Submitted:
22 September 2023
Posted:
25 September 2023
You are already at the latest version
Abstract
Pathophysiology of depression is related with reduced volume of the hippocampus and amygdala and hypertrophy of the nucleus accumbens. The mechanism of these changes is not well under-stood, but clinical studies have shown that administration of the fast-acting antidepressant keta-mine reversed the decrease in hippocampus and amygdala volume in depressed patients, and the magnitude of this effect correlated with the reduction of depressive symptoms. In the present study, we attempted to find out whether the psychedelic substance psilocybin affects neurotransmission in the limbic system in comparison to ketamine. Psilocybin and ketamine increased the release of dopamine (DA) and serotonin (5-HT) in the nucleus accumbens of naive rats as demonstrated using microdialysis. Both drugs influenced glutamate and GABA release in the nucleus accumbens, hippocampus and amygdala and increased ACh levels in the hippocampus. The changes in D2, 5-HT1A and 5-HT2A receptor density in the nucleus accumbens and hippocampus was observed as a long-lasting effect. A marked anxiolytic effect of psilocybin in acute phase and 24 h post-treatment was shown in the open field test. These data provide the neurobiological background for psilocybin effect on stress, anxiety and structural changes in the limbic system and translate into antidepres-sant effect of psilocybin in depressed patients.
Keywords:
neurotransmitter release
; dopamine D2 receptors
; serotonin 5-HT1A
; 5-HT2A receptors
; limbic system
1. Introduction
As reported by the World Health Organization, major depressive disorder can affect up to 5% of adult population worldwide [1], while 10 to 15% of adults experience a depressive episode during their lifetime, though this statistics can be underestimated because many individuals suffering from affective disorders do not seek professional help [2]. Even though the pathophysiology of depression is still not clearly understood, a number of key factors relevant to this problem have been proposed. Firstly, numerous risk factors have been mentioned, with the most significant being either traumatic or chronic stress [3]. Moreover, depression-related changes have been observed in various brain structures, most importantly in the frontal cortex and the limbic system [4], though the direction of changes was opposite, namely hypoactivity was shown in the cortex while hyperactivity in the limbic structures [5]. The prefrontal cortex appears to be a center that integrates many sensory inputs from the structures like hippocampus, amygdala and nucleus accumbens, assigning them either rewarding or aversive properties [3].
The most important component of the limbic system that is affected in major depressive disorder is the hippocampus [4]. Like in the case of the prefrontal cortex, studies showed a clear correlation between atrophy of the hippocampus and severity and length of the disorder [3]. Patients with greater hippocampal volume seem to respond better to the treatment, while those with a smaller volume are more prone to being treatment resistant [4]. Those structural changes translate into functional ones, as the hippocampus plays a crucial role in processes of learning and memory [5], and is one of a few places where neurogenesis can occur in adults.
The amygdala plays a central role in processes associated with emotional states and fear processing. As with the hippocampus, its reduced volume correlates with the severity of the depression [6] moreover, its hyperactivity correlates with the severity of the disorder [5].
In contrast to the two aforementioned brain areas, the nucleus accumbens exhibits hypertrophy both in preclinical models and in individuals suffering from depression, which probably arises from the observed increase in the length of the dendrites and spine density [7]. Furthermore, according to DSM-V [8] anhedonia is one of the two core symptoms of depression, while inability to experience pleasure is a clear manifestation of disorders in the functioning of the reward system, where the nucleus accumbens plays a crucial role [9].
Recent studies showed that ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist proposed as a prototypical fast-acting antidepressant drug [10,11] exerts its beneficial effect not only in the frontal cortex, but also by targeting areas in the limbic system. Administration of ketamine promotes neural plasticity in the hippocampus in preclinical models [12] and increases hippocampal volume in depressed patients [13], while significantly improving their mood [13,14], which is an effect correlated with the volume of the hippocampus. Zhou et al. [15] observed that ketamine administration increased the amygdala volume in depressed patients, and the magnitude of the effect correlated with the reduction of depressive symptoms. Furthermore, ketamine treatment seems to reverse the depression-induced hypertrophy of the nucleus accumbens [7]. Altogether, the research cited above leads to the conclusion that ketamine affects not only the prefrontal cortex, but also the limbic structures, inducing a complex effect that normalizes its functioning and increases its connectivity with the prefrontal cortex [3].
Despite opening a new chapter in the treatment of affective disorders and becoming the prototypical fast-acting antidepressant drug, ketamine is far from perfect. Its effects fade around the second week after the drug administration, leading to the need of repeated dosing [16]. Furthermore, it possesses a plethora of adverse effects, of which drug abuse is the most significant one. Similarly to ketamine, psychedelics seem to induce synaptic plasticity and neurogenesis in preclinical models [16,17], while being devoid of the drawbacks of ketamine. This phenomenon translates further into antidepressant effects observed in preclinical models after administration of psychedelics [18,19]. Among serotonergic hallucinogens, psilocybin is currently the most studied compound, as it has proven to be at least as effective as common antidepressant drugs [20]. In contrast to ketamine, reports on the possible effects exerted by psychedelics on the limbic system are scarce, justifying the need for thorough studies.
To address the questions regarding the response of the limbic system to the administration of psilocybin and to compare it to ketamine, we have examined the effects of both drugs on neurotransmission in the hippocampus, amygdala and nucleus accumbens using microdialysis in freely-moving rats. Furthermore, Western blot analysis was performed to assess the long-lasting effects of the chosen drugs on protein levels of selected receptors. As the limbic system plays a key role in locomotion and fear response, the rat behavior was studied in the open field test both directly and 24h after administration of the drugs, to examine both acute and possible prolonged effects.
2. Materials and Methods
2.1. Animals
Adult male Wistar-Han rats (280-350 g; Charles River, Germany) were used in all experiments. The animals were initially acclimatized and housed (6 per cage) in environmentally controlled rooms (ambient temperature 23 ± 1°C, humidity 55 ± 10%, and 12:12 light: dark cycle). Rats were handled once daily before the beginning of the experiments; an enriched environment was not applied. The animals had free access to tap water and typical laboratory food (VRF 1, Special Diets Services, Witham, UK). All animal use procedures were conducted in strict accordance with European regulations for animal experimentation (EU Directive 2010/63/EU on the Protection of Animals Used for Scientific Purposes). The 2nd Local Institutional Animal Care and Use Committee (IACUC) in Kraków, Poland, approved the experimental protocols for Experimentation on Animals (permit numbers: 112/2021, 324/2021 and 79/2022).
2.2. Drugs and reagents
Ketamine hydrochloride was purchased from Tocris/Bio-Techne (Poland) and psilocybin was synthesized at the Department of Medicinal Chemistry of the Maj Institute of Pharmacology using the method described by Shirota et al. [21]; both were dissolved in sterile water. All solutions were made fresh on the day of experiment. The dose of ketamine (10 mg/kg) was based on a report by Popik et al. [22], while doses of psilocybin (2 and 10 mg/kg) on work by Jefsen et al. [23]. Psilocybin was given subcutaneously (sc) while ketamine intraperitoneally (ip) in the volume of 2 ml/kg. The control group was treated with 0.9% NaCl solution in the same way. Ketamine, xylasine hydrochlorides and sodium pentobarbital used for anesthetizing the animals came from Biowet Puławy (Puławy, Poland). All necessary chemicals of the highest purity used for analysis by high-performance liquid chromatography (HPLC) were obtained from Merck (Warszawa, Poland). O-phthalaldehyde (OPA) from Sigma-Aldrich (Poznań, Poland) was used for the derivatization of glutamate and GABA to electroactive compounds. The reagents used in immunohistochemistry were purchased from Sigma-Aldrich (Poznań, Poland), Vector Laboratories (Burlingame, CA, USA), and Proteintech (Manchester, UK).
2.3. Brain microdialysis
Ketamine and xylazine (75 and 10 mg/kg, respectively) were injected intramuscularly to anesthetize the animals. Microdialysis probes (MAB 4.15.3Cu and MAB 4.15.2Cu, AgnTho’s AB, Sweden) were implanted into the following brain structures using the determined coordinates (mm): nucleus accumbens AP +1.6, L +1.0, V -8.0, hippocampus AP -5.8, L 4.5, V -5.0 and amygdala AP -3.1, L +4.5, V -8.0 from the dura [24]. Seven days after implantation, probe inlets were connected to a syringe pump (BAS, West Lafayette, IN, USA) which delivered artificial cerebrospinal fluid composed of (mM): 147 NaCl, 4 KCl, 2.2 CaCl2·2H2O, 1.0 MgCl2 at a flow rate of 2 µL/min. Five baseline samples were collected every 20 minutes after the washout period of 2 hours. The respective drugs were administered, and dialysate fractions were collected for the next 240 minutes. As the experiment ended, the rats were terminated, and their brains underwent histological examination to validate probe placement.
2.4. Extracellular concentration of DA, 5-HT, glutamate, GABA and acetylcholine
Extracellular DA and 5-HT levels were analyzed using an Ultimate 3000 System (Dionex, USA), electrochemical detector Coulochem III (model 5300; ESA, USA) with a 5020 guard cell, a 5040 amperometric cell, and a Hypersil Gold C18 analytical column (3 μm, 100 × 3 mm; Thermo Fisher Scientific, USA). The details of the method have been described elsewhere [25,26]. The chromatographic data were processed by Chromeleon v.6.80 (Dionex, USA) software package run on a personal computer.
Glutamate and GABA levels in the extracellular fluid were measured by HPLC with electrochemical detection after derivatization of samples with OPA/sulfite reagent to form isoindole-sulfonate derivatives as previously described [25,26]. The data were processed using Chromax 2005 (Pol-Lab, Warszawa, Poland) software on a personal computer.
Extracellular levels of ACh were analyzed by UHPLC with electrochemical detection. The ACh analysis is based on ion-pairing HPLC separation, followed by on-line enzymatic conversion of ACh to hydrogen peroxide and detection on a Pt working electrode (SenCell with 2 mm Pt working electrode) and HyREF reference electrode at the potential of 200 mV. Chromatography was performed using the ALEXYS Neurotransmitter Analyzer, a DECADE Elite electrochemical detector, AS 110 Autosampler, and LC 110 pump (Antec Leyden B. V., Zoeterwoude, The Netherlands). ACh as positively charged was separated on Acquity UPLC HSS T3 analytical column (1.8 μm, 1 × 50 mm; Waters, Milford, MA, USA). After separation, ACh passed through an immobilized enzyme reactor AChE/ChOx IMER (AC-ENZYM II, 1 × 4 mm, Eicom, Kyoto, Japan). The mobile phase was composed of 50 mM monosodium orthophosphate buffer adjusted to pH 7.8, 0.5 mM Na2EDTA, 2.8 g/L 1-octanesulfonic acid sodium salt and 0.5 mM tetramethylammonium chloride. The flow rate during analysis was set to 0.05 mL/min. The chromatographic data were processed by CLARITY v.6.2.0.208 (DataApex Ltd.) chromatography software run on a personal computer.
