar energy for sustainable and low-carbon hydrogen production. | [61] |
| 2. | 2009 | The work conducted by Yilanci et al. involving a PV/fuel cell system that harnesses concentrated solar energy to produce hydrogen is indicative of advancements in sustainable hydrogen production. | [56] |
| 3. | 2011 | The study conducted by González et al., which investigates hydrogen generation using various methods, including photochemical, electrochemical, and thermochemical processes driven by solar energy, is indicative of comprehensive research into sustainable hydrogen production. | [51] |
| 4. | 2012 | The work conducted by Ngoh and Njomo, which involved a hydrogen generation experiment using hybrid thermochemical cycles and concentrated solar energy, addresses an important aspect of solar-driven hydrogen production. Their findings, particularly the emphasis on solar-to-electricity conversion efficiency as a key factor in overall hydrogen generation efficiency, highlight a crucial consideration in the development of sustainable hydrogen production technologies. | [62] |
| 5. | 2012 | The research conducted by Xiao et al., which focuses on solar energy-driven hydrogen production through a thermochemical cycle, sheds light on the challenges and potential advancements in this field. One of the key takeaways from their work is the importance of developing a reactor that can simultaneously perform the synthesis and hydrolysis of reduced metal/oxide nanoparticles, along with the capacity to recover the regenerated oxide particles. | [63] |
| 6. | 2013 | The work conducted by Chaubey et al., which involves the development of a hydrogen-based method utilizing concentrated solar energy in biological, electrochemical, and thermochemical processes, showcases a multidisciplinary approach to sustainable hydrogen production. Their research likely explores various methods for harnessing concentrated solar energy to generate hydrogen while emphasizing the use of biomass as a clean and renewable resource. | [64] |
| 7. | 2015 | The research by Agrafiotis et al. contributes to the understanding of concentrated solar energy applications in thermochemical hydrogen and syngas production. Such processes are essential for developing clean and sustainable pathways for hydrogen and syngas production, aligning with the goals of reducing carbon emissions and advancing renewable energy technologies. | [65] |
| 8. | 2016 | The work by Dan Zhao et al., involving the use of TiO2 and CdS nanocomposites, is related to the development of materials with applications in solar energy storage, photocatalysis, and particularly in photocatalytic hydrogen production and water splitting. | [66] |
| 9. | 2017 | The review conducted by Villafán-Vidales et al. on the thermochemical process of hydrogen production using concentrated solar energy likely provides a comprehensive overview of the various steps and considerations involved in this innovative and sustainable approach. | [66] |
| 10. | 2017 | By addressing the challenge of TiO2's wide bandgap and enhancing its light-absorbing capabilities, Wang et al.'s research likely contributes to the advancement of photocatalytic processes for solar-driven hydrogen production, which is a critical component of sustainable and clean energy systems. Blackening TiO2 can improve solar absorption and thus contribute to the improvement of solar-hydrogen efficiency. | [67] |
| 11. | 2018 | The review conducted by Jun Chi et al., which focuses on different water electrolysis processes and their various operating characteristics, is likely a comprehensive examination of electrolysis methods for hydrogen production. | [35] |
| 12. | 2019 | The research conducted by Kuan-Yeow Show et al., involving the use of laboratory-scale systems to produce biohydrogen from microalgae with the assistance of solar energy, is a significant contribution to the field of renewable energy and sustainable hydrogen production. | [68] |
| 13. | 2020 | The work conducted by Chao Zhou et al., which focuses on niobium-based semiconductors for photocatalysis, represents an important area of research within the field of solar-driven hydrogen production. | [69] |
| 14. | 2021 | The review conducted by Chen and Hainin focuses on the topic of photoelectrochemical (PEC) water splitting, specifically delving into the principles and development of Sb2Se3 photocathodes within this context. | [70] |
| 15. | 2021 | Jung Eun Lee et al. addresses the use of metal oxides, metal halides, and sulfur oxides in thermochemical processes for hydrogen production, with a focus on mitigating excessive temperature rise and stress on metal oxides during redox reactions. | [71] |
| 16. | 2022 | Rengui Li et al. appears to focus on the concept of a water splitting panel and a hydrogen farm, particularly in the context of photocatalyst particulate and the photocatalytic water splitting method. | [72] |
| 17. | 2022 | Muhammad Usman et al. appears to focus on the discussion of composites involving Metal-Organic Frameworks (MOFs) and graphitic carbon nitride (g-C3N4) for sustainable hydrogen production and portable energy harvesting applications. | [73] |
Table 4.
Selected nanomaterials used for PEC hydrogen generation including experimental conditions, corresponding performance, and key advantages.