2.5. Western blotting
The Western blot procedure was performed as previously described in Wojtas et al. [27]. The hippocampus and nucleus accumbens were homogenized (TissueLyser, Retsch, Munich, Germany) in lysis buffer ( PathScan Sandwich ELISA Lysis Buffer, Cell Signaling, Denver, CO, USA). Bicinchoninic Acid Kit ( Sigma-Aldrich, Poznań, Poland) was used to determine protein concentrations. Protein extracts (20 μg of protein per lane for 5-HT1A, 40 μg protein per lane for 5-HT2A and 40 μg protein per lane for D2 analysis ) were separated on a 7.5% SDS-PAGE gel and transferred to nitrocellulose membranes using an electrophoretic transfer system (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were then stained with Ponceau S to confirm gel transfer and cut into three parts: the lower portion was used for GAPDH protein assessment, and the proteins with molecular weights greater than 37 kDa were determined from the next portions of the membrane. The blots were washed, and non-specific binding sites were blocked with 5% albumin (Bovine Serum Albumin; Sigma- Aldrich) and blocking reagent (Lumi Light Western Blotting kit, Roche, Basil Switzerland) in Tris-buffered saline (TBS) for 1 h at room temperature. Then the blots were incubated overnight at 4 °C with the following primary antibodies: rabbit anti-5-HT1A (1:1000; ab85615, Abcam), rabbit anti-5-HT2A (1:500; ab216959, Abcam), rabbit anti-D2 (1:500; AB5084P, Sigma-Aldrich) and rabbit anti-GAPDH (1:10000; 14C10, 2118, Cell Signaling Technology). The peroxidase-conjugated secondary anti-rabbit IgG antibody (1:1000, Roche) was used to detect immune complexes (incubation for 1h at room temperature). Blots were visualized using enhanced chemiluminescence (ECL, Lumi-LightPlus Western Blotting Kit, Roche) and scanned using a luminescent image analyzer (LAS-4000, Fujifilm, Boston, MA, USA). The molecular weights of immunoreactive bands were calculated on the basis of the migration of molecular weight markers (Bio-Rad Laboratories) using Multi Gauge V3.0 (Fujifilm) software. The levels of analyzed proteins were normalized to GAPDH protein.
2.6. Open Field Test
The open field test was performed according to modification of the procedure described by Rogóż and Skuza [28]. A round black arena (1 m in diameter) was virtually divided into eight radiant sections formed by lines intersecting the center of the field. The test was conducted in a dimly lit room, except for the middle of the arena, which was illuminated by a 75 W light bulb placed 75 cm above. Rats were placed in the middle of the arena 60 minutes after drug injection. Their behavior was recorded for 5 minutes. The exploration was quantified with the following parameters: time of walking, number of line crossings reflecting ambulatory distance, episodes of looking under the edge of the field (peeping), number of grooming events, number of rearings as vertical activity and time spent in the central zone.
2.7. Statistical analysis
Drug effects on DA, 5-HT, ACh, GABA and glutamate release in the brain regions were analyzed with repeated measures ANOVA on normalized responses followed by Tukey’s post hoc test. All obtained data were presented as a percentage of the basal level, assumed to be 100%. Total effects expressed as area under the curve (AUC) and GABA/GLU ratio were analyzed with one-way ANOVA followed by Tukey’s post hoc test. The results obtained in the open field test and western blotting data were analyzed with one-way ANOVA followed by Tukey’s post hoc test. The differences were considered significant if p < 0.05. The detected outliers were removed from the data set using Grubb’s test. All statistical analyses were carried out using STATISTICA v.10 StatSoft Inc. 1984-2011 (USA) and GraphPad Prism v.5.00 GraphPad Software Inc. (USA).
3. Results
3.1. The effect of psilocybin and ketamine on extracellular levels of DA and 5-HT in the rat nucleus accumbens
Psilocybin at doses of 2 and 10 mg/kg significantly increased extracellular levels of DA up to ca. 180% of baseline in the rat nucleus accumbens (Figure 1A). Ketamine (10 mg/kg) was more potent in increasing (up to ca. 250% of baseline) DA extracellular level (Figure 1A). Repeated measures ANOVA showed a significant effect of treatment groups (F3,23 = 123, p < 0.0001), sampling period (F11,253 = 15.2, p < 0.0001), and the interaction between treatment groups and sampling period (F33,253 = 15.9, p < 0.0001). Total effects expressed as AUC shown in Figure 1B were significantly increased for psilocybin 2 and 10 mg/kg and ketamine (F3,23 = 124, p < 0.001, one-way ANOVA).
The extracellular 5-HT level was increased in the rat nucleus accumbens by both doses of psilocybin (up to 200-250 % of baseline) and less potently by ketamine (up to 200 % of baseline) (Figure 1C). Repeated measures ANOVA showed a significant effect of treatment groups (F3,20 = 267, p < 0.0001), sampling period (F11,220 = 23, p < 0.0001), and the interaction between treatment groups and sampling period (F33,220 = 16.1, p < 0.0001). Total effects expressed as AUC shown in Figure 1D were significantly increased for both psilocybin doses, 2 and 10 mg/kg, and for ketamine (F3,20 = 268, p < 0.0001, one-way ANOVA).
3.2. The effect of psilocybin and ketamine on extracellular levels of glutamate and GABA in the rat nucleus accumbens, hippocampus and amygdala
The extracellular glutamate (GLU) level in the nucleus accumbens was slightly but significantly decreased by both psilocybin doses (to ca. 80 % of baseline), but was markedly increased by ketamine (up to 160 % of baseline) (Figure 2A). Repeated measures ANOVA showed a significant effect of treatment groups (F3,26 = 99, p < 0.0001), sampling period (F11,286= 32, p < 0.0001), and the interaction between treatment groups and sampling period (F33,286 = 8.7, p < 0.0001). Total effects expressed as AUC shown in Figure 2B were significantly decreased by psilocybin 2 mg/kg and 10 mg/kg and increased by ketamine (F3,26 = 99, p < 0.0001, one-way ANOVA).
The extracellular level of GABA in the nucleus accumbens was slightly and not significantly increased by psilocybin dose of 2 mg/kg (up to ca. 120 % of baseline), more potently by the higher dose of 10 mg/kg (up to ca. 150% of baseline) and by ketamine (up to 180 % of baseline) (Figure 2C). Repeated measures ANOVA showed a significant effect of treatment groups (F3,25 = 62, p < 0.0001), sampling period (F11,275 = 12.8, p < 0.0001), and the interaction between treatment groups and sampling period (F33,275 = 7.9, p < 0.0001). Total effects expressed as AUC shown in Figure 2D were significantly increased by the higher dose of psilocybin and ketamine (F3,35 = 276, p < 0.0001, one-way ANOVA).
The extracellular glutamate (GLU) level in the hippocampus was significantly decreased by psilocybin dose of 2 mg/kg (up to ca. 50 % of baseline) but was markedly increased by the higher dose of 10 mg/kg (up to ca. 150% of baseline) and by ketamine (up to 140 % of baseline) (Figure 2E). Repeated measures ANOVA showed a significant effect of treatment groups (F3,22 = 365, p < 0.0001), sampling period (F11,242 = 19.4, p < 0.0001), and the interaction between treatment groups and sampling period (F33,242 = 8.7, p < 0.0001). Total effects expressed as AUC shown in Figure 2F were significantly decreased for psilocybin 2 mg/kg and significantly increased for psilocybin 10 mg/kg and ketamine (F3,22 = 366, p < 0.0001, one-way ANOVA).
The extracellular level of GABA in the hippocampus was significantly increased by psilocybin dose of 2 mg/kg (up to ca. 120 % of baseline), more potently by the higher dose of 10 mg/kg (up to ca. 140% of baseline) and by ketamine (up to 140 % of baseline) (Figure 2G). Repeated measures ANOVA showed a significant effect of treatment groups (F3,22 = 223, p < 0.0001), sampling period (F11,242 = 16.8, p < 0.0001), and the interaction between treatment groups and sampling period (F33,242 = 8.5, p < 0.0001). Total effects expressed as AUC shown in Figure 2H were significantly increased by both psilocybin doses and ketamine (F3,22 = 224, p < 0.0001, one-way ANOVA).
The extracellular glutamate (GLU) level in the amygdala was significantly increased by psilocybin dose of 2 mg/kg (up to ca. 140 % of baseline) and by ketamine (up to ca. 160% of baseline) between 20 to 100 min of collection period and was not affected by both psilocybin doses and ketamine for the rest of time (Figure 2I, M). Repeated measures ANOVA showed a significant effect of treatment groups (F3,18 = 20.5, p < 0.0001), sampling period (F4,12 = 15.4, p < 0.0001), and the interaction between treatment groups and sampling period (F4,72 = 11.6, p < 0.0001). Total effects expressed as AUC between 20 – 100 min of collection period and shown in Figure 2N were significantly increased for psilocybin 2 mg/kg and ketamine and not changed for psilocybin 10 mg/kg (F3,18 = 20.5, p < 0.0001, one-way ANOVA). Total effects expressed as AUC for the whole collection period did not differ from the control group (Figure 2J).
The extracellular level of GABA in the amygdala was slightly but not significantly increased by psilocybin dose of 2 mg/kg and ketamine and more potently by higher psilocybin dose of 10 mg/kg (up to ca. 120% of baseline) (Figure 2O) between 20 to 100 min of collection period and was not affected by both psilocybin doses and ketamine for the rest of time (Figure 2K). Repeated measures ANOVA showed a significant effect of treatment groups (F3,18 = 3.2, p < 0.05), sampling period (F4,12 = 2.9, p < 0.03), and the interaction between treatment groups and sampling period (F4,72 = 4.9, p < 0.001). Total effects expressed as AUC between 20 – 100 min of collection period and shown in Figure 2P were significantly increased for psilocybin 10 mg/kg and were not changed for psilocybin 2 mg/kg and ketamine (F3,18 = 3.2, p < 0.05, one-way ANOVA). Total effects expressed as AUC for the whole collection period did not differ from the control group in spite of ketamine (F3,18 = 6.5, p < 0.004, one-way ANOVA) (Figure 2L).
3.3. The effect of psilocybin and ketamine on GABA/glutamate ratio in the rat nucleus accumbens, hippocampus and amygdala
To find out the net effect of psilocybin and ketamine on GABA and glutamate release in rat brain regions, the GABA/GLU ratio was calculated. The mean of GABA/GLU index of AUC values for each group is presented in Figure 3A. There was an increase in GABA/GLU ratio for the whole collection period for psilocybin 2 and 10 mg/kg but not for ketamine in the nucleus accumbens (F3,25 = 31, p < 0.0001, one-way ANOVA). The increase in GABA/GLU ratio of AUC values for psilocybin 2 mg/kg but not for its higher dose and ketamine was observed in the hippocampus (Figure 3A) (F3,25 = 255, p < 0.0001, one-way ANOVA). No change was found in GABA/GLU ratio of AUC values for the whole collection period in the amygdala (Figure 3A) (F3,18 = 0.96, p < 0.43, one-way ANOVA). However, the decrease in GABA/GLU ratio of AUC values for psilocybin 2 mg/kg and ketamine was observed between 20 – 100 min of collection period (Figure 3B) (F3,18 = 8.3, p < 0.001, one-way ANOVA).
3.4. The effect of psilocybin and ketamine on extracellular levels of ACh in the rat hippocampus
The extracellular ACh level was increased in the rat hippocampus by both doses of psilocybin and ketamine (up to 700 % , 200 % and 250 % of baseline, respectively) (Figure 4A). Repeated measures ANOVA showed a significant effect of treatment groups (F3,18 = 74, p < 0.0001), sampling period (F5,90 = 92, p < 0.0001), and the interaction between treatment groups and sampling period (F15,90 = 35, p < 0.0001). Total effects expressed as AUC shown in Figure 4B were significantly increased for both psilocybin doses, 2 and 10 mg/kg, and for ketamine (F3,18 = 74, p < 0.0001, one-way ANOVA).
3.5. The effect of psilocybin and ketamine on 5-HT1A and 5-HT2A receptors level in the rat hippocampus
Both doses of psilocybin significantly decreased the 5-HT1A protein level by ca. 12-13 % while ketamine significantly increased it by 18 % over control (F3,28 = 60, p < 0.0001, one-way ANOVA) as measured 7 days after drug administration (Figure 5A,B). 5-HT2A protein level was decreased by psilocybin 2 mg/kg dose, but was increased by the higher one and ketamine by 11 and 35 %, respectively in comparison to control (F3,28 = 57, p < 0.001, one-way ANOVA) (Figure 5C,D).
3.6. The effect of psilocybin and ketamine on D2 and 5-HT2A receptors level in the rat nucleus accumbens
In the nucleus accumbens, the dopamine D2 protein receptor level was significantly increased by the higher dose of psilocybin by ca. 30 %, but not by its lower dose or ketamine (F3,28 = 11.6, p < 0.0001, one-way ANOVA) as measured 7 days after drug administration (Figure 6C,D). 5-HT2A protein level was not affected by any treatment (F3,28 = 0.24, p < 0.87, one-way ANOVA)(Figure 6A,B).
3.7. The effect of psilocybin and ketamine on activity of rats in the Open Field Test
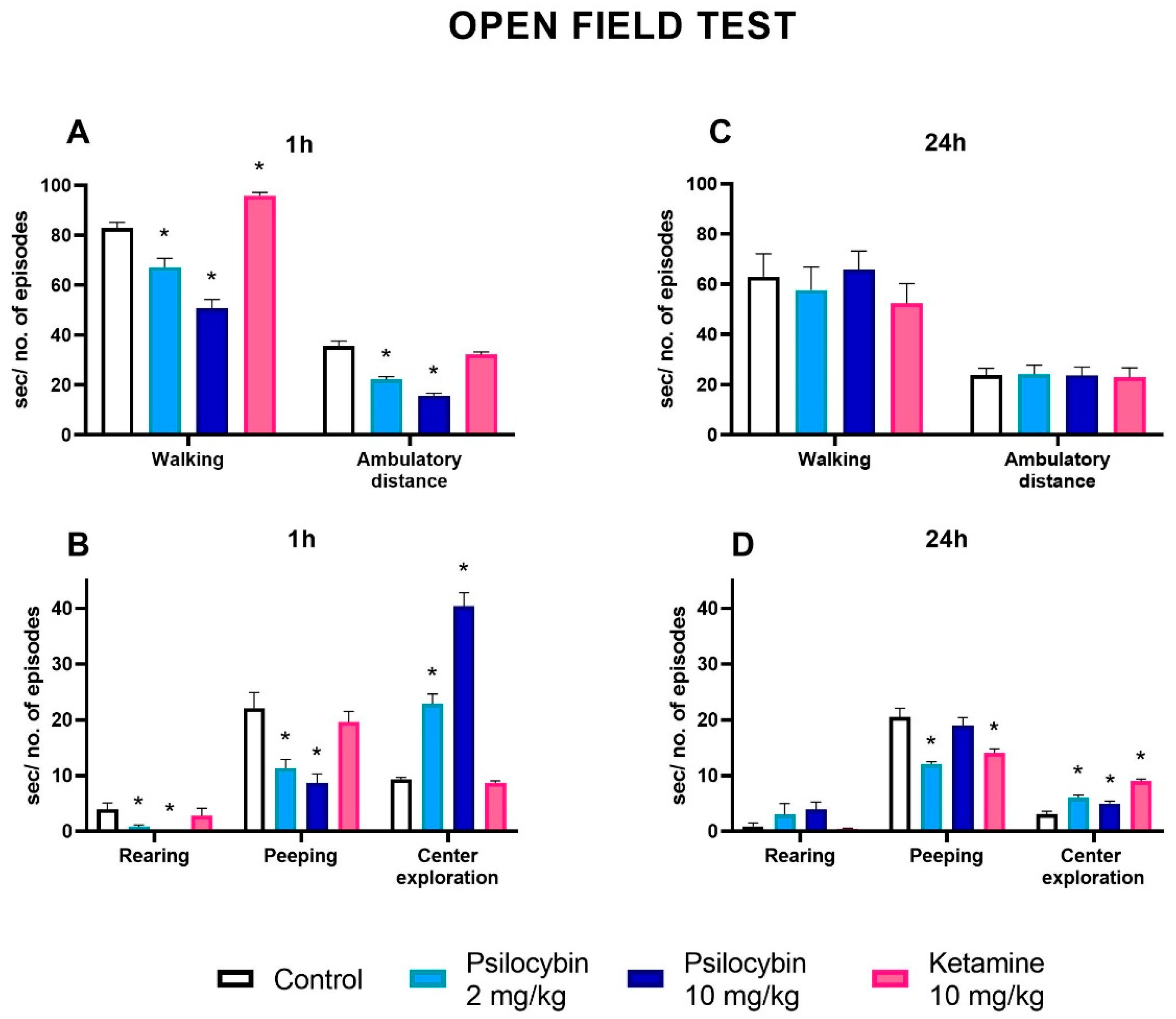
Psilocybin at doses of 2 and 10 mg/kg significantly decreased the time of walking while ketamine (10 mg/kg) increased it above the control level one hour after drug administration (Figure 7A) (F3,36 = 47, p < 0.001, one-way ANOVA). The number of crossings reflecting ambulatory distance was significantly decreased by both doses of psilocybin, but was not changed by ketamine (Figure 7A)(F3,36 = 44, p < 0.0001, one-way ANOVA). The number of episodes of peeping and rearing reflecting vertical activity was decreased by both doses of psilocybin and not changed by ketamine one hour after administration (Figure 7B) (F3,36 = 10.1, p < 0.001; F3,36 = 4.7, p < 0.01, one-way ANOVA, respectively). The center exploration was significantly increased by both doses of psilocybin but no difference between control and ketamine was observed one hour after drug administration (Figure 7B)(F3,36 = 98, p < 0.0001, one-way ANOVA).
The time of walking and the number of crossings reflecting ambulatory distance were not changed by both doses of psilocybin and ketamine 24 h after drug administration (Figure 7C) (F3,36 = 0.49, p < 0.69; F3,36 = 0.02, p < 0.99, one-way ANOVA, respectively). Similarly, the number of episodes of rearing was not changed by both doses of psilocybin and ketamine 24 h after drug administration (Figure 7D) (F3,36 = 1.87, p < 0.15, one-way ANOVA). However, the number of episodes of peeping was decreased by psilocybin dose of 2 mg/kg and ketamine 24 h after drug administration (Figure 7D)(F3,36 = 12.2, p < 0.0001, one-way ANOVA). The center exploration was significantly increased by both doses of psilocybin and ketamine 24 h after drug administration (Figure 7D)(F3,36 = 29, p < 0.0001, one-way ANOVA).
4. Discussion
The data from numerous empirical studies support the idea that fast-acting psychedelics enable signaling within anatomical networks essential for a range of cognitive and affective tasks. Antidepressant properties of psilocybin are mediated via modulation of the prefrontal and limbic regions [29]. The molecular target of psilocybin in the brain was identified as serotonin 5-HT2A receptor expressed in the subset of deep pyramidal cells in layer V of the prefrontal cortex [30]. The interaction of psilocybin with 5-HT2A receptors produces psychomimetic effects and rise in glutamate levels as confirmed in clinical studies and animal models [27,31]. Acute activation of glutamate neurotransmission was associated with upregulation of BDNF and subsequent synaptic plasticity [3,29]. Intriguingly, depression and chronic stress increase cortical and hippocampal extracellular glutamate and excitotoxicity, subsequently precipitating neuronal atrophy [32]. Thus, the question arises whether antidepressant effects of rapid-acting psychedelics are due to the inhibition of glutamate neurotransmission. Psilocybin also interacts with 5-HT1A receptors although with lower affinity [33,34]. Excitatory 5-HT2A and inhibitory 5-HT1A receptors colocalize in both cortical pyramidal neurons and GABAergic interneurons, thus the cellular response is determined by the summation of 5-HT1A inhibition and 5-HT2A excitation [35]. Through the modulation of cellular excitability, psilocybin may impact cortical projections to other brain circuits such as the nucleus accumbens, hippocampus and amygdala [36]. Thus, glutamatergic and GABAergic signaling would be implicated in cortico-limbic function.
Psilocybin exhibits no affinity for dopamine D2 receptors [29], but it interacts indirectly with mesolimbic dopaminergic pathways, which play a significant role in the brain reward system [37]. This proposed indirect mechanism of action is suggested by a low addictive potential of psilocybin. Furthermore, there is a positive correlation between depression and dopamine deficiency [38].
In our work, we demonstrated a dose-dependent increase in extracellular levels of DA elicited by psilocybin. The regulation of mesocortical DA by psilocybin may involve cortical glutamatergic fibers expressing 5-HT2A receptors projecting to the nucleus accumbens or ventral tegmental area (VTA), thus indirectly increasing DA release. On the other hand, also 5-HT1A receptors seem indirectly alter DA release due to a low density of 5-HT1A receptors in the nucleus accumbens. However, 5-HT1A receptors may control DA release by reducing the 5-HT neuron activity as a consequence of the stimulation of 5-HT1A autoreceptors in the dorsal raphe nucleus or by reducing pyramidal cell activity projecting to VTA neurons [39]. A similar effect on DA release in the nucleus accumbens exerted by psilocin, an active metabolite of psilocybin, was demonstrated by Sakashita et al. [40].
Ketamine, a fast-acting antidepressant used in our study for comparison, also elevated DA release with a potency similar to the higher dose of psilocybin. However, the mechanism of ketamine action in the regulation DA release in the nucleus accumbens differs from that of psilocybin since it is mediated through disinhibition of glutamatergic fibers projecting to the VTA by blocking NMDA receptors located in cortical GABAergic interneurons [41].
The excitatory effect of psilocybin on cortical pyramidal neurons projecting to the dorsal raphe may be responsible for a dose-dependent increase in 5-HT release in the nucleus accumbens. However, 5-HT release from serotonin terminals may be also modulated by 5-HT2A receptors located in GABAergic interneurons in the dorsal raphe cells, but this mechanism seems to be less pronounced since the levels of 5-HT2A receptor mRNA in the dorsal raphe cells are low [42]. The weaker effect of ketamine on serotonin release as compared to psilocybin may involve NMDA receptors in GABAergic neurons disinhibiting glutamatergic innervation of dorsal raphe cells [35]. In addition, AMPA receptors might be also involved in the behavioral and neurochemical effects of ketamine [43]. 2016). What is more, ketamine might enhance serotonergic transmission by the inhibition of SERT activity [44].
Intriguingly, preclinical studies demonstrated hypertrophy of the nucleus accumbens in models of depression [32]. It is suggested that the stress-induced nucleus accumbens hypertrophy may be related to dopaminergic neurotransmission abnormalities in the VTA pathway to the nucleus accumbens [45]. Furthermore, stress and depression are believed to precipitate phasic activation of the VTA – nucleus accumbens pathway, leading to DA and BDNF release in the nucleus accumbens [46]. Subsequently, the stress-induced BDNF release results in nucleus accumbens hypertrophy and in depressive-like behavior [47]. Our experiments showing the stimulation of DA release in the nucleus accumbens by psilocybin do not seem to explain the abovementioned observations. However, the effect of psilocybin in naive rats may differ from its action in animals exposed to stress. Further studies are necessary to find out whether fast-acting antidepressants are able to normalize stress-induced abnormalities in the nucleus accumbens observed in rodents and depressive patients [7].
Our data show another pattern of psilocybin action on extracellular glutamate levels in limbic regions. Direct stimulation of 5-HT1A receptors located on pyramidal cells or 5-HT2A receptors on GABAergic interneurons by psilocybin, reducing the prefrontal cortex output to the nucleus accumbens [35,36], may be responsible for the decrease in glutamate release in the nucleus accumbens. A lack of dose-response linearity in this effect stems from differences in density of receptor subtypes in both locations [33,35]. Ketamine used in our study as a comparator significantly increased glutamate levels in the nucleus accumbens. The ketamine-induced enhancement of glutamate release is likely mediated via its blocking activity on NMDA receptors within GABAergic interneurons resulting in disinhibition of pyramidal cells projecting to the nucleus accumbens [48].