Table 4.
Selected nanomaterials used for PEC hydrogen generation including experimental conditions, corresponding performance, and key advantages.
| Photoanode material | Electrolyte | Illumination | Performance | Advantage | Application | Ref. |
|---|---|---|---|---|---|---|
| Surface modified TiO2 nanotube arrays |
NaOH | 300 W xenon lamp (100 mW.cm-2) equipped with an AM 1.5 G filter | Incident photon-current conversion efficiency = 36% at 350 nm |
Reduced charge carrier recombination and improved hole injection efficiency | H2 production | [117] |
| TiO2 films prepared by sol–gel dip-coating |
KCl, MgCl2, CaCl2, Phenol and Na2SO4 (simulated wastewater) or oilfield-produced wastewater | 150 W metal-halide lamp with UV filter | rH2 = 12.36 mmol/h and 80% phenol degradation (simulated wastewater) rH2 = 9.11 mmol/h (oil field-produced wastewater) |
Low-cost, reduced toxicity, remarkable stability | H2 production and wastewater treatment | [142] |
| Photoanode material | Electrolyte | Illumination | Performance | Advantage | Application | Ref. |
| TiO2/Pt/FTO | Methyl orange, methanol and Na2SO4 |
400 W xenon lamp (100 mW.cm-2) equipped with optical filters |
Quantum Yield = 3.70% at λ=420nm rH2 = 11.4 mmol/h |
Enhanced charge separation | H2 production and wastewater treatment | [142] |
| TiNT/WO3/Ti | RB5 dye and Na2SO4 | 125 W high-pressure Hg lamp (12.55 mWcm-2) | H2 generation efficiency = 46% | Enhanced charge separation | H2 production and wastewater treatment | [143] |
| TiO2-1 wt% Au@TiO2/ Al2O3/Cu2O |
Humic acid and Na2SO4 | 150 W xenon lamp (100 mW.cm-2) equipped with an AM 1.5G filter |
Solar-to-hydrogen conversion efficiency = 0.5% and 87% humic acid degradation |
Self-biased PEC. Enhanced charge separation. Wide spectral absorption | H2 production and wastewater treatment | [144] |
Table 5.
Summary of metal-doped photocatalyst for H2 production.
| Photocatalyst | Light source | H2 Production Efficiency | References |
|---|---|---|---|
| TiO2 | UV irradiation | 200 μmol.h-1 | [161] |
| Au/TiO2 | 150W metal halide lamp | 120 μmol.min-1 | [148] |
| Ag/TiO2 | UV irradiation | 147.9 ± 35.5 μmol.h-1.g-1 for anatase Ag/TiO24.65 ± 0.39 μmol.h-1.g-1 for amorphous TiO2 | [11] |
| Co/TiO2 | 300 W Xe lamp | 11, 021 μmol.h-1.g-1 | [96] |
| Co/TiO2 | 8500 μmol.h-1.g-1 | [150] | |
| Ni/TiO2 | visible light irradiation (λ > 420 nm) | 21 712 μmol.h-1.g-1 | [152] |
| Al/TiO2 | UV irradiation | 312 μmol.h-1 | [161] |
| Cu/TiO2 | UV irradiation | 900 μmol.h-1 | [161] |
| Ni/ZCS QDs | 18.87 mmol.h−1.g−1 | [153] | |
| Ni/TiO2 | 300W high pressure Hg lamp | 185 μmol.h-1 | [155] |
| Fe/TiO2 | 250 W tungsten halogen lamp | 15.5 μmol.h-1 | [156] |
| Cr/TiO2 | 250 W tungsten halogen lamp | 5.3 μmol.h-1 | [156] |
| CdS0.9Se0.1 QDs | simulated solar irradiation (320–780 nm) | 29.12 mmol.h−1.g−1 | [157] |
| Ni-Mo/g-C3N4 | 1785 μmol.h-1.g-1 | [158] | |
| NiCd/CdS | under visible light irradiation (λ>410 nm) | 11.57 mmol·h−1·g−1 | [159] |
| NiCu-TiO2 | filtered illumination (λ>420nm) | 0.02μL.h-1cm-2 | [160] |
| Al-Cu/TiO2 | UV irradiation | 1093 μmol.h-1 | [161] |
| Fe-Ni/TiO2 | 500W Xenon lamp | 361.64 μmol.h-1.g-1 | [162] |
| Fe-Ag/TiO2 | Hg lamp (λ=254 nm) and 500W Xenon lamp | 515.45 μmol.h-1.g-1 | [163] |
| CoNi-ZIS | under visible light irradiation (λ>420 nm) | 100.1 μmol.h-1 | [164] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