Moreover, psilocybin and ketamine increased GABA release in this region. GABAergic neurons are widely distributed in the shell and core of the nucleus accumbens. They bear 5-HT1A and 5-HT2A receptor subtypes although differ in their density [49]. Direct activation of inhibitory or excitatory subtypes may depend on the psilocybin dose. In addition, cortical projections to the nucleus accumbens may also influence excitability of GABAergic neurons regulating GABA levels via other receptor types [50]. The effect on cortical projection by NMDA receptor blockade may explain a possible mechanism of ketamine action on GABA release in the nucleus accumbens [48].
Diverse psilocybin effect on glutamate release was found in the hippocampus. Its low dose decreased while the higher one increased glutamate levels. In contrast, GABA release was dose-dependently enhanced by psilocybin. The observed changes in glutamate and GABA release depend on the stimulation of hippocampal 5-HT1A and 5-HT2A receptors by psilocybin. Both receptor subtypes have various distribution and density patterns in the hippocampus. 5-HT1A receptors are highly expressed on both principal glutamatergic cells and GABAergic interneurons [51]. Activation of 5-HT1A receptors primarily leads to the inhibition of hippocampal pyramidal cells [52]. This may be the cause of the decrease in glutamate release by low-dose psilocybin in our study. However, activation of 5-HT1A receptors expressed on GABAergic interneurons would disinhibit principal glutamatergic cells and thus would counteract the direct effect of 5-HT1A receptors expressed on principal neurons. Additionally, 5-HT2A receptors are expressed on both principal glutamatergic cells and on different subtypes of hippocampal interneurons, though in a lower density than 5-HT1A receptors [53]. Thus, since 5-HT2A receptors are stimulatory and are expressed on both principal cells and GABAergic interneurons, it could be expected that they would have mixed effects on the both cell types. This mechanism may underlie the stimulatory effect of the higher psilocybin dose on glutamate release and it’s both doses on GABA release in the hippocampus. However, it has to be noted that the resultant effect of the lower psilocybin dose on amino acid neurotransmission is inhibitory. Activation of corticolimbic excitatory projections, either through selective antagonism of inhibitory interneurons or cortical disinhibition by ketamine is a probable mechanism of the increase in glutamate and GABA extracellular levels in the hippocampus [43]. Considering reduced hippocampal volume in depressed patients resulting from hippocampal stress-induced neuronal atrophy and low levels of GABA and glutamate demonstrated in depressed patients [7,32,54], normalization of amino acid neurotransmission and hippocampal volume by psilocybin may be expected.
Additional beneficial effect of psilocybin in depressed patients with cognitive impairments may be related to its stimulatory effect on ACh levels as found in our study. ACh appears to act as a neuromodulator in the brain and its role is to change neuronal excitability, alter presynaptic release of neurotransmitters and coordinate the firing of groups of neurons. ACh contributes also to synaptic plasticity [55]. The primary source of cholinergic innervation of the hippocampus derives from the basal forebrain cholinergic system [56]. Generally 5-HT exerts stimulatory influence on the release of ACh; however, the effect depends on mediation via particular serotonin receptor subtypes. 5-HT1A receptor subtype mediates stimulatory effect on ACh release in the hippocampus as shown in the hippocampal perfusate of conscious freely moving rats [57]. However, systemic administration of the 5-HT2A receptor agonist DOI in a high dose also increased ACh release in the hippocampus, but mescaline, a potent 5-HT2A agonist, did not affect ACh release[58]. This findings are in line with our data and support the idea that both receptor subtypes seem to be involved in the stimulatory effect of psilocybin on ACh release in the hippocampus. However, the lack of linearity in the dose-response effect needs further studies. The recent data of Pacheco et al. [59] showing enhancement of cognitive flexibility in rats by psilocybin may correlate with the psilocybin effect on ACh release in the hippocampus. The role of NMDA receptors in the regulation of ACh release cannot be excluded as ketamine also increased ACh release with the strength similar to the psilocybin higher dose.
Abnormally high amygdala reactivity to negative affective stimuli has been implicated in the pathophysiology of depression [60]. Negative affect and amygdala response was reduced by psilocybin suggesting that psilocybin may increase emotional and brain plasticity [61]. In our study, psilocybin and ketamine effects on extracellular glutamate and GABA levels were of short duration and the overall effect shown as GABA/glutamate index was stimulatory. Considering the fact that prolonged anxiety induced by chronic stress in mice causes dysfunction of basolateral amygdala projection neurons [62] and that reduced amygdala volume correlated with the severity of depression [6], our data indicate that facilitation of synaptic transmission from the prefrontal cortex to the amygdala and especially predominant role of glutamatergic pathways may be the underlying mechanism of the beneficial effect of psilocybin and ketamine in treatment of anxiety and depressive states.
In order to assess the long-lasting effects of psilocybin, we examined the density level of several receptor subtypes in the limbic system. The increase in dopamine D2 receptor density in the nucleus accumbens by the higher psilocybin dose may be subsequent effect to the increased DA release in the nucleus accumbens and the regulatory mechanism restoring balance in dopaminergic nerve terminals. The 5-HT2A receptor mRNA levels were intermediate in the nucleus accumbens [63] and were mostly observed in spiny projecting neurons responsible principally for movement control [64]. Their density was not changed by acute administration either of psilocybin or ketamine in our study.
Instead, long term density changes were detected for 5-HT1A and 5-HT2A receptors in the hippocampus. The decreased density of 5-HT1A receptors induced by psilocybin may be beneficial due to sensitization of this receptors in depression, while the increased levels of 5-HT2A receptors after the higher dose of psilocybin may reduce functional deficit of this subtype observed in depression [65] and related to increased synaptogenesis found in the hippocampus of the pig brain [66]. Interestingly, decreased density of 5-HT2A receptors in the hippocampus and prefrontal cortex of the pig brain found by Raval et al. [66] supports our findings after the lower dose of psilocybin. Ketamine-induced increase in 5-HT1A and 5-HT2A receptor density in the hippocampus observed in our work as was also evidenced for other non-competitive antagonists NMDA receptor [67].
The changes in the levels of neurotransmitters in the limbic system provide the neurobiological background for psilocybin effect on stress and anxiety. In our study, psilocybin affected animal behavior in the open field test. The data demonstrate marked anxiolytic effect of psilocybin in acute phase and 24 h post-treatment as shown by increased center penetration and decreased exploration of peripheral zone of the open field. Instead, ketamine was not effective or its weak anxiolytic effect was observed only 24 h after treatment. Furthermore, in contrast to psilocybin, ketamine significantly affected exploration resulting probably from stronger as compared to psilocybin stimulation of DA levels in the nucleus accumbens. The psilocybin effect observed in the open field test linking behavioral symptoms with modest elevation of DA and 5-HT in the nucleus accumbens and predominance of GABAergic neurotransmission in other regions of the limbic system has therapeutic implication for treating anxiety and mood disorders.
5. Conclusions
In conclusion, the presented data have mechanistic significance and show the implications of the psilocybin impact on neurotransmitter levels for the therapy of depression and anxiolytic disorders. The increased dopaminergic and serotonergic neurotransmission in the nucleus accumbens and predominant GABAergic signaling in the studied brain regions link the current findings with limbic system abnormalities observed in clinical studies.
Supplementary Materials
Original Western blot details are presented in supplementary material 1, elevated plus maze data are presented in supplementary material 2 (Figure 1S), extracellular basal levels of neurotransmitters in respective animal groups are presented in supplementary material 3 (Table 1S).
Author Contributions
Conceptualization, A.W., K.G.; Methodology, A.W., K.G., M.M.; Investigation A.W., A.B., A.W.-B., M.M., K.G.; Resources K.G., M.M.; Data Curation K.G., M.M.; Writing Original Draft Preparation A.W., K.G., M.M.; Writing—Review & Editing A.W., K.G., M.M., A.B., A.W.-B.; Visualization A.B., A.W.-B.; Supervision K.G., M.M.; Project Administration K.G.; Funding Acquisition K.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Science Centre grant no. 2020/37/B/NZ7/03753 and statutory funds of the Maj Institute of Pharmacology, Polish Academy of Sciences
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Local Ethical Commission for Experimentations on Animals in Kraków (permit no. 112/2021, 324/2021, 79/2022). The experimental procedures were conducted according to recommendations of the National Institutes of Health and were approved by the Ethical Committee of the 2nd Local Institutional Animal Care and Use Committee (IACUC) in Kraków, Poland in conformity with institutional guidelines and in compliance with national and international laws and policies (permit numbers: 112/2021, 324/2021 and 79/2022). According to the 3R policy, the number of animals was reduced to an essential minimum. All the procedures regarding the study design, animal experiments, statistical analysis, and data reporting fulfill the criteria of the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (https://www.nc3rs.org.uk/arrive-guidelines, access date: 14 May 2022).
Informed Consent Statement
Not applicable.
Data Availability Statement
All data is contained within the article and Supplementary Material.
Acknowledgments
We thank Klara Szweda for technical support in performing the experiments.
Conflicts of Interest
The authors declare no conflict of interest
References
- Steffen, A.; Nübel, J.; Jacobi, F.; Bätzing, J.; Holstiege, J. Mental and Somatic Comorbidity of Depression: A Comprehensive Cross-Sectional Analysis of 202 Diagnosis Groups Using German Nationwide Ambulatory Claims Data. BMC psychiatry. 2020, 20.1, 1-15. [CrossRef]
- Chand, S.P.; Arif, H. Depression. Treasure Island, FL: StatPearls Publishing. 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK430847 (accessed on January 2023).
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. 2016, Mar;22(3):238-49. [CrossRef] [PubMed] [PubMed Central]
- Pandya, M.; Altinay, M.; Malone, D.A., Jr.; Anand, A. Where in the brain is depression? Curr Psychiatry Rep. 2012, Dec;14(6):634-42. [CrossRef] [PubMed] [PubMed Central]
- Trifu, S.C.; Trifu, A.C.; Aluaş, E.; Tătaru, M.A.; Costea, R.V. Brain changes in depression. Rom J Morphol Embryol. 2020, Apr-Jun;61(2):361-370. [CrossRef] [PubMed] [PubMed Central]
- Sheline, Y.I.; Gado, M.H.; Price, J.L. Amygdala core nuclei volumes are decreased in recurrent major depression. NeuroReport. 1998, 9(9):2023–2028, PubMed: 9674587. [CrossRef]
- Abdallah, C.G.; Jackowski, A.; Salas, R.; Gupta, S.; Sato, J.R.; Mao, X.; Coplan, J.D.; Shungu, D.C.; Mathew, SJ. The Nucleus Accumbens and Ketamine Treatment in Major Depressive Disorder. Neuropsychopharmacology. 2017, Jul;42(8):1739-1746. Epub 2017 Mar 8, PMID: 28272497; PMCID: PMC5518908. [CrossRef]
- American Psychiatric Association. (2013). Diagnostic and Statistical Manual of Mental Disorders. Arlington. [CrossRef]
- Xu, L.; Nan, J.; Lan, Y. The Nucleus Accumbens: A Common Target in the Comorbidity of Depression and Addiction. Front Neural Circuits. 2020, Jun 30;14:37. [CrossRef] [PubMed] [PubMed Central]
- Maeng, S.; Zarate, C.A. The Role of Glutamate in Mood Disorders: Results from the Ketamine in Major Depression Study and the Presumed Cellular Mechanism Underlying Its Antidepressant Effects. Curr. Psychiatry Rep. 2007, 9, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.C.; Henter, I.D.; Zarate, C.A. Targeting the Glutamatergic System to Treat Major Depressive Disorder: Rationale and Progress to Date. Drugs. 2012, 72, 1313–1333. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hu, Y.M.; Zhou, Z.Q.; Zhang, G.F.; Yang, J.J. Acute administration of ketamine in rats increases hippocampal BDNF and mTOR levels during forced swimming test. Ups J Med Sci. 2013, Mar;118(1):3-8. Epub 2012 Sep 13, PMID: 22970723; 13, PMCID: PMC3572668. [CrossRef]
- Zhou, Y.; Wu, F.; Liu, W.; Zheng, W.; Wang, C.; Zhan, Y.; Lan, X.; Ning, Y. Volumetric changes in subcortical structures following repeated ketamine treatment in patients with major depressive disorder: A longitudinal analysis. Transl. Psychiatry. 2020, 10:264. [CrossRef]
- Abdallah, C.G.; Salas, R.; Jackowski, A.; Baldwin, P.; Sato, J.R.; Mathew, S.J. Hippocampal volume and the rapid antidepressant effect of ketamine. J. Psychopharmacol. 2015, May;29(5):591-5. Epub 2014 Aug 13. [CrossRef] [PubMed] [PubMed Central]
- Zhou, Y.L.; Wu, F.C.; Wang, C.Y.; Zheng, W.; Lan, X.F.; Deng, X.R.; Ning, Y.P. Relationship between hippocampal volume and inflammatory markers following six infusions of ketamine in major depressive disorder. J Affect Disord. 2020, Nov 1;276:608-615. Epub 2020 Jul 16, PMID: 32871692. [CrossRef]
- Hibicke, M.; Landry, A.N.; Kramer, H.M.; Talman, Z.K.; Nichols, C.D. Psychedelics, but Not Ketamine, Produce Persistent Antidepressant-like Effects in a Rodent Experimental System for the Study of Depression. ACS Chem Neurosci. 2020, Mar 18;11(6):864-871. Epub 2020 Mar 5. [CrossRef] [PubMed]
- Ly, C.; Greb, A.C.; Cameron, L.P.; Wong, J.M.; Barragan, E.V.; Wilson, P.C.; Burbach, K.F.; Soltanzadeh Zarandi, S.; Sood, A.; Paddy, M.R.; et al. Psychedelics Promote Structural and Functional Neural Plasticity. Cell Rep. 2018, Jun 12;23(11):3170-3182. [CrossRef] [PubMed] [PubMed Central]
- Cameron, L.P.; Benson, C.J.; Dunlap, L.E.; Olson, D.E. Effects of N, N-Dimethyltryptamine on Rat Behaviors Relevant to Anxiety and Depression. ACS Chem. Neurosci. 2018, Jul 18;9(7):1582-1590. Epub 2018 Apr 24. [CrossRef] [PubMed] [PubMed Central]
- Hesselgrave, N.; Troppoli, T.A.; Wulff, A.B.; Cole, A.B.; Thompson, S.M. ; Harnessing psilocybin: antidepressant-like behavioral and synaptic actions of psilocybin are independent of 5-HT2R activation in mice. Proc Natl Acad Sci U S A. 2021, Apr 27;118(17):e2022489118. [CrossRef] [PubMed] [PubMed Central]
- Carhart-Harris, R.; Giribaldi, B.; Watts, R.; Baker-Jones, M.; Murphy-Beiner, A.; Murphy, R.; Martell, J.; Blemings, A.; Erritzoe, D.; Nutt, D.J. Trial of Psilocybin versus Escitalopram for Depression. N. Engl. J. Med. 2021, Apr 15;384(15):1402-1411. [CrossRef] [PubMed]
- Shirota, O.; Hakamata, W.; Goda, Y. Concise Large-Scale Synthesis of Psilocin and Psilocybin, Principal Hallucinogenic Constituents of “Magic Mushroom. ” J. Nat. Prod. 2003, 66, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Popik, P.; Hogendorf, A.; Bugno, R.; Khoo, S.Y.S.; Zajdel, P.; Malikowska-Racia, N.; Nikiforuk, A.; Golebiowska, J. Effects of Ketamine Optical Isomers, Psilocybin, Psilocin and Norpsilocin on Time Estimation and Cognition in Rats. Psychopharmacology (Berl). 2022. [CrossRef] [PubMed]
- Jefsen, O.; Højgaard, K.; Christiansen, S.L.; Elfving, B.; Nutt, D.J.; Wegener, G.; Müller, H.K. Psilocybin Lacks Antidepressant-like Effect in the Flinders Sensitive Line Rat. Acta Neuropsychiatr. 2019. [CrossRef] [PubMed]
- Paxinos, G.; Watson, G. The Rat Brain in Stereotaxic Coordinates. 1998, Academic Press: San Diego.
- Herian, M.; Skawski, M.; Wojtas, A.; Sobocińska, M.K.; Noworyta, K.; Gołembiowska, K. Tolerance to Neurochemical and Behavioral Effects of the Hallucinogen 25I-NBOMe. Psychopharmacology (Berl). 2021, 238, 2349–2364. [Google Scholar] [CrossRef] [PubMed]
- Wojtas, A.; Herian, M.; Skawski, M.; Sobocińska, M.; González-Marín, A.; Noworyta-Sokołowska, K.; Gołembiowska, K. Neurochemical and Behavioral Effects of a New Hallucinogenic Compound 25B-NBOMe in Rats. Neurotox. Res. 2021, 39, 305–326. [Google Scholar] [CrossRef] [PubMed]
- Wojtas, A.; Bysiek, A.; Wawrzczak-Bargiela, A.; Szych, Z.; Majcher-Maślanka, I.; Herian, M.; Maćkowiak, M. ; Gołembiowska,K. Effect of Psilocybin and Ketamine on Brain Neurotransmitters, Glutamate Receptors, DNA and Rat Behavior. Int. J. Mol. Sci. 2022, 23, 6713. [Google Scholar] [CrossRef] [PubMed]
- Rogóz, Z.; Skuza, G. Anxiolytic-like Effects of Olanzapine, Risperidone and Fluoxetine in the Elevated plus-Maze Test in Rats. Pharmacol. Reports 2011, 63, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, F.X.; Kometer, M. The neurobiology of psychedelic drugs: implications for the treatment of mood disorders. Nature Neurosciences. 2010, 11, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Beique, J.C.; Imad, M.; Mladenovic, L.; Gingrich, J.A.; Andrade, R. Mechanism of 5-hydroxytryptamine 2A receptor-mediated facilitation of synaptic activity in prefrontal cortex. Proc. Natl. Acad. Sci. USA. 2007, 104, 9870-5. [Google Scholar] [CrossRef] [PubMed]
- Mason, N.L.; Kuypers, K.P.; Muller, F.; Reckweg, J.; Tse, D.H.Y.; Toennes, S.W.; Hutten, N.R.P.W.; Jansen, J.F.A.; Stiers, P.; Feilding, A.; Ramaekers, J.G. Me, myself, bye: regional alterations in glutamate and the experience of ego dissolution with psilocybin. Neuropsychopharmacology. 2020, 45:2003–2011. [CrossRef]
- Abdallah, C.G.; Sanacora, G.; Duman, R.S.; Krystal, J.H. The Neurobiology of Depression, Ketamine and Rapid-Acting Antidepressants: Is it Glutamate Inhibition or Activation? Pharmacol. Ther. 2018, October ; 190: 148–158. [CrossRef]
- Passie, T.; Seifert, J.; Schneider, U.; Emrich, M.H. The pharmacology of psilocybin. Addict. Biol. 2002, 7, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Rickli, A.; Moning, O.D.; Hoener, M.C.; Liechti, M.E. Receptor interaction profiles of novel psychoactive tryptamines compared with classic hallucinogens. European Neuropsychopharmacology. 2016, 26, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Celada, P.; Puig, M.V.; Artigas, F. Serotonin modulation of cortical neurons and networks. Front. Integr. Neurosci. 2013, 7:25. [CrossRef]
- Meccia, J.; Lopez, J.; Bagot, R.C. Probing the antidepressant potential of psilocybin: integrating insight from human research and animal models towards an understanding of neural circuit mechanisms. Psychopharmacology. 2023, 240:27–40. [CrossRef]
- Malenka, R.C.; Nestler, E.J.; Hyman, S.E. (2009). "Chapter 6: Widely Projecting Systems: Monoamines, Acetylcholine, and Orexin". In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York: McGraw-Hill Medical. pp. 147–148, 154–157, ISBN 9780071481274.
- Dailly, E.; Chenu, F.; Renard, C.E.; Bourin, M. Dopamine, depression and antidepressants. Fundam. Clin. Pharmacol. 2004, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- De Deurwaerdère, P.; Di Giovanni, G. Serotonergic modulation of the activity of mesencephalic dopaminergic systems: Therapeutic implications. Progress in Neurobiology. 2017, 151: 175–236. [CrossRef]
- Sakashita, Y.; Abe, K.; Katagiri, N.; Kambe, T.; Saitoh, T.; Utsunomiya, I.; Horiguchi, Y.; Taguchi, K. Effect of psilocin on extracellular dopamine and serotonin levels in the mesoaccumbens and mesocortical pathway in awake rats. Biological and Pharmaceutical Bulletin, 2015, 38(1), 134-138. [CrossRef]
- Del Arco, A.; Mora, F. Prefrontal cortex–nucleus accumbens interaction: In vivo modulation by dopamine and glutamate in the prefrontal cortex. Pharmacology, Biochemistry and Behavior. 2008, 90.2: 226–235. [CrossRef]
- Moulédous, L.; Roullet, P.; Guiard, B.P. Brain Circuits Regulated by the 5-HT2A Receptor: Behavioural Consequences on Anxiety and Fear Memory. 5-HT2A Receptors in the Central Nervous System. 2018, 231-258. [CrossRef]
- Miller, O.H.; Moran, J.T.; Hall, B.J. Two cellular hypotheses explaining the initiation of ketamine's antidepressant actions: Direct inhibition and disinhibition. Neuropharmacology. 2016, 100: 17-26. [CrossRef]
- Ago, Y.; Tanabe, W.; Higuchi, M.; Tsukada, S.; Tanaka, T.; Yamaguchi, T.; Igarashi, H.; Yokoyama, R.; Seiriki, K.; Kasai, A.; et al. (R)-Ketamine Induces a Greater Increase in Prefrontal 5-HT Release Than (S)-Ketamine and Ketamine Metabolites via an AMPA Receptor-Independent Mechanism. Int. J. Neuropsychopharm. 2019, 22(10): 665–674. [CrossRef]
- Chaudhury, D.; Walsh, J.J.; Friedman, A.K.; Juarez, B.; Ku, S.M.; Koo, J.W.; Ferguson, D.; Tsai, H.-C.; Pomeranz, L.; Christoffel, D.J.; et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013, 493: 532–536. [CrossRef]
- Walsh, J.J.; Friedman, A.K.; Sun, H.; Heller, E.A.; Ku, S.M.; Juarez, B.; Burnham, V.L.; Mazei-Robison, M.S.; Ferguson, D.; Golden, S.A.; et al. Stress and CRF gate neural activation of BDNF in the mesolimbic reward pathway. Nat Neurosci. 2014, 17: 27–29. [CrossRef]
- Wook Koo, J.; Labonte, B.; Engmann, O.; Calipari, E.S.; Juarez, B.; Lorsch, Z.; Walsh, J.J.; Friedman, A.K.; Yorgason, J.T.; Han, M.-H.; et al. Essential role of mesolimbic brain-derived neurotrophic factor in chronic social stress-induced depressive behaviors. Biol Psychiatry. 2016, 80.6: 469–478. [CrossRef]
- Vollenweider, F.X.; Smallridge, J.W. Classic Psychedelic Drugs: Update on Biological Mechanisms. Pharmacopsychiatry. 2022, 55: 121–138. [CrossRef]
- Beliveau, V.; Ganz, M.; Feng, L.; Ozenne, B.; Højgaard, L.; Fisher, PM.; Svarer, C.; Greve, D.N.; Knudsen, G.M. A High-Resolution In Vivo Atlas of the Human Brain’s Serotonin System. The Journal of Neuroscience. 2017, January 4, 37(1):120 –128. [CrossRef]
- Zhu, Z.; Wang, G.; Ma, K.; Cui, S.; Wang, J.-H. GABAergic neurons in nucleus accumbens are correlated to resilience and vulnerability to chronic stress for major depression. Oncotarget. 2017, 8: 35933-35945. [CrossRef]
- Dale, E.; Pehrson, A.L.; Jeyarajah, T.; Li, Y.; Leiser, S.C.; Smagin, G.; Olsen, C.K.; Sanchez, C. Effects of serotonin in the hippocampus: how SSRIs and multimodal antidepressants might regulate pyramidal cell function. CNS Spectrums. 2016, 21, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.; Nicoll, R.A. Pharmacologically distinct actions of serotonin on single pyramidal neurons of the rat hippocampus recorded in vitro. J Physiol. 1987, 394: 99–124. [CrossRef]
- Bombardi, C. Neuronal localization of 5-HT2A receptor immunoreactivity in the rat hippocampal region. Brain Res. Bull. 2012, 87(2–3): 259–273. [CrossRef]
- Abdallah, C.G.; Jackowski, A.; Sato, J.R.; Mao, X.; Kang, G.; Cheema, R.; Coplan, J.D.; Mathew, S.J.; Shungu, D.C. Prefrontal Cortical GABA Abnormalities Are Associated With Reduced Hippocampal Volume In Major Depressive Disorder. Eur. Neuropsychopharmacol. 2015, August ; 25(8): 1082–1090. [CrossRef]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012, 76.1: 116-129. [CrossRef]
- Gielow, M.R.; Zaborszky, L. The Input-Output Relationship of the Cholinergic Basal Forebrain. Cell Rep. 2017, 18: 1817-1830. [CrossRef]
- Fujii, T.; Yoshizawa, M.; Nakai, K.; Fujimoto, K.; Suzuki, T.; Kawashima, K. Demonstration of the facilitatory role of 8-OH-DPAT on cholinergic transmission in the rat hippocampus using in vivo microdialysis. Brain Res. 1997, Jul 4;761(2):244-9. [CrossRef]
- Nair, S.G.; Gudelsky, G.A. Activation of 5-HT2 receptors enhances the release of acetylcholine in the prefrontal cortex and hippocampus of the rat. Synapse. 2004, 53.4:202–207. [CrossRef]
- Pacheco, A.T.; Olson, R.J.; Garza, G.; Moghaddam, B. Acute psilocybin enhances cognitive flexibility in rats. Neuropsychopharmacology. 2023, 48:1011–1020. [CrossRef]
- Almeida, J.R.C.; Versace, A.; Hassel, S.; Kupfer, D.J.; Phillips, M.L. Elevated amygdala activity to sad facial expressions: a state marker of bipolar but not unipolar depression. Biol. Psychiatry. 2010, 67, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Barrett, F.S.; Doss, M.K.; Sepeda, N.D.; James, J.; Pekar, J.J.; Griffiths, R.R. Emotions and brain function are altered up to one month after a single high dose of psylocybin. Sci. Rep. 2020, 10:2214. [CrossRef]
- Liu, W.Z.; Huang, S.H.; Wang, Y.; Wang, C.Y.; Pan, H.Q.; Zhao, K.; Hu, P.; Pan, B.X.; Zhang, W.H. Medial prefrontal cortex input to basolateral amygdala controls acute stress-induced short-term anxiety-like behavior in mice. Neuropsychopharmacology. 2023, 48:734–744. [CrossRef]
- Pompeiano, M.; Palacios, J.M.; Mengod, G. Distribution of serotonin 5-HT2 receptor family mRNAs: comparison between 5-HT2A and 5-HT2C receptors. Mol Brain Res. 1994, 23:163–178. [CrossRef]
- Cornea-Hebert, V.; Riad, M.; Wu, C.; Singh, S.K.; Descarries, L. Cellular and subcellular distribution of the serotonin 5-HT2A receptor in the central nervous system of adult rat. Journal of Comparative Neurology. 1999, 409.2: 187-209. [CrossRef]
- Nikolaus, S.; Müller, H.-W.; Hautzel, H. Different patterns of 5-HT receptor and transporter dysfunction in neuropsychiatric disorders – a comparative analysis of in vivo imaging findings. Rev. Neurosci. 2016, 27(1): 27–59. [CrossRef]
- Raval, N.R.; Johansen, A.; Donovan, L.L.; Ros, N.F.; Ozenne, B.; Hansen, H.D.; Knudsen, G.M. A Single Dose of Psilocybin Increases Synaptic Density and Decreases 5-HT2A Receptor Density in the Pig Brain. Int. J. Mol. Sci. 2021, 22, 835. [Google Scholar] [CrossRef] [PubMed]
- Wedzony, K.; Maćkowiak, M.; Czyrak, A.; Fijał, K.; Michalska, B. Single doses of MK-801, a non-competitive antagonist of NMDA receptors, increase the number of 5-HT1A serotonin receptors in the rat brain. Brain Res. 1997, 756: 84-91. [CrossRef]
Figure 1.
The time-course (A, C) and total (B, D) effect of psilocybin (2 and 10 mg/kg) and ketamine (10 mg/kg) on the dopamine (DA) and serotonin (5-HT) levels in the rat nucleus accumbens. The total effect was calculated as an area under the concentration-time curve (AUC) and is expressed as a percentage of the basal level. Values are the mean ± SEM (n = 6-8) for each neurotransmitter. The drug injection is indicated with an arrow. Filled symbols or * show statistically significant differences (p < 0.001) between control and drug treatment groups as estimated by repeated measures ANOVA (time-course) or one-way ANOVA (total effect) followed by Tukey’s post hoc test.
Figure 1.
The time-course (A, C) and total (B, D) effect of psilocybin (2 and 10 mg/kg) and ketamine (10 mg/kg) on the dopamine (DA) and serotonin (5-HT) levels in the rat nucleus accumbens. The total effect was calculated as an area under the concentration-time curve (AUC) and is expressed as a percentage of the basal level. Values are the mean ± SEM (n = 6-8) for each neurotransmitter. The drug injection is indicated with an arrow. Filled symbols or * show statistically significant differences (p < 0.001) between control and drug treatment groups as estimated by repeated measures ANOVA (time-course) or one-way ANOVA (total effect) followed by Tukey’s post hoc test.
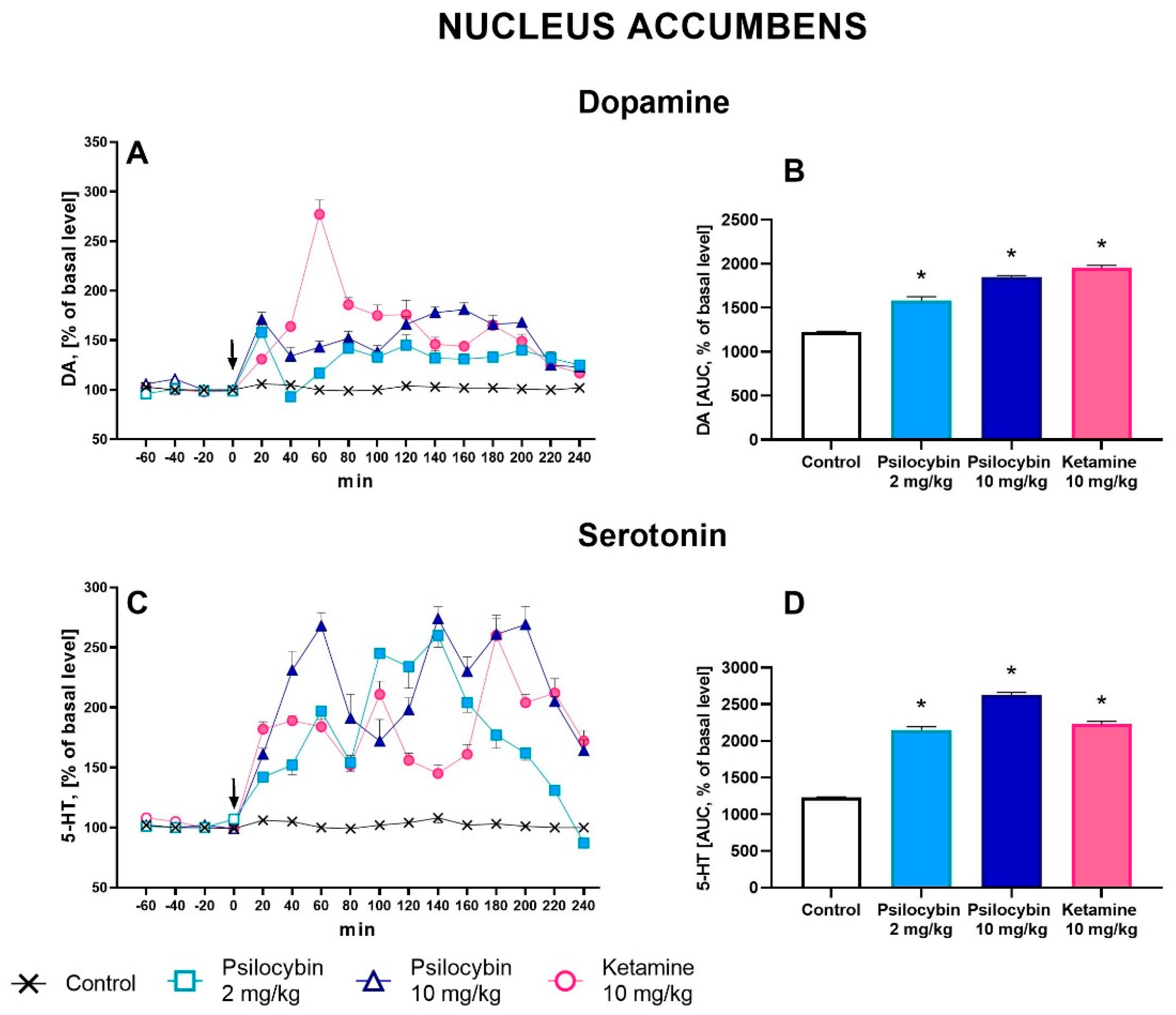
Figure 2.
The time-course (A, C, E, G, I, K, M, O) and total (B, D, F, H, J ,L, N, P) effect of psilocybin (2 and 10 mg/kg) and ketamine (10 mg/kg) on the extracellular levels of glutamate (GLU) and γ-aminobutyric acid (GABA) in the rat nucleus accumbens, hippocampus and amygdala. The total effect was calculated as an area under the concentration-time curve (AUC) and is expressed as a percentage of the basal level. Values are the mean ± SEM (n = 6-9 in nucleus accumbens groups, n = 6-7 in hippocampus groups, n = 5-6 in amygdala groups) for each neurotransmitter. The drug injection is indicated with an arrow. Filled symbols or * (p < 0.001) show statistically significant differences between control and drug treatment groups as estimated by repeated measures ANOVA (time-course) or one-way ANOVA (total effect) followed by Tukey’s post hoc test.
Figure 2.
The time-course (A, C, E, G, I, K, M, O) and total (B, D, F, H, J ,L, N, P) effect of psilocybin (2 and 10 mg/kg) and ketamine (10 mg/kg) on the extracellular levels of glutamate (GLU) and γ-aminobutyric acid (GABA) in the rat nucleus accumbens, hippocampus and amygdala. The total effect was calculated as an area under the concentration-time curve (AUC) and is expressed as a percentage of the basal level. Values are the mean ± SEM (n = 6-9 in nucleus accumbens groups, n = 6-7 in hippocampus groups, n = 5-6 in amygdala groups) for each neurotransmitter. The drug injection is indicated with an arrow. Filled symbols or * (p < 0.001) show statistically significant differences between control and drug treatment groups as estimated by repeated measures ANOVA (time-course) or one-way ANOVA (total effect) followed by Tukey’s post hoc test.
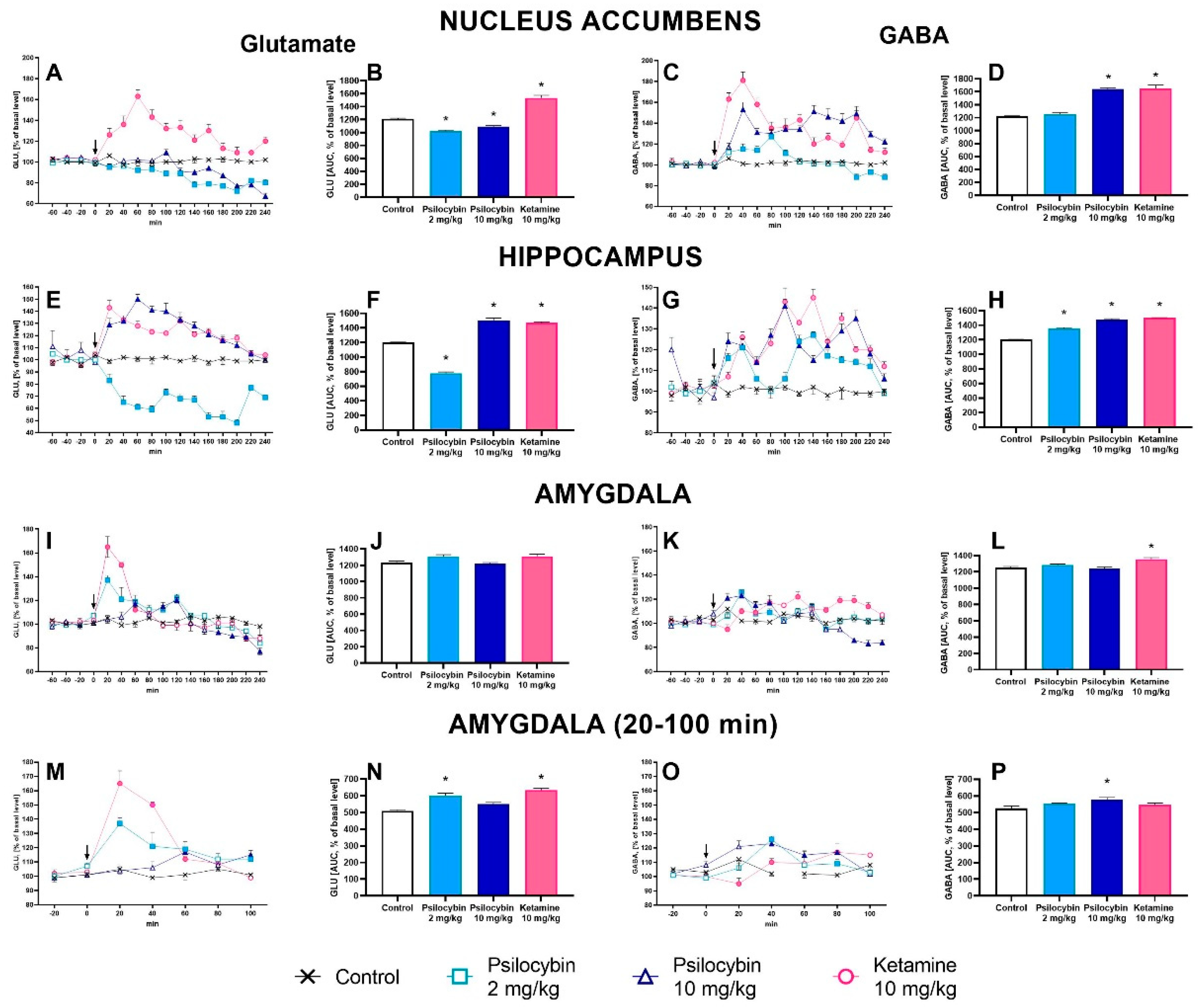
Figure 3.
The AUC GABA/GLU ratio during 20-240 min of collection period after administration of psilocybin (2 and 10 mg/kg) and ketamine (10 mg/kg) in the rat nucleus accumbens, hippocampus and amygdala (A) and during 20-100 min of collection period in the amygdala (B). Values are the mean ± SEM (n = 6-9 in the nucleus accumbens groups, n= 6-7 in hippocampus groups and n=5-6 in amygdala groups) as estimated by one-way ANOVA followed by Tukey’s post hoc test. * p < 0.02, * p < 0.001 indicate statistically significant differences between control and drug treatment groups.
Figure 3.
The AUC GABA/GLU ratio during 20-240 min of collection period after administration of psilocybin (2 and 10 mg/kg) and ketamine (10 mg/kg) in the rat nucleus accumbens, hippocampus and amygdala (A) and during 20-100 min of collection period in the amygdala (B). Values are the mean ± SEM (n = 6-9 in the nucleus accumbens groups, n= 6-7 in hippocampus groups and n=5-6 in amygdala groups) as estimated by one-way ANOVA followed by Tukey’s post hoc test. * p < 0.02, * p < 0.001 indicate statistically significant differences between control and drug treatment groups.
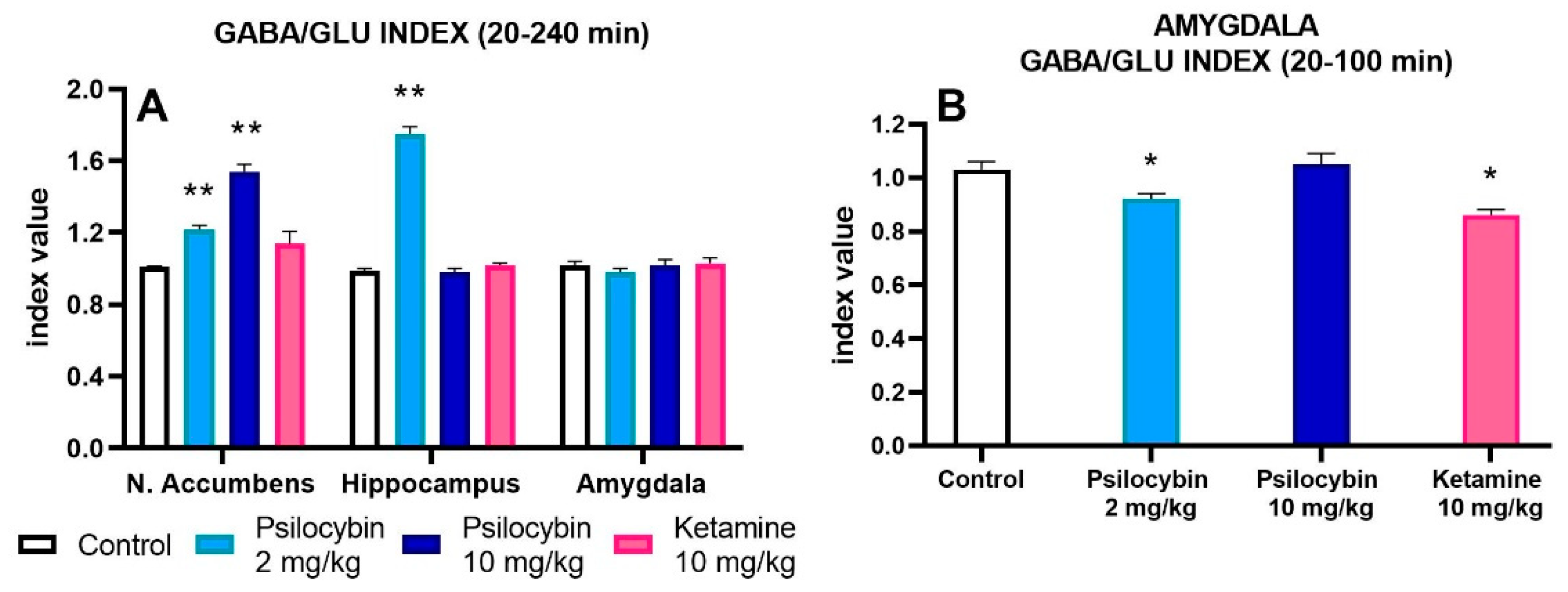
Figure 4.
The time-course (A) and total (B) effect of psilocybin (2 and 10 mg/kg) and ketamine (10 mg/kg) on the extracellular acetylcholine (ACh) levels in the rat hippocampus. The total effect was calculated as an area under the concentration-time curve (AUC) and is expressed as a percentage of the basal level. Values are the mean ± SEM (n = 5-7). The drug injection is indicated with an arrow. Filled symbols or * show statistically significant differences (p < 0.001) between control and drug treatment groups as estimated by repeated measures ANOVA (time-course) or one-way ANOVA (total effect) followed by Tukey’s post hoc test.
Figure 4.
The time-course (A) and total (B) effect of psilocybin (2 and 10 mg/kg) and ketamine (10 mg/kg) on the extracellular acetylcholine (ACh) levels in the rat hippocampus. The total effect was calculated as an area under the concentration-time curve (AUC) and is expressed as a percentage of the basal level. Values are the mean ± SEM (n = 5-7). The drug injection is indicated with an arrow. Filled symbols or * show statistically significant differences (p < 0.001) between control and drug treatment groups as estimated by repeated measures ANOVA (time-course) or one-way ANOVA (total effect) followed by Tukey’s post hoc test.
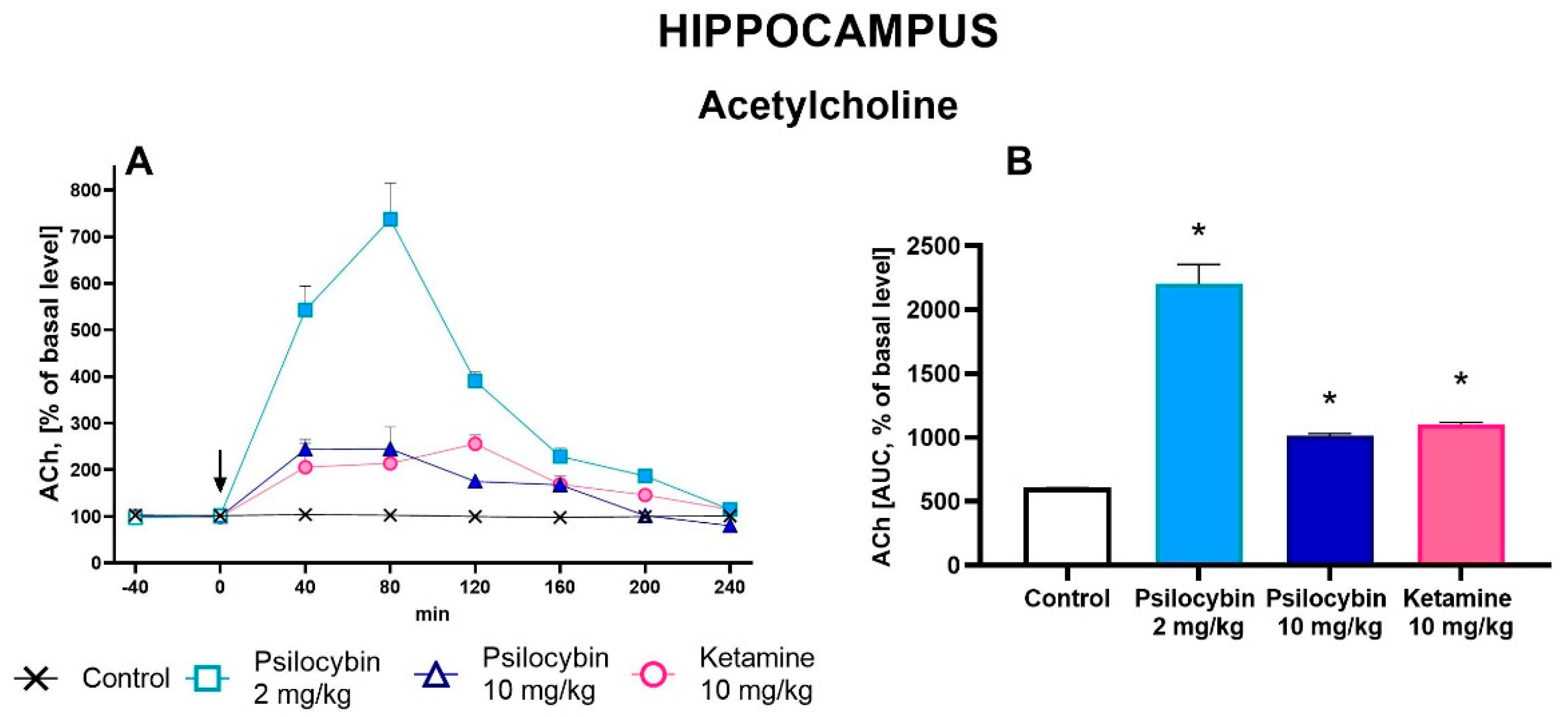
Figure 5.
The levels of 5-HT1A receptor (A) and 5-HT2A receptor (C) in the rat hippocampus estimated 7 days after psilocybin (2 and 10 mg/kg) or ketamine (10 mg/kg) administration. The data are shown as percentages of the levels of the appropriate control groups. Each data point represents the mean ± SEM (n = 10). * p < 0.001 vs. appropriate control group (one-way ANOVA followed by Tukey’s post hoc test). Representative examples of photomicrographs of the immunoblots using 5-anti-HT1A and anti-5-HT2A antibodies (B and D, respectively).
Figure 5.
The levels of 5-HT1A receptor (A) and 5-HT2A receptor (C) in the rat hippocampus estimated 7 days after psilocybin (2 and 10 mg/kg) or ketamine (10 mg/kg) administration. The data are shown as percentages of the levels of the appropriate control groups. Each data point represents the mean ± SEM (n = 10). * p < 0.001 vs. appropriate control group (one-way ANOVA followed by Tukey’s post hoc test). Representative examples of photomicrographs of the immunoblots using 5-anti-HT1A and anti-5-HT2A antibodies (B and D, respectively).
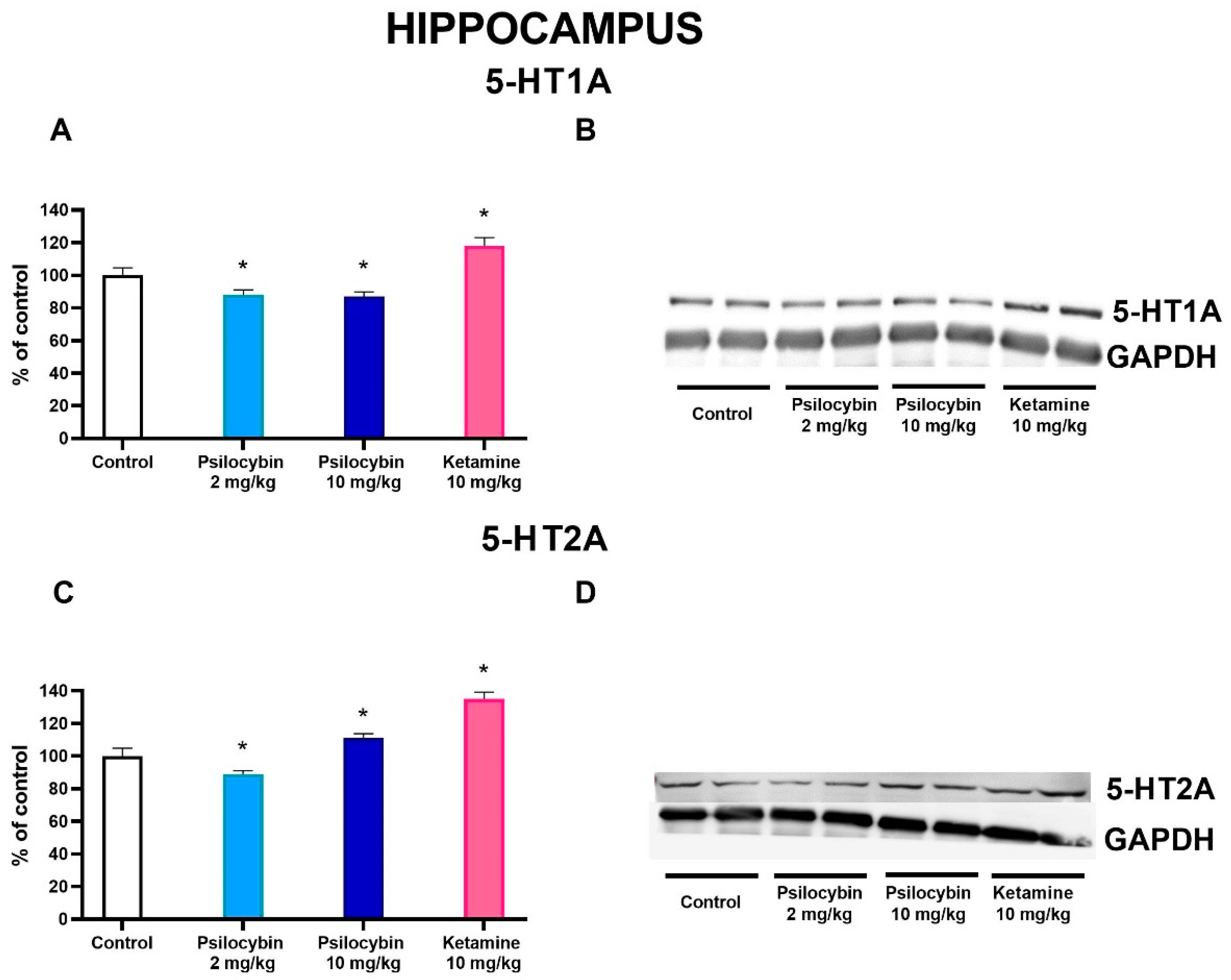
Figure 6.
The levels of 5-HT2A receptor (A) and D2 receptor (C) in the rat nucleus accumbens estimated 7 days after psilocybin (2 and 10 mg/kg) or ketamine (10 mg/kg) administration. The data are shown as percentages of the levels of the appropriate control groups. Each data point represents the mean ± SEM (n = 10). * p < 0.0001 vs. appropriate control group (one-way ANOVA followed by Tukey’s post hoc test). Representative examples of photomicrographs of the immunoblots using 5-anti-HT2A and anti-D2 antibodies (B and D, respectively).
Figure 6.
The levels of 5-HT2A receptor (A) and D2 receptor (C) in the rat nucleus accumbens estimated 7 days after psilocybin (2 and 10 mg/kg) or ketamine (10 mg/kg) administration. The data are shown as percentages of the levels of the appropriate control groups. Each data point represents the mean ± SEM (n = 10). * p < 0.0001 vs. appropriate control group (one-way ANOVA followed by Tukey’s post hoc test). Representative examples of photomicrographs of the immunoblots using 5-anti-HT2A and anti-D2 antibodies (B and D, respectively).
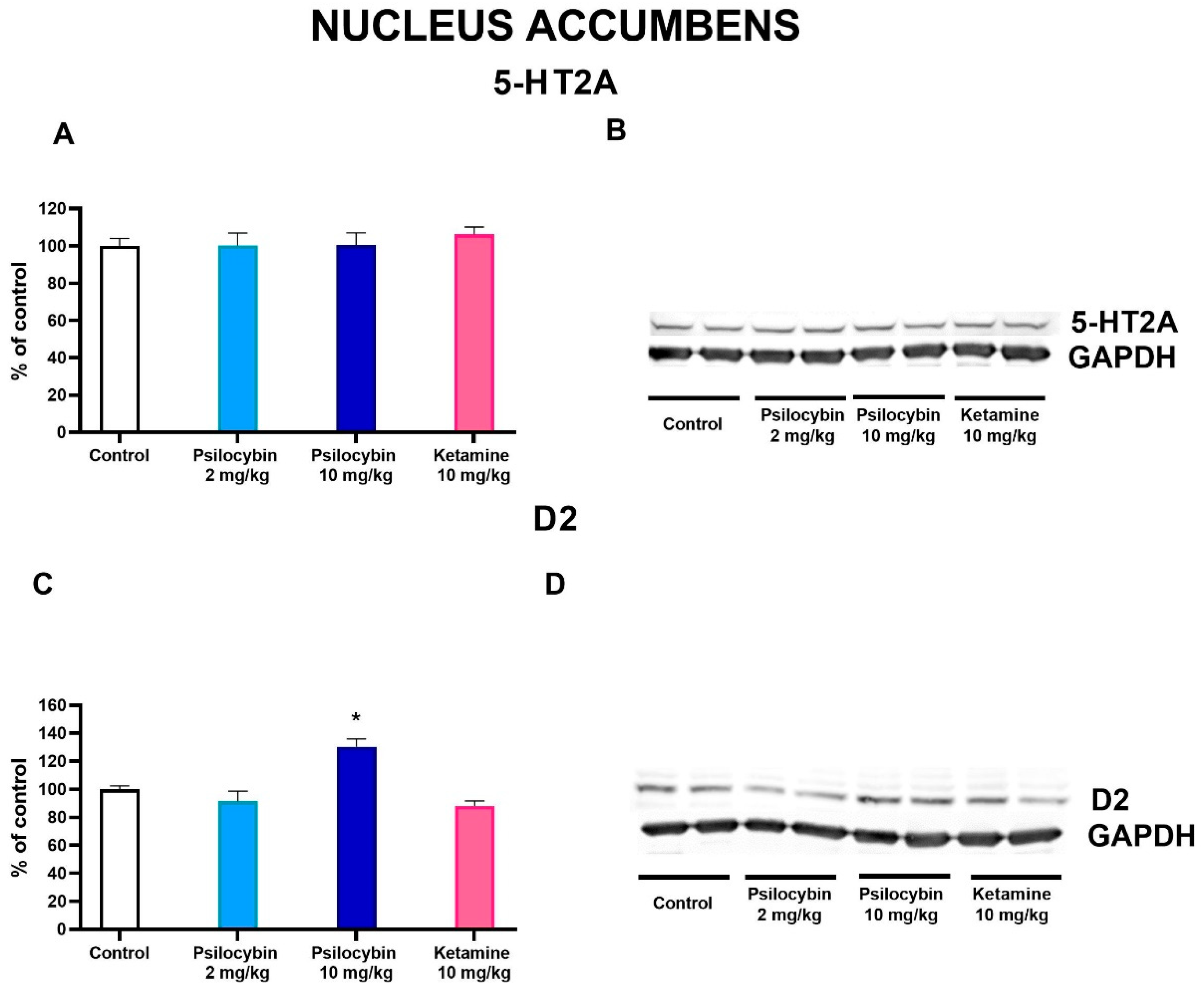
Figure 7.
The effect of psilocybin (2 and 10 mg/kg) and ketamine (10 mg/kg) on rat behavior in the open field test. The time spent on walking and ambulatory distance at 1h (A) and 24 h (C) after administration, the number of rearing and peeping episodes and time spent in the center at 1 h (B) and 24 h (D), respectively. Values are the mean ± SEM (n = 10). * p < 0.01 compared to the control (one-way ANOVA followed by Tukey’s post hoc test).
Figure 7.
The effect of psilocybin (2 and 10 mg/kg) and ketamine (10 mg/kg) on rat behavior in the open field test. The time spent on walking and ambulatory distance at 1h (A) and 24 h (C) after administration, the number of rearing and peeping episodes and time spent in the center at 1 h (B) and 24 h (D), respectively. Values are the mean ± SEM (n = 10). * p < 0.01 compared to the control (one-way ANOVA followed by Tukey’s post hoc test).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



